 Masoumeh Tangestani Fard
Masoumeh Tangestani Fard Con Stough*
Con Stough*- Centre for Human Psychopharmacology, Swinburne University of Technology, Melbourne, VIC, Australia
Age is associated with increased risk for several disorders including dementias, cardiovascular disease, atherosclerosis, obesity, and diabetes. Age is also associated with cognitive decline particularly in cognitive domains associated with memory and processing speed. With increasing life expectancies in many countries, the number of people experiencing age-associated cognitive impairment is increasing and therefore from both economic and social terms the amelioration or slowing of cognitive aging is an important target for future research. However, the biological causes of age associated cognitive decline are not yet, well understood. In the current review, we outline the role of inflammation in cognitive aging and describe the role of several inflammatory processes, including inflamm-aging, vascular inflammation, and neuroinflammation which have both direct effect on brain function and indirect effects on brain function via changes in cardiovascular function.
Introduction
Life expectancies have increased considerably since the 1950s in developed countries. For example, the population of octogenarians in developed countries has increased four-fold, the population of nonagenarians eight-fold, and the population of centenarians 20-fold. Aging is a progressive decline in the physiological integrity of different organs of the human body, which leads to impaired body function and enhanced vulnerability to death. This deterioration is a crucial risk factor for the main human pathologies, including neurodegenerative diseases, cardiovascular disorders, cancer and diabetes (López-Otín et al., 2013). To date, several variations of innate and acquired immunity have been observed in the elderly. These alterations have been generally, explained as a deterioration of immunity, which has been referred to as immunosenescence and is characterized via chronic inflammatory conditions. Immunosenescence refers to increased susceptibility of the elderly to infection and may be explained in terms of molecular and cellular mechanisms responsible for inflammatory age-associated disorders (Larbi et al., 2008; Caruso et al., 2009). A better understanding of immunosenescence and the development of novel strategies to counteract it are necessary, not only for anti-aging strategies aimed at preventing or slowing down cognitive aging but, more notably with the aim of prolonging healthy life, through preventing infectious and age-associated disorders and improving the quality of life in later years (Candore et al., 2008; Jirillo et al., 2008; Larbi et al., 2008; Caruso et al., 2009; Holmes et al., 2009; Trollor et al., 2010; Barrientos et al., 2015; Di Benedetto et al., 2017). Cognitive aging is characterized by a decline in memory and other cognitive processes, changes in behaviors and impaired ability to live an independent and high functioning life (Cunningham and Hennessy, 2015). In the current review article, we bring together different biological processes related to inflammation within the context of cognitive aging. There have been few theoretical models of the molecular and cellular mechanisms of cognitive decline, with most of the literature focusing on abnormal aging and cognitive disorders of aging such as Alzheimer’s Dementia (AD; Changeux and Dehaene, 1989; Miller and Cohen, 2001; Zlokovic, 2005; Bishop et al., 2010). The cytokine model of cognitive function explained by McAfoose and Baune (2009) emphasized the important role of cytokines in cognitive process at the molecular level such as in synaptic plasticity, neurogenesis, and neuromodulation, which may subserve learning, memory, and other cognitive processes. This cytokine-mediated model of cognitive processes has been proposed to be causative in terms of longer-term pathogenesis related to some neuropsychiatric disorders such as AD and Major Depression (McAfoose and Baune, 2009) but there is a lack of clarity in terms of how some of these processes may affect cognitive aging. In this review, we outline the involvement of three main aging features of the central nervous system (CNS) that underpin cognitive decline (Figure 1). Specifically, we present a model of cognitive aging that comprises three main aging features of the CNS, including immunosenescence, vascular aging, and brain aging and we briefly review the role of each of these components in terms of changes in cognition with increasing age.
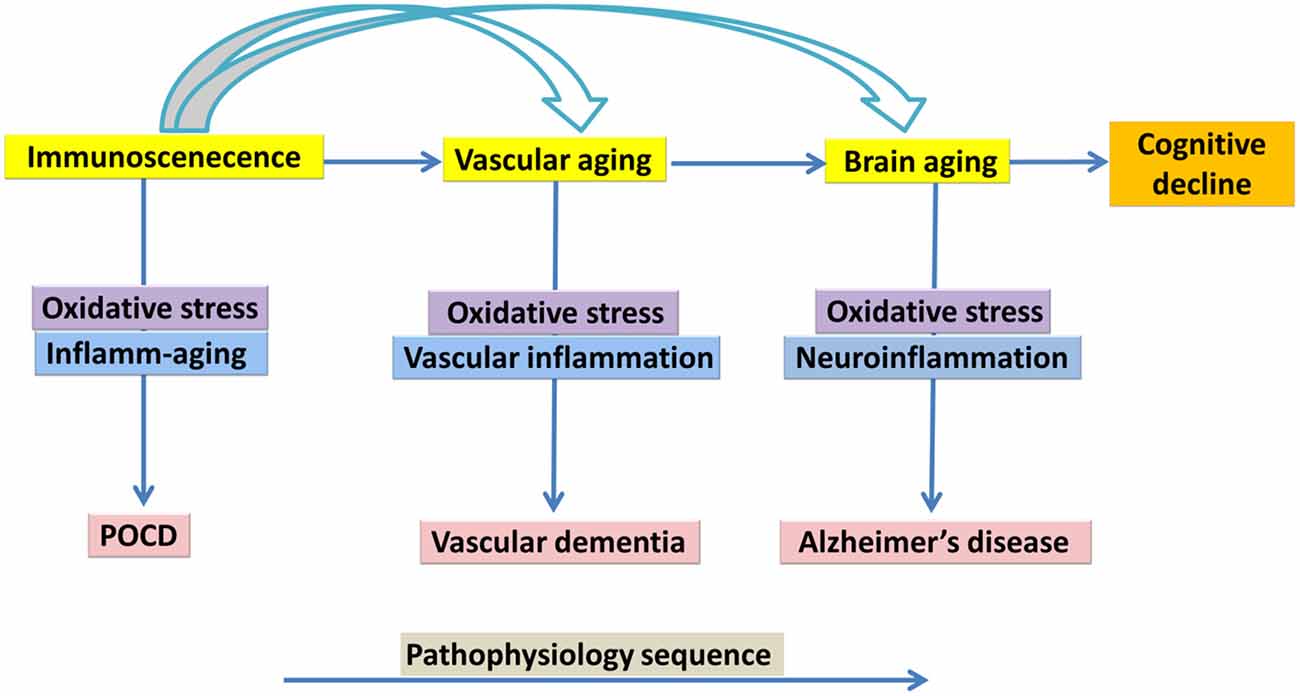
Figure 1. Immunosenescence, vascular aging, brain aging in association with cognitive decline, a suggested model of underlying mechanism.
Cognition
Cognition refers to mental processes that are often measured in terms of our ability to allocate attention, recall information, to perceive relationships as well as the ability to think locally and abstractly amongst other cognitive domains. Some of these cognitive domains decrease, as we get older (Christensen, 2001; Singh-Manoux et al., 2012). In particular, memory and processing speed appear to be more sensitive to age than other cognitive domains (Salthouse, 1996; Christensen, 2001). A reduction in cognitive function affects more than 50% of people over 60 years of age (Skaper et al., 2014). Dementia is a generic term that encompasses several diseases with different pathologies such as AD, vascular dementia (VD), frontotemporal dementia, and dementia with Lewy bodies. Their common characteristic is a progressive reduction in cognitive performance, which leads to functional dependency and death (Gao et al., 2016). However, it is unclear which biological processes underpin these changes. Some researchers have proposed a linkage between inflammatory processes and cognition. Although most of this research has been derived from animal studies, the results of which could also be applied to understanding human conditions such as cognitive aging. These investigations have emphasized a close association between some aspects of the immune system, processes at the level of the neuron and vascular systems (Zlokovic, 2005; McAfoose and Baune, 2009; Grammas, 2011; Broussard et al., 2012; Davenport et al., 2012; Kousik et al., 2012; Barrientos et al., 2015; Di Benedetto et al., 2017; Tarantini et al., 2017). Interestingly, a recent review by Gauthier et al. (2018) argued for the importance of considering the interaction of several factors involved in age-associated cognitive decline (particularly AD) such as vascular small vessel disease, neuroinflammation and Lewy body pathology (Gauthier et al., 2018). Vijayan and Reddy (2016) also argued that stroke was a major risk factor contributing to AD and VD through several cellular and molecular changes including inflammation, oxidative stress, mitochondrial dysfunction, vascular changes and marked changes in brain proteins.
Immunosenescence
The remarkable development of human survival and lifespan to well beyond childbearing ages has been completely “unpredicted” via evolution (Baylis et al., 2013). As a result, the human immune system is exposed to significant additional antigenic exposure outside the forces of natural selection (De Martinis et al., 2005; Franceschi, 2007; Franceschi et al., 2007). The immune system is highly effective for the first 40 years of life and after that, similar to all other organs and systems of the body undergoes a process of senescence and certain features begin to reveal effectual reduction (Piazza et al., 2010; Salvioli et al., 2013). The immunity begins to exhibit negative effects on human aging (antagonistic pleiotropy) leading to gradual systemic failures (De Martinis et al., 2005; Franceschi, 2007; Franceschi et al., 2007). The process of senescence causes a progressive reshaping of its functions in a pervasive process, which influences almost all the compartments of the immune system, particularly the branch in control of the acquired immunity (Salvioli et al., 2013). The immune system during aging declines in efficiency and reliability resulting in greater susceptibility to pathological conditions as a consequence of chronic inflammatory responses, for instance, Alzheimer’s disease, cardiovascular disease, auto-reactivity as well as an enhanced vulnerability to infectious disease (Baylis et al., 2013). These variations are further compounded by a reduction in responsiveness-impaired communication among all cells of the immune system. The overall alteration of the immune system during aging is termed “immunosenescence” and has a multifactorial etiology (Weiskopf et al., 2009). Immunosenescence of the acquired immune system includes the involvement of the thymus and reduced responsiveness to new antigen load, due to reduced naïve: memory cell ratio and expansion of mature cell clones (Baylis et al., 2013; Müller and Pawelec, 2015). Thymic output decreases with age resulting in reduced T-cell repertoire and enhanced oligoclonal expansion of memory and effector-memory cells (Pawelec, 2012). This imbalance leads to a reduced ability to clear novel pathogens (prolonging infection duration) as well as an elevation in functionally distinct T-cell populations that have an amplified pro-inflammatory phenotype (Weiskopf et al., 2009). Immunosenescence of the innate immune system is mainly defined by the reduction in cellular superoxide production and capability for phagocytosis (Pawelec, 2012). Of the innate immune system, monocytes and macrophages are assumed to lead to inflamm-aging (cellular exhaustion) more than any other cell type. Monocyte variations with age can cause inflamm-aging via reduced function and a functional shift against a proinflammatory phenotype (Shaw et al., 2010).
What Is Inflamm-Aging?
Another striking characteristic feature of immunosenescence is an increase in cellular production of proinflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL)-6 and IL-1β in serum’s individuals (Zanni et al., 2003; Salvioli et al., 2013) and has been indicated as inflamm-aging the chronic sub-clinical elevated production of pro-inflammatory mediators typical of elderly (Blagosklonny and Hall, 2009; Blagosklonny, 2010). Inflamm-aging is a consequence of a cumulative lifetime exposure to antigenic load due to both clinical and sub-clinical infections as well as non-infective antigens (Baylis et al., 2013). It is believed to be related to several age-associated disorders sharing a similar inflammatory basis. However, recent research indicates that inflamm-aging is at least in part independent from immunological stimuli. In addition, centenarians who prevented or delayed major inflammatory disorders display elevation in inflammatory mediators (Salvioli et al., 2013). The result of the inflammatory response is tissue damage and the release of reactive oxygen species (ROS), which can lead to oxidative damage and in turn stimulate the production of increased levels of cytokines, principally from cells of the innate immune system (Cannizzo et al., 2011) as well as the acquired immune system. These events initiate a vicious cycle in which the immune system is remodeled favoring a chronic pro-inflammatory response where, healing responses, pathophysiological variations, and tissue damage occur at the same time. Irreversible molecular and cellular damage, which is not clinically noticeable, gradually accumulates over decades (Baylis et al., 2013). Theoretically, the process of inflamm-aging could account for the increased frequency of inflammation-based pathologies, which occur with increased age (e.g., neurodegeneration, cardiovascular diseases, arthritis, type II diabetes, and several types of cancers). The hypothesis that aging is driven via unnecessary inflammatory responses stems from the concept of inflamm-aging and is consistent with the recent theory of aging as a quasi-program (Blagosklonny and Hall, 2009; Blagosklonny, 2010).
Another cellular phenomena, relevant to immunosenescence and inflammation is “cellular senescence” which is a hallmark of aging and has been reported in a large number of studies in which the mechanisms have been extensively reviewed (Campisi and d’Adda di Fagagna, 2007; Collado et al., 2007; Collado and Serrano, 2010; Kuilman et al., 2010; Salama et al., 2014; van Deursen, 2014; Walters et al., 2016). Cellular senescence can occur in almost all cell types that are capable of cell division (Bitto et al., 2010; Coppé et al., 2010b; Chinta et al., 2015). Recent evidence in epithelial cells and fibroblasts have revealed that cellular senescence is mediated through a large increase in the production of 40–80 factors that play a vital role in intercellular signaling (Coppé et al., 2008, 2010a; Young and Narita, 2009; Chinta et al., 2015). The release of these series of factors has been termed “senescence-associated secretory phenotype,” or SASP and they generally increase at mRNA levels (Coppé et al., 2008) and enhance a wide array of proteases, chemokines, growth factors, and cytokines. SASP proteins that are characterized as inflammatory stimulators, include IL-8, IL-6, IL-1, granulocyte macrophage colony stimulating factor, monocyte chemotactic protein-2 (MCP-2), MCP-3, matrix metalloproteinase-1 (MMP-1), MMP-3, growth regulated oncogene-α, and several Insulin-like growth factor-binding proteins (Kumar et al., 1992; Wang et al., 1996; Coppé et al., 2008) which are among the most largely secreted SASP factors and potentially can cause or aggravate, age-related pathology, both degenerative and hyperplastic (Chinta et al., 2015). Therefore, senescent cells are a source of chronic inflammation senescence/inflamm-aging during the aging process (Freund et al., 2010). Some other studies also indicated that inflammation could exacerbate the biological aging and cellular senescence, which may be a cause for the loss of cognitive function (Panossian et al., 2003; Honig et al., 2006; Ma et al., 2013).
