 Emmanuelle Duron1,2,3,4*
Emmanuelle Duron1,2,3,4* Jean-Sébastien Vidal1,5Dominique Grousselle2
Jean-Sébastien Vidal1,5Dominique Grousselle2 Audrey Gabelle6
Audrey Gabelle6 Sylvain Lehmann7Florence Pasquier8Stéphanie Bombois8
Sylvain Lehmann7Florence Pasquier8Stéphanie Bombois8 Luc Buée9Bernadette Allinquant2Susanna Schraen-Maschke9,10Christiane Baret10
Luc Buée9Bernadette Allinquant2Susanna Schraen-Maschke9,10Christiane Baret10 Anne-Sophie Rigaud1,5Olivier Hanon1,5
Anne-Sophie Rigaud1,5Olivier Hanon1,5 Jacques Epelbaum2,11
Jacques Epelbaum2,11- 1AP-HP, Hôpital Broca, Service de Gériatrie, Paris, France
- 2Université Sorbonne Paris Cité, UMR-S894, INSERM Université Paris Descartes, Centre de Psychiatrie et Neuroscience, Paris, France
- 3APHP, Hôpital Paul Brousse, Service de Gériatrie du Dr Karoubi, Villejuif, France
- 4Université Paris-Sud 11, Centre de Recherche en Épidemiologie et Santé des Population- Depression et Antidépresseurs, INSERM UMR-1178, Le Kremlin-Bicêtre, France
- 5Université Paris Descartes, Sorbonne Paris Cité, Paris, France
- 6Memory Research and Resources Center, Gui de Chauliac Hospital, University of Montpellier, Montpellier, France
- 7Laboratoire de Biochimie Protéomique Clinique, CHU Montpellier, University of Montpellier, INSERM, Montpellier, France
- 8University of Lille, INSERM 1171, CHU, Centre Mémoire (CMRR) Distalz, Lille, France
- 9Univ Lille, Inserm, UMRS 1172, LabEx DISTALZ, Lille, France
- 10UF de Neurobiologie, Centre Biologie Pathologie du CHU-Lille, Lille, France
- 11MECADEV UMR 7179 CNRS, Muséum National d’Histoire Naturelle, Paris, France
A combination of low cerebrospinal fluid (CSF) Amyloid β1–42 (Aβ1–42) and high Total-Tau (T-Tau) and Phosphorylated-Tau (P-Tau) occurs at a prodromal stage of Alzheimer’s disease (AD) and recent findings suggest that network abnormalities and interneurons dysfunction contribute to cognitive deficits. Somatostatin (SOM) and Neuropeptide Y (NPY) are two neuropeptides which are expressed in GABAergic interneurons with different fates in AD the former only being markedly affected. The aim of this study was to analyze CSF SOM, NPY and CSF Aβ1–42; T-Tau, P-Tau relationships in 43 elderly mild cognitively impairment (MCI) participants from the Biomarker of AmyLoïd pepTide and AlZheimer’s disease Risk (BALTAZAR) cohort. In these samples, CSF SOM and CSF Aβ1–42 on the one hand, and CSF NPY and CSF T-Tau and P-Tau on the other hand are positively correlated. CSF SOM and NPY concentrations should be further investigated to determine if they can stand for early AD biomarkers.
Clinical Trial Registration: www.ClinicalTrials.gov, identifier #NCT01315639.
Introduction
Alzheimer’s disease (AD) neuropathology is characterized by intraneuronal protein clusters of hyperphosphorylated Tau protein (neurofibrillary tangles) and extracellular amyloid beta (Aβ) protein aggregation that start decades before the occurrence of clinical symptoms (Bateman et al., 2012; Epelbaum et al., 2017). Research criteria for AD includes cerebrospinal fluid (CSF) pathophysiological markers (Dubois et al., 2014): low CSF Aβ1–42 concentration reflects the brain amyloid burden, high Total-Tau (T-Tau) CSF concentration reflects the intensity of neuronal loss and high Phosphorylated-Tau (P-Tau) is believed to be a direct marker of tangle pathology. The combination of low Aβ1–42 and high T-Tau or p-Tau, which occurs at a prodromal stage of AD (Hansson et al., 2006) and even before clinical impairment (Sutphen et al., 2015) has a sensitivity of 90%–95% and a specificity of about 90% in AD diagnosis (de Souza et al., 2011; Gabelle et al., 2013; Lehmann et al., 2014). In this way, an individual patient’s risk estimation of any-type dementia can be improved by adding CSF biomarker information to clinical and imaging tests as recommended in routine care (Handels et al., 2017; Albert et al., 2018).
Recent findings suggest that network abnormalities and interneuron dysfunction contribute to cognitive deficits (Palop and Mucke, 2016). Somatostatin (SOM) and Neuropeptide Y (NPY) are two neuropeptides, co-expressed in GABAergic interneurons in cortex and hippocampus and in brain areas implicated in learning and memory (Epelbaum et al., 2009; Borbély et al., 2013). SOM concentration and expression levels are decreased in the cortex and hippocampus of AD patients (Davies et al., 1980; Gahete et al., 2010) while NPY-positive neurons are reduced in AD hippocampus (Kowall and Beal, 1988). In experimental AD models, increasing expression of Aβ1–42 with aging is associated with an early fall of SOM interneurons in the hippocampus followed by NPY-positive ones (Saiz-Sanchez et al., 2016) or a reverse pattern (Wilcock et al., 2008; Albuquerque et al., 2015). Remarkably, SOM (in its 14 amino-acid moiety) was recently observed to be the most selectively enriched oligomeric Aβ1–42 binder (Wang et al., 2017). In addition, a low level of CSF SOM correlates with cognitive deficits in AD (Tamminga et al., 1987) while CSF NPY concentrations changes are not as well documented (Gabriel et al., 1993).
To date, the relationships between CSF Aβ1–42, T-Tau, P-Tau and CSF SOM and NPY concentrations have never been reported especially in subjects suffering from cognitive impairment. The aim of this study was to decipher the concentrations of SOM and NPY in elderly subjects with cognitive impairment and analyze these relationships with the other AD biomarkers.
