 Katie Irwin
Katie Irwin Claire Sexton2,3
Claire Sexton2,3 Tarun Daniel
Tarun Daniel Lorina Naci
Lorina Naci- 1Department of Neuroscience, University of Georgia, Athens, GA, United States
- 2Memory and Aging Center, Global Brain Health Institute, University of California, San Francisco, San Francisco, CA, United States
- 3Oxford Centre for Human Brain Activity, Wellcome Centre for Integrative Neuroimaging, Department of Psychiatry, University of Oxford, Oxford, United Kingdom
- 4Mercer’s Institute for Successful Ageing, St. James’s Hospital, Dublin, Ireland
- 5The Global Brain Health Institute, Trinity College Dublin, Dublin, Ireland
- 6School of Psychology, Trinity College Dublin, Dublin, Ireland
Dementia, particularly Alzheimer’s disease (AD), is a growing pandemic that presents profound challenges to healthcare systems, families, and societies throughout the world. By 2050, the number of people living with dementia worldwide could almost triple, from 47 to 132 million, with associated costs rising to $3 trillion. To reduce the future incidence of dementia, there is an immediate need for interventions that target the disease process from its earliest stages. Research programs are increasingly starting to focus on midlife as a critical period for the beginning of AD-related pathology, yet the indicators of the incipient disease process in asymptomatic individuals remain poorly understood. We address this important knowledge gap by examining evidence for cognitive and structural brain changes that may differentiate, from midlife, healthy aging and pathological AD-related processes. This review crystallizes emerging trends for divergence between the two and highlights current limitations and opportunities for future research in this area.
Introduction
As more individuals are living longer than ever before, the older population is growing exponentially across the globe. In 2015, approximately 8.5% of the world’s population was over the age of 65, and by 2050, this number is expected to almost double to 16.7% (He et al., 2016). Understanding how to effectively promote healthy aging and prevent neurodegenerative diseases that present symptoms in older adults thus is becoming increasingly important.
One of the biggest challenges associated with the aging population is dementia. The World Alzheimer Report has suggested that the number of people living with dementia could triple to 132 million people worldwide by 2050 (Prince et al., 2015). Dementia not only dramatically alters the lives of those with the condition, but it also confers a severe burden on families, friends, caregivers, and the healthcare systems at large. The worldwide cost of dementia was estimated at $818 billion in 2016 and could increase to approximately $2 trillion by 2030 (Prince et al., 2015).
What Is Dementia?
Although dementia predominantly affects older people, it is neither a normal part of aging nor an exaggeration of it. It is not a single disease, but instead a clinical syndrome formed by a range of recognizable symptoms. The Diagnostic and Statistical Manual of Mental Disorders (2013) Fifth Edition (DSM-5,), replaces the term “dementia” with “major neurocognitive disorder” and defines the condition as significant cognitive decline that, critical to the diagnosis, impairs independent living. Cognitive domains that can be impacted include learning and memory, executive function, language, complex attention, perceptual-motor function, and social cognition (Diagnostic and Statistical Manual of Mental Disorders, 2013). Dementia involves a myriad of adverse neurocognitive changes, and the precise pathological processes underlying these changes are not well understood (Sacuiu, 2016). This review focuses on AD, the most common type of dementia (Barker et al., 2002).
Alzheimer’s Disease
Alzheimer’s disease accounts for around 60–80% of dementia cases, and its symptoms are projected to affect greater numbers of people every year (Alzheimer’s Association, 2017). Insidious and irreversible memory decline is the most recognized feature of AD, beginning with initial short-term memory deficits that make learning new information difficult, but other areas of cognition such as word-finding, visuoconstructional, and executive abilities can also decline (Sacuiu, 2016). As a patient progresses through mild, moderate, and severe stages of AD, greater memory deficits, increased confusion, and personality and behavioral changes, among other symptoms, are frequently observed and lead to round-the-clock assistance needs with everyday activities (Logiudice and Watson, 2014).
The precise brain mechanism affected by neural degeneration in the earliest stages of AD is still largely hypothesized (Sacuiu, 2016). Recent evidence suggests that various subcortical brain nuclei may show the first AD-related pathology (Stratmann et al., 2016). The transentorhinal region is thought to be the first affected site in the cerebral cortex, and in later stages of the disease, atrophy spreads throughout cerebral cortex association areas (Braak et al., 2006).
Alzheimer’s disease can be divided into two types: familial and sporadic, both of which are associated with similar symptoms. Less than 1% of cases are attributable to familial AD (Bateman et al., 2010), which is passed on from parents to children through mutations in specific genes (Bateman et al., 2010) and is associated with an earlier onset compared with sporadic AD (typically between ages 50 and 65, compared with after age 65 in sporadic AD). In contrast, sporadic AD does not have a specific genetic foundation (Piaceri et al., 2013). Instead, “susceptibility genes” that do not directly cause AD but increase one’s susceptibility of developing the disease are implicated. The most potent of the susceptibility genes is the APOE 𝜀4 allele (Corder et al., 1993). Carrying the 𝜀4 allele imposes a greater risk of developing AD compared to the 𝜀3 allele, increasing in a dose dependent manner from 20 to 90% in homozygous carriers (Corder et al., 1993), whereas the 𝜀2 allele has been suggested to provide a protective effect, lowering the risk of AD (Corder et al., 1994). For this reason, the APOE 𝜀4 allele is frequently used in studies as a proxy for the eventual diagnosis of AD, due to the difficulty of following up participants until the clinical diagnosis of AD is confirmed. However, because the APOE 𝜀4 allele is merely a risk factor, individuals that carry the allele may never develop AD, and, conversely, individuals without the allele can develop the disease (Michaelson, 2014). Paradoxically, the APOE 𝜀4 allele has been associated with better episodic memory in young adults, and the underlying reasons for this switch are poorly understood (Mondadori et al., 2006). In addition to susceptibility genes, an array of genetic, environmental, and lifestyle factors can contribute to the development of AD (Livingston et al., 2017), but the exact etiology of the disease remains unknown (Piaceri et al., 2013).
The question of when and in what ways healthy aging diverges from the incipient AD remains poorly understood and the focus of active research (Ritchie et al., 2015; Mortamais et al., 2017), with very recent research suggesting that this divergence may be observed as early as midlife (Ritchie et al., 2017). The identification of pathological aging in midlife could be transformational. The brain is thought to be modifiable in neural and cognitive ways (Lachman et al., 2015), so early detection and intervention could lead to improved treatment and, ultimately, prevention of Alzheimer’s dementia.
Divergence in Midlife
Before dementia’s symptoms occur, an intermediate stage of MCI, formally termed minor neurocognitive disorder by DSM-5 (Diagnostic and Statistical Manual of Mental Disorders, 2013), may occur. MCI can be a transitional stage between normal aging and dementia, but not all people who experience it will develop dementia. MCI is characterized by observable cognitive deficits that resemble, but are less severe than, those typical of different dementias. In order to be labeled MCI, cognitive deficits must be greater than would be expected from the individual’s age and education level but not significantly impair daily functioning as in dementia (Albert et al., 2011; Diagnostic and Statistical Manual of Mental Disorders, 2013). However, particularly in AD, pathophysiological processes leading to the disorder may have already begun an irreversible trajectory of neurodegeneration by the stage of MCI, as several studies suggest that dementia’s pathology may be present years or even decades before its clinical diagnosis (Braak and Braak, 1997; Jack et al., 2009; Ritchie et al., 2015). Intervention prior to the development of MCI thus may be necessary to significantly reduce dementia incidence. Consequently, research efforts are increasingly focusing on preclinical AD – those at risk based on genetic and/or family history factors, but not yet showing symptoms – in order to detect early biomarkers, monitor progression, and employ disease-modifying agents as they become available (Dubois et al., 2016). However, the early divergence of healthy and pathological aging remains elusive. To address this critical knowledge gap, this review examines current evidence for significant cognitive and neural changes in midlife that may distinguish healthy and pathological brain aging indicative of AD (Tables 1–3).
