Abstract
Frontotemporal dementia includes a large spectrum of neurodegenerative disorders. SQSTM1, coding for p62 protein, plays a vital role in the pathogenesis of FTD. Here, we report a case of a female patient with SQSTM1 mutation S224X, who was 59 years old when she initially exhibited memory decline, mild personality changes, and subtle atrophy of frontal/temporal lobes in magnetic resonance imaging (MRI). Genetic testing revealed a nonsense mutation of the SQSTM1 gene (S224X), resulting in premature termination of protein synthesis and a predicted truncated protein 217 amino acids shorter than the normal protein. Moreover, neither intact nor truncated SQSTM1 proteins was detectable in SQSTM1 S224X mutant overexpressing HEK-293T cells. We assayed for SQSTM1 cDNA in samples from the patient's peripheral leucocytes, and did not detect its mutation. The test of quantitative PCR showed significant decreased level of SQSTM1 mRNA from peripheral leucocytes of the patient compared to five dementia controls. Our results identify a novel pathogenic SQSTM1 S224X mutation in an atypical FTD patient accompanied with loss of SQSTM1/p62 protein expression probably due to SQSTM1 gene haploinsufficiency.
Introduction
Frontotemporal dementia is the second common form of neurodegenerative dementia in presenile population, characterized by atrophy of frontal and/or temporal lobes, and is frequently linked to genetic mutations (Miller et al., 2015; Luis et al., 2016). The three primary clinical FTD subtypes include behavioral variant FTD (bvFTD), nonfluent variant primary progressive aphasia (nfvPPA), and semantic variant primary progressive aphasia (svPPA) (Miller et al., 2015; O'Connor et al., 2016). As the most common presentation, bvFTD presents as a progressive change in personality with abnormalities in social-emotional behavior, and its diagnosis remains difficult, with patients being erroneously considered as having Alzheimer's disease or psychiatric disorders (Pottier et al., 2016; Tosun et al., 2016). The definite diagnosis is based on three major pathological subtypes characterized by the presence of 43 kDa TAR DNA-binding protein (TDP43), tau, or fused in sarcoma (FUS) positive neuronal inclusions (Boutoleau-Bretonniere et al., 2015). Approximately 30–50% of FTD patients have a positive family history, and 10% exhibit an autosomal dominant mode of inheritance. Mutations in the genes that encode microtubule associated protein tau (MAPT), progranulin and C9orf72 are the most common causes of FTD (Sun et al., 2017). Sequestosome 1 (SQSTM1), coding for p62 protein, is an adaptor protein that contains several protein-protein interaction motifs and serves as a signaling hub in a variety of key cellular processes including cell differentiation, transcriptional regulation, apoptosis, and oxidative stress response (Rea et al., 2014). SQSTM1, which has been identified in FTD in 2012 (Rubino et al., 2012), suggesting a role in the pathogenesis of neurodegenerative disease (Boutoleau-Bretonniere et al., 2015), is initially considered as a monogenic cause of Paget disease of bone (PDB) in 2002 (Laurin et al., 2002) and amyotrophic lateral sclerosis in 2011 (Fecto et al., 2011). Mutation in the SQSTM1 gene is a rare cause of FTD and ALS (van der Zee et al., 2014). Rubino et al. only identified 3 missense mutations in the SQSTM1 gene in 3 of 170 Italian patients with FTD, and 3 missense variants in 3 of 124 Italian patients with ALS (Rubino et al., 2012).
Here we report an atypical bvFTD patient with memory decline as an initial symptom and mild personality change emerging gradually, carried a novel pathogenic variant of the SQSTM1 gene causing absent expression of SQSTM1/p62 protein.
Materials and methods
Genetic procedures
Total genomic DNA was prepared and amplified from peripheral blood according to standard procedures. The quality of DNA was assessed by Qubit 3.0 (Thermo Fisher, USA) and agarose gel electrophoresis. Then the sequencing library was prepared according to the SureSelectXT Target Enrichment System Manual (Agilent, USA), and whole exome sequencing was performed by HiSeq X Ten (Illumina, USA). After this next-generation sequencing and bio-information analysis of the sequencing data, especially AD and FTD related genes including APP, PSEN1, PSEN2, MAPT, GRN, CHMP2B, C9ORF72, VCP, FUS, SQSTM1, TREM2, TYROBP etc., we found that there was a nonsense mutation in SQSTM1 gene. The nonsense mutation of SQSTM1 gene were analyzed by Sanger sequencing (forward primer: 5′-AGCGTCTGCCCAGACTACGA-3′ and reverse primer: 5′-CAGGCACTTAGGCACCTCAG-3′, the values of Tm were 63.4 and 61.0 respectively, and the length of amplified product is 547 bp). Moreover, we detected APOE genotyping at locus of rs429358 and rs7412. The software of Mutation Taster was applied to predict the pathogenicity of the detected mutations (http://www.mutationtaster.org/).
