 Xian-Si Zeng
Xian-Si Zeng Wen-Shuo Geng
Wen-Shuo Geng Jin-Jing Jia
Jin-Jing Jia Lei Chen
Lei Chen Peng-Peng Zhang
Peng-Peng Zhang- College of Life Sciences, Institute for Conservation and Utilization of Agro-Bioresources in Dabie Mountains, Xinyang Normal University, Xinyang, China
It has been 200 years since Parkinson disease (PD) was described by Dr. Parkinson in 1817. The disease is the second most common neurodegenerative disease characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta. Although the pathogenesis of PD is still unknown, the research findings from scientists are conducive to understand the pathological mechanisms. It is well accepted that both genetic and environmental factors contribute to the onset of PD. In this review, we summarize the mutations of main seven genes (α-synuclein, LRRK2, PINK1, Parkin, DJ-1, VPS35 and GBA1) linked to PD, discuss the potential mechanisms for the loss of dopaminergic neurons (dopamine metabolism, mitochondrial dysfunction, endoplasmic reticulum stress, impaired autophagy, and deregulation of immunity) in PD, and expect the development direction for treatment of PD.
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disease after Alzheimer disease. In 1817, an English physician, Dr. James Parkinson, firstly described the pathological characteristic of paralysis agitans (shaking palsy) (Schnabel, 2010), and the pattern was eventually called “PD” by Charcot and Vulpian in 1862. In 1912, Friederich Lewy first described the intraneuronal inclusions, namely “Lewy bodies” which define shaking palsy at a neuropathological level, and provided a detailed histopathological analysis of the nervous system to further understanding of shaking palsy. Now PD is known as a progressive, neurodegenerative disorder characterized by severe motor symptoms, including static tremor, postural imbalance, bradykinesia and muscle rigidity. α-synuclein is the main component of Lewy bodies and has a central role in PD (Spillantini et al., 1997). Epidemiological investigations have disclosed that few of PD patients are hereditable forms, while the overwhelming majority are sporadic forms (Schapira, 2008). The prevalence of PD varies with age, from 1–2% at ages of 55–65 years to 3.5% at 85–89 years(Ibanez et al., 2017). Unfortunately, the ultimate cause of PD is not well understood yet.
Although the pathological mechanisms are largely unknown, both genetic and environmental factors are considered to involve in potential pathogenesis of PD (Gan-Or et al., 2015). Polymeropoulos et al identified the first PD-linked mutation, α-synuclein (A53T), in Italian and Greek origin families in Polymeropoulos et al. (1997). Langston et al. (1983) found that four persons with notable parkinsonism after intravenous injection of an illicit drug, containing 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) in 1983. Afterward, many PD-linked gene mutations and several environmental factors were found. The potential mechanisms for loss of dopaminergic neurons were also well studied. In this review, we summarize the mutations of main seven genes (α-synuclein, LRRK2, PINK1, Parkin, DJ-1, VPS35 and GBA1) associated to PD and potential mechanisms for loss of dopaminergic neurons (dopamine metabolism, mitochondrial dysfunction, endoplasmic reticulum stress, impaired autophagy, and deregulation of immunity), and also expect the development direction for treatment of PD.
Genetic Basis of PD
There is an increasing list of mutations related to the onset of PD. They account for more than 50% of early-onset cases and about 2–3% of late-onset forms of PD. Findings of gene mutations linked to the onset of familial PD cases have confirmed their critical roles in the progress of PD and made it easy to understand the molecular pathogenesis for hereditary PD.
α-synuclein
A missense mutation in α-synuclein (A53T), encoded by the SNCA/PARK1 gene, was firstly identified to cause a familial form of PD (Polymeropoulos et al., 1997). Soon afterward, series of missense point mutations in the N-terminal of α-synuclein, including A30P, E46K, H50Q, G51D and A53E, were also identified. SNCA-linked PD is often of early onset and usually progresses rapidly. α-synuclein is the main component in Lewy bodies and most is phosphorylated at Ser129 of α-synuclein, which facilitated α-synuclein fibril uptake by neurons and exacerbated the pathology progression of PD (Sato et al., 2011; Karampetsou et al., 2017). Rodriguez et al have revealed the structure of toxic core of α-synuclein as pairs of face-to-face b-sheets by using micro-electron diffraction (Rodriguez et al., 2015). The α-synuclein variants forming oligomers were more toxic than that forming fibrils, so the former triggered more severe loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) in animals (Winner et al., 2011). The dosage of α-synuclein gene(SNCA) is responsible for parkinsonism (Ross et al., 2008). Point mutation and triplication carriers show almost complete penetrance, whereas penetrance in duplication carriers ranges between 30% and 50%. What’s more, SNCA triplication carriers also tend to have even earlier onset and more severe phenotype than duplication carriers (Schneider and Alcalay, 2017). Triplication of SNCA gene triggered unfolded protein response in induced pluripotent stem cells (iPSCs) generated from a patient with PD (Heman-Ackah et al., 2017).
Mutations of α-synuclein are toxic to dopaminergic neurons because they alter a series of intracellular signal programs. α-synuclein A53T mutation could suppress autophagy in the brain of transgenic mice at early stage and lead to synucleinopathy (Pupyshev et al., 2017). A53T also induces mitochondrial dysfunction- and endoplasmic reticulum stress-mediated cell death pathways in PC12 cells (Smith et al., 2005a). Proteasomal protein cleavage is significantly impaired by overexpression of mutant α-synuclein in dopaminergic cells, such as SH-SY5Y and PC12 (Zondler et al., 2017). Matsumoto et al. (2017) have reported that erythrocyte-derived extracellular vesicles containing α-synuclein could transmit across the blood brain barrier; this may be a new mechanism by which the brain and periphery communicate throughout the initiation and development of PD. α-synuclein specifically binds to TrkB receptors and inhibits BDNF/TrkB signaling pathway, resulting in the death of dopaminergic neurons (Fang et al., 2017; Kang et al., 2017). Another mutation A30P could promote the neurodegeneration of dopaminergic neurons via activating microglia and upregulating the expression of PHOX and Mac-1 (Zhang et al., 2007). The mutations of α-synuclein linked to PD and the pathogenesis are summarized in Table 1.
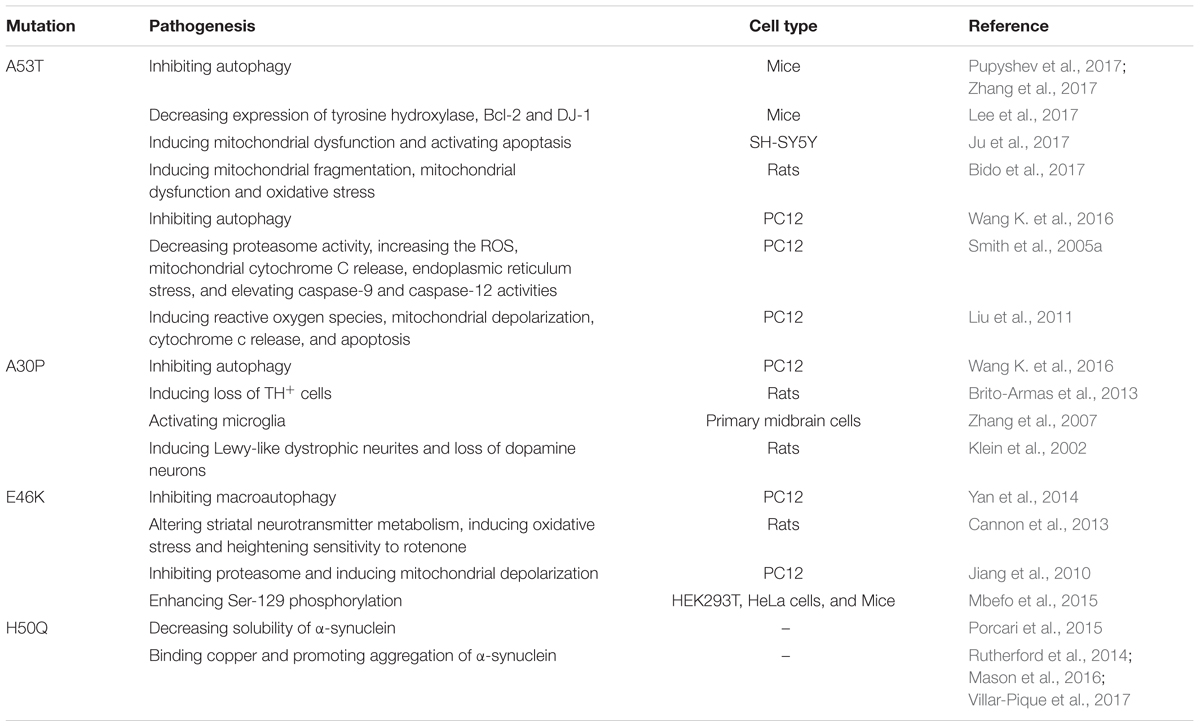
TABLE 1. The mutations of α-synuclein linked to PD and the Pathogenesis.
Both mRNA and protein levels of α-synuclein showed a double increase in PD (Farrer et al., 2004), therefore, reducing α-synuclein expression may provide a potential therapeutic approach (Lewis et al., 2008). Zharikov et al. (2015) have clarified that knockdown of α-synuclein by using a shRNA attenuates the progressive motor deficits and the degeneration of dopaminergic neurons in SNc in rotenone-induced rats model of PD. Intranasal delivery of inhibitory oligonucleotides, siRNA or antisense oligonucleotide combined with the triple monoamine reuptake inhibitor indatraline selectively decreases α-synuclein expression in the brainstem of mice and reduces levels of endogenous α-synuclein mRNA and protein in SNc (Alarcon-Aris et al., 2017). Systemic exosomal siRNA delivery also significantly reduced the aggregation of α-synuclein in SNc dopaminergic neurones of S129D α-synuclein transgenic mice (Cooper et al., 2014).
LRRK2
Leucine-rich repeat kinase 2(LRRK2) is a multi-domain protein, containing both Rab GTPase and kinase enzymatic activities (Steger et al., 2017). LRRK2 mutations are linked with “classical” late-onset PD and accounts for 4% of hereditary PD (Ferreira and Massano, 2017). The penetrance is incomplete and age-dependent. Mutations of LRRK2/PARK8 gene represent the highest risk of familial PD, causing autosomal dominant PD (Zimprich et al., 2004). Morbidity of PD is significantly elevating among LRRK2 G2019S mutation carriers as aging (Healy et al., 2008). LRRK2 G2019S mutation elevates mobility of α-synuclein and enhances α-synuclein accumulation in cultured primary neurons and in dopaminergic neurons in SNc of PD brain (Volpicelli-Daley et al., 2016). The mutation also promotes the transmission of Tau in the mouse brain neurons, implicating for understanding the development of tau protein neuropathology in LRRK2-linked PD (Nguyen et al., 2018). Pathogenic mutations in LRRK2 increase its kinase activities in cells and tissues, so small molecule inhibitors of LRRK2 kinase can be used to block its activity to provide neuroprotection in some PD models (West, 2017). Intracerebral injection of LRRK2 antisense oligonucleotides also decreases endogenous protein levels of normal LRRK2 and inhibits the formation of fibril-induced α-synuclein inclusions (Zhao et al., 2017).
LRRK2 not only inhibits neuronal survival but also impairs dopamine signal. LRRK2 played important roles in the death of neurons via directly phosphorylating apoptosis signal-regulating kinase 1 at Thr832 site and activating the kinase activity (Yoon et al., 2017). In model organisms, it has numerous roles within the secretory pathway and may contribute to dopaminergic signaling (Smith et al., 2005b). LRRK2 could promote neuronal death via inhibiting myocyte-specific enhancer factor 2D activity, which is required for neuronal survival (Han et al., 2017). LRRK2 G2019S mutation impairs dopamine receptor D1 internalization, leading to an alteration in signal transduction (Rassu et al., 2017). In addition, LRRK2 G2019S mutation enhances the kinase activity and results in the impairment of synaptic vesicle trafficking selectively in ventral midbrain neurons, including dopaminergic neurons (Pan et al., 2017).