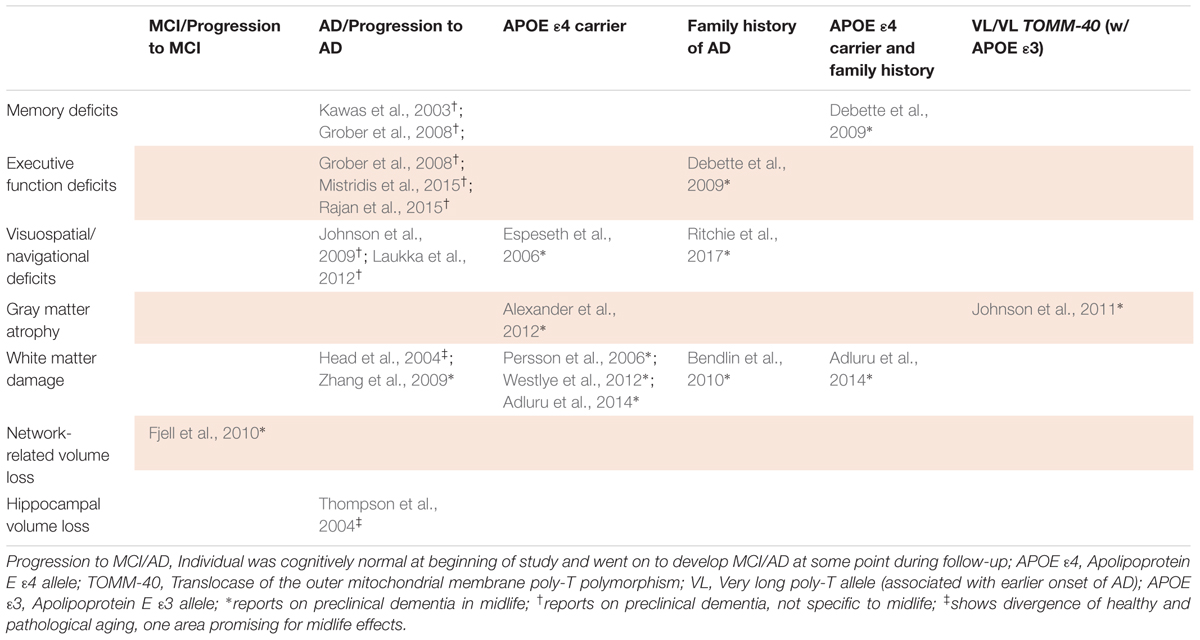
TABLE 1. Studies of healthy versus pathological aging.
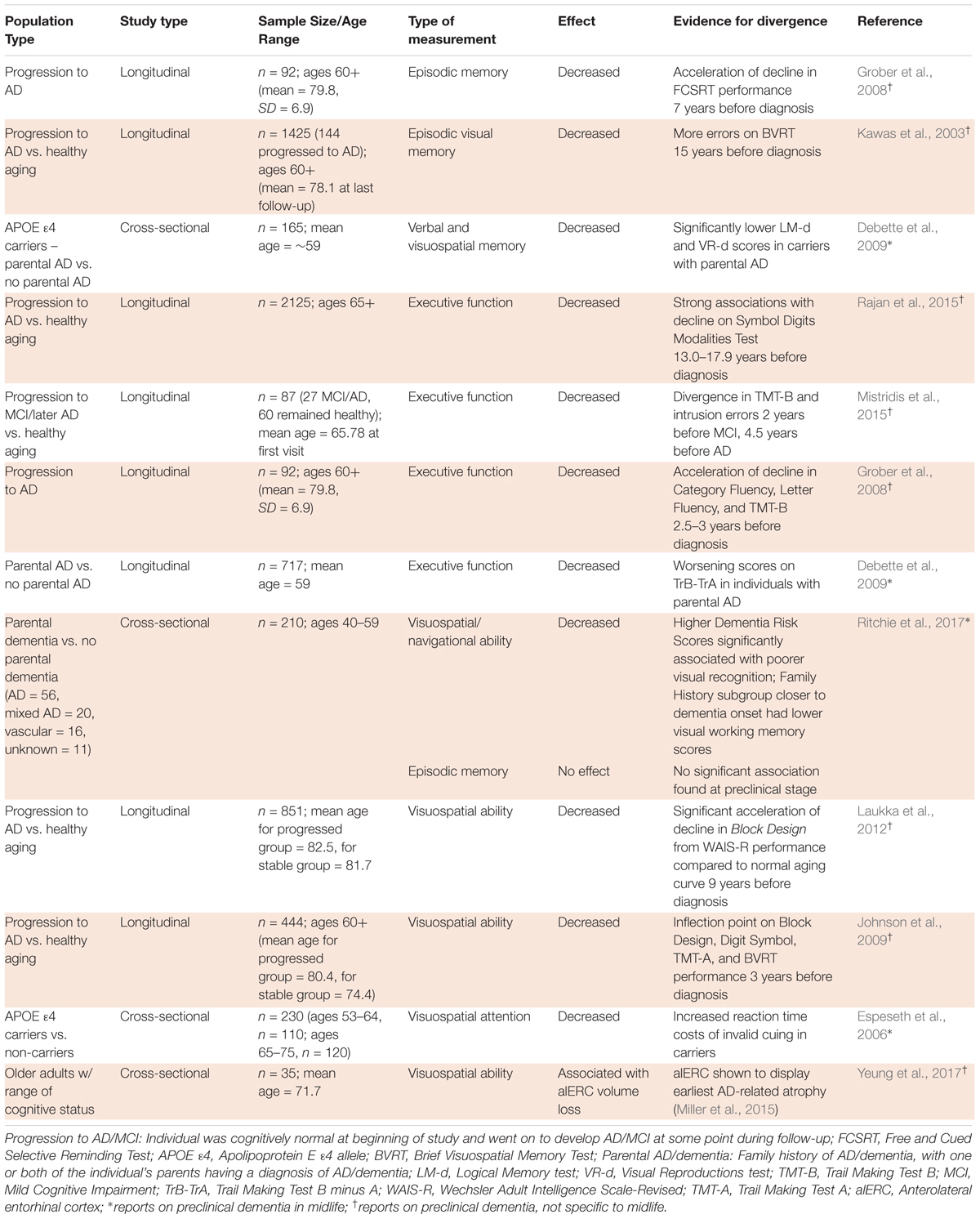
TABLE 2. Divergence of healthy and pathological aging in cognitive studies.
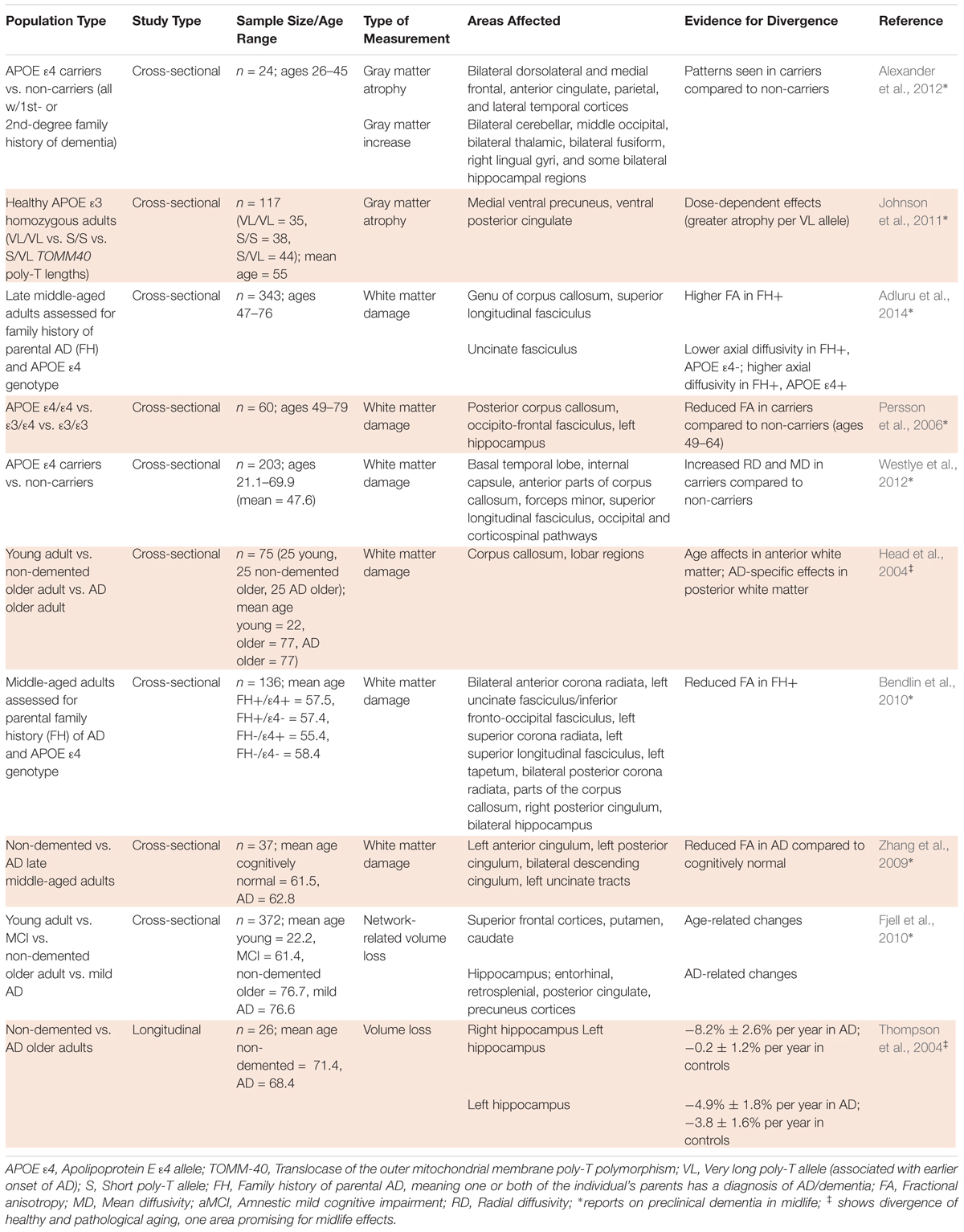
TABLE 3. Divergence of healthy and pathological aging in structural neuroimaging studies.