Inflamm-Aging, Cellular Senescence and Cognitive Aging
The relationship between inflamm-aging and cognitive aging has been reported in several human studies (Dik et al., 2005; de Rooij et al., 2007; Schram et al., 2007; Marioni et al., 2009; Kim et al., 2015). For instance, Trollor et al. (2010) argued for a relationship between inflamm-aging and mild cognitive impairment (MCI), in the Sydney memory and aging study cohort, a longitudinal study of 1,037 Australians aged 70–90 years. The findings showed that the concentration of TNF-α and serum amyloid A, were higher in individuals with MCI compared with cognitively normal participants (Trollor et al., 2010). Three hundred community-dwelling individuals with mild to severe Alzheimer’s disease were tested on cognitive measures and inflamm-aging. An increase in the serum level of TNF-α and a two-fold increase in the rate of cognitive decline over 6 months were observed in around half of all study participants. In the baseline group, high levels of TNF-α was associated with a four-fold enhancement in the rate of cognitive decline. Individuals with low serum levels of TNF-α showed no cognitive decline over the 6 months (Holmes et al., 2009). In another study by Yaffe et al. (2003), a group of 3,031 White and African-Americans with mean age of 74 years were assessed in terms of cognitive function and inflamm-aging. Individuals in the highest tertile for C-reactive protein (CRP) or IL-6 had nearly two points lower scores on the Modified Mini-Mental State Examination (3MS) at baseline. These scores then declined further over the 2 years of the study in comparison with those with the lowest tertile for CRP or IL-6. Participants with the highest inflammatory mediators tertile were also more likely to have cognitive decline compared with participants with the lowest tertile for IL-6 and for CRP but not for TNF-α (Yaffe et al., 2003).
While research on the cellular senescence in brain aging is at early stages, the role of cellular senescence in peripheral tissues during several age-related pathologies has been more frequently explored (Chinta et al., 2015). Animal studies have been published on brain cellular senescence and cognitive aging (Cho et al., 2015; Tarantini et al., 2017), for example a study by Li et al. (2018) reported on D-galactose-induced aging in mouse model and showed that Zicao (Acetyl shikonin) treatment significantly reduced hippocampus senescence and cognitive impairments through upregulating the expression of SIRT1 and suppressing inflammatory cytokines such as IL-1β and TNF-α (Li et al., 2018). Ungvari et al. (2017) reported that whole brain irradiation-induced accelerated brain senescence is notably associated with cerebromicrovascular dysfunction and cognitive decline. Parisotto et al. (2016) showed that Melatonin treatment reduced cellular senescence and oxidative damage in the hippocampus of a mouse model of down syndrome. Studies on human brain samples have also indicated an increase in DNA-protein kinase catalytic subunit and incorporating phosphorylated histone, γH2AX (which exhibited a neuronal DNA damage response) is correlated with cognitive impairment (Simpson et al., 2015) (Figure 2).
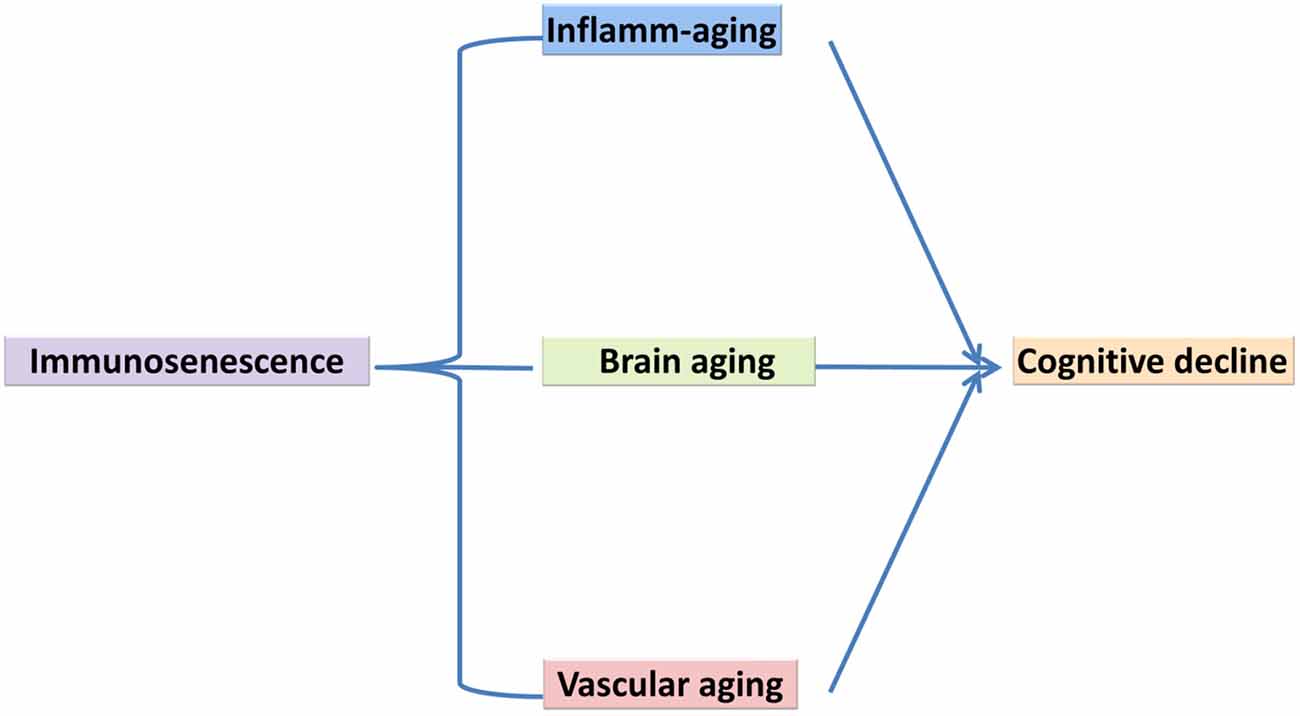
Figure 2. Immunosenescence is a key mechanism underlying cognitive decline.
Immunosenescence and Oxidative Stress
The majority of age-related disorders are associated with a low level of chronic inflammation (De la Fuente and Miquel, 2009; Cannizzo et al., 2011). Currently, it is not yet, fully understood whether inflammatory responses lead to the development of degenerative chronic disorders or whether the chronic pathologies can lead to inflammatory response observed during aging. Regardless of the cause-effect relationship among age-dependent disorders and inflammation, oxidative stress is known to play a main role in maintaining the chronic inflammation or inflamm-aging observed in aging and age-dependent disorders (De la Fuente and Miquel, 2009). Toll-like-receptors and the Nalp-3 inflammasome are two main molecular pathways which develop inflammatory responses through oxidative damage produced by free radicals (Cannizzo et al., 2011). Moreover, increases in oxidative stress can lead to telomere length attrition and cellular senescence which may be associated with cognitive decline (Ma et al., 2013).
The idea of ROS involvement in aging process, dates back to 1995 when Harman suggested the “Free Radical Theory of Aging” or “oxidative stress theory of aging,” referring to accumulation of free radicals during aging process which could cause biomolecule damage and the development of pathological condition (Harman, 1992, 2006; Harvey et al., 2015; Black et al., 2017). Free radicals are molecules with unpaired electrons which are mainly unstable (Cannizzo et al., 2011) and increase the non-enzymatic oxidation of biomolecules (proteins, carbohydrates, lipids and nucleic acids; Halliwell, 2009; Buonocore et al., 2010; Hamanaka and Chandel, 2010). Two main biochemical mechanisms link immunosenescence to oxidative stress: (i) a decline in cellular functions because of oxidative damage in protein, lipid and carbohydrate; and (ii) cellular apoptosis followed by accumulation of oxidized molecular aggregates. The elevation in free radicals volume in several aging cells has also been observed in immunological cells (Nomellini et al., 2008). Additionally, the amount of catalase, superoxide dismutase, and glutathione peroxidase which are the enzymes responsible for free radical clearance in the cytosol are reduced in aged cells (Cannizzo et al., 2011). Similarly, the amount of manganese superoxide dismutase which is an antioxidant enzyme situated in the mitochondria, and defenses macrophages from apoptosis increased through oxidized low-density lipoprotein is also decreased in aging macrophages (Fujimoto et al., 2010) all of which contribute to the elevated level of cellular oxidative stress (Nomellini et al., 2008; Cannizzo et al., 2011).
Vascular Aging
Vascular aging has been described in terms of changes in structure and function of the endothelium and smooth muscle cells and the communication routes between these two cell layers that form the vascular wall (Younger, 2004; Pase et al., 2012; El Assar et al., 2013). Impaired endothelial vasodilation is a virtual manifestation of arterial aging and a clinical indicator of vascular dysfunction, which may occur over a long period of time (Younger, 2004; Seals et al., 2006; El Assar et al., 2013). Moreover, impaired endothelial vasodilation is the first stage in changes in vascular outcomes and cardiovascular disease in elderly people (Seals et al., 2006; El Assar et al., 2013; van Buchem et al., 2014). With advancing age, there are changes that occur in the vasculature, including endothelial dysfunction, vascular remodeling, increased vascular stiffness and vascular inflammation, which contributes to hypertension (Grammas, 2011; Baierle et al., 2015; Harvey et al., 2015). In terms of hypertension, large and small arteries undergo mechanical structural, and functional changes which contribute to vascular complication and elevated cardiovascular risk (Savoia and Schiffrin, 2006; van Buchem et al., 2014). Moreover, the renin–angiotensin system exhibit a key role in the pathophysiology and development of hypertension and cardiovascular disease (Marchesi et al., 2008). Hypertension-enhanced vascular changes include low-grade inflammatory processes in which inflammation contributes to the pathophysiology of high blood pressure (Savoia and Schiffrin, 2006). One crucial hallmark of vascular aging is an increased arterial stiffness, resulting in the loss of arterial elasticity, compromising vascular adaptation to blood flow and pressure changes (El Assar et al., 2013; van Buchem et al., 2014; Pase et al., 2015). This increased arterial stiffness is often revealed via increases in the speed of propagation pressure/flow waves (Pase et al., 2010, 2012; El Assar et al., 2013). Arterial stiffness causes impaired endothelial vasodilation leading to endothelial dysfunction (Scuteri et al., 2008; Pase et al., 2010, 2015). Besides changes in the structure and function of the endothelium, endothelial dysfunction also has a crucial role in age-associated microvascular dysfunction (Rodríguez-Mañas et al., 2009). Small and large arteries are not isolated systems but are involved in crosstalk so that changes in the integrity and function of small arteries impact on the functionality of larger arteries, a condition that prevents small artery remodeling and organ damage (Laurent et al., 2009). This crosstalk between large artery changes and small cerebral arteries may be important in terms of cognitive aging (El Assar et al., 2013; van Buchem et al., 2014).
The functionality of the brain depends on a constant blood supply and disruptions in cerebral blood flow can lead to brain diseases and death (Moskowitz et al., 2010; van Buchem et al., 2014). Because of this, cerebrovascular control mechanisms function to ensure that the brain blood supply is sufficient for its energy requirements (Iadecola and Nedergaard, 2007). Increased neuronal activity is associated with an elevation in cerebral blood flow (functional hyperemia) which is thought to generate energy substrates and remove the toxic component that is derived from brain cellular activity (Paulson et al., 2010; Zlokovic, 2011). Cerebrovascular autoregulation manages cerebral blood flow to some degree ensuring some consistency in the range of blood pressure, and protecting the brain tissue from unwanted swings in perfusion pressure (van Beek et al., 2008). Specialized receptors on endothelial cells surface transduce mechanical (shear stress) and chemical stimuli, for instance endothelin, nitric oxide and prostanoids (Wolburg et al., 2009). Compelling evidence suggest that the health of brain is also dependent on the overall health of the cardiovascular system, and the existence of parenchymal and vascular inflammation which has often been offered as a link among atherosclerosis and AD (Grammas, 2011; Tzikas et al., 2014). To date, atherosclerosis and AD are known to share similar vascular risk factors for instance hypercholesterolemia, hypertension, as well as heart failure (Helzner et al., 2009; Roselli et al., 2009; Gorelick et al., 2016) and arterial stiffness (Triantafyllidi et al., 2009; Gorelick et al., 2016). The treatment of vascular risk factors has been shown to reduce the risk of developing Alzheimer’s disease (Babarskiene et al., 2002) and dementia (Jellinger, 2013) and to slow cognitive decline in AD patients (Deschaintre et al., 2009; Helzner et al., 2009). Compelling data also establishes a relationship between aortic stiffening and cognitive dysfunction (Pase et al., 2012; de la Torre, 2012) with greater cognitive impairment associated with poorer cerebral microcirculation and increased aortic stiffness (Triantafyllidi et al., 2009; Pase et al., 2010, 2012).
One of the key variations during vascular aging is the formation and development of inflame-aging (Franceschi, 2007). Inflamm-aging is not only independent of traditional cardiovascular disease risk factors but also accelerates arterial thickening and arterial stiffness independently from processes associated with cardiovascular disease (Scuteri et al., 2011). There is accumulating evidence for an increased systemic inflammatory mediators (inflamm-aging) for example IL-1β, TNF-α, members of the superfamily of IL-6, as well as, elevated amount of CRP in plasma of older adults while compared with young adults (Ferrucci et al., 2005). This increase in inflammatory markers is associated with age, and independent of other cardiovascular disease (Miles et al., 2008). The pleiotropic proinflammatory IL-6 has been importantly associated with age-dependent vascular disorders (Ungvari et al., 2004). Moreover, elevated plasma amount of IL-6 has been associated with larger disability and mortality in older people (Cesari et al., 2012). CRP levels are also correlated with elevated arterial stiffness in middle-aged and elderly (Mattace-Raso et al., 2004; Nakhai-Pour et al., 2007). However, expressions of MCP-1 and MMP are higher in the thickened arterial intima of vessels taken from autopsies of older people in comparison with those from young adult (El Assar et al., 2013). Vascular endothelial cells play a key role in the pathobiology of vascular inflammatory processes due to their potential interaction with elements related to systemic inflammation. They are potentially active participants during vasculitis, not only passive targets of injury (Younger, 2004). This is consistent with arterial changes that are associated with endothelial dysfunction (Scuteri et al., 2008; Pase et al., 2010, 2015). Several chronic vascular disorders are part of a progressive process, initiating and developing through local inflammation of large and medium sized arteries (Renna et al., 2013). It is scientifically relevant in this regard that pro-inflammatory signaling mechanism in the vascular wall has been well characterized and the risk of progressing age-associated neurodegenerative disease is related to increased circulatory inflammatory cytokines, such as IL-6 and TNF-α (Simen et al., 2011). Serum levels of homocysteine has an associated with atherosclerosis and can damage blood vessels. Crucially, AD disease data have demonstrated that higher levels of homocysteine independently and strongly predict the development of dementia (Fiolaki et al., 2014; Kim et al., 2018). VD is a heterogeneous group of brain disorders in which cognitive decline is attributable to cerebrovascular pathologies, and includes a large component of dementia prevalence (Gorelick et al., 2011; Iadecola, 2013). The level of some circulatory inflammatory proteins such as CRP and 1-antichymotrypsin have been observed to increase before the onset of VD (Engelhart et al., 2004). The level of CRP has also been shown to increase 25 years before the onset of VD (Schmidt et al., 2002). In the Conselice study of brain aging, with 4 years of follow-up, the combination of high levels of IL-6 and CRP led to a nearly three-fold increased risk of VD (Ravaglia et al., 2007). Yano et al. (2010) also reported that pentraxin 3, a circulatory inflammatory biomarker predicts cognitive decline in elderly hypertensive patients.
What Is Vascular Inflammation (Vasculitis)?