Materials and Methods
The Biomarker of AmyLoïd pepTide and AlZheimer’s disease Risk (BALTAZAR) study was a longitudinal, multicenter study including 23 French memory clinics. It was approved by the local ethics committee (ClinicalTrials.gov Identifier #NCT01315639). The BALTAZAR study aimed to study the relationships between plasmatic biomarkers (Aβ and soluble amyloid precursor protein α, sAPPα) and determine the relevance of these biomarkers on the risk of conversion of patient aged 70 years and older with mild cognitive impairment (MCI) according to international criteria (Petersen, 2004; Portet et al., 2006) towards AD stage. All study participants or their legal guardians provided written informed consents. The study protocol was approved by local ethics comity (Comité de Protection des Personnes, Ile de France IV Saint Louis Hospital) and conducted in accordance with the Declaration of Helsinki.
In a subsample ancillary study, we prospectively included consecutive subjects attending a geriatric day care hospital at the Broca hospital, suffering from MCI, who underwent a lumbar puncture, with CSF AD biomarkers measurements (Aβ1–42, T-Tau and P-Tau) and Magnetic Resonance Imaging (MRI) scans at baseline. Subjects with geriatric depression scale (GDS) score over 20/30 (a score over 15/30 indicating a risk of depression; Yesavage, 1988) were excluded.
Clinical and Biological Data
Demographic data, vascular risks factors: hypertension defined as self-reported diagnosis of hypertension or use of antihypertensive medications or blood pressure >140/90 mmHg, diabetes defined as history of diabetes or use of glucose-modifying medications, cardio-vascular diseases (coronary heart disease, stroke, heart failure) and treatments were recorded by a physician. The educational level was scored as elementary school, secondary school and high school diploma and above. Each participant had a physical examination performed by a physician including the calculation of the body mass index (BMI).
Fasting blood samples were collected for all subjects (complete blood count, measurement of electrolytes, total cholesterol, thyroid stimulating hormone, folate and vitamin B12, fasting blood glucose, albumin, C-Reactive Protein (CRP). APOE was genotyped in a single centralized laboratory (Centre de Biologie-Pathologie, Lille University Hospital, France) using a classical polymerase chain reaction (PCR) digestion method.
CSF SOM and NPY concentrations were measured by radioimmunoassay as previously reported for SOM (Grouselle et al., 1998) and using a NPY radioimmunoassay kit (Phoenix, Strasbourg, France). CSF T-Tau, P-Tau and Aβ1–42 concentrations were measured in the same laboratory (team 33, Biochimie Hôpital St Eloi—IRB, Montpellier) using the commercial kits: INNOTEST®hTAU Ag, INNOTEST®β-Amyloid(1–42) (Andreasen et al., 2001) and INNOTEST®Phospho-Tau(181P) (Vanderstichele et al., 2006; Fujirebio®, Belgium). The quality of the results was ensured by using validated standard operating procedures (del Campo et al., 2012) and internal quality controls (QCs). The QC coefficients of variation obtained on the CSF analyses within each lot and between lots ranged consistently below 15%. In addition, external QC procedures were used to confirm the quality of our results.
Cognitive Evaluation
All subjects underwent comprehensive cognitive assessment performed by trained neuropsychologists (Philippi et al., 2016). Episodic memory was tested by the Free and Cued Selective Reminding Test (FCSRT; Grober et al., 1988) which has be shown to predicts AD (Grober et al., 2000). Verbal fluency (Thurstone and Thurstone, 1964), executive function (Trail Making Test A and B; Tombaugh, 2004) and language production (Deloche and Hannequin, 1997) were evaluated. Depression was screened using the GDS (Yesavage, 1988). Disability was evaluated using Instrumental Activities of Daily Living (Lawton and Brody, 1969) and Activities of Daily Living (Katz et al., 1963) scales. Participants were then categorized as MCI according to international criteria (Petersen, 2004; Portet et al., 2006), amnestic MCI (aMCI) and non-aMCI (naMCI) subtypes (Petersen, 2004).
MRI Protocol
The MRI protocol included 3D volumetric T1 weighted (W) an axial FLAIR T2W, an axial EG T2W and an axial T2W FSE. MRI analysis was centralized and analyzed by the Centre d’acquisition et de traitement d’images (CATI; Operto et al., 2016). Right and left hippocampal volume was obtained for each participant using automatic segmentation of the hippocampus (Chupin et al., 2009).The hippocampal volume was normalized using the following calculation: hippocampal volume/total brain volume × mean total brain volume.
Statistical Analysis
General, medical and cognitive characteristics were first presented in the whole sample with percentages and numbers for categorical variables and means and standard deviations (SD) for continuous variables. Means and SD of variables of interest for the different values of each categorical variable were compared by analysis of variance. Pearson’s correlation coefficients and p-value were calculated for variables of interest and each continuous variable. Two multivariate linear regression models were performed with CSF SOM and NPY as dependent variables and biological variables significant associated with SOM and NPY in univariate analysis as independent variables. All Statistical analyses were performed using R Software version 3.1.1 (R Core Team (2014). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria1. In all analyses, the 2-sided α-level of 0.05 was used for significance testing.
Results
Demographic, medical and cognitive characteristics of the 43 participants included are summarized in Table 1. The mean age (years (SD)) was 78.6 (5.4) with 53.5% of women. Fifty-six percent of the sample received at least a high school diploma, 20.9% had diabetes, 72.1% hypertension. The mean mini mental state examination (MMSE; SD) was: 27.2 (1.7)/30. MCI subtypes were aMCI and naMCI, in 69.8% and 30.2% of cases, respectively. APOE ε4 allele was present among 25% of the 43 tested participants. As shown in Table 1, these characteristics did not differ from the others participants suffering from MCI included in the BALTAZAR study at the Broca Hospital which were not tested for CSF SOM and NPY.