Literature Search Criteria
Online research for studies evaluating potential midlife cognitive and neurobiological markers of AD was conducted in PubMed, and a report of the group’s age range was required. Midlife was considered 40–65 years, inclusive. Populations representing pathological aging were defined as those who developed AD in longitudinal studies that recruited healthy populations and tracked who developed dementia, as well as at-risk groups in cross-sectional studies that compared low- and high-risk individuals, based on genetic and/or family history factors. Behavioral studies of cognition and structural neuroimaging studies investigating the underlying brain mechanisms were included. Task-based functional neuroimaging studies were excluded due to wide variation between tasks across studies. Studies of uncommon groups, e.g., women with diabetes, were also excluded. Table 1 shows common cognitive and neuroimaging-based deviations from age-matched healthy controls reported in midlife populations at risk for AD pathology.
Cognition
Cognitive assessments that can detect subtle changes before noticeable clinical symptoms appear may provide robust markers of pathological aging. However, little consensus has been reached regarding the best methods for detecting cognitive symptoms of preclinical AD in individuals who do not meet the threshold for MCI diagnosis (Albert et al., 2011). This difficulty is not only attributable to the subtlety of these changes (Mortamais et al., 2017), but also to the tendency of cognitive assessments to implicate more than one aspect of cognition (Mortamais et al., 2017) and to discrepancies between cross-sectional and longitudinal study procedures (Salthouse, 2014). Despite these obstacles, early deficits in some aspects of cognition have been pinpointed by multiple studies as potential indicators of divergence from healthy aging. Table 2 shows a summary of the evidence for cognitive divergence between healthy and pathological aging in midlife populations.
Normal aging involves many cognitive changes, including variations in memory, executive function, reasoning, spatial abilities, attention, and language abilities (Blazer et al., 2015). Most aspects of memory show age-related decline, with decrements of varying intensity (Healey and Kahana, 2016). Semantic memory has been shown to improve until age 60, while episodic memory abilities stay roughly the same until 60–65 years (Rönnlund et al., 2005; Nyberg et al., 2012). A longitudinal study of midlife women ages 42–52 at entry showed that they experienced a mean decline in verbal episodic memory of 0.02 per year, or 2% of the mean baseline score in 10 years (Karlamangla et al., 2017). After about 60 years, both semantic and episodic memory face a sharp decrease (Rönnlund et al., 2005).
As episodic memory is thought to remain relatively steady until 60 years (Rönnlund et al., 2005), significant decline before the age of 60 could be indicative of pathological aging. Studies have proposed that episodic memory is a strong predictor of dementia, with decline more sizeable than that associated with the normal age-related processes detectable 7–15 years before diagnosis of AD (Kawas et al., 2003; Grober et al., 2008; Mortamais et al., 2017). Multiple studies have suggested that episodic memory decline is a marker of preclinical AD, but few studies have focused on preclinical AD specifically in midlife. One study that assessed middle-aged offspring of individuals with sporadic AD (mean age, ∼59) showed that the interaction of parental AD (one or more parents diagnosed with AD), with APOE 𝜀4 status was significantly associated with declines in verbal and visuospatial memory (Debette et al., 2009). More studies assessing middle-aged participants are needed to support these associations.
Executive function – an umbrella term for many high-order cognitive processes including memory, inhibition, task switching, etc. (Turner and Spreng, 2012) and, broadly speaking, the ability to self-regulate behavior – also shows an age-related decline. Rönnlund et al. (2007) measured executive function performance with the Tower of Hanoi task and showed a decline after 65 years that prevailed after adjustments for education and test-retest effects. Reasoning, one facet of executive function and self-regulation of behavior and actions, similarly declines with age (Blazer et al., 2015). Cognitive decline in executive function has also been linked more broadly with cognitive decline in intelligence (Salthouse et al., 2003).
Studies have not reached a consensus on when executive function declines in dementia. Rajan et al. (2015) suggested that executive dysfunction precedes memory loss 13 or more years before AD diagnosis, based on effect on the Symbol Digits Modalities Test, while Mistridis et al. (2015) posited that executive function declines 4.5 years before diagnosis, according to Trail Making Test B and intrusion errors. The Baltimore Longitudinal Study of Aging agreed more with the latter figure, showing acceleration of decline in executive function, as tested by Category Fluency, Letter Fluency, and Trail Making Test B, 2.5–3 years before diagnosis (Grober et al., 2008). Despite these inconsistencies, evidence suggests that executive dysfunction is detectable in preclinical AD and that significant impairment in executive function before the age of 65 would indicate a divergence from healthy aging. Similarly to episodic memory decline, executive dysfunction’s relevance to preclinical AD specifically in midlife has been less well studied. Debette et al. (2009) showed that middle-aged participants with parental AD (mean age, 59) were more likely to experience executive function decline, but additional studies are needed to provide a strong evidence base for this association.
Spatial ability — the ability to comprehend spatial relations between objects or images —also declines with age. Studies that compare older to younger participants have historically found that older individuals take longer to perform tasks that require a good grasp of spatial information (Bruin et al., 2016). Although spatial abilities are known to decline with age (Bruin et al., 2016), this decline is exacerbated in pathological aging. Ritchie et al. (2017), used COGNITO, an extensive neuropsychological battery, to evaluate various aspects of cognition in 40–59 year-olds and found that individuals at higher risk for AD, based on family history, showed significantly larger deficits in visuospatial and navigational tasks, but not episodic memory as may be expected. A study by Laukka et al. (2012) similarly found visuospatial ability to show significant acceleration of decline before episodic or semantic memory, at 9 years prior to AD diagnosis. A longitudinal study by Johnson et al. (2009) that analyzed cognitive decline from the age of 60 showed that visuospatial performance exhibited a sharp inflection point 3 years before AD diagnosis and declined with increasing rates afterward, whereas individuals who did not develop AD remained relatively stable in this domain. These studies provide evidence that differences in visuospatial abilities detected by Ritchie et al. (2017) in middle-aged individuals at higher risk for dementia hold when participants are studied until clinical diagnosis.
Also using a risk-related proxy for progression to AD, Espeseth et al. (2006) found slower reaction times in an attention task following invalid location cues in healthy middle-aged APOE 𝜀4 carriers compared to non-carriers (𝜀3/𝜀3 genotype), further associating deficits in visuospatial processing with a higher risk for AD. More longitudinal studies that assess cognition in middle-aged individuals and follow participants until older age to confirm progression/no progression to AD are needed, so that midlife data is not restricted to high risk vs. low risk comparisons. A recent study by Yeung et al. (2017) used an eye-tracking approach to measure decline in the visuospatial ability of configural processing, or attending to the arrangement of an object’s features, which the group found was strongly associated with volume loss in the alERC, a region shown to display early AD-related atrophy (Miller et al., 2015). The eye-tracking cognitive task was proposed as a sensitive and practical way to detect changes in alERC volume (Yeung et al., 2017), which has been suggested as a valuable biomarker of preclinical dementia in midlife (see neuroimaging section), but the eye-tracking task has not yet been employed for a middle-aged study group. Overall, decline of visuospatial ability on both memory and attention task performance has shown promise as a midlife indicator of pathological aging.
