 Qing Wang1
Qing Wang1 Jie Zhang
Jie Zhang- 1Wenzhou Seventh People’s Hospital, Wenzhou, China
- 2Department of Pathology, Hangzhou Normal University, Hangzhou, China
- 3Independent Researcher, Hangzhou, China
Objective: Preclinical studies have found both hyperactivity of hypothalamic- pituitary- adrenal (HPA) axis and synaptic degeneration are involved in the pathogenesis of Alzheimer’s disease (AD). However, the data on the relationship of activity of HPA axis and synaptic degeneration in humans are limited.
Methods: We compared CSF cortisol levels in 310 subjects, including 92 cognitively normal older people, 149 patients with mild cognitive impairment (MCI), and 69 patients with mild AD. Several linear and logistic regression models were conducted to investigate associations between CSF cortisol and synaptosomal-associated protein 25 (SNAP-25, reflecting synaptic degeneration) and other AD-related biomarkers.
Results: We found that levels of cortisol in CSF were associated with SNAP-25 levels and tau pathologies but not amyloid-β protein. However, there were no significant differences in CSF cortisol levels among the three diagnostic groups.
Conclusion: The HPA axis may play a crucial role in synaptic degeneration in AD pathogenesis.
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder characterized by a progressive decline of memory and cognitive function (Scheltens et al., 2016). Although it is well known that amyloid plagues and neurofibrillary tangles are two key neuropathological features in AD, the mechanisms underlying the pathogenesis of AD are not fully understood. A growing body of evidence support the idea that the hyperactivity of hypothalamic- pituitary- adrenal (HPA) axis with excess cortisol may be involved in cognitive impairment and dementia (Csernansky et al., 2006; Lee et al., 2007; Gil-Bea et al., 2010). In addition, it has been reported that the degree of hyperactivity of the HPA-axis was associated with the severity of hippocampal atrophy and cognitive impairment in patients with dementia (de Leon et al., 1988;Gurevich et al., 1990). Animal studies reported that chronic stress and glucocorticoids can accelerate memory decline and increase amyloid-β and tau pathologies in mouse models of AD (Green et al., 2006; Jeong et al., 2006). Further, the authors also found that elevated glucocorticoid levels increase Aβ production by increasing levels of amyloid precursor protein (APP) and β-APP cleaving enzyme (BACE) (Jeong et al., 2006). Taken together, these findings highlight an important role of cortisol in the pathogenesis of AD.
Synaptic function has been regarded as a critical mechanism underlying cognitive impairment in the pathogenesis of AD (Selkoe, 2002). A previous study suggested that presynaptic proteins (e.g., synaptobrevin, synaptotagmin, and Rab3a) were significantly decreased in AD brains (Sze et al., 2000). Synaptosomal-associated protein 25 (SNAP-25) is a presynaptic protein which plays an important role in neuronal survival and cognition (Toonen et al., 2017). One previous study showed that SNAP-25 was elevated in the CSF of patients with mild cognitive impairment (MCI) and AD, indicating that SNAP-25 may be a useful biomarker reflecting synapse degeneration (Brinkmalm et al., 2014; Zhang et al., 2018). Importantly, it has been demonstrated that corticosterone can cause a substantial loss of synapses in rodents, indicating a potential role of corticosterone in the maintenance of synapse function (Tata et al., 2006). To our knowledge, no human studies on the association between CSF cortisol levels and synapse degeneration have been reported.
Here, the levels of cortisol in CSF among normal controls, patients with MCI and AD had been compared. Second, we examined the relationship of CSF cortisol levels and synapse degeneration. Finally, we investigated the associations between CSF cortisol levels and typical AD-related biomarkers, including hippocampal volumes, CSF Aβ42, t-tau, and p-tau levels.
Materials and Methods
Alzheimer’s Disease Neuroimaging Initiative
Data used in the preparation of this study were extracted from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. The primary goal of ADNI has been to investigate whether neuropsychological testing, neuroimaging data, blood, and CSF biomarkers could be integrated to assess the progression of MCI and mild AD. This study was approved by local ethical committees. All subjects or authorized representatives provides written informed consent. Further information can be found at the website: http://www.adni-info.org.
Participants
Demographics were obtained from the ADNI database. In this analysis, there were 92 normal controls, 149 patients with MCI, and 69 patients with mild AD.
APOE Genotyping, Clinical and Memory Assessments
APOE genotypes of the study participants were extracted from the ADNI database1. Participants underwent comprehensive cognitive assessments, while we only selected Mini-Mental State Exam (MMSE) and Rey Auditory Verbal Learning Test (RAVLT immediate recall and delayed recall) in the current analysis.