Analysis of SQSTM1 mutant protein expression
Plasmid DNA for wild type SQSTM1 was prepared by inserting coding sequence of the human SQSTM1 gene (NM_003900) into the pcDNA3.1/Myc-His vector, and the SQSTM1 S224X mutation was obtained by PCR-based site-directed mutagenesis with c.671C>A. All constructs were verified by sequencing (Minbo Biotech, Xiamen, China). Human embryonic kidney cells (HEK 293T) were grown to 80% confluence and transfected with vectors by Turbofect Transfection Reagent (Thermo Fisher, USA) according to the manufacturer's instructions. Media were then replaced with fresh DMEM containing 10% FBS. Cells were then collected 24 h later for western blotting. Equal amounts of total proteins (20 μg) were subjected to SDS-PAGE and transferred to PVDF membrane (Millipore, USA). Membranes were incubated with antibodies specific for SQSTM1 Gly162 (Cell Signaling Technology, 8025, USA), SQSTM1 Gly410 (Cell Signaling Technology, 5114, USA), Myc (Proteintech, 16282-1-AP, USA), or GAPDH (Abcam, ab181602, USA). Proteins were quantified using ImageJ Software.
Characterization of SQSTM1 gene expression
The mutation S224X (c.671 C>A) of SQSTM1 cDNA amplicons were obtained from the patient's mRNA sample by RT-PCR reaction. The cycling parameters of RT-PCR were 3 min at 95°C, followed by 30 s at 94°C, followed by 35 cycles of 60°C for 30 s, 72°C for 30 s, and a final extension of 72°C for 5 min. The pair of PCR primer sequence included the forward primer 5′-CTGTCTGAGGGCTCTCGC-3′ and reverse primer 5′-TCAACTTAATGCCCAGAGG-3′. The pyrosequencing and Sanger sequencing were performed following the manufacturer's protocols (Sangon, China). The analysis was used through PyroMark Software 1.0.11 software environment (Sangon, China).
Total cellular RNA was extracted from cell culture using TRIzol (Invitrogen, USA) according to manufacturer's procedures. Real-time PCR analysis was performed using 7900HT PCR instrument (ABI, USA). PCR conditions were at 95°C for 1 min, followed by 40 cycles at 95°C for 15 s, and 60°C for 30 s. For each biological replicate, three technical replicates were performed. The pair of PCR primer sequence included the forward primer 5′-TGGCGGAGCAGATG AGGAAG-3′ and reverse primer 5′-GGACTGGAGTTCACCTGTAGACG-3′.
Statistical analysis
All data are presented as mean ± standard error of mean. The data were analyzed by one-way analysis of variance (ANOVA). Results were considered to be statistically different when p < 0.05.
Ethics and patient consent
We received approval from the regional ethical standards committee on human experimentation for our experiments using human materials. We also received written informed consent for research from the participants and guardians.
Results
Case report
The patient underwent a clinical evaluation at our institution and was then enrolled in the Foundation of China Alzheimer's disease and related disorders study. Additional data from the proband and her relatives were collected and analyzed.
A 65-year-old, right-handed female with 15 years of school education was first seen in our geriatric psychiatry department in December 2016 for memory difficulties over 6 years combined with mild personality change over 1 year. She received surgical treatment for oophoroma in 2005 and drug treatment for hyperthyroidism in 2015, and gained full control of both diseases.
