 Wanlin Yang
Wanlin Yang Kijung Sung2†
Kijung Sung2† Robert A. Rissman
Robert A. Rissman Chengbiao Wu
Chengbiao Wu- 1Department of Neurology and Institute of Neurology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Neurosciences, University of California, San Diego, San Diego, CA, United States
- 3Department of Respiratory Medicine, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
- 4Veterans Affairs San Diego Health Care System, San Diego, CA, United States
Nerve growth factor (NGF) exerts multifaceted functions through different stages of life. A missense mutation (R100W) in the beta-NGF gene was found in hereditary sensory autonomic neuropathy V (HSAN V) patients with severe loss of pain perception but without overt cognitive impairment. To better understand the pathogenesis of HSAN V, we generated the first NGFR100W knock in mouse model for HSAN V. We found that the homozygotes exhibited a postnatal lethal phenotype. A majority of homozygous pups died within the first week. Some homozygous pups could ingest more milk and survived up to 2 months by reducing litter size. Whole mount in situ hybridization using E10.5 embryos revealed that, compared to wild type, R100W mutation did not alter the gene expression patterns of TrkA and P75NTR in the homozygotes. We also found that the homozygotes displayed normal embryonic development of major organs (heart, lung, liver, kidney, and spleen). Furthermore, the homozygotes exhibited severe loss of PGP9.5-positive intra-epidermal sensory fibers. Taken together, our results suggest that, as with HSAN V patients, the R100W mutation primarily affects the peripheral sensory nervous system in the mouse model. This novel mouse model makes it possible to further study in vivo how NGFR100W uncouple trophic function from nociception of NGF.
Introduction
Nerve growth factor (NGF), a member of the neurotrophin family, exerts multiple actions through different stages of life. During the embryogenesis, NGF, as a potent trophic factor, is necessary for the development and differentiation of sensory and sympathetic neurons (Levi-Montalcini, 1952, 1987). During the adulthood, NGF is important for the survival and functional modulation of basal forebrain cholinergic neurons (BFCNs) and sensory neurons (Hefti, 1986; Mobley et al., 1986; Ritter et al., 1991; Lewin and Mendell, 1993; Pezet and McMahon, 2006; Budni et al., 2015). NGF binds to two distinctive receptors, tyrosine kinase receptor A (TrkA) and the 75 kD neurotrophin receptor (p75NTR) (Chao and Hempstead, 1995). Signaling through TrkA mainly exerts trophic function to modulate the neuronal survival and differentiation, while p75NTR may promote cell survival and differentiation by interacting with TrkA or facilitate neuronal apoptosis by interacting with sortilin (Chao and Hempstead, 1995; Bibel and Barde, 2000; Nykjaer et al., 2005; Chen et al., 2009).
Hereditary sensory autonomic neuropathy (HSAN) is a heterogeneous group (type I to V) of neurodegenerative disorders of peripheral nervous system characterized by different degrees of sensory and autonomic dysfunctions (Hilz, 2002). HSAN IV (OMIM # 256800) (Indo, 2001, 2002) and HSAN V (OMIM # 608654) (Einarsdottir et al., 2004; Carvalho et al., 2011) are caused by the mutations in the genes coding for the TrkA and NGF, respectively. HSAN IV patients display mental retardation and severe congenital pain insensitivity, which leads to severe joint destruction and multiple bone fractures (Indo, 2001, 2002). Interestingly, unlike HSAN IV patients, HSAN V patients show similar profound loss of pain sensitivity, but without overt mental retardation (Einarsdottir et al., 2004; Minde et al., 2004). Einarsdottir et al. (2004) first identified the R100W mutation in HSAN V patients located in the Norrbottnian region of Sweden in 2004. The substitution of C to T at nucleotide position 661 (661C > T) in exon 3 of NGF gene located on chromosome 1p11.2-p13.2 result in a substitution of tryptophan (W) for arginine (R) at position 221 in the proNGF polypeptide corresponding to residue R100W in mature NGF (Einarsdottir et al., 2004; Minde et al., 2004). HSAN V patients appear to suffer from loss of pain sensation, but without overt mental retardation, suggesting that the NGFR100W mutation may selectively cause loss of pain function while retaining its trophic function. In a previous study, we already demonstrated that NGFR100W maintained the ability of binding to and activating the TrkA to support neuronal survival and differentiation, but failed to bind and engage p75NTR signaling pathways (Sung et al., 2018).
In this study, we generated a knock in mouse model carrying the naturally occurring NGF mutation, NGFR100W, to model HSAN V. This research is the first to directly investigate a mice model for NGFR100W and HSAN V. We found that the beta-NGFR100W knock-in homozygous mice had a pre-mature death within 2 months. Developmental impairments in heart, lung, liver, kidney, and spleen were not observed in homozygous mice. More importantly, there is a severe loss of PGP9.5-positive intra-epidermal sensory fibers in the homozygotes.
Materials and Methods
Ethics Statement
All experimental procedures were approved by the Institutional Animal Care and Use Committee of University of California San Diego, Shanghai Jiao Tong University School of Medicine and performed in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals. All tests were carried out using sex- and age-matched mice. In this study, we used heterozygotes and wild type mice that were littermates as control. All mice were housed in individual cages receiving 12 h light/dark cycle at 22°C with access to standard chow and water ad libitum.
Generation of a NGFR100W Knock-in Mouse Model
NGFR100W knock-in mice on a C57BL/6 background were produced by the Model Animal Research Center of Nanjing University (Nanjing, Jiangsu, China). Briefly, a targeting construct containing the A-to-T mutation at the 661 position in exon 3 was generated using classical bacterial artificial chromosome (BAC) recombineering and transfected into ES cells by electroporation. Gene-targeted homologous recombinant clones were identified by Southern blot analysis of genomic DNA isolated from individual neomycin resistant ES cell colonies. Thereafter, appropriately targeted ES cells were microinjected into donor blastocysts followed by implantation into pseudopregnant foster mothers to obtain chimeric mice. Chimeric males were crossed with C57BL/6 females to get heterozygote mice. Heterozygous were interbred to obtain homozygous. Mice were genotyped by PCR. The following PCR primers were used: forward primer 5-GGGGAAGGAGGGAAGACATA-3; reverse primer 5-GATTCCCTTAGGAAGGTTCTGG-3. Band sizes: wild-type band 305 bp; mutant band 435 bp.
Whole-Mount RNA in situ Hybridization to Mouse Embryos
Whole-mount in situ hybridization was performed according to previously described procedures (Pizard et al., 2004). Briefly, E10.5 embryos were dissected in cold 1× PBS and immediately fixed with 4% paraformaldehyde (PFA) overnight at 4°C. Embryos were then rinsed in PBT (PBS with 0.1% Triton X-100) to remove fixative, dehydrated by an ascending methanol series and stored in 100% methanol at -20°C. Embryos were rehydrated through a graded methanol series into PBT, digested with 10 μg/ml protease K for 10 min and post-fixed in 4% PFA plus 0.2% glutaraldehyde for 20 min. After prehybridization at 65°C for 2 h’ embryos were incubated overnight in hybridization buffer containing antisense probe at 65°C. Embryos were then washed with a series of washing buffer and blocked in 1× TBST containing 20% sheep serum for 3 h at room temperature. Embryos were incubated overnight with anti-digoxygenin-labeled (DIG)-probes (1:5000; Roche). Following probe incubation, embryos were washed eight times with 1× TBST and rinsed twice for 15 min in fresh NTMT solution. When hybridization signal is optimal, embryos were refixed in 4% PFA.