Cerebrospinal Fluid Data
Quantification of Cortisol in CSF
The levels of CSF cortisol were determined using Luminex xMAP immunoassay technology, which measures a range of inflammatory, lipid, metabolic, and other indices (Olsson et al., 2013). Further information can be found at the website: http://www.adni-info.org. The CSF cortisol data were extracted from the ADNI database. Value are given in ng/ml.
Quantification of SNAP- 25 in CSF
Levels of SNAP-25 in CSF were determined at the departments of pathology and neurology at Washington University, St. Louis, United States. A sandwich ELISA that was developed using the Erenna® immunoassay system to determine the levels of SNAP-25 in CSF. All samples were run in triplicates. Values are given as pg/ml. The detailed protocol can be found at http://www.adni-info.org.
Quantification of Aβ42, t-tau, and p-tau in CSF
Levels of Aβ42, t-tau, and p-tau in CSF were determined by the ADNI Core Biomarkers Team using xMAP Luminex platform and Innogenetics/Fujirebio AlzBio3 immunoassay kits. The detailed procedure for this determination is described elsewhere (Shaw et al., 2009). Using a previously proposed CSF Aβ42 cutoff < 192 pg/ml (Shaw et al., 2009), participants were classified as abnormal or normal. The ROC analysis found that this cutoff value can detect the autopsy-confirmed AD cases with a high sensitivity (96.4%) (Shaw et al., 2009). Tau phosphorylated at the threonine 18 was measured.
Hippocampal Volumes
The hippocampal volumes data were extracted from the ADNI database. The detailed neuroimaging methods used by ADNI have been described elsewhere (Zhang et al., 2011). Briefly, structural MRI brain scans were obtained using 1.5T MRI scanners with a standardized protocol, which is described in detail at adni.loni.usc.edu. Hippocampal volume measurements were performed using FreeSurfer software2.
Statistical Analysis
First, the F-test and x2 test were performed to investigate the mean differences of continuous variables and the distributions of categorical variables, respectively. Second, the Pearson correlation tests were used to investigate the correlations of CSF cortisol levels and other AD-related biomarkers in all participants. Finally, linear and logistic regression models were used to examine the association between cortisol and other biomarkers after adjusting several potential confounding variables. More specifically, logistic regression models were used to examine the association between amyloid status (<192 pg/ml vs. >192 pg/ml; dependent variable) and CSF cortisol levels (independent variable). Model 1 was unadjusted. Model 2 was adjusted for age, education, age, APOE genotype, MMSE scores and diagnosis. All statistical analyses were performed using R software. A two tailed P < 0.05 was considered to be statistically significant.
Results
Sample Characteristics
Table 1 describes the demographic and clinical information: 92 normal controls, 149 patients with MCI, and 69 patients with AD. No significant differences in age and education were found across the three diagnostic groups. Expectedly, a significant difference in MMSE scores across the three diagnostic groups was detected (p < 0.001). In addition, there were significant differences in RAVLT immediate and delayed recall scores across the three diagnostic groups (p < 0.001). Consistent with the previous data, more than 50% of patients with MCI and AD carried at least one APOE 4 allele (Farrer et al., 1997). There were significant differences in CSF SNAP-25, Aβ42, t-tau, p-tau, and hippocampal volumes across the three diagnostic groups (P < 0.005). However, no significant differences in CSF cortisol levels were found between the three groups (P > 0.05).
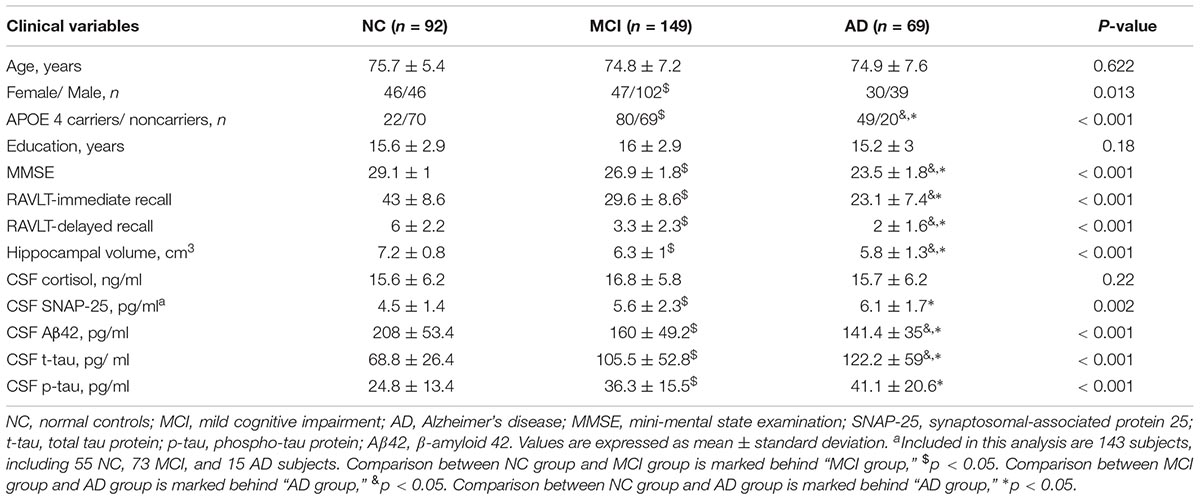
TABLE 1. Demographic and clinical variables.