Her caregiver described the patient's forgetfulness at age 59, which had developed insidiously. She was referred to hospital because of memory decline and depression, and was prescribed antidepressant drugs. The patient refused to take the medications, and went on to care for her sick mother. At 5 years post-onset of symptoms, she began to lose her way and fall occasionally. In June 2015, she fell, resulting in cracking her head and bleeding. Her family brought her to hospital for testing. The brain computed tomography (CT) revealed cerebral atrophy, and electromyogram/ nerve conduction velocity (EMG/NCV) showed no positive finding. Subsequently, therapy with huperzine A was initiated. However, the patient took the medication irregularly, and her condition gradually aggravated. In May 2016, her family noticed her daytime somnolence, sluggishness, and reticence. Magnetic resonance imaging (MRI) showed mild atrophy of the cerebral cortex. Subsequently, combination therapy with Exelon and Escitalopram was started. In December 2016, the patient was brought to our department, and standard blood tests were normal. The neurological evaluation showed slow gait, normal muscular tension, brisk tendon reflexes, positive sign of bilateral palm-chin reflex, and negative Babinski sign. Her Mini Mental State Examination (MMSE) score was 20/30 and Montreal Cognitive Assessment (MoCA) score 15/30 (Figures 1A,B). Brain MRI in Dec 2016 revealed subtle atrophy in frontal and temporal lobes (Figure 1C). Although these findings did not lead to a definitive diagnosis, Memantine, Exelon, and Sertraline were administered as therapy. After 10 months, she was referred to our department again, and complained more serious memory decline. Her MMSE score was 12/30, and MoCA 10/30 (Figures 1A,B). However, brain MRI in Oct 2017 revealed no significant difference when compared with the last MRI (Figure 1D).
Figure 1

Summary of MMSE and MoCA, and imaging data. Summary of MMSE (A) and MoCA (B) scores in Dec 2016 and Oct 2017 displayed the significant damage in cognitive function and decline trend with time. The brain MRI in Dec 2016 (C) and Oct 2017 (D) showed subtle atrophy of frontal and temporal lobes on transverse FLAIR weighted and sagittal T1 weighted sequences.
The family history is summarized in Figure 2B. There are no similar manifestations in the relatives of the patient.
Figure 2
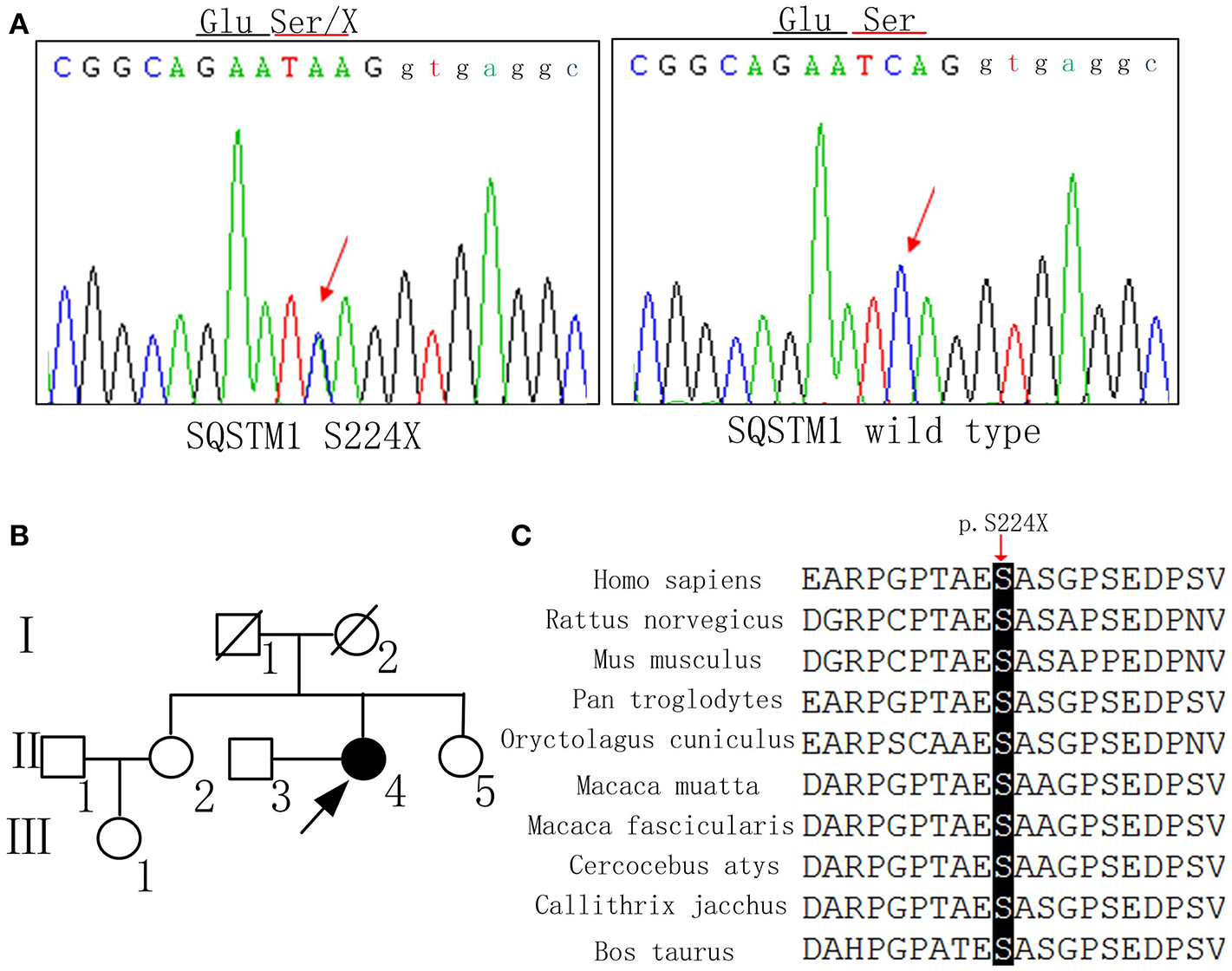
DNA sequence, pedigree of the family, and gene conservation. (A) DNA sequence at codon 224 of SQSTM1 gene from the patient and a control. The arrow indicates a mutated hemizygous site and a normal homozygous site, respectively. (B) The proband is indicated by an arrow (II-4). Her younger sister (II-5) also received the gene test and did not carry the same mutation. (C) The p.S224X heterozygous nonsense mutation occurs at highly conserved position, as shown by a comparison of the corresponding sequences of 10 vertebrates.
