 Gen Shinozaki1*
Gen Shinozaki1* Patricia R. Braun2Benjamin W. Q. Hing1Andrew Ratanatharathorn3Mason J. Klisares1Gabrielle N. Duncan1Sydney S. Jellison1 Jonathan T. Heinzman1
Patricia R. Braun2Benjamin W. Q. Hing1Andrew Ratanatharathorn3Mason J. Klisares1Gabrielle N. Duncan1Sydney S. Jellison1 Jonathan T. Heinzman1 Yasunori Nagahama4Liesl Close4Sayeh Sabbagh1
Yasunori Nagahama4Liesl Close4Sayeh Sabbagh1 Brian J. Dlouhy4Matthew A. Howard4Hiroto Kawasaki4Hyunkeun R. Cho5
Brian J. Dlouhy4Matthew A. Howard4Hiroto Kawasaki4Hyunkeun R. Cho5- 1Department of Psychiatry, Carver College of Medicine, University of Iowa, Iowa City, IA, United States
- 2Department of Psychiatry and Behavioral Sciences, Johns Hopkins University, Baltimore, MD, United States
- 3Department of Epidemiology, School of Public Health, Columbia University Medical Center, Columbia University, New York, NY, United States
- 4Department of Neurosurgery, University of Iowa, Carver College of Medicine, Iowa City, IA, United States
- 5Department of Biostatistics, College of Public Health, University of Iowa, Iowa City, IA, United States
Background: Delirium in elderly patients is common and dangerous. Major risk factors include aging and exogenous insults, such as infection or surgery. In animal models, aging enhances pro-inflammatory cytokine release from microglia in response to exogenous insults. The epigenetic mechanism DNA methylation (DNAm) regulates gene expression and changes with age. Older individuals may have methylation changes that influence the increased cytokine upon insult, but the degree to which aging affects DNAm of cytokine genes is not fully understood.
Methods: The relationship between DNAm and aging of pro-inflammatory cytokine genes (TNF-alpha, IL1-beta, IL-6) was investigated using methylation array data in two cohorts. Brain and blood samples were collected from a neurosurgery cohort (NSG) of 21 subjects who underwent brain resection. A second cohort, the Grady Trauma Project (GTP), included blood samples from 265 subjects.
Results: In the NSG cohort, a significant negative correlation between age and DNAm in brain was found at a CpG in IL-6. With the GTP dataset, significant negative correlations between age and DNAm were seen at most of the CpGs in TNF-alpha. Also, TNF-Alpha expression increases with age. These GTP DNAm correlations were also nominally significant in NSG blood samples. In neuronal negative NSG brain tissue, a similar negative trend was observed.
Conclusions: With aging, a decrease in DNAm of cytokines gene CpGs in glia and blood was seen. As this can affect their expression, additional research is needed to fully elucidate the role of DNAm in aging and how it may influence the pathogenesis of delirium.
Introduction
Although delirium in hospitalized elderly patients is common and dangerous, it is underdiagnosed and undertreated (Inouye, 2006; Inouye et al., 2014). It is estimated that 11.8 million patients over the age of 65 who are hospitalized annually have a minimal 15–20% chance of developing delirium (2–3 million cases a year). Delirium can add over $60,000 in healthcare costs per patient, this leads to costs to the healthcare system of over $150 billion per year in the U.S. alone (Inouye et al., 2014). Extensive efforts have been made to develop screening tools that can be easily administered [e.g., the Confusion Assessment Method (CAM) (Inouye et al., 1990), CAM-ICU (Ely et al., 2001, 2004), and the Delirium Rating Scale-revised 98 (DRS-R-98; Trzepacz et al., 2001)]. Nevertheless, delirium remains seriously underdiagnosed and undertreated (Ely et al., 2004; Spronk et al., 2009). In busy hospitals, the current screening methods have been shown to have suboptimal sensitivity (38–47%) in intensive care units (Van Eijk et al., 2011; Nishimura et al., 2016). Given that our society is aging, it is becoming increasingly important to predict which patients are at risk for delirium. This will require the identification of biomarkers of delirium risk.
Elevated Cytokines Among Delirium Patients: Human Data
Because infection or surgical insult commonly trigger delirium, it has been hypothesized that delirium pathophysiology is a consequence of inflammation and inflammatory cytokines. Dillon et al. showed an association between levels of an inflammatory marker, C-reactive protein (CRP), and delirium (Dillon et al., 2017), and they also reported that the duration and severity of delirium can be predicted by high CRP levels (Vasunilashorn et al., 2017). Other groups reported associations of delirium with high levels of Galectin-3 and CRP among women in postpartum intensive care units (Zhu et al., 2017). A pro-inflammatory cytokine, IL-6, was elevated after surgery among patients with delirium compared to controls (Liu et al., 2013; Vasunilashorn et al., 2015). Thus, accumulating evidence from human studies suggests that inflammation may play key roles in the pathogenesis of delirium, but an understanding of why delirium is associated with an enhanced inflammatory response is lacking.
Enhanced Cytokines Among Aged Animals in Response to Exogenous Insults: Animal Model
A role for cytokines in cognitive disturbance is also supported by animal studies. Behavioral studies have suggested that aged rats experience cognitive disturbance after exposure to lipopolysaccharide [LPS; (Chen et al., 2008; Henry et al., 2009)] or surgical insult (Hovens et al., 2013; Wang et al., 2015). This is thought to be caused in part by an elevation of inflammatory cytokines, including IL-1beta, IL-6, and TNF-alpha, both in blood and in cerebrospinal fluid (Hovens et al., 2013; Wang et al., 2015), especially among aged animals. Such a mechanism is supported by the observation that the addition of agents that block these cytokine pathways prevents the effects of cognitive disturbances among aged animals after infection (Barrientos et al., 2012) or surgical insult (Frank et al., 2010). These animal models support roles for aging in conjunction with exogenous insults, such as infection (LPS) or surgical incision, in the pathogenesis of “delirium-like” cognitive disturbance that is mediated in party by inflammatory responses. Indeed, this combination is consistent with clinical characteristics of delirium, where the prevalence of delirium is higher in elderly patients in the hospital who have experienced infection or surgical intervention (Figure 1).