Vasculitis is inflammation of the blood vessel wall, which causes different pathologies, depending on the type of impaired organ (Jennette and Falk, 1997; Hoffman and Calabrese, 2014). Vasculitis includes a heterogeneous class of disorders recognized via inflammation and necrosis of the blood vessel wall. Based on the Chapel Hill Consensus Conference, the primary systemic vasculitis could be characterized into three major groups influencing large-sized, medium- and small sized vessels, respectively (Jennette and Falk, 2007). Small sized vessel vasculitis is categorized under several names including Wegener’s granulomatosis, Churg–Strauss syndrome, Microscopic polyangiitis, Henoch–Schönlein purpura, Essential cryoglobulinemic Vasculitis, Cutaneous leukocytoclastic angiitis (Jennette and Falk, 1997). Medium-size arteries are involved in Kawasaki syndrome of childhood and in classic polyarteritis nodosa (Berlit, 2010). Large vessel vasculitis is giant-cell arteritis and Takayasu arteritis (Prieto-González et al., 2015). The cellular and molecular characterizations of vasculitis are complex and diverse, depending on the type of disorder and organ. Extensive reviews have been published on this topic (Younger, 2004; Rosenberg, 2009; Pantoni, 2010; Zuccoli et al., 2011; Hoffman and Calabrese, 2014; Pipitone et al., 2018).
Vascular Inflammation and Cognitive Aging
The blood-brain barrier (BBB) is a dynamic and active barrier, selectively allowing the entrance of molecules (oxygen and nutrients) from blood into the brain and concomitantly protecting the brain form infections and blood toxins (Zlokovic, 2011). It consists of a monolayer of brain endothelial cells sealed with “tight junctions” which are proteinaceous transmembrane complexes made of members for instance; claudins, occluding and junctional molecule-1, and sub membrane molecules connecting to the actin-network (Jia et al., 2013). Endothelial cells of the BBB manage the neuronal environment through controlling the transport of numerous molecules from the blood to the brain parenchyma and vice versa. They also manage fluctuations via the synthesis of mediators able to impact on nerve cells function and vascular endothelial growth factor (Marchesi et al., 2008; Attems et al., 2011; Sanchez et al., 2013). Moreover, the BBB is permeable to pro-inflammatory mediators from systemic inflammation and permits leucocyte migration in to the brain (Lyman et al., 2014). Interestingly, vascular endothelial cells have a critical role in the pathobiology of vascular inflammation (Younger, 2004) and the variations in the metabolism of endothelial cells can cause neurodegenerative disorders. The neurovascular unit, which strongly influences neuronal cell activity of the brain (Bertini et al., 2013), also consist of astrocytes, and microglia which are the main neuroinflammatory principle cells (Seth and Koul, 2008; Ransohoff and Perry, 2009; Ransohoff and Cardona, 2010; Grammas, 2011), as well as pericytes and neurons (Grammas, 2011). Astrocytes connect to endothelial cells, providing support to those cells and as a result are able to regulate BBB maintenance they, act as mediators between BBB and neurons (Hawkins and Davis, 2005). Deregulation of the BBB, and neurovascular dysfunction has been observed to lead to neurodegeneration and cognitive decline (Iadecola, 2004; Zlokovic, 2011; Leung et al., 2013). Signaling cascade are correlated with angiogenesis and vascular activation and are up regulated in the micro vessels in brains of patients with AD leading to a complicated neuroinflammatory response, and neuronal synaptic disconnection, which can damage or kill the nerve cells (Zlokovic, 2008; Grammas, 2011; Sanchez et al., 2013), and consequently exhibit cognitive impairments (Ryan and Nolan, 2016). In AD human studies, there has been a significant elevation in inflammatory mediators in the cerebral microcirculation. Brain endothelial cells in AD express high level of inflammatory adhesion molecules, for instance cationic antimicrobial protein 37 kDa, MCP-1 and intercellular adhesion molecule-1 (ICAM-1; Frohman et al., 1991; Pereira et al., 1996; Grammas and Ovase, 2001). Moreover, AD brain micro-vessels express significantly higher amount and range of inflammatory mediators such as thrombin, nitric oxide, transforming growth factor-β, TNF-α, IL-1β, IL-6, IL-8 and MMPs (Dorheim et al., 1994; Grammas and Ovase, 2001, 2002; Thirumangalakudi et al., 2006). Crucially, inflammation has a key role in linking several vascular and neuronal damage to cardiovascular risk factors (Gorelick et al., 2011) such as arterial stiffness and hypertension (Gorelick et al., 2016). Inflammation is actively involved in cerebral vasculature, although the role of inflammation in vasculopathy is poorly understood (Klohs et al., 2014).
Unfortunately, most of the data available on vascular inflammation in relation to cognitive aging are based on animal models (Takeda et al., 2010; Yu et al., 2012; Won et al., 2013; Kaiser et al., 2014; Acharya et al., 2015) and available human studies mainly assess the role of systemic inflammatory biomarkers or symptomatic changes of cardiovascular risk factors in relation to cognitive aging. We describe here some of the studies that assist us in better understanding of the relationship between vascular inflammation and cognition. Several human studies have reported that platelet activity is significantly, correlated with dementia severity, supporting the role of vascular inflammation in the pathogenesis and progression of dementia (Laske et al., 2008, 2012; Stellos et al., 2010, 2014). Laske et al. (2008) suggested a correlation among soluble glycoprotein VI as markers of platelet activity with the pathogenesis of AD. Yano et al. (2010) reported that pentraxin 3, is a useful inflammatory biomarker for predicting cognitive decline in elderly hypertensive patients. Some human studies have also reported evidence that statins, cholesterol-lowering drugs have therapeutic application in AD (Jick et al., 2000; Wolozin et al., 2000) and patients in these studies exhibited a slower cognitive decline (Sparks et al., 2006). To investigate the relation among midlife hypertension and onset of AD later in life, Kruyer et al. (2015) chemically enhanced chronic hypertension in the transgenic Swedish-Dutch-Iowa mutation mouse model of AD. Hypertension increased cognitive impairments on the Barnes maze test and led to an increase in microvascular deposition of Aβ, vascular inflammation, BBB leakage, and pericyte loss. Moreover, hypertension enhanced hippocampal neurodegeneration at an early age in this mouse line, establishes this as a useful research model of AD with mixed amyloid and vascular pathologies (Kruyer et al., 2015). Grammas and colleagues reported that the vascular activation inhibitor, sunitinib, reduces Aβ as well as cerebrovascular expression of inflammatory proteins improved cognitive function in 3xTg-AD murine models and AD2576APP Swe (Grammas et al., 2014). Blocking angiotensin II signaling helps reduce neurodegeneration and to develop longevity in rodents (Benigni et al., 2010).
Vascular Aging and Oxidative Stress
Inflammation and oxidative stress are underlying factors in the development of vascular aging and it is difficult to determine the effects of these two factors independently as several interplays co-exist between oxidative stress and inflammation and vice versa (Csiszar et al., 2008; Montezano and Touyz, 2014). Oxidative stress is crucially involved in several molecular and cellular interactions of vascular aging, which include: (1) elevated amount of pro-inflammatory responses in vascular cells; (2) vascular dysfunction among oxidative modification of structural and functional proteins which regulates vascular contraction/relaxation fibrosis and calcification; (3) variations in calcium homeostasis in vascular cells; (4) activation of redox-sensitive pro-inflammatory and profibrotic transcription factors; and (5) activation of molecular mechanisms causing senescence and autophagy in endothelial and vascular smooth muscle cells (Tatchum-Talom and Martin, 2004; Harvey et al., 2015).
Brain Aging
To date, numerous functional and structural changes related to normal brain aging have been reported and indicated that brain mass reduces in the order of 2%–3% per decade after the age of 50, and in participants 80 years or older brain mass decreases 10% in comparison to young adults (Drachman, 2006). Voxel-based morphometry and magnetic resonance imaging has revealed that age particularly impacts on the volume of white and gray matter at parietal, prefrontal and temporal areas (Salat et al., 2004; Samanez-Larkin and Knutson, 2015; Von Bernhardi et al., 2015). Complex learning abilities, for instance, dual tasks (e.g., memorizing a word list while walking), show a progressive decline in the elderly (Salat et al., 2005; Von Bernhardi et al., 2015). However, cognitive decline during aging varies considerably, with some older people reporting normal cognitive abilities (Shock et al., 1984). Consistent with brain tissue and neuropsychological changes there are some brain changes that occur as a consequence of molecular and cellular changes in the body such as increases in the permeability of the BBB, enhancement in systemic inflammation, degeneration of neurons and other brain cells which could also lead to the production of ROS. It has been suggested that BBB permeability increases in aged animals (Blau et al., 2012; Enciu et al., 2013) facilitating infiltration via monocytes producing mitochondria-generated ROS (Zlokovic, 2008; Lyons et al., 2009). Microarray analysis of brain tissue provided from aged and young rodents have reported that the upregulated genes in the aged rodents are correlated with oxidative stress and inflammation (Lyons et al., 2009). In other research, enhancement in both major histocompatibility complexes (MHC)-II and glial fibrillary acidic protein (GFAP) were demonstrated in the brain of aged mice strengthening the hypothesis that activation of microglia and astrocytes are indicators of brain aging (Godbout et al., 2005) and all these changes could lead to chronic “neuroinflammation” (Lynch, 2009; Dong et al., 2014; Lyman et al., 2014).
What Is Neuroinflammation?
Neuroinflammation is a complex cellular and molecular cascade in brain where immunological cells have a key role in its initiation and development (Glass et al., 2010; Skaper et al., 2014; Rizzo et al., 2014; Arulselvan et al., 2016). The BBB is one of several key players during neuroinflammation, where non-neuronal cells (e.g., microglia, asterocytes and pericytes) together with neurons form a functional unit, often referred to as a neurovascular unit to process a neuroinflammatory response (Iadecola, 2004; Hawkins and Davis, 2005; Zlokovic, 2005, 2008, 2011). BBB breakdown can occur under different conditions such as due to inflammatory responses, neurodegenerative processes, and vascular disorders. It can cause a neuroinflammatory response and generate neurotoxic products that cause a progressive synaptic disconnection, neuronal dysfunction and cell loss leading to a vicious circle in disorders such as AD, Parkinson’s disease (PD), multiple sclerosis (MS), and other disorders (Zlokovic, 2005, 2008, 2011). During the neuroinflammatory process, microglia and astrocytes are the main active immunological cells, which regulate both the enhancement as well as reduction of inflammatory production (Seth and Koul, 2008; Ransohoff and Perry, 2009; Ransohoff and Cardona, 2010). This condition is achievable via the synthesis of cytokines, up- or downregulation of several cell surface receptors such as pathogen recognition receptors, cytokine receptors, and several other receptors vital for antigen presentation (Ransohoff and Perry, 2009; Ransohoff and Cardona, 2010). Chronic neuroinflammation or upregulated neuroinflammation is commonly known via an enhancement in microglia activation and elevated level of inflammatory cytokines such as IL-1β, which are prevalent in almost all neurodegenerative diseases such as AD, MS, PD and amyotrophic lateral sclerosis (Lynch, 2009; Glass et al., 2010). Interestingly, these variations are similar, though less dramatic in the non-disease aging brain (Lynch, 2009). Supporting cells in the CNS (microglia and astrocytes) during neuroinflammation have a high turnover and exacerbate the process of brain cellular aging (Ma et al., 2013).
Research has also characterized the role of microglia during brain aging indicating a role of morphological, biological, physiological, anatomical, and molecular changes in aging microglia which also exhibit a comprehensive depiction of the senescent microglia phenotype (Norden and Godbout, 2013; Wong, 2013). This is due to the dynamic role of microglia cells in the CNS which have a crucial role in the development, plasticity and immune surveillance of the brain (Norden and Godbout, 2013). As such, we focus on the role of the microglia during neuroinflammation and brain aging. Microglia account for 10% of the total glial cell population in the brain. As a consequence of damage to the brain, microglia cells transform their morphology dramatically, migrating to the lesion sites and proliferate (Cerbai et al., 2012; Jurgens and Johnson, 2012; Norden and Godbout, 2013). Proliferated microglia cells phagocytose debris and dying cells and/or generate cytokines to support injured neurons and to keep the microenvironment homeostasis and are therefore useful for neuronal survival. Data has also shown a neurotoxic role of microglia whenever- they are activated in severe injury or neurodegenerative disorders (Jurgens and Johnson, 2012; Norden and Godbout, 2013). The two-way communication between neurons and microglia is vital for maintaining homeostasis during the physiological and chronic inflammatory response in the CNS. While microglia activation is crucial and beneficial in response to disorders and injury, unrestricted or prolonged activation could have harmful impacts on brain performance and behavior. To avoid inflammation-associated damage, microglia reactivity is modulated via neurons in the healthy brain (Jurgens and Johnson, 2012). Overall density and numbers of microglia have been shown to enhance notably with advancing age in several CNS compartments, which include the hippocampus (Wong, 2013) auditory and visual cortex (Tremblay et al., 2012) and the retina (Damani et al., 2011). These elevations might lead a low rate of basal microglia proliferation or in other respects i.e., to slow incremental recruitment of macrophages or monocytes from the periphery (Cerbai et al., 2012; Wong, 2013). From histopathology studies, some evidence has been provided that in the aged brain, microglia morphologies exhibit a perinuclear cytoplasm hypertrophy and retracted processes, almost similar to activated microglia (Miller and Streit, 2007; Wong, 2013). Aged microglia immunophenotypes also exhibit those of activated microglia, with enhanced expression of MHC II and complement receptor 3 (Frank et al., 2006; Ziv et al., 2006; Wong, 2013). In activated microglia, molecular markers are often observed to be up regulated, (such as ionized calcium-binding adaptor molecule1). Moreover, these markers are enhanced in aged microglia with no-evidence of injury or disease (Frank et al., 2006; Wong, 2013). During healthy aging, in human in vivo positron emission tomography (PET) using [11C]-PK11195 (TSPO radioligands) has shown an enhancement in ligand binding in several cortical and subcortical areas, demonstrating an elevated amount of basal microglia activation (Schuitemaker et al., 2012). Moreover, aged microglia express elevated amounts of effector molecules similar to activated microglia which is known to be the primary cell source of cytokines release such as TNF-α, IL-6, IL1-β (Lynch, 2009; Wong, 2013). Elevated expression of inflammatory cytokines (such as IL1-β, TNF-α, IL-6) are also elevated in aged microglia in situ (Wong, 2013), isolated ex vivo (Sierra et al., 2007; Njie et al., 2012) or when cultured in vitro (Ye and Johnson, 1999). Additionally, older brains show increased interaction among T cells and microglia (Lynch, 2009). Microglia senescence has an underlying role in the switching of microglia from neuroprotection in the young brain to neurotoxic in the aged (Norden and Godbout, 2013). Targeting some of these features of microglia senescence might constitute a feasible therapeutic strategy for some disorders and even cognitive aging itself (Jurgens and Johnson, 2012; Norden and Godbout, 2013; Wong, 2013). Overall, there is general agreement on the enhancement of basal state microglia activity in healthy aging, such that aged microglia may lead to chronic states of “para-inflammation” (Medzhitov, 2008) that is correlated with the enhanced vulnerability of the aged CNS to neurodegenerative disorders in which there is a crucial role of chronic neuroinflammation (Wong, 2013).