Genetic analysis
The mutation c.671 C>A, p. S224X of SQSTM1 was detected by whole exome sequencing, and validated in gDNA by Sanger Sequencing (Figure 2A). This mutation has not been reported as pathogenic elsewhere, and is predicted to lead to premature termination of protein synthesis. The pathogenicity prediction of the nonsense mutation by Mutation Taster software was disease causing with a probability equal to 1. The Combined Annotation Dependent Depletion (CADD) predicted that Raw score was 6.91 and PHRED was 33. The mutation was not found in the dbSNP, 1000G, HGMD, or ExAC database. At the same time, we didn't detect the same mutation in 200 normal Chinese individuals. The mutation site of the SQSTM1 gene was also assessed by evaluating the patient's unaffected younger sister, who did not carry the same mutation. Because the patient's father and mother were deceased, we could not determine whether the mutation was hereditary. The p.S224X heterozygous nonsense mutation is located at highly conserved position, as shown by a comparison of the corresponding sequences of 10 vertebrates (Figure 2C). The APOE genotype of the patient was ε3/ε4.
Analysis of SQSTM1 mutant protein expression
HEK 293T cells were transfected with an empty-vector control or plasmids encoding either wild type or S224X mutant Myc-His tagged SQSTM1 for 24 h. SQSTM1 was recognized in western blotting by different antibodies, which were SQSTM1 Gly162 and SQSTM1 Gly410 directed upstream and downstream, respectively, of the SQSTM1 mutation site (S224X). The SQSTM1 Gly162 antibody was used to detect truncated and intact SQSTM1 protein, and Gly410 to detect intact SQSTM1 protein (Figure 3A). Western blotting demonstrated that the levels of intact or truncated SQSTM1 protein in the S224X mutation group were all significantly reduced compared to wild type, similar to the vehicle (p < 0.01) (Figure 3B). Western blotting of Myc protein demonstrated no detectable Myc protein in the vehicle or S224X mutation group, only in the SQSTM1 wild type group (Figure 3A).