Different facets of attention exhibit distinct age-related patterns of change. Selective attention abilities deteriorate with age (McDowd and Shaw, 2000), but recent research suggests that this reduction is due to increased distractibility in other senses rather than mere decline of attention abilities (Hugenschmidt et al., 2009). Divided attention shows age-related deficits after 60 years, as attention-switching tasks tend to take longer as people age (Tsang and Shaner, 1998; Glisky, 2007). Changes in sustained attention with age are still debated, however, as sustained attention has been shown to increase, decrease, and stay the same in different studies likely due to methodological disparities across studies (Staub et al., 2013). Variances from these patterns that are clearly indicative of pathological aging have not yet been identified, besides the interaction between APOE 𝜀4 and visuospatial attention shown by Espeseth et al. (2006).
Finally, the effects of healthy aging on language abilities vary between components of language processing. When presented with word-finding tasks, older participants report more cases of the TOT effect, and the proportion of TOTs is reported to linearly increase with age (Shafto and Tyler, 2014). In addition to TOT states, picture-naming tasks have provided evidence for decline in language production abilities with increasing age (Nicholas et al., 1985; Ramsay et al., 1999; Goral, 2004). Older participants also, on average, produce simpler sentences and speak at a slower rate than younger participants (Shafto and Tyler, 2014). In contrast to these age-related declines in speech production, speech comprehension, including syntax and semantics, appears more robust due to distinct patterns of gray matter preservation/atrophy relative to those involved in speech production (Shafto and Tyler, 2014). Further research is needed to determine whether deviations from these established patterns can serve as early indicators of AD.
Structural Neuroimaging
The structure of the brain undergoes significant changes with age. Postmortem studies have documented an array of age-related changes in brain morphology, including reduced brain weight and volume, ventricular and sulcal expansion, decreased synaptic density, loss of neuronal bodies and dendritic spines, loss of myelination, accumulation of lipofuscin, and thinning of cerebral vasculature (Raz and Rodrigue, 2006; Lindenberger, 2014).
In vivo structural neuroimaging plays an important role in identifying typical, age-related changes to brain structure and significant differences stemming from pathological processes. Evidence for differences in brain structure between healthy and pathological aging in midlife populations is summarized in Table 3. Below, we discuss current evidence for significant differences with respect to gray matter atrophy, white matter damage, network-related volume loss, and hippocampal volume loss.
Cortical gray matter density reductions over dorsal frontal and parietal association cortices have been shown to occur at a steady pace between the ages of 7 and 60 but slow down afterward (Sowell et al., 2003). Cortical gray matter volume in the posterior temporal region, however, appears to increase up to the age of 30 years and then rapidly decline (Sowell et al., 2003). Reduction of gray matter density relatively early in life may indirectly measure an increase in cerebral myelination, as brain weight is fairly stable until age 40 (Dekaban, 1978). Because gray matter density continues to decline into the 50s and white matter volume has peaked around the mid-40s (Bartzokis et al., 2001; Sowell et al., 2003), studies have suggested that the later loss may be due to neurodegeneration rather than an increase in myelination. A recent longitudinal study in healthy Chinese adults above the age of 55 found annual rates of gray matter decline (cm3/year) of -0.57/-0.29/-0.39/-0.16 in the frontal/parietal/temporal/occipital lobes, respectively (Leong et al., 2017). The frontal and parietal measurements generally agree with Western studies of adults aged 30–99 in one study, 59–85 in another, and 64–86 in a third, which have reported rates of -0.56 to -1.05/-0.21 to -0.90 in frontal/parietal lobes, but the temporal/occipital measurements were lower than the Western estimates of -0.43 to -0.55/-0.33 to -0.36 for these regions (Jernigan et al., 2001; Resnick et al., 2003; Driscoll et al., 2009). Together, these studies have suggested that annual decline is fastest in frontal lobes, with rates suggested to decelerate with increasing age, initially slowing around the age of 40 (Storsve et al., 2014).
Different patterns of gray matter atrophy are found in AD. In general, as aforementioned, healthy age-related processes in midlife manifest rapid gray matter reductions in frontal regions (Terribilli et al., 2011), but young adult to early middle-aged APOE 𝜀4 carriers (26–45 years old) have shown greater gray matter reductions in dorsolateral and medial prefrontal, parietal, and lateral temporal regions, concomitant with increases in other brain regions compared to non-carriers (Table 3) (Alexander et al., 2012). An interpretation of these relative increases is that they may serve to sustain normal cognition despite the gray matter reductions of the prefrontal, parietal, and temporal regions in APOE 𝜀4 carriers (Alexander et al., 2012).
Another study used a TOMM-40 (translocase of the outer mitochondrial membrane) poly-T polymorphism, shown to predict age of onset of AD, instead of APOE as a genetic indicator of increased AD risk. APOE 𝜀3/𝜀4 heterozygotes have shown an association between a short (S) poly-T allele and later onset of AD and between a very long (VL) poly-T allele and earlier onset of AD (Johnson et al., 2011). This study found that healthy middle-aged adults (mean age, 55) with APOE 𝜀3 homozygous, VL/VL TOMM-40 genotypes showed decreasing gray matter volume in the medial ventral precuneus and ventral posterior cingulate, with dose-dependent effects compared to S/VL and S/S TOMM-40 groups (Johnson et al., 2011). These studies suggest that, in relation to AD risk-related genotypes, structural effects on gray matter are already observable by middle age, decades before potential diagnosis of AD.
White matter has also been found to decrease during healthy aging and to show distinct patterns of change in pathological aging. DTI studies that have investigated white matter FA and mean diffusivity measures of structural brain connectivity have suggested that decline in white matter microstructure begins between 18 and 50 years (Westlye et al., 2010; Bartzokis et al., 2012; Kochunov et al., 2012; Lebel et al., 2012; Sexton et al., 2014). The recent study of healthy Chinese adults (Leong et al., 2017) found similar patterns for the rate of white matter decline as for gray matter, with atrophy greatest in the frontal regions, followed by temporal, parietal, and occipital regions. Several studies have reported similar findings of age-related white matter reduction being predominant in frontal regions (Gunning-Dixon et al., 2009; Barrick et al., 2010; Bennett et al., 2010; Burzynska et al., 2010; Sexton et al., 2014; Bender and Raz, 2015; Bender et al., 2016), and it is thought to accelerate beginning by about age 50 (Sexton et al., 2014).
However, in pathological compared to healthy aging in late middle-aged adults, loss of white matter integrity is not foremost in anterior regions, but instead appears most pronounced in regions including the cingulum, corpus callosum, and superior longitudinal fasciculus (Persson et al., 2006; Gunning-Dixon et al., 2009; Adluru et al., 2014). A DTI study found significant FA differences between APOE 𝜀4 carriers and non-carriers ages 49–64 in the occipito-frontal fasciculus and the posterior portion of the corpus callosum, suggesting white matter integrity is compromised in APOE 𝜀4 carriers before the onset of AD (Persson et al., 2006). Another DTI study comparing white matter in middle-aged APOE 𝜀4 carriers and non-carriers (mean age, 47.6) significantly associated risk for AD with increased radial and mean diffusivity likewise in the corpus callosum and occipital pathways, among other regions (see Table 3 for full results) (Westlye et al., 2012). Another study found significantly lower FA levels associated with parental family history of AD in cognitively healthy, middle-aged adults (mean age, 56.99), rather than with only APOE status. Effects of family history were shown in regions known to be affected in AD, such as the medial temporal lobe, with larger effects found on the left side of the brain (Bendlin et al., 2010). Some studies have supported the value of these susceptibility proxies by yielding similar patterns. For example, Head et al. (2004) demonstrated a dissociation between normal and pathological aging in a DTI study of young adults, cognitively healthy older adults, and older adults with AD, where age-associated decline in white matter integrity was apparent in anterior brain regions, while AD-related effects were observed on posterior fiber tracts. One study that compared late middle-aged cognitively normal (mean age, 61.5) and AD (mean age, 62.8) individuals, instead of using susceptibility proxies, found similar results, with AD significantly associated with lower FA values in the cingulum and left uncinate tracts (Zhang et al., 2009). Taken together the above findings offer some evidence that topographical distributions of gray and white matter atrophy provide insights into deviations from healthy aging in midlife, yet more studies that follow participants to clinical diagnosis of MCI or AD are needed to validate these potential early neurobiological markers (Sexton et al., 2011).
