 Harald Lund
Harald Lund Melanie Pieber
Melanie Pieber Robert A. Harris
Robert A. Harris- Department of Clinical Neuroscience, Karolinska Institutet, Centre for Molecular Medicine, Karolinska Hospital at Solna, Solna, Sweden
While bone marrow-derived Ly6Chi monocytes can infiltrate the central nervous system (CNS) they are developmentally and functionally distinct from resident microglia. Our understanding of the relative importance of these two populations in the distinct processes of pathogenesis and resolution of inflammation during neurodegenerative disorders was limited by a lack of tools to specifically manipulate each cell type. During recent years, the development of experimental cell-specific depletion models has enabled this issue to be addressed. Herein we compare and contrast the different depletion approaches that have been used, focusing on the respective functionalities of microglia and monocyte-derived macrophages in a range of neurodegenerative disease states, and discuss their prospects for immunotherapy.
Peripheral and Resident Myeloid Cell Populations
“Myeloid cell” is a collective term that encompasses many innate immune cells including macrophages and monocytes. Resident macrophages are specialized subsets defined by their organ location (e.g., central nervous system (CNS) microglia, liver Kupffer cells), having been differentiated in these specialized microenvironments during development from (in the majority of cases) embryonic precursors. They primarily conduct homeostatic functions in the tissues, but their numbers and properties can change during a local inflammatory process, and they can proliferate within tissues in order to maintain homeostasis. In contrast, circulating monocytes are derived from hematopoietic stem cells in the bone marrow and are generally divided into two subsets in the blood of mice based on their expression of Ly6C. Ly6Clow monocytes have endothelium patrolling and scavenging functions whereas Ly6Chi monocytes infiltrate tissue in response to inflammation and differentiate into macrophages. Monocytes are constantly turned over and generally have limited life spans in the circulation.
Microglia Depletion Models
Depletion of CNS-resident or infiltrating microglia/macrophage cell populations has been used to dissect the role of these cell types during neurodegenerative disorders. The major advances in developing microglia/macrophage depletion tools are summarized below, but for a more detailed review the reader is referred to Waisman et al. (2015), Wieghofer et al. (2015) and Jäkel and Dimou (2017). Depletion of macrophage populations first became a readily available technique through the development of clodronate-encapsulated liposomes in the 1980s (van Rooijen, 1989). Intravenous administration efficiently depleted circulating monocytes (Sunderkötter et al., 2004) and since they did not pass the blood brain barrier this technique could be used to ablate circulating monocytes without affecting CNS-resident macrophage populations (Zattoni et al., 2011). More specific targeting of Ly6Chi monocytes can be accomplished by antibody-mediated depletion (Mack et al., 2001), or by the use of CCR2−/− mice in which Ly6Chi monocytes are unable to leave the bone marrow (Serbina and Pamer, 2006). Intracerebroventricular (i.c.v) delivery of clodronate liposomes has also been used to deplete microglia (Lee et al., 2012; Hanafy, 2013; Asai et al., 2015). In addition, i.c.v injection of mannosylated liposomes is an established technique to specifically target mannose receptor (CD206)-expressing perivascular macrophages without affecting resident microglia numbers in both rats (Polfliet et al., 2001; Newman et al., 2005) and mice (Galea et al., 2005; Hawkes and McLaurin, 2009).
A major breakthrough in the development of efficient microglia depletion tools was the development of mice expressing the herpes-simplex virus encoded suicide-gene thymidine kinase (HSVTK) under the CD11b-promoter (Heppner et al., 2005). Administration of ganciclovir via an osmotic pump-connected i.c.v cannula resulted in up to 95% depletion of microglia (Grathwohl et al., 2009) with the only drawback being that drug administration became toxic after extended delivery, thereby limiting this approach to a period of 4 weeks. In subsequent studies, it was demonstrated that cessation of ganciclovir delivery resulted in complete exchange of the microglial pool by peripheral myeloid cells (Varvel et al., 2012, 2015; Prokop et al., 2015). A major step forward in achieving more specific microglia targeting was the development of CX3CR1CreER mice (Goldmann et al., 2013; Parkhurst et al., 2013). When bred with Rosa26DTR mice and subsequent to peripheral administration of the drug tamoxifen followed by diphtheria toxin, efficient depletion of microglia resulted without affecting bone marrow-derived CX3CR1+ cells (Parkhurst et al., 2013; Bruttger et al., 2015). Since other CNS-associated (perivascular, meningeal and choroid plexus) macrophages also express CX3CR1 (Goldmann et al., 2016), these cells are most likely also depleted in CX3CR1CreERRosa26DTR mice. Transcriptional profiling has revealed genes uniquely expressed by microglia such as Sall1, and this has been successfully utilized in Sall1CreER mice which target microglia while sparing both peripheral and CNS-associated macrophage populations (Buttgereit et al., 2016). However, Sall1CreER mice have currently not been used to deplete microglia.
Microglia require CSF1R during development, since CSF1R−/− mice completely lack microglia (Ginhoux et al., 2010). Microglia can use both ligands of CSF1R for their survival (CSF1 and IL-34) since mice mutant for either cytokine display reduction but not complete loss of microglia (Ginhoux et al., 2010; Greter et al., 2012; Wang et al., 2012). Elmore et al. (2014) recently demonstrated that microglia remain dependent on CSF1R for survival in adult animals. Pharmacological inhibition of CSF1R yields complete ablation (>99%) of microglia within 21 days. This approach is practical because it requires no mouse breeding and microglial depletion can be maintained as long as the drug is administered. Depletion of the microglial pool disturbs CNS homeostasis, and while neurons do not regenerate, microglia have significant potential to self-renew through proliferation. The incomplete depletion (80%) accomplished in CX3CR1CreERRosa26DTR mice is quickly recuperated by hyper-proliferation of surviving microglia (Bruttger et al., 2015). Even more efficient depletion (99%) using the CSF1R-inhibitor is followed by such rapid repopulation that the existence of a microglia-progenitor has been suggested (Elmore et al., 2014). A recent report, however, demonstrates the relatively high turnover of microglia in the steady-state, arguing against the existence of a microglia progenitor (Tay et al., 2017). Conversely, using the CD11b-HSVTK model repopulation occurs from peripheral myeloid cells (Varvel et al., 2012; Prokop et al., 2015).
Given this historic perspective of microglia depletion research, we review below the lessons learned using these different strategies, focusing on models of neuroinflammation (experimental autoimmune encephalomyelitis, EAE), acute neurodegeneration (stroke, toxin-induced neurodegeneration) or chronic neurodegeneration (Amyotrophic Lateral Sclerosis (ALS), Alzheimer’s disease, Prion disease). We do not review the role of microglia in development or homeostasis and refer the reader to separate reviews addressing these areas of research (Greter and Merad, 2012; Nayak et al., 2014).
Amyotrophic Lateral Sclerosis
ALS is a generally fatal neurodegenerative disease characterized by progressive paralysis of skeletal muscles associated with motor neuron death and glial activation in the lumbar spinal cord. The most commonly used mouse model of ALS is based on the over-expression of human SOD1 carrying the G93A mutation (mSOD1 mice). Mice accumulate mutant SOD1 aggregates in the spinal cord, leading to motor neuron death and recapitulation of the central aspects of ALS pathology including an age-dependent accumulation of CD11b+ (Alexianu et al., 2001; Gowing et al., 2008), CD68+ (Henkel et al., 2006; Beers et al., 2011), MHC II+ (Hall et al., 1998), CD11c+ (Beers et al., 2011) microglia/macrophages in the spinal cord of mSOD1 mice.
That mutant SOD1 expression in microglia/macrophages contribute to disease progression has been convincingly demonstrated in the slow progressing SOD1G37R line in which CD11b-Cre-mediated removal of SOD1 from myeloid cells slowed progression of disease and extended survival (Boillée et al., 2006). Similarly, repopulating the empty microglial niche in mSOD1/PU.1−/− mice with wildtype bone marrow extends survival compared to mice transplanted with mSOD1 bone marrow (Beers et al., 2006; Lee et al., 2012). It should be noted that one study reported no increase in motorneuron loss following partial (40%) elimination of microglia/macrophages during the symptomatic disease stage in CD11b-TK/mSOD1 mice (Gowing et al., 2008; Table 1).