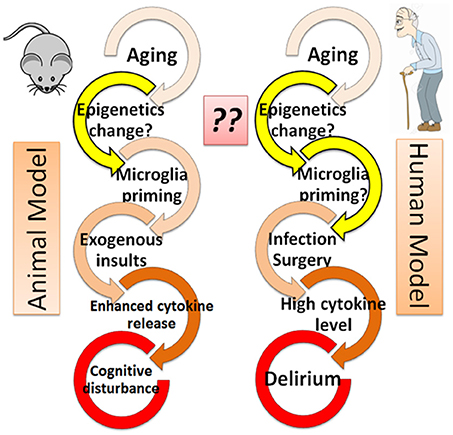
Figure 1. Epigenetics hypothesis–the missing link in the cascade from aging to delirium? Both animal and human models are shown. The role of epigenetics (yellow arrows) is not well understood and thus warrants further investigation.
Heightened Microglial Activation in Response to Exogenous Insults Among Aged Animals: Similarity to Delirium
In response to exogenous insults, microglia cells have been shown to release more cytokines in the brain of aged animals than in young animals, indicating activated microglia may contribute to the pathogenesis of cognitive decline in aged animals (Dilger and Johnson, 2008; Schreuder et al., 2017). Although the microglia response is not demonstrated in human studies, the similarity of cognitive decline in both animal models and human patients who experience exogenous insults (Figure 1) suggests that an enhanced microglial response to surgical or infectious insult may play a role in the pathophysiology of delirium (Dilger and Johnson, 2008; Schreuder et al., 2017).
Epigenetics Hypothesis: The Missing Link?
Although animal models support the notion that microglia are activated in response to exogenous insult (infection or surgery) in the context of aging, why aged animals have an enhanced cytokine release remains unclear. It is possible that the microglia response is molecularly preprogrammed prior to exposure to exogenous insult, and that this programming changes with aging. Aging is known to greatly influence gene expression in the brain (Kang et al., 2011), and such changes in gene expression are tightly regulated by epigenetic modifications, including DNA methylation (DNAm; Zampieri et al., 2015), histone modifications (Gong et al., 2015), and micro RNAs (Danka Mohammed et al., 2017). Indeed, DNAm pattens in brain have been shown to change dynamically throughout the human lifespan (Numata et al., 2012), and epigenetic processes involved in aging have been well studied (Day et al., 2013; Sen et al., 2016).
It is possible that aging-related epigenetic modifications in microglia enable an increased cytokine release in aged animals by altering the control of gene expression, and that this contributes to the pathophysiology of delirium. These epigenetic changes associated with aging could also occur in blood, where cytokines are released by monocytes and other blood cell types. Because delirium can be triggered by exogenous insults such as infection or surgery that cause inflammation in peripheral sites outside of the brain, epigenetic control of cytokine release from peripheral blood cells could also be partially responsible in the pathophysiology of delirium. Models with animals and cell cultures have shown that aging and LPS exposure affect cytokine production associated with DNAm changes (Green et al., 2015; Matt et al., 2016). Aged mice had decreased methylation in the promoter of IL-1beta in microglia at baseline and after LPS exposure in comparison to young mice. This decrease in DNAm was associated with heightened mRNA, IL-1β production, and prolonged sickness behavior (Matt et al., 2016). In another study, fibroblast taken from cows at different ages (5 or 16 months of age) were exposed to LPS, and the older cows showed an enhanced pro-inflammatory response with an increase in IL-6, IL-8, and TNF-alpha production, together with hypomethylation at promoter regions of those genes (Green et al., 2015).
To the best of our knowledge, however, no human study has investigated the role of epigenetics in delirium. Thus, it is crucial to investigate the role of epigenetics in delirium in human subjects. Our central hypothesis is that age-associated DNAm change in cytokine genes occurs in the general population, but in patients susceptible to delirium, DNAm is more significantly altered. Thus in response to exogenous insults in individuals who have differential methylation, cytokine release is more enhanced both peripherally (i.e., from monocytes in blood) and centrally (i.e., from microglia in brain), leading to delirium. Before investigating this broad hypothesis, it is necessary to determine if and in which specific CpGs/genes the aging process alters DNAm among cytokine genes in the general population.
In the present report, we used samples from two different cohorts to examine the association between aging and DNAm of cytokine genes. One cohort was a set of patients with medically intractable epilepsy from whom neurosurgically resected brain tissue and blood samples were collected simultaneously [neurosurgery cohort (NSG)]. Additionally, blood samples with DNAm and expression data were used from an independent, large cohort of 265 subjects from the Grady Trauma Project (GTP) (Smith et al., 2011).
Methods and Materials
Study Subjects and Sample Collection for NSG Cohort
A more detailed overview of study participants and sample collection process has been described previously (Braun et al., 2017; Shinozaki et al., 2017). Briefly, 21 subjects with medically intractable epilepsy undergoing neurosurgery were recruited for this study between March 2014 and April 2017 at the University of Iowa Hospitals and Clinics. This study was approved by the University of Iowa's Human Subjects Research Institutional Review Board. Written informed consent was obtained, and whole blood samples were collected in EDTA tubes, saliva with the Oragene DISCOVER™ kit (DNA Genotek Inc., OGR-500), and buccal tissue with swabs (Puritan, 25-1506 1PF TT MC). Resected brain tissue samples were immediately stored and transported on dry ice, and a portion of each brain region was sent to pathology. All samples were stored at −80°C. Typically, blood samples were taken at the end of surgery in the operating room, and saliva and buccal swabs were collected within 2 days after the operation. FACS was performed as previously described (Braun et al., 2017; Shinozaki et al., 2017).
Sample Processing and Epigenetics Platform
Methylome assays were performed as previously described. Briefly, genomic DNA from saliva, buccal, and whole blood were isolated with the MasterPureTM DNA extraction kit (Epicenter, MCD85201). DNA was bisulfite-converted using the EZ DNA Methylation™ Kit (Zymo Research, D5002). The Infinium HumanMethylationEPIC BeadChip™ Kit (Illumina, WG-317-1002) was used to analyze DNAm of 21 subjects with brain, blood, saliva, and buccal samples. Raw data was processed using the R packages RnBeads and Minfi (Aryee et al., 2014; Fortin et al., 2017); this enables quality control checks, data filtering, and normalization of the data in addition to differential methylation analyses (Aryee et al., 2014; Assenov et al., 2014; Fortin et al., 2017).