Epidermal Innervation
For the detection of epidermal innervation, skin samples were processed as described before (Arcourt et al., 2017). Skins from hind paw footpad of P0 pups were dissected, fixed in methanol/acetone (1:1) for 30 min at -20°C, and then cryoprotected in 30% sucrose overnight at 4°C. Samples were embedded in Tissue-Tek O.C.T compound (Electron Microscopy Sciences, Hatfield, PA, United States) and sectioned into 45 μm. Sections were incubated in 50 mM Glycine for 45 min at room temperature, blocked in 10% goat serum, 1% BSA, and 0.5% Triton X in PBS for 1 h at room temperature, and incubated with rabbit anti-PGP9.5 (Proteintech Group, Inc, Rosemont, IL, United States) with a dilution of 1/300 to 1/500 overnight at 4°C. Sections were then incubated with anti-rabbit Alexa Fluor 594 (Invitrogen, Carlsbad, CA, United States) with a dilution of 1:500 at room temperature for 1 h and counterstained with Hoechst for nuclei for 5 min at room temperature. Images were acquired by confocal microscopy (Leica TCS SP8 Confocal Microscope). For each mouse, 10–15 sections throughout the section thickness (45 μm.) and entire length (200 μm) were analyzed to identify all of the fibers and the intensity of PGP9.5-labeled fibers in the epidermis region was quantified using ImageJ.
Histopathology
Heart, lung, liver, kidney, and spleen were dissected and fixed in 4% paraformaldehyde for overnight. Samples were then dehydrated in a graded series of alcohol, embedded in paraffin, and sectioned into 4 μm slices. Serial sections were stained with hematoxylin and eosin (HE) and observed under a light microscope (Leica DMi8 Live Imaging Microscope).
Statistical Analysis
All data were analyzed with GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, United States). The results were presented as mean values ± SEM. Student’s t-test was performed for two group comparisons. One-way ANOVA or Two-way ANOVA was performed for multiple comparisons. Differences with P < 0.05 was considered statistically significant.
Results
Homozygous Mice Are Born Live but Fail to Thrive
To shed light on the role of NGFR100W mutation in the pathogenesis of HSAN V, we generate NGFR100W knock-in mice on C57BL/6 background through the knock-in strategy (Figure 1A). Briefly, a targeting construct containing the A-to-T mutation at the 661 position, which leading to the amino acid substitution R to W at 100 position of mature NGF, was electroporated into ES cells. The positive ES clones containing the mutation were selected and microinjected into donor blastocysts followed by implantation into pseudopregnant foster mothers to obtain male chimeric mice. Chimeric males were mated with C57BL/6 females to obtain heterozygotes (+/fln). +/fln mice were interbred to obtain homozygous (fln/fln). The +/fln and fln/fln mice were genotyped by PCR using tail DNA (Figure 1B).
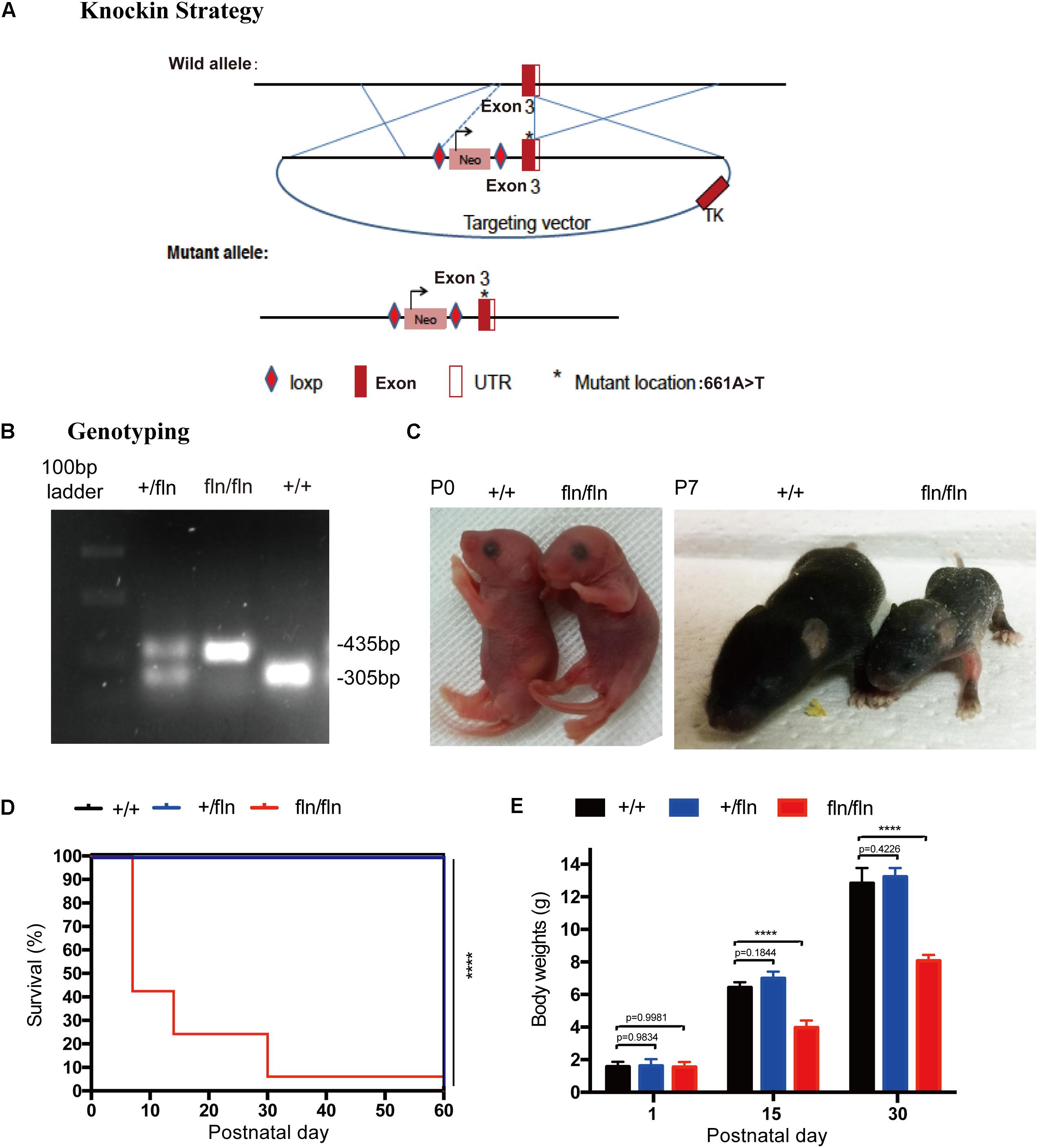
FIGURE 1. Gene-targeting strategy for generation of NGFR100W knock-in mice model. (A) Diagram illustrating the gene-targeting strategy used to produce the NGFR100W knock-in mice in C57BL/6 background. Starred exon 3 represents the mutated exon harboring the A-to-T mutation at the 661 position. (B) PCR genotyping of wild-type (+/+), the heterozygote (+/fln) and the homozygote (fln/fln): wild-type band: 305 bp, mutant band: 435 bp. (C) fln/fln mice appeared normal at birth (P0), but failed to grow during early postnatal life (e.g., at P7) compared with +/+ littermates. (D) Survival curves for +/+ (n = 20), +/fln (n = 20), and fln/fln (n = 20) mice. fln/fln mice died within 2 months. (E) The body weight growth curves for +/+ (n = 6), +/fln (n = 6), and fln/fln (n = 6) mice. fln/fln mice gained weight more slowly than their littermate. ∗∗∗∗p < 0.0001, one way ANOVA test followed by Tukey’s multiple comparisons test.