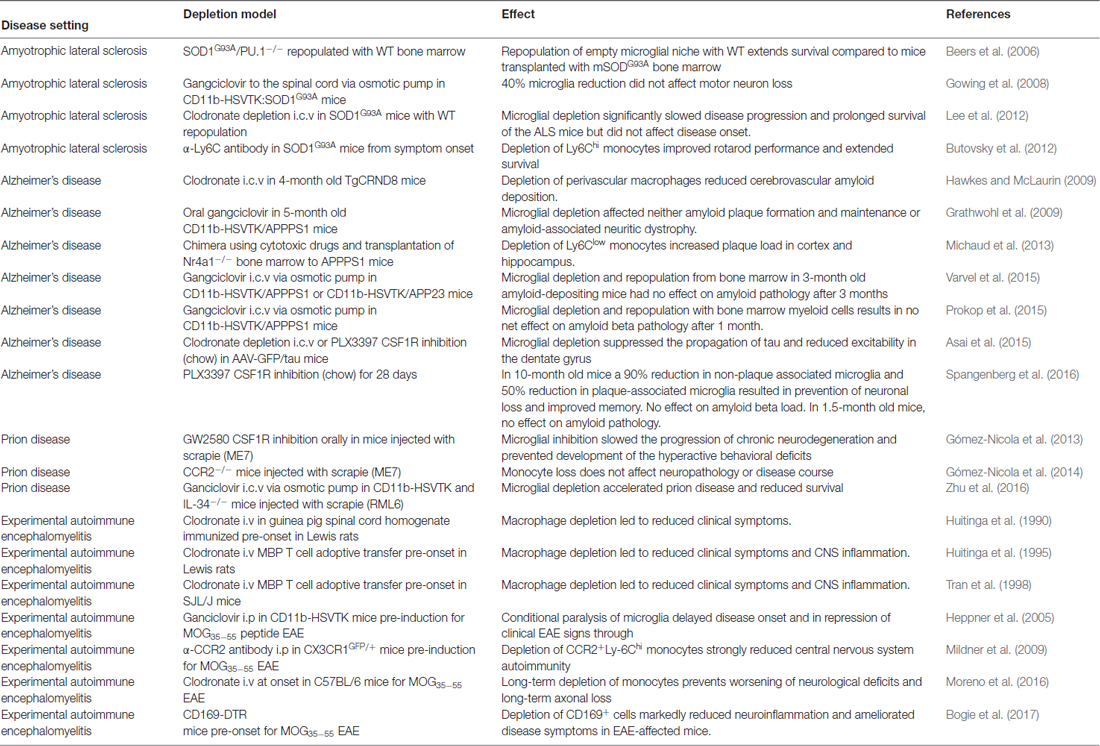
Table 1. Chronic neurodegeneration models.
Whether microglia or monocytes make up the majority of the activated macrophage population in the spinal cord is a matter of controversy. One elegant study using parabiotically connected GFP+ and mSOD1 mice recorded no parenchymal GFP+Iba-1+ cells in the spinal cord at the late stage of disease when microgliosis is extensive (Ajami et al., 2007). Similarly, a more recent study did not detect any Ly6C+ cells within the CD11b+CD45+ “microglial” compartment (Chiu et al., 2013). However, Butovsky et al. (2012) reported that Ly6Chi monocytes progressively accumulate in the spinal cord (but not brain) of mSOD1 mice, reaching as much as 30% of the CD11b+ compartment in late stage animals, and that their antibody-mediated depletion attenuated motor neuron death and delayed disease onset and mortality. Such monocyte-specific contribution to disease progression is very interesting, since most previous studies have used targeting systems that do not discriminate between microglia and monocytes. However, this study would benefit from confirmation in other laboratories since the presence of spinal cord monocyte-derived macrophages is contradicted by parabiosis experiments (Ajami et al., 2007).
Alzheimer’S Disease
Alzheimer’s disease is the most prevalent form of neurodegeneration and is characterized by the presence of amyloid beta plaques and neurofibrillary tangles. The recognition of a glial reaction as a hallmark of Alzheimer’s disease neuropathology was, despite being described by Alois Alzheimer himself (Alzheimer et al., 1995) long disregarded as an epiphenomenon. However, the recent discoveries that several immune and microglia/myeloid-expressed genes (i.e., CD33, CR1, TREM2) are genetically linked to Alzheimer’s disease has established the role of microglia in disease progression (Lambert et al., 2009; Griciuc et al., 2013; Guerreiro et al., 2013; Jonsson et al., 2013).
Amyloid Beta
The in vivo microglial response to amyloid beta has been well characterized in amyloid-depositing mouse models. Microglia rapidly migrate to newly formed plaques (Meyer-Luehmann et al., 2008) where they display an altered morphology (Frautschy et al., 1998; Stalder et al., 1999) and upregulate a vast array of surface molecules (Bornemann et al., 2001; Frank et al., 2008). Whether microglia actively internalize amyloid beta fibrils in naive APP mice has been a matter of debate (Stalder et al., 2001; Bolmont et al., 2008), but the increasing amyloid load observed with age indicates that microglia are ultimately unable to control the amyloid burden. Direct proof that microglia do not limit either amyloid plaque formation or growth was provided by Grathwohl et al. (2009) who demonstrated that complete ablation of microglia for 4 weeks in either young or aged APPPS1 mice resulted in no net effect on amyloid beta burden or plaque-associated neuritic pathology (Table 1). This finding was replicated in the 5XFAD disease model using CSF1R inhibition to deplete microglia, which resulted in no difference in amyloid pathology in either young or old 5XFAD mice. Interestingly, however, this model is characterized by development of substantial neuronal loss with age and microglial depletion both prevented this neuronal loss and improved contextual memory. Microglial depletion was also accompanied by an attenuation of disease-driven inflammatory gene expression (Spangenberg et al., 2016).
These studies are important because they explain the observation that amyloid deposits progressively increase in APP mice despite the association of microglia. This is supported by evidence that microglia in the vicinity of amyloid beta plaques progressively lose phagocytic capacity (Hickman et al., 2008; Krabbe et al., 2013). However, it is important to understand that microglia can be stimulated into removing existing amyloid plaques by a variety of means (Boissonneault et al., 2009; Leinenga and Götz, 2015; Iaccarino et al., 2016; Daria et al., 2017).
Whether monocytes take part in the response to amyloid deposits is a matter of long-standing debate. Previous studies claimed that monocytes/peripheral myeloid cells possessed superior ability to remove amyloid deposits compared to microglia (Malm et al., 2005; Stalder et al., 2005; Simard et al., 2006; Butovsky et al., 2007). However, these studies were all based on whole body irradiation chimeras and exposure of the brain to irradiation has subsequently been elucidated to condition the brain for monocyte infiltration (Mildner et al., 2007). More recent studies using brain-protected irradiation chimeras (Mildner et al., 2011) or chemotherapy-induced myeloablation (Lampron et al., 2012; Michaud et al., 2013) have demonstrated very little engraftment of monocytes during disease progression in amyloid-depositing mice. Furthermore, complete exchange of the microglial compartment with peripheral monocytic cells did not affect amyloid beta burden (Prokop et al., 2015; Varvel et al., 2015), which was final proof that monocytes do not confer better amyloid beta removal ability than do microglia.
The possibility remains that monocytes take part in the removal of amyloid deposits in the cerebrovasculature, so-called cerebral amyloid angiopathy. Using two-photon in vivo imaging patrolling Ly6Clow monocytes have been observed to actively crawl on amyloid beta-laden cerebral blood vessels (Michaud et al., 2013). Ly6Clow monocytes are dependent on the transcription factor Nr4a1 for their survival (Hanna et al., 2012) and specific elimination of Ly6Clow monocytes by transplanting Nr4a1−/− bone marrow into chemotherapy-myeloablated APP/PS1 mice significantly increased the build up of amyloid deposits (Michaud et al., 2013).