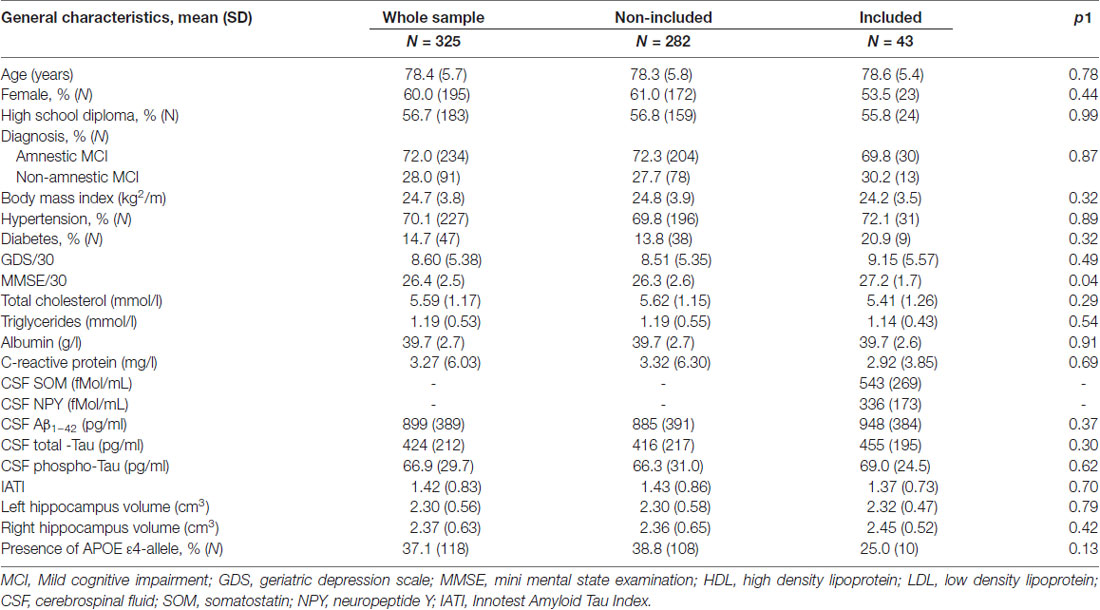
Table 1. General characteristics of the included MCI subjects compared to the non-included MCI in the Biomarker of AmyLoïd pepTide and AlZheimer’s disease Risk (BALTAZAR) study at the Broca hospital site.
The CSF SOM and NPY concentrations according to demographic, clinical, cognitive, biological (including APOE ε4 genotype) and imaging characteristics are summarized in Tables 2, 3, respectively. Factors associated with high CSF SOM concentrations were hypertension (p = 0.004), diabetes (p < 0.0001), low plasma total cholesterol (p = 0.004) and LDL cholesterol (p = 0.002), high CSF NPY concentrations (p = 0.03) and high CSF Aβ1–42 concentrations (p = 0.03; Table 2).
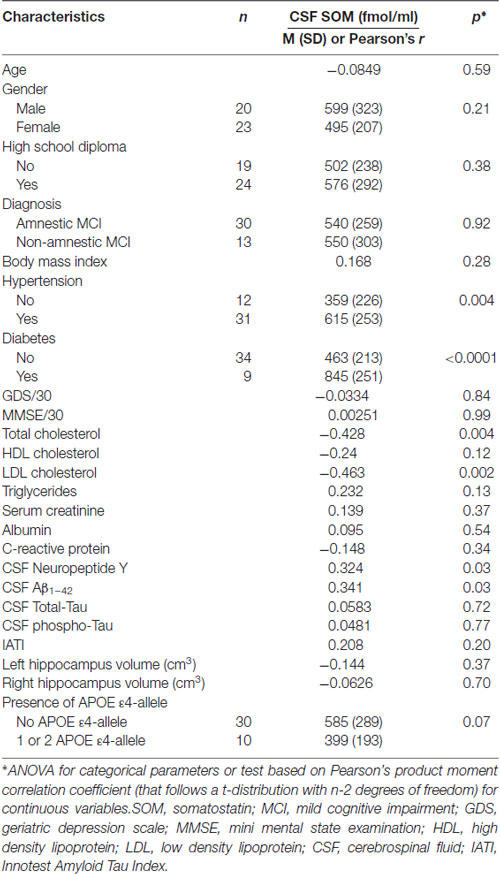
Table 2. CSF SOM according to demographic, clinical, cognitive, biological (including APOE ε4 genotype) and Magnetic Resonance Imaging (MRI) characteristics in MCI subjects.
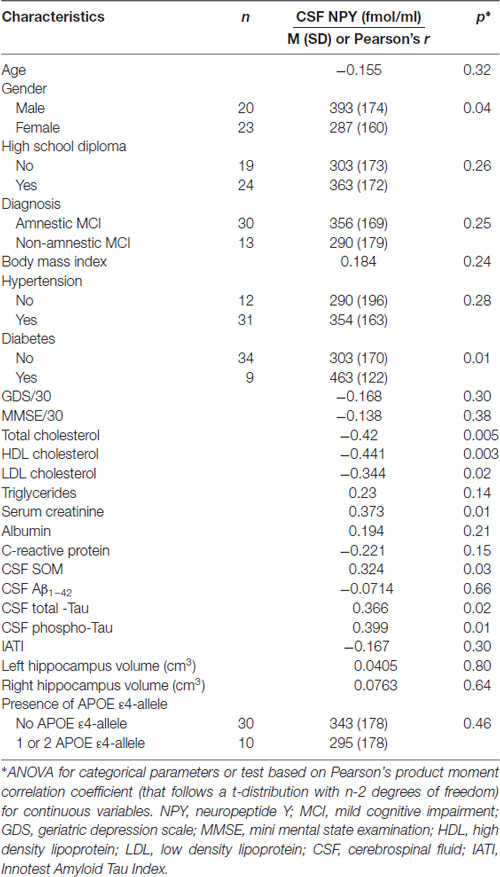
Table 3. CSF NPY according to demographic, clinical, cognitive, biological (including APOE ε4 genotype) and MRI characteristics in MCI subjects.
Factors associated with high CSF NPY concentrations were male gender (p = 0.04), diabetes (p = 0.01), low plasma total cholesterol (p = 0.005), HDL cholesterol (p = 0.003) and LDL cholesterol (p = 0.02), high serum creatinine (p = 0.01); high CSF T-Tau (p = 0.02) and P-Tau concentrations (p = 0.01) and high CSF SOM concentrations (p = 0.03; Table 3).
The results of the multivariate analysis are presented in Tables 4A,B including significant biological variables in univariate analysis. CSF SOM concentrations remained independently associated with CSF Aβ1–42 concentrations (p = 0.01) and blood total cholesterol concentrations (p = 0.03) and CSF NPY remained significantly associated with CSF T-Tau (p = 0.03). CSF SOM and CSF NPY were no more significantly associated. The Figure 1 illustrates the correlations between CSF SOM, NPY, Aβ1–42 and T-Tau in included subjects.

Table 4A. Factors associated with CSF SOM.
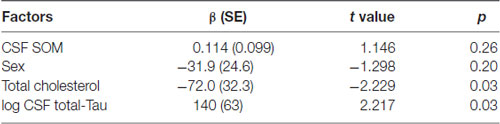
Table 4B. Factors associated with CSF NPY.