Statistical Analysis
All statistical analyses were performed in R (R Core Team, 2013). DNAm correlation was calculated with Pearson's correlation using the average methylation for each tissue. The correlation of cytokine genes was performed with CpGs from those genes present on the array. The degree of correlation between aging and DNAm was calculated for each CpG in the cytokine genes tested. The correlation coefficient and its significance level was calculated with Spearman's test.
Data From GTP Cohort
Detailed sample collection for the Grady Trauma Project can be found in Smith et al. (2011). Illumina HumanMethylation450 DNAm data was processed according to a previously described pipeline (Ratanatharathorn et al., 2017). Briefly, the Infinium protocol was assessed at each step by visual inspection of control probes, after which background normalized beta values, methylated signals, unmethylated signals, and detection p-values were exported to R (Ihaka and Gentleman, 1996). Based on the R package CpGassoc, low-intensity samples (probe detection call rates < 90% and an average intensity value less than half the overall median or 2,000 arbitrary units) and proves with detection p-values > 0.001 were removed (Barfield et al., 2012). Sex chromosome cross-hybridizing probes were also removed (Chen et al., 2013). Type I and Type II probes were normalized with Beta Mixture Quantile Normalization after which the ComBat procedure was run twice to remove chip then position effects while controlling for gender and PTSD status (Leek et al., 2012; Teschendorff et al., 2013). Prior to analysis, β-values were logit transformed into M-values (Du et al., 2010).
For the gene expression analysis, RNA was extracted from whole blood collected in Tempus tubes at about 8:30 a.m., for all participants. All samples had Bioanalyzer RNA Integrity Number (RIN) 6. Probes were considered sufficiently expressed if they had a detection p-value of < 0.01 in 5% of the samples. In total 15,877 probes met these criteria. The array was log2 transformed and normalized using the Supervised Normalization Method (Mecham et al., 2010).
Results
Patient Demographics
NSG Study
We analyzed brain and peripheral tissue samples from 21 NSG subjects, and among them, seven were female and the average age was 31 years (SD ± 16.4). The resected and analyzed brain regions included the temporal cortex (11 individuals), the frontal cortex (four individuals), the hippocampus (four individuals), and the occipital area (two individuals). Fluorescence-activated cell sorting (FACS) was performed on brain tissues to separate cells positive for a neuronal marker, resulting in six neuronal-positive (neuron) samples and 13 neuronal-negative (glia) ones with sufficient DNA quantity for analysis.
GTP Study
Detailed information about the GTP cohort has been described previously (Smith et al., 2011). Briefly, the total sample size with available DNAm and expression data is 265, 71% were female, and the average age was 42 years (SD ± 12) at the time of collection. Ninety-four percent of samples were received from African American individuals and 5% were received from Caucasian individuals.
DNA Methylation From NSG Brain and Blood Samples
We first used our dataset from the NSG study (Braun et al., 2017; Shinozaki et al., 2017) to see if DNAm patterns among cytokine genes are correlated with aging in the brain. We looked at DNAm levels and their association with age across subjects (age rage 5–62 years) in brain tissue resected during neurosurgery. We chose the pro-inflammatory cytokine genes, IL-1beta, IL-6, and TNF-alpha, to test, which includes a total of 52 CpGs. Six CpGs correlated with aging at nominal significance (p < 0.05). Given that one in 20 tests would be significant by chance alone, in 52 CpGs we expected to have 2.6 that are significant by chance. Our top hit was in IL-6 (cg23731304), which had a rho of −0.73 and a p-value of 0.00060 (Figure 2A). This was significant even after correction for multiple testing at the level of 0.05/52 = 0.00096. We then looked at this specific CpG and its DNAm trend in blood, and we found that its rho was −0.42 and the p-value was 0.075 (Figure 2B). The correlation of DNAm between brain and blood at this CpG was also significant (rho = 0.65; p-value = 0.0026) (Figure 2C).
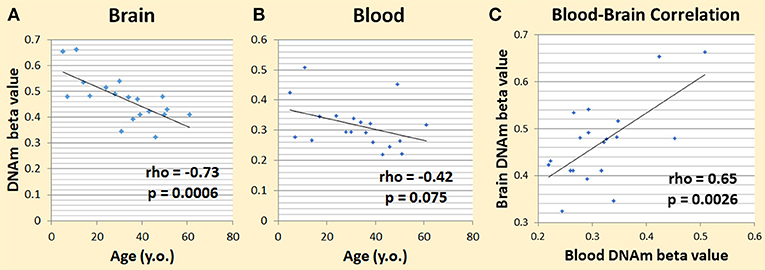
Figure 2. Preliminary data showing DNAm changes with age. (A) Changes in brain IL-6 cg23731304 DNAm and age correlated significantly. (B) Changes in blood IL-6 cg23731304 DNAm showed a similar trend as brain with aging. (C) Blood and brain DNA.m at IL-6 cg23731304 correlated significantly.
DNA Methylation and Expression From GTP Cohort, a Total of 265 Subjects' Blood Samples
Similar to the NSG preliminary analysis, we sought to investigate the association between DNAm of IL-1beta, IL-6, IL-8, and TNF-alpha and aging among the GTP cohort. The dataset included 74 CpGs, and among them DNAm levels at 14 CpGs was associated with aging at nominal significance (p < 0.05). There were 38 CpGs that were significantly associated at nominal levels with aging, predominantly with TNF-alpha represented. In fact, all 27 CpGs in TNF-alpha were at least nominally significant (Table 2). Eight CpGs were significant at genome-wide significant levels (p < 5 × E-8), all in TNF-alpha (Table 2). The top hit was at cg10650821 (r = −0.41; p-value of 2.85 × E-12) (Table 1, and Figure 3). Moreover, when expression levels of these cytokines were tested for a subgroup of 215 subjects with available expression data, TNF-alpha expression was significantly positively associated with aging as expected (r = 0.37, p = 3.84 × E-7) (Figure 4).