CSF Cortisol Levels in the Three Groups
As Figure 1 shows, no significant differences in CSF cortisol levels were found across three groups (p > 0.05). In this analysis, we included 310 subjects, including 92 normal controls, 149 patients with MCI, and 69 patients with mild AD.
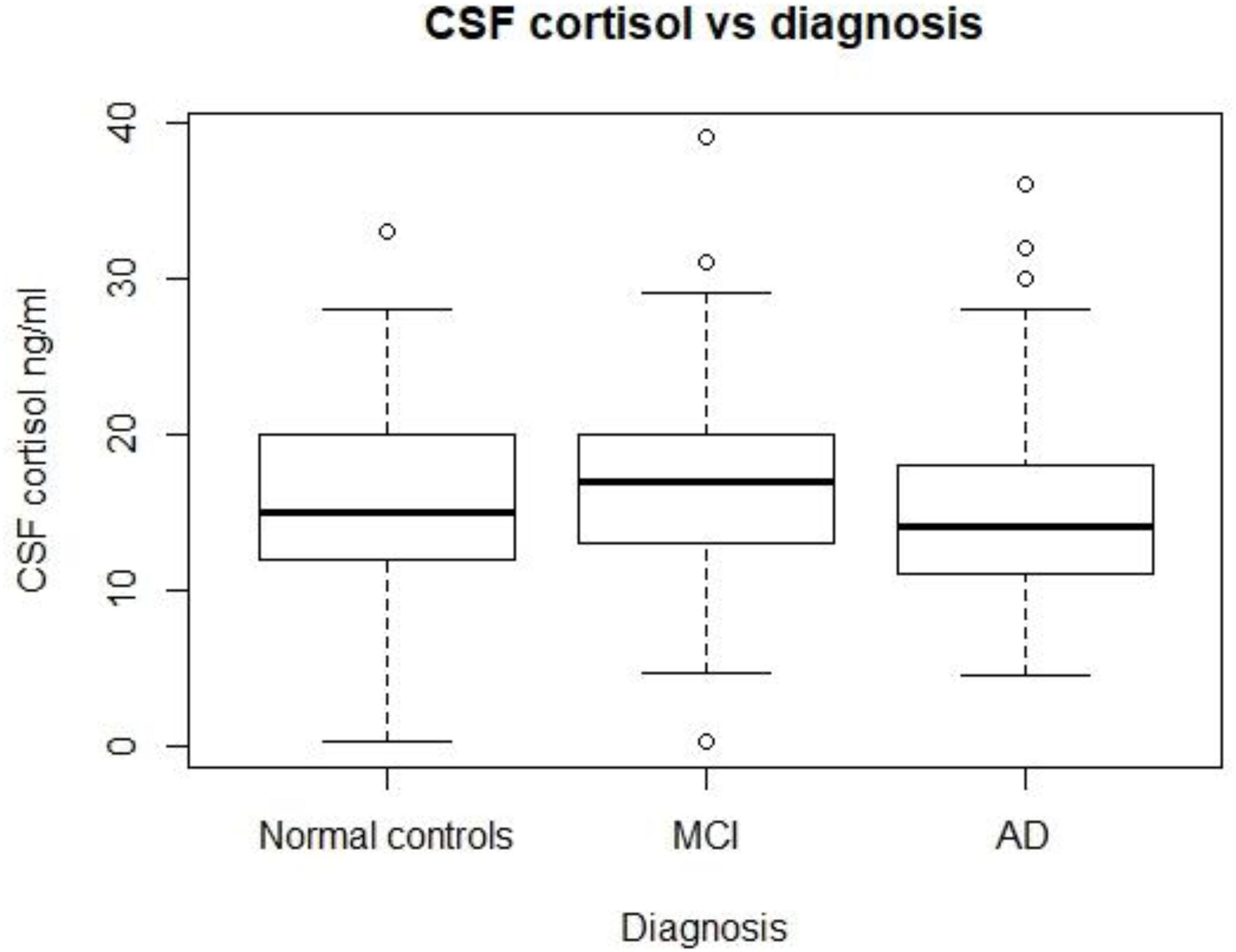
FIGURE 1. CSF cortisol levels in three diagnostic groups. There are no significant differences in CSF cortisol between the three diagnostic groups (p > 0.05). MCI, mild cognitive impairment; AD, Alzheimer’s disease.