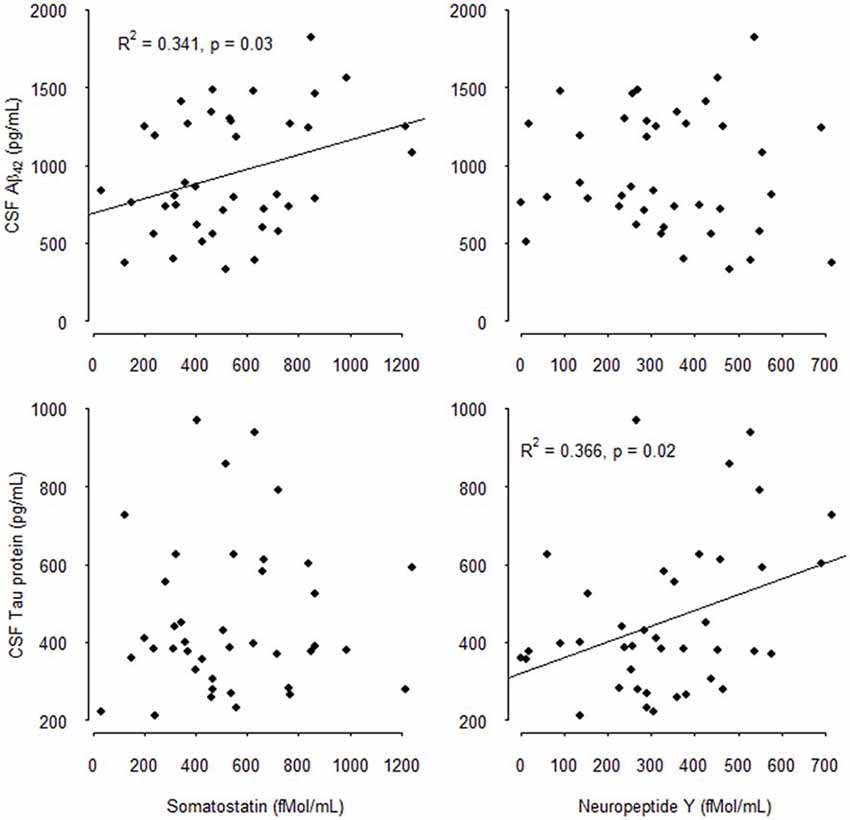
Figure 1. Correlations between cerebrospinal fluid (CSF) Somatostatin (SOM), Neuropeptide Y (NPY), Amyloid β1–42 (Aβ1–42), and total-Tau (T-Tau) in mild cognitively impairment (MCI) subjects.
Discussion
The present results indicate that high CSF SOM and high CSF Aβ1–42 concentrations on the one hand and high CSF NPY and high CSF Tau and P-Tau concentrations on the other hand, are correlated in a small population of elderly subjects suffering from MCI. Moreover, high CSF SOM concentrations are also related to diabetes and hypertension and low plasma cholesterol level. High CSF NPY concentrations are related to male gender, diabetes, and low plasma cholesterol.
Tau and P-Tau are released in the CSF very early in the AD process when neuronal damage occurs (Sperling et al., 2011). The correlation between CSF NPY and CSF T-Tau and P-Tau may reflect an early loss of a subset of hippocampal NPY/SOM neurons containing neurofibrillary tangles (Chan-Palay, 1987). However, the majority of cortical NPY-containing neurons are spared in AD (Gaspar et al., 1989; Unger and Lange, 1992). NPY may also stimulate autophagy of neurons containing neurofibrillary tangles to allow T-Tau and P-Tau clearance (Duarte-Neves et al., 2016). NPY is contained in GABA neurons (Aoki and Pickel, 1989) mainly in the amygdala, the hippocampus and the striatum (Duarte-Neves et al., 2016). NPY is neurotrophic, regulates calcium homeostasis, stimulates autophagy and attenuates inflammation processes involved in AD pathology (Duarte-Neves et al., 2016). In the APP23 mouse model, NPY is increased in mice cortex and hippocampus (Diez et al., 2000, 2003) which can reflect NPY up regulation (Duarte-Neves et al., 2016). In another AD transgenic mouse model, presenting both Presenilin 1 and APP mutations (PS1 × APP), figuring a later AD stage, hippocampal NPY neuronal densities in hippocampus are decreased (Ramos et al., 2006). Taken together, it may be suggested that, in earlier states, NPY levels are increased, and then disease progression results in NPY-interneurons loss leading to the decrease in NPY levels (Duarte-Neves et al., 2016). In AD brains, morphology of NPY-positive neurons were initially reported as altered in the cerebral cortex and in the hippocampus (Chan-Palay, 1987) and the density of NPY binding sites is decreased in those regions (Martel et al., 1990). Nevertheless, studies about NPY levels in AD CSF provided controversial results: some of them report that CSF NPY is decreased in AD compared than controls (Martignoni et al., 1992; Edvinsson et al., 1993) whereas this was not replicated in others (Atack et al., 1988; Heilig et al., 1995).
In contrast to NPY, the SOM neuronal systems of the hippocampus and cortical areas appear to be especially sensitive to aging (Martel et al., 2012; Stanley et al., 2012; French et al., 2017; Rozycka and Liguz-Lecznar, 2017; ) and cortex and hippocampal SOM concentrations are further decreased in brains of demented patients (Epelbaum et al., 1983). In frontal and temporal cortical areas, a marked decrease in the density of SOM containing neurons is observed in AD, whereas density of NPY-interneurons is unchanged (Gaspar et al., 1989). Accordingly, the proportion of single-labeled SOM neurons decreases and correlates with senile plaques, indicating that single SOM-interneurons, in cortical layers II-III and V, are preferentially affected relative to co-localized SOM-NPY neurons (Gaspar et al., 1989). More recent neuro-pathological studies show that SOM-interneurons, in the anterior olfactory nucleus, the piriform cortex (with co-localization of Aβ1–42; Saiz-Sanchez et al., 2015), the amygdala and entorhinal cortex are early involved in AD process (Saiz-Sanchez et al., 2016). The association between CSF Aβ1–42 and SOM concentrations may be explained by the fact that Aβ seems to specially affect SOM neurons. SOM and Aβ1–42 have complex physical interactions (Wang et al., 2017). Moreover SOM increases neprilysin activity involved in Aβ degradation (Saito et al., 2005; Epelbaum et al., 2009). SOM also regulates Aβ1–42 catabolism by modulating insulin degrading enzyme proteolytic activity (Tundo et al., 2012). Furthermore, Aβ intra-hippocampal injections in rats induced aberrant inhibitory network activity associated with an impairment of hippocampal memory processes (Villette et al., 2010). This effect can be explained by the selective disappearance of hippocampal projecting neurons containing SOM (Villette et al., 2012).