Lack of Ly6Clow monocytes could also explain why CCR2−/−APP mice have increased levels of amyloid deposition in cerebral blood vessels (El Khoury et al., 2007; Mildner et al., 2011). While the authors attributed increased amyloid buildup to loss of CCR2 in the perivascular myeloid compartment, involvement of Ly6Clow monocytes cannot be excluded. In fact, perivascular macrophages are not lost in CCR2−/− mice (Goldmann et al., 2016), in contrast to Ly6Clow monocytes which are directly derived from Ly6Chi monocytes and therefore are also significantly reduced in CCR2−/− mice (Yona et al., 2013). Another study has attempted to more specifically address the role of perivascular macrophages in buildup of vascular amyloid deposits through i.c.v injection of clodronate to deplete CD163+ perivascular macrophages while sparing parenchymal Iba-1+ microglia, this procedure resulting in a 5-fold increase in cerebral amyloid angiopathy load (Hawkes and McLaurin, 2009).
Tau
Microtubule-associated protein tau (MAPT, tau) is a microtubule-stabilizing protein that during the course of AD becomes hyperphosphorylated, a process leading to dissociation from microtubules and aggregation into paired helical filaments and formation of neurofibrillary tangles (Wang and Mandelkow, 2016). Tau aggregates follow a predictable pattern of neuron-to-neuron spreading in the brain (Braak et al., 2011) and a recent study addressed the possible role of microglia in this process. Asai et al. (2015) developed a simple but elegant model of tau propagation in wildtype mice by injecting tau-expressing adeno-associated virus into the entorhinal cortex and observed how tau aggregates spread to the nearby hippocampus in only 4 weeks. Depletion of microglia using either clodronate liposomes or CSF1R inhibition dramatically halted the propagation of tau into the hippocampus. Similar results were obtained by inhibiting exosome synthesis, suggesting microglia could seed tauopathy (Asai et al., 2015).
There is also evidence that microgliosis can precede deposition of insoluble tau in transgenic mice (Yoshiyama et al., 2007; Maphis et al., 2015), indicating that microglia could drive spatiotemporal tau propagation through production of neuroinflammatory mediators. Loss of CX3CR1 amplifies tau pathology in hTau mice in an IL-1β-dependent manner (Bhaskar et al., 2010) and tau hyperphosphorylation could be induced in wildtype mice by intracerebral transplantation of CX3CR1−/− hTau microglia (Maphis et al., 2015). This was the proof-of-concept that reactive microglia could drive tau pathology.
Prion Disease
Prion disease is a transmissible neurodegenerative disorder affecting both animals and humans. The contribution of myeloid subsets to disease progression has been characterized using mouse models in which prion disease is produced by intracerebral inoculation of scrapie protein strains. While microglial proliferation and activation occurs irrespective of the inoculated prion strain (Cunningham et al., 2005) and correlates with onset of neurological deficits (Boche et al., 2006) there is little evidence of monocyte infiltration during disease progression. Preventing monocytes from entering the circulation using CCR2−/− mice does not exacerbate neuropathology or disease course in prion-infected mice (Gómez-Nicola et al., 2014; Table 1). Whether microglial proliferation observed during prion disease is beneficial or deleterious is a matter of conflicting data. Using a CSF1R inhibitor Gómez-Nicola et al. (2013) limited microglial proliferation which prevented neurodegeneration, improved behavioral impairments and extended survival in prion mice. Conversely, Zhu et al. (2016) reported that microglial depletion (CD11b-HSVTK) or reduction (IL-34−/−) exacerbated prion disease and reduced survival, suggesting a neuroprotective role for microglia. Since the two studies have used different scrapie strains, different strategies to limit the microglial response and different timing of the microglial targeting, it is difficult to consolidate the data sets. However, it could indicate strain-specific microglial responses or suggest that microglia possess neuroprotective and neurotoxic reactions to prions at different time points during the disease course.
Experimental Autoimmune Encephalomyelitis
The development of demyelinating lesions that is characteristic of multiple sclerosis (MS) and its mouse model EAE is initiated by autoreactive, myelin-specific CD4+ T cells. These are reactivated within the CNS (Goverman, 2009), secreting factors that open the blood-brain barrier and recruit a heterogeneous population of myeloid cells that drive demyelination. The primary importance of myeloid cells in pathogenesis is indicated by the strong genetic association of MHC II and MS (Sawcer et al., 2011). While infiltrating monocyte-derived macrophages drive disease pathogenesis (as detailed below), the contribution of resident microglia during the different disease phases is still not completely understood.
It has been demonstrated that EAE severity correlates with numbers of monocytes infiltrating the spinal cord and that prevention of this infiltration protects against EAE development (Fife et al., 2000; Mildner et al., 2009; Ajami et al., 2011). Importantly, the presence of the infiltrating cells in the CNS is transient, their numbers decreasing significantly during the remission phase of EAE (Hesske et al., 2010; Ajami et al., 2011), leaving the niche occupied by resident microglia. It is evident from our review (Table 1), that the majority of depletion studies in EAE have focused on preventing monocyte infiltration into the CNS from the periphery, with EAE pathology and symptoms being abrogated irrespective of the manner in which this is accomplished. Compared to infiltrating monocytes, microglia display low levels of MHCII and co-stimulatory molecules during EAE (Vainchtein et al., 2014) and their MHCII expression is dispensable for EAE induction (Greter et al., 2005). While preventing microglial expansion (referred to as microglial paralysis) has been demonstrated to substantially ameliorate the clinical signs of EAE and to strongly reduce CNS inflammation (Heppner et al., 2005), recent data indicate an alternative role of microglia compared to monocytes. Using electron-microscopy to study microglia and macrophage-specific interactions with axons, monocytes were shown to initiate demyelination at nodes of Ranvier, whereas microglia appeared to scavenge myelin debris (Yamasaki et al., 2014).
Given that EAE is primarily a disease state caused by infiltrating monocytes, the role of microglia in driving pathogenesis is not major. However, the role of microglia in restoring CNS during remission (healing) phases, or their lack of achieving this role during chronic disease states, warrants further investigation.
Stroke
Stroke is caused by severe occlusion or stenosis of a cerebral artery and is the second leading cause of death and disability worldwide. Brain resident microglia become activated by various cytokines and damage-associated molecular patterns which are released by necrotic or apoptotic tissues (Shichita et al., 2012). An inflammatory response is triggered during the early stages of brain injury, leading to an influx of peripheral immune cells including macrophages and neutrophils (Tanaka et al., 2003; Gelderblom et al., 2009). Different studies have been performed to characterize the role of resident microglia and infiltrating peripheral macrophages in various mouse and rat models of stroke, but their roles are still under debate and vary depending on the experimental model employed (Table 2).
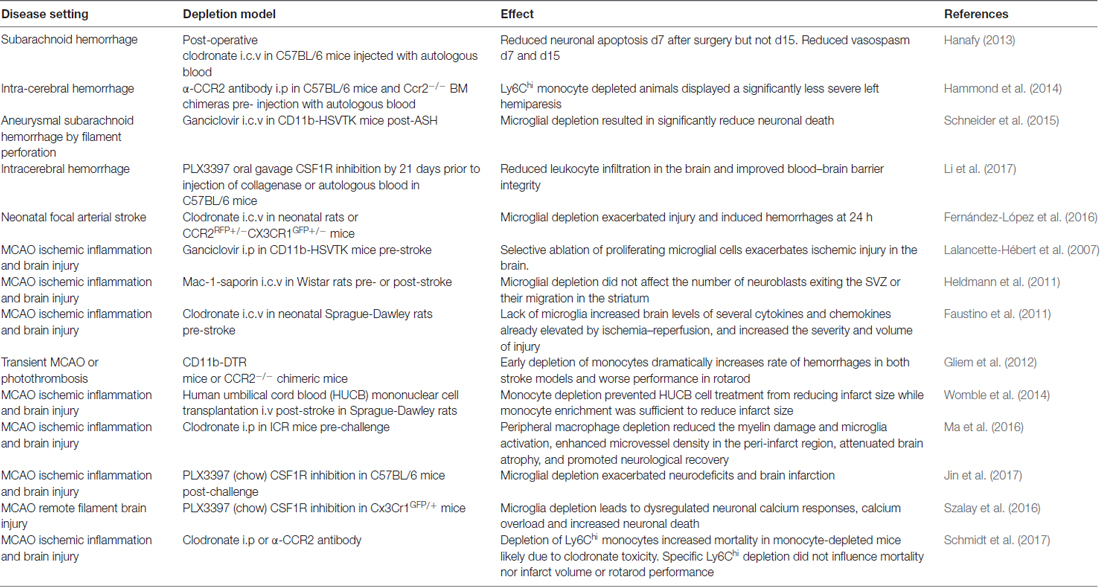
Table 2. Acute neurodegeneration models—stroke and related conditions.