Figure 3
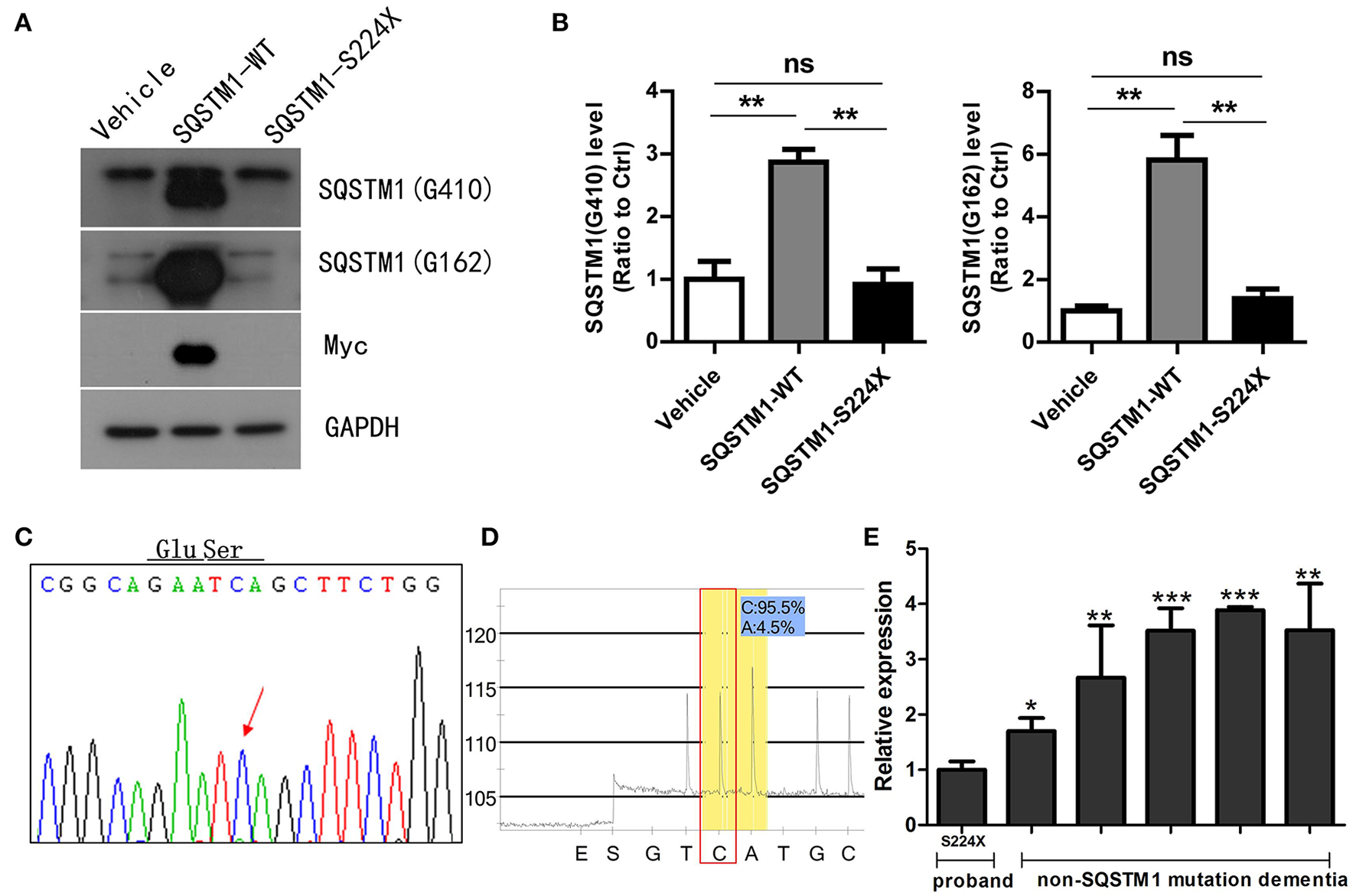
Mutant SQSTM1 protein and cDNA levels. (A) HEK 293T cells were transfected with an empty vector or plasmids encoding either wild type or mutant Myc-His-tagged SQSTM1 (S224X) for 24 h. Intact and truncated SQSTM1 proteins were recognized by western blotting with SQSTM1 (G410) and SQSTM1 (G162) antibodies, respectively. (B) The protein levels of SQSTM1 were quantified by densitometry and expressed as ratios to GAPDH. There were significantly reduced levels of intact or truncated SQSTM1 proteins in the S224X mutation group compared to wild type, similar to the vehicle. There was no expression of Myc protein in the vehicle or S224X mutation group, only in wild type. Data were plotted as mean ± SEM (n = 3). **p < 0.01, One-way ANOVA. (C,D)SQSTM1 cDNA from peripheral blood leucocytes of the patient was analyzed by Sanger sequencing and pyrosequencing. The mutation was not detected in SQSTM1 cDNA through Sanger sequencing, and only 4% of mutation was detected through pyrosequencing. The arrow indicates the mutant site. (E)SQSTM1 gene expression, measured by quantitative real-time PCR, normalized to β-actin, in peripheral blood leucocytes from the patient and five dementia controls without SQSTM1 mutation. The expression of SQSTM1 mRNA from the patient was significantly reduced compared to controls. Data were plotted as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA.
Analysis of SQSTM1 gene expression
SQSTM1 cDNA was amplified by RT-PCR from mRNA extracted from peripheral blood leucocytes of the patient. We did not find the same mutation in SQSTM1 cDNA (Figures 3C,D) through pyrosequencing or Sanger sequencing, which was inconsistent with DNA sequencing result. For analyzing the gene transcription differences between the SQSTM1 S224X mutation and SQSTM1 wild type, we included another five dementia controls (female: 3, male: 2, and age matched) without the SQSTM1 mutation. The relative transcription of SQSTM1 normalized to β-actin was analyzed using the concept of the threshold cycle (Ct) comparative method. Although the small sized of the samples adopted in the present study, it seemed that all dementia controls without SQSTM1 mutation presented an increased relative transcription level of SQSTM1 mRNA (up to 3.06 ± 0.88-fold) with regards to the patient (p < 0.05) (Figure 3E).