The fln/fln mice exhibited a postnatal lethal phenotype (Figure 1D). A huge majority of fln/fln pups died within the first week of life. fln/fln mice exhibited smaller milk spot in the stomach by visual inspection, indicating they could not ingest normal quantities of milk. By moving littermates that has bigger milk spot to a wild-type (+/+) surrogate mother to reduce litter size, some fln/fln mice could ingest more milk and survived up to 2 months. The fln/fln mice were born with normal body weight and size, but grew more slowly than their +/+ and +/fln littermates. As shown in Figure 1C, newborn fln/fln pup had comparable body size to the +/+ littermates, but became significantly smaller on P7. By 15 and 30 days of age, the body weight of fln/fln pups was remarkably lower, only 61.6 and 62.8% of that of +/+ littermates, respectively (Figure 1E). Whereas, the +/fln mice exhibited normal body weight and were indistinguishable from the wild-type (+/+) littermates by observing gross morphology or spontaneous behavior (Figure 1E). These results indicated that fln/fln mice were born live with normal weight, but failed to thrive.
NGFR100W Mutation Do Not Affect the Gene Expression Patterns of TrkA and P75NTR
The NGF signaling is mediated by two known receptors (Chao and Hempstead, 1995), TrkA and p75NTR, we next assessed whether the gene expression patterns of TrkA and P75NTR in +/fln and fln/fln E10.5 embryos exhibited defects using whole mount in situ hybridization. As shown in Figure 2, TrkA was highly expressed in cranial and spinal sensory ganglia, including dorsal root ganglions, trigeminal, facial ganglion, and superior ganglion. Whereas, P75NTR was expressed in wider range including the peripheral nerve trunks, dorsal root ganglia, and spinal cords. There was no obvious difference in gene expression patterns of TrkA and P75NTR among +/+, +/fln, and fln/fln embryos mice do not display gross developmental defects.
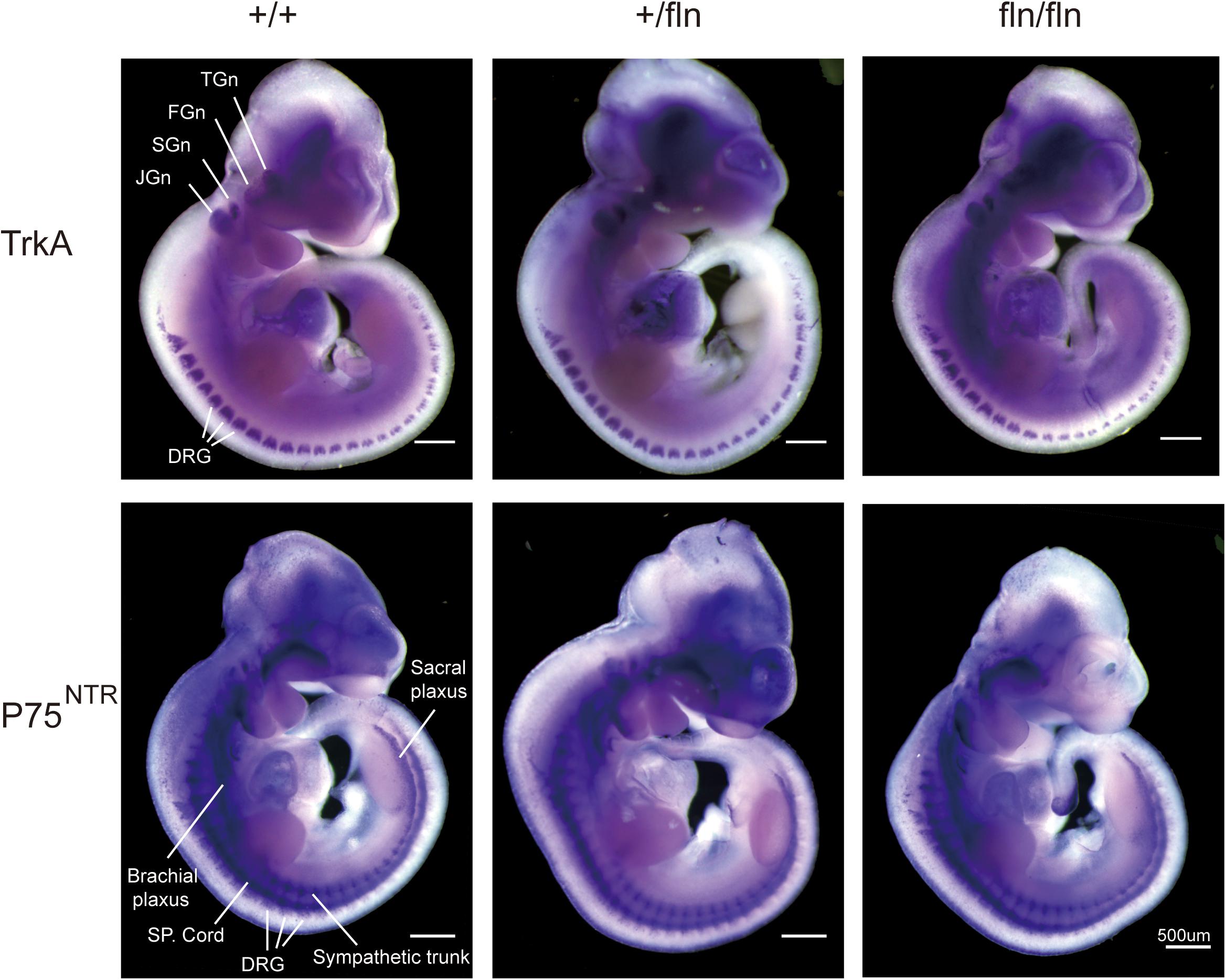
FIGURE 2. Expression of TrkA and P75NTR is normal in homozygous. Whole-mount in situ hybridization using antisense riboprobe for TrkA and P75NTR mRNA was performed on +/+ (n = 3), +/fln (n = 3), and fln/fln (n = 3) embryos at E10.5. TGn, trigeminal ganglion; FGn, facial ganglion; SGn, superior ganglion; JGn, jugular ganglion; DRG, dorsal root ganglion. Bar = 500 μm.
Homozygous Embryos Do Not Display Gross Developmental Defects
Although P0 fln/fln pups were born live and indistinguishable from their +/+ littermates in size and weight, a huge majority of fln/fln pups died within the first week of life. As the fln/fln mice exhibited a postnatal lethal phenotype, we then analyzed the tissue morphology more closely to assess whether fln/fln exhibited embryonic developmental defects. H&E staining of heart, lung, liver, and spleen tissues from +/+, +/fln, and fln/fln embryos at E18.5 was performed (Figure 3). Histological comparison failed to reveal any striking abnormalities in major organs of +/fln and fln/fln embryos.