In addition to regional loss of gray and white matter integrity, network-level patterns of brain volume loss have been shown to differentiate between healthy and pathological aging. The study of healthy Chinese adults (Leong et al., 2017) showed cerebral volume loss of 0.56%/year, similarly to Western longitudinal studies’ rates of -0.5% (Jack et al., 2005)/0.55% (Enzinger et al., 2005). Although cerebral shrinkage is normal in aging, a study by Fjell et al. (2010) concluded that while volumetric reductions in the fronto-striatal executive network are typical of healthy aging, greater changes in the medial temporo-parietal episodic memory network are representative of AD or progression to AD (see Table 3 for full results). This study used CSF tau measures of, on average, late middle-aged MCI individuals (mean age, 61.4) to validate MRI patterns gathered from healthy young (mean age, 22.2), healthy elderly (mean age, 76.7), and mild AD (mean age, 76.6) participants; only volume loss in the medial temporo-parietal network was associated with pathological levels of tau in the MCI patients. These results provide evidence for a detectable divergence between patterns of morphometric change in healthy vs. pathological aging. Because the AD-associated network atrophy resembles the spread of pathological tau in AD, this atrophy could be related to disease progression mechanisms, further suggesting the potential for network-related atrophy as a sensitive marker of midlife progression toward AD (Fjell et al., 2010).
Within the medial temporal lobe, volume loss associated with AD has been shown to begin in the alERC and progress medially through the rest of the entorhinal cortex to the hippocampus (Miller et al., 2015). In these regions normal aging can involve atrophy of 0.2–3.8% per year, whereas individuals with AD can experience rates of 4.9–8.2% (Thompson et al., 2004). Some variability has been reported within this range (Fraser et al., 2015; Leong et al., 2017), yet the age-related hippocampal volume reductions shown in several longitudinal studies (Raz et al., 2005; Fraser et al., 2015; Leong et al., 2017) fail to reach the high rates reported in AD (Lindenberger, 2014). Studies extending the analysis of such diverging atrophy rates to midlife are still needed. Interestingly, studies have shown that reduced hippocampal volume, a hallmark of AD, is associated with decreased vestibular function (Brandt et al., 2005). Compared to age-matched controls, individuals with AD have been shown to display worse vestibular function, which is related to visuospatial abilities, a cognitive domain mentioned above to be impacted more strongly in pathological than healthy aging (Harun et al., 2016). The vestibular system could thus be investigated as a link between these two neurobiological and cognitive markers of AD.
Conclusion
In summary, associations have been found between higher risk for AD and greater midlife decline in episodic memory and executive function (Debette et al., 2009). Other evidence may suggest, however, that trends in visuospatial ability deficits more strongly differentiate healthy vs. pathological aging in midlife (Ritchie et al., 2017). Other cognitive domains such as attention and language abilities have not yet displayed substantial differences in middle-aged individuals of varying dementia risk.
In addition to cognitive markers, structural neuroimaging has shown diverging trends in gray matter reduction and loss of white matter integrity in healthy vs. pathological aging. Healthy aging is more strongly associated with decline in frontal regions (Terribilli et al., 2011), while middle-aged individuals more likely to develop AD have shown greater gray matter reductions in dorsolateral and medial prefrontal, parietal, and lateral temporal regions (Alexander et al., 2012) and have shown loss of white matter integrity in regions including the cingulum, corpus callosum, superior longitudinal fasciculus, and left uncinate fasciculus (Persson et al., 2006; Gunning-Dixon et al., 2009; Adluru et al., 2014). Additionally, midlife volumetric reductions in the fronto-striatal executive network seem to be a normal part of aging, while reductions in the medial temporo-parietal episodic memory network seem to indicate pathological aging (Fjell et al., 2010). Finally, entorhinal cortex and hippocampal atrophy rates appear to diverge in healthy and pathological brain aging (Raz et al., 2005; Lindenberger, 2014; Fraser et al., 2015; Leong et al., 2017), but it is not yet known if this divergence is relevant to midlife.
Limitations and Future Directions
Various limitations hinder the identification of midlife cognitive and neurobiological markers of preclinical AD. Selection bias can interfere with the accurate interpretation of findings, as individuals who are very healthy may decline to participate in aging studies because they are too busy and individuals who are too unhealthy may not be able to participate (Minder et al., 2002). Socially isolated people and people without financial means are also likely underrepresented in studies (Ford et al., 2008). Additionally, the methodology of aging studies is challenged by individuals who enter a study as a cognitively normal participant but develop (unnoticed) cognitive impairment associated with preclinical AD during the study (Lezak et al., 2012). Further, cross-sectional and longitudinal studies present unique difficulties that make comparison of findings across them problematic. Cross-sectional studies are prone to cohort effects, in which potentially vast differences between cohorts can confound findings assumed to be the result of aging (Hedden and Gabrieli, 2004; Salthouse, 2014), and longitudinal studies are prone to practice effects, in which cognitive decline can be hidden by individuals’ improvement on tests that they have completed several times (Salthouse, 2010; Abner et al., 2012). Attrition that results in the healthiest, wealthiest, or most motivated participants remaining in the study to its conclusion can also bias longitudinal studies (Van Beijsterveldt et al., 2002). Additionally, as aforementioned, many studies use the presence of the APOE 𝜀4 allele as a proxy for the future diagnosis of dementia due to the difficulty of following up study participants over many years, possibly decades, to clinical diagnosis. Differences between measures gathered from high- and low-risk participants, such as gray matter volume, for example, thus are not synonymous with differences between individuals who progress to AD diagnosis and those who do not.
The development of early cognitive and neurobiological markers for preclinical AD in midlife would be greatly facilitated by standardization of cognitive and neuroimaging measures used across research groups. Although some cognitive trends have been found, these often do not accurately capture the individual aging process, which can differ considerably across people and fluctuate within the individual (Lindenberger, 2014). The development of assessments that have predictive value at the individual level is crucially important for the identification of individuals who can benefit from early interventions and preventative measures. Until more sensitive and specific cognitive assessments are developed, neurobiological markers may provide more promise for predicting AD development at an individual level. Longitudinal studies that confirm AD diagnosis rather than relying on risk-related proxies are more challenging and costlier to conduct, but they may be necessary for more finely pinpointing markers differentiating healthy and pathological brain aging in midlife. Additionally, as AD-related pathology has been shown to appear in subcortical nuclei before the hippocampus as previously mentioned (Braak et al., 2006; Stratmann et al., 2016), future neuroimaging studies could focus on diverging rates of volumetric reduction in these regions instead of only in the entorhinal cortex and hippocampus.
Although many challenges surround the study of healthy aging and AD, significant progress has been made in understanding potential neurobiological and cognitive markers of preclinical AD. Further research is needed in regards to midlife, however. The significance of developing sensitive markers for detecting healthy vs. AD-related changes in midlife remains paramount, as this period constitutes a highly favorable therapeutic window for reducing risk factors and introducing disease-modifying agents once they are developed.
Author Contributions
KI and TD wrote the initial manuscript. CS and BL provided critical feedback. LN provided feedback, edited the manuscript, and oversaw the project.
Funding
This research was supported by a L’Oréal-UNESCO for Women in Science Research Excellence Grant to LN, and a Project Grant from the Global Brain Health Institute to LN and BL. CES is supported by the Global Brain Health Institute and NIHR Oxford Health Biomedical Research Centre.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MG-R and handling Editor declared their shared affiliation at the time of the review.
Abbreviations
AD, Alzheimer’s disease; alERC, anterolateral entorhinal cortex; APOE, Apolipoprotein E; DTI, diffusion tensor imaging; FA, fractional anisotropy; MCI, mild cognitive impairment; S/VL, short/very long poly-T allele; TOMM, translocase of the outer mitochondrial membrane; TOT, tip-of-the-tongue.