Discussion
The patient exhibited early-onset dementia combined with the initial symptom of memory decline, a gradual appearance of mild personality change, subtle atrophy of frontal/temporal lobes in MRI, negative appearance in EMG/NCV, and rapidly progressive course. The data of whole exome sequencing showed that AD related genes were all negative. These findings suggested an atypical bvFTD. The SQSTM1 mutation (S224X) was found in the patient, which produced a stop codon and resulted in a predicted truncated protein 217 amino acids shorter than the normal SQSTM1 protein. SQSTM1 has been reported to be associated with FTD-ALS type 3 (MIM: 616437). There isn't positive finding of EMG/NCV at present, but it still needs time to follow up the patient. The mutation is novel, not appearing in the dbSNP, 1000G, HGMD, ExAC database, and 200 normal Chinese individuals. The pathogenicity prediction by the Mutation-Taster application was disease causing, with a probability equal to 1. To determine the pathogenicity of this mutation, we overexpressed SQSTM1 S224X mutant comparable to that of wild type SQSTM1 in cell culture. Western blot of SQSTM1 recognized by Gly 410 and Gly 162 antibodies showed significant decrease of fully intact protein and truncated protein of SQSTM1. There was no expressional difference between mutant protein and vehicle. Western blotting of the Myc tag demonstrated that the S224X mutation led to loss of SQSTM1 protein expression after eliminating the effect of endogenous SQSTM1 protein from HEK 293T cells. Furthermore, in SQSTM1 cDNA from peripheral blood leucocytes of the patient, we didn't detect the mutation (S224X) by Sanger sequencing and pyrosequencing, and found significantly decreased level of SQSTM1 mRNA compared to five dementia controls without SQSTM1 mutation. The above results suggested absent expression of SQSTM1/p62 protein in the S224X mutant overexpressing HEK-293T cells and significant decrease of SQSTM1 mRNA level in the patient, which was possibly caused by nonsense-mediated mRNA decay (NMD), an mRNA degradation pathway regulating gene expression and mRNA quantity (Lopez-Perrote et al., 2016).
The SQSTM1 gene encodes SQSTM1/p62 protein, a scaffolding protein, which regulates a variety of biological processes, including nuclear factor kappa B (NF-κB) signaling, apoptosis, transcription regulation, and ubiquitin-mediated autophagy (Rea et al., 2014). Collet et al. demonstrated that PDB patients with SQSTM1 mutation (P392L, A381V, A390X, and L413F) had an increased level of SQSTM1/p62, and overproduction of the protein probably was involved in the pathophysiology of PDB (Collet et al., 2007). In FTD patients, SQSTM1 mutations (E396X and R212C) are reportedly associated with p62 and TDP43 inclusions in brain (Kovacs et al., 2016). However, in the present study, we demonstrated that a novel SQSTM1 mutation (S224X) causing loss of SQSTM1/p62 protein expression, not protein overproduction. Haack et al. identified three different biallelic loss-of-function variants (c.2T>A, p.?; R96X; E104Vfs*48) in SQSTM1 gene in nine patients with neurodegenerative disorder, and confirmed absence of SQSTM1/p62 protein in these patients (Haack et al., 2016). In mice, the knock out of SQSTM1 led to obesity and impaired glucose tolerance (Rodriguez et al., 2006). Furthermore, the chronic absence of SQSTM1/p62 promotes neurodegeneration with neurofibrillary tangles in hippocampal and cortical neurons manifesting with depression and short-term memory decline (Haack et al., 2016), which is similar to the clinical presentations of the present patient. There are multiple variants of SQSTM1 gene that cause diverse patterns of protein expression. Generally speaking, the imbalance of SQSTM1/p62 expression induced by SQSTM1 mutations is probably involved in the pathological mechanisms of FTD.