The relationships between CSF neuropeptide concentrations, hypertension, diabetes and fasting blood cholesterol levels are particularly interesting in view of the metabolic and non-clinical abnormalities, such as alterations in body weight and neuroendocrine functions, which often precede the cognitive decline (Low and Singer, 2008; McGrath et al., 2017). A polymorphism in the NPY gene resulting in a change of leucine 7 to proline in the signal peptide is associated with elevated cholesterol levels, higher alcohol consumption, and may be a risk factor for various metabolic and cardiovascular diseases (de Luis et al., 2016). Another one, the IVSI-100G/T polymorphism, influences cholesterol levels in the CSF and this effect is more pronounced in a German population of AD patients than in their controls (Kolsch et al., 2006). However, it does not influence plasma cholesterol levels. In the case of SOM, fasting plasma concentration of N-Terminal proSOM, a surrogate marker of SOM —given the very short half-life of the bioactive peptide— independently predicts development of coronary artery disease and both all-cause and cardiovascular mortality in the 5,389 fasting participants of the population-based study Malmö Preventive Project (Hedback et al., 2016) and in the 8,134 participants from the Prevention of Renal and Vascular End-stage Disease (PREVEND) study in Groningen (Abbasi et al., 2017). Moreover, higher levels of circulating N-terminal-prosomatostatin are associated with increased incidence of vascular dementia in the prospective population-based Malmö Preventive Project (Holm et al., 2017). The association between high CSF SOM concentrations and low plasma cholesterol levels in MCI subjects may be associated with weight loss and lower cholesterol level as observed in the early stages of AD (Duron and Hanon, 2008). Nevertheless, CSF SOM concentrations do not necessarily reflect plasma SOM concentrations.
SOM and its receptors are potential pharmacological targets for AD. FK962, which promotes SOM production, in the brain, co-administered with donepezil, enhances cognition in rats (McCarthy et al., 2011). In AD patients (Craft et al., 1999) as in cognitively normal subjects (Watson et al., 2009), intravenous administration of octreotide, a SOM 1 receptor agonist, facilitates memory.
The present study is limited by its monocentric design, the small sample size and neuropsychological diagnosis heterogeneity. Moreover, in the subsample studied, mean MMSE was slightly higher than in non included subjects’ mean MMSE. It should be noted that MMSE provides only a global assessment of cognition, not precise enough to detect slight differences, At any rate, MMSE was neither correlated with CSF SOM nor CSF NPY. Nevertheless, it would be interesting to know whether the concentrations of both peptides in MCI cases are altered as compared to a control population but this raises a number of ethical issues.
Nevertheless, this is the first report of a relationship between CSF SOM and NPY, two peptides produced by subsets of GABergic interneurons, and the CSF AD markers (CSF Aβ1–42 and T-Tau) in an exhaustively phenotyped population.
We report for the first time an independent relationship between CSF SOM and NPY, two peptides known to be affected in AD pathophysiology, and CSF markers of the two pathophysiological processes in AD: Aβ1–42 deposition and hyper phosphorylated Tau aggregation. The present study needs to be replicated in a larger sample size. If those results are confirmed additional interest of CSF SOM and NPY to already known AD biomarkers could be tested.
Author Contributions
ED, DG, AG, CB, BA, and SL performed measurements and collected data. J-SV performed statistical analysis. FP, LB, SS-M, A-SR, OH, and JE supervised the study. SB collected data revised the manuscript.
Funding
This research was supported by grants from ANR (ANR-10-MALZ-0003), Gerond’IF (Gerontopôle d’ile de France), the Sorbonne Paris Cité Dynamique du vieillir programme and the Programme Hospitalier de Recherche Clinique 2009 BALTAZAR (Biomarker of AmyLoïd pepTide and AlZheimer’s disease Risk), French ministry of health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Cecile Viollet for support in technical realization.
Footnotes
References
Abbasi, A., Kieneker, L. M., Corpeleijn, E., Gansevoort, R. T., Gans, R. O. B., Struck, J., et al. (2017). Plasma N-terminal prosomatostatin and risk of incident cardiovascular disease and all-cause mortality in a prospective observational cohort: the PREVEND study. Clin. Chem. 63, 278–287. doi: 10.1373/clinchem.2016.259275
Albert, M., Zhu, Y., Moghekar, A., Mori, S., Miller, M. I., Soldan, A., et al. (2018). Predicting progression from normal cognition to mild cognitive impairment for individuals at 5 years. Brain 141, 877–887. doi: 10.1093/brain/awx365
Albuquerque, M. S., Mahar, I., Davoli, M. A., Chabot, J., Mechawar, N., Quirion, R., et al. (2015). Regional and sub-regional differences in hippocampal GABAergic neuronal vulnerability in the TgCRND8 mouse model of Alzheimer’s disease. Front. Aging Neurosci. 7:30. doi: 10.3389/fnagi.2015.00030
Andreasen, N., Minthon, L., Davidsson, P., Vanmechelen, E., Vanderstichele, H., Winblad, B., et al. (2001). Evaluation of CSF-tau and CSF-Aβ42 as diagnostic markers for Alzheimer disease in clinical practice. Arch. Neurol. 58, 373–379. doi: 10.1001/archneur.58.3.373
Aoki, C., and Pickel, V. M. (1989). Neuropeptide Y in the cerebral cortex and the caudate-putamen nuclei: ultrastructural basis for interactions with GABAergic and non-GABAergic neurons. J. Neurosci. 9, 4333–4354. doi: 10.1523/JNEUROSCI.09-12-04333.1989