Correlations Between CSF Cortisol Levels and SNAP- 25, Aβ42, t-tau and p-tau Levels
To explore the mechanism underlying the role of cortisol in the pathogenesis of AD, the correlations between CSF cortisol and other AD-related biomarkers in all participants were analyzed using Pearson correlation tests (Figure 2). A positive correlation between CSF cortisol and SNAP-42 levels was observed (r = 0.22, p = 0.009). Additionally, the correlations between CSF cortisol between tau protein were analyzed. Levels of cortisol in CSF were associated with CSF t-tau (r = 0.13, p = 0.025) and p-tau (r = 0.13, p = 0.018) levels. However, no correlations between CSF cortisol and Aβ42 levels were found (p > 0.05, Figure 2).
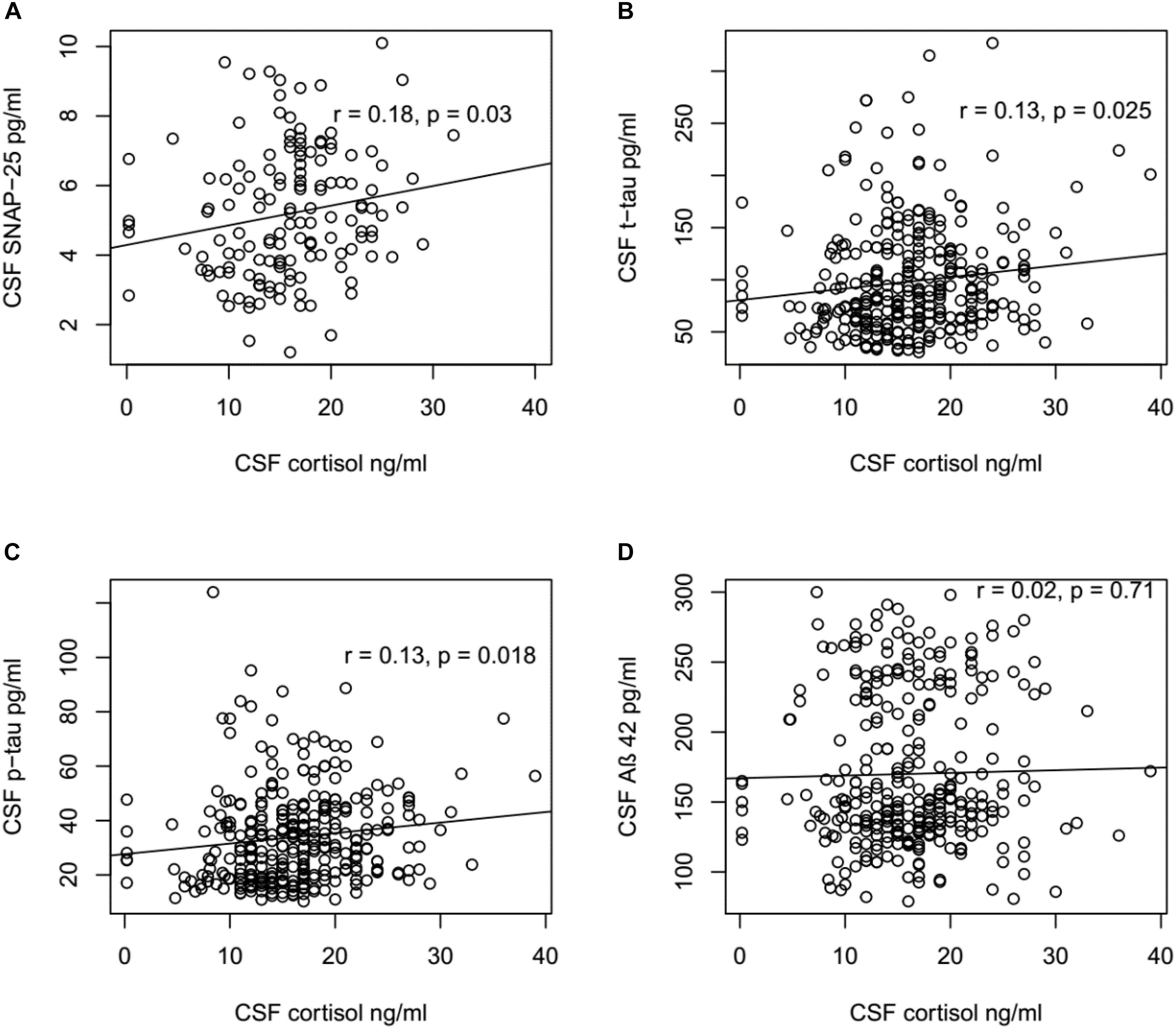
FIGURE 2. Correlation between CSF cortisol levels and SNAP-25, Aβ42, t-tau, and p-tau levels. (A) CSF cortisol levels were positively associated with SNAP-25 in all participants (r = 0.18, p = 0.03). (B) CSF cortisol levels were positively associated with t-tau in all participants (r = 0.13, p = 0.025). (C) CSF cortisol levels were positively associated with p-tau in all participants (r = 0.13, p = 0.018). (D) No significant relationship existed between CSF cortisol levels and Aβ42 in all participants (r = 0.02, p = 0.71). (A) Included in this analysis are 143 subjects, including 55 NC, 73 MCI, and 15 AD subjects. (B–D) Included in this analysis are 310 subjects, including 92 NC, 149 MCI, and 69 AD subjects.