References
Abner, E. L., Dennis, B. C., Mathews, M. J., Mendiondo, M. S., Caban-Holt, A., Kryscio, R. J., et al. (2012). Practice effects in a longitudinal, multi-center Alzheimer’s disease prevention clinical trial. Trials 13:217. doi: 10.1186/1745-6215-13-217
Adluru, N., Destiche, D. J., Lu, S. Y., Doran, S. T., Birdsill, A. C., Melah, K. E., et al. (2014). White matter microstructure in late middle-age: effects of apolipoprotein E4 and parental family history of Alzheimers disease. Neuroimage 4, 730–742. doi: 10.1016/j.nicl.2014.04.008
Albert, M. S., Dekosky, S. T., Dickson, D., Dubois, B., Feldman, H. H., Fox, N. C., et al. (2011). The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimers disease. Alzheimers Dement. 7, 270–279. doi: 10.1016/j.jalz.2011.03.008
Alexander, G. E., Bergfield, K. L., Chen, K., Reiman, E. M., Hanson, K. D., Lin, L., et al. (2012). Gray matter network associated with risk for Alzheimers disease in young to middle-aged adults. Neurobiol. Aging 33, 2723–2732. doi: 10.1016/j.neurobiolaging.2012.01.014
Alzheimer’s Association (2017). 2017 Alzheimers disease facts and figures. Alzheimers Dement. 13, 325–373. doi: 10.1016/j.jalz.2017.02.001
Barker, W. W., Luis, C. A., Kashuba, A., Luis, M., Harwood, D. G., Loewenstein, D., et al. (2002). Relative frequencies of alzheimer disease, lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the state of Florida brain bank. Alzheimer Dis. Assoc. Disord. 16, 203–212. doi: 10.1097/00002093-200210000-00001
Barrick, T. R., Charlton, R. A., Clark, C. A., and Markus, H. S. (2010). White matter structural decline in normal ageing: a prospective longitudinal study using tract-based spatial statistics. Neuroimage 51, 565–577. doi: 10.1016/j.neuroimage.2010.02.033
Bartzokis, G., Beckson, M., Lu, P., Nuechterlein, K., Edwards, N., and Mintz, J. (2001). Age-related changes in frontal and temporal lobe volumes in men. Arch. Gen. Psychiatry 58, 461–465. doi: 10.1001/archpsyc.58.5.461
Bartzokis, G., Lu, P. H., Heydari, P., Couvrette, A., Lee, G. J., Kalashyan, G., et al. (2012). Multimodal magnetic resonance imaging assessment of white matter aging trajectories over the lifespan of healthy individuals. Biol. Psychiatry 72, 1026–1034. doi: 10.1016/j.biopsych.2012.07.010
Bateman, R. J., Aisen, P. S., Strooper, B. D., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2010). Autosomal-dominant Alzheimers disease: a review and proposal for the prevention of Alzheimers disease. Alzheimers Res. Ther. 3:1. doi: 10.1186/alzrt59
Bender, A. R., and Raz, N. (2015). Normal-appearing cerebral white matter in healthy adults: mean change over 2 years and individual differences in change. Neurobiol. Aging 36, 1834–1848. doi: 10.1016/j.neurobiolaging.2015.02.001
Bender, A. R., Völkle, M. C., and Raz, N. (2016). Differential aging of cerebral white matter in middle-aged and older adults: a seven-year follow-up. Neuroimage 125, 74–83. doi: 10.1016/j.neuroimage.2015.10.030
Bendlin, B. B., Ries, M. L., Canu, E., Sodhi, A., Lazar, M., Alexander, A. L., et al. (2010). White matter is altered with parental family history of Alzheimers disease. Alzheimers Dement. 6, 394–403. doi: 10.1016/j.jalz.2009.11.003
Bennett, I. J., Madden, D. J., Vaidya, C. J., Howard, D. V., and Howard, J. H. (2010). Age-related differences in multiple measures of white matter integrity: a diffusion tensor imaging study of healthy aging. Hum. Brain Mapp. 31, 378–390. doi: 10.1002/hbm.20872
Blazer, D. G., Yaffe, K., and Liverman, C. T. (2015). Cognitive Aging: Progress in Understanding and Opportunities for Action. Washington, DC: The National Academies Press.
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., and Braak, E. (1997). Frequency of stages of alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357. doi: 10.1016/s0197-4580(97)00056-0
Brandt, T., Schautzer, F., Hamilton, D. A., Brüning, R., Markowitsch, H. J., Kalla, R., et al. (2005). Vestibular loss causes hippocampal atrophy and impaired spatial memory in humans. Brain 128, 2732–2741. doi: 10.1093/brain/awh617
Bruin, N. D., Bryant, D. C., Maclean, J. N., and Gonzalez, C. L. (2016). Assessing visuospatial abilities in healthy aging: a novel visuomotor task. Front. Aging Neurosci. 8:7. doi: 10.3389/fnagi.2016.00007
Burzynska, A. Z., Preuschhof, C., Bäckman, L., Nyberg, L., Li, S. C., Lindenberger, U., et al. (2010). Age-related differences in white matter microstructure: region-specific patterns of diffusivity. Neuroimage 49, 2104–2112. doi: 10.1016/j.neuroimage.2009.09.041
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer’s disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Debette, S., Wolf, P. A., Beiser, A., Au, R., Himali, J. J., Pikula, A., et al. (2009). Association of parental dementia with cognitive and brain MRI measures in middle-aged adults. Neurology 73, 2071–2078. doi: 10.1212/wnl.0b013e3181c67833
Dekaban, A. (1978). Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann. Neurol. 4, 345–356. doi: 10.1002/ana.410040410
Diagnostic and Statistical Manual of Mental Disorders (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Washington, DC: American Psychiatric Association.
Driscoll, I., Davatzikos, C., An, Y., Wu, X., Shen, D., Kraut, M., et al. (2009). Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology 72, 1906–1913. doi: 10.1212/wnl.0b013e3181a82634
Dubois, B., Hampel, H., Feldman, H. H., Scheltens, P., Aisen, P., Andrieu, S., et al. (2016). Preclinical Alzheimers disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 12, 292–323. doi: 10.1016/j.jalz.2016.02.002
Enzinger, C., Fazekas, F., Matthews, P. M., Ropele, S., Schmidt, H., Smith, S., et al. (2005). Risk factors for progression of brain atrophy in aging: six-year follow-up of normal subjects. Neurology 64, 1704–1711. doi: 10.1212/01.wnl.0000161871.83614.bb
Espeseth, T., Greenwood, P. M., Reinvang, I., Fjell, A. M., Walhovd, K. B., Westlye, L. T., et al. (2006). Interactive effects of APOE and CHRNA4 on attention and white matter volume in healthy middle-aged and older adults. Cogn. Affect. Behav. Neurosci. 6, 31–43. doi: 10.3758/cabn.6.1.31
Fjell, A. M., Amlien, I. K., Westlye, L. T., Stenset, V., Fladby, T., Skinningsrud, A., et al. (2010). CSF biomarker pathology correlates with a medial temporo-parietal network affected by very mild to moderate Alzheimers disease but not a fronto-striatal network affected by healthy aging. Neuroimage 49, 1820–1830. doi: 10.1016/j.neuroimage.2009.09.029
Ford, J. G., Howerton, M. W., Lai, G. Y., Gary, T. L., Bolen, S., Gibbons, M. C., et al. (2008). Barriers to recruiting underrepresented populations to cancer clinical trials: a systematic review. Cancer 112, 228–242. doi: 10.1002/cncr.23157
Fraser, M. A., Shaw, M. E., and Cherbuin, N. (2015). A systematic review and meta-analysis of longitudinal hippocampal atrophy in healthy human ageing. Neuroimage 112, 364–374. doi: 10.1016/j.neuroimage.2015.03.035
Glisky E. L. (2007). “Changes in Cognitive Function in Human Aging,” in Brain Aging: Models, Methods, and Mechanisms. Boca Raton, FL: CRC Press/Taylor & Francis.
Goral, M. (2004). First-language decline in healthy aging: implications for attrition in bilingualism. J. Neurolinguistics 17, 31–52. doi: 10.1016/s0911-6044(03)00052-6
Grober, E., Hall, C. B., Lipton, R. B., Zonderman, A. B., Resnick, S. M., and Kawas, C. (2008). Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. J. Int. Neuropsychol. Soc. 14, 266–278. doi: 10.1017/S1355617708080302
Gunning-Dixon, F. M., Brickman, A. M., Cheng, J. C., and Alexopoulos, G. S. (2009). Aging of cerebral white matter: a review of MRI findings. Int. J. Geriatr. Psychiatry 24, 109–117. doi: 10.1002/gps.2087
Harun, A., Oh, E. S., Bigelow, R. T., Studenski, S., and Agrawal, Y. (2016). Vestibular impairment in dementia. Otol. Neurotol. 37, 1137–1142. doi: 10.1097/MAO.0000000000001157
He, W., Goodkind, D., and Kowal, P. (2016). An Aging World: 2015. Suitland, MD: United States Census Bureau.