Neuroinflammation and Cognitive Aging
Cognitive aging is characterized by a reduction in cognitive abilities in the elderly. Although the underlying mechanism involved in this process is not fully understood, neuroinflammation appears to be a significant contributor (Ownby, 2010). Several research studies have attempted to describe the relationship between neuroinflammation and cognition. To date, several animal studies (Hovens et al., 2014; Czerniawski et al., 2015; Elmore et al., 2015; Michels et al., 2015; Wei et al., 2015; Flannery et al., 2016; Laurent et al., 2017; Reis et al., 2017; Sanchez-Marin et al., 2017; Wang et al., 2018) have addressed this issue. For instance, a study by Sun et al. (2015) in male Sprague-Dawley rats showed that LPS could induce IL-17A, TNF-α, IL-6, iNOS, and COX-2 expression in the hippocampus when followed by IL-17A-neutralizing antibody treatment, and it significantly eliminated neuroinflammatory response through suppression of microglia activation and improved memory. Tian et al. (2015) also found that in a mice surgery model, partial hepatectomy enhanced the amount of IL-17A in the hippocampus and increased cognitive impairment, while vitamin D intervention reduced cognitive deficits by inhibiting Th17 cells and increasing T reg cell numbers. Tan et al. (2015) study also showed that transfusion of old red blood cells in Sprague-Dawley rats increased IL-6 in the hippocampus and enhanced ionized calcium-binding adapter molecule 1, in the cerebral cortex and hippocampus (neuroinflammatory response), and impaired memory and learning. Cognition and behavior were evaluated by Barnes maze and fear conditioning tests (Tan et al., 2015). A study by Hajiluian et al. (2017) with male Wistar rats model, showed that vitamin D reversed obesity-induced cognitive impairments via a reduction in nuclear factor kappa B (NFκB) amount (a key neuro-inflammatory factor) and an increase in brain derived neurotrophic factor concentration and modulation of the BBB permeability in the hippocampus. Cognitive function was examined by the Morris water maze test and the BBB permeability was evaluated by Evans blue dye in the hippocampus (Hajiluian et al., 2017).
The most widely applied transgenic Alzheimer animal models do not demonstrate the degree of inflammation in neurodegeneration and cognitive decline comparable to human disease. Therefore, a more suitable animal model, is required which closely mimics the resulting cognitive decline and memory loss in humans in order for us to be able to better understand the impact of neuroinflammation on neurodegenerative disorders (Millington et al., 2014). A few animal studies have specifically focused on the relationship between neuroinflammation and cognitive aging. For example, in the age-related Alzheimer mouse model, head injury can cause a chronic neuroinflammatory response, which initiates and causes cognitive impairment. A study by Webster et al. (2015) found that a single mild traumatic brain injury in the APP/PS1 knock-in mouse led to a delayed onset of neuroinflammatory response and a more persistent glia cells activation (microglia and astrocyte) in comparison to injured wild-type mice who consequently developed cognitive impairment (Webster et al., 2015). Fonken et al. (2016) demonstrated that high mobility group box 1 (HMGB1) mediates neuroinflammatory response priming in the aged brain of rats by blocking the HMGB1 action using a competitive antagonist Box-A. Aged-microglia became desensitized to an immunological challenge and therefore were prevented from an exaggerated neuroinflammatory response and sickness behavior following infection (Fonken et al., 2016). In the mice model of dementia 6-Shogaol (an active constituent of ginger) eliminated neuroinflammatory processes by inhibiting microgliosis and astrogliosis and consequently improved cognitive process (Moon et al., 2014).
PET has been frequently used in human studies to quantify microglia activity by labeling the translocator protein 18 kDa (TSPO), which becomes over expressed upon activation of microglia cells (Knezevic and Mizrahi, 2018). Studies that focus on neuroinflammation in MCI reported a similar association between cognitive impairment and TSPO binding (Okello et al., 2009; Yasuno et al., 2012; Schuitemaker et al., 2013). The only research with participants who had MCI reported a significant correlation among cognition and neuroinflammation was on the combined AD and MCI participants in the analysis (Kreisl et al., 2013a,b). There was a significant association among PET related inflammatory binding and impaired performance on the MMSE, logical memory immediate, clinical dementia rating, trail making part B tasks, and block design, with the strongest associations among [11C]-PBR28 (TSPO radioligands) binding in the inferior parietal lobule and performance on block design and clinical dementia rating score (Kreisl et al., 2013b). Three relevant studies have reported on relationships between neuropsychological assessments in AD patients and neuroinflammation (Kreisl et al., 2013b; Schuitemaker et al., 2013; Suridjan et al., 2015). In the study by Schuitemaker et al. (2013), there was no significant correlation between cognition and [11C]-PK11195 (TSPO radioligands) binding (Schuitemaker et al., 2013). However, two investigations with second-generation radioligands reported a significant association between performance on cognitive scales and TSPO binding. Kreisl et al. (2013b) reported a strong negative association between performance on the block design and [11C]-PBR28 (TSPO radioligands) binding in the inferior parietal lobule. Similarly, Suridjan et al. (2015) identified a negative correlation between performance on the visuo-spatial tasks in the repeatable battery for the assessment of neuropsychological status and [18F]-FEPPA (TSPO radioligands) binding in the parietal and prefrontal cortices. The former research additionally identified a negative association between [11C]-PBR28 binding and trail making part B task and performance on the logical memory immediate recall assesses executive function and memory, respectively. Three studies have reported that lower performance on the MMSE significantly correlated with higher levels of TSPO binding (Edison et al., 2008; Yokokura et al., 2011; Kreisl et al., 2013b). However, there are other studies that have demonstrated no association between these two variables (Yasuno et al., 2008; Schuitemaker et al., 2013; Varrone and Nordberg, 2015), and another study reported a positive association among MMSE score and the global cortical index (Hamelin et al., 2016). Based on the in vivo PET data, it can be concluded that chronic neuroinflammatory response plays a key role in the pathology of AD. Whether this chronic neuroinflammatory response is an initial factor or it happens consequentially during the development of disease is not known. The contradictory results across different studies using TSPO variables may be explained by methodological differences in the studies and how cognitive impairment was assessed. Future research should be conducted to include larger populations with longer observation, categorizing population via genetic, cognitive and behavioral phenotypes to study other factors, which could influence microglia activation (Figure 3).
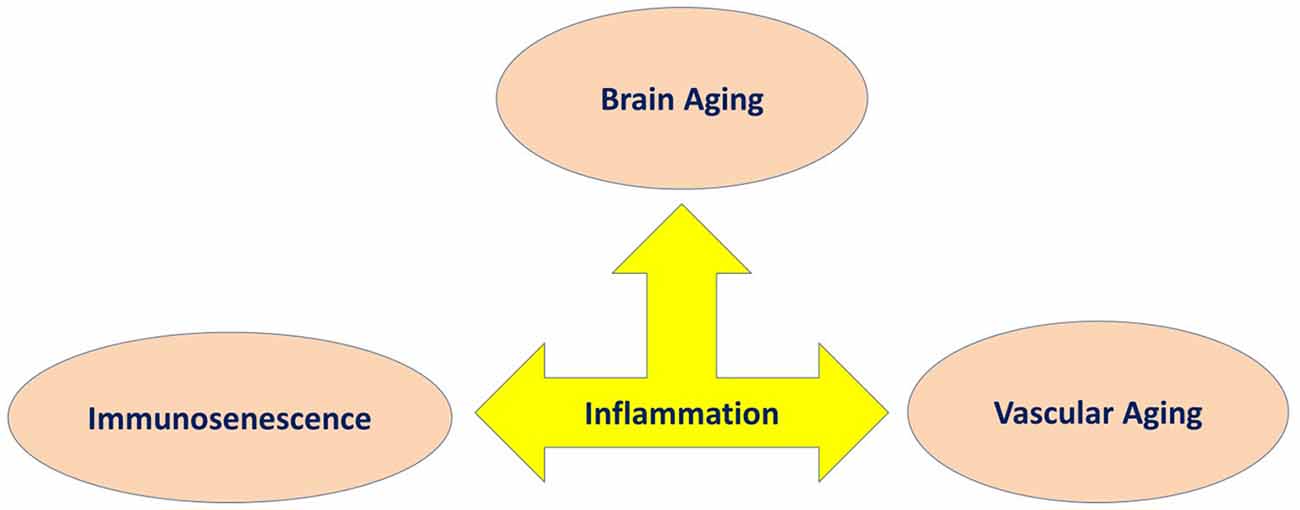
Figure 3. Inflammation as a common underlying mechanism in three main features of aging involved in cognitive decline.
Discussion
Although, cognitive aging is a complex, multifactorial process, there are a number of key biological processes involved in this process. As such, we have proposed the involvement of three main processes that may explain cognitive aging, which includes immunosenescence, vascular aging and brain aging. Within this model, we described how they share common pathological processes such as inflame-aging, vascular inflammation and neuroinflammation, respectively (Figure 1). Capturing these three key processes in any single study is difficult for a number of reasons; however, their utility taken together should be better explored (Piazza et al., 2010; Cannizzo et al., 2011; Salvioli et al., 2013; Di Benedetto et al., 2017). We have also argued that immunosenescence is a critical key mechanism involved in cognitive aging (Figures 2, 3). Better understanding the development of immunosenescence in each individual should be further considered, and appears to be sensitive to both genetic and environmental factors (Weiskopf et al., 2009; Baylis et al., 2013; Di Benedetto et al., 2017). Immunosenescence affects how body regulate and manage different cellular and molecular interactions in almost all body organs (Markiewski and Lambris, 2007; Ownby, 2010; Fard et al., 2015; Arulselvan et al., 2016; Di Benedetto et al., 2017), particularly the brain during chronic neuroinflammatory processes. Neuroinflammation serves several fundamental roles in the brain structure and function, such as ion homeostasis, involvement in the regulation of metabolic function, the production of anti-oxidant species, synaptic levels of glutamate, modulation of neuroplasticity, maintenance of BBB, as well as protection from both endogenous and exogenous factors (Benarroch, 2005; Shih et al., 2006; Wang and Bordey, 2008; Broussard et al., 2012; Di Benedetto et al., 2017). We also propose that neuroinflammation is an underlying mechanism underpinning cognitive performance playing a vital role in learning and memory as we grow older (McAfoose and Baune, 2009; Ownby, 2010; Ryan and Nolan, 2016; Di Benedetto et al., 2017; Hajiluian et al., 2017). It is important to bear in mind that, there are normal or physiological levels of inflammation however, chronic events (Markiewski and Lambris, 2007; Fard et al., 2015; Sun et al., 2015; Tian et al., 2015; Arulselvan et al., 2016) may reshape a normal inflammatory response into an exaggerated response which could then be characterized in terms of inflamm-aging, vascular inflammation and chronic neuroinflammation. We therefore propose, that cognitive aging is a consequence of these three inflammatory processes which interact in a synergistic manner (Figures 2, 3). As such, treatments that ameliorate any or all aspects of exaggerated inflammatory responses may be beneficial in improving cognition or even ameliorating cognitive decline in the elderly. For instance it is commonly accepted that anti-inflammatory drugs are able to block important inflammatory pathways such as NF-κB, and mitogen-activated protein kinase (MAPK; Fard et al., 2015; Arulselvan et al., 2016) and these could be explored as treatments to ameliorate cognitive decline with age. Notably, whilst, nerve cells remain the most vital functional unit of the nervous system, immune cells acting directly or indirectly on brain function are more easily manipulated targets than nerve cells. Better understanding of the complex neuro-immune interactions will pave the way for the development of new therapies that target the immune system for the benefit of brain (Kipnis and Filiano, 2018), and cardiovascular function.
In general, immunosenescence is a pathological process, which manifests as a gradual decline in the functionality of our immune system over the lifespan at the molecular and cellular level and as such is hard to evaluate its initiation and development (Piazza et al., 2010; Cannizzo et al., 2011; Salvioli et al., 2013; Di Benedetto et al., 2017). Environmental factors such as life style and genetical factors directly influence immunosenescence in each induvial (Weiskopf et al., 2009; Baylis et al., 2013; Di Benedetto et al., 2017). Interestingly, a review by Kennedy et al. (2017) described the impact of exercise on improving cognitive functioning via several parallel pathways such as the modulation of inflammation and oxidative stress during brain aging. Another interesting report by Windham et al. (2014) on the associations between inflammation and cognitive function among two different population groups of African Americans and European Americans, demonstrated that population with high vascular risk, higher inflammation and poorer cognition were associated with markers of TNF-α activity and especially apparent in African Americans.
We have argued for the importance of three distinct, but inter-related inflammatory processes linked to cognitive aging such as inflame-aging, vascular inflammation and neuroinflammation (Figure 1). This model can be applied across different cognitive processes, however, not all older individuals show pathological inflammatory processes across these three domains at one time point. Chronic neuroinflammation may be the most directly relevant inflammatory domain in terms of cognitive aging, however inflamm-aging may be an important initiator of prolonged neuroinflammation. Finally, vascular inflammation may be more transient and specifically be related to cardiovascular disorders (Wilson et al., 2002; Zlokovic, 2005; Xie et al., 2009; Iadecola, 2010; Grammas, 2011; Broussard et al., 2012; Davenport et al., 2012; Kousik et al., 2012; Barrientos et al., 2015; Chi et al., 2016; Di Benedetto et al., 2017; Tarantini et al., 2017). In support of our model, there is evidence that some disorders show these relationships. For instance, in postoperative cognitive dysfunction (POCD) there is a significant relationship between systemic inflammatory mediators and neuroinflammation. A major operation can activate specific homeostatic reactions, which can generate an inflammatory response with the production of several inflammatory mediators (Hu et al., 2010; Terrando et al., 2011; Elwood et al., 2017). Damaged tissues activate monocytes, macrophages, fibroblasts and endothelial cells, and release various inflammatory products including oxidative radicals, complement split products and cytokines, such as IL-1, IL-6, TNF-α. These circulatory mediators can directly penetrate the BBB (Wilson et al., 2002). Cytokines bind to their receptors in the CNS, activate vascular endothelial cells and microglia cells, and subsequently, enhance neuroinflammatory response and a series of molecular reactions (Wilson et al., 2002; Xie et al., 2009). This neuroinflammatory response can lead to cognitive decline, which could impact on the production and activity of neurotransmitters, decrease neural plasticity, and increase neurotoxicity (Hu et al., 2010). Interestingly, a review by Cunningham and Hennessy (2015) noted that a growing body of clinical, preclinical and epidemiological evidence shows that chronic co-morbidities and systemic inflammatory contributes to the progression of dementia. VD is known to be correlated with cerebrovascular pathologies (Gorelick et al., 2011; Iadecola, 2013). Recent data, has revealed a role of cerebrovascular disorders, not only as initial cause of cognitive decline, but also as an adjuvant to the expression of dementia in AD and other neurodegenerative pathologies (Iadecola, 2013). Vascular risk factors impair the structure and function of cerebral blood vessels and the neurovascular unit, and these pathological changes are then mediated via vascular oxidative stress and inflammation (Iadecola, 2010). In the review by Sharp et al. (2011), hypertension was shown to be an important risk factor in cerebrovascular disease, cognitive impairment and AD as well as, for VD. Moreover, VD is associated with microinfarcts of cerebral blood vessels preventing oxygen supply to neurons which is caused to some extent because inflammation of the arterial wall affects the accumulation of thrombotic factor (Libby et al., 2009). Accumulating evidence also suggests that neuroinflammation is a hallmark of AD and several studies have shown the existence of biomarkers of neuroinflammation, in brain tissue of AD patients (e.g., inflammatory cytokines, chemokines, and activated microglia (Eikelenboom et al., 2010; Lee et al., 2010; Sudduth et al., 2013; Klohs et al., 2014; Morales et al., 2014; Hoeijmakers et al., 2017). Epidemiological studies have also shown that long-term consumption of non-steroidal anti-inflammatory drugs inhibits the progression of AD and delay its onset, suggesting that there is a significant relation among AD pathogenesis and neuroinflammation (Holmes et al., 2009; Lee et al., 2010) (Figure 1).