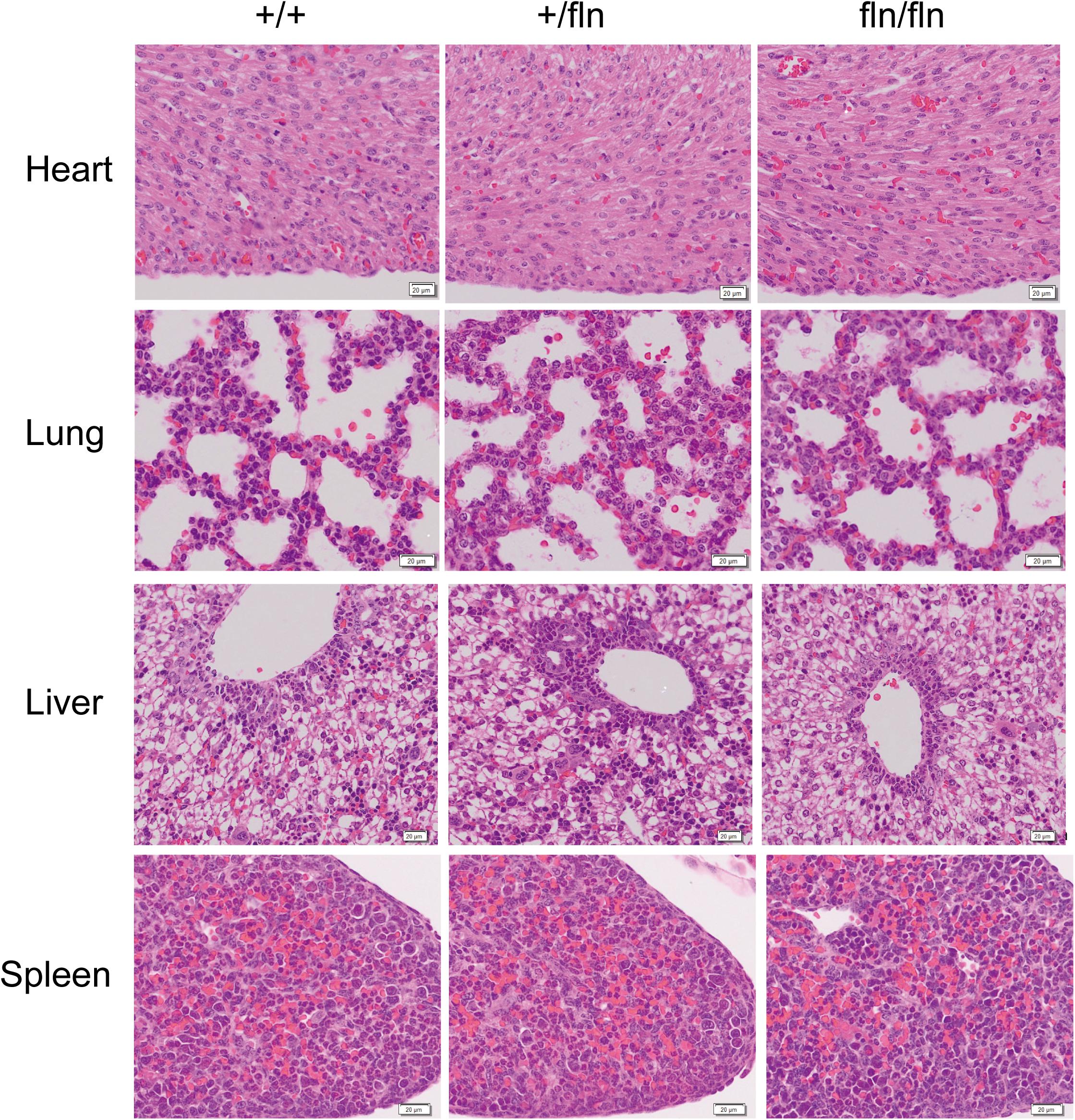
FIGURE 3. No obvious histological abnormalities were observed in heart, lung, liver, and spleen of homozygous. Representative sections of H&E staining on heart, lung, liver, and spleen samples from +/+ (n = 3), +/fln (n = 3), and fln/fln (n = 3) embryos at E18.5. Bar = 20 μm.
Previous research on NGFR100W revealed that R100W mutation selectively disrupted binding of NGF to p75NTR, while it retained the ability of binding to TrkA (Covaceuszach et al., 2010; Sung et al., 2018). The p75NTR is transiently synthesized in embryonic kidney (Sariola et al., 1991). A study reported that anti-sense oligonucleotide inhibition of p75NTR expression in organ culture inhibited kidney morphogenesis, indicating that p75NTR signaling played an important role in embryonic development of the kidney (Sariola et al., 1991). We next detected whether fln/fln embryos displayed developmental defects of the kidney. As illustrated in Figure 4, fln/fln embryos did not show obviously difference from their +/+ and +/fln littermates.
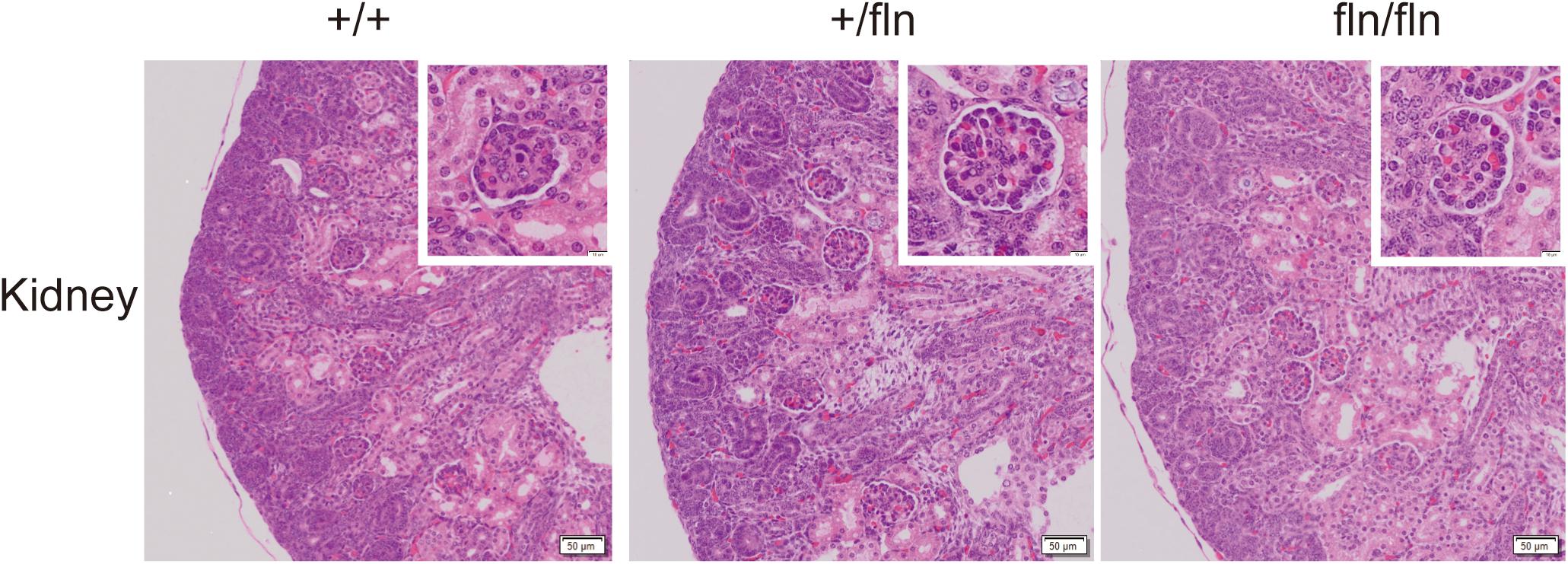
FIGURE 4. No kidney defects were detected in homozygous. Representative sections of H&E staining on kidney samples from +/+ (n = 3), +/fln (n = 3), and fln/fln (n = 3) embryos at E18.5. Bar = 50 μm at low magnification, Bar = 10 μm at higher magnification.