Head, D., Buckner, R., Shimony, J., Williams, L., and Akbudak, E. (2004). Differential vulnerability of anterior white matter in nondemented aging with minimal acceleration in dementia of the alzheimer type: evidence from diffusion tensor imaging. Cereb. Cortex 14, 410–423. doi: 10.1093/cercor/bhh003
Healey, M. K., and Kahana, M. J. (2016). A four-component model of age-related memory change. Psychol. Rev. 123, 23–69. doi: 10.1037/rev0000015
Hedden, T., and Gabrieli, J. D. (2004). Insights into the ageing mind: a view from cognitive neuroscience. Nat. Rev. Neurosci. 5, 87–96. doi: 10.1038/nrn1323
Hugenschmidt, C. E., Peiffer, A. M., Mccoy, T. P., Hayasaka, S., and Laurienti, P. J. (2009). Preservation of crossmodal selective attention in healthy aging. Exp. Brain Res. 198, 273–285. doi: 10.1007/s00221-009-1816-3
Jack, C. R., Lowe, V. J., Weigand, S. D., Wiste, H. J., Senjem, M. L., Knopman, D. S., et al. (2009). Serial PIB and MRI in normal, mild cognitive impairment and Alzheimers disease: implications for sequence of pathological events in Alzheimers disease. Brain 132, 1355–1365. doi: 10.1093/brain/awp062
Jack, C. R., Shiung, M. M., Weigand, S. D., Obrien, P. C., Gunter, J. L., Boeve, B. F., et al. (2005). Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 65, 1227–1231. doi: 10.1212/01.wnl.0000180958.22678.91
Jernigan, T. L., Archibald, S. L., Fennema-Notestine, C., Gamst, A. C., Stout, J. C., Bonner, J., et al. (2001). Effects of age on tissues and regions of the cerebrum and cerebellum. Neurobiol. Aging 22, 581–594. doi: 10.1016/s0197-4580(01)00217-2
Johnson, D. K., Storandt, M., Morris, J. C., and Galvin, J. E. (2009). Longitudinal study of the transition from healthy aging to alzheimer disease. Arch. Neurol. 66, 1254–1259. doi: 10.1001/archneurol.2009.158
Johnson, S. C., Rue, A. L., Hermann, B. P., Xu, G., Koscik, R. L., Jonaitis, E. M., et al. (2011). The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE 𝜀3/𝜀3 genotype. Alzheimers Dement. 7, 456–465. doi: 10.1016/j.jalz.2010.11.012
Karlamangla, A. S., Lachman, M. E., Han, W., Huang, M., and Greendale, G. A. (2017). Evidence for cognitive aging in midlife women: study of women’s health across the nation. PLoS One 12:e0169008. doi: 10.1371/journal.pone.0169008
Kawas, C. H., Corrada, M. M., Brookmeyer, R., Morrison, A., Resnick, S. M., Zonderman, A. B., et al. (2003). Visual memory predicts Alzheimer’s disease more than a decade before diagnosis. Neurology 60, 1089–1093. doi: 10.1212/01.WNL.0000055813.36504.BF
Kochunov, P., Williamson, D. E., Lancaster, J., Fox, P., Cornell, J., Blangero, J., et al. (2012). Fractional anisotropy of water diffusion in cerebral white matter across the lifespan. Neurobiol. Aging 33, 9–20. doi: 10.1016/j.neurobiolaging.2010.01.014
Lachman, M. E., Teshale, S., and Agrigoroaei, S. (2015). Midlife as a pivotal period in the life course: balancing growth and decline at the crossroads of youth and old age. Int. J. Behav. Dev. 39, 20–31. doi: 10.1177/0165025414533223
Laukka, E. J., Macdonald, S. W., Fratiglioni, L., and Bäckman, L. (2012). Preclinical cognitive trajectories differ for Alzheimer’s disease and vascular dementia. J. Int. Neuropsychol. Soc. 18, 191–199. doi: 10.1017/S1355617711001718
Lebel, C., Gee, M., Camicioli, R., Wieler, M., Martin, W., and Beaulieu, C. (2012). Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage 60, 340–352. doi: 10.1016/j.neuroimage.2011.11.094
Leong, R. L., Lo, J. C., Sim, S. K., Zheng, H., Tandi, J., Zhou, J., et al. (2017). Longitudinal brain structure and cognitive changes over 8 years in an East Asian cohort. Neuroimage 147, 852–860. doi: 10.1016/j.neuroimage.2016.10.016
Lezak, M., Howieson, D., Bigler, E., and Tranel, D. (2012). Neuropsychological Assessment, 5 Edn. New York, NY: Oxford University Press.
Lindenberger, U. (2014). Human cognitive aging: corriger la fortune? Science 346, 572–578. doi: 10.1126/science.1254403
Livingston, G., Sommerlad, A., Orgeta, V., Costafreda, S. G., Huntley, J., Ames, D., et al. (2017). Dementia prevention, intervention, and care. Lancet 390, 2673–2734. doi: 10.1016/s0140-6736(17)31363-6
Logiudice, D., and Watson, R. (2014). Dementia in older people: an update. Intern. Med. J. 44, 1066–1073. doi: 10.1111/imj.12572
McDowd, J. M., and Shaw, R. J. (2000). “Attention and aging: a functional perspective,” in The Handbook of Aging and Cognition, eds T. A. Salthouse and F. I. M. Craik (Mahwah, NJ: Erlbaum).
Michaelson, D. M. (2014). APOE 𝜀4: the most prevalent yet understudied risk factor for Alzheimers disease. Alzheimers Dement. 10, 861–868. doi: 10.1016/j.jalz.2014.06.015
Miller, M. I., Ratnanather, J. T., Tward, D. J., Brown, T., Lee, D. S., Ketcha, M., et al. (2015). Network neurodegeneration in Alzheimer’s disease via MRI based shape diffeomorphometry and high-field atlasing. Front. Bioeng. Biotechnol. 3:54. doi: 10.3389/fbioe.2015.00054
Minder, C. E., Müller, T., Gillmann, G., Beck, J. C., and Stuck, A. E. (2002). Subgroups of refusers in a disability prevention trial in older adults: baseline and follow-up analysis. Am. J. Public Health 92, 445–450. doi: 10.2105/ajph.92.3.445
Mistridis, P., Krumm, S., Monsch, A. U., Berres, M., and Taylor, K. I. (2015). The 12 years preceding mild cognitive impairment due to Alzheimer’s disease: the temporal emergence of cognitive decline. J. Alzheimers Dis. 48, 1095–1107. doi: 10.3233/jad-150137
Mondadori, C. R. A., De Quervain, D. J.-F., Buchmann, A., Mustovic, H., Wollmer, M. A., Schmidt, C. F., et al. (2006). Better memory and neural efficiency in young apolipoprotein E 4 carriers. Cereb. Cortex 17, 1934–1947. doi: 10.1093/cercor/bhl103
Mortamais, M., Ash, J. A., Harrison, J., Kaye, J., Kramer, J., Randolph, C., et al. (2017). Detecting cognitive changes in preclinical Alzheimers disease: a review of its feasibility. Alzheimers Dement. 13, 468–492. doi: 10.1016/j.jalz.2016.06.2365
Nicholas, M., Obler, L., Albert, M., and Goodglass, H. (1985). Lexical retrieval in healthy aging. Cortex 21, 595–606. doi: 10.1016/s0010-9452(58)80007-6
Nyberg, L., Lovden, M., Riklund, K., Lindenberger, U., and Backman, L. (2012). Memory aging and brain maintenance. Trends Cogn. Sci. 16, 292–305. doi: 10.1016/j.tics.2012.04.005
Persson, J., Lind, J., Larsson, A., Ingvar, M., Cruts, M., Broeckhoven, C. V., et al. (2006). Altered brain white matter integrity in healthy carriers of the APOE 4 allele: a risk for AD? Neurology 66, 1029–1033. doi: 10.1212/01.wnl.0000204180.25361.48
Piaceri, I., Nacmias, B., and Sorbi, S. (2013). Genetics of familial and sporadic Alzheimer s disease. Front. Biosci. E5:167–177. doi: 10.2741/E605
Prince, M. J., Wu, F., Guo, Y., Gutierrez Robledo, L. M., O’Donnell, M., Sullivan, R., et al. (2015). The burden of disease in older people and implications for health policy and practice. Lancet 385, 549–562. doi: 10.1016/S0140-6736(14)61347-7