Using macrophage depletion via clodronate liposomes, Gliem et al. (2012) reported that the early influx of peripheral macrophages prevents hemorrhagic infarct transformation in a model of middle cerebral artery occlusion (MCAO), with mice treated at early time points having increased peri-lesional hemorrhage. Similar results were obtained using CD11b-DTR mice for macrophage depletion in the same study. In contrast, Ma et al. (2016) demonstrated a positive effect of peripheral macrophage depletion on the stroke lesion following clodronate treatment, with decreased myelin damage and microglial activation as well as decreased brain atrophy and increased neurological recovery following MCAO. Likewise, antibody blockade of CCR2-infiltrating macrophages reduced early motor deficits following intracerebral hemorrhage (Hammond et al., 2014), again indicating a negative role of myeloid cells in this model. Yet another recently published study contradicts all the aforementioned studies and concludes that targeting monocytes/macrophages (using clodronate, anti-CCR2 antibody or M1/M2 transfer) had no therapeutic value in acute ischemic stroke but only on mortality (Schmidt et al., 2017). The discrepancies in these studies, even though the same disease model was used, can be explained due to the different time points of monocyte depletion. Both studies that reported a detrimental effect of monocyte depletion (Hammond et al., 2014; Ma et al., 2016) depleted monocytes before inducing MCAO whereas in the first study (Gliem et al., 2012) monocyte depletion was performed after stroke induction, suggesting a differing role of monocytes during the course of disease.
Numerous stroke studies have been performed using selective depletion of microglia instead of peripheral monocytes. Microglia seem to have a supporting role in various models of stroke, as exemplified by studies of neonatal focal arterial stroke. The i.c.v injection of clodronate liposomes before induction of the lesion yielded specific microglia depletion without affecting the periphery, resulting in increased local inflammation and injury severity as well as reduced vessel coverage that triggers hemorrhages in the injured brain regions (Faustino et al., 2011; Fernández-López et al., 2016). Microglia are therefore thought to contribute to the endogenous protection mechanisms of the brain during early stages after injury in neonates. Similarly, depletion of proliferating microglia using adult CD11b-HSVTK mice resulted in exacerbation of the stroke lesion, increased neuronal death and pro-inflammatory cytokine levels (Lalancette-Hébert et al., 2007). Depletion of microglia using a CSF1R inhibitor prior and subsequent to MCAO also exacerbated brain infarction and neurological deficits by promoting leukocyte infiltration into the brain and increased inflammatory cytokine levels in the area of the lesion, supporting the beneficial role of microglia in MCAO (Jin et al., 2017). Interestingly, using the same depletion method in the intracerebral hemorrhage model, microglia were concluded to have the completely opposite role in that model of brain injury. Depletion of microglia thus led to reduced lesion size, brain edema and neurodeficits, a lack of microglia attenuated leukocyte infiltration, decreased inflammatory cytokine levels and preserved the integrity of the blood brain barrier (Li et al., 2017). Most studies to date have focused on the effect of microglia/monocyte depletion during the acute phase of brain injury. However, the roles of microglia/monocytes can differ at various time points and between injury models, underlining the many potential roles of inflammatory monocytes and microglia in the pathogenic and regenerative CNS (Hammond et al., 2014).
Toxin-Induced Neurodegeneration
Microglia can have multiple roles during acute hippocampal neurodegeneration (Table 3), as demonstrated using a model of diphtheria toxin-inducible neuronal loss. Microglia numbers increase dramatically after lesion formation, without evidence of peripheral myeloid infiltration. Elimination of microglia during this procedure exacerbated neuronal loss (Rice et al., 2015). However, elimination subsequent to lesion formation improved cognitive recovery and reduced inflammatory signaling (Rice et al., 2015), demonstrating a deleterious response of microglia after as opposed to during lesion formation, again stressing how important accurate timing in microglia depletion studies is to obtain comparable results between studies. In a subsequent study, the elimination and repopulation of microglia after the procedure was reported to similarly reduce neuroinflammation, improve behavioral recovery and synaptic densities (Rice et al., 2017). The role of microglia in these toxin-induced models appears to be time-dependent, with an acute pathogenic role becoming a return-to-homeostasis (healing) role at later timepoints.
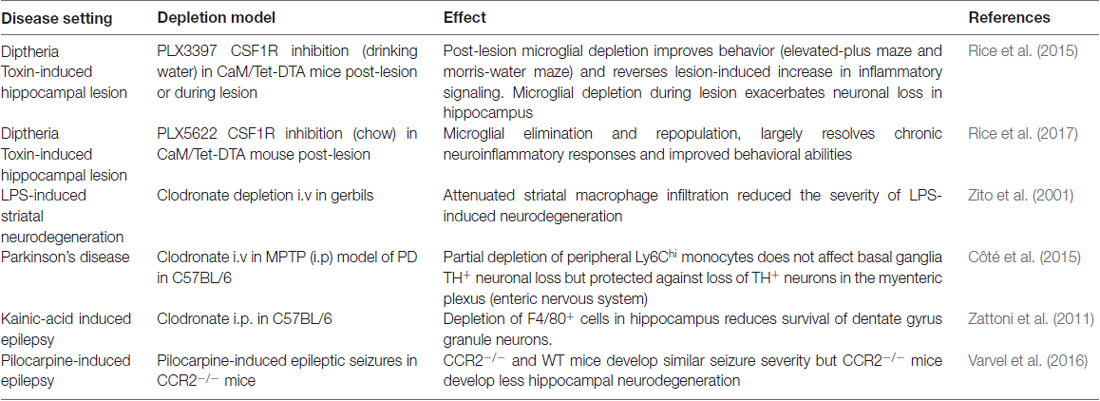
Table 3. Acute neurodegeneration models—toxin-induced neurodegeneration.
While partial depletion of circulating monocytes did not affect MPTP-induced neuronal loss in the basal ganglia (Côté et al., 2015; a model of Parkinson’s disease), LPS-induced striatal neurodegeneration in gerbils was attenuated by similar means (Zito et al., 2001). Kainic acid- or pilocarpine-induced eplieptic seizures in mice provoke microglial activation in the hippocampus and a delayed entry of monocytes into the parenchyma (Varvel et al., 2016). Varvel et al. (2016) elegantly showed that monocytopenic CCR2−/− mice developed less hippocampal neurodegeneration, suggesting a detrimental role of monocytes. However, peripheral clodronate administration reduced accumulation of CNS-infiltrating F4/80+ macrophages and reduced survival of dentate gyrus granule cells (Zattoni et al., 2011) indicating a beneficial role of monocytes.
Injury
Acute injury can be inflicted on the CNS in different ways, be it by crush or cut in the spinal cord, or traumatic brain injury (Table 4) as typified by traffic accidents and more recently by professional American sportsmen (Cherry et al., 2016). What is often characteristic of these injuries is a chronic state of neuroinflammation that can be experienced years after the initial injury, and this is associated with permanent microglial dysfunction. Despite this variation in the type of injury model and in the method of microglial depletion that have been reported there are few effects of reduced microglia numbers during the responses in these settings.
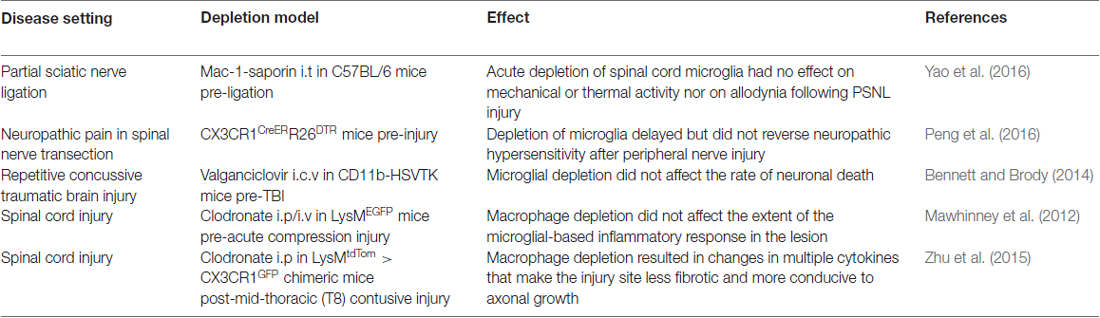
Table 4. Acute neurodegeneration models—injury.