Nerve growth factor is produced in the hippocampus throughout life and essential for hippocampal plasticity and learning (Conner et al., 2009). We next examined whether fln/fln embryos displayed developmental defects of hippocampus. As illustrated in Figure 5, no obvious differences in the pyramidal cell layer in the hippocampus are discernable between +/+, +/fln, and fln/fln littermates at E18.5 of age. These findings indicated that homozygous embryos display normal embryonic development of major organs were indistinguishable from those of wild-type littermates in size, weight, and general appearance.
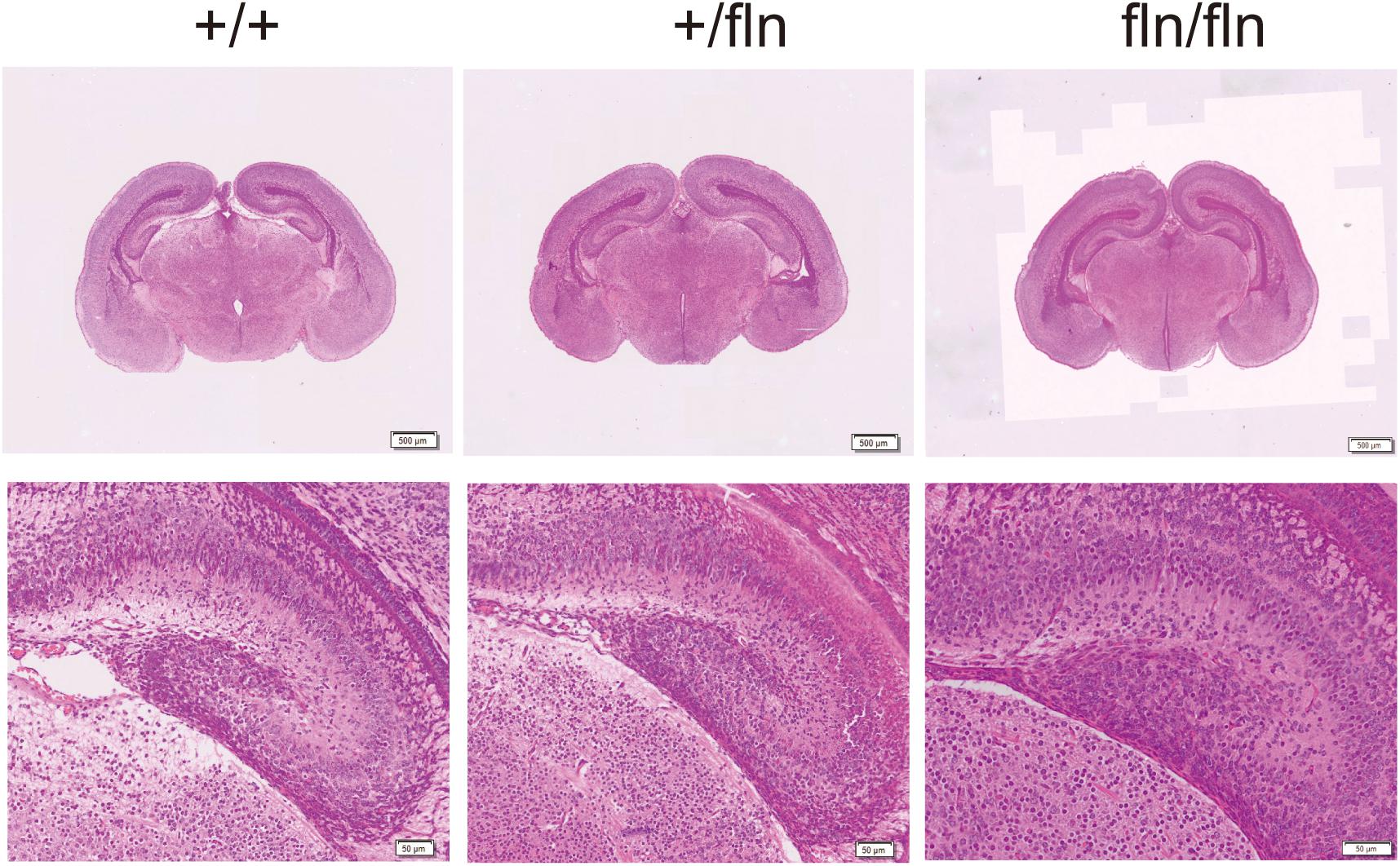
FIGURE 5. No obvious histological abnormality was detected in homozygous. Representative sections of H&E staining on hippocampus samples from +/+ (n = 3), +/fln (n = 3), and fln/fln (n = 3) embryos at E18.5. Bar = 500 μm at low magnification, Bar = 50 μm at higher magnification.
Homozygous Mice Exhibited Severe Loss of PGP9.5-Positive Intra-Epidermal Sensory Fibers
Nerve growth factor is important for the development and differentiation of sensory neurons during embryogenesis (Levi-Montalcini, 1987; Li et al., 2017; Sun et al., 2018). Painful signals on the skin were transmited to the spinal cord and brain by myelinated Aδ and unmyelinated C sensory nerve fibers (Adrienne and Dubin, 2010). Aδ and C nerve fibers are significantly decreased in HSAN V patients (Einarsdottir et al., 2004; Minde et al., 2004, 2006). We then investigated whether +/fln and fln/fln pus displayed severe sensory nerve fibers loss in hind paw skin. The PGP9.5 antibody was used to examine intra-epidermal sensory fibers (IENFs), including Aδ and C nerve fiber. The staining of hind paw skins revealed that the IENFs appeared significantly reduced in fln/fln pups compared with +/+ and +/fln pups (Figure 6). There was no significant differrence between +/+ and +/fln pups.