SQSTM1/p62 plays a key role in a variety of vital cellular processes, but it is unexpected that its absence is compatible with survival above age of 40 years (Haack et al., 2016), and mice lacking SQSTM1/p62 were fertile and lived more than 1 year inspite of adult-onset obesity and diabetes (Komatsu et al., 2007). This phenomenon probably argues for a redundancy of involved pathways or effective compensatory mechanisms (Haack et al., 2016). The present patient likely reflected SQSTM1 gene haploinsufficiency due to a combination of protein instability and NMD. Meanwhile, the patient displayed the APOE ε3/ε4 genotype, which confers an increased risk of developing Alzheimer's disease (AD). Whether APOE ε4 is a risk factor for FTD remains controversial. Ji et al. examined 432 patients with AD, 62 with FTD, and 381 controls. APOE ε4 allele frequency was significantly increased in late-onset AD (24.86), early-onset AD (18.02), and FTD (16.13) patients compared with controls (7.34), which suggested that the ApoE ε4 genotype is a risk factor for AD and FTD (Ji et al., 2013). However, Gustafson et al. and Verpillat et al. reported no correlation between the ε4 allele and FTD, but a larger increase in the ε2 allele in FTD compared with controls (Gustafson et al., 1997; Verpillat et al., 2002). Whether the APOE ε3/ε4 genotype in this case is promoting FTD requires further scrutiny.
In summary, we firstly identified a novel SQSTM1 mutation (S224X) in an atypical bvFTD patient, and the mutation caused absence of SQSTM1/p62 protein, which was consistent with a reduced level of SQSTM1 mRNA from peripheral leucocytes of the patient. The mechanisms underlying these observations are possibly associated with SQSTM1 gene haploinsufficiency. Different variants of the SQSTM1 gene result in diverse expression patterns, and imbalance of SQSTM1/p62 protein induces different pathogenic processes. In addition, there was a factor that limited the findings of the present study. Without a large family showing dementia and enough gene samples from family members, it is difficult to demonstrate that SQSTM1 S224X was fully responsible for FTD. However, we have provided a clue for discussing the pathogenicity significance of SQSTM1 S224X mutation in FTD, which should foster an understanding of the effect of SQSTM1 mutation on FTD when the mutation can be verified in a large family showing dementia.
Statements
Ethics statement
This study was carried out in accordance with the recommendations of “Shanghai Mental Health Center ethical standards committee on human experimentation” with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the “Shanghai Mental Health Center ethical standards committee”. All subjects also gave written informed consent for the publication of this case report.
Author contributions
LS and ZR: performed the experiments; LS: wrote the paper; WL: collected the samples; XL, SX, and HZ: supervised the experiments.
Acknowledgments
This study was supported by grants of National Key R&D Program of China (2017YFC1310501500), Western medical guidance project of Shanghai science and Technology Commission (17411970100), Natural Science Foundation of China (81301139 and 81771164), Precision medical research project of Shanghai Jiaotong University School of Medicine (15ZH4010), Natural Science Foundation of Guangdong Province of China (2016A030313821 and 2017A030313604), and the Educational Department of Fujian Province of China (JZ160403).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Boutoleau-Bretonnière C. Camuzat A. Le Ber I. Bouya-Ahmed K. Guerreiro R. Deruet A. et al . (2015). A phenotype of atypical apraxia of speech in a family carrying SQSTM1 mutation. J. Alzheimers. Dis.43, 625–630. 10.3233/JAD-141512
2
Collet C. Michou L. Audran M. Chasseigneaux S. Hilliquin P. Bardin T. et al . (2007). Paget's disease of bone in the French population: novel SQSTM1 mutations, functional analysis, and genotype-phenotype correlations. J. Bone Miner. Res.22, 310–317. 10.1359/jbmr.061106
3
Fecto F. Yan J. Vemula S. P. Liu E. Yang Y. Chen W. et al . (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol.68, 1440–1446. 10.1001/archneurol.2011250
4
Gustafson L. Abrahamson M. Grubb A. Nilsson K. Fex G. (1997). Apolipoprotein-E genotyping in Alzheimer's disease and frontotemporal dementia. Dement. Geriatr. Cogn. Disord.8, 240–243. 10.1159/000106637
5