Thus in a model of repetitive brain concussion microglia did not contribute to acute axon degeneration after multiple concussive injuries, although there is still a possibility of longer-term effects on axon functionality (Bennett and Brody, 2014). However, in a mouse model of spine transection, resident microglia and peripheral monocytes were concluded to act synergistically to initiate hypersensitivity and to promote the transition from acute to chronic pain following peripheral nerve injury (Peng et al., 2016).
It is possible that the severity of the extensive trauma in many of these models affects too large an area to make the microglial loss significantly synergistic, or alternatively that the ensuing chronic microglial dysfunction only gradually develops over time.
Other In Vivo and In Vitro Models of Depletion
Given the proven propensity of pharmacological agents to efficiently deplete microglia and macrophages, it is not surprising that these have been applied in additional in vivo and in vitro settings (Table 5). It is apparent that clinical cognitive decline is a long-term condition induced by both cranial irradiation (Acharya et al., 2016) and peripheral surgery (Degos et al., 2013), and in both settings the depletion of myeloid cells increases cognitive ability. While in the former setting an effect on microglia would be expected, and has recently been reported to differ depending on the age of the mice (Han et al., 2016), the latter is intriguing as a peripheral skeletal injury is seemingly a long distance away from the CNS. The interplay between peripheral macrophages and CNS-resident microglia is thus probably more intricate and extensive than we currently understand. Organotypic CNS slice cultures provide an alternative to live mice for study of CNS homeostasis and disease, and microglia depletion agents can be applied to these.
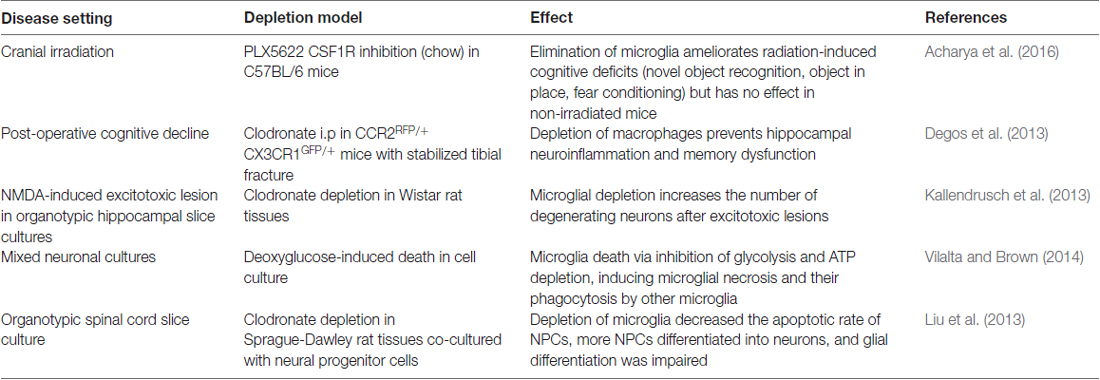
Table 5. Other in vivo and in vitro models.
Generally the findings suggest that tissues devoid of microglia have less ability to protect neurons.
Points of Perspective
Given the vast expansion in the field of microglia research during recent years it will be important to build on previous interpretations in the light of new knowledge. For example, the absence of any effect on amyloid beta burden by microglia depletion in Alzheimer’s disease models could be explained by recent results suggesting that the key function of amyloid plaque-associated microglia is to form a barrier around it (to compact the amyloid fibrils into a dense plaque) rather than solely their previously perceived role in amyloid phagocytosis (Yuan et al., 2016).
It has proven difficult to permanently deplete microglia in a specific manner and there are caveats with all the depletion models employed. The nature of the cell death of depleted microglia within the CNS is also an issue that has not been addressed, and this might not only vary between depletion systems, but might also trigger downstream (e.g., epigenetic) programmes that we are currently unaware of but which have significant bearing on the interpretation of the studies.
The enormous propensity of the CNS to repopulate with myeloid cells, be they surviving microglia colonies that hyperproliferate, or CNS-adapted infiltrating monocytes that fill up the available niche, indicates that a myeloid-deficient CNS is a highly non-physiological condition. While the infiltrating monocytes co-occupy the CNS and begin to express microglia-specific proteins, it still remains to be proven if they develop full microglial functionality. Exactly how the CNS regulates the repopulation process and senses when the niche is replenished are important unanswered questions.
What is clear from our review is that there is some disparity between the results of different microglia depletion systems in the same disease model. Discerning the underlying molecular mechanisms that lead to these different outcomes will serve to further our knowledge of the pathogenesis that we wish to target therapeutically. In particular, the relative timing of microglial depletion in relation to the insult might explain some of the disparity, and deserves further consideration.
While the homeostatic functions of microglia are well understood, these cells always having activity in surveying the CNS, the molecular basis for these functions are less well characterized. TGFβ appears to be a key cytokine in preventing microglia and other infiltrating myeloid cells from pro-inflammatory (pathogenic) activation (Buttgereit et al., 2016; Parsa et al., 2016). The existence of sub-populations in different regions of the CNS has been demonstrated (Grabert et al., 2016), and so it is plausible that even homeostatic microglia function varies between different CNS microenvironments (Harris, 2014). A very recent study using a novel fate-mapping strategy has described such regional differences of self-organization of mature microglial subpopulations during both health and disease in the CNS (Tay et al., 2017). If and when subpopulation-specific microglial markers can be defined then it might even be possible to deplete individual subpopulations in the future. The more refined our understanding of these issues becomes, the more likely we will be able to design efficient therapeutic paradigms.
As it is clear that microglia are implicated as a part of all the neurodegenerative diseases described in this review article, modulation of microglial functionality (suppression of pro-inflammatory, pathogenic properties) or replacement with CNS-adapting macrophages are intriguing prospects for immunotherapy. We have previously reported that immunosuppressive macrophages can down-modulate pro-inflammatory macrophage and T cell activities in settings of experimental MS (Zhang et al., 2014), indicating that immunomodulation with favorable clinical outcome is possible. With the knowledge that monocytes can become CNS-adapted during repopulation of a microglia depleted CNS, further effort in manipulating this phenomenon to a therapeutic end is warranted.
Author Contributions
All authors have had similar roles in defining the literature to be reviewed, preparing individual sections of the text and working up the final version of the manuscript.
Funding
Funding provided by the Swedish Medical Research Council, AlzheimerFond and Karolinska Institutet (KID).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acharya, M. M., Green, K. N., Allen, B. D., Najafi, A. R., Syage, A., Minasyan, H., et al. (2016). Elimination of microglia improves cognitive function following cranial irradiation. Sci. Rep. 6:31545. doi: 10.1038/srep31545
Ajami, B., Bennett, J. L., Krieger, C., McNagny, K. M., and Rossi, F. M. V. (2011). Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–1149. doi: 10.1038/nn.2887
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W., and Rossi, F. M. V. (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543. doi: 10.1038/nn2014
Alexianu, M. E., Kozovska, M., and Appel, S. H. (2001). Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 57, 1282–1289. doi: 10.1212/wnl.57.7.1282
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Beers, D. R., Henkel, J. S., Xiao, Q., Zhao, W., Wang, J., Yen, A. A., et al. (2006). Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A 103, 16021–16026. doi: 10.1073/pnas.0607423103
Beers, D. R., Zhao, W., Liao, B., Kano, O., Wang, J., Huang, A., et al. (2011). Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav. Immun. 25, 1025–1035. doi: 10.1016/j.bbi.2010.12.008
Bennett, R. E., and Brody, D. L. (2014). Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury. J. Neurotrauma 31, 1647–1663. doi: 10.1089/neu.2013.3320
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Boche, D., Cunningham, C., Docagne, F., Scott, H., and Perry, V. H. (2006). TGFβ1 regulates the inflammatory response during chronic neurodegeneration. Neurobiol. Dis. 22, 638–650. doi: 10.1016/j.nbd.2006.01.004
Bogie, J. F., Boelen, E., Louagie, E., Delputte, P., Elewaut, D., van Horssen, J., et al. (2017). CD169 is a marker for highly pathogenic phagocytes in multiple sclerosis. Mult. Scler. 101:1352458517698759. doi: 10.1177/1352458517698759 [Epub ahead of print].