LRRK2 G2019S mutation shows a close relationship with mitochondrial dysfunction. Howlett et al. (2017) have reported that this mutation induces mitochondrial DNA damage in cultured primary neurons from rat midbrain, which is dependent on LRRK2 kinase activity, and pharmacological inhibition restores the integrity of mitochondrial DNA. PD-associated LRRK2 mutations upregulate the expression of mitochondrial calcium uniporter, a mitochondrial calcium transporter and then promote the uptake of dendritic and mitochondrial calcium in cortical neurons and familial PD patient fibroblasts (Verma et al., 2017). LRRK2 G2019S binds to and enhances its interaction with mitochondrial dynamin-like protein 1(DLP1, a fission protein), which increases mitochondrial DLP1 levels and induces neuronal toxicity, mitochondrial fragmentation, and mitochondrial dysfunction (Wang et al., 2012). LRRK2 G2019S mutation also impairs mitochondrial function by decreasing the activity of respiratory chain complex IV, whereas siRNA-mediated knockdown of LRRK2 restores the mitochondrial morphology (Mortiboys et al., 2015). Ursocholanic acid and ursodeoxycholic acid improves the LRRK2 G2019-decreased mitochondrial function in vitro and in vivo, suggesting that ursocholanic acid and ursodeoxycholic acid may be promising drugs for future neuroprotective trials (Mortiboys et al., 2013, 2015). The mutations of LRRK2 linked to PD and the pathogenesis are summarized in Table 2.
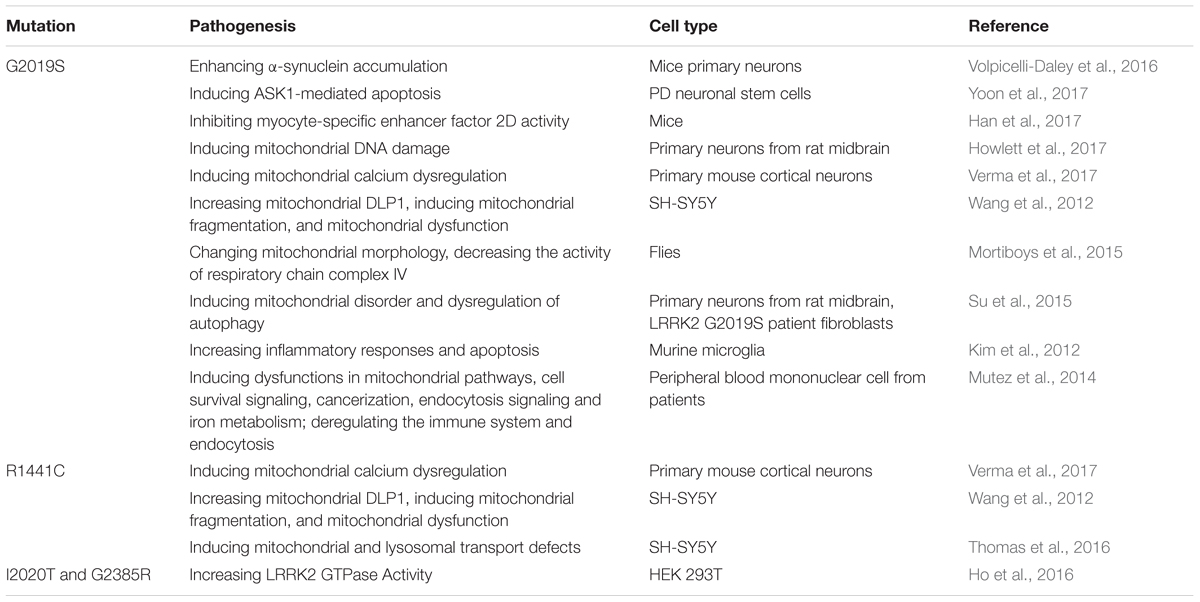
TABLE 2. The mutations of LRRK2 linked to PD and the Pathogenesis.
PINK1 and Parkin
PTEN-induced putative kinase (PINK) 1, a mitochondrial protein kinase, is a critical protein linked with the pathogenesis of PD. PINK1 deficiency causes the autosomal recessive PARK6 variant of PD. PINK1 mutations have been reported to account for approximately 1–3% of early onset PD in populations of European ancestry, 8.9% of autosomal recessive PD in a sample of Japanese families, and 2.5% of early onset PD in a sample of ethnic Chinese, Malays, and Indians (Bekris et al., 2010). PINK1 loss is associated with mitochondrial dysfunction and mitochondrial Ca2+ mishandling via directly interacting with and phosphorylating LETM1 at Thr192 site to increase Ca2+ release and facilitate its transport (Huang et al., 2017). In a Caenorhabditis elegans Model of PD, non-apoptotic neurodegeneration of dopaminergic cells is induced by the dysregulation of mitochondrial unfolded protein response, which is dependent on the PINK1 homolog (Martinez et al., 2017). Heterozygous PINK1 G411S mutation shows a significant decrease in kinase activity of PINK1 and could increase the risk of PD via a dominant-negative mechanism (Puschmann et al., 2017). Mutation of PINK1 show an accumulation of dysfunctional mitochondria with age, leading to activation of the mitochondrial unfolded protein response, which promotes longevity and dopamine neuron survival in PD models (Cooper et al., 2017). I368N and Q456X mutations of PINK1 reduce either its stability/protein levels or kinase activity, increasing the risk of PD (Ando et al., 2017). Other mutations, including A168P, H271Q, L347P and G309D, weaken the recruitment of Parkin to depolarized mitochondria (Narendra et al., 2010).
Parkin, encoded by PARK2, is originally considered to serve as a ubiquitin E3 ligase, which can be activated by autophosphorylated PINK1 (Zhuang et al., 2016) and is required for the ubiquitin-proteasome degradation of target substrates (Kumar et al., 2017). Mutations in the Parkin gene are a common cause of early onset PD in many countries and account for 10–25% of cases (Sanyal et al., 2015). PINK1 accumulates on the membrane of dysfunctional mitochondria, activates the E3 ubiquitin ligase activity of Parkin and recruits them to damaged mitochondria from cytoplasm to promote their mitophagic degradation (Narendra et al., 2009). Reactive oxygen species also induces PINK1/Parkin pathway-dependent mitophagy by promoting Parkin recruitment to mitochondria (Xiao et al., 2017). Mutations of Parkin, including R42P, R46P, K211N, C212Y, C253Y, C289G, and C441R, cause a deficit in its recruitment to depolarized mitochondria and further inhibit mitophagy (Narendra et al., 2010). Silence of PINK1/Parkin by siRNA promotes mitochondrial fragmentation in cultured primary neurons of mouse (Lutz et al., 2009). The mutations of PINK1 and Parkin linked to PD and the pathogenesis is summarized in Table 3.
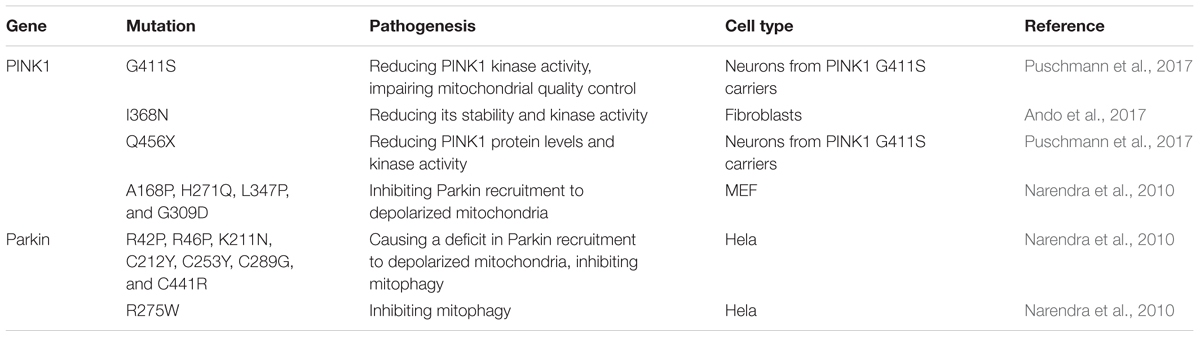
TABLE 3. The mutations of PINK1 and Parkin linked to PD and the Pathogenesis.
DJ-1
Mutations of DJ-1, encoded by the PARK7 gene, have been linked with early onset of recessive PD, which were first identified in 2 European families with an age of onset of 20–40 years (Bonifati et al., 2003). Several PD-associated DJ-1 mutations have been identified, including L166P, M26I, L10P and P158Δ. Recently, genetic analysis from a single case research reveals a novel mutation of L172Q in the PARK7 gene, which is the first time to report the neuropathology from a DJ-1-linked PD brain (Taipa et al., 2016). DJ-1 could interact with and steady PINK1 in cells overexpressing PINK1 (Tang et al., 2006). DJ-1 has been reported to also interact with α-synuclein, modulate its aggregation by weak hydrophobic interaction (Zhu et al., 2017) and restore α-synuclein-induced cellular toxicity (Zondler et al., 2014).
The neuroprotective roles mediated by DJ-1 are summarized as anti-oxidant properties, anti-apoptotic effects, effects on mitochondrial respiratory function, morphology, turnover and biogenesis (Mukherjee et al., 2015). Currently, the most acknowledged role of DJ-1 in the pathophysiology of PD is the neuroprotective function against oxidative stress (Biosa et al., 2017). DJ-1 mutations mainly affect a protein involved in intracellular oxidation-reduction (Ishikawa et al., 2009). Mutant DJ-1(L166P and M26I) increases the sensitivity of SHSY5Y cells toward oxidative stress, because it abolishes the neuroprotection against H2O2 and the induction of thioredoxin-1 through inhibiting nuclear factor erythroid-2 related factor 2 signal pathway (Im et al., 2012). D149A mutation also abolishes its protection (Giaime et al., 2010). The three mutations, L172Q, L10P and P158Δ, are involved in reduction of protein stability (Ramsey and Giasson, 2010; Taipa et al., 2016). DJ-1 deficiency in neurons shows a decrease of glutamine influx and serine biosynthesis, which attenuates cellular antioxidant response and leads to the degeneration of dopaminergic neurons (Meiser et al., 2016). DJ-1-Deficiecy in embryonic stem cells-derived dopaminergic neurons also increases the sensitivity to toxins-induced oxidative stress (Martinat et al., 2004). Drosophila carrying a mutant DJ-1β gene, the homologue of human DJ-1, show a reduced climbing ability reflecting locomotor defects (Sanz et al., 2017). DJ-1 overexpression ameliorates mitochondrial function, including the mitochondrial quantity and activity of respiratory chain complex I, by phosphorylating Akt at Thr308 site in both SH-SY5Y and PC-12 cells (Zhang et al., 2016). Recombinant DJ-1 can also reverse dopamine depletion in α-synuclein or 6-hydroxydopamine (6-OHDA)-triggered dopaminergic degeneration in vivo (Batelli et al., 2015). Some natural compounds protect against neurotoxin-induced Parkinsonism by upregulating DJ-1 expression. A marine compound exerts neuroprotective effects through upregulating DJ-1 expression and activating DJ-1-targeted pathway in 6-OHDA-induced zebrafish and rat models of PD (Feng et al., 2016). A standardized flavonoid extract from Safflower restores the expression of DJ-1, as well as the expression of tyrosine hydroxylase and dopamine transporter in rotenone-induced Parkinson rats (Ablat et al., 2016). The mutations of DJ-1 linked to PD and the pathogenesis is summarized in Table 4.
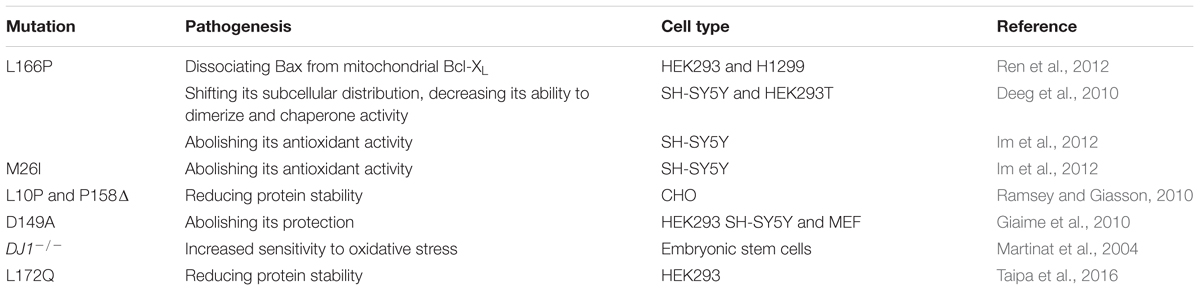
TABLE 4. The mutations of DJ-1 linked to PD and the Pathogenesis.