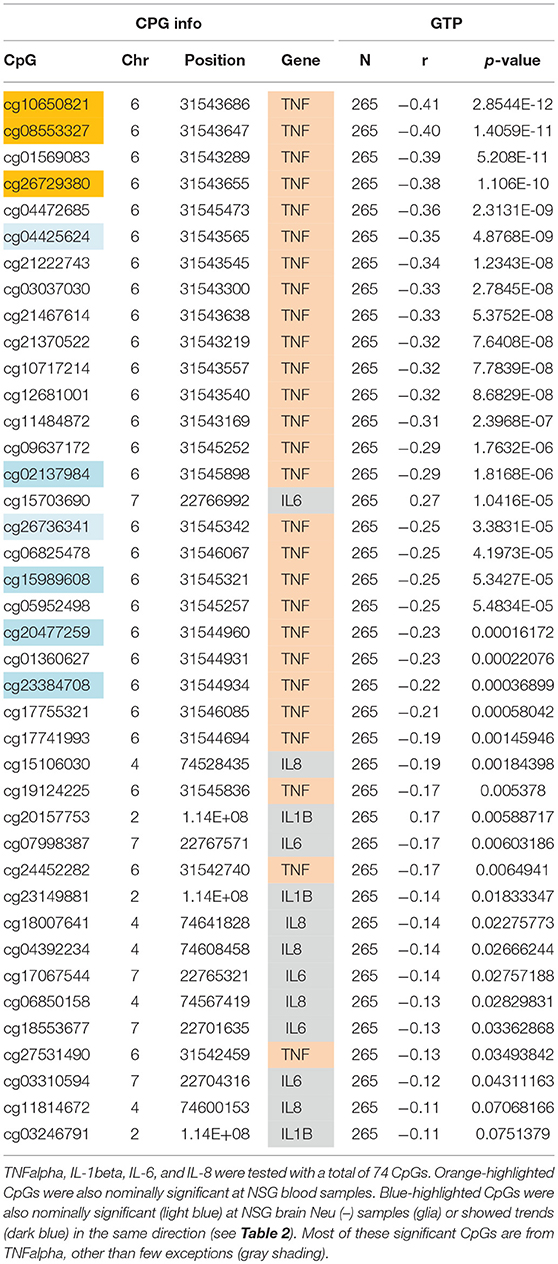
Table 1. Correlations of age and DNAm levels from blood samples obtained from the independent GTP cohort.
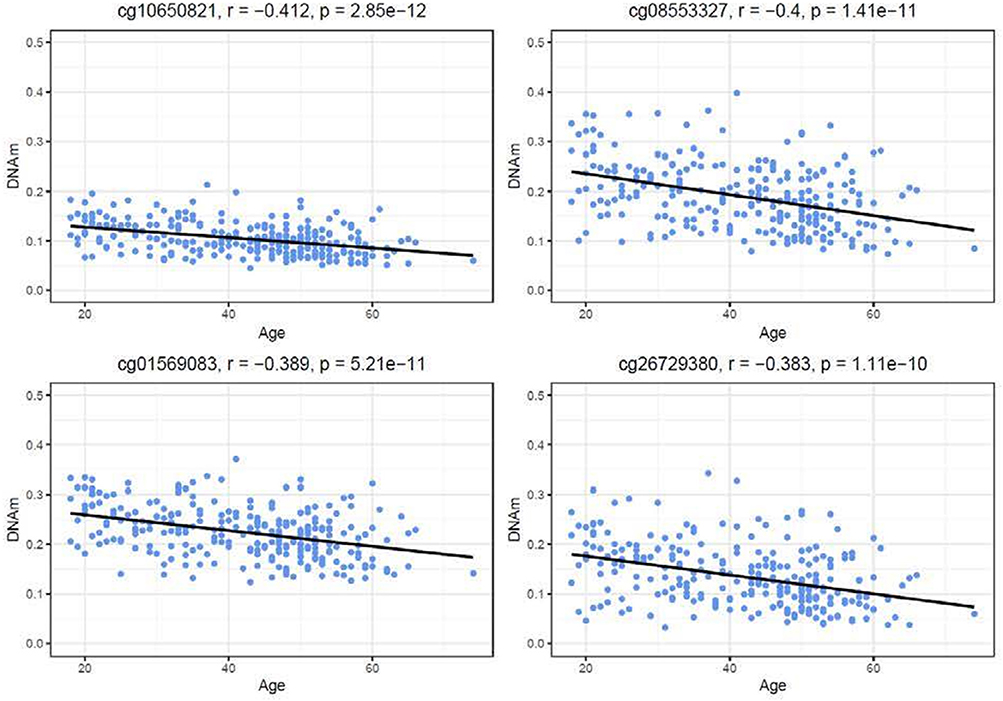
Figure 3. Correlations of age and DNAm levels from blood samples obtained from the GTP cohort. Top 4 hit CpG sites in TNFalpha and their DNAm level (beta value) in correlation with age (years) are shown.
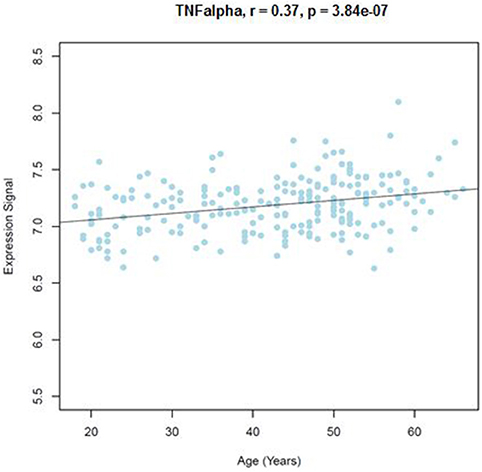
Figure 4. Correlations of age and TNFalpha expression level from blood samples obtained from the GTP cohort. TNFalpha expression level in correlation with age (years) is shown.
NSG Samples Revisited, DNA Methylation from Blood, and FACS-Sorted Brain
We sought to determine if the GTP top findings were also represented in the NSG study. Three of the top four hits (cg10650821, cg08553327, and cg26729380, all from TNF-alpha, highlighted in orange in Table 2) were correlated with aging at nominal significance in blood (N = 21), resulting in an independent cohort replicating the finding.