Rajan, K. B., Wilson, R. S., Weuve, J., Barnes, L. L., and Evans, D. A. (2015). Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology 85, 898–904. doi: 10.1212/wnl.0000000000001774
Ramsay, C. B., Nicholas, M., Au, R., Obler, L. K., and Albert, M. L. (1999). Verb naming in normal aging. Appl. Neuropsychol. 6, 57–67. doi: 10.1207/s15324826an0602_1
Raz, N., Lindenberger, U., Rodrigue, K. M., Kennedy, K. M., Head, D., Williamson, A., et al. (2005). Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb. Cortex 15, 1676–1689. doi: 10.1093/cercor/bhi044
Raz, N., and Rodrigue, K. M. (2006). Differential aging of the brain: patterns, cognitive correlates and modifiers. Neurosci. Biobehav. Rev. 30, 730–748. doi: 10.1016/j.neubiorev.2006.07.001
Resnick, S. M., Pham, D. L., Kraut, M. A., Zonderman, A. B., and Davatzikos, C. (2003). Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J. Neurosci. 23, 3295–3301. doi: 10.1523/jneurosci.23-08-03295.2003
Ritchie, K., Carrière, I., Su, L., Obrien, J. T., Lovestone, S., Wells, K., et al. (2017). The midlife cognitive profiles of adults at high risk of late-onset Alzheimers disease: the PREVENT study. Alzheimers Dement. 13, 1089–1097. doi: 10.1016/j.jalz.2017.02.008
Ritchie, K., Ritchie, C. W., Yaffe, K., Skoog, I., and Scarmeas, N. (2015). Is late-onset Alzheimers disease really a disease of midlife? Alzheimers Dement. 1, 122–130. doi: 10.1016/j.trci.2015.06.004
Rönnlund, M., Lövdén, M., and Nilsson, L. (2007). Cross-sectional versus longitudinal age gradients of tower of Hanoi performance: the role of practice effects and cohort differences in education. Aging Neuropsychol. Cogn. 15, 40–67. doi: 10.1080/13825580701533751
Rönnlund, M., Nyberg, L., Bäckman, L., and Nilsson, L. (2005). Stability, growth, and decline in adult life span development of declarative memory: cross-sectional and longitudinal data from a population-based study. Psychol. Aging 20, 3–18. doi: 10.1037/0882-7974.20.1.3
Sacuiu, S. F. (2016). Dementias. Handb. Clin. Neurol. 138, 123–151. doi: 10.1016/B978-0-12-802973-2.00008-2
Salthouse, T. A. (2010). Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology 24, 563–572. doi: 10.1037/a0019026
Salthouse, T. A. (2014). Why are there different age relations in cross-sectional and longitudinal comparisons of cognitive functioning? Curr. Dir. Psychol. Sci. 23, 252–256. doi: 10.1177/0963721414535212
Salthouse, T. A., Atkinson, T. M., and Berish, D. E. (2003). Executive functioning as a potential mediator of age-related cognitive decline in normal adults. J. Exp. Psychol. Gen. 132, 566–594. doi: 10.1037/0096-3445.132.4.566
Sexton, C. E., Kalu, U. G., Filippini, N., Mackay, C. E., and Ebmeier, K. P. (2011). A meta-analysis of diffusion tensor imaging in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 32, 2322.e5-2322.e18. doi: 10.1016/j.neurobiolaging.2010.05.019
Sexton, C. E., Walhovd, K. B., Storsve, A. B., Tamnes, C. K., Westlye, L. T., Johansen-Berg, H., et al. (2014). Accelerated changes in white matter microstructure during aging: a longitudinal diffusion tensor imaging study. J. Neurosci. 34, 15425–15436. doi: 10.1523/jneurosci.0203-14.2014
Shafto, M. A., and Tyler, L. K. (2014). Language in the aging brain: the network dynamics of cognitive decline and preservation. Science 346, 583–587. doi: 10.1126/science.1254404
Sowell, E. R., Peterson, B. S., Thompson, P. M., Welcome, S. E., Henkenius, A. L., and Toga, A. W. (2003). Mapping cortical change across the human life span. Nat. Neurosci. 6, 309–315. doi: 10.1038/nn1008
Staub, B., Doignon-Camus, N., Després, O., and Bonnefond, A. (2013). Sustained attention in the elderly: what do we know and what does it tell us about cognitive aging? Ageing Res. Rev. 12, 459–468. doi: 10.1016/j.arr.2012.12.001
Storsve, A. B., Fjell, A. M., Tamnes, C. K., Westlye, L. T., Overbye, K., Aasland, H. W., et al. (2014). Differential longitudinal changes in cortical thickness, surface area and volume across the adult life span: regions of accelerating and decelerating change. J. Neurosci. 34, 8488–8498. doi: 10.1523/jneurosci.0391-14.2014
Stratmann, K., Heinsen, H., Korf, H., Del Turco, D., Ghebremedhin, E., Seidel, K., et al. (2016). Precortical phases of AD. Brain Pathol. 26, 371–386. doi: 10.1111/bpa.12289
Terribilli, D., Schaufelberger, M. S., Duran, F. L., Zanetti, M. V., Curiati, P. K., Menezes, P. R., et al. (2011). Age-related gray matter volume changes in the brain during non-elderly adulthood. Neurobiol. Aging 32, 354–368. doi: 10.1016/j.neurobiolaging.2009.02.008
Thompson, P. M., Hayashi, K. M., De Zubicaray, G. I., Janke, A. L., Rose, S. E., Semple, J., et al. (2004). Mapping hippocampal and ventricular change in Alzheimer disease. Neuroimage 22, 1754–1766. doi: 10.1016/j.neuroimage.2004.03.040
Tsang, P. S., and Shaner, T. L. (1998). Age, attention, expertise, and time-sharing performance. Psychol. Aging 13, 323–347. doi: 10.1037/0882-7974.13.2.323
Turner, G. R., and Spreng, R. N. (2012). Executive Functions and Neurocognitive Aging: Dissociable Patterns of Brain Activity. doi: 10.1016/j.neurobiolaging.2011.06.005
Van Beijsterveldt, C. E., van Boxtel, M. P., Bosma, H., Houx, P. J., Buntinx, F., and Jolles, J. (2002). Predictors of attrition in a longitudinal cognitive aging study: the Maastricht Aging Study (MAAS). J. Clin. Epidemiol. 55, 216–223. doi: 10.1016/S0895-4356(01)00473-5
Westlye, L. T., Reinvang, I., Rootwelt, H., and Espeseth, T. (2012). Effects of APOE on brain white matter microstructure in healthy adults. Neurology 79, 1961–1969. doi: 10.1212/wnl.0b013e3182735c9c
Westlye, L. T., Walhovd, K. B., Dale, A. M., Bjørnerud, A., Due-Tønnessen, P., Engvig, A., et al. (2010). Life-span changes of the human brain white matter: diffusion tensor imaging (DTI) and volumetry. Cereb. Cortex 20, 2055–2068. doi: 10.1093/cercor/bhp280
Yeung, L., Olsen, R. K., Bild-Enkin, H. E., Dangelo, M. C., Kacollja, A., Mcquiggan, D. A., et al. (2017). Anterolateral entorhinal cortex volume predicted by altered intra-item configural processing. J. Neurosci. 37, 5527–5538. doi: 10.1523/jneurosci.3664-16.2017
Keywords: aging, early markers, midlife, Alzheimer’s disease, dementia, cognition and brain structure
Citation: Irwin K, Sexton C, Daniel T, Lawlor B and Naci L (2018) Healthy Aging and Dementia: Two Roads Diverging in Midlife? Front. Aging Neurosci. 10:275. doi: 10.3389/fnagi.2018.00275
Received: 04 July 2018; Accepted: 27 August 2018;
Published: 19 September 2018.
Edited by:
Daniel Ortuño-Sahagún, Universidad de Guadalajara, MexicoReviewed by:
Eric Anson, University of Rochester, United StatesMarisol Godínez-Rubí, Universidad de Guadalajara, Mexico
Copyright © 2018 Irwin, Sexton, Daniel, Lawlor and Naci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorina Naci, bmFjaWxAdGNkLmll