Atack, J. R., Beal, M. F., May, C., Kaye, J. A., Mazurek, M. F., Kay, A. D., et al. (1988). Cerebrospinal fluid somatostatin and neuropeptide Y. Concentrations in aging and in dementia of the Alzheimer type with and without extrapyramidal signs. Arch. Neurol. 45, 269–274. doi: 10.1001/archneur.1988.00520270043019
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Borbély, E., Scheich, B., and Helyes, Z. (2013). Neuropeptides in learning and memory. Neuropeptides 47, 439–450. doi: 10.1016/j.npep.2013.10.012
Chan-Palay, V. (1987). Somatostatin immunoreactive neurons in the human hippocampus and cortex shown by immunogold/silver intensification on vibratome sections: coexistence with neuropeptide Y neurons and effects in Alzheimer-type dementia. J. Comp. Neurol. 260, 201–223. doi: 10.1002/cne.902600205
Chupin, M., Hammers, A., Liu, R. S. N., Colliot, O., Burdett, J., Bardinet, E., et al. (2009). Automatic segmentation of the hippocampus and the amygdala driven by hybrid constraints: method and validation. Neuroimage 46, 749–761. doi: 10.1016/j.neuroimage.2009.02.013
Craft, S., Asthana, S., Newcomer, J. W., Wilkinson, C. W., Matos, I. T., Baker, L. D., et al. (1999). Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Arch. Gen. Psychiatry 56, 1135–1140. doi: 10.1001/archpsyc.56.12.1135
Davies, P., Katzman, R., and Terry, R. D. (1980). Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature 288, 279–280. doi: 10.1038/288279a0
de Luis, D. A., Izaola, O., de la Fuente, B., Primo, D., and Aller, R. (2016). Association of neuropeptide Y gene rs16147 polymorphism with cardiovascular risk factors, adipokines, and metabolic syndrome in patients with obesity. J. Nutrigenet. Nutrigenomics 9, 213–221. doi: 10.1159/000452131
de Souza, L. C., Lamari, F., Belliard, S., Jardel, C., Houillier, C., De Paz, R., et al. (2011). Cerebrospinal fluid biomarkers in the differential diagnosis of Alzheimer’s disease from other cortical dementias. J. Neurol. Neurosurg. Psychiatry 82, 240–246. doi: 10.1136/jnnp.2010.207183
del Campo, M., Mollenhauer, B., Bertolotto, A., Engelborghs, S., Hampel, H., Simonsen, A. H., et al. (2012). Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: an update. Biomark. Med. 6, 419–430. doi: 10.2217/bmm.12.46
Deloche, G., and Hannequin, D. (1997). DO 80 Epreuve de Dénomination Orale d’images. Paris: Centre de Psychologie Appliquée.
Diez, M., Danner, S., Frey, P., Sommer, B., Staufenbiel, M., Wiederhold, K., et al. (2003). Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing β-amyloid precursor protein (APP) with the Swedish double mutation (APP23). Neurobiol. Dis. 14, 579–594. doi: 10.1016/j.nbd.2003.08.003
Diez, M., Koistinaho, J., Kahn, K., Games, D., and Hökfelt, T. (2000). Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F β-amyloid precursor protein—initial observations. Neuroscience 100, 259–286. doi: 10.1016/s0306-4522(00)00261-x
Duarte-Neves, J., Pereira de Almeida, L., and Cavadas, C. (2016). Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 95, 210–224. doi: 10.1016/j.nbd.2016.07.022
Dubois, B., Feldman, H. H., Jacova, C., Hampel, H., Molinuevo, J. L., Blennow, K., et al. (2014). Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629. doi: 10.1016/S1474-4422(14)70090-0
Duron, E., and Hanon, O. (2008). Vascular risk factors, cognitive decline, and dementia. Vasc. Health Risk Manag. 4, 363–381. doi: 10.2147/VHRM.S1839
Edvinsson, L., Minthon, L., Ekman, R., and Gustafson, L. (1993). Neuropeptides in cerebrospinal fluid of patients with Alzheimer’s disease and dementia with frontotemporal lobe degeneration. Dementia 4, 167–171. doi: 10.1159/000107318
Epelbaum, S., Genthon, R., Cavedo, E., Habert, M. O., Lamari, F., Gagliardi, G., et al. (2017). Preclinical Alzheimer’s disease: a systematic review of the cohorts underlying the concept. Alzheimers Dement. 13, 454–467. doi: 10.1016/j.jalz.2016.12.003
Epelbaum, J., Guillou, J., Gastambide, F., Hoyer, D., Duron, E., and Viollet, C. (2009). Somatostatin, Alzheimer’s disease and cognition: an old story coming of age? Prog. Neurobiol. 89, 153–161. doi: 10.1016/j.pneurobio.2009.07.002
Epelbaum, J., Ruberg, M., Moyse, E., Javoy-Agid, F., Dubois, B., and Agid, Y. (1983). Somatostatin and dementia in Parkinson’s disease. Brain Res. 278, 376–379. doi: 10.1016/0006-8993(83)90277-9
French, L., Ma, T., Oh, H., Tseng, G. C., and Sibille, E. (2017). Age-related gene expression in the frontal cortex suggests synaptic function changes in specific inhibitory neuron subtypes. Front. Aging Neurosci. 9:162. doi: 10.3389/fnagi.2017.00162
Gabelle, A., Dumurgier, J., Vercruysse, O., Paquet, C., Bombois, S., Laplanche, J., et al. (2013). Impact of the 2008–2012 French Alzheimer Plan on the use of cerebrospinal fluid biomarkers in research memory center: the PLM study. J. Alzheimers Dis. 34, 297–305. doi: 10.3233/JAD-121549
Gabriel, S. M., Bierer, L. M., Harotunian, V., Purohit, D. P., Perl, D. P., and Davis, K. L. (1993). Widespread deficits in somatostatin but not neuropeptide Y concentrations in Alzheimer’s disease cerebral cortex. Neurosci. Lett. 155, 116–120. doi: 10.1016/0304-3940(93)90686-f
Gahete, M. D., Rubio, A., Durán-Prado, M., Avila, J., Luque, R. M., and Castaño, J. P. (2010). Expression of Somatostatin, cortistatin and their receptors, as well as dopamine receptors, but not of neprilysin, are reduced in the temporal lobe of Alzheimer’s disease patients. J. Alzheimers Dis. 20, 465–475. doi: 10.3233/jad-2010-1385