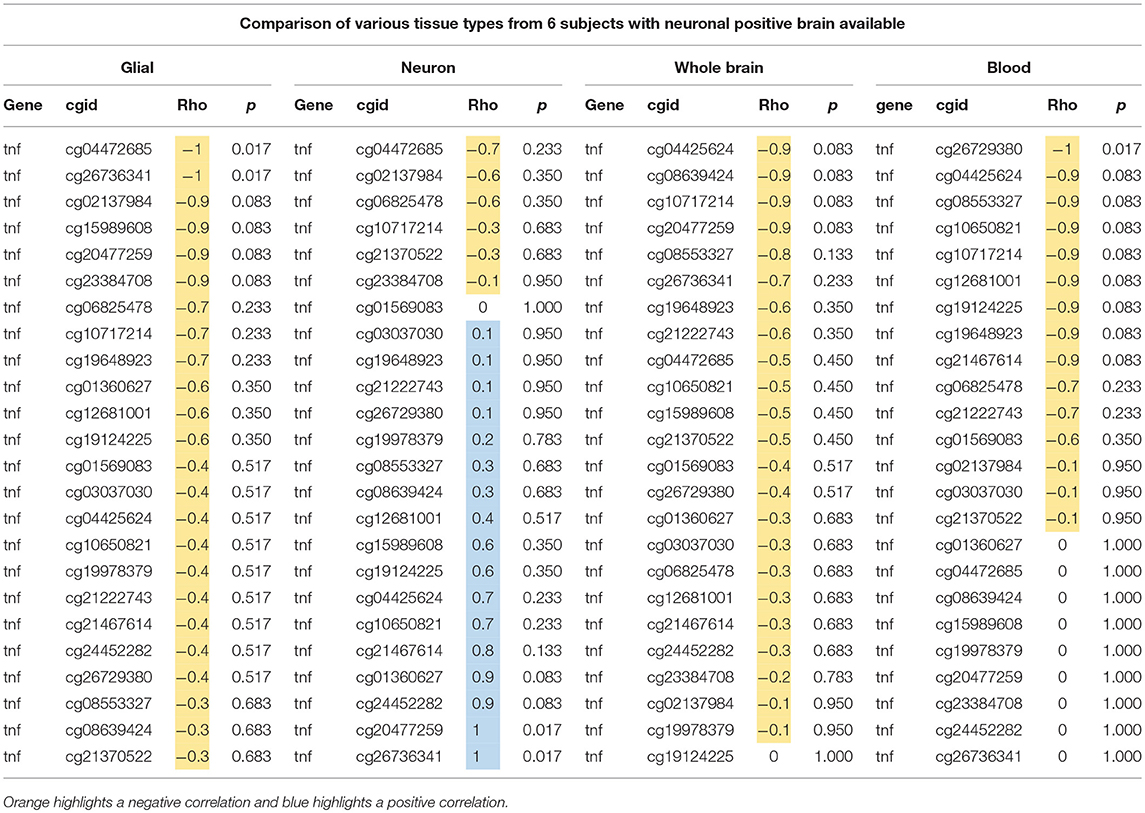
Table 2. Comparison of age and DNAm at 24 CpGs in TNF-alpha gene using glial, neuron, whole brain, and blood from six individuals.
Next, we tested if these associations of DNAm in TNF-alpha with aging in the GTP cohort were similarly present in our NSG brain tissues. We used data from just the six subjects who have samples from each tissue type (whole brain, neuronal-positive, neuronal-negative, and blood) to better compare the trend in correlation between age and DNAm at all 24 CpGs tested in the TNF-alpha gene. In both whole brain tissue and FACS-sorted neuronal-positive (neuron) cells, no trends were identified, but in FACS-sorted neuronal-negative (glial) cells nominal significant associations with aging were seen at cg04472685 and cg26736341 (p = 0.017, light blue highlight in Tables 1, 2), and similar trends were seen in next four top CpG sites (dark blue highlight in Tables 1, 2).
All 24 CpGs showed a negative correlation in glia (rho = −1~–0.3), whole brain (rho = −0.9~0), and blood (rho = −1~0), whereas neuronal-positive cells showed variable rho value ranges from negative to positive (rho = −0.7~1) (Table 2). This shows a contrast between glia and neuron cell types in trends of DNAm associating with aging, as well as the similarity in trends between glia and blood. Of note, though, the sample size was very small, which could limit the ability to detect statistically significant trends.
Discussion
The NSG data from 21 samples showed that in human brain the DNAm level of one CpG at a promotor region of the pro-inflammatory cytokine gene (IL-6) decreases with age, which could potentially associate with an increase in IL-6 expression. Also, the DNAm level of this CpG in IL-6 is correlated between brain and blood, and in blood, the DNAm shows a similar trend with aging. These findings suggest that blood DNAm of this IL-6 CpG has the utility of an epigenetic biomarker of age-related changes in brain. In fact, there is also a report that shows a low DNAm at a single CpG in IL-6 is associated with heightened expression from blood obtained from human subjects (Nile et al., 2008).
The data from an independent cohort in the GTP cohort revealed an age-associated DNAm decrease along with aging at multiple CpGs from blood samples. The pattern is very consistent with the NSG cohort. The top signal showed a genome-wide significant p-value of 2.85 × E−12. Adjusting for multiple comparison correction of the 74 CpG sites tested, 28 CpGs were significant at 0.05/74 = 0.00068, as listed in Table 2 (24 CpGs from GTP). The majority of these findings from blood DNAm were in TNF-alpha and showed a decrease with aging, which is consistent with literature (Gowers et al., 2011). Moreover, expression of TNF-alpha was positively correlated with aging as expected based on negative correlation of DNAm with age. On the contrary, expression of other pro-inflammatory cytokines except TNF-alpha was not correlated with aging from this cohort. This could indicate that other cytokines are stabilized in certain range when human body is in stable condition. However, because of underline DNAm change along with aging, when individuals are exposed to exogenous insults, they are more prone to enhanced expression, which, after a certain threshold, could put them more at risk for delirium. In fact, relationship between direct measurement of TNF-alpha level and delirium has been controversial. In patients with delirium, multiple study reported that TNF-alpha remain not significant (De Rooij et al., 2007; Çinar et al., 2014; Brum et al., 2015). Of note, sample sizes of delirium cases in these reports are less than 100 (64 cases, 15 cases, and 17 cases, respectively), thus the result might have been underpowered. Also, controlling for the level of exogenous insults can be difficult confounding factors to control. These conflicting data suggests that cytokine level itself may not work as a reliable biomarker of the risk of delirium, and DNAm may potentially provide a better underline risk factor for delirium.