Linear and Logistic Regression Results
We examined the associations between CSF cortisol and memory outcomes, synapse degeneration (SNAP-25), and AD-related biomarkers (hippocampal volumes, Aβ42, tau protein). Higher levels of cortisol in CSF were significantly associated with greater SNAP-25, t-tau, p-tau levels (p < 0.05, Table 2). After controlling for covariates in model 2, the associations between cortisol and SNAP -25, t-tau, p-tau levels remained (Table 2). However, CSF cortisol levels were not significantly associated with performance on tests of memory and hippocampal volumes (Table 2). In addition, we evaluated the relationship between cortisol and amyloid status (<192 pg/ml vs. >192 pg/ml) using logistic regression model, but the results did not reach significance.
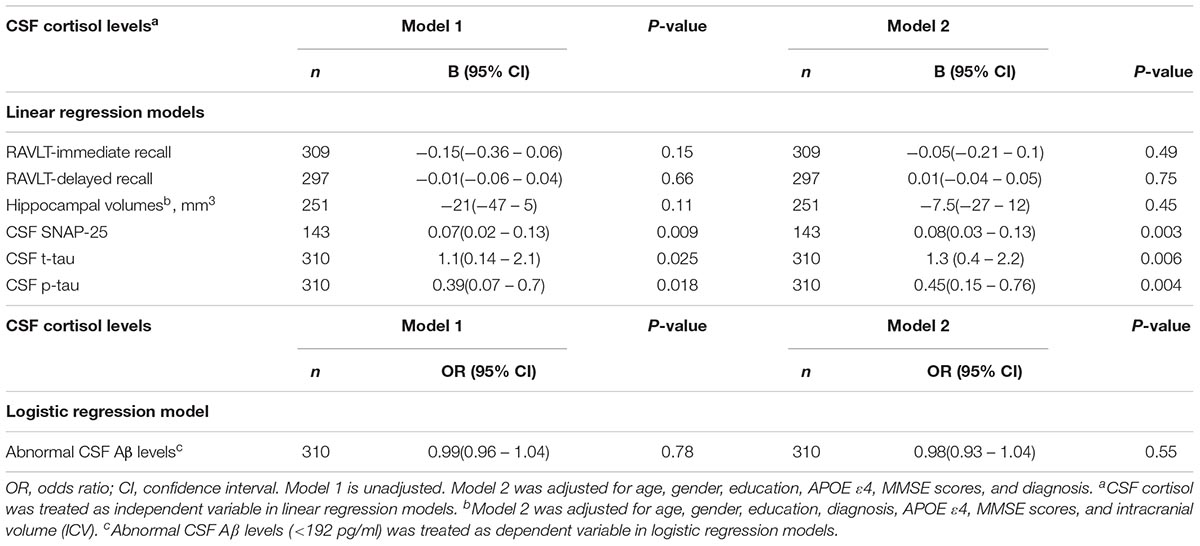
TABLE 2. Association between CSF cortisol and cognition and other biomarkers.
Discussion
To the best of our knowledge, the current study was the first to investigate the relationship of CSF cortisol levels and synapse degeneration among cognitively normal older people, patients with MCI and AD. In addition, we also found that levels of cortisol in CSF were associated with tau pathologies but not amyloid-β protein. However, no significant differences in CSF cortisol levels were found among the three diagnostic groups.