Inflammation and Cognitive Impairment in Dementia
Cognitive aging is a generic concept, with more than 100 diseases leading to dementia (Vijayan and Reddy, 2016), while we discussed the three specific inflammatory domains including neuroinflammation supported by three types of dementia such as POCD, VD, and AD. Our proposed model can be applied to other types of pathological cognitive aging, as well. It also seems plausible to hypothesize high commonality in the three inflammatory domains outlined across cognitive aging (particularly chronic neuroinflammation which is common feature of all dementias). As we discussed earlier, the specific inflammatory hallmark in each dementia varies depending on the type of dementia. We now focus on some of these issues in more detail.
The involvement of cellular senescence and inflammatory process via complex molecular and cellular processes occur in neurons and gilal cell in PD and MS patients have been reviewed previously by Kritsilis et al. (2018). Neuroinflammation is one of the mechanisms involved in PD-cognitive impairment. Elevation in cortical microglia activation has been reported in 11 PD-dementia patients, using biological parametric mapping analysis (Fan et al., 2015). Cerebrospinal fluid level of cytokines was observed to be correlated with PD-cognitive impairment (Lindqvist et al., 2013). Petrou et al. (2016) found correlations among diabetes, gray matter loss and PD-cognitive impairment using magnetic resonance imaging in 36 patients, possibly due to neuroinflammation caused by mitochondrial dysfunction (Aviles-Olmos et al., 2013; Petrou et al., 2016). In a human study by Hall et al. (2018), inflammatory biomarkers such as CRP and serum amyloid A in cerebrospinal fluid were higher in PD-dementia elderly individuals compared to elderly individuals without PD. In addition, inflammation was associated with more motor symptoms and cognitive decline (Hall et al., 2018).
MS is an inflammatory neurodegenerative disease of the CNS mainly effecting young adults. In this condition, infiltrating myelin-reactive lymphocytes (mainly T-cells but also B-cells) attack axon antigens and myelin sheaths on oligodendrocytes and neurons in the CNS. These insults cause a neuroinflammatory cascade, formation of large demyelinating plaques in the white matter and gliosis, and synaptopathy, leading to an impairment of the neuronal signaling and, later on to neurodegeneration (Dendrou et al., 2015; Mandolesi et al., 2015; Musella et al., 2018). Clinical characteristics include motor impairments, sensory and visual disturbances, pain, fatigue, mood disturbances and cognitive deficits (Dendrou et al., 2015). MS patients frequently suffer from cognitive impairment and it estimated between 40% and 65% of MS patients tend to progress cognitive impairment gradually over time. Cognitive impairment is present in MS patient with progressive clinical onset compered to patients in relapsing remitting phase (Huijbregts et al., 2004; Ruet et al., 2013; Planche et al., 2016; Matias-Guiu et al., 2017), although some heterogeneous results have been reported as well (Rao, 1990; Potagas et al., 2008). In MS patients during immunosenescence and possibly due to inflame-aging, additional inflammatory responses cause chronic neuroinflammatory responses which therefore significantly accelerate CNS aging (Dendrou et al., 2015; Musella et al., 2018). Several studies have also suggested that an exacerbation of common neuropathological aspects of aging brain and MS such as neuroinflammatory processes, synaptic dysfunction, cellular loss (synaptopathy) and synaptic plasticity impairment (Di Filippo et al., 2008; Weiss et al., 2014; Mandolesi et al., 2015; Stampanoni Bassi et al., 2017; Musella et al., 2018), may explain the effect of aging on MS disability. A recent study by Surendranathan et al. (2018) reported that both neuroinflammatory responses with microglia activation and peripheral inflammatory changes are observed in dementia with Lewy bodies. A review by Swardfager et al. (2018) recently indicated that an increased risk of dementia in severe psychological disorders such as major depressive disorder and bipolar disorder is associated with inflammation, oxidative stress and variations in metabolic pathways. Moreover, those pathways are involved in the premature development of metabolic and vascular comorbidities (Swardfager et al., 2018).
Patients who survive a stroke are also at high risk of recurrent microvascular changes. The mechanisms that underpin this process is not fully understood. A recent data showed that stroke-enhanced atherosclerosis is induced by brain-released biochemical which lead to vascular inflammation and plaque formation (Rust et al., 2018). Inflammation in stroke appear to have both beneficial and detrimental effects (Jin et al., 2010). Chronic inflammatory response may trigger neurotoxic pathways leading to progressive degeneration. Damaged neurons also may exacerbate neuroinflammation-mediated disorders by producing chemokines and activation of microglia (Zhang and Yang, 2014). Numerous recent longitudinal studies have examined the correlation among inflammatory biomarkers and post-stroke dementia but a relationship is not yet established. Erythrocyte sedimentation rate, CRP, IL-6, IL-12 were also suggested as predictors of post-stroke cognitive impairment (Rothenburg et al., 2010; Narasimhalu et al., 2015). Kliper et al. (2013) showed strong relationship between cognitive performance and erythrocyte sedimentation rate between stroke survivors, where higher erythrocyte sedimentation rate levels were correlated with poorer performance in cognitive tests, particularly memory scores. Preliminary data has also demonstrated that cortical amyloid deposition and post-stroke white matter neuroinflammation contribute to post-stroke dementia (Arboix et al., 2014). Interestingly, Malojcic et al. (2017) argued that cerebral hypoperfusion is linked to cognitive decline either as an aggravating factor or risk factor. Hypoperfusion as a consequence of macroangiopathy, microangiopathy, or cardiac dysfunction may also promote or accelerate neuroinflammation, BBB disruption and neurodegeneration.
Obesity is correlated with low grade systemic inflammation, peripheral insulin resistance and high oxidative stress (Chunchai et al., 2016; Saiyasit et al., 2018). Moreover, obesity can cause neurodegeneration and cognitive impairment via induction of hippocampal inflammation, hippocampal mitochondrial dysfunction, hippocampal insulin resistance, and hippocampal oxidative stress (Chunchai et al., 2016; Stranahan et al., 2016; Saiyasit et al., 2018). These changes also are associated with a decline in hippocampal synaptic plasticity and the number of dendritic spines (Sa-Nguanmoo et al., 2018; Saiyasit et al., 2018). Moreover, obesity induced through high-fat-diet consumption, leads to an elevation in amyloid plaque formation and neuronal cell death (Kothari et al., 2017; Pintana et al., 2017). Growing evidence demonstrates a close relationship among type 2 diabetes Mellitus and neurodegenerative diseases such as AD. They share several pathological characteristics comprising inflammation, oxidative stress, brain vasculopathy, impaired insulin sensitivity, tau hyper-phosphorylation, and amyloid accumulation (Tumminia et al., 2018). Riederer et al. (2017) posited that peripheral inflammatory biomarkers in diabetes mellitus can pass BBB and initiate neuroinflammation and microglia activity, thus contributing to the pathophysiology of AD and VD.
There is also data that supports the notion that a reduction in vascular functions and brain metabolism occur decades before the onset of cognitive impairments and these reductions are highly associated with chronic neuroinflammation that develop over time. Crucially, recent findings suggest that the gut microbiota (GMB) play a vital role in modulating immune reactions in the brain through the brain-gut axis (Hoffman et al., 2017). Some recent data has indicated that a specific subset of the GMB can stimulate neuroinflammation in rodents (Erny et al., 2015; Palm et al., 2015; Petra et al., 2015) and influence brain function and behavior in rodents and humans (Li et al., 2009; Bercik et al., 2011; Diaz Heijtz et al., 2011). A study by Hoffman et al. (2017) found that inflammation influences neurovascular function, brain metabolism, gut microbiome, memory and anxiety in aging mice. Interestingly, a study by Cattaneo et al. (2017) in elderly humans indicated that an elevation of a proinflammatory GMB taxon, Escherichia/Shigella, and a decrease in the abundance of an anti-inflammatory taxon, E. rectale, are correlated with systemic inflammatory biomarkers in patients with cognitive impairment and brain amyloidosis. This finding is in line with the hypothesis that GMB composition may drive peripheral inflammation, leading chronic neuroinflammatory responses, brain amyloidosis and probably, neurodegeneration and cognitive decline in AD (Cattaneo et al., 2017).
Pharmacological Approaches
Current treatment paradigms for neurodegenerative disorders such as dementia are limited by their significant side-effects and poor long-term efficacy, creating an essential need to develop preventative therapies that target common pre-symptomatic risk factors such as inflammation and oxidative stress. There are a large number of pharmacological agents applied to improve cognitive decline by managing inflammation and oxidative stress; however, in this section we briefly summarize the key published studies on this topic (Herman et al., 2018).
Phytochemicals are able to interfere with the NF-κB pathway and manage inflammation. They suppress the ubiquitination or phosphorylation of signaling molecules, and therefore, supress the degradation of IκB. The translocation of NF-κB to the nucleus and subsequent transcription of pro-inflammatory cytokines are suppressed via the actions of phytochemicals. In addition, natural bio-compounds that prevent the interaction of NF-κB can block NF-κB’s transcriptional activity by suppressing its binding to target DNA. Several polyphenols such as curcumin, pterostilbene, resveratrol, macranthoin G, punicalagin, salidroside, 4-O-methylhonokiol, genistein, lycopene, gallic acid and obovatol have been reviewed as potent NF-κB inhibitors for AD treatment by Seo et al. (2018). Several alkaloids including galantamine, tetrandrine, glaucocalyxin B, oridonin, berberine, anatabine have also shown anti-inflammatory properties in AD models in vitro as well as in vivo. Moreover, vitamins (such as vitamin-D, alfa-Tocopherol quinine, Retinoic acid), artemisinin, tanshinone IIA, geniposide, dihydroasparagusic acid, xanthoceraside, 1,8-cineole, L-theranine, and paeoniflorin were posited as promising NF-κB inhibitors (Seo et al., 2018).
An interesting review by Skvarc et al. (2018) has summarized several novel therapies with common anti-inflammatory properties in different stages of preclinical and clinical levels as therapeutic targets to deal with POCD such as Parecoxib/COX-II inhibitors, Statins, Pregabalin, Dexmedetomidine, Lidocaine, Ketamine, Minocycline, and N-Acetylcysteine.
Vinpocetine [14-ethoxycarbonyl-(3a,16a-ethyl)-14,15-eburnamine] is a synthetic derivative of vinca alkaloid vincamine which is an alkaloid extracted from the periwinkle plant, Vinca minor (Gulyás et al., 2002a,b). Currently, vinpocetine is available in many countries as a dietary supplement to improve cognition and memory. Moreover, it has been clinically used in several countries for treatment of cerebrovascular disorders such as stroke and dementia. Zhang et al. (2017) has argued that vinpocetine is a potent anti-inflammatory agent based on different in vitro cell culture models. By directly suppressing IKK activity, and enhancing the stability of IκB, that leads to binding of IκB with NF-κB and subsequent suppression of NF-κB dependent inflammatory molecule expression (Jeon et al., 2010; Zhang et al., 2017). Vinpocetine can penetrate the BBB and enter the brain (Gulyás et al., 2002a,b). The anti-inflammatory effects of vinpocetine has also been demonstrated in several animal models in vivo. In a rat cerebral ischemia-reperfusion injury model, NF-κB and TNF-α level were found to be correlated with changes in brain edema and infarct volume. Vinpocetine suppressed NF-κB and TNF-α expression and reduced the inflammatory response after cerebral ischemia-reperfusion (Wang et al., 2014). More importantly, the anti-inflammatory effect of vinpocetine was recently reported in a study involving 60 patients with anterior cerebral circulation occlusion and onset of stroke (Zhang et al., 2018). Participants treated with vinpocetine not only had a better recovery of psychoneurological function and improved clinical outcomes, but also had reduced NF-κB signaling activation and pro-inflammatory biomarker expression (Zhang et al., 2017).
A recent review by Masoumi et al. (2018) has also summarized evidence for the hypothesis that Apelin neuropeptide is an effective and comprehensive therapeutic agent to improve cognitive function in AD, and unlike current therapies, can influence a broad range of molecular mechanisms involved in AD pathogenesis. Apelin reduces the accumulation of Aβ and decreases phosphorylation and accumulation of tau protein. Apelin also prevents neurodegeneration by suppressing production of inflammatory mediators, especially TNF-α, IL-6 and IL-1β, which plays an important role during neuroinflammatory process and the pathogenesis of AD. Apelin can modulate N-methyl D-aspartate receptors and therefore decrease excitotoxicity and death of neurons and prevents neuronal apoptosis. In addition, it exhibited high antioxidant properties preventing free radicals and ROS productions. Apelin also enhances synaptic plasticity of the neurons, and improves cognitive function and memory by increasing factors such as endothelial nitric oxide, angiotensin converting enzyme-2 and Glucagon-like peptide-1 (Masoumi et al., 2018). Also see other reviews on this topic (Rodriguez-Grande et al., 2017; Solas et al., 2017; Meeusen and Decroix, 2018; Zhong et al., 2018; Fish et al., 2019).
Limitations and Future Research
Studies examining the involvement of the three main inflammatory types posited by us in this review (inflame-aging, vascular inflammation, and neuroinflammation) in cognitive processes are mainly hampered by the lack of direct studies involving humans. While animal studies help us understand the cellular and molecular mechanism of cognition, human clinical studies are still needed given the differences in complexity and range of cognition that can be studies between animals and humans. Another major limitation of the current literature is that there are only a few sensitive biomarkers for inflammation in humans with the available markers specifically reflecting molecular and cellular processes rather than brain related inflammation. Another challenge in studying the biological processes that underpin cognitive aging are that processes such immunosenescence and chronic inflammation have a progressive and time dependent nature, which require observation over a long time. This is obviously, expensive from a clinical perspective. Cognitive aging itself is a complex and multifactorial disorder, which requires the investigation of diverse molecular and cellular processes.
Conclusion
In developed countries, there is a significant increase in the proportion of older citizens. Increasing number of older citizens pose a number of critical issues for our societies including increased cognitive aging, increased numbers of patients with AD and a loss of independence and reliance on social security. Therefore, a better understanding of the role of processes such as inflammation in cognitive and brain aging is important. Immunosenescence is a pathological phenomenon and a central concept that brings together our understanding of age-associated chronic disorders, functional decline and aging across the lifetime. A better understanding of the cellular and molecular mechanisms underlying brain aging, as well as their potential interactions, provides a growing list of factors that can be targeted for specific interventions aimed to prevent or delay cognitive decline associated with aging. Future studies on the role of inflammation during cognitive aging are necessary and would benefit from directly addressing the three types of inflammation described in this review article.
Author Contributions
Both authors were involved in all aspects of the conceptualization and writing of the manuscript.