VPS35
Mutations of the Vacuolar protein sorting 35 (VPS35) gene were first discovered in an Austrian family with late-onset PD. It accounts for 1% of familial PD. VPS35, a major component of the retromer cargo-recognition complex, regulates the process of transmembrane protein sorting between endosomes and the Golgi (Bonifacino and Hurley, 2008). It has been reported that VPS35 D620N mutation is pathogenic in patients with PD in Asian, European and American families (Chen et al., 2017). VPS35 D620N mutation led to a decrease of enzymatic activity in complex I and II in PD patient fibroblasts (Zhou et al., 2017) and caused mitochondrial dysfunction by recycling DLP1 complexes (Wang W. et al., 2016). What’s more, VPS35 deficiency also led to mitochondrial dysfunction and finally resulted in the loss of dopaminergic neurons (Tang et al., 2015b). VPS35 mutant D620N disrupted the interaction of VPS35 with aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2), a Parkin substrate, and lysosome-associated membrane protein-2a (LAMP2A). Overexpression of VPS35 inhibited AIMP2-enhanced cell death in SH-SY5Y cells; In contrast, downregulation of VPS35 resulted in AIMP2 dependent-cell death (Yun et al., 2017). VPS35-deficient DA neurons exhibit impaired endosome-to-Golgi retrieval of Lamp2a, which may contribute to the reduced α-synuclein degradation through chaperone-mediated autophagy (Tang et al., 2015a). Co-expression of high copy numbers of VPS35 D686N (the yeast equivalent of D620N) and low copy numbers of α-synuclein caused toxicity. Another PD-linked VPS35 mutation R524W impaired the endosomal association of retromer and also induced the aggregation of α-synuclein (Follett et al., 2016). In a Drosophila model, VPS35 P316S mutation also produced some of PD phenotypes, including reduction of climbing ability and loss of dopaminergic neurons, enhanced the susceptibility of Drosophila to rotenone (Wang et al., 2014). The mutations of VPS35 linked to PD and the pathogenesis is summarized in Table 5.
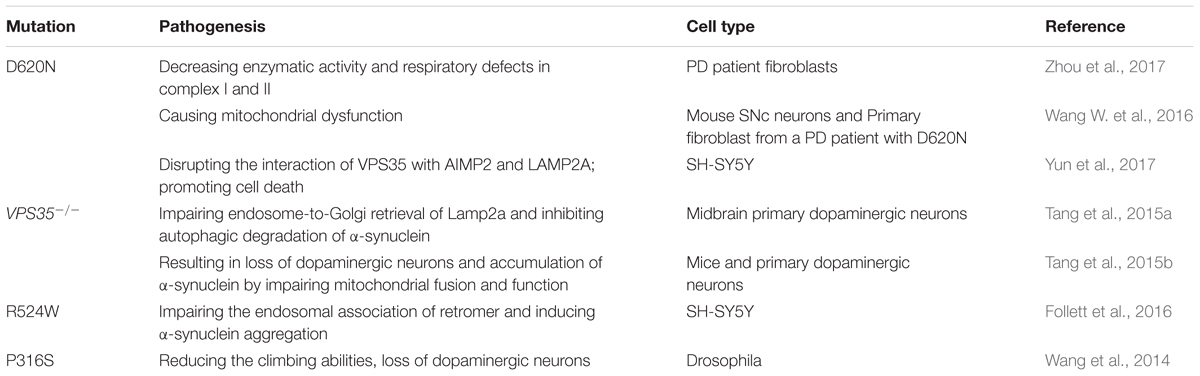
TABLE 5. The mutations of VPS35 linked to PD and the Pathogenesis.
GBA1
Glucocerebrosidase (GBA1) gene mutations, encoding gluco-cerebrosidase 1 (GCase 1) which catabolizes glycolipid glucocerebroside to ceramide and glucose in lysosome (Bae et al., 2015) and responsible for Gaucher disease, are the most common genetic risk factor for PD (Taguchi et al., 2017). It is recently reported that both heterozygous and homozygous mutations of the GBA1 gene have been linked to PD, with more than 10% of individuals carrying GBA1 mutations developing PD by the age of 80 (Migdalska-Richards et al., 2016). A significant and progressive reduction in GCase 1 activity and protein levels is also observed in the brains and cerebrospinal fluid of PD patients (Murphy et al., 2014; Rocha et al., 2015; Parnetti et al., 2017). Activation of GCase 1 by using a small-molecule modulator restored the function of lysosome and then cleared the accumulation of pathological α-synuclein in midbrain neurons of PD patients (Mazzulli et al., 2016), indicating the potential importance of GCase 1 in the development of idiopathic PD. It is estimated that about 10–25% of PD cases carry a GBA1 mutation, in which N370S and L444P are the most common mutations (Sidransky et al., 2009; McNeill et al., 2012). GBA1 knockout inhibited macroautophagy and chaperone-mediated autophagy in MEFs and downregulation of GCase 1 increased α-synuclein protein levels in SH-SY5Y cells (Magalhaes et al., 2016). GBA1 deficiency also resulted in the accumulation of glycosphingolipids (GSL), then decreased α-synuclein tetramers resisting aggregation and related multimers and increased α-synuclein monomers in CRISPR-GBA1 knockout SH-SY5Y cells. What’s more, α-synuclein tetramers and related multimers were also reduced in PD induced pluripotent stem cell (iPSC)-derived human dopaminergic neurons carrying N370S GBA1 mutation (Kim et al., 2018). In contrast, inhibition of GSL accumulation and overexpression of GBA1 to enhance GCase 1 activity reverse the destabilization of α-synuclein tetramers and protect against α-synuclein preformed fibril-induced toxicity in human dopaminergic neurons. N370S mutation produced GCase 1 retention within the endoplasmic reticulum, interrupting its traffic to the lysosome, leading to endoplasmic reticulum stress activation and triggering unfolded protein response and Golgi apparatus fragmentation (Garcia-Sanz et al., 2017, 2018). Decreased α-synuclein tetramers and related multimers were also observed in murine neurons carrying the heterozygous L444P GBA1 mutation (Kim et al., 2018) and L444P GBA1 mutation aggravated α-synuclein-induced loss of dopaminergic neurons in SNc of 24-month-old mice (Migdalska-Richards et al., 2017). The mutations of GBA1 linked to PD and the pathogenesis is summarized in Table 6.
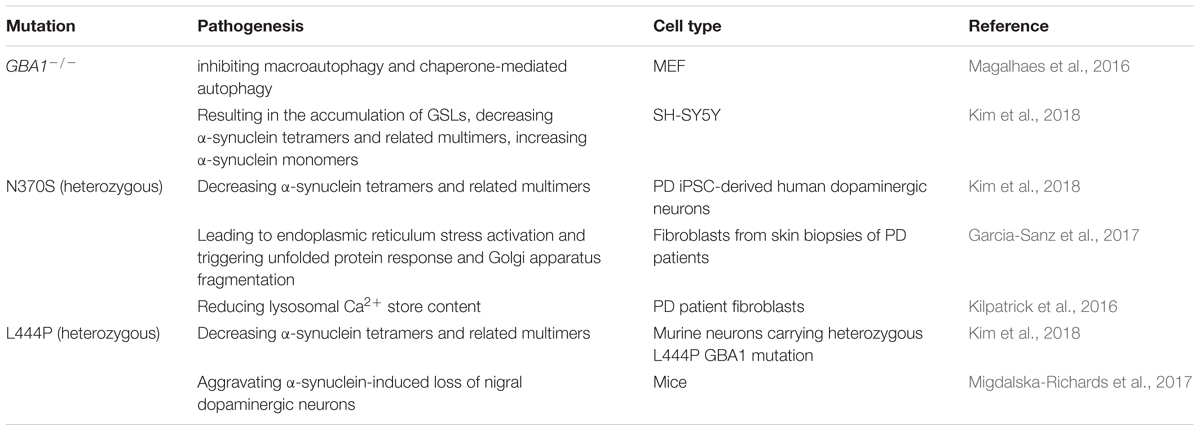
TABLE 6. The mutations of GBA1 linked to PD and the Pathogenesis.
Mechanisms of Neuronal Cell Death in PD
The most obvious progress has been made in understanding the proximate causes, especially the deaths of dopaminergic neurons that explain the classic motor symptoms of the disease. The deaths mainly occur in the SNc, in which the neurotransmitter dopamine is normally synthesized and pumped into movement-regulating brain regions. Though up to 70% of these dopaminergic neurons in SNc will be killed during the development of PD, symptoms are seldom noticed because the striatum, immediately downstream of the SNc, can compensate to some extent. However, massive neurons in the striatum also start to die eventually. Currently, it is widely accepted that dopamine metabolism, oxidative stress and mitochondrial dysfunction, endoplasmic reticulum stress, impairment of protein degradation systems, and neuroinflammation are involved in the death of dopaminergic neurons in SNc (Table 7).

TABLE 7. Main mechanisms involving the death of the dopaminergic neuron.
Dopamine Metabolism
Parkinson disease is a movement disorder and the first neurodegenerative disease, which is confirmed severe depletion of dopamine (regulates Parkinson’s tremor) in the striatum (Dirkx et al., 2017). Oxidation of dopamine could induce oxidative stress and is harmful to dopaminergic neurons. Mitochondrial oxidative stress results in the accumulation of oxidized dopamine and ultimately leads to reduced activity of glucocerebrosidase, lysosomal dysfunction, and α-synuclein accumulation in human and PD neurons (Burbulla et al., 2017; Garcia-Sanz et al., 2017). Increased levels of dopamine in cytoplasm and its metabolites are neurotoxic and lead to selective death of neurons in SNc, administration that decreases cytoplasmic dopamine levels provide neuroprotection in cultured midbrain neurons (Mosharov et al., 2009). Aminochrome, which is formed within the cytoplasm during the auto-oxidation of dopamine to neuromelanin, could induce neurotoxicity in dopaminergic neurons when DT-diaphorase and glutathione transferase mutate (Herrera et al., 2017). Auto-oxidation of dopamine aggravates overexpression of human α-synuclein A53T mutation-induced non-apoptotic cell death in PC12 cells (Zhou et al., 2009). Dopamine not only spontaneously oxidize to form melanin and neuromelanin, but also is catalyzed to the immediate products being hydrogen peroxide and aldehydes by monoamine oxidase (Goldstein et al., 2014). Monoamine oxidase in the outer membrane of mitochondria catalyzes conversion of cytosolic dopamine to 3,4-dihydroxyphenylacetaldehyde, which is an endogenous neurotoxin implicated in PD (Panneton et al., 2010), cause extensive aggregation of glyceraldehyde-3-phosphate dehydrogenase and irreversibly inhibit the enzyme activity, resulting in the production of reactive oxygen species to damage neurons (Vanle et al., 2017). Dopamine also induces the production of soluble A53T α-synuclein oligomers and promotes the degeneration of neurons in SNc (Mor et al., 2017). Vesicular uptake inhibition of dopamine with Reserpine rapidly increases the cytosolic concentration of 3,4-dihydroxyphenylacetaldehyde, contributing to the dimerization of α-synuclein and apoptosis (Goldstein et al., 2012). Dopamine induces mitochondrial permeability transition pore opening via production of reactive oxygen species in mouse neurons with PINK1 defect (Gandhi et al., 2012). Reuptake of extracellular dopamine to cytoplasm leads to accumulation of dopamine, which triggers neurotoxicity in dopaminergic neurons. Disrupting the interaction between dopamine D2 receptor and dopamine transporter provides a protective role against dopamine neurotoxicity (Su and Liu, 2017).
Mitochondrial Dysfunction
Mitochondrial disorders have been implicated in almost all neurodegenerative diseases, including PD (Subramaniam and Chesselet, 2013; Connolly et al., 2017). PD-linked mutations could induce mitochondrial dysfunction. Soluble prefibrillar α-synuclein oligomers showed several phenotypes of mitochondrial dysfunction observed previously in cell and animal models of PD: dysfunction of complex I, change in membrane potential, disruption of Ca2+ homeostasis, and enhanced release of cytochrome c (Luth et al., 2014). PD-associated mutation of VPS35 gene, encoding a key component of the retromer complex, leaded to mitochondrial fragmentation and neuronal death in vitro and in vivo by enhanced interactions with DLP1 (Wang W. et al., 2016). Inhibiting the enzymatic activity of the GCase 1, or silencing the GBA1 gene in SH-SY5Y, also caused mitochondrial dysfunction, showing reduced activity of mitochondrial respiratory chain and mitochondrial depolarization and fragmentation associated with increased levels of ROS (Cleeter et al., 2013). In the SNc of idiopathic PD patients, the expression of mitochondrial DNA transcription factors (TFAM and TFB2M) is decreased. Consistent with this, real-time PCR analysis exhibits a reduced copy number of mitochondrial DNA and fewer molecules associated with transcription/replication in PD patients (Grunewald et al., 2016). 4-hydroxynonenal, a lipid peroxidation product, promotes the intracellular accumulation, extrusion of extracellular vesicles containing toxic α-synuclein, and then internalization into neighboring neurons, finally results in the neurodegenerative development of PD (Zhang et al., 2018).