Epidemiological studies suggested that chronic environmental stress plays an important role in the pathogenesis of AD (Pardon, 2011; Zverova et al., 2013). Additionally, preclinical studies have found that glucocorticoids and corticotrophin-releasing hormone (CRH) were involved in AD pathogenesis by manipulating the levels of Aβ and tau pathologies (Kulstad et al., 2005; Green et al., 2006; Jeong et al., 2006; Filipcik et al., 2012; Rothman et al., 2012). Conversely, Baglietto-Vargas et al. (2013) reported that mifepristone, a glucocorticoid receptor antagonist, can reduce levels of Aβ and tau and ameliorate cognitive deficits in mice models of AD. Taken together, these findings highlight an important role of chronic stress and stress-related hormones in affecting AD pathogenesis. In the present study, we found that levels of cortisol in CSF were positively correlated with tau pathology and synapse degeneration in normal controls, subjects with MCI and AD. Our findings were consistent with the results from previous studies (Green et al., 2006; Sotiropoulos et al., 2011). For instance, it has been reported that chronic stress can trigger tau hyperphosphorylation, a core mechanism in AD, and lead to cognitive impairment (Green et al., 2006; Sotiropoulos et al., 2011). Importantly, tau hyperphosphorylation plays a key role in synaptic function and neuronal survival related with AD due to the fact that phosphorylated tau destabilizes microtubules and results in tau accumulation and cytoskeletal damage (Fitzpatrick et al., 2017). In addition, tau mislocalization to synapses may be associated with synaptic dysfunction in AD (Hoover et al., 2010; Tai et al., 2012). These findings could possibly explain how higher levels of cortisol in brain lead to synapse dysfunction, and ultimately cognitive deficits. Although numerous evidence suggested an association of cortisol with synaptic function in preclinical studies, the data in human are limited. Our data found that CSF cortisol was associated with synaptic degeneration in normal controls, patients with MCI and AD. These findings strengthen the idea that CSF cortisol may be associated with synapse degeneration. However, further studies are needed to explore the mechanisms that underlie the associations of cortisol and synaptic degeneration.
In the present study, no significant differences in CSF cortisol levels were observed among three diagnostic groups. It has been reported that there were no differences in cortisol levels in plasma (Csernansky et al., 2006), salivary (Wolf et al., 2002; Souza-Talarico et al., 2010) or CSF (Popp et al., 2009) between cognitively normal subjects and patients with MCI. In contrast, others found greater levels of cortisol in salivary in patients with MCI (Arsenault-Lapierre et al., 2010). However, in patients with AD, previous studies reported elevated levels of cortisol in plasma, salivary, urinary and (Greenwald et al., 1986; Peskind et al., 2001; Rasmuson et al., 2001; Hoogendijk et al., 2006; Curto et al., 2017). These inconsistencies in the published literature on cortisol levels in MCI or AD may result from several differences between studies, including: (1) the sample, which can be salivary, plasma, urinary and CSF; (2) sample size, which can be vary from a handful of participants to many 100’s of participants; and (3) methodological differences in the assay procedure which may have effects on sensitivity.
In present study, we did not find significant correlations between CSF cortisol and Aβ-42 levels, which is consistent with a previous study (Laske et al., 2009). However, in the same study, Laske et al. (2009) found that cortisol levels in serum, but not CSF, were associated with CSF Aβ-42 levels, suggesting serum and CSF cortisol levels appear to be independent in AD patients. Recently, one study investigating the relationship of serum cortisol and amyloid metabolism in patients with major depressive disorder (MDD) found that serum cortisol levels were not associated with serum Aβ levels at baseline. However, cortisol levels in serum at baseline were found to be correlated with serum Aβ levels 1 year later (Ishijima et al., 2018). The precise association of cortisol and amyloid metabolism needs further investigation.
Several limitations should be noted. First, cross-sectional design limits our ability to explore the temporal relationship of cortisol and synapse degeneration and AD-related biomarker. Second, the findings of this study may not be generalizable to other populations. Finally, this study included only subjects with mild AD. Further study should also include moderate and severe dementia patients to clarify the relationship of severity of cognitive impairment and CSF cortisol levels.
Conclusion
In conclusion, we found that CSF cortisol levels were associated with tau pathologies and synapse degeneration but not amyloid- beta, highlighting a potential role of cortisol in synapse degeneration in AD patients.
Author Contributions
JZ conceived and designed the studies. QW, WZ, and JZ performed the research, analyzed the data, and wrote the manuscript.