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Boissonneault, V., Filali, M., Lessard, M., Relton, J., Wong, G., and Rivest, S. (2009). Powerful beneficial effects of macrophage colony-stimulating factor on β-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain 132, 1078–1092. doi: 10.1093/brain/awn331
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C. A., Klunk, W. E., et al. (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292. doi: 10.1523/jneurosci.4814-07.2008
Bornemann, K. D., Wiederhold, K.-H., Pauli, C., Ermini, F., Stalder, M., Schnell, L., et al. (2001). Aβ-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am. J. Pathol. 158, 63–73. doi: 10.1016/s0002-9440(10)63945-4
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Bruttger, J., Karram, K., Wörtge, S., Regen, T., Marini, F., Hoppmann, N., et al. (2015). Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106. doi: 10.1016/j.immuni.2015.06.012
Butovsky, O., Kunis, G., Koronyo-Hamaoui, M., and Schwartz, M. (2007). Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur. J. Neurosci. 26, 413–416. doi: 10.1111/j.1460-9568.2007.05652.x
Butovsky, O., Siddiqui, S., Gabriely, G., Lanser, A. J., Dake, B., Murugaiyan, G., et al. (2012). Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Invest. 122, 3063–3087. doi: 10.1172/jci62636
Buttgereit, A., Lelios, I., Yu, X., Vrohlings, M., Krakoski, N. R., Gautier, E. L., et al. (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406. doi: 10.1038/ni.3585
Cherry, J. D., Tripodis, Y., Alvarez, V. E., Huber, B., Kiernan, P. T., Daneshvar, D. H., et al. (2016). Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 4:112. doi: 10.1186/s40478-016-0382-8
Chiu, I. M., Morimoto, E. T. A., Goodarzi, H., Liao, J. T., O’Keeffe, S., Phatnani, H. P., et al. (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401. doi: 10.1016/j.celrep.2013.06.018
Côté, M., Poirier, A. A., Aubé, B., Jobin, C., Lacroix, S., and Soulet, D. (2015). Partial depletion of the proinflammatory monocyte population is neuroprotective in the myenteric plexus but not in the basal ganglia in a MPTP mouse model of Parkinson’s disease. Brain Behav. Immun. 46, 154–167. doi: 10.1016/j.bbi.2015.01.009
Cunningham, C., Deacon, R. M. J., Chan, K., Boche, D., Rawlins, J. N. P., and Perry, V. H. (2005). Neuropathologically distinct prion strains give rise to similar temporal profiles of behavioral deficits. Neurobiol. Dis. 18, 258–269. doi: 10.1016/j.nbd.2004.08.015
Daria, A., Colombo, A., Llovera, G., Hampel, H., Willem, M., Liesz, A., et al. (2017). Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 36, 583–603. doi: 10.15252/embj.201694591
Degos, V., Vacas, S., Han, Z., Van Rooijen, N., Gressens, P., Su, H., et al. (2013). Depletion of bone marrow-derived macrophages perturbs the innate immune response to surgery and reduces postoperative memory dysfunction. Anesthesiology 118, 527–536. doi: 10.1097/aln.0b013e3182834d94
El Khoury, J., Toft, M., Hickman, S. E., Means, T. K., Terada, K., Geula, C., et al. (2007). Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438. doi: 10.1038/nm1555
Elmore, M. R. P., Najafi, A. R., Koike, M. A., Dagher, N. N., Spangenberg, E. E., Rice, R. A., et al. (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397. doi: 10.1016/j.neuron.2014.02.040
Faustino, J. V., Wang, X., Johnson, C. E., Klibanov, A., Derugin, N., Wendland, M. F., et al. (2011). Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 31, 12992–13001. doi: 10.1523/jneurosci.2102-11.2011
Fernández-López, D., Faustino, J., Klibanov, A. L., Derugin, N., Blanchard, E., Simon, F., et al. (2016). Microglial cells prevent hemorrhage in neonatal focal arterial stroke. J. Neurosci. 36, 2881–2893. doi: 10.1523/jneurosci.0140-15.2016
Fife, B. T., Huffnagle, G. B., Kuziel, W. A., and Karpus, W. J. (2000). CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 192, 899–906. doi: 10.1084/jem.192.6.899
Frank, S., Burbach, G. J., Bonin, M., Walter, M., Streit, W., Bechmann, I., et al. (2008). TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. doi: 10.1002/glia.20710
Frautschy, S. A., Yang, F., Irrizarry, M., Hyman, B., Saido, T. C., Hsiao, K., et al. (1998). Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 152, 307–317.
Galea, I., Palin, K., Newman, T. A., Van Rooijen, N., Perry, V. H., and Boche, D. (2005). Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia 49, 375–384. doi: 10.1002/glia.20124
Gelderblom, M., Leypoldt, F., Steinbach, K., Behrens, D., Choe, C.-U., Siler, D. A., et al. (2009). Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40, 1849–1857. doi: 10.1161/strokeaha.108.534503
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gliem, M., Mausberg, A. K., Lee, J.-I., Simiantonakis, I., van Rooijen, N., Hartung, H.-P., et al. (2012). Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 71, 743–752. doi: 10.1002/ana.23529
Goldmann, T., Wieghofer, P., Jordão, M. J. C., Prutek, F., Hagemeyer, N., Frenzel, K., et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805. doi: 10.1038/ni.3423
Goldmann, T., Wieghofer, P., Müller, P. F., Wolf, Y., Varol, D., Yona, S., et al. (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618–1626. doi: 10.1038/nn.3531
Gómez-Nicola, D., Fransen, N. L., Suzzi, S., and Perry, V. H. (2013). Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 33, 2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013
Gómez-Nicola, D., Schetters, S. T. T., and Perry, V. H. (2014). Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 62, 1041–1052. doi: 10.1002/glia.22660
Goverman, J. (2009). Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 9, 393–407. doi: 10.1038/nri2550
Gowing, G., Philips, T., Van Wijmeersch, B., Audet, J. N., Dewil, M., Van Den Bosch, L., et al. (2008). Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J. Neurosci. 28, 10234–10244. doi: 10.1523/JNEUROSCI.3494-08.2008
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Grathwohl, S. A., Kälin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., et al. (2009). Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363. doi: 10.1038/nn.2432
Greter, M., Heppner, F. L., Lemos, M. P., Odermatt, B. M., Goebels, N., Laufer, T., et al. (2005). Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 11, 328–334. doi: 10.1038/nm1197
Greter, M., Lelios, I., Pelczar, P., Hoeffel, G., Price, J., Leboeuf, M., et al. (2012). Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37, 1050–1060. doi: 10.1016/j.immuni.2012.11.001
Greter, M., and Merad, M. (2012). Regulation of microglia development and homeostasis. Glia 61, 121–127. doi: 10.1002/glia.22408
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Hall, E. D., Oostveen, J. A., and Gurney, M. E. (1998). Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23, 249–256. doi: 10.1002/(SICI)1098-1136(199807)23:3<249::AID-GLIA7>3.0.CO;2-#
Hammond, M. D., Taylor, R. A., Mullen, M. T., Ai, Y., Aguila, H. L., Mack, M., et al. (2014). CCR2+Ly6Chi inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J. Neurosci. 34, 3901–3909. doi: 10.1523/JNEUROSCI.4070-13.2014
Han, W., Umekawa, T., Zhou, K., Zhang, X.-M., Ohshima, M., Dominguez, C. A., et al. (2016). Cranial irradiation induces transient microglia accumulation, followed by long-lasting inflammation and loss of microglia. Oncotarget 7, 82305–82323. doi: 10.18632/oncotarget.12929
Hanafy, K. A. (2013). The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflammation 10:83. doi: 10.1186/1742-2094-10-83
Hanna, R. N., Shaked, I., Hubbeling, H. G., Punt, J. A., Wu, R., Herrley, E., et al. (2012). NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res. 110, 416–427. doi: 10.1161/CIRCRESAHA.111.253377
Harris, R. A. (2014). Spatial, temporal, and functional aspects of macrophages during “the good, the bad, and the ugly” phases of inflammation. Front. Immunol. 5:612. doi: 10.3389/fimmu.2014.00612