Numerous studies have repeatedly demonstrated that the expression and activity of respiratory chain complex I are markedly reduced in PD (Yin et al., 2014). The initial evidence is that exposure to environmental 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) caused degeneration of dopaminergic neurons and parkinsonism (Langston et al., 1983). MPTP, a PD neurotoxin, is metabolized to its active metabolite MPP+ by monoamine oxidase and then is transferred into dopaminergic neurons by dopamine transporter (Storch et al., 2004). MPP+ is considered as an inhibitor of mitochondrial complex I, inhibits ATP production, then results in the generation of superoxide and peroxynitrite, and finally ablates dopaminergic neurons (Khaldy et al., 2003; Yan et al., 2013). Partial deficiency of mitochondrial complex I in dopaminergic neurons increases the vulnerability of mice toward MPTP (Sterky et al., 2012). Other neurotoxins can also induce degeneration of dopaminergic neurons and parkinsonism. The herbicide paraquat, reduced by NADPH oxidase on microglia, is also transported into dopaminergic neurons by dopamine transporter, where it induces mitochondrial dysfunction and neurotoxicity (Rappold et al., 2011). Injection of PD neurotoxin 6-OHDA into the right SNc also reduces activity of mitochondrial oxidative phosphorylation enzymes, particularly selective decrease of complex I activity (Dabbeni-Sala et al., 2001). Chronic treatment of rodents with pesticide rotenone, a well-established complex I inhibitor, selectively induces toxicity to dopaminergic neurons (Betarbet et al., 2000; Choi et al., 2011).
There is a potential interaction between environmental neurotoxin-induced mitochondrial dysfunction and genetic risk factors in PD pathogenesis. Injection of sub-toxic MPTP enhances the neurotoxicity of mutant α-synuclein and induces vulnerability of dopamine neurons, which may be due to the downregulation of DJ-1 (Lee et al., 2017). The higher level of neurotoxicity in SNc than VTA neurons is due to SNc neuron-specific MPP+-induced increase in cytosolic dopamine and Ca2+, followed by an elevation of mitochondrial Ca2+ and mitochondrial oxidation, which is dependent on the expression of α-synuclein (Lieberman et al., 2017). A long non-coding RNA promotes MPTP-induced parkinsonism via upregulating the expression of LRRK2 (Liu et al., 2016). Mice expressing LRRK2 G2019S mutation exhibit elevated susceptibility to MPTP-induced neurotoxicity (Karuppagounder et al., 2016). Downregulation of PINK1 enhances the sensitivity of dopaminergic neurons to MPTP-induced neurotoxicity, which can be inhibited by overexpression of Parkin or DJ-1 (Haque et al., 2012).
Endoplasmic Reticulum Stress
Parkinson disease is a disease classified as protein-misfolding disorders. Endoplasmic reticulum stress is observed in the SNc of PD patient brain and results in the death of dopaminergic neurons (Selvaraj et al., 2012). Increasing experimental evidence have also indicated that the accumulation of misfolded or unfolded proteins results in endoplasmic reticulum stress, which contributes to the neuronal death or apoptosis (Ryu et al., 2002; Bai et al., 2007; Colla et al., 2012; Fouillet et al., 2012; Valdes et al., 2014). In the central nervous system, the unfolded protein response(UPR) is initiated by the activation of three transmembrane proteins: (1) PRKR-like ER kinase (PERK), (2) inositol-requiring enzyme 1α (IRE1α) and (3) activating transcription factor 6 (ATF6) (Kaufman, 2002; Schroder and Kaufman, 2005). Under stressed conditions, the three transmembrane proteins dissociate from glucose regulated protein 78(GRP78) and activate series of intracellular signals to alleviate the burden of endoplasmic reticulum, such as inhibiting transcription and translation to reduce the amount of proteins entering the endoplasmic reticulum, increasing the levels of molecular chaperones to enhance the folding capacity of endoplasmic reticulum (Walter and Ron, 2011).
Mild endoplasmic reticulum stress is considered to have a neuroprotective role. However, severe endoplasmic reticulum stress usually leads to apoptosis or death of neurons. IRE1α/XBP1 is the most conserved UPR signaling pathway and a key regulator of UPR. IRE1α/XBP1 signal pathway of UPR is activated in SNCA triplication iPSC neurons, resulting in culminating neurodegeneration (Heman-Ackah et al., 2017). Conditional knockout of XBP1 in the nervous system inhibits the death of dopaminergic neurons induced by stereotaxic injections of 6-OHDA (Valdes et al., 2014). Our previous study also demonstrates that MPP+/MPTP activates IRE1α pathway, leading to the loss of dopaminergic neurons in vitro and in vivo; Thioredoxin-1 exerts a neuroprotective effect via inhibiting the activation of IRE1α (Zeng et al., 2014). Overexpression of human α-synuclein activates endoplasmic reticulum stress via activation of PERK and ATF6 signaling pathways as well as the upregulation of proaoptotic CHOP in dopaminergic neurons in SNc, which is inhibited by overexpression of glucose regulated protein 78 (Gorbatyuk et al., 2012). Rifampicin protects PC12 cells against the cytotoxicity of PD neurotoxin rotenone by upregulating GRP78 expression via PERK/eIF2α/ATF4 pathway (Jing et al., 2014). Endoplasmic reticulum stress signaling is enhanced in PINK1 and Parkin mutants model of PD, and genetic or pharmacological inhibition of PERK is protective (Celardo et al., 2016). The N370S mutation of GBA1 gene produces a significant reduction in GCase 1 protein and enzyme activity, and retention within the endoplasmic reticulum, which interrupted its traffic to the lysosome and further led to endoplasmic reticulum stress activation and triggered UPR and Golgi apparatus fragmentation(Garcia-Sanz et al., 2017). Sustained activation of ATF4 does not repress human alpha-synuclein overexpression-induced neurodegeneration and induces severe apoptosis of dopaminergic neurons in a rat model of PD (Gully et al., 2016). Recently,Coppola-Segovia et al. (2017) have presented a new model of PD, intranigral injection of Tunicamycin, in which the endoplasmic reticulum stressor recapitulates some of the phenotypic characteristics observed in rodent models of PD (such as locomotor impairment, α-synuclein oligomerization, and loss of dopaminergic neurons), reinforcing that endoplasmic reticulum stress could be an important contributor to the development of PD (Coppola-Segovia et al., 2017).
Impaired Autophagy
Autophagy plays a critical role in the clearance of accumulated misfolded proteins and degradation of damaged organelles. Resembling endoplasmic reticulum stress, the initial autophagy is also widely considered to be cyto-protective. Overexpression of autophagy-related gene 5 inhibits the apoptosis of dopaminergic neurons in MPTP-induced zebrafish model of PD (Hu et al., 2017). It can be achieved to inhibit the accumulation of α-synuclein by activating mTOR-dependent macroautophagy (Sheng et al., 2017). DJ-1 reduced the accumulation and aggregation of α-synuclein via chaperone-mediated autophagy in both SH-SY5Y cells and PD animal models (Xu et al., 2017). Interaction between PINK-1 and α-synuclein in the cytoplasm, which is dependent on the kinase activity of PINK1 and is abolished by point mutation of G309D in its kinase domain, enhanced the degradation of toxic α-synuclein via activation of autophagy (Liu et al., 2017). Endoplasmic reticulum stressors, including thapsigargin, tunicamycin, 2-mercaptoethanol and Brefeldin A, activate PERK and then recruit MKK4 to lysosomes, accompanied by the activation of p38 MAPK which directly phosphorylates the receptor LAMP2A at sites T211 and T213, and further triggers chaperone-mediated autophagy to provide a pro-survival mechanism (Li et al., 2017).
However, autophagy is usually defective or inhibited in the pathology of PD. Impairment of autophagy has been implicated as a causative mechanism in the development of PD (Cai et al., 2016). The autophagy levels in dopaminergic brain areas of α-synuclein mutant A53T transgenic mice are reduced when compared to wide-type mice (Pupyshev et al., 2017). Inhibition of autophagy boosts α-synuclein release and transfer from cell to cell via extracellular vesicles, and consequent progression of synucleinopathies (Minakaki et al., 2017). Cuervo et al. (2004) have demonstrated that pathogenic α-synuclein mutations, such as A53T and A30P, could bind to the receptors on the lysosomal membrane and inhibit chaperone-mediated autophagy, resulting in accumulation of abnormal α-synuclein in the Lewy bodies and subsequent parkinsonism. Inhibition of chaperone-mediated autophagy results in progressive loss of dopaminergic neurons in SNc, severe decrease of dopamine levels in striatum, and motor deficits (Xilouri et al., 2016). The mitochondria of neurons, suffering from oxidative stress and dysfunction, can’t be removed by impaired mitophagy in PD, leading to the accumulation of dysfunctional mitochondria (Gao et al., 2017). The formation of S-Nitrosylated PINK1 reduces the translocation of Parkin to mitochondrial membranes and inhibits mitophagy in human iPSC-derived cells as well as in α-synuclein transgenic PD mice, which contributes to neuronal cell death (Oh et al., 2017). GBA1 mutation produces an accumulation of cholesterol, which alters autophagy-lysosome function and impairs autophagy, rendering the neurons more vulnerable and sensitive to apoptosis(Garcia-Sanz et al., 2017, 2018). Elevated RTP801, product of DNA damage inducible transcript 4, represses autophagy, then increases the accumulation of oligomeric α-synuclein and further aggravates ER stress-induced neuronal apoptosis (Zhang et al., 2017).
Deregulation of Immunity
Increasing evidence indicates that the immune system plays a role in the development of PD. Several previous studies have demonstrated that pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6 and IFN-γ, are upregulated in both cerebrospinal fluid and post-mortem brain of PD patients (Mogi et al., 1994a,b; Blum-Degen et al., 1995; Brodacki et al., 2008). Massive studies have demonstrated that microglia are activated in neurotoxins-induced PD models, including MPTP, 6-OHDA and rotenone (Akiyama and McGeer, 1989; McGeer et al., 2003; Sherer et al., 2003).
There is an intimate relation between α-synuclein and immune system (Bandres-Ciga and Cookson, 2017). In a rat α-synuclein overexpressing model of PD, microglia are also activated in the striatum, accompanying the upregulation of TNF-α, IL-1β, and IFN-γ (Chung et al., 2009). Lack of IFN-β function shows an increase of α-synuclein-containing Lewy bodies, a reduction of dopaminergic neurons and impairment of dopamine signaling in SNc. In contrast, overexpression of IFN-β inhibits the loss of dopaminergic neuron in a model of familial PD (Ejlerskov et al., 2015). A recent study has showed that T cells from PD patients recognize α-synuclein peptides as antigenic epitopes which may explain the association of PD with specific major histocompatibility complex alleles (Sulzer et al., 2017). Major histocompatibility complex I is expressed in human SNc neurons and is inducible in human stem cell-derived dopaminergic neurons. Dopaminergic neurons internalize foreign ovalbumin and display antigen derived from this protein by major histocompatibility complex I to activate T cells, resulting in autoimmune responses and dopaminergic neurons death (Cebrian et al., 2014). α-synuclein induces the entry of pro-inflammatory peripheral CCR2+ monocytes into SNc, promotes the expression of major histocompatibility complex II and the subsequent degeneration of dopaminergic neurons (Harms et al., 2018). Genetic allelic variants of Mhc2ta, the major regulator of major histocompatibility complex II expression, regulates α-synuclein-induced microglial activation and the neurodegeneration of dopaminergic neurons (Jimenez-Ferrer et al., 2017).