Funding
This review was supported by an Australian Research Council Discovery grant to CS, Prof. Andrew Scholey and Prof. Kevin Croft (DP1093825).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acharya, N. K., Goldwaser, E. L., Forsberg, M. M., Godsey, G. A., Johnson, C. A., Sarkar, A., et al. (2015). Sevoflurane and Isoflurane induce structural changes in brain vascular endothelial cells and increase blood-brain barrier permeability: Possible link to postoperative delirium and cognitive decline. Brain Res. 1620, 29–41. doi: 10.1016/j.brainres.2015.04.054
Arboix, A., Blanco-Rojas, L., and Martí-Vilalta, J. L. (2014). Advancements in understanding the mechanisms of symptomatic lacunar ischemic stroke: translation of knowledge to prevention strategies. Expert Rev. Neurother. 14, 261–276. doi: 10.1586/14737175.2014.884926
Arulselvan, P., Fard, M. T., Tan, W. S., Gothai, S., Fakurazi, S., Norhaizan, M. E., et al. (2016). Role of antioxidants and natural products in inflammation. Oxid. Med. Cell. Longev. 2016:5276130. doi: 10.1155/2016/5276130
Attems, J., Jellinger, K., Thal, D., and Van Nostrand, W. (2011). Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 37, 75–93. doi: 10.1111/j.1365-2990.2010.01137.x
Aviles-Olmos, I., Limousin, P., Lees, A., and Foltynie, T. (2013). Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain 136, 374–384. doi: 10.1093/brain/aws009
Babarskiene, M., Babeanu, S., Bossini, A., Fagard, R., Gil-Extremera, B., Laks, T., et al. (2002). The prevention of dementia with antihypertensive treatment. Arch. Intern. Med. 162, 2046–2052. doi: 10.1001/archinte.162.18.2046
Baierle, M., Nascimento, S. N., Moro, A. M., Brucker, N., Freitas, F., Gauer, B., et al. (2015). Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxid. Med. Cell. Longev. 2015:804198. doi: 10.1155/2015/804198
Barrientos, R., Kitt, M., Watkins, L., and Maier, S. (2015). Neuroinflammation in the normal aging hippocampus. Neuroscience 309, 84–99. doi: 10.1016/j.neuroscience.2015.03.007
Baylis, D., Bartlett, D. B., Patel, H. P., and Roberts, H. C. (2013). Understanding how we age: insights into inflammaging. Longev. Healthspan 2:8. doi: 10.1186/2046-2395-2-8
Benarroch, E. E. (2005). “Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system,” in Mayo Clinic Proceedings, ed. M. D. P. Hugues Berry (Amsterdam: Elsevier), 1326–1338.
Benigni, A., Cassis, P., and Remuzzi, G. (2010). Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol. Med. 2, 247–257. doi: 10.1002/emmm.201000080
Bercik, P., Denou, E., Collins, J., Jackson, W., Lu, J., Jury, J., et al. (2011). The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 141, 599–609.e3. doi: 10.1053/j.gastro.2011.04.052
Berlit, P. (2010). Diagnosis and treatment of cerebral vasculitis. Ther. Adv. Neurol. Disord. 3, 29–42. doi: 10.1177/1756285609347123
Bertini, G., Bramanti, P., Constantin, G., Pellitteri, M., Radu, B. M., Radu, M., et al. (2013). New players in the neurovascular unit: insights from experimental and clinical epilepsy. Neurochem. Int. 63, 652–659. doi: 10.1016/j.neuint.2013.08.001
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535. doi: 10.1038/nature08983
Bitto, A., Sell, C., Crowe, E., Lorenzini, A., Malaguti, M., Hrelia, S., et al. (2010). Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 316, 2961–2968. doi: 10.1016/j.yexcr.2010.06.021
Black, C. N., Bot, M., Révész, D., Scheffer, P. G., and Penninx, B. (2017). The association between three major physiological stress systems and oxidative DNA and lipid damage. Psychoneuroendocrinology 80, 56–66. doi: 10.1016/j.psyneuen.2017.03.003
Blagosklonny, M. V. (2010). Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle 9, 3171–3176. doi: 10.4161/cc.9.16.13120
Blagosklonny, M. V., and Hall, M. N. (2009). Growth and aging: a common molecular mechanism. Aging 1, 357–362. doi: 10.18632/aging.100040
Blau, C. W., Cowley, T. R., O’Sullivan, J., Grehan, B., Browne, T. C., Kelly, L., et al. (2012). The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol. Aging 33, 1005.e23–1005.e35. doi: 10.1016/j.neurobiolaging.2011.09.035
Broussard, G. J., Mytar, J., Li, R.-C., and Klapstein, G. J. (2012). The role of inflammatory processes in Alzheimer’s disease. Inflammopharmacology 20, 109–126. doi: 10.1007/s10787-012-0130-z
Buonocore, G., Perrone, S., and Tataranno, M. L. (2010). Oxygen toxicity: chemistry and biology of reactive oxygen species. Semin. Fetal Neonatal. Med. 15, 186–190. doi: 10.1016/j.siny.2010.04.003
Campisi, J., and d’Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. doi: 10.1038/nrm2233
Candore, G., Balistreri, C. R., Colonna-Romano, G., Grimaldi, M. P., Lio, D., Listi’, F., et al. (2008). Immunosenescence and anti-immunosenescence therapies: the case of probiotics. Rejuv. Res. 11, 425–432. doi: 10.1089/rej.2008.0662
Cannizzo, E. S., Clement, C. C., Sahu, R., Follo, C., and Santambrogio, L. (2011). Oxidative stress, inflamm-aging and immunosenescence. J. Proteom. 74, 2313–2323. doi: 10.1016/j.jprot.2011.06.005
Caruso, C., Buffa, S., Candore, G., Colonna-Romano, G., Dunn-Walters, D., Kipling, D., et al. (2009). Mechanisms of immunosenescence. Immun. Ageing 6:10. doi: 10.1186/1742-4933-6-10
Cattaneo, A., Cattane, N., Galluzzi, S., Provasi, S., Lopizzo, N., Festari, C., et al. (2017). Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68. doi: 10.1016/j.neurobiolaging.2016.08.019
Cerbai, F., Lana, D., Nosi, D., Petkova-Kirova, P., Zecchi, S., Brothers, H. M., et al. (2012). The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS One 7:e45250. doi: 10.1371/journal.pone.0045250
Cesari, M., Kritchevsky, S. B., Nicklas, B., Kanaya, A. M., Patrignani, P., Tacconelli, S., et al. (2012). Oxidative damage, platelet activation and inflammation to predict mobility disability and mortality in older persons: results from the health aging and body composition study. J. Gerontol. A Biol. Sci. Med. Sci. 67, 671–676. doi: 10.1093/gerona/glr246
Changeux, J.-P., and Dehaene, S. (1989). Neuronal models of cognitive functions. Cognition 33, 63–109. doi: 10.1016/0010-0277(89)90006-1
Chi, G. C., Fitzpatrick, A. L., Sharma, M., Jenny, N. S., Lopez, O. L., and DeKosky, S. T. (2016). Inflammatory biomarkers predict domain-specific cognitive decline in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 72, 796–803. doi: 10.1093/gerona/glw155
Chinta, S. J., Woods, G., Rane, A., Demaria, M., Campisi, J., and Andersen, J. K. (2015). Cellular senescence and the aging brain. Exp. Gerontol. 68, 3–7. doi: 10.1016/j.exger.2014.09.018
Cho, S.-H., Chen, J. A., Sayed, F., Ward, M. E., Gao, F., Nguyen, T. A., et al. (2015). SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 35, 807–818. doi: 10.1523/JNEUROSCI.2939-14.2015
Christensen, H. (2001). What cognitive changes can be expected with normal ageing? Aust. N. Z. J. Psychiatry 35, 768–775. doi: 10.1046/j.1440-1614.2001.00966.x
Chunchai, T., Samniang, B., Sripetchwandee, J., Pintana, H., Pongkan, W., Kumfu, S., et al. (2016). Vagus nerve stimulation exerts the neuroprotective effects in obese-insulin resistant rats, leading to the improvement of cognitive function. Sci. Rep. 6:26866. doi: 10.1038/srep26866
Collado, M., Blasco, M. A., and Serrano, M. (2007). Cellular senescence in cancer and aging. Cell 130, 223–233. doi: 10.1016/j.cell.2007.07.003
Collado, M., and Serrano, M. (2010). Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57. doi: 10.1038/nrc2772
Coppé, J.-P., Desprez, P.-Y., Krtolica, A., and Campisi, J. (2010a). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
Coppé, J.-P., Patil, C. K., Rodier, F., Krtolica, A., Beauséjour, C. M., Parrinello, S., et al. (2010b). A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One 5:e9188. doi: 10.1371/journal.pone.0009188
Coppé, J.-P., Patil, C. K., Rodier, F., Sun, Y., Muñoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. doi: 10.1371/journal.pbio.0060301
Csiszar, A., Wang, M., Lakatta, E. G., and Ungvari, Z. (2008). Inflammation and endothelial dysfunction during aging: role of NF-κB. J. Appl. Physiol. 105, 1333–1341. doi: 10.1152/japplphysiol.90470.2008
Cunningham, C., and Hennessy, E. (2015). Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res. Ther. 7:33. doi: 10.1186/s13195-015-0117-2
Czerniawski, J., Miyashita, T., Lewandowski, G., and Guzowski, J. F. (2015). Systemic lipopolysaccharide administration impairs retrieval of context-object discrimination, but not spatial, memory: evidence for selective disruption of specific hippocampus-dependent memory functions during acute neuroinflammation. Brain Behav. Immun. 44, 159–166. doi: 10.1016/j.bbi.2014.09.014
Damani, M. R., Zhao, L., Fontainhas, A. M., Amaral, J., Fariss, R. N., and Wong, W. T. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276. doi: 10.1111/j.1474-9726.2010.00660.x
Davenport, M. H., Hogan, D. B., Eskes, G. A., Longman, R. S., and Poulin, M. J. (2012). Cerebrovascular reserve: the link between fitness and cognitive function? Exerc. Sport Sci. Rev. 40, 153–158. doi: 10.1097/JES.0b013e3182553430
De la Fuente, M., and Miquel, J. (2009). An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 15, 3003–3026. doi: 10.2174/138161209789058110
de la Torre, J. C. (2012). Cardiovascular risk factors promote brain hypoperfusion leading to cognitive decline and dementia. Cardiovasc. Psychiatry Neurol. 2012:367516. doi: 10.1155/2012/367516
De Martinis, M., Franceschi, C., Monti, D., and Ginaldi, L. (2005). Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 579, 2035–2039. doi: 10.1016/j.febslet.2005.02.055
de Rooij, S. E., van Munster, B. C., Korevaar, J. C., and Levi, M. (2007). Cytokines and acute phase response in delirium. J. Psychosom. Res. 62, 521–525. doi: 10.1016/j.jpsychores.2006.11.013
Dendrou, C. A., Fugger, L., and Friese, M. A. (2015). Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558. doi: 10.1038/nri3871
Deschaintre, Y., Richard, F., Leys, D., and Pasquier, F. (2009). Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology 73, 674–680. doi: 10.1212/WNL.0b013e3181b59bf3
Diaz Heijtz, R., Wang, S., Anuar, F., Qian, Y., Björkholm, B., Samuelsson, A., et al. (2011). Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U S A 108, 3047–3052. doi: 10.1073/pnas.1010529108
Di Benedetto, S., Müller, L., Wenger, E., Düzel, S., and Pawelec, G. (2017). Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 75, 114–128. doi: 10.1016/j.neubiorev.2017.01.044
Di Filippo, M., Sarchielli, P., Picconi, B., and Calabresi, P. (2008). Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol. Sci. 29, 402–412. doi: 10.1016/j.tips.2008.06.005
Dik, M., Jonker, C., Hack, C., Smit, J., Comijs, H., and Eikelenboom, P. (2005). Serum inflammatory proteins and cognitive decline in older persons. Neurology 64, 1371–1377. doi: 10.1212/01.WNL.0000158281.08946.68
Dong, H., Zhang, X., and Qian, Y. (2014). Mast cells and neuroinflammation. Med. Sci. Monit. Basic Res. 20, 200–206. doi: 10.12659/MSMBR.893093
Dorheim, M.-A., Tracey, W. R., Pollock, J. S., and Grammas, P. (1994). Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 205, 659–665. doi: 10.1006/bbrc.1994.2716
Drachman, D. A. (2006). Aging of the brain, entropy and Alzheimer disease. Neurology 67, 1340–1352. doi: 10.1212/01.wnl.0000240127.89601.83
Edison, P., Archer, H. A., Gerhard, A., Hinz, R., Pavese, N., Turkheimer, F. E., et al. (2008). Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R) PK11195-PET and [11C] PIB-PET study. Neurobiol. Dis. 32, 412–419. doi: 10.1016/j.nbd.2008.08.001
Eikelenboom, P., van Exel, E., Hoozemans, J. J., Veerhuis, R., Rozemuller, A. J., and Van Gool, W. A. (2010). Neuroinflammation-an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener. Dis. 7, 38–41. doi: 10.1159/000283480
El Assar, M., Angulo, J., and Rodríguez-Mañas, L. (2013). Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 65, 380–401. doi: 10.1016/j.freeradbiomed.2013.07.003
Elmore, M. R., Lee, R. J., West, B. L., and Green, K. N. (2015). Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology and neuroinflammation. PLoS One 10:e0122912. doi: 10.1371/journal.pone.0122912
Elwood, E., Lim, Z., Naveed, H., and Galea, I. (2017). The effect of systemic inflammation on human brain barrier function. Brain Behav. Immun. 62, 35–40. doi: 10.1016/j.bbi.2016.10.020
Enciu, A.-M., Gherghiceanu, M., and Popescu, B. O. (2013). Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid. Med. Cell. Longev. 2013:297512. doi: 10.1155/2013/297512
Engelhart, M. J., Geerlings, M. I., Meijer, J., Kiliaan, A., Ruitenberg, A., van Swieten, J. C., et al. (2004). Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch. Neurol. 61, 668–672. doi: 10.1001/archneur.61.5.668
Erny, D., Hrabě de Angelis, A. L., Jaitin, D., Wieghofer, P., Staszewski, O., David, E., et al. (2015). Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977. doi: 10.1038/nn.4030
Fan, Z., Aman, Y., Ahmed, I., Chetelat, G., Landeau, B., Chaudhuri, K. R., et al. (2015). Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement. 11, 608–621.e7. doi: 10.1016/j.jalz.2014.06.016
Fard, M. T., Arulselvan, P., Karthivashan, G., Adam, S. K., and Fakurazi, S. (2015). Bioactive extract from Moringa oleifera inhibits the pro-inflammatory mediators in lipopolysaccharide stimulated macrophages. Pharmacogn. Mag. 11, S556–S563. doi: 10.4103/0973-1296.172961
Ferrucci, L., Corsi, A., Lauretani, F., Bandinelli, S., Bartali, B., Taub, D. D., et al. (2005). The origins of age-related proinflammatory state. Blood 105, 2294–2299. doi: 10.1182/blood-2004-07-2599