Gaspar, P., Duyckaerts, C., Febvret, A., Benoit, R., Beck, B., and Berger, B. (1989). Subpopulations of somatostatin 28-immunoreactive neurons display different vulnerability in senile dementia of the Alzheimer type. Brain Res. 490, 1–13. doi: 10.1016/0006-8993(89)90424-1
Grober, E., Buschke, H., Crystal, H., Bang, S., and Dresner, R. (1988). Screening for dementia by memory testing. Neurology 38, 900–903. doi: 10.1212/wnl.38.6.900
Grober, E., Lipton, R. B., Hall, C., and Crystal, H. (2000). Memory impairment on free and cued selective reminding predicts dementia. Neurology 54, 827–832. doi: 10.1212/wnl.54.4.827
Grouselle, D., Winsky-Sommerer, R., David, J. P., Delacourte, A., Dournaud, P., and Epelbaum, J. (1998). Loss of somatostatin-like immunoreactivity in the frontal cortex of Alzheimer patients carrying the apolipoprotein epsilon 4 allele. Neurosci. Lett. 255, 21–24. doi: 10.1016/s0304-3940(98)00698-3
Handels, R. L. H., Vos, S. J. B., Kramberger, M. G., Jelic, V., Blennow, K., van Buchem, M., et al. (2017). Predicting progression to dementia in persons with mild cognitive impairment using cerebrospinal fluid markers. Alzheimers Dement. 13, 903–912. doi: 10.1016/j.jalz.2016.12.015
Hansson, O., Zetterberg, H., Buchhave, P., Londos, E., Blennow, K., and Minthon, L. (2006). Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 5, 228–234. doi: 10.1016/s1474-4422(06)70355-6
Hedback, T., Almgren, P., Nilsson, P. M., and Melander, O. (2016). N-terminal prosomatostatin as a risk marker for cardiovascular disease and diabetes in a general population. J. Clin. Endocrinol. Metab. 101, 3437–3444. doi: 10.1210/jc.2016-1736
Heilig, M., Sjögren, M., Blennow, K., Ekman, R., and Wallin, A. (1995). Cerebrospinal fluid neuropeptides in Alzheimer’s disease and vascular dementia. Biol. Psychiatry 38, 210–216. doi: 10.1016/0006-3223(94)00239-y
Holm, H., Nägga, K., Nilsson, E. D., Ricci, F., Cinosi, E., Melander, O., et al. (2017). N-terminal prosomatostatin and risk of vascular dementia. Cerebrovasc. Dis. 44, 259–265. doi: 10.1159/000479940
Katz, S., Ford, A. B., Moscowitz, R. W., Jackson, B. A., and Jaffe, M. W. (1963). Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychosocial function. JAMA 185, 914–919. doi: 10.1001/jama.1963.03060120024016
Kolsch, H., Lutjohann, D., Jessen, F., Urbach, H., von Bergmann, K., Maier, W., et al. (2006). Polymorphism in neuropeptide Y influences CSF cholesterol levels but is no major risk factor of Alzheimer’s disease. J. Neural Transm. 113, 231–238. doi: 10.1007/s00702-005-0319-z
Kowall, N. W., and Beal, M. F. (1988). Cortical somatostatin, neuropeptide Y, and NADPH diaphorase neurons: normal anatomy and alterations in Alzheimer’s disease. Ann. Neurol. 23, 105–114. doi: 10.1002/ana.410230202
Lawton, M. P., and Brody, E. M. (1969). Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 9, 179–186. doi: 10.1093/geront/9.3_part_1.179
Lehmann, S., Dumurgier, J., Schraen, S., Wallon, D., Blanc, F., Magnin, E., et al. (2014). A diagnostic scale for Alzheimer’s disease based on cerebrospinal fluid biomarker profiles. Alzheimers Res. Ther. 6:38. doi: 10.1186/alzrt267
Low, P. A., and Singer, W. (2008). Management of neurogenic orthostatic hypotension: an update. Lancet Neurol. 7, 451–458. doi: 10.1016/S1474-4422(08)70088-7
Martel, J. C., Alagar, R., Robitaille, Y., and Quirion, R. (1990). Neuropeptide Y receptor binding sites in human brain. Possible alteration in Alzheimer’s disease. Brain Res. 519, 228–235. doi: 10.1016/0006-8993(90)90082-m
Martel, G., Dutar, P., Epelbaum, J., and Viollet, C. (2012). Somatostatinergic systems: an update on brain functions in normal and pathological aging. Front. Endocrinol. 3:154. doi: 10.3389/fendo.2012.00154
Martignoni, E., Blandini, F., Petraglia, F., Pacchetti, C., Bono, G., and Nappi, G. (1992). Cerebrospinal fluid norepinephrine, 3-methoxy-4-hydroxyphenylglycol and neuropeptide Y levels in Parkinson’s disease, multiple system atrophy and dementia of the Alzheimer type. J. Neural Transm. Park Dis. Dement. Sect. 4, 191–205. doi: 10.1007/bf02260903
McCarthy, A. D., Owens, I. J., Bansal, A. T., McTighe, S. M., Bussey, T. J., and Saksida, L. M. (2011). FK962 and donepezil act synergistically to improve cognition in rats: potential as an add-on therapy for Alzheimer’s disease. Pharmacol. Biochem. Behav. 98, 76–80. doi: 10.1016/j.pbb.2010.11.019
McGrath, E. R., Beiser, A. S., DeCarli, C., Plourde, K. L., Vasan, R. S., Greenberg, S. M., et al. (2017). Blood pressure from mid- to late life and risk of incident dementia. Neurology 89, 2447–2454. doi: 10.1212/WNL.0000000000004741
Operto, G., Chupin, M., Batrancourt, B., Habert, M., Colliot, O., Benali, H., et al. (2016). CATI: a large distributed infrastructure for the neuroimaging of cohorts. Neuroinformatics 14, 253–264. doi: 10.1007/s12021-016-9295-8
Palop, J. J., and Mucke, L. (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17, 777–792. doi: 10.1038/nrn.2016.141
Petersen, R. C. (2004). Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256, 183–194. doi: 10.1111/j.1365-2796.2004.01388.x
Philippi, N., Noblet, V., Duron, E., Cretin, B., Boully, C., Wisniewski, I., et al. (2016). Exploring anterograde memory: a volumetric MRI study in patients with mild cognitive impairment. Alzheimers Res. Ther. 8:26. doi: 10.1186/s13195-016-0190-1
Portet, F., Ousset, P. J., Visser, P. J., Frisoni, G. B., Nobili, F., Scheltens, P., et al. (2006). Mild cognitive impairment (MCI) in medical practice: a critical review of the concept and new diagnostic procedure. Report of the MCI Working Group of the European Consortium on Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 77, 714–718. doi: 10.1136/jnnp.2005.085332