Other PD-linked gene mutations also exhibit an association with immune system. Immune system and endocytosis are involved in the pathology of sporadic PD forms and LRRK2 G2019S carrying patients (Mutez et al., 2014). LRRK2 G2019S could alter the differentiation of bone marrow myelopoiesis and immune homeostasis (Park et al., 2017). PINK1 deficiency serves as an early modulator of innate immunity in neurons(Torres-Odio et al., 2017), because PINK1/Parkin could suppress inflammatory conditions triggered immune-response-eliciting pathway by inhibiting mitochondrial antigen presentation (Matheoud et al., 2016). At last, we may draw a conclusion that PD is an autoimmune disorder and deregulation of immunity is responsible for parkinsonism.
Conclusion and Expectation
In summary, pathology of PD is compositive and due to the combined action of heredity and environmental factors. Mutations of single-gene or multi-genes could aggravate the effects of environmental neurotoxins. The pathogenesis of mutations linked to PD are associated with dysfunctions of mitochondria and endoplasmic reticulum, impairment of autophagy, and deregulation of immunity. Conversely, environmental neurotoxins could also deregulate the expression of PD-linked genes.
Although PD is widely researched and the therapy is positively explored, it is still incurable and persecutes tens of million sufferers worldwide. Many scientists are unremittingly searching for potential treating approaches. Mao et al. (2016) have demonstrated that lymphocyte-activation gene 3 interact with misfolded preformed fibrils of α-synuclein, initiates its neuron-to-neuron transmission and then induces loss of dopaminergic neurons (Mao et al., 2016). Using an unbiased screen targeting endogenous gene expression, Mittal et al. (2017) have discovered that the β2-adrenoreceptor (β2AR) is a regulator of the α-synuclein gene. Salbutamol, a β2AR agonist, reduced the risk of developing PD. On the other hand, a β2AR antagonist correlated with increased risk. What’s more, activation of β2AR protected PD patient-derived cells and model mice (Mittal et al., 2017). Gut bacteria are required for motor deficits, microglia activation, and α-synuclein pathology, suggesting that alterations in the human microbiome may increase the risk for PD (Sampson et al., 2016). Targeting these aspects may provide effective and safe treatments to halt or slow the progression of PD.
Shields et al. (2017) have developed a DART (drugs acutely restricted by tethering), which antagonizes the a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor (AMPAR, a postsynaptic glutamate receptor), and applied the technique to a mouse model of PD. Human induced pluripotent stem cells-derived dopaminergic progenitor cells could function as midbrain dopaminergic neurons after transplantation in a monkey model of PD (Kikuchi et al., 2017). Using three transcription factors (NEUROD1, LMX1A, and ASCL1), miR218 and collectively designated NeAL218, dopamine neurons are generated by directly reprogramming human astrocytes in vitro and mouse astrocytes in vivo (Rivetti di Val Cervo et al., 2017). Ambroxol, which was initially used to treat airway mucus hypersecretion and hyaline membrane disease in infants and was then recognized as a pH-dependent, mixed-type inhibitor of GCase1, acts as a chaperone for GCase1 and enhances lysosomal function and autophagy. Clinical trials of ambroxol are currently ongoing in PD and PD dementia (Balestrino and Schapira, 2018). These studies might also provide new and alternative strategies to treat PD.
Author Contributions
X-SZ and J-JJ designed the article contents and wrote the original paper. X-SZ, W-SG, J-JJ, LC, and P-PZ searched the related literature and discussed the writing. X-SZ, W-SG, and J-JJ revised the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (31600837, 81502312, 31601167), Henan Science and Technology Project (172102310622), Key Scientific Research Projects of University in Henan Province (16A180017, 16A180018), the Ph.D. Early Development Program, and Nanhu Scholars Program for Young Scholars of XYNU.
References
Ablat, N., Lv, D., Ren, R., Xiaokaiti, Y., Ma, X., Zhao, X., et al. (2016). Neuroprotective effects of a standardized flavonoid extract from safflower against a rotenone-induced rat model of Parkinson’s disease. Molecules 21:E1107. doi: 10.3390/molecules21091107
Akiyama, H., and McGeer, P. L. (1989). Microglial response to 6-hydroxydopamine-induced substantia nigra lesions. Brain Res. 489, 247–253. doi: 10.1016/0006-8993(89)90857-3
Alarcon-Aris, D., Recasens, A., Galofre, M., Carballo-Carbajal, I., Zacchi, N., Ruiz-Bronchal, E., et al. (2017). Selective alpha-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson’s disease. Mol. Ther. 26, 550–567. doi: 10.1016/j.ymthe.2017.11.015
Ando, M., Fiesel, F. C., Hudec, R., Caulfield, T. R., Ogaki, K., Gorka-Skoczylas, P., et al. (2017). The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 12:32. doi: 10.1186/s13024-017-0174-z
Bae, E. J., Yang, N. Y., Lee, C., Lee, H. J., Kim, S., Sardi, S. P., et al. (2015). Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp. Mol. Med. 47:e153. doi: 10.1038/emm.2014.128
Bai, J., Nakamura, H., Kwon, Y. W., Tanito, M., Ueda, S., Tanaka, T., et al. (2007). Does thioredoxin-1 prevent mitochondria- and endoplasmic reticulum-mediated neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine? Antioxid. Redox Signal. 9, 603–608. doi: 10.1089/ars.2006.1513
Balestrino, R., and Schapira, A. H. V. (2018). Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neuroscientist doi: 10.1177/1073858417748875 [Epub ahead of print].
Bandres-Ciga, S., and Cookson, M. R. (2017). Alpha-synuclein triggers T-cell response. Is Parkinson’s disease an autoimmune disorder?. Mov. Disord. 32:1327. doi: 10.1002/mds.27116
Batelli, S., Invernizzi, R. W., Negro, A., Calcagno, E., Rodilossi, S., Forloni, G., et al. (2015). The Parkinson’s disease-related protein DJ-1 protects dopaminergic neurons in vivo and cultured cells from alpha-synuclein and 6-hydroxydopamine toxicity. Neurodegener. Dis. 15, 13–23. doi: 10.1159/000367993
Bekris, L. M., Mata, I. F., and Zabetian, C. P. (2010). The genetics of Parkinson disease. J. Geriatr. Psychiatry Neurol. 23, 228–242. doi: 10.1177/0891988710383572
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Bido, S., Soria, F. N., Fan, R. Z., Bezard, E., and Tieu, K. (2017). Mitochondrial division inhibitor-1 is neuroprotective in the A53T-alpha-synuclein rat model of Parkinson’s disease. Sci. Rep. 7:7495. doi: 10.1038/s41598-017-07181-0
Biosa, A., Sandrelli, F., Beltramini, M., Greggio, E., Bubacco, L., and Bisaglia, M. (2017). Recent findings on the physiological function of DJ-1: beyond Parkinson’s disease. Neurobiol. Dis. 108, 65–72. doi: 10.1016/j.nbd.2017.08.005
Blum-Degen, D., Muller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20. doi: 10.1016/0304-3940(95)12192-7
Bonifacino, J. S., and Hurley, J. H. (2008). Retromer. Curr. Opin. Cell Biol. 20, 427–436. doi: 10.1016/j.ceb.2008.03.009
Bonifati, V., Rizzu, P., Squitieri, F., Krieger, E., Vanacore, N., van Swieten, J. C., et al. (2003). DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 24, 159–160. doi: 10.1007/s10072-003-0108-0
Brito-Armas, J. M., Baekelandt, V., Castro-Hernandez, J. R., Gonzalez-Hernandez, T., Rodriguez, M., and Castro, R. (2013). Melatonin prevents dopaminergic cell loss induced by lentiviral vectors expressing A30P mutant alpha-synuclein. Histol. Histopathol. 28, 999–1006. doi: 10.14670/HH-28.999
Brodacki, B., Staszewski, J., Toczylowska, B., Kozlowska, E., Drela, N., Chalimoniuk, M., et al. (2008). Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 441, 158–162. doi: 10.1016/j.neulet.2008.06.040
Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261. doi: 10.1126/science.aam9080
Cai, Y., Arikkath, J., Yang, L., Guo, M. L., Periyasamy, P., and Buch, S. (2016). Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12, 225–244. doi: 10.1080/15548627.2015.1121360
Cannon, J. R., Geghman, K. D., Tapias, V., Sew, T., Dail, M. K., Li, C., et al. (2013). Expression of human E46K-mutated alpha-synuclein in BAC-transgenic rats replicates early-stage Parkinson’s disease features and enhances vulnerability to mitochondrial impairment. Exp. Neurol. 240, 44–56. doi: 10.1016/j.expneurol.2012.11.007
Cebrian, C., Zucca, F. A., Mauri, P., Steinbeck, J. A., Studer, L., Scherzer, C. R., et al. (2014). MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 5:3633. doi: 10.1038/ncomms4633
Celardo, I., Costa, A. C., Lehmann, S., Jones, C., Wood, N., Mencacci, N. E., et al. (2016). Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 23:e2271. doi: 10.1038/cddis.2016.173
Chen, Y. F., Chang, Y. Y., Lan, M. Y., Chen, P. L., and Lin, C. H. (2017). Identification of VPS35 p.D620N mutation-related Parkinson’s disease in a Taiwanese family with successful bilateral subthalamic nucleus deep brain stimulation: a case report and literature review. BMC Neurol. 17:191. doi: 10.1186/s12883-017-0972-5
Choi, W. S., Palmiter, R. D., and Xia, Z. (2011). Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 192, 873–882. doi: 10.1083/jcb.201009132
Chung, C. Y., Koprich, J. B., Siddiqi, H., and Isacson, O. (2009). Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 29, 3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009
Cleeter, M. W., Chau, K. Y., Gluck, C., Mehta, A., Hughes, D. A., Duchen, M., et al. (2013). Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 62, 1–7. doi: 10.1016/j.neuint.2012.10.010
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012). Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
Connolly, N. M. C., Theurey, P., Adam-Vizi, V., Bazan, N. G., Bernardi, P., Bolanos, J. P., et al. (2017). Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 25, 542–572. doi: 10.1038/s41418-017-0020-4
Cooper, J. F., Machiela, E., Dues, D. J., Spielbauer, K. K., Senchuk, M. M., and Van Raamsdonk, J. M. (2017). Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 7:16441. doi: 10.1038/s41598-017-16637-2
Cooper, J. M., Wiklander, P. B., Nordin, J. Z., Al-Shawi, R., Wood, M. J., Vithlani, M., et al. (2014). Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 29, 1476–1485. doi: 10.1002/mds.25978
Coppola-Segovia, V., Cavarsan, C., Maia, F. G., Ferraz, A. C., Nakao, L. S., Lima, M. M., et al. (2017). ER stress induced by tunicamycin triggers alpha-synuclein oligomerization, dopaminergic neurons death and locomotor impairment: a new model of Parkinson’s disease. Mol. Neurobiol. 54, 5798–5806. doi: 10.1007/s12035-016-0114-x
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Dabbeni-Sala, F., Di Santo, S., Franceschini, D., Skaper, S. D., and Giusti, P. (2001). Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity. FASEB J. 15, 164–170. doi: 10.1096/fj.00-0129com
Deeg, S., Gralle, M., Sroka, K., Bahr, M., Wouters, F. S., and Kermer, P. (2010). BAG1 restores formation of functional DJ-1 L166P dimers and DJ-1 chaperone activity. J. Cell Biol. 188, 505–513. doi: 10.1083/jcb.200904103
Dirkx, M. F., den Ouden, H. E., Aarts, E., Timmer, M. H., Bloem, B. R., Toni, I., et al. (2017). Dopamine controls Parkinson’s tremor by inhibiting the cerebellar thalamus. Brain 140, 721–734. doi: 10.1093/brain/aww331
Ejlerskov, P., Hultberg, J. G., Wang, J., Carlsson, R., Ambjorn, M., Kuss, M., et al. (2015). Lack of neuronal IFN-beta-IFNAR causes lewy body- and Parkinson’s disease-like dementia. Cell. 163, 324–339. doi: 10.1016/j.cell.2015.08.069