Funding
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Arsenault-Lapierre, G., Chertkow, H., and Lupien, S. (2010). Seasonal effects on cortisol secretion in normal aging, mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 31, 1051–1054. doi: 10.1016/j.neurobiolaging.2008.07.011
Baglietto-Vargas, D., Medeiros, R., Martinez-Coria, H., Laferla, F. M., and Green, K. N. (2013). Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol. Psychiatry 74, 357–366. doi: 10.1016/j.biopsych.2012.12.003
Brinkmalm, A., Brinkmalm, G., Honer, W. G., Frolich, L., Hausner, L., Minthon, L., et al. (2014). SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol. Neurodegener. 9:53. doi: 10.1186/1750-1326-9-53
Csernansky, J. G., Dong, H., Fagan, A. M., Wang, L., Xiong, C., Holtzman, D. M., et al. (2006). Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry 163, 2164–2169. doi: 10.1176/ajp.2006.163.12.2164
Curto, M., Martocchia, A., Ferracuti, S., Comite, F., Scaccianoce, S., Girardi, P., et al. (2017). Increased Total Urinary Cortisol (tUC) and serum Brain-derived Neurotrophic Factor (BDNF) ratio in Alzheimer Disease (AD)-affected Patients. Alzheimer Dis. Assoc. Disord. 31, 173–176. doi: 10.1097/WAD.0000000000000156
de Leon, M. J., Mcrae, T., Tsai, J. R., George, A. E., Marcus, D. L., Freedman, M., et al. (1988). Abnormal cortisol response in Alzheimer’s disease linked to hippocampal atrophy. Lancet 2, 391–392. doi: 10.1016/S0140-6736(88)92855-3
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease meta analysis consortium. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Filipcik, P., Novak, P., Mravec, B., Ondicova, K., Krajciova, G., Novak, M., et al. (2012). Tau protein phosphorylation in diverse brain areas of normal and CRH deficient mice: up-regulation by stress. Cell. Mol. Neurobiol. 32, 837–845. doi: 10.1007/s10571-011-9788-9
Fitzpatrick, A. W. P., Falcon, B., He, S., Murzin, A. G., Murshudov, G., Garringer, H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. doi: 10.1038/nature23002
Gil-Bea, F. J., Aisa, B., Solomon, A., Solas, M., Del Carmen Mugueta, M., Winblad, B., et al. (2010). HPA axis dysregulation associated to apolipoprotein E4 genotype in Alzheimer’s Disease. J. Alzheimers Dis. 22, 829–838. doi: 10.3233/JAD-2010-100663
Green, K. N., Billings, L. M., Roozendaal, B., Mcgaugh, J. L., and Laferla, F. M. (2006). Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 26, 9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006
Greenwald, B. S., Mathe, A. A., Mohs, R. C., Levy, M. I., Johns, C. A., and Davis, K. L. (1986). Cortisol and Alzheimer’s disease, II: dexamethasone suppression, dementia severity, and affective symptoms. Am. J. Psychiatry 143, 442–446. doi: 10.1176/ajp.143.4.442
Gurevich, D., Siegel, B., Dumlao, M., Perl, E., Chaitin, P., Bagne, C., et al. (1990). HPA axis responsivity to dexamethasone and cognitive impairment in dementia. Prog. Neuropsychopharmacol. Biol. Psychiatry 14, 297–308.
Hoogendijk, W. J., Meynen, G., Endert, E., Hofman, M. A., and Swaab, D. F. (2006). Increased cerebrospinal fluid cortisol level in Alzheimer’s disease is not related to depression. Neurobiol. Aging 27, 780.e1–780.e2. doi: 10.1016/j.neurobiolaging.2005.07.017
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Ishijima, S., Baba, H., Maeshima, H., Shimano, T., Inoue, M., Suzuki, T., et al. (2018). Glucocorticoid may influence amyloid beta metabolism in patients with depression. Psychiatry Res. 259, 191–196. doi: 10.1016/j.psychres.2017.10.008
Jeong, Y. H., Park, C. H., Yoo, J., Shin, K. Y., Ahn, S. M., Kim, H. S., et al. (2006). Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer’s disease model. FASEB J. 20, 729–731. doi: 10.1096/fj.05-4265fje
Kulstad, J. J., Mcmillan, P. J., Leverenz, J. B., Cook, D. G., Green, P. S., Peskind, E. R., et al. (2005). Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J. Neuropathol. Exp. Neurol. 64, 139–146. doi: 10.1093/jnen/64.2.139
Laske, C., Stransky, E., Fritsche, A., Eschweiler, G. W., and Leyhe, T. (2009). Inverse association of cortisol serum levels with T-tau, P-tau 181 and P-tau 231 peptide levels and T-tau/Abeta 1-42 ratios in CSF in patients with mild Alzheimer’s disease dementia. Eur. Arch. Psychiatry Clin. Neurosci. 259, 80–85. doi: 10.1007/s00406-008-0838-3
Lee, B. K., Glass, T. A., Mcatee, M. J., Wand, G. S., Bandeen-Roche, K., Bolla, K. I., et al. (2007). Associations of salivary cortisol with cognitive function in the Baltimore memory study. Arch. Gen. Psychiatry 64, 810–818. doi: 10.1001/archpsyc.64.7.810