The TNF-alpha results from the GTP were further interrogated in the NSG cohort, and it was shown that DNAm in almost all CpGs in TNF-alpha decrease with advanced age in blood. Furthermore, DNAm in TNF-alpha CpGs in FACS-sorted neuronal-negative tissue showed a similar decrease with aging. One CpG (cg15989608) was found to be nominally significantly associated with aging even with a limited sample size of 13 cases. This CpG was also significantly associated with aging from blood samples among the GTP cohort with a p-value 5.34 × E−5. When the NSG data was compared among six subjects who have samples from whole brain, glia, neuron and blood, a negative correlation was seen for most of TNF-alpha's 24 CpGs in glial, whole brain, and blood tissues, whereas neuronal-positive cells showed a wider range of correlations. This similar trend in glia and blood in TNF-alpha supports our hypothesis that glial cells (including microglia) and blood cells (including monocytes) could have similar DNAm changes in association with aging among pro-inflammatory cytokine genes.
It is possible that the trends we saw in DNAm decreasing with aging were due to the fact that DNAm in general tends to decrease with aging; however, as different tissues varied in their aging trends, this indicates the findings were not generalized. Thus, it is possible that age-associated decreases in DNAm level among pro-inflammatory cytokine genes are more dominant among glia and blood cells, as compared to neuronal cells.
Strengths of this report include its unique use of both blood and brain samples, in addition to FACS-sorted brain tissue, to investigate epigenetic changes in cytokine genes associated with aging. This approach using FACS-sorted brain could then compare neuron and glia cell types to find their distinct patterns of DNAm change with aging. Another strength is the use of two independent cohorts for the analysis, which allowed us to identify a persistent decrease of DNAm in TNF-alpha in two distinct cohorts. In fact, the data indicates that the result presented here can be potentially useful not only for delirium research, but for investigation of other age-related disorders, especially those where inflammatory process is playing a role.
Limitations of this report include the brain DNAm investigation is limited to 21 samples from the NSG project, with an even smaller sample size of FACS-sorted brain tissue. However, our correlation results between brain and blood at specific cytokine genes suggest that DNAm from blood could be potentially a good surrogate for that of brain, especially from glia. Unfortunately, we also did not have specific blood cell type DNAm data, such as monocytes, or specific glial cell types, such as microglia, which warrants further investigation because of our hypothesis about the role of microglia. As there is a lack of research in the epigenetics of delirium to guide the investigation of specific gene targets, we tested pro-inflammatory cytokine genes based on their potential role in delirium. In addition to these genes, however, a genome-wide approach would be of use for future studies that investigate epigenetic mechanisms specific to delirium instead of aging more broadly.
The data presented here provide evidence of DNAm-associated changes in cytokine genes with aging in blood and brain tissues, especially among glial cells. As it is possible that individuals susceptible to delirium have exacerbated or dysregulated changes in DNAm in these genes, this research provides a basis for further testing our hypothesis of epigenetic change in delirium. For this goal, we have initiated a study to collect samples from subjects with and without delirium to test genome-wide DNAm difference.
An identification of epigenetic biomarkers associated with delirium could potentially improve current practice of medicine and surgery. For example, where possible, patients who are identified to be at high risk for delirium may postpone surgery until the risk diminishes, or preventative measure could be employed to have patients closely monitored after surgery to minimize dangerous outcomes. This would allow for limited resources in the hospital to be allocated more efficiently.
In summary, we propose a hypothesis for the role DNAm on cytokine genes in delirium pathophysiology. Specifically, we hypothesize that with aging, there is a decrease in DNAm in pro-inflammatory cytokine genes, which could make them more prone to be expressed, especially in response to exogenous insults, such as infection or surgery. Thus, such inflammatory response with heightened cytokine levels could potentially lead to delirium. This preliminary investigation of DNAm associated with aging in pro-inflammatory cytokines showed that DNAm in TNF-alpha and IL-6 CpGs are negatively correlated with aging both in brain and blood tissues. For TNF-alpha, glia and blood showed similar DNAm trends with aging in contrast to neuron tissues. Importantly, this study shows that DNAm levels in cytokine genes are associated with aging, but it is necessary to determine if aged individuals susceptible to delirium have different methylation in contrast to aged individuals who do not develop delirium after exogenous insults.
Ethics Statement
This study was carried out in accordance with the recommendations of the University of Iowa IRB with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the University of Iowa IRB.
Author Contributions
GS conceptualized the study and designed the study. PB, BH, JH, MK, GD and SJ processed samples. PB, BH, AR and HC analyzed the data. YN, LC, BD, MH, and HK obtained the samples. SS organized the samples and data. GS drafted the initial manuscript. PB, BH, AR and HC revised the manuscript. All authors reviewed the manuscript and approved the content.
Funding
Authors would like to thank Drs. Kelly Ressler and Alicia Smith for their generosity in sharing data from the Grady Trauma Project. Support for GS was through a National Institutes of Health Research Career Development K Award K23MH107654. PB received training funding from the National Institutes of Health Predoctoral Training Grant T32GM008629, PI Daniel Eberl. The FACS data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran's Administration Medical Center. The use of the Becton Dickinson FACS fusion high-speed cell sorter reported in this publication was supported by the National Center for Research Resources of the National Institutes of Health under Award Number 1 S10 OD016199-01A1.
Conflict of Interest Statement
GS has disclosed that he is a founder of Predelix Medical LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AK and handling Editor declared their shared affiliation at the time of the review.