Fang, F., Yang, W., Florio, J. B., Rockenstein, E., Spencer, B., Orain, X. M., et al. (2017). Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Sci. Rep. 7:3868. doi: 10.1038/s41598-017-04232-4
Farrer, M., Kachergus, J., Forno, L., Lincoln, S., Wang, D. S., Hulihan, M., et al. (2004). Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 55, 174–179. doi: 10.1002/ana.10846
Feng, C. W., Hung, H. C., Huang, S. Y., Chen, C. H., Chen, Y. R., Chen, C. Y., et al. (2016). Neuroprotective effect of the marine-derived compound 11-Dehydrosinulariolide through DJ-1-related pathway in in vitro and in vivo models of Parkinson’s disease. Mar. Drugs 14:E187. doi: 10.3390/md14100187
Ferreira, M., and Massano, J. (2017). An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 135, 273–284. doi: 10.1111/ane.12616
Follett, J., Bugarcic, A., Yang, Z., Ariotti, N., Norwood, S. J., Collins, B. M., et al. (2016). Parkinson disease-linked Vps35 R524W mutation impairs the endosomal association of retromer and induces α-synuclein aggregation. J. Biol. Chem. 291, 18283–18298. doi: 10.1074/jbc.M115.703157
Fouillet, A., Levet, C., Virgone, A., Robin, M., Dourlen, P., Rieusset, J., et al. (2012). ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. doi: 10.4161/auto.19716
Gandhi, S., Vaarmann, A., Yao, Z., Duchen, M. R., Wood, N. W., and Abramov, A. Y. (2012). Dopamine induced neurodegeneration in a PINK1 model of Parkinson’s disease. PLoS One 7:e37564. doi: 10.1371/journal.pone.0037564
Gan-Or, Z., Dion, P. A., and Rouleau, G. A. (2015). Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 11, 1443–1457. doi: 10.1080/15548627.2015.1067364
Gao, F., Yang, J., Wang, D., Li, C., Fu, Y., Wang, H., et al. (2017). Mitophagy in Parkinson’s disease: pathogenic and therapeutic implications. Front. Neurol. 8:527. doi: 10.3389/fneur.2017.00527
Garcia-Sanz, P., Orgaz, L., Bueno-Gil, G., Espadas, I., Rodriguez-Traver, E., Kulisevsky, J., et al. (2017). N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 32, 1409–1422. doi: 10.1002/mds.27119
Garcia-Sanz, P., Orgaz, L., Fuentes, J. M., Vicario, C., and Moratalla, R. (2018). Cholesterol and multilamellar bodies: lyosomal dysfunction in GBA-Parkinson disease. Autophagy 25, 1–7. doi: 10.1080/15548627.2018.1427396
Giaime, E., Sunyach, C., Druon, C., Scarzello, S., Robert, G., Grosso, S., et al. (2010). Loss of function of DJ-1 triggered by Parkinson’s disease-associated mutation is due to proteolytic resistance to caspase-6. Cell Death Differ. 17, 158–169. doi: 10.1038/cdd.2009.116
Goldstein, D. S., Kopin, I. J., and Sharabi, Y. (2014). Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol. Ther. 144, 268–282. doi: 10.1016/j.pharmthera.2014.06.006
Goldstein, D. S., Sullivan, P., Cooney, A., Jinsmaa, Y., Sullivan, R., Gross, D. J., et al. (2012). Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 cells: relevance to the pathogenesis of Parkinson’s disease. J. Neurochem. 123, 932–943. doi: 10.1111/j.1471-4159.2012.07924.x
Gorbatyuk, M. S., Shabashvili, A., Chen, W., Meyers, C., Sullivan, L. F., Salganik, M., et al. (2012). Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 20, 1327–1337. doi: 10.1038/mt.2012.28
Grunewald, A., Rygiel, K. A., Hepplewhite, P. D., Morris, C. M., Picard, M., and Turnbull, D. M. (2016). Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann. Neurol. 79, 366–378. doi: 10.1002/ana.24571
Gully, J. C., Sergeyev, V. G., Bhootada, Y., Mendez-Gomez, H., Meyers, C. A., Zolotukhin, S., et al. (2016). Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 627, 36–41. doi: 10.1016/j.neulet.2016.05.039
Han, K. A., Shin, W. H., Jung, S., Seol, W., Seo, H., Ko, C., et al. (2017). Leucine-rich repeat kinase 2 exacerbates neuronal cytotoxicity through phosphorylation of histone deacetylase 3 and histone deacetylation. Hum. Mol. Genet. 26, 1–18. doi: 10.1093/hmg/ddw363
Haque, M. E., Mount, M. P., Safarpour, F., Abdel-Messih, E., Callaghan, S., Mazerolle, C., et al. (2012). Inactivation of Pink1 gene in vivo sensitizes dopamine-producing neurons to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson disease genes, Parkin or DJ-1. J. Biol. Chem. 287, 23162–23170. doi: 10.1074/jbc.M112.346437
Harms, A. S., Thome, A. D., Yan, Z., Schonhoff, A. M., Williams, G. P., Li, X., et al. (2018). Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp. Neurol. 300, 179–187. doi: 10.1016/j.expneurol.2017.11.010
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. doi: 10.1016/S1474-4422(08)70117-0
Heman-Ackah, S. M., Manzano, R., Hoozemans, J. J. M., Scheper, W., Flynn, R., Haerty, W., et al. (2017). Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 26, 4441–4450. doi: 10.1093/hmg/ddx331
Herrera, A., Munoz, P., Steinbusch, H. W. M., and Segura-Aguilar, J. (2017). Are dopamine oxidation metabolites involved in the loss of dopaminergic neurons in the nigrostriatal system in Parkinson’s disease? ACS Chem. Neurosci. 8, 702–711. doi: 10.1021/acschemneuro.7b00034
Ho, D. H., Jang, J., Joe, E. H., Son, I., Seo, H., and Seol, W. (2016). G2385R and I2020T mutations increase LRRK2 GTPase activity. Biomed. Res. Int. 2016:7917128. doi: 10.1155/2016/7917128
Howlett, E. H., Jensen, N., Belmonte, F., Zafar, F., Hu, X., Kluss, J., et al. (2017). LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 26, 4340–4351. doi: 10.1093/hmg/ddx320
Hu, Z. Y., Chen, B., Zhang, J. P., and Ma, Y. Y. (2017). Up-regulation of autophagy-related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson’s disease. J. Biol. Chem. 292, 18062–18074. doi: 10.1074/jbc.M116.764795
Huang, E., Qu, D., Huang, T., Rizzi, N., Boonying, W., Krolak, D., et al. (2017). PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 8:1399. doi: 10.1038/s41467-017-01435-1
Ibanez, L., Dube, U., Saef, B., Budde, J., Black, K., Medvedeva, A., et al. (2017). Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha-synuclein cerebrospinal fluid levels. BMC Neurol. 17:198. doi: 10.1186/s12883-017-0978-z
Im, J. Y., Lee, K. W., Woo, J. M., Junn, E., and Mouradian, M. M. (2012). DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 21, 3013–3024. doi: 10.1093/hmg/dds131
Ishikawa, S., Taira, T., Niki, T., Takahashi-Niki, K., Maita, C., Maita, H., et al. (2009). Oxidative status of DJ-1-dependent activation of dopamine synthesis through interaction of tyrosine hydroxylase and 4-dihydroxy-L-phenylalanine (L-DOPA) decarboxylase with DJ-1. J. Biol. Chem. 284, 28832–28844. doi: 10.1074/jbc.M109.019950
Jiang, M., Porat-Shliom, Y., Pei, Z., Cheng, Y., Xiang, L., Sommers, K., et al. (2010). Baicalein reduces E46K alpha-synuclein aggregation in vitro and protects cells against E46K alpha-synuclein toxicity in cell models of familiar Parkinsonism. J. Neurochem. 114, 419–429. doi: 10.1111/j.1471-4159.2010.06752.x
Jimenez-Ferrer, I., Jewett, M., Tontanahal, A., Romero-Ramos, M., and Swanberg, M. (2017). Allelic difference in Mhc2ta confers altered microglial activation and susceptibility to alpha-synuclein-induced dopaminergic neurodegeneration. Neurobiol. Dis. 106, 279–290. doi: 10.1016/j.nbd.2017.07.016
Jing, X., Shi, Q., Bi, W., Zeng, Z., Liang, Y., Wu, X., et al. (2014). Rifampicin protects PC12 cells from rotenone-induced cytotoxicity by activating GRP78 via PERK-eIF2alpha-ATF4 pathway. PLoS One 9:e92110. doi: 10.1371/journal.pone.0092110
Ju, C., Gao, J., Hou, L., Wang, L., Zhang, F., Sun, F., et al. (2017). Neuroprotective effect of chondroitin sulfate on SHSY5Y cells overexpressing wild-type or A53T mutant alphasynuclein. Mol. Med. Rep. 16, 8721–8728. doi: 10.3892/mmr.2017.7725
Kang, S. S., Zhang, Z., Liu, X., Manfredsson, F. P., Benskey, M. J., Cao, X., et al. (2017). TrkB neurotrophic activities are blocked by alpha-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, 10773–10778. doi: 10.1073/pnas.1713969114
Karampetsou, M., Ardah, M. T., Semitekolou, M., Polissidis, A., Samiotaki, M., Kalomoiri, M., et al. (2017). Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 7:16533. doi: 10.1038/s41598-017-15813-8
Karuppagounder, S. S., Xiong, Y., Lee, Y., Lawless, M. C., Kim, D., Nordquist, E., et al. (2016). LRRK2 G2019S transgenic mice display increased susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated neurotoxicity. J. Chem. Neuroanat. 76, 90–97. doi: 10.1016/j.jchemneu.2016.01.007
Kaufman, R. J. (2002). Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 110, 1389–1398. doi: 10.1172/JCI0216886
Khaldy, H., Escames, G., Leon, J., Bikjdaouene, L., and Acuna-Castroviejo, D. (2003). Synergistic effects of melatonin and deprenyl against MPTP-induced mitochondrial damage and DA depletion. Neurobiol. Aging 24, 491–500. doi: 10.1016/S0197-4580(02)00133-1
Kikuchi, T., Morizane, A., Doi, D., Magotani, H., Onoe, H., Hayashi, T., et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548, 592–596. doi: 10.1038/nature23664
Kilpatrick, B. S., Magalhaes, J., Beavan, M. S., McNeill, A., Gegg, M. E., Cleeter, M. W., et al. (2016). Endoplasmic reticulum and lysosomal Ca(2)(+) stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium 59, 12–20. doi: 10.1016/j.ceca.2015.11.002
Kim, B., Yang, M. S., Choi, D., Kim, J. H., Kim, H. S., Seol, W., et al. (2012). Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One 7:e34693. doi: 10.1371/journal.pone.0034693
Kim, S., Yun, S. P., Lee, S., Umanah, G. E., Bandaru, V. V. R., Yin, X., et al. (2018). GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. U.S.A. 115, 798–803. doi: 10.1073/pnas.1700465115
Klein, R. L., King, M. A., Hamby, M. E., and Meyer, E. M. (2002). Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum. Gene Ther. 13, 605–612. doi: 10.1089/10430340252837206
Kumar, A., Tamjar, J., Waddell, A. D., Woodroof, H. I., Raimi, O. G., Shaw, A. M., et al. (2017). Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife 6:e29985. doi: 10.7554/eLife.29985
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
Lee, S., Oh, S. T., Jeong, H. J., Pak, S. C., Park, H. J., Kim, J., et al. (2017). MPTP-induced vulnerability of dopamine neurons in A53T alpha-synuclein overexpressed mice with the potential involvement of DJ-1 downregulation. Korean J. Physiol. Pharmacol. 21, 625–632. doi: 10.4196/kjpp.2017.21.6.625
Lewis, J., Melrose, H., Bumcrot, D., Hope, A., Zehr, C., Lincoln, S., et al. (2008). In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 3:19. doi: 10.1186/1750-1326-3-19
Li, W., Zhu, J., Dou, J., She, H., Tao, K., Xu, H., et al. (2017). Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 8:1763. doi: 10.1038/s41467-017-01609-x
Lieberman, O. J., Choi, S. J., Kanter, E., Saverchenko, A., Frier, M. D., Fiore, G. M., et al. (2017). alpha-synuclein-dependent calcium entry underlies differential sensitivity of cultured SN and VTA dopaminergic neurons to a Parkinsonian neurotoxin. eNeuro 4:ENEURO.0167-17.2017. doi: 10.1523/ENEURO.0167-17.2017
Liu, J., Wang, X., Lu, Y., Duan, C., Gao, G., Lu, L., et al. (2017). Pink1 interacts with alpha-synuclein and abrogates alpha-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 8:e3056. doi: 10.1038/cddis.2017.427
Liu, S., Cui, B., Dai, Z. X., Shi, P. K., Wang, Z. H., and Guo, Y. Y. (2016). Long Non-coding RNA HOTAIR promotes Parkinson’s disease induced by MPTP through up-regulating the expression of LRRK2. Curr. Neurovasc. Res. 13, 115–120. doi: 10.2174/1567202613666160316155228
Liu, Z., Yu, Y., Li, X., Ross, C. A., and Smith, W. W. (2011). Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol. Res. 63, 439–444. doi: 10.1016/j.phrs.2011.01.004
Luth, E. S., Stavrovskaya, I. G., Bartels, T., Kristal, B. S., and Selkoe, D. J. (2014). Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 289, 21490–21507. doi: 10.1074/jbc.M113.545749