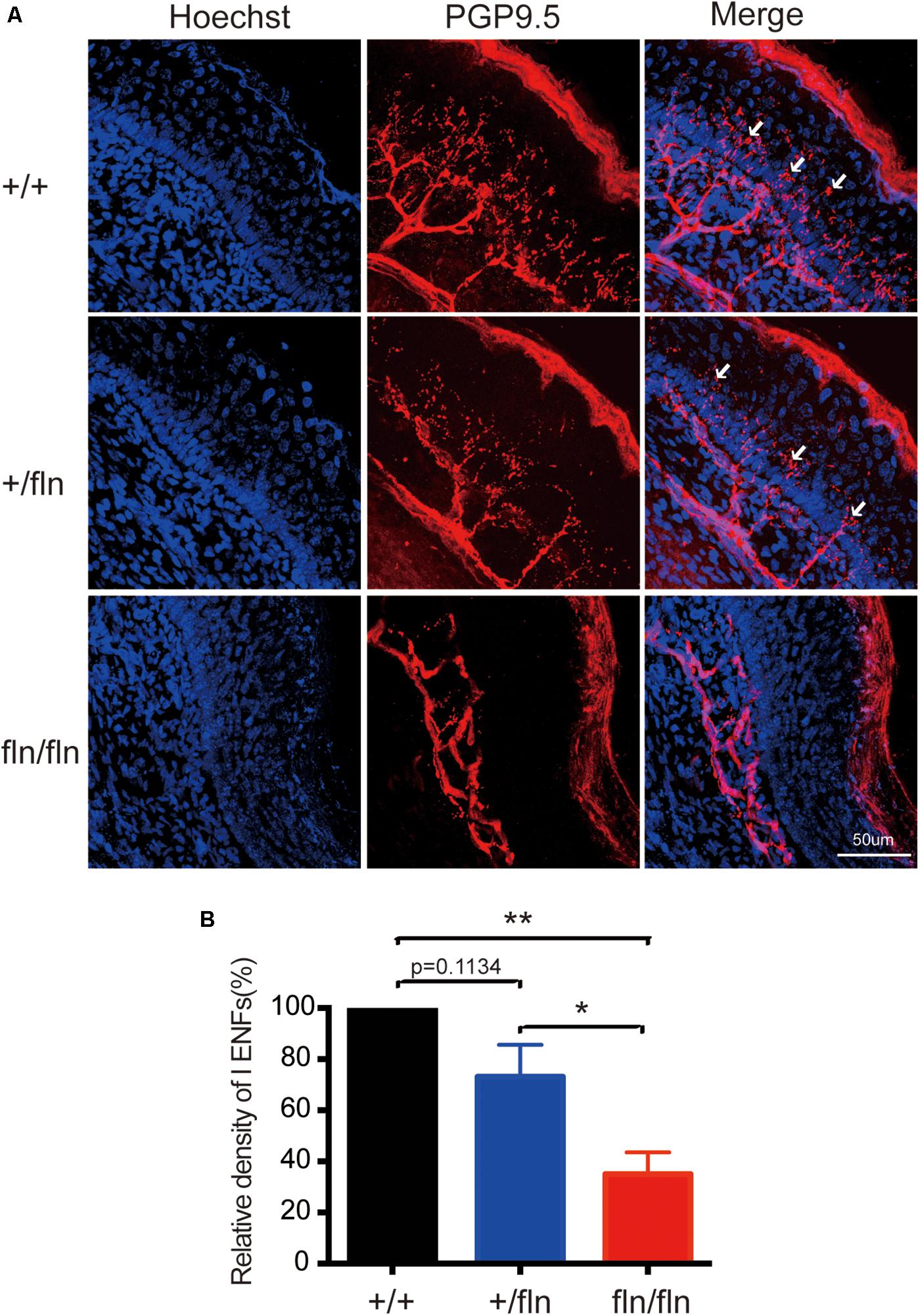
FIGURE 6. Loss of PGP9.5-positive intra-epidermal sensory fibers (IENF) in the footpad skin in homozygous. (A) Confocal microscopic images from sections through the hindpaw footpad skin from +/+ (n = 3), +/fln (n = 3), and fln/fln (n = 3) P1 mice stained with pan-nerve fiber marker PGP9.5 (red) and nuclear marker Hoechst (blue) to visualize skin cells. The arrowhead indicates the free nerve endings, (B) Quantification of PGP9.5-positive IENF intensities. Mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, one way ANOVA test followed by Tukey’s multiple comparisons test.
Discussion
HSAN is a heterogeneous group (type I to V) of rare, clinically and genetically peripheral neuropathies characterized by different degrees of sensory and autonomic dysfunctions (Capsoni, 2014). Genetic mutations responsible for HSANs have been demonstrated. In the cases of HSAN IV (OMIM # 256800) (Indo et al., 2001; Indo, 2002) and HSAN V (OMIM # 608654) (Einarsdottir et al., 2004; Minde et al., 2004), which are caused by mutations in the genes coding for the TrkA and NGF respectively, both of them are all involved in NGF signaling pathway abnormality. Interestingly, HSAN IV patients display severe congenital pain insensitivity and mental retardation, while HSAN V patients show similar pain insensitivity, but without overt mental retardation (Capsoni, 2014). The mutation 661C > T in NGF gene of HSAN V patients located in the Norrbottnian region of Sweden was first identified by Einarsdottir et al in 2004 (Einarsdottir et al., 2004). This mutation causes a substitution of tryptophan (W) for arginine (R) at position 221 in the proNGF polypeptide corresponding to residue R100 in mature NGF. The different clinical manifestations between HSAN IV and HSAN V patients motivated us to generate NGFR100W knock-in mice to better define the pathogenesis of HSAN V. This research is the first to directly investigate a mice model for NGFR100W. We found that the homozygous mice exhibited a postnatal lethal phenotype. Most homozygous pups died within the first week of life. Some homozygous pups could ingest more milk and survived up to 2 months by reducing litter size. The homozygous and wild type embryos showed similar gene expression patterns of TrkA and P75NTR. The homozygous mice displayed normal embryonic development of major organs. Furthermore, the homozygous mice exhibited severe loss of PGP9.5-positive intra-epidermal sensory fibers.
In 2004, Einarsdottir et al first described the R100W mutation in HSAN V patients located in the Norrbottnian region of Sweden Minde et al. (2004). It has been shown previously that HSAN V homozygous patients showed a small reduction of myelinated Aδ nerve fibers and a severe reduction of unmyelinated C nerve fibers, while this reduction was milder in heterozygous carriers (Einarsdottir et al., 2004; Larsson et al., 2009). Consistent with HSAN V patients, we confirmed that homozygous mice exhibited severe loss of intra-epidermal sensory fibers in hind paw skin, including Aδ and C nerve fibers. In many ways, our NGFR100W homozygotes resemble the NGF knockout homozygotes mice in their severe sensory phenotypic presentation. Crowley et al. generated the first transgenic NGF knockout mice model in 1994 and found that homozygotes appeared to reveal somewhat loss of sensory fibers in the skin (Crowley et al., 1994). The similarity between NGFR100W knock-in and NGF knockout homozygotes indicated the deficits in the NGFR100W protein synthesis and secretion or function and structure. Indeed, Larsson et al already demonstrated that proNGFR100W was predominantly accumulated in the cell lysate and the secretion of mature NGFR100W was markedly reduced (Larsson et al., 2009). The accumulation of proNGFR100W did not appear to be due to an inability of the convertases, such as furin and plasmin, to cleave the pro-protein to mature protein, but was related to the intracellular localization that would prevent processing and secretion of mature NGFR100W (Larsson et al., 2009). The reduced secretion of mature NGFR100W may lead to deficiency of mature NGF in innervated targets, which could explain the severe loss of sensory fibers in HSAN V patients and NGFR100W knock-in mice. One the other hand, structural insights into the R100W mutation in NGF protein also found that residue R100 is not directly participated in the interface of TrkA, but it involved in the p75NTR interaction surface, suggesting that R100 mutation selectively disrupted the binding of NGF to p75NTR (Covaceuszach et al., 2010). Further studies demonstrated that R100W mutation did not affect NGF binding to TrkA, while it abolished NGF binding to p75NTR (Capsoni et al., 2011). Our previous study also demonstrated that NGFR100W maintained the ability of binding to and activating the TrkA to support neuronal survival and differentiation (Sung et al., 2018), which could explain why the cognitive impairment of HSAN V patients appeared very limited or absent.