References
Aryee, M. J., Jaffe, A. E., Corrada-Bravo, H., Ladd-Acosta, C., Feinberg, A. P., Hansen, K. D., et al. (2014). Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. doi: 10.1093/bioinformatics/btu049
Assenov, Y., Müller, F., Lutsik, P., Walter, J., Lengauer, T., and Bock, C. (2014). Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138–1140. doi: 10.1038/nmeth.3115
Barfield, R. T., Kilaru, V., Smith, A. K., and Conneely, K. N. (2012). CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics 28, 1280–1281. doi: 10.1093/bioinformatics/bts124
Barrientos, R. M., Hein, A. M., Frank, M. G., Watkins, L. R., and Maier, S. F. (2012). Intracisternal interleukin-1 receptor antagonist prevents postoperative cognitive decline and neuroinflammatory response in aged rats. J. Neurosci. 32, 14641–14648. doi: 10.1523/JNEUROSCI.2173-12.2012
Braun, P., Hafner, M., Nagahama, Y., Hing, B., Mckane, M., Grossbach, A., et al. (2017). Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Eur. Neuropsychopharmacol. 27:S506. doi: 10.1016/j.euroneuro.2016.09.612
Brum, C., Stertz, L., Borba, E., Rumi, D., Kapczinski, F., and Camozzato, A. (2015). Association of serum brain-derived neurotrophic factor (BDNF) and tumor necrosis factor-alpha (TNF-alpha) with diagnosis of delirium in oncology inpatients. Rev. Bras. Psiquiatr. 37, 197–202. doi: 10.1590/1516-4446-2014-1450
Chen, J., Buchanan, J. B., Sparkman, N. L., Godbout, J. P., Freund, G. G., and Johnson, R. W. (2008). Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 22, 301–311. doi: 10.1016/j.bbi.2007.08.014
Chen, Y. A., Lemire, M., Choufani, S., Butcher, D. T., Grafodatskaya, D., Zanke, B. W., et al. (2013). Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209. doi: 10.4161/epi.23470
Çinar , M. A., Balikçi, A., Sertoglu, E., Mehmet, A. K., Serdar, M. A., and Özmenler, K. N. (2014). Role of CRP, TNF-a, and IGF-1 in delirium pathophysiology. Noro Psikiyatr. Ars. 51, 376–382. doi: 10.5152/npa.2014.6999
Danka Mohammed, C. P., Park, J. S., Nam, H. G., and Kim, K. (2017). MicroRNAs in brain aging. Mech. Ageing Dev. 168, 3–9. doi: 10.1016/j.mad.2017.01.007
Day, K., Waite, L. L., Thalacker-Mercer, A., West, A., Bamman, M. M., Brooks, J. D., et al. (2013). Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 14:R102. doi: 10.1186/gb-2013-14-9-r102
De Rooij, S. E., Van Munster, B. C., Korevaar, J. C., and Levi, M. (2007). Cytokines and acute phase response in delirium. J. Psychosom. Res. 62, 521–525. doi: 10.1016/j.jpsychores.2006.11.013
Dilger, R. N., and Johnson, R. W. (2008). Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J. Leukoc. Biol. 84, 932–939. doi: 10.1189/jlb.0208108
Dillon, S. T., Vasunilashorn, S. M., Ngo, L., Otu, H. H., Inouye, S. K., Jones, R. N., et al. (2017). Higher C-reactive protein levels predict postoperative delirium in older patients undergoing major elective surgery: a longitudinal nested case-control study. Biol. Psychiatry 81, 145–153. doi: 10.1016/j.biopsych.2016.03.2098
Du, P., Zhang, X., Huang, C. C., Jafari, N., Kibbe, W. A., Hou, L., et al. (2010). Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11:587. doi: 10.1186/1471-2105-11-587
Ely, E. W., Inouye, S. K., Bernard, G. R., Gordon, S., Francis, J., May, L., et al. (2001). Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 286, 2703–2710. doi: 10.1001/jama.286.21.2703
Ely, E. W., Stephens, R. K., Jackson, J. C., Thomason, J. W., Truman, B., Gordon, S., et al. (2004). Current opinions regarding the importance, diagnosis, and management of delirium in the intensive care unit: a survey of 912 healthcare professionals. Crit. Care Med. 32, 106–112. doi: 10.1097/01.CCM.0000098033.94737.84
Fortin, J. P., Triche, T. J. Jr., and Hansen, K. D. (2017). Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 33, 558–560. doi: 10.1093/bioinformatics/btw691
Frank, M. G., Barrientos, R. M., Hein, A. M., Biedenkapp, J. C., Watkins, L. R., and Maier, S. F. (2010). IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav. Immun. 24, 254–262. doi: 10.1016/j.bbi.2009.10.005
Gong, H., Qian, H., Ertl, R., Astle, C. M., Wang, G. G., Harrison, D. E., et al. (2015). Histone modifications change with age, dietary restriction and rapamycin treatment in mouse brain. Oncotarget 6, 15882–15890. doi: 10.18632/oncotarget.4137
Gowers, I. R., Walters, K., Kiss-Toth, E., Read, R. C., Duff, G. W., and Wilson, A. G. (2011). Age-related loss of CpG methylation in the tumour necrosis factor promoter. Cytokine 56, 792–797. doi: 10.1016/j.cyto.2011.09.009
Green, B. B., Mckay, S. D., and Kerr, D. E. (2015). Age dependent changes in the LPS induced transcriptome of bovine dermal fibroblasts occurs without major changes in the methylome. BMC Genomics 16:30. doi: 10.1186/s12864-015-1223-z
Henry, C. J., Huang, Y., Wynne, A. M., and Godbout, J. P. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Hovens, I. B., Schoemaker, R. G., Van Der Zee, E. A., Heineman, E., Nyakas, C., and Van Leeuwen, B. L. (2013). Surgery-induced behavioral changes in aged rats. Exp. Gerontol. 48, 1204–1211. doi: 10.1016/j.exger.2013.07.011
Ihaka, R., and Gentleman, R. (1996). R: a language for data analysis and graphics. J. Comput. Graphical Stat. 5, 299–314.