Fiolaki, A., Tsamis, K. I., Milionis, H. J., Kyritsis, A. P., Kosmidou, M., and Giannopoulos, S. (2014). Atherosclerosis, biomarkers of atherosclerosis and Alzheimer’s disease. Int. J. Neurosci. 124, 1–11. doi: 10.3109/00207454.2013.821988
Fish, P. V., Steadman, D., Bayle, E. D., and Whiting, P. (2019). New approaches for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 29, 125–133. doi: 10.1016/j.bmcl.2018.11.034
Flannery, B. M., Bruun, D. A., Rowland, D. J., Banks, C. N., Austin, A. T., Kukis, D. L., et al. (2016). Persistent neuroinflammation and cognitive impairment in a rat model of acute diisopropylfluorophosphate intoxication. J. Neuroinflammation 13:267. doi: 10.1186/s12974-016-0744-y
Fonken, L. K., Frank, M. G., Kitt, M. M., D’Angelo, H. M., Norden, D. M., Weber, M. D., et al. (2016). The alarmin HMGB1 mediates age-induced neuroinflammatory priming. J. Neurosci. 36, 7946–7956. doi: 10.1523/JNEUROSCI.1161-16.2016
Franceschi, C. (2007). Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr. Rev. 65, S173–S176. doi: 10.1111/j.1753-4887.2007.tb00358.x
Franceschi, C., Capri, M., Monti, D., Giunta, S., Olivieri, F., Sevini, F., et al. (2007). Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 128, 92–105. doi: 10.1016/j.mad.2006.11.016
Frank, M. G., Barrientos, R. M., Biedenkapp, J. C., Rudy, J. W., Watkins, L. R., and Maier, S. F. (2006). mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 27, 717–722. doi: 10.1016/j.neurobiolaging.2005.03.013
Freund, A., Orjalo, A. V., Desprez, P.-Y., and Campisi, J. (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246. doi: 10.1016/j.molmed.2010.03.003
Frohman, E. M., Frohman, T. C., Gupta, S., de Fougerolles, A., and van den Noort, S. (1991). Expression of intercellular adhesion molecule 1 (ICAM-1) in Alzheimer’s disease. J. Neurol. Sci. 106, 105–111. doi: 10.1016/0022-510X(91)90202-I
Fujimoto, H., Kobayashi, H., and Ohno, M. (2010). Age-induced reduction in mitochondrial manganese superoxide dismutase activity and tolerance of macrophages against apoptosis induced by oxidized low density lipoprotein. Circ. J. 74, 353–360. doi: 10.1253/circj.cj-09-0491
Gao, Q., Camous, X., Lu, Y.-X., Lim, M.-L., Larbi, A., and Ng, T.-P. (2016). Novel inflammatory markers associated with cognitive performance: singapore longitudinal ageing studies. Neurobiol. Aging 39, 140–146. doi: 10.1016/j.neurobiolaging.2015.12.002
Gauthier, S., Zhang, H., Ng, K., Pascoal, T., and Rosa-Neto, P. (2018). Impact of the biological definition of Alzheimer’s disease using amyloid, tau and neurodegeneration (ATN): what about the role of vascular changes, inflammation, Lewy body pathology? Transl. Neurodegener. 7:12. doi: 10.1186/s40035-018-0117-9
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Godbout, J., Chen, J., Abraham, J., Richwine, A., Berg, B., Kelley, K., et al. (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 19, 1329–1331. doi: 10.1096/fj.05-3776fje
Gorelick, P. B., Counts, S. E., and Nyenhuis, D. (2016). Vascular cognitive impairment and dementia. Biochim. Biophys. Acta 1862, 860–868. doi: 10.1016/j.bbadis.2015.12.015
Gorelick, P. B., Scuteri, A., Black, S. E., DeCarli, C., Greenberg, S. M., Iadecola, C., et al. (2011). Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42, 2672–2713. doi: 10.1161/STR.0b013e3182299496
Grammas, P. (2011). Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 8:26. doi: 10.1186/1742-2094-8-26
Grammas, P., Martinez, J., Sanchez, A., Yin, X., Riley, J., Gay, D., et al. (2014). A new paradigm for the treatment of Alzheimer’s disease: targeting vascular activation. J. Alzheimers Dis. 40, 619–630. doi: 10.3233/jad-2014-132057
Grammas, P., and Ovase, R. (2001). Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 22, 837–842. doi: 10.1016/s0197-4580(01)00276-7
Grammas, P., and Ovase, R. (2002). Cerebrovascular transforming growth factor-β contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 160, 1583–1587. doi: 10.1016/s0002-9440(10)61105-4
Gulyás, B., Halldin, C., Sandell, J., Karlsson, P., Sóvágó, J., Kárpáti, E., et al. (2002a). PET studies on the brain uptake and regional distribution of [11C] vinpocetine in human subjects. Acta Neurol. Scand. 106, 325–332. doi: 10.1034/j.1600-0404.2002.01302.x
Gulyás, B., Halldin, C., Sóvágó, J., Sandell, J., Cselényi, Z., Vas, Á., et al. (2002b). Drug distribution in man: a positron emission tomography study after oral administration of the labelled neuroprotective drug vinpocetine. Eur. J. Nucl. Med. Mol. Imaging 29, 1031–1038. doi: 10.1007/s00259-002-0823-4
Hajiluian, G., Nameni, G., Shahabi, P., Mesgari-Abbasi, M., Sadigh-Eteghad, S., and Farhangi, M. A. (2017). Vitamin D administration, cognitive function, BBB permeability and neuroinflammatory factors in high-fat diet-induced obese rats. Int. J. Obes. 41, 639–644. doi: 10.1038/ijo.2017.10
Hall, S., Janelidze, S., Surova, Y., Widner, H., Zetterberg, H., and Hansson, O. (2018). Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci. Rep. 8:13276. doi: 10.1038/s41598-018-31517-z
Halliwell, B. (2009). The wanderings of a free radical. Free Radic. Biol. Med. 46, 531–542. doi: 10.1016/j.freeradbiomed.2008.11.008
Hamanaka, R. B., and Chandel, N. S. (2010). Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35, 505–513. doi: 10.1016/j.tibs.2010.04.002
Hamelin, L., Lagarde, J., Dorothée, G., Leroy, C., Labit, M., Comley, R. A., et al. (2016). Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264. doi: 10.1093/brain/aww017
Harman, D. (1992). Free radical theory of aging. Mutat. Res. 275, 257–266. doi: 10.1016/0921-8734(92)90030-S
Harman, D. (2006). Free radical theory of aging: an update. Ann. N Y Acad. Sci. 1067, 10–21. doi: 10.1196/annals.1354.003
Harvey, A., Montezano, A. C., and Touyz, R. M. (2015). Vascular biology of ageing—implications in hypertension. J. Mol. Cell. Cardiol. 83, 112–121. doi: 10.1016/j.yjmcc.2015.04.011
Hawkins, B. T., and Davis, T. P. (2005). The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 57, 173–185. doi: 10.1124/pr.57.2.4
Helzner, E. P., Luchsinger, J. A., Scarmeas, N., Cosentino, S., Brickman, A. M., Glymour, M. M., et al. (2009). Contribution of vascular risk factors to the progression in Alzheimer disease. Arch. Neurol. 66, 343–348. doi: 10.1001/archneur.66.3.343
Herman, F., Westfall, S., Brathwaite, J., and Pasinetti, G. M. (2018). Suppression of presymptomatic oxidative stress and inflammation in neurodegeneration by grape-derived polyphenols. Front. Pharmacol. 9:867. doi: 10.3389/fphar.2018.00867
Hoeijmakers, L., Ruigrok, S. R., Amelianchik, A., Ivan, D., van Dam, A.-M., Lucassen, P. J., et al. (2017). Early-life stress lastingly alters the neuroinflammatory response to amyloid pathology in an Alzheimer’s disease mouse model. Brain Behav. Immun. 63, 160–175. doi: 10.1016/j.bbi.2016.12.023
Hoffman, G. S., and Calabrese, L. H. (2014). Vasculitis: determinants of disease patterns. Nat. Rev. Rheumatol. 10, 454–462. doi: 10.1038/nrrheum.2014.89
Hoffman, J. D., Parikh, I., Green, S. J., Chlipala, G., Mohney, R. P., Keaton, M., et al. (2017). Age drives distortion of brain metabolic, vascular and cognitive functions, and the gut microbiome. Front. Aging Neurosci. 9:298. doi: 10.3389/fnagi.2017.00298
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., et al. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. doi: 10.1212/WNL.0b013e3181b6bb95
Honig, L. S., Schupf, N., Lee, J. H., Tang, M. X., and Mayeux, R. (2006). Shorter telomeres are associated with mortality in those with APOE ε4 and dementia. Ann. Neurol. 60, 181–187. doi: 10.1002/ana.20894
Hovens, I. B., Schoemaker, R. G., van der Zee, E. A., Absalom, A. R., Heineman, E., and van Leeuwen, B. L. (2014). Postoperative cognitive dysfunction: involvement of neuroinflammation and neuronal functioning. Brain Behav. Immun. 38, 202–210. doi: 10.1016/j.bbi.2014.02.002
Hu, Z., Ou, Y., Duan, K., and Jiang, X. (2010). Inflammation: a bridge between postoperative cognitive dysfunction and Alzheimer’s disease. Med. Hypotheses 74, 722–724. doi: 10.1016/j.mehy.2009.10.040
Huijbregts, S., Kalkers, N., De Sonneville, L., de Groot, V., Reuling, I., and Polman, C. (2004). Differences in cognitive impairment of relapsing remitting, secondary, and primary progressive MS. Neurology 63, 335–339. doi: 10.1212/01.wnl.0000129828.03714.90
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iadecola, C. (2010). The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 120, 287–296. doi: 10.1007/s00401-010-0718-6
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376. doi: 10.1038/nn2003
Jellinger, K. A. (2013). Pathology and pathogenesis of vascular cognitive impairment—a critical update. Front. Aging Neurosci. 5:17. doi: 10.3389/fnagi.2013.00017
Jennette, J. C., and Falk, R. J. (1997). Small-vessel vasculitis. N. Engl. J. Med. 337, 1512–1523. doi: 10.1056/NEJM199711203372106
Jennette, J. C., and Falk, R. J. (2007). Nosology of primary vasculitis. Curr. Opin. Rheumatol. 19, 10–16. doi: 10.1097/bor.0b013e3280119877
Jeon, K.-I., Xu, X., Aizawa, T., Lim, J. H., Jono, H., Kwon, D.-S., et al. (2010). Vinpocetine inhibits NF-κB-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proc. Natl. Acad. Sci. U S A 107, 9795–9800. doi: 10.1073/pnas.0914414107
Jia, W., Martin, T. A., Zhang, G., and Jiang, W. G. (2013). Junctional adhesion molecules in cerebral endothelial tight junction and brain metastasis. Anticancer Res. 33, 2353–2359.
Jick, H., Zornberg, G. L., Jick, S. S., Seshadri, S., and Drachman, D. A. (2000). Statins and the risk of dementia. Lancet 356, 1627–1631. doi: 10.1016/s0140-6736(00)03155-x
Jin, R., Yang, G., and Li, G. (2010). Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol. 87, 779–789. doi: 10.1189/jlb.1109766
Jirillo, E., Candore, G., Magrone, T., and Caruso, C. (2008). A scientific approach to anti-ageing therapies: state of the art. Curr. Pharm. Des. 14, 2637–2642. doi: 10.2174/138161208786264070
Jurgens, H. A., and Johnson, R. W. (2012). Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp. Neurol. 233, 40–48. doi: 10.1016/j.expneurol.2010.11.014
Kaiser, D., Weise, G., Möller, K., Scheibe, J., Pösel, C., Baasch, S., et al. (2014). Spontaneous white matter damage, cognitive decline and neuroinflammation in middle-aged hypertensive rats: an animal model of early-stage cerebral small vessel disease. Acta Neuropathol. Commun. 2:169. doi: 10.1186/s40478-014-0169-8
Kennedy, G., Hardman, R. J., Macpherson, H., Scholey, A. B., and Pipingas, A. (2017). How does exercise reduce the rate of age-associated cognitive decline? A review of potential mechanisms. J. Alzheimers Dis. 55, 1–18. doi: 10.3233/JAD-160665
Kim, J., Kim, H., Roh, H., and Kwon, Y. (2018). Causes of hyperhomocysteinemia and its pathological significance. Arch. Pharm. Res. 41, 372–383. doi: 10.1007/s12272-018-1016-4
Kim, H., Shin, A., and Lee, K. J. (2015). Differences in C-reactive protein level in patients with Alzheimers disease and mild cognitive impairment. J. Psychiatry 18:194. doi: 10.4172/psychiatry.1000194
Kliper, E., Bashat, D. B., Bornstein, N. M., Shenhar-Tsarfaty, S., Hallevi, H., Auriel, E., et al. (2013). Cognitive decline after stroke: relation to inflammatory biomarkers and hippocampal volume. Stroke 44, 1433–1435. doi: 10.1161/STROKEAHA.111.000536
Kipnis, J., and Filiano, A. J. (2018). Neuroimmunology in 2017: the central nervous system: privileged by immune connections. Nat. Rev. Immunol. 18, 83–84. doi: 10.1038/nri.2017.152
Klohs, J., Rudin, M., Shimshek, D. R., and Beckmann, N. (2014). Imaging of cerebrovascular pathology in animal models of Alzheimer’s disease. Front. Aging Neurosci. 6:32. doi: 10.3389/fnagi.2014.00032
Knezevic, D., and Mizrahi, R. (2018). Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 80, 123–131. doi: 10.1016/j.pnpbp.2017.05.007
Kothari, V., Luo, Y., Tornabene, T., O’Neill, A. M., Greene, M. W., Geetha, T., et al. (2017). High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 499–508. doi: 10.1016/j.bbadis.2016.10.006
Kousik, S. M., Napier, T. C., and Carvey, P. M. (2012). The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front. Pharmacol. 3:121. doi: 10.3389/fphar.2012.00121
Kreisl, W. C., Jenko, K. J., Hines, C. S., Lyoo, C. H., Corona, W., Morse, C. L., et al. (2013a). A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J. Cereb. Blood Flow Metab. 33, 53–58. doi: 10.1038/jcbfm.2012.131
Kreisl, W. C., Lyoo, C. H., McGwier, M., Snow, J., Jenko, K. J., Kimura, N., et al. (2013b). In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 136, 2228–2238. doi: 10.1093/brain/awt145
Kritsilis, M., V Rizou, S., Koutsoudaki, P., Evangelou, K., Gorgoulis, V., and Papadopoulos, D. (2018). Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19:E2937. doi: 10.3390/ijms19102937
Kruyer, A., Soplop, N., Strickland, S., and Norris, E. H. (2015). Chronic hypertension leads to neurodegeneration in the TgSwDI mouse model of Alzheimer’s disease. Hypertension 66, 175–182. doi: 10.1161/hypertensionaha.115.05524
Kuilman, T., Michaloglou, C., Mooi, W. J., and Peeper, D. S. (2010). The essence of senescence. Genes Dev. 24, 2463–2479. doi: 10.1101/gad.1971610
Kumar, S., Millis, A., and Baglioni, C. (1992). Expression of interleukin 1-inducible genes and production of interleukin 1 by aging human fibroblasts. Proc. Natl. Acad. Sci. U S A 89, 4683–4687. doi: 10.1073/pnas.89.10.4683