Hereditary sensory autonomic neuropathy V homozygous patients can survive to adulthood, but they do acquire multiple fractures and being burned during childhood due to the lack of deep pain sensations and increased cold and heat thresholds (Einarsdottir et al., 2004; Minde et al., 2004; Larsson et al., 2009). However, none of those patients displayed systemic diseases related to heart and lungs (Einarsdottir et al., 2004). Whereas, in the cases of our NGFR100W knock-in mice, a huge majority of homozygous mice died within the first week of life, which led us to examine whether the homozygous mice exhibited embryonic developmental defects. Histological comparison of heart, lung, liver, kidney, and spleen tissues failed to reveal any striking abnormalities in major organs of homozygous embryos, indicating that postnatal lethality of homozygous mice was not caused by embryonic developmental defects. Observations during the early postnatal days showed that homozygous mice displayed smaller milk spot in the stomach, indicating they could not ingest normal quantities of milk. Some homozygous mice could ingest more milk and survived up to 2 months by reducing litter size. Therefore, we speculated that postnatal lethality of homozygous mice might be related in part to ineffective ingestion. Furthermore, as homozygous mice displayed severe loss of IENFs in skin, the deficits in the sensory nervous system might be one of the leading causes of death of homozygous mice. Before the birth, embryos are isolated in the uterus and obtain nutrients from their mothers. After birth, the environment is drastically change and the newborn pups start to receive external sensory stimuli and need to interact with other littermates and mother to keep warm and obtain milk (Weber et al., 2013; Toda and Kawasaki, 2014). The sensory nervous system deficits may affect the mother-pup and pup–pup interactions (Toda and Kawasaki, 2014). During the first week of life, we also found that homozygous mice often stayed outside the nest. After birth, the newborn pups are totally dependent on their mother for thermoregulation and nutrition, and could die from hypothermia or starvation (Terrien et al., 2011). The maternal behavior in mouse and human is different. Mouse mother sometimes tend to cull if a pup is too weak and likely to die, while human are the opposite and will try their best to keep the babies healthy, which may explain why the HSAN V homozygous patients can survive to adulthood and our homozygous mice exhibit postnatal lethality. Furthermore, as lack of more specific or detailed histological analysis, we cannot rule out the possibility that other pathological changes outside the nervous system contribute to the postnatal lethality of homozygous. Further studies need to be performed to investigate the pathological changes in other tissues.
Taken together, we generated the first NGFR100W knock in mouse model to study the pathogenesis of HSAN V. We found that NGFR100W knock-in homozygotes had a pre-mature death within 2 months. However, The developmental impairments in heart, lung, liver, kidney, and spleen were not observed in homozygous mice. More importantly, we demonstrated that, as with HSAN V patients, the homozygous mice displayed severe loss of sensory fibers in the skin. This novel mouse model makes it possible to further investigate the pathogenesis for HSAN V. More importantly, it provides a better tool to further study in vivo how NGFR100W uncouple trophic function from nociception of NGF.
Author Contributions
WY, KS, JD, and CW designed and performed the study and wrote the manuscript. RAR helped with the design and histological analysis. WX and FZ performed analysis of PGP9.5/IENFs staining experiments.
Funding
This research was supported by project grants from grants from Tau Consortium (CW); NIH grants AG051848, BX003040, AG051839, and AG018440 (RAR). Dr. J. Ding was supported by 1: National Natural Science Foundation of China (Grant# 81630029); 2: by Program for Outstanding Medical Academic Leader of Shanghai Municipality; 3: National Natural Science Foundation of China (Grant# 81171200); 4: the Ministry of Science and Technology of the People’s Republic of China (2014CB965002, 2012BAI10B03); 5: Science and Technology Commission of Shanghai Municipality (13JC1401502, 13140904000) and 6: Shanghai Municipal Education Commission (12ZZ115). Dr. X. Wei was supported by National Natural Science Foundation of China (Grant# 81600920).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Ms. Pauline Hu for her technical assistance. We would also like to thank Ms. Angela Zeng and Mr. Simon Kim for helping with quantitation of IENFs. We would also like to thank Alexander Pope for helping correct grammatical mistakes and typo errors in this paper.
References
Adrienne, E., and Dubin, A. P. (2010). Nociceptors: the sensors of the pain pathway. J. Clin. Invest. 120, 3760–3772. doi: 10.1172/JCI42843
Arcourt, A., Gorham, L., Dhandapani, R., Prato, V., Taberner, F. J., Wende, H., et al. (2017). Touch receptor-derived sensory information alleviates acute pain signaling and fine-tunes nociceptive reflex coordination. Neuron 93, 179–193. doi: 10.1016/j.neuron.2016.11.02
Bibel, M., and Barde, Y. A. (2000). Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 14, 2919–2937. doi: 10.1101/gad.841400
Budni, J., Bellettini-Santos, T., Mina, F., Garcez, M. L., and Zugno, A. I. (2015). The involvement of BDNF. NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 6, 331–341. doi: 10.14336/AD.2015.0825
Capsoni, S. (2014). From genes to pain: nerve growth factor and hereditary sensory and autonomic neuropathy type V. Eur. J. Neurosci. 39, 392–400. doi: 10.1111/ejn.12461
Capsoni, S., Covaceuszach, S., Marinelli, S., Ceci, M., Bernardo, A., Minghetti, L., et al. (2011). Taking pain out of NGF: a “painless” NGF mutant, linked to hereditary sensory autonomic neuropathy type V, with full neurotrophic activity. PLoS One 6:e17321. doi: 10.1371/journal.pone.0017321
Carvalho, O. P., Thornton, G. K., Hertecant, J., Houlden, H., Nicholas, A. K., Cox, J. J., et al. (2011). A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5 neuropathy. J. Med. Genet. 48, 131–135. doi: 10.1136/jmg.2010.081455
Chao, M. V., and Hempstead, B. L. (1995). p75 and Trk: a two-receptor system. Trends Neurosci. 18, 321–326. doi: 10.1016/0166-2236(95)93922-K
Chen, Y., Zeng, J., and Cen, L. (2009). Multiple roles of the p75 neurotrophin receptor in the nervous system. J. Int. Med. Res. 37, 281–288. doi: 10.1177/147323000903700201
Conner, J. M., Franks, K. M., Titterness, A. K., Russell, K., Merrill, D. A., Christie, B. R., et al. (2009). NGF is essential for hippocampal plasticity and learning. J. Neurosci. 29, 10883–10889. doi: 10.1523/JNEUROSCI.2594-09.2009
Covaceuszach, S., Capsoni, S., Marinelli, S., Pavone, F., Ceci, M., Ugolini, G., et al. (2010). In vitro receptor binding properties of a “painless” NGF mutein, linked to hereditary sensory autonomic neuropathy type V. Biochem. Biophys. Res. Commun. 391, 824–829. doi: 10.1016/j.bbrc.2009.11.146
Crowley, C., Spencer, S. D., Nishimura, M. C., Chen, K. S., Pitts-Meek, S., Armanini, M. P., et al. (1994). Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 76, 1001–1011. doi: 10.1016/0092-8674(94)90378-6
Einarsdottir, E., Carlsson, A., Minde, J., Toolanen, G., Svensson, O., Solders, G., et al. (2004). A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum. Mol. Genet. 13, 799–805. doi: 10.1093/hmg/ddh096
Hefti, F. (1986). Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 6, 2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986
Hilz, M. J. (2002). Assessment and evaluation of hereditary sensory and autonomic neuropathies with autonomic and neurophysiological examinations. Clin. Auton. Res. 12(Suppl. 1), I33–I43. doi: 10.1007/s102860200017
Indo, Y. (2001). Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Hum. Mutat. 18, 462–471. doi: 10.1002/humu.1224
Indo, Y. (2002). Genetics of congenital insensitivity to pain with anhidrosis (CIPA) or hereditary sensory and autonomic neuropathy type IV. Clinical, biological and molecular aspects of mutations in TRKA(NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Clin. Auton. Res. 12(Suppl. 1), I20–I32.