Hawkes, C. A., and McLaurin, J. (2009). Selective targeting of perivascular macrophages for clearance of β-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. U S A 106, 1261–1266. doi: 10.1073/pnas.0805453106
Heldmann, U., Mine, Y., Kokaia, Z., Ekdahl, C. T., and Lindvall, O. (2011). Selective depletion of Mac-1-expressing microglia in rat subventricular zone does not alter neurogenic response early after stroke. Exp. Neurol. 229, 391–398. doi: 10.1016/j.expneurol.2011.03.005
Henkel, J. S., Beers, D. R., Siklós, L., and Appel, S. H. (2006). The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol. Cell. Neurosci. 31, 427–437. doi: 10.1016/j.mcn.2005.10.016
Heppner, F. L., Greter, M., Marino, D., Falsig, J., Raivich, G., Hövelmeyer, N., et al. (2005). Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 11, 146–152. doi: 10.1038/nm1177
Hesske, L., Vincenzetti, C., Heikenwalder, M., Prinz, M., Reith, W., Fontana, A., et al. (2010). Induction of inhibitory central nervous system-derived and stimulatory blood-derived dendritic cells suggests a dual role for granulocyte-macrophage colony-stimulating factor in central nervous system inflammation. Brain 133, 1637–1654. doi: 10.1093/brain/awq081
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective -amyloid clearance pathways in aging alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Huitinga, I., Ruuls, S. R., Jung, S., Van Rooijen, N., Hartung, H. P., and Dijkstra, C. D. (1995). Macrophages in T cell line-mediated, demyelinating, and chronic relapsing experimental autoimmune encephalomyelitis in Lewis rats. Clin. Exp. Immunol. 100, 344–351. doi: 10.1111/j.1365-2249.1995.tb03675.x
Huitinga, I., van Rooijen, N., de Groot, C. J., Uitdehaag, B. M., and Dijkstra, C. D. (1990). Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J. Exp. Med. 172, 1025–1033. doi: 10.1084/jem.172.4.1025
Iaccarino, H. F., Singer, A. C., Martorell, A. J., Rudenko, A., Gao, F., Gillingham, T. Z., et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235. doi: 10.1038/nature20587
Jäkel, S., and Dimou, L. (2017). Glial cells and their function in the adult brain: a journey through the history of their ablation. Front. Cell. Neurosci. 11:81. doi: 10.3389/fncel.2017.00024
Jin, W.-N., Shi, S. X.-Y., Li, Z., Li, M., Wood, K., Gonzales, R. J., et al. (2017). Depletion of microglia exacerbates postischemic inflammation and brain injury. J. Cereb. Blood Flow Metab. 37, 2224–2236. doi: 10.1177/0271678x17694185
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kallendrusch, S., Kremzow, S., Nowicki, M., Grabiec, U., Winkelmann, R., Benz, A., et al. (2013). The G protein-coupled receptor 55 ligand l-α-lysophosphatidylinositol exerts microglia-dependent neuroprotection after excitotoxic lesion. Glia 61, 1822–1831. doi: 10.1002/glia.22560
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 8:e60921. doi: 10.1371/journal.pone.0060921
Lalancette-Hébert, M., Gowing, G., Simard, A., Weng, Y. C., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lampron, A., Lessard, M., and Rivest, S. (2012). Effects of myeloablation, peripheral chimerism, and whole-body irradiation on the entry of bone marrow-derived cells into the brain. Cell Transplant. 21, 1149–1159. doi: 10.3727/096368911x593154
Lee, J. C., Seong, J., Kim, S. H., Lee, S. J., Cho, Y. J., An, J., et al. (2012). Replacement of microglial cells using Clodronate liposome and bone marrow transplantation in the central nervous system of SOD1G93A transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 418, 359–365. doi: 10.1016/j.bbrc.2012.01.026
Leinenga, G., and Götz, J. (2015). Scanning ultrasound removes amyloid-β and restores memory in an Alzheimer’s disease mouse model. Sci. Transl. Med. 7:278ra33. doi: 10.1126/scitranslmed.aaa2512
Li, M., Li, Z., Ren, H., Jin, W.-N., Wood, K., Liu, Q., et al. (2017). Colony stimulating factor 1 receptor inhibition eliminates microglia and attenuates brain injury after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 37, 2383–2395. doi: 10.1177/0271678x16666551
Liu, X., Chu, T.-H., Su, H., Guo, A., and Wu, W. (2013). Neural progenitor cell apoptosis and differentiation were affected by activated microglia in spinal cord slice culture. Neurol. Sci. 35, 415–419. doi: 10.1007/s10072-013-1532-4
Ma, Y., Li, Y., Jiang, L., Wang, L., Jiang, Z., Wang, Y., et al. (2016). Macrophage depletion reduced brain injury following middle cerebral artery occlusion in mice. J. Neuroinflammation 13:38. doi: 10.1186/s12974-016-0504-z
Mack, M., Cihak, J., Simonis, C., Luckow, B., Proudfoot, A. E., Plachý, J., et al. (2001). Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol. 166, 4697–4704. doi: 10.4049/jimmunol.166.7.4697
Malm, T. M., Koistinaho, M., Pärepalo, M., Vatanen, T., Ooka, A., Karlsson, S., et al. (2005). Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to β-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol. Dis. 18, 134–142. doi: 10.1016/j.nbd.2004.09.009
Maphis, N., Xu, G., Kokiko-Cochran, O. N., Jiang, S., Cardona, A., Ransohoff, R. M., et al. (2015). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755. doi: 10.1093/brain/awv081
Mawhinney, L. A., Thawer, S. G., Lu, W.-Y., Rooijen, N. V., Weaver, L. C., Brown, A., et al. (2012). Differential detection and distribution of microglial and hematogenous macrophage populations in the injured spinal cord of lys-EGFP-ki transgenic mice. J. Neuropathol. Exp. Neurol. 71, 180–197. doi: 10.1097/NEN.0b013e3182479b41
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., et al. (2008). Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724. doi: 10.1038/nature06616
Michaud, J.-P., Bellavance, M.-A., Préfontaine, P., and Rivest, S. (2013). Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 5, 646–653. doi: 10.1016/j.celrep.2013.10.010
Mildner, A., Mack, M., Schmidt, H., Brück, W., Djukic, M., Zabel, M. D., et al. (2009). CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132, 2487–2500. doi: 10.1093/brain/awp144
Mildner, A., Schlevogt, B., Kierdorf, K., Bottcher, C., Erny, D., Kummer, M. P., et al. (2011). Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. 31, 11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011
Mildner, A., Schmidt, H., Nitsche, M., Merkler, D., Hanisch, U.-K., Mack, M., et al. (2007). Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10, 1544–1553. doi: 10.1038/nn2015
Moreno, M. A., Burns, T., Yao, P., Miers, L., Pleasure, D., and Soulika, A. M. (2016). Therapeutic depletion of monocyte-derived cells protects from long-term axonal loss in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 290, 36–46. doi: 10.1016/j.jneuroim.2015.11.004
Nayak, D., Roth, T. L., and McGavern, D. B. (2014). Microglia development and function. Annu. Rev. Immunol. 32, 367–402. doi: 10.1146/annurev-immunol-032713-120240
Newman, T. A., Galea, I., van Rooijen, N., and Perry, V. H. (2005). Blood-derived dendritic cells in an acute brain injury. J. Neuroimmunol. 166, 167–172. doi: 10.1016/j.jneuroim.2005.04.026
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Parsa, R., Lund, H., Tosevski, I., Zhang, X.-M., Malipiero, U., Beckervordersandforth, J., et al. (2016). TGFβ regulates persistent neuroinflammation by controlling Th1 polarization and ROS production via monocyte-derived dendritic cells. Glia 64, 1925–1937. doi: 10.1002/glia.23033
Peng, J., Gu, N., Zhou, L., B Eyo, U., Murugan, M., Gan, W.-B., et al. (2016). Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat. Commun. 7:12029. doi: 10.1038/ncomms12029
Polfliet, M. M., Goede, P. H., van Kesteren-Hendrikx, E. M., van Rooijen, N., Dijkstra, C. D., and van den Berg, T. K. (2001). A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J. Neuroimmunol. 116, 188–195. doi: 10.1016/s0165-5728(01)00282-x