Larbi, A., Franceschi, C., Mazzatti, D., Solana, R., Wikby, A., and Pawelec, G. (2008). Aging of the immune system as a prognostic factor for human longevity. Physiology 23, 64–74. doi: 10.1152/physiol.00040.2007
Laske, C., Leyhe, T., Stransky, E., Eschweiler, G. W., Bueltmann, A., Langer, H., et al. (2008). Association of platelet-derived soluble glycoprotein VI in plasma with Alzheimer’s disease. J. Psychiatr. Res. 42, 746–751. doi: 10.1016/j.jpsychires.2007.07.017
Laske, C., Sopova, K., and Stellos, K. (2012). Platelet activation in Alzheimer’s disease: from pathophysiology to clinical value. Curr. Vasc. Pharmacol. 10, 626–630. doi: 10.2174/157016112801784657
Laurent, S., Briet, M., and Boutouyrie, P. (2009). Large and small artery cross-talk and recent morbidity-mortality trials in hypertension. Hypertension 54, 388–392. doi: 10.1161/hypertensionaha.109.133116
Laurent, C., Dorothée, G., Hunot, S., Martin, E., Monnet, Y., Duchamp, M., et al. (2017). Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140, 184–200. doi: 10.1093/brain/aww270
Lee, Y.-J., Han, S. B., Nam, S.-Y., Oh, K.-W., and Hong, J. T. (2010). Inflammation and Alzheimer’s disease. Arch. Pharm. Res. 33, 1539–1556. doi: 10.1007/s12272-010-1006-7
Leung, K. K., Bartlett, J. W., Barnes, J., Manning, E. N., Ourselin, S., Fox, N. C., et al. (2013). Cerebral atrophy in mild cognitive impairment and Alzheimer disease rates and acceleration. Neurology 80, 648–654. doi: 10.1212/wnl.0b013e318281ccd3
Li, Q., Zeng, J., Su, M., He, Y., and Zhu, B. (2018). Acetylshikonin from Zicao attenuates cognitive impairment and hippocampus senescence in D-galactose-induced aging mouse model via upregulating the expression of SIRT1. Brain Res. Bull. 137, 311–318. doi: 10.1016/j.brainresbull.2018.01.007
Li, M., Zhou, H., Hua, W., Wang, B., Wang, S., Zhao, G., et al. (2009). Molecular diversity of Bacteroides spp. in human fecal microbiota as determined by group-specific 16S rRNA gene clone library analysis. Syst. Appl. Microbiol. 32, 193–200. doi: 10.1016/j.syapm.2009.02.001
Libby, P., Ridker, P. M., and Hansson, G. K. (2009). Inflammation in atherosclerosis: from pathophysiology to practice. J. Am. Coll. Cardiol. 54, 2129–2138. doi: 10.1016/j.jacc.2009.09.009
Lindqvist, D., Hall, S., Surova, Y., Nielsen, H. M., Janelidze, S., Brundin, L., et al. (2013). Cerebrospinal fluid inflammatory markers in Parkinson’s disease-associations with depression, fatigue and cognitive impairment. Brain Behav. Immun. 33, 183–189. doi: 10.1016/j.bbi.2013.07.007
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lyman, M., Lloyd, D. G., Ji, X., Vizcaychipi, M. P., and Ma, D. (2014). Neuroinflammation: the role and consequences. Neurosci. Res. 79, 1–12. doi: 10.1016/j.neures.2013.10.004
Lynch, M. A. (2009). Age-related neuroinflammatory changes negatively impact on neuronal function. Front. Aging Neurosci. 1:6. doi: 10.3389/neuro.24.006.2009
Lyons, A., McQuillan, K., Deighan, B. F., O’Reilly, J.-A., Downer, E. J., Murphy, A. C., et al. (2009). Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. Brain Behav. Immun. 23, 1020–1027. doi: 10.1016/j.bbi.2009.05.060
Ma, S. L., Lau, E. S., Suen, E. W., Lam, L. C. W., Leung, P. C., Woo, J., et al. (2013). Telomere length and cognitive function in southern Chinese community-dwelling male elders. Age Ageing 42, 450–455. doi: 10.1093/ageing/aft036
Malojcic, B., Giannakopoulos, P., Sorond, F. A., Azevedo, E., Diomedi, M., Oblak, J. P., et al. (2017). Ultrasound and dynamic functional imaging in vascular cognitive impairment and Alzheimer’s disease. BMC Med. 15:27. doi: 10.1186/s12916-017-0799-3
Mandolesi, G., Gentile, A., Musella, A., Fresegna, D., De Vito, F., Bullitta, S., et al. (2015). Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 11, 711–724. doi: 10.1038/nrneurol.2015.222
Marchesi, C., Paradis, P., and Schiffrin, E. L. (2008). Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 29, 367–374. doi: 10.1016/j.tips.2008.05.003
Marioni, R. E., Stewart, M. C., Murray, G. D., Deary, I. J., Fowkes, F. G. R., Lowe, G. D., et al. (2009). Peripheral levels of fibrinogen, C-reactive protein, and plasma viscosity predict future cognitive decline in individuals without dementia. Psychosom. Med. 71, 901–906. doi: 10.1097/psy.0b013e3181b1e538
Markiewski, M. M., and Lambris, J. D. (2007). The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 171, 715–727. doi: 10.2353/ajpath.2007.070166
Masoumi, J., Abbasloui, M., Parvan, R., Mohammadnejad, D., Pavon-Djavid, G., Barzegari, A., et al. (2018). Apelin, a promising target for Alzheimer disease prevention and treatment. Neuropeptides 70, 76–86. doi: 10.1016/j.npep.2018.05.008
Matias-Guiu, J. A., Cortés-Martínez, A., Valles-Salgado, M., Oreja-Guevara, C., Pytel, V., Montero, P., et al. (2017). Functional components of cognitive impairment in multiple sclerosis: a cross-sectional investigation. Front. Neurol. 8:643. doi: 10.3389/fneur.2017.00643
Mattace-Raso, F. U., van der Cammen, T. J., van der Meer, I. M., Schalekamp, M. A., Asmar, R., Hofman, A., et al. (2004). C-reactive protein and arterial stiffness in older adults: the Rotterdam Study. Atherosclerosis 176, 111–116. doi: 10.1016/j.atherosclerosis.2004.04.014
McAfoose, J., and Baune, B. (2009). Evidence for a cytokine model of cognitive function. Neurosci. Biobehav. Rev. 33, 355–366. doi: 10.1016/j.neubiorev.2008.10.005
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. doi: 10.1038/nature07201
Meeusen, R., and Decroix, L. (2018). Nutritional supplements and the brain. Int. J. Sport Nutr. Exerc. Metab. 28, 200–211. doi: 10.1123/ijsnem.2017-0314
Michels, M., Danieslki, L. G., Vieira, A., Florentino, D., Dall’Igna, D., Galant, L., et al. (2015). CD40-CD40 ligand pathway is a major component of acute neuroinflammation and contributes to long-term cognitive dysfunction after sepsis. Mol. Med. 21, 219–226. doi: 10.2119/molmed.2015.00070
Miles, E. A., Rees, D., Banerjee, T., Cazzola, R., Lewis, S., Wood, R., et al. (2008). Age-related increases in circulating inflammatory markers in men are independent of BMI, blood pressure and blood lipid concentrations. Atherosclerosis 196, 298–305. doi: 10.1016/j.atherosclerosis.2006.11.002
Miller, E. K., and Cohen, J. D. (2001). An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 24, 167–202. doi: 10.1146/annurev.neuro.24.1.167
Miller, K. R., and Streit, W. J. (2007). The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 3, 245–253. doi: 10.1017/s1740925x08000136
Millington, C., Sonego, S., Karunaweera, N., Rangel, A., Aldrich-Wright, J. R., Campbell, I. L., et al. (2014). Chronic neuroinflammation in Alzheimer’s disease: new perspectives on animal models and promising candidate drugs. Biomed Res. Int. 2014:309129. doi: 10.1155/2014/309129
Montezano, A. C., and Touyz, R. M. (2014). Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid. Redox Signal. 20, 164–182. doi: 10.1089/ars.2013.5302
Moon, M., Kim, H. G., Choi, J. G., Oh, H., Lee, P. K., Ha, S. K., et al. (2014). 6-Shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia. Biochem. Biophys. Res. Commun. 449, 8–13. doi: 10.1016/j.bbrc.2014.04.121
Morales, I., Guzmán-Martínez, L., Cerda-Troncoso, C., Farías, G. A., and Maccioni, R. B. (2014). Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 8:112. doi: 10.3389/fncel.2014.00112
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Müller, L., and Pawelec, G. (2015). As we age: does slippage of quality control in the immune system lead to collateral damage? Ageing Res. Rev. 23, 116–123. doi: 10.1016/j.arr.2015.01.005
Musella, A., Gentile, A., Rizzo, F. R., De Vito, F., Fresegna, D., Bullitta, S., et al. (2018). Interplay between age and neuroinflammation in multiple sclerosis: effects on motor and cognitive functions. Front. Aging Neurosci. 10:238. doi: 10.3389/fnagi.2018.00238
Nakhai-Pour, H., Grobbee, D., Bots, M., Muller, M., and Van Der Schouw, Y. (2007). C-reactive protein and aortic stiffness and wave reflection in middle-aged and elderly men from the community. J. Hum. Hypertens. 21, 949–955. doi: 10.1038/sj.jhh.1002255
Narasimhalu, K., Lee, J., Leong, Y.-L., Ma, L., De Silva, D. A., Wong, M.-C., et al. (2015). Inflammatory markers and their association with post stroke cognitive decline. Int. J. Stroke 10, 513–518. doi: 10.1111/ijs.12001
Njie, E. G., Boelen, E., Stassen, F. R., Steinbusch, H. W., Borchelt, D. R., and Streit, W. J. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008
Nomellini, V., Gomez, C. R., and Kovacs, E. J. (2008). “Aging and impairment of innate immunity,” in Trends in Innate Immunity, eds A. Egesten, A. Schmidt and H. Herwald (Switzerland: Karger Publishers), 188–205.
Norden, D. M., and Godbout, J. P. (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34. doi: 10.1111/j.1365-2990.2012.01306.x
Okello, A., Edison, P., Archer, H., Turkheimer, F., Kennedy, J., Bullock, R., et al. (2009). Microglial activation and amyloid deposition in mild cognitive impairment a PET study. Neurology 72, 56–62. doi: 10.1212/01.wnl.0000338622.27876.0d
Ownby, R. L. (2010). Neuroinflammation and cognitive aging. Curr. Psychiatry Rep. 12, 39–45. doi: 10.1007/s11920-009-0082-1
Palm, N. W., de Zoete, M. R., and Flavell, R. A. (2015). Immune-microbiota interactions in health and disease. Clin. Immunol. 159, 122–127. doi: 10.1016/j.clim.2015.05.014
Panossian, L., Porter, V., Valenzuela, H., Zhu, X., Reback, E., Masterman, D., et al. (2003). Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiol. Aging 24, 77–84. doi: 10.1016/s0197-4580(02)00043-x
Pantoni, L. (2010). Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 9, 689–701. doi: 10.1016/s1474-4422(10)70104-6
Parisotto, E. B., Vidal, V., García-Cerro, S., Lantigua, S., Wilhelm Filho, D., Sanchez-Barceló, E. J., et al. (2016). Chronic melatonin administration reduced oxidative damage and cellular senescence in the hippocampus of a mouse model of Down syndrome. Neurochem. Res. 41, 2904–2913. doi: 10.1007/s11064-016-2008-8
Pase, M. P., Grima, N., Cockerell, R., Stough, C., Scholey, A., Sali, A., et al. (2015). The effects of long-chain omega-3 fish oils and multivitamins on cognitive and cardiovascular function: a randomized, controlled clinical trial. J. Am. Coll. Nutr. 34, 21–31. doi: 10.1080/07315724.2014.880660
Pase, M. P., Herbert, A., Grima, N., Pipingas, A., and O’Rourke, M. (2012). Arterial stiffness as a cause of cognitive decline and dementia: a systematic review and meta-analysis. Intern. Med. J. 42, 808–815. doi: 10.1111/j.1445-5994.2011.02645.x
Pase, M. P., Pipingas, A., Kras, M., Nolidin, K., Gibbs, A. L., Wesnes, K. A., et al. (2010). Healthy middle-aged individuals are vulnerable to cognitive deficits as a result of increased arterial stiffness. J. Hypertens. 28, 1724–1729. doi: 10.1097/hjh.0b013e32833b1ee7
Paulson, O. B., Hasselbalch, S. G., Rostrup, E., Knudsen, G. M., and Pelligrino, D. (2010). Cerebral blood flow response to functional activation. J. Cereb. Blood Flow Metab. 30, 2–14. doi: 10.1038/jcbfm.2009.188
Pawelec, G. (2012). Hallmarks of human “immunosenescence”: adaptation or dysregulation? Immun. Ageing 9:15. doi: 10.1186/1742-4933-9-15
Pereira, H. A., Kumar, P., and Grammas, P. (1996). Expression of CAP37, a novel inflammatory mediator, in Alzheimer’s disease. Neurobiol. Aging 17, 753–759. doi: 10.1016/s0197-4580(96)00118-2
Petra, A. I., Panagiotidou, S., Hatziagelaki, E., Stewart, J. M., Conti, P., and Theoharides, T. C. (2015). Gut-microbiota-brain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin. Ther. 37, 984–995. doi: 10.1016/j.clinthera.2015.04.002
Petrou, M., Davatzikos, C., Hsieh, M., Foerster, B., Albin, R., Kotagal, V., et al. (2016). Diabetes, gray matter loss, and cognition in the setting of Parkinson disease. Acad. Radiol. 23, 577–581. doi: 10.1016/j.acra.2015.07.014
Piazza, J. R., Almeida, D. M., Dmitrieva, N. O., and Klein, L. C. (2010). Frontiers in the use of biomarkers of health in research on stress and aging. J. Gerontol. B Psychol. Sci. Soc. Sci. 65, 513–525. doi: 10.1093/geronb/gbq049
Pintana, H., Apaijai, N., Kerdphoo, S., Pratchayasakul, W., Sripetchwandee, J., Suntornsaratoon, P., et al. (2017). Hyperglycemia induced the Alzheimer’s proteins and promoted loss of synaptic proteins in advanced-age female Goto-Kakizaki (GK) rats. Neurosci. Lett. 655, 41–45. doi: 10.1016/j.neulet.2017.06.041
Pipitone, N. A., Versari, A., and Salvarani, C. (2018). Usefulness of PET in recognizing and managing vasculitides. Curr. Opin. Rheumatol. 30, 24–29. doi: 10.1097/bor.0000000000000459
Planche, V., Gibelin, M., Cregut, D., Pereira, B., and Clavelou, P. (2016). Cognitive impairment in a population-based study of patients with multiple sclerosis: differences between late relapsing-remitting, secondary progressive and primary progressive multiple sclerosis. Eur. J. Neurol. 23, 282–289. doi: 10.1111/ene.12715
Potagas, C., Giogkaraki, E., Koutsis, G., Mandellos, D., Tsirempolou, E., Sfagos, C., et al. (2008). Cognitive impairment in different MS subtypes and clinically isolated syndromes. J. Neurol. Sci. 267, 100–106. doi: 10.1016/j.jns.2007.10.002
Prieto-González, S., Arguis, P., and Cid, M. C. (2015). Imaging in systemic vasculitis. Curr. Opin. Rheumatol. 27, 53–62. doi: 10.1097/BOR.0000000000000130
Ransohoff, R. M., and Cardona, A. E. (2010). The myeloid cells of the central nervous system parenchyma. Nature 468, 253–262. doi: 10.1038/nature09615
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Rao, S. M. (1990). A Manual for the Brief Repeatable Battery of Neuropsychological Tests