Olsson, B., Hertze, J., Ohlsson, M., Nägga, K., Höglund, K., Basun, H., et al. (2013). Cerebrospinal fluid levels of heart fatty acid binding protein are elevated prodromally in Alzheimer’s disease and vascular dementia. J. Alzheimers Dis. 34, 673–679. doi: 10.3233/JAD-121384
Pardon, M. C. (2011). Therapeutic potential of some stress mediators in early Alzheimer’s disease. Exp. Gerontol. 46, 170–173. doi: 10.1016/j.exger.2010.09.006
Peskind, E. R., Wilkinson, C. W., Petrie, E. C., Schellenberg, G. D., and Raskind, M. A. (2001). Increased CSF cortisol in AD is a function of APOE genotype. Neurology 56, 1094–1098. doi: 10.1212/WNL.56.8.1094
Popp, J., Schaper, K., Kolsch, H., Cvetanovska, G., Rommel, F., Klingmuller, D., et al. (2009). CSF cortisol in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 30, 498–500. doi: 10.1016/j.neurobiolaging.2007.07.007
Rasmuson, S., Andrew, R., Nasman, B., Seckl, J. R., Walker, B. R., and Olsson, T. (2001). Increased glucocorticoid production and altered cortisol metabolism in women with mild to moderate Alzheimer’s disease. Biol. Psychiatry 49, 547–552. doi: 10.1016/S0006-3223(00)01015-5
Rothman, S. M., Herdener, N., Camandola, S., Texel, S. J., Mughal, M. R., Cong, W. N., et al. (2012). 3xTgAD mice exhibit altered behavior and elevated Abeta after chronic mild social stress. Neurobiol. Aging 33, 830.e1–830.e12. doi: 10.1016/j.neurobiolaging.2011.07.005
Scheltens, P., Blennow, K., Breteler, M. M., De Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Shaw, L. M., Vanderstichele, H., Knapik-Czajka, M., Clark, C. M., Aisen, P. S., Petersen, R. C., et al. (2009). Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413. doi: 10.1002/ana.21610
Sotiropoulos, I., Catania, C., Pinto, L. G., Silva, R., Pollerberg, G. E., Takashima, A., et al. (2011). Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 31, 7840–7847. doi: 10.1523/JNEUROSCI.0730-11.2011
Souza-Talarico, J. N., Chaves, E. C., Lupien, S. J., Nitrini, R., and Caramelli, P. (2010). Relationship between cortisol levels and memory performance may be modulated by the presence or absence of cognitive impairment: evidence from healthy elderly, mild cognitive impairment and Alzheimer’s disease subjects. J. Alzheimers Dis. 19, 839–848. doi: 10.3233/JAD-2010-1282
Sze, C. I., Bi, H., Kleinschmidt-Demasters, B. K., Filley, C. M., and Martin, L. J. (2000). Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J. Neurol. Sci. 175, 81–90. doi: 10.1016/S0022-510X(00)00285-9
Tai, H. C., Serrano-Pozo, A., Hashimoto, T., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. doi: 10.1016/j.ajpath.2012.06.033
Tata, D. A., Marciano, V. A., and Anderson, B. J. (2006). Synapse loss from chronically elevated glucocorticoids: relationship to neuropil volume and cell number in hippocampal area CA3. J. Comp. Neurol. 498, 363–374. doi: 10.1002/cne.21071
Toonen, R. F., Verhage, M., Noor, A., and Zahid, S. (2017). A review of the role of synaptosomal-associated protein 25 (SNAP-25) in neurological disorders. J. Neurosci. 127, 805–811.
Wolf, O. T., Convit, A., Thorn, E., and De Leon, M. J. (2002). Salivary cortisol day profiles in elderly with mild cognitive impairment. Psychoneuroendocrinology 27, 777–789. doi: 10.1016/S0306-4530(01)00079-8
Zhang, D., Wang, Y., Zhou, L., Yuan, H., and Shen, D. (2011). Multimodal classification of Alzheimer’s disease and mild cognitive impairment. Neuroimage 55, 856–867. doi: 10.1016/j.neuroimage.2011.01.008
Zhang, H., Therriault, J., Kang, M. S., Ng, K. P., Pascoal, T. A., Rosa-Neto, P., et al. (2018). Cerebrospinal fluid synaptosomal-associated protein 25 is a key player in synaptic degeneration in mild cognitive impairment and Alzheimer’s disease. Alzheimers Res. Ther. 10:80. doi: 10.1186/s13195-018-0407-6
Keywords: cortisol, SNAP-25, Alzheimer’s disease, tau pathology, synapse degeneration, mild cognitive impairment
Citation: Wang Q, Zhou W, Zhang J and The Alzheimer’s Disease Neuroimaging Initiative (2018) Levels of Cortisol in CSF Are Associated With SNAP-25 and Tau Pathology but Not Amyloid-β. Front. Aging Neurosci. 10:383. doi: 10.3389/fnagi.2018.00383
Received: 27 August 2018; Accepted: 01 November 2018;
Published: 20 November 2018.
Edited by:
Nibaldo C. Inestrosa, Pontificia Universidad Católica de Chile, ChileReviewed by:
Ramesh Kandimalla, Texas Tech University Health Sciences Center, United StatesAyodeji A. Asuni, New York University, United States
Copyright © 2018 Wang, Zhou, Zhang and The Alzheimer’s Disease Neuroimaging Initiative. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Zhang, amllemhhbmdAcG9zdC5jb20=
†Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.