Prokop, S., Miller, K. R., Drost, N., Handrick, S., Mathur, V., Luo, J., et al. (2015). Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 212, 1811–1818. doi: 10.1084/jem.20150479
Rice, R. A., Pham, J., Lee, R. J., Najafi, A. R., West, B. L., and Green, K. N. (2017). Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65, 931–944. doi: 10.1002/glia.23135
Rice, R. A., Spangenberg, E. E., Yamate-Morgan, H., Lee, R. J., Arora, R. P. S., Hernandez, M. X., et al. (2015). Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J. Neurosci. 35, 9977–9989. doi: 10.1523/JNEUROSCI.0336-15.2015
Sawcer, S., Hellenthal, G., Pirinen, M., Spencer, C. C. A., Patsopoulos, N. A., Moutsianas, L., et al. (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. doi: 10.1038/nature10251
Schmidt, A., Strecker, J.-K., Hucke, S., Bruckmann, N.-M., Herold, M., Mack, M., et al. (2017). Targeting different monocyte/macrophage subsets has no impact on outcome in experimental stroke. Stroke 48, 1061–1069. doi: 10.1161/STROKEAHA.116.015577
Schneider, U. C., Davids, A.-M., Brandenburg, S., Müller, A., Elke, A., Magrini, S., et al. (2015). Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 130, 215–231. doi: 10.1007/s00401-015-1440-1
Serbina, N. V., and Pamer, E. G. (2006). Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317. doi: 10.1038/ni1309
Shichita, T., Sakaguchi, R., Suzuki, M., and Yoshimura, A. (2012). Post-ischemic inflammation in the brain. Front. Immunol. 3:132. doi: 10.3389/fimmu.2012.00132
Simard, A. R., Soulet, D., Gowing, G., Julien, J.-P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49, 489–502. doi: 10.1016/j.neuron.2006.01.022
Spangenberg, E. E., Lee, R. J., Najafi, A. R., Rice, R. A., Elmore, M. R. P., Blurton-Jones, M., et al. (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139, 1265–1281. doi: 10.1093/brain/aww016
Stalder, M., Deller, T., Staufenbiel, M., and Jucker, M. (2001). 3D-reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol. Aging 22, 427–434. doi: 10.1016/s0197-4580(01)00209-3
Stalder, A. K., Ermini, F., Bondolfi, L., Krenger, W., Burbach, G. J., Deller, T., et al. (2005). Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J. Neurosci. 25, 11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005
Stalder, M., Phinney, A., Probst, A., Sommer, B., Staufenbiel, M., and Jucker, M. (1999). Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 154, 1673–1684. doi: 10.1016/s0002-9440(10)65423-5
Sunderkötter, C., Nikolic, T., Dillon, M. J., Van Rooijen, N., Stehling, M., Drevets, D. A., et al. (2004). Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 172, 4410–4417. doi: 10.4049/jimmunol.172.7.4410
Szalay, G., Martinecz, B., Lénárt, N., Környei, Z., Orsolits, B., Judák, L., et al. (2016). Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 7:11499. doi: 10.1038/ncomms11499
Tanaka, R., Komine-Kobayashi, M., Mochizuki, H., Yamada, M., Furuya, T., Migita, M., et al. (2003). Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience 117, 531–539. doi: 10.1016/s0306-4522(02)00954-5
Tay, T. L., Mai, D., Dautzenberg, J., Fernández-Klett, F., Lin, G., Sagar, et al. (2017). A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 20, 793–803. doi: 10.1038/nn.4547
Tran, E. H., Hoekstra, K., van Rooijen, N., Dijkstra, C. D., and Owens, T. (1998). Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 161, 3767–3775.
Vainchtein, I. D., Vinet, J., Brouwer, N., Brendecke, S., Biagini, G., Biber, K., et al. (2014). In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia 62, 1724–1735. doi: 10.1002/glia.22711
van Rooijen, N. (1989). The liposome-mediated macrophage “suicide” technique. J. Immunol. Methods 124, 1–6. doi: 10.1016/0022-1759(89)90178-6
Varvel, N. H., Grathwohl, S. A., Baumann, F., Liebig, C., Bosch, A., Brawek, B., et al. (2012). Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc. Natl. Acad. Sci. U S A 109, 18150–18155. doi: 10.1073/pnas.1210150109
Varvel, N. H., Grathwohl, S. A., Degenhardt, K., Resch, C., Bosch, A., Jucker, M., et al. (2015). Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 212, 1803–1809. doi: 10.1084/jem.20150478
Varvel, N. H., Neher, J. J., Bosch, A., Wang, W., Ransohoff, R. M., Miller, R. J., et al. (2016). Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. U S A 113, E5665–E5674. doi: 10.1073/pnas.1604263113
Vilalta, A., and Brown, G. C. (2014). Deoxyglucose prevents neurodegeneration in culture by eliminating microglia. J. Neuroinflammation 11:58. doi: 10.1186/1742-2094-11-58
Waisman, A., Ginhoux, F., Greter, M., and Bruttger, J. (2015). Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol. 36, 625–636. doi: 10.1016/j.it.2015.08.005
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi: 10.1038/nrn.2015.1
Wang, Y., Szretter, K. J., Vermi, W., Gilfillan, S., Rossini, C., Cella, M., et al. (2012). IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13, 753–760. doi: 10.1038/ni.2360
Wieghofer, P., Knobeloch, K.-P., and Prinz, M. (2015). Genetic targeting of microglia. Glia 63, 1–22. doi: 10.1002/glia.22727
Womble, T. A., Green, S., Shahaduzzaman, M., Grieco, J., Sanberg, P. R., Pennypacker, K. R., et al. (2014). Monocytes are essential for the neuroprotective effect of human cord blood cells following middle cerebral artery occlusion in rat. Mol. Cell. Neurosci. 59, 76–84. doi: 10.1016/j.mcn.2014.01.004
Yamasaki, R., Lu, H., Butovsky, O., Ohno, N., Rietsch, A. M., Cialic, R., et al. (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–1549. doi: 10.1084/jem.20132477
Yao, Y., Echeverry, S., Shi, X. Q., Yang, M., Yang, Q. Z., Wang, G. Y. F., et al. (2016). Dynamics of spinal microglia repopulation following an acute depletion. Sci. Rep. 6:22839. doi: 10.1038/srep22839
Yona, S., Kim, K.-W., Wolf, Y., Mildner, A., Varol, D., Breker, M., et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. doi: 10.1016/j.immuni.2012.12.001
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S.-M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 92, 252–264. doi: 10.1016/j.neuron.2016.09.016
Zattoni, M., Mura, M. L., Deprez, F., Schwendener, R. A., Engelhardt, B., Frei, K., et al. (2011). Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 31, 4037–4050. doi: 10.1523/JNEUROSCI.6210-10.2011
Zhang, X.-M., Lund, H., Mia, S., Parsa, R., and Harris, R. A. (2014). Adoptive transfer of cytokine-induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia 62, 804–817. doi: 10.1002/glia.22643
Zhu, C., Herrmann, U. S., Falsig, J., Abakumova, I., Nuvolone, M., Schwarz, P., et al. (2016). A neuroprotective role for microglia in prion diseases. J. Exp. Med. 213, 1047–1059. doi: 10.1083/jcb.2134oia109
Zhu, Y., Soderblom, C., Krishnan, V., Ashbaugh, J., Bethea, J. R., and Lee, J. K. (2015). Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol. Dis. 74, 114–125. doi: 10.1016/j.nbd.2014.10.024
Zito, M. A., Koennecke, L. A., McAuliffe, M. J., McNally, B., Van Rooijen, N., and Heyes, M. P. (2001). Depletion of systemic macrophages by liposome-encapsulated clodronate attenuates striatal macrophage invasion and neurodegeneration following local endotoxin infusion in gerbils. Brain Res. 892, 13–26. doi: 10.1016/s0006-8993(00)03135-8
Keywords: microglia, monocyte, neurodegeneration, depletion, experimental models in neuroscience
Citation: Lund H, Pieber M and Harris RA (2017) Lessons Learned about Neurodegeneration from Microglia and Monocyte Depletion Studies. Front. Aging Neurosci. 9:234. doi: 10.3389/fnagi.2017.00234
Received: 01 May 2017; Accepted: 05 July 2017;
Published: 28 July 2017.
Edited by:
Yu Tang, University of Texas Southwestern Medical Center, United StatesReviewed by:
Caihong Zhu, Institute of Neuropathology, SwitzerlandKnut Biber, Albert Ludwig University of Freiburg, Germany
Copyright © 2017 Lund, Pieber and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert A. Harris, cm9iZXJ0LmhhcnJpc0BraS5zZQ==