Indo, Y., Mardy, S., Miura, Y., Moosa, A., Ismail, E. A., Toscano, E., et al. (2001). Congenital insensitivity to pain with anhidrosis (CIPA): novel mutations of the TRKA (NTRK1) gene, a putative uniparental disomy, and a linkage of the mutant TRKA and PKLR genes in a family with CIPA and pyruvate kinase deficiency. Hum. Mutat. 18, 308–318. doi: 10.1002/humu.1192
Larsson, E., Kuma, R., Norberg, A., Minde, J., and Holmberg, M. (2009). Nerve growth factor R221W responsible for insensitivity to pain is defectively processed and accumulates as proNGF. Neurobiol. Dis. 33, 221–228. doi: 10.1016/j.nbd.2008.10.012
Levi-Montalcini, R. (1952). Effects of mouse tumor transplantation on the nervous system. Ann. N. Y. Acad. Sci. 55, 330–344. doi: 10.1111/j.1749-6632.1952.tb26548.x
Levi-Montalcini, R. (1987). The nerve growth factor 35 years later. Science 237, 1154–1162. doi: 10.1126/science.3306916
Lewin, G. R., and Mendell, L. M. (1993). Nerve growth factor and nociception. Trends Neurosci. 16, 353–359. doi: 10.1016/0166-2236(93)90092-Z
Li, R., Ma, J., Wu, Y., Nangle, M., Zou, S., Li, Y., et al. (2017). Dual delivery of NGF and bFGF coacervater ameliorates diabetic peripheral neuropathy via inhibiting schwann cells apoptosis. Int. J. Biol. Sci. 13, 640–651. doi: 10.7150/ijbs.18636
Minde, J., Svensson, O., Holmberg, M., Solders, G., and Toolanen, G. (2006). Orthopedic aspects of familial insensitivity to pain due to a novel nerve growth factor beta mutation. Acta Orthop. 77, 198–202. doi: 10.1080/17453670610045911
Minde, J., Toolanen, G., Andersson, T., Nennesmo, I., Remahl, I. N., Svensson, O., et al. (2004). Familial insensitivity to pain (HSAN V) and a mutation in the NGFB gene. A neurophysiological and pathological study. Muscle Nerve 30, 752–760. doi: 10.1002/mus.20172
Mobley, W. C., Rutkowski, J. L., Tennekoon, G. I., Gemski, J., Buchanan, K., and Johnston, M. V. (1986). Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res. 387, 53–62. doi: 10.1016/0169-328X(86)90020-3
Nykjaer, A., Willnow, T. E., and Petersen, C. M. (2005). p75NTR–live or let die. Curr. Opin. Neurobiol. 15, 49–57. doi: 10.1016/j.conb.2005.01.004
Pezet, S., and McMahon, S. B. (2006). Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci. 29, 507–538. doi: 10.1146/annurev.neuro.29.051605.112929
Pizard, A., Haramis, A., Carrasco, A. E., Franco, P., Lopez, S., and Paganelli, A. (2004). Whole-mount in situ hybridization and detection of RNAs in vertebrate embryos and isolated organs. Curr. Protoc. Mol. Biol. 66, 14.9.1–14.9.24. doi: 10.1002/0471142727.mb1409s66
Ritter, A. M., Lewin, G. R., Kremer, N. E., and Mendell, L. M. (1991). Requirement for nerve growth factor in the development of myelinated nociceptors in vivo. Nature 350, 500–502. doi: 10.1038/350500a0
Sariola, H., Saarma, M., Sainio, K., Arumae, U., Palgi, J., Vaahtokari, A., et al. (1991). Dependence of kidney morphogenesis on the expression of nerve growth factor receptor. Science 254, 571–573. doi: 10.1126/science.1658930
Sun, Q., Tang, D. D., Yin, E. G., Wei, L. L., Chen, P., Deng, S. P., et al. (2018). Diagnostic significance of serum levels of nerve growth factor and brain derived neurotrophic factor in diabetic peripheral neuropathy. Med. Sci. Monit. 24, 5943–5950. doi: 10.12659/MSM.909449
Sung, K., Ferrari, L. F., Yang, W., Chung, C., Zhao, X., Gu, Y., et al. (2018). Swedish nerve growth factor mutation (NGF(R100W)) defines a role for TrkA and p75(NTR) in nociception. J. Neurosci. 38, 3394–3413. doi: 10.1523/JNEUROSCI.1686-17.2018
Terrien, J., Perret, M., and Aujard, F. (2011). Behavioral thermoregulation in mammals: a review. Front. Biosci. 16, 1428–1444. doi: 10.2741/3797
Toda, T., and Kawasaki, H. (2014). The development of suckling behavior of neonatal mice is regulated by birth. Mol. Brain 7:8. doi: 10.1186/1756-6606-7-8
Keywords: hereditary sensory autonomic neuropathy V, nerve growth factor, TrkA, p75 neurotrophic factor receptor, Intraepidermal Nerve Fiber
Citation: Yang W, Sung K, Zhou F, Xu W, Rissman RA, Ding J and Wu C (2018) Targeted Mutation (R100W) of the Gene Encoding NGF Leads to Deficits in the Peripheral Sensory Nervous System. Front. Aging Neurosci. 10:373. doi: 10.3389/fnagi.2018.00373
Received: 27 September 2018; Accepted: 26 October 2018;
Published: 13 November 2018.
Edited by:
Jiawei Zhou, Shanghai Institutes for Biological Sciences (CAS), ChinaReviewed by:
Dennis Qing Wang, Southern Medical University, ChinaGuanghui Wang, Soochow University, China
Copyright © 2018 Yang, Sung, Zhou, Xu, Rissman, Ding and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianqing Ding, anFkaW5nMThAMTYzLmNvbQ== Chengbiao Wu, Y2h3MDQ5QHVjc2QuZWR1
†These authors have contributed equally to this work