Ramos, B., Baglietto-Vargas, D., del Rio, J. C., Moreno-Gonzalez, I., Santa-Maria, C., Jimenez, S., et al. (2006). Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging 27, 1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022
Rozycka, A., and Liguz-Lecznar, M. (2017). The space where aging acts: focus on the GABAergic synapse. Aging Cell 16, 634–643. doi: 10.1111/acel.12605
Saito, T., Iwata, N., Tsubuki, S., Takaki, Y., Takano, J., Huang, S., et al. (2005). Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat. Med. 11, 434–439. doi: 10.1038/nm1206
Saiz-Sanchez, D., De la Rosa-Prieto, C., Ubeda-Banon, I., and Martinez-Marcos, A. (2015). Interneurons, tau and amyloid-β in the piriform cortex in Alzheimer’s disease. Brain Struct. Funct. 220, 2011–2025. doi: 10.1007/s00429-014-0771-3
Saiz-Sanchez, D., Flores-Cuadrado, A., Ubeda-Bañon, I., de la Rosa-Prieto, C., and Martinez-Marcos, A. (2016). Interneurons in the human olfactory system in Alzheimer’s disease. Exp. Neurol. 276, 13–21. doi: 10.1016/j.expneurol.2015.11.009
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.007
Stanley, E. M., Fadel, J. R., and Mott, D. D. (2012). Interneuron loss reduces dendritic inhibition and GABA release in hippocampus of aged rats. Neurobiol. Aging 33, 431.e1–431.e13. doi: 10.1016/j.neurobiolaging.2010.12.014
Sutphen, C. L., Jasielec, M. S., Shah, A. R., Macy, E. M., Xiong, C., Vlassenko, A. G., et al. (2015). Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 72, 1029–1042. doi: 10.1001/jamaneurol.2015.1285
Tamminga, C. A., Foster, N. L., Fedio, P., Bird, E. D., and Chase, T. N. (1987). Alzheimer’s disease: low cerebral somatostatin levels correlate with impaired cognitive function and cortical metabolism. Neurology 37, 161–165. doi: 10.1212/wnl.37.1.161
Thurstone, L., and Thurstone, T. (1964). Manuel d’application de la Batterie Factorielle P.M.A (Primary Mental Activities). Adaptation française du Centre de Psychologie Appliquée. Paris: Centre de Psychologie Appliquée.
Tombaugh, T. N. (2004). Trail making test A and B: normative data stratified by age and education. Arch. Clin. Neuropsychol. 19, 203–214. doi: 10.1016/s0887-6177(03)00039-8
Tundo, G., Ciaccio, C., Sbardella, D., Boraso, M., Viviani, B., Coletta, M., et al. (2012). Somatostatin modulates insulin-degrading-enzyme metabolism: implications for the regulation of microglia activity in AD. PLoS One 7:e34376. doi: 10.1371/journal.pone.0034376
Unger, J. W., and Lange, W. (1992). NADPH-diaphorase-positive cell populations in the human amygdala and temporal cortex: neuroanatomy, peptidergic characteristics and aspects of aging and Alzheimer’s disease. Acta Neuropathol. 83, 636–646. doi: 10.1007/bf00299414
Vanderstichele, H., De Vreese, K., Blennow, K., Andreasen, N., Sindic, C., Ivanoiu, A., et al. (2006). Analytical performance and clinical utility of the INNOTEST PHOSPHO-TAU181P assay for discrimination between Alzheimer’s disease and dementia with Lewy bodies. Clin. Chem. Lab. Med. 44, 1472–1480. doi: 10.1515/cclm.2006.258
Villette, V., Poindessous-Jazat, F., Bellessort, B., Roullot, E., Peterschmitt, Y., Epelbaum, J., et al. (2012). A new neuronal target for β-amyloid peptide in the rat hippocampus. Neurobiol. Aging 33, 1126.e1–1126.e14. doi: 10.1016/j.neurobiolaging.2011.11.024
Villette, V., Poindessous-Jazat, F., Simon, A., Léna, C., Roullot, E., Bellessort, B., et al. (2010). Decreased rhythmic GABAergic septal activity and memory-associated theta oscillations after hippocampal amyloid-β pathology in the rat. J. Neurosci. 30, 10991–11003. doi: 10.1523/JNEUROSCI.6284-09.2010
Wang, H., Muiznieks, L. D., Ghosh, P., Williams, D., Solarski, M., Fang, A., et al. (2017). Somatostatin binds to the human amyloid β peptide and favors the formation of distinct oligomers. Elife 6:e28401. doi: 10.7554/elife.28401
Watson, G. S., Baker, L. D., Cholerton, B. A., Rhoads, K. W., Merriam, G. R., Schellenberg, G. D., et al. (2009). Effects of insulin and octreotide on memory and growth hormone in Alzheimer’s disease. J. Alzheimers Dis. 18, 595–602. doi: 10.3233/jad-2009-1165
Wilcock, D. M., Lewis, M. R., Van Nostrand, W. E., Davis, J., Previti, M. L., Gharkholonarehe, N., et al. (2008). Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J. Neurosci. 28, 1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008
Keywords: somatostatin, neuropeptide Y, peptides Aβ1–42, tau proteins, cerebrospinal fluid, mild cognitive impairment
Citation: Duron E, Vidal J-S, Grousselle D, Gabelle A, Lehmann S, Pasquier F, Bombois S, Buée L, Allinquant B, Schraen-Maschke S, Baret C, Rigaud A-S, Hanon O and Epelbaum J (2018) Somatostatin and Neuropeptide Y in Cerebrospinal Fluid: Correlations With Amyloid Peptides Aβ1–42 and Tau Proteins in Elderly Patients With Mild Cognitive Impairment. Front. Aging Neurosci. 10:297. doi: 10.3389/fnagi.2018.00297
Received: 11 July 2018; Accepted: 10 September 2018;
Published: 01 October 2018.
Edited by:
Changiz Geula, Northwestern University, United StatesReviewed by:
Franc Llorens, Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), SpainIllana Gozes, Tel Aviv University, Israel
Copyright © 2018 Duron, Vidal, Grousselle, Gabelle, Lehmann, Pasquier, Bombois, Buée, Allinquant, Schraen-Maschke, Baret, Rigaud, Hanon and Epelbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emmanuelle Duron, ZW1tYW51ZWxsZS5kdXJvbkBhcGhwLmZy