Inouye, S. K. (2006). Delirium in older persons. N. Engl. J. Med. 354, 1157–1165. doi: 10.1056/NEJMra052321
Inouye, S. K., Van Dyck, C. H., Alessi, C. A., Balkin, S., Siegal, A. P., and Horwitz, R. I. (1990). Clarifying confusion: the confusion assessment method. a new method for detection of delirium. Ann. Intern. Med. 113, 941–948. doi: 10.7326/0003-4819-113-12-941
Inouye, S. K., Westendorp, R. G., and Saczynski, J. S. (2014). Delirium in elderly people. Lancet 383, 911–922. doi: 10.1016/S0140-6736(13)60688-1
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. doi: 10.1038/nature10523
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E., and Storey, J. D. (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. doi: 10.1093/bioinformatics/bts034
Liu, P., Li, Y. W., Wang, X. S., Zou, X., Zhang, D. Z., Wang, D. X., et al. (2013). High serum interleukin-6 level is associated with increased risk of delirium in elderly patients after noncardiac surgery: a prospective cohort study. Chin. Med. J. 126, 3621–3627. doi: 10.3760/cma.j.issn.0366-6999.20130211
Matt, S. M., Lawson, M. A., and Johnson, R. W. (2016). Aging and peripheral lipopolysaccharide can modulate epigenetic regulators and decrease IL-1beta promoter DNA methylation in microglia. Neurobiol. Aging 47, 1–9. doi: 10.1016/j.neurobiolaging.2016.07.006
Mecham, B. H., Nelson, P. S., and Storey, J. D. (2010). Supervised normalization of microarrays. Bioinformatics 26, 1308–1315. doi: 10.1093/bioinformatics/btq118
Nile, C. J., Read, R. C., Akil, M., Duff, G. W., and Wilson, A. G. (2008). Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 58, 2686–2693. doi: 10.1002/art.23758
Nishimura, K., Yokoyama, K., Yamauchi, N., Koizumi, M., Harasawa, N., Yasuda, T., et al. (2016). Sensitivity and specificity of the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU) and the Intensive Care Delirium Screening Checklist (ICDSC) for detecting post-cardiac surgery delirium: A single-center study in Japan. Heart Lung 45, 15–20. doi: 10.1016/j.hrtlng.2015.11.001
Numata, S., Ye, T., Hyde, T. M., Guitart-Navarro, X., Tao, R., Wininger, M., et al. (2012). DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 90, 260–272. doi: 10.1016/j.ajhg.2011.12.020
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available Online at: http://www.R-project.org/
Ratanatharathorn, A., Boks, M. P., Maihofer, A. X., Aiello, A. E., Amstadter, A. B., Ashley-Koch, A. E., et al. (2017). Epigenome-wide association of PTSD from heterogeneous cohorts with a common multi-site analysis pipeline. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 619–630. doi: 10.1002/ajmg.b.32568
Schreuder, L., Eggen, B. J., Biber, K., Schoemaker, R. G., Laman, J. D., and De Rooij, S. E. (2017). Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: a systematic review. Brain Behav. Immun. 62, 362–381. doi: 10.1016/j.bbi.2017.01.010
Sen, P., Shah, P. P., Nativio, R., and Berger, S. L. (2016). Epigenetic mechanisms of longevity and aging. Cell 166, 822–839. doi: 10.1016/j.cell.2016.07.050
Shinozaki, G., Braun, P., Hing, B., Nagahama, Y., Gaul, L., Heinzman, J., et al. (2017). Genome-wide DNA methylation comparison by illumina epic array between live human brain and peripheral tissues within individuals. Biol. Psychiatry 81:S24. doi: 10.1016/j.biopsych.2017.02.068
Smith, A. K., Conneely, K. N., Kilaru, V., Mercer, K. B., Weiss, T. E., Bradley, B., et al. (2011). Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 700–708. doi: 10.1002/ajmg.b.31212
Spronk, P. E., Riekerk, B., Hofhuis, J., and Rommes, J. H. (2009). Occurrence of delirium is severely underestimated in the ICU during daily care. Intens. Care Med. 35, 1276–1280. doi: 10.1007/s00134-009-1466-8
Teschendorff, A. E., Marabita, F., Lechner, M., Bartlett, T., Tegner, J., Gomez-Cabrero, D., et al. (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196. doi: 10.1093/bioinformatics/bts680
Trzepacz, P. T., Mittal, D., Torres, R., Kanary, K., Norton, J., and Jimerson, N. (2001). Validation of the Delirium Rating Scale-revised-98: comparison with the delirium rating scale and the cognitive test for delirium. J. Neuropsychiatry Clin. Neurosci. 13, 229–242. doi: 10.1176/jnp.13.2.229
Van Eijk, M. M., Van Den Boogaard, M., Van Marum, R. J., Benner, P., Eikelenboom, P., Honing, M. L., et al. (2011). Routine use of the confusion assessment method for the intensive care unit: a multicenter study. Am. J. Respir. Crit. Care Med. 184, 340–344. doi: 10.1164/rccm.201101-0065OC
Vasunilashorn, S. M., Dillon, S. T., Inouye, S. K., Ngo, L. H., Fong, T. G., Jones, R. N., et al. (2017). High C-reactive protein predicts delirium incidence, duration, and feature severity after major noncardiac surgery. J. Am. Geriatr. Soc. 65, e109–e116. doi: 10.1111/jgs.14913
Vasunilashorn, S. M., Ngo, L., Inouye, S. K., Libermann, T. A., Jones, R. N., Alsop, D. C., et al. (2015). Cytokines and postoperative delirium in older patients undergoing major elective surgery. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1289–1295. doi: 10.1093/gerona/glv083
Wang, H. L., Ma, R. H., Fang, H., Xue, Z. G., and Liao, Q. W. (2015). Impaired spatial learning memory after isoflurane anesthesia or appendectomy in aged mice is associated with microglia activation. J. Cell Death 8, 9–19. doi: 10.4137/JCD.S30596
Zampieri, M., Ciccarone, F., Calabrese, R., Franceschi, C., Bürkle, A., and Caiafa, P. (2015). Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 151, 60–70. doi: 10.1016/j.mad.2015.02.002
Keywords: delirium, epigenetics, DNA methylation, cytokine, tNF-alpha, aging
Citation: Shinozaki G, Braun PR, Hing BWQ, Ratanatharathorn A, Klisares MJ, Duncan GN, Jellison SS, Heinzman JT, Nagahama Y, Close L, Sabbagh S, Dlouhy BJ, Howard MA, Kawasaki H and Cho HR (2018) Epigenetics of Delirium and Aging: Potential Role of DNA Methylation Change on Cytokine Genes in Glia and Blood Along With Aging. Front. Aging Neurosci. 10:311. doi: 10.3389/fnagi.2018.00311
Received: 18 July 2018; Accepted: 14 September 2018;
Published: 23 October 2018.
Edited by:
Ashok Kumar, University of Florida, United StatesReviewed by:
Aida Karachi, University of Florida, United StatesAlexander V. Favorov, Johns Hopkins University, United States
Copyright © 2018 Shinozaki, Braun, Hing, Ratanatharathorn, Klisares, Duncan, Jellison, Heinzman, Nagahama, Close, Sabbagh, Dlouhy, Howard, Kawasaki and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gen Shinozaki, Z2VuLXNoaW5vemFraUB1aW93YS5lZHU=