Lutz, A. K., Exner, N., Fett, M. E., Schlehe, J. S., Kloos, K., Lammermann, K., et al. (2009). Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951. doi: 10.1074/jbc.M109.035774
Magalhaes, J., Gegg, M. E., Migdalska-Richards, A., Doherty, M. K., Whitfield, P. D., and Schapira, A. H. (2016). Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum. Mol. Genet. 25, 3432–3445. doi: 10.1093/hmg/ddw185
Mao, X., Ou, M. T., Karuppagounder, S. S., Kam, T. I., Yin, X., Xiong, Y., et al. (2016). Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. doi: 10.1126/science.aah3374
Martinat, C., Shendelman, S., Jonason, A., Leete, T., Beal, M. F., Yang, L., et al. (2004). Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES- derived cell model of primary Parkinsonism. PLoS Biol. 2:e327. doi: 10.1371/journal.pbio.0020327
Martinez, B. A., Petersen, D. A., Gaeta, A. L., Stanley, S. P., Caldwell, G. A., and Caldwell, K. A. (2017). Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 37, 11085–11100. doi: 10.1523/JNEUROSCI.1294-17.2017
Mason, R. J., Paskins, A. R., Dalton, C. F., and Smith, D. P. (2016). Copper binding and subsequent aggregation of alpha-synuclein are modulated by N-terminal acetylation and ablated by the H50Q missense mutation. Biochemistry 55, 4737–4741. doi: 10.1021/acs.biochem.6b00708
Matheoud, D., Sugiura, A., Bellemare-Pelletier, A., Laplante, A., Rondeau, C., Chemali, M., et al. (2016). Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 166, 314–327. doi: 10.1016/j.cell.2016.05.039
Matsumoto, J., Stewart, T., Sheng, L., Li, N., Bullock, K., Song, N., et al. (2017). Transmission of alpha-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: another mechanism for initiation and progression of Parkinson’s disease? Acta Neuropathol. Commun. 5:71. doi: 10.1186/s40478-017-0470-4
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016). Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
Mbefo, M. K., Fares, M. B., Paleologou, K., Oueslati, A., Yin, G., Tenreiro, S., et al. (2015). Parkinson disease mutant E46K enhances alpha-synuclein phosphorylation in mammalian cell lines, in yeast, and in vivo. J. Biol. Chem. 290, 9412–9427. doi: 10.1074/jbc.M114.610774
McGeer, P. L., Schwab, C., Parent, A., and Doudet, D. (2003). Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann. Neurol. 54, 599–604. doi: 10.1002/ana.10728
McNeill, A., Duran, R., Hughes, D. A., Mehta, A., and Schapira, A. H. (2012). A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 83, 853–854. doi: 10.1136/jnnp-2012-302402
Meiser, J., Delcambre, S., Wegner, A., Jager, C., Ghelfi, J., d’Herouel, A. F., et al. (2016). Loss of DJ-1 impairs antioxidant response by altered glutamine and serine metabolism. Neurobiol. Dis. 89, 112–125. doi: 10.1016/j.nbd.2016.01.019
Migdalska-Richards, A., Daly, L., Bezard, E., and Schapira, A. H. (2016). Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann. Neurol. 80, 766–775. doi: 10.1002/ana.24790
Migdalska-Richards, A., Wegrzynowicz, M., Rusconi, R., Deangeli, G., Di Monte, D. A., Spillantini, M. G., et al. (2017). The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 140, 2706–2721. doi: 10.1093/brain/awx221
Minakaki, G., Menges, S., Kittel, A., Emmanouilidou, E., Schaeffner, I., Barkovits, K., et al. (2017). Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14, 98–119. doi: 10.1080/15548627.2017.1395992
Mittal, S., Bjornevik, K., Im, D. S., Flierl, A., Dong, X., Locascio, J. J., et al. (2017). beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 357, 891–898. doi: 10.1126/science.aaf3934
Mogi, M., Harada, M., Kondo, T., Riederer, P., Inagaki, H., Minami, M., et al. (1994a). Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 180, 147–150.
Mogi, M., Harada, M., Riederer, P., Narabayashi, H., Fujita, K., and Nagatsu, T. (1994b). Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165, 208–210. doi: 10.1016/0304-3940(94)90746-3
Mor, D. E., Tsika, E., Mazzulli, J. R., Gould, N. S., Kim, H., Daniels, M. J., et al. (2017). Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 20, 1560–1568. doi: 10.1038/nn.4641
Mortiboys, H., Aasly, J., and Bandmann, O. (2013). Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain 136, 3038–3050. doi: 10.1093/brain/awt224
Mortiboys, H., Furmston, R., Bronstad, G., Aasly, J., Elliott, C., and Bandmann, O. (2015). UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology 85, 846–852. doi: 10.1212/WNL.0000000000001905
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Mukherjee, U. A., Ong, S. B., Ong, S. G., and Hausenloy, D. J. (2015). Parkinson’s disease proteins: novel mitochondrial targets for cardioprotection. Pharmacol. Ther. 156, 34–43. doi: 10.1016/j.pharmthera.2015.10.005
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848. doi: 10.1093/brain/awt367
Mutez, E., Nkiliza, A., Belarbi, K., de Broucker, A., Vanbesien-Mailliot, C., Bleuse, S., et al. (2014). Involvement of the immune system, endocytosis and EIF2 signaling in both genetically determined and sporadic forms of Parkinson’s disease. Neurobiol. Dis. 63, 165–170. doi: 10.1016/j.nbd.2013.11.007
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2009). Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5, 706–708. doi: 10.4161/auto.5.5.8505
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Nguyen, A. P. T., Daniel, G., Valdes, P., Islam, M. S., Schneider, B. L., and Moore, D. J. (2018). G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum. Mol. Genet. 27, 120–134. doi: 10.1093/hmg/ddx389
Oh, C. K., Sultan, A., Platzer, J., Dolatabadi, N., Soldner, F., McClatchy, D. B., et al. (2017). S-Nitrosylation of PINK1 attenuates PINK1/parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep. 21, 2171–2182. doi: 10.1016/j.celrep.2017.10.068
Pan, P. Y., Li, X., Wang, J., Powell, J., Wang, Q., Zhang, Y., et al. (2017). Parkinson’s disease-associated LRRK2 hyperactive kinase mutant disrupts synaptic vesicle trafficking in ventral midbrain neurons. J. Neurosci. 37, 11366–11376. doi: 10.1523/JNEUROSCI.0964-17.2017
Panneton, W. M., Kumar, V. B., Gan, Q., Burke, W. J., and Galvin, J. E. (2010). The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One 5:e15251. doi: 10.1371/journal.pone.0015251
Park, J., Lee, J. W., Cooper, S. C., Broxmeyer, H. E., Cannon, J. R., and Kim, C. H. (2017). Parkinson disease-associated LRRK2 G2019S transgene disrupts marrow myelopoiesis and peripheral Th17 response. J. Leukoc. Biol. 102, 1093–1102. doi: 10.1189/jlb.1A0417-147RR
Parnetti, L., Paciotti, S., Eusebi, P., Dardis, A., Zampieri, S., Chiasserini, D., et al. (2017). Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 32, 1423–1431. doi: 10.1002/mds.27136
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Porcari, R., Proukakis, C., Waudby, C. A., Bolognesi, B., Mangione, P. P., Paton, J. F., et al. (2015). The H50Q mutation induces a 10-fold decrease in the solubility of alpha-synuclein. J. Biol. Chem. 290, 2395–2404. doi: 10.1074/jbc.M114.610527
Pupyshev, A. B., Korolenko, T. A., Akopyan, A. A., Amstislavskaya, T. G., and Tikhonova, M. A. (2017). Suppression of autophagy in the brain of transgenic mice with overexpression of capital A, Cyrillic53capital TE, Cyrillic-mutant alpha-synuclein as an early event at synucleinopathy progression. Neurosci. Lett. 672, 140–142. doi: 10.1016/j.neulet.2017.12.001
Puschmann, A., Fiesel, F. C., Caulfield, T. R., Hudec, R., Ando, M., Truban, D., et al. (2017). Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 140, 98–117. doi: 10.1093/brain/aww261
Ramsey, C. P., and Giasson, B. I. (2010). L10p and P158DEL DJ-1 mutations cause protein instability, aggregation, and dimerization impairments. J. Neurosci. Res. 88, 3111–3124. doi: 10.1002/jnr.22477
Rappold, P. M., Cui, M., Chesser, A. S., Tibbett, J., Grima, J. C., Duan, L., et al. (2011). Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc. Natl. Acad. Sci. U.S.A. 108, 20766–20771. doi: 10.1073/pnas.1115141108
Rassu, M., Del Giudice, M. G., Sanna, S., Taymans, J. M., Morari, M., Brugnoli, A., et al. (2017). Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS One 12:e0179082. doi: 10.1371/journal.pone.0179082
Ren, H., Fu, K., Mu, C., Zhen, X., and Wang, G. (2012). L166P mutant DJ-1 promotes cell death by dissociating Bax from mitochondrial Bcl-XL. Mol. Neurodegener. 7:40. doi: 10.1186/1750-1326-7-40
Rivetti di Val Cervo, P., Romanov, R. A., Spigolon, G., Masini, D., Martin-Montanez, E., Toledo, E. M., et al. (2017). Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 35, 444–452. doi: 10.1038/nbt.3835
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hallett, P., et al. (2015). Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2, 433–438. doi: 10.1002/acn3.177
Rodriguez, J. A., Ivanova, M. I., Sawaya, M. R., Cascio, D., Reyes, F. E., Shi, D., et al. (2015). Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 525, 486–490. doi: 10.1038/nature15368
Ross, O. A., Braithwaite, A. T., Skipper, L. M., Kachergus, J., Hulihan, M. M., Middleton, F. A., et al. (2008). Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 63, 743–750. doi: 10.1002/ana.21380
Rutherford, N. J., Moore, B. D., Golde, T. E., and Giasson, B. I. (2014). Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J. Neurochem. 131, 859–867. doi: 10.1111/jnc.12806
Ryu, E. J., Harding, H. P., Angelastro, J. M., Vitolo, O. V., Ron, D., and Greene, L. A. (2002). Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci. 22, 10690–10698.
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e12. doi: 10.1016/j.cell.2016.11.018
Sanyal, J., Jana, A., Ghosh, E., Banerjee, T. K., Chakraborty, D. P., and Rao, V. R. (2015). Evaluation of PARKIN gene variants in West Bengal Parkinson’s disease patients. J. Hum. Genet. 60, 485–492. doi: 10.1038/jhg.2015.49
Sanz, F. J., Solana-Manrique, C., Munoz-Soriano, V., Calap-Quintana, P., Molto, M. D., and Paricio, N. (2017). Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic. Biol. Med. 108, 683–691. doi: 10.1016/j.freeradbiomed.2017.04.364
Sato, H., Arawaka, S., Hara, S., Fukushima, S., Koga, K., Koyama, S., et al. (2011). Authentically phosphorylated alpha-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 31, 16884–16894. doi: 10.1523/JNEUROSCI.3967-11.2011
Schapira, A. H. (2008). Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 7, 97–109. doi: 10.1016/S1474-4422(07)70327-7
Schneider, S. A., and Alcalay, R. N. (2017). Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov. Disord. 32, 1504–1523. doi: 10.1002/mds.27193
Schroder, M., and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789. doi: 10.1146/annurev.biochem.73.011303.074134
Selvaraj, S., Sun, Y., Watt, J. A., Wang, S., Lei, S., Birnbaumer, L., et al. (2012). Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 122, 1354–1367. doi: 10.1172/JCI61332
Sheng, Y. L., Chen, X., Hou, X. O., Yuan, X., Yuan, B. S., Yuan, Y. Q., et al. (2017). Urate promotes SNCA/alpha-synuclein clearance via regulating mTOR-dependent macroautophagy. Exp. Neurol. 297, 138–147. doi: 10.1016/j.expneurol.2017.08.007
Sherer, T. B., Betarbet, R., Kim, J. H., and Greenamyre, J. T. (2003). Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 341, 87–90. doi: 10.1016/S0304-3940(03)00172-1
Shields, B. C., Kahuno, E., Kim, C., Apostolides, P. F., Brown, J., Lindo, S., et al. (2017). Deconstructing behavioral neuropharmacology with cellular specificity. Science 356:aaj2161. doi: 10.1126/science.aaj2161
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., et al. (2005a). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811.