Haack T. B. Ignatius E. Calvo-Garrido J. Iuso A. Isohanni P. Maffezzini C. et al . (2016). Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am. J. Hum. Genet.99, 735–743. 10.1016/j.ajhg.2016.06.026
6
Ji Y. Liu M. Huo Y. R. Liu S. Shi Z. Liu S. et al . (2013). Apolipoprotein Epsilon epsilon4 frequency is increased among Chinese patients with frontotemporal dementia and Alzheimer's disease. Dement. Geriatr. Cogn. Disord.36, 163–170. 10.1159/000350872
7
Komatsu M. Waguri S. Koike M. Sou Y. S. Ueno T. Hara T. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell131, 1149–1163. 10.1016/j.cell.2007.10.035
8
Kovacs G. G. van der Zee J. Hort J. Kristoferitsch W. Leitha T. Höftberger R. et al . (2016). Clinicopathological description of two cases with SQSTM1 gene mutation associated with frontotemporal dementia. Neuropathology36, 27–38. 10.1111/neup.12233
9
Laurin N. Brown J. P. Morissette J. Raymond V. (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet.70, 1582–1588. 10.1086/340731
10
López-Perrote A. Castaño R. Melero R. Zamarro T. Kurosawa H. Ohnishi T. et al . (2016). Human nonsense-mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res.44, 1909–1923. 10.1093/nar/gkv1527
11
Luis E. Ortiz A. Eudave L. Ortega-Cubero S. Borroni B. van der Zee J. et al . (2016). Neuroimaging correlates of frontotemporal dementia associated with SQSTM1 mutations. J. Alzheimers. Dis.53, 303–313. 10.3233/JAD-160006
12
Miller L. Rollinson S. Callister J. B. Young K. Harris J. Mann D. M. et al . (2015). Pickering-brown: p62/SQSTM1 analysis in frontotemporal lobar degeneration. Neurobiol. Aging36, 1603.e5-1603.e9. 10.1016/j.neurobiolaging.2014.08.035
13
O'Connor C. M. Clemson L. Hornberger M. Leyton C. E. Hodges E. Mioshi J. R. et al . (2016). Longitudinal change in everyday function and behavioral symptoms in frontotemporal dementia. Neurol. Clin. Pract.6, 419–428. 10.1212/CPJ.0000000000000264
14
Pottier C. Ravenscroft T. A. Sanchez-Contreras M. Rademakers R. (2016). Genetics of FTLD: overview and what else we can expect from genetic studies. J. Neurochem.138(Suppl. 1), 32–53. 10.1111/jnc.13622
15
Rea S. L. Majcher V. Searle M. S. Layfield R. (2014). SQSTM1 mutations–bridging Paget disease of bone and ALS/FTLD. Exp. Cell Res.325, 27–37. 10.1016/j.yexcr.2014.01.020
16
Rodriguez A. Durán A. Selloum M. Champy M. F. Diez-Guerra F. Moscat J. et al . (2006). Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab.3, 211–222. 10.1016/j.cmet.2006.01.011
17
Rubino E. Rainero I. Chiò A. Rogaeva E. Galimberti D. Fenoglio P. et al . (2012). SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology79, 1556–1562. 10.1212/WNL.0b013e31826e25df
18
Sun L. Chen K. Li X. Xiao S. (2017). Rapidly progressive frontotemporal dementia associated with MAPT Mutation G389R. J. Alzheimers Dis.55, 777–785. 10.3233/JAD-160802
19
Tosun D. Schuff N. Rabinovici G. D. Ayakta N. Miller B. L. Rosen L. et al . (2016). Diagnostic utility of ASL-MRI and FDG-PET in the behavioral variant of FTD and AD. Ann. Clin. Transl. Neurol. 3, 740–751. 10.1002/acn3.330
20
van der Zee J. Van Langenhove T. Kovacs G. G. Dillen L. Deschamps W. Van Broeckhoven C. et al . (2014). Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol.128, 397–410. 10.1007/s00401-014-1298-7
21
Verpillat P. Camuzat A. Hannequin D. Thomas-Anterion C. Puel M. Belliard S. et al . (2002). Apolipoprotein E gene in frontotemporal dementia: an association study and meta-analysis. Eur. J. Hum. Genet.10, 399–405. 10.1038/sj.ejhg.5200820
Summary
Keywords
Frontotemporal dementia, FTD, SQSTM1 , p62, S224X, nonsense mutation
Citation
Sun L, Rong Z, Li W, Zheng H, Xiao S and Li X (2018) Identification of a Novel Hemizygous SQSTM1 Nonsense Mutation in Atypical Behavioral Variant Frontotemporal Dementia. Front. Aging Neurosci. 10:26. doi: 10.3389/fnagi.2018.00026
Received
25 November 2017
Accepted
22 January 2018
Published
06 February 2018
Volume
10 - 2018
Edited by
Athanasios Alexiou, Novel Global Community Educational Foundation (NGCEF), Hebersham, Australia
Reviewed by
Daniel Hesselson, Garvan Institute of Medical Research, Australia; Melissa Calegaro Nassif, Universidad Mayor, Chile
Updates
Copyright
© 2018 Sun, Rong, Li, Zheng, Xiao and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honghua Zheng honghua@xmu.edu.cn
†These authors have contributed equally to this work.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.