Smith, W. W., Pei, Z., Jiang, H., Moore, D. J., Liang, Y., West, A. B., et al. (2005b). Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 18676–18681.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Steger, M., Diez, F., Dhekne, H. S., Lis, P., Nirujogi, R. S., Karayel, O., et al. (2017). Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6:e31012. doi: 10.7554/eLife.31012
Sterky, F. H., Hoffman, A. F., Milenkovic, D., Bao, B., Paganelli, A., Edgar, D., et al. (2012). Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum. Mol. Genet. 21, 1078–1089. doi: 10.1093/hmg/ddr537
Storch, A., Ludolph, A. C., and Schwarz, J. (2004). Dopamine transporter: involvement in selective dopaminergic neurotoxicity and degeneration. J. Neural Transm. (Vienna). 111, 1267–1286. doi: 10.1007/s00702-004-0203-2
Su, P., and Liu, F. (2017). A peptide disrupting the D2R-DAT interaction protects against dopamine neurotoxicity. Exp. Neurol. 295, 176–183. doi: 10.1016/j.expneurol.2017.05.010
Su, Y. C., Guo, X., and Qi, X. (2015). Threonine 56 phosphorylation of Bcl-2 is required for LRRK2 G2019S-induced mitochondrial depolarization and autophagy. Biochim. Biophys. Acta 1852, 12–21. doi: 10.1016/j.bbadis.2014.11.009
Subramaniam, S. R., and Chesselet, M. F. (2013). Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 106-107, 17–32. doi: 10.1016/j.pneurobio.2013.04.004
Sulzer, D., Alcalay, R. N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., et al. (2017). T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546, 656–661. doi: 10.1038/nature22815
Taguchi, Y. V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., et al. (2017). Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 37, 9617–9631. doi: 10.1523/JNEUROSCI.1525-17.2017
Taipa, R., Pereira, C., Reis, I., Alonso, I., Bastos-Lima, A., Melo-Pires, M., et al. (2016). DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 139, 1680–1687. doi: 10.1093/brain/aww080
Tang, B., Xiong, H., Sun, P., Zhang, Y., Wang, D., Hu, Z., et al. (2006). Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson’s disease. Hum. Mol. Genet. 15, 1816–1825. doi: 10.1093/hmg/ddl104
Tang, F. L., Erion, J. R., Tian, Y., Liu, W., Yin, D. M., Ye, J., et al. (2015a). VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35, 10613–10628. doi: 10.1523/JNEUROSCI.0042-15.2015
Tang, F. L., Liu, W., Hu, J. X., Erion, J. R., Ye, J., Mei, L., et al. (2015b). VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 12, 1631–1643. doi: 10.1016/j.celrep.2015.08.001
Thomas, J. M., Li, T., Yang, W., Xue, F., Fishman, P. S., and Smith, W. W. (2016). 68 and FX2149 attenuate mutant LRRK2-R1441C-induced neural transport impairment. Front. Aging Neurosci. 8:337. doi: 10.3389/fnagi.2016.00337
Torres-Odio, S., Key, J., Hoepken, H. H., Canet-Pons, J., Valek, L., Roller, B., et al. (2017). Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflammation 14:154. doi: 10.1186/s12974-017-0928-0
Valdes, P., Mercado, G., Vidal, R. L., Molina, C., Parsons, G., Court, F. A., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. U.S.A. 111, 6804–6809. doi: 10.1073/pnas.1321845111
Vanle, B. C., Florang, V. R., Murry, D. J., Aguirre, A. L., and Doorn, J. A. (2017). Inactivation of glyceraldehyde-3-phosphate dehydrogenase by the dopamine metabolite, 3,4-dihydroxyphenylacetaldehyde. Biochem. Biophys. Res. Commun. 492, 275–281. doi: 10.1016/j.bbrc.2017.08.067
Verma, M., Callio, J., Otero, P. A., Sekler, I., Wills, Z. P., and Chu, C. T. (2017). Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 37, 11151–11165. doi: 10.1523/JNEUROSCI.3791-16.2017
Villar-Pique, A., Rossetti, G., Ventura, S., Carloni, P., Fernandez, C. O., and Outeiro, T. F. (2017). Copper(II) and the pathological H50Q alpha-synuclein mutant: environment meets genetics. Commun. Integr. Biol. 10:e1270484. doi: 10.1080/19420889.2016.1270484
Volpicelli-Daley, L. A., Abdelmotilib, H., Liu, Z., Stoyka, L., Daher, J. P., Milnerwood, A. J., et al. (2016). G2019S-LRRK2 expression ments alpha-synuclein sequestration into inclusions in neurons. J. Neurosci. 36, 7415–7427. doi: 10.1523/JNEUROSCI.3642-15.2016
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, H. S., Toh, J., Ho, P., Tio, M., Zhao, Y., and Tan, E. K. (2014). In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol. Brain 7:73. doi: 10.1186/s13041-014-0073-y
Wang, K., Huang, J., Xie, W., Huang, L., Zhong, C., and Chen, Z. (2016). Beclin1 and HMGB1 ameliorate the alpha-synuclein-mediated autophagy inhibition in PC12 cells. Diagn. Pathol. 11:15. doi: 10.1186/s13000-016-0459-5
Wang, W., Wang, X., Fujioka, H., Hoppel, C., Whone, A. L., Caldwell, M. A., et al. (2016). Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63. doi: 10.1038/nm.3983
Wang, X., Yan, M. H., Fujioka, H., Liu, J., Wilson-Delfosse, A., Chen, S. G., et al. (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 21, 1931–1944. doi: 10.1093/hmg/dds003
West, A. B. (2017). Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 298, 236–245. doi: 10.1016/j.expneurol.2017.07.019
Winner, B., Jappelli, R., Maji, S. K., Desplats, P. A., Boyer, L., Aigner, S., et al. (2011). In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199. doi: 10.1073/pnas.1100976108
Xiao, B., Goh, J. Y., Xiao, L., Xian, H., Lim, K. L., and Liou, Y. C. (2017). Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 292, 16697–16708. doi: 10.1074/jbc.M117.787739
Xilouri, M., Brekk, O. R., Polissidis, A., Chrysanthou-Piterou, M., Kloukina, I., and Stefanis, L. (2016). Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 12, 2230–2247. doi: 10.1080/15548627.2016.1214777
Xu, C. Y., Kang, W. Y., Chen, Y. M., Jiang, T. F., Zhang, J., Zhang, L. N., et al. (2017). DJ-1 inhibits alpha-synuclein aggregation by regulating chaperone-mediated autophagy. Front. Aging Neurosci. 9:308. doi: 10.3389/fnagi.2017.00308
Yan, J. Q., Yuan, Y. H., Gao, Y. N., Huang, J. Y., Ma, K. L., Gao, Y., et al. (2014). Overexpression of human E46K mutant alpha-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 50, 685–701. doi: 10.1007/s12035-014-8738-1
Yan, M. H., Wang, X., and Zhu, X. (2013). Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 62, 90–101. doi: 10.1016/j.freeradbiomed.2012.11.014
Yin, F., Boveris, A., and Cadenas, E. (2014). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. 20, 353–371. doi: 10.1089/ars.2012.4774
Yoon, J. H., Mo, J. S., Kim, M. Y., Ann, E. J., Ahn, J. S., Jo, E. H., et al. (2017). LRRK2 functions as a scaffolding kinase of ASK1-mediated neuronal cell death. Biochim. Biophys. Acta 1864, 2356–2368. doi: 10.1016/j.bbamcr.2017.09.001
Yun, S. P., Kim, H., Ham, S., Kwon, S. H., Lee, G. H., Shin, J. H., et al. (2017). VPS35 regulates parkin substrate AIMP2 toxicity by facilitating lysosomal clearance of AIMP2. Cell Death Dis. 8:e2741. doi: 10.1038/cddis.2017.157
Zeng, X. S., Jia, J. J., Kwon, Y., Wang, S. D., and Bai, J. (2014). The role of thioredoxin-1 in suppression of endoplasmic reticulum stress in Parkinson disease. Free Radic. Biol. Med. 67, 10–18. doi: 10.1016/j.freeradbiomed.2013.10.013
Zhang, S., Eitan, E., Wu, T. Y., and Mattson, M. P. (2018). Intercellular transfer of pathogenic alpha-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol. Aging 61, 52–65. doi: 10.1016/j.neurobiolaging.2017.09.016
Zhang, W., Dallas, S., Zhang, D., Guo, J. P., Pang, H., Wilson, B., et al. (2007). Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 55, 1178–1188. doi: 10.1002/glia.20532
Zhang, Y., Gong, X. G., Wang, Z. Z., Sun, H. M., Guo, Z. Y., Hu, J. H., et al. (2016). Overexpression of DJ-1/PARK7, the Parkinson’s disease-related protein, improves mitochondrial function via Akt phosphorylation on threonine 308 in dopaminergic neuron-like cells. Eur. J. Neurosci. 43, 1379–1388. doi: 10.1111/ejn.13216
Zhang, Z., Chu, S. F., Wang, S. S., Jiang, Y. N., Gao, Y., Yang, P. F., et al. (2017). RTP801 is a critical factor in the neurodegeneration process of A53T alpha-synuclein in a mouse model of Parkinson’s disease under chronic restraint stress. Br. J. Pharmacol. 175, 590–605. doi: 10.1111/bph.14091
Zhao, H. T., John, N., Delic, V., Ikeda-Lee, K., Kim, A., Weihofen, A., et al. (2017). LRRK2 antisense oligonucleotides ameliorate alpha-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol. Ther. Nucleic Acids 8, 508–519. doi: 10.1016/j.omtn.2017.08.002
Zharikov, A. D., Cannon, J. R., Tapias, V., Bai, Q., Horowitz, M. P., Shah, V., et al. (2015). shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest. 125, 2721–2735. doi: 10.1172/JCI64502
Zhou, L., Wang, W., Hoppel, C., Liu, J., and Zhu, X. (2017). Parkinson’s disease-associated pathogenic VPS35 mutation causes complex I deficits. Biochim. Biophys. Acta 1863, 2791–2795. doi: 10.1016/j.bbadis.2017.07.032
Zhou, Z. D., Kerk, S. Y., Xiong, G. G., and Lim, T. M. (2009). Dopamine auto-oxidation aggravates non-apoptotic cell death induced by over-expression of human A53T mutant alpha-synuclein in dopaminergic PC12 cells. J. Neurochem. 108, 601–610. doi: 10.1111/j.1471-4159.2008.05795.x
Zhu, M. S., H Patel, S., and Han, S. (2017). DJ-1, a Parkinson’s disease related protein, aggregates under denaturing conditions and co-aggregates with alpha-synuclein through hydrophobic interaction. Biochim. Biophys. Acta 1861, 1759–1769. doi: 10.1016/j.bbagen.2017.03.013
Zhuang, N., Li, L., Chen, S., and Wang, T. (2016). PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 7:e2501. doi: 10.1038/cddis.2016.396
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. doi: 10.1016/j.neuron.2004.11.005
Zondler, L., Kostka, M., Garidel, P., Heinzelmann, U., Hengerer, B., Mayer, B., et al. (2017). Proteasome impairment by alpha-synuclein. PLoS One 12:e0184040. doi: 10.1371/journal.pone.0184040
Keywords: Parkinson disease, neurodegeneration, gene mutation, neuronal death, mechanism
Citation: Zeng X-S, Geng W-S, Jia J-J, Chen L and Zhang P-P (2018) Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 10:109. doi: 10.3389/fnagi.2018.00109
Received: 16 January 2018; Accepted: 03 April 2018;
Published: 17 April 2018.
Edited by:
Nibaldo C. Inestrosa, Pontificia Universidad Católica de Chile, ChileReviewed by:
Nicola B. Mercuri, Università degli Studi di Roma “Tor Vergata”, ItalyRosario Moratalla, Consejo Superior de Investigaciones Científicas (CSIC), Spain
Copyright © 2018 Zeng, Geng, Jia, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian-Si Zeng, enhzLTIwMDVAMTYzLmNvbQ== Jin-Jing Jia, amlhamluamluZzE5ODZAMTI2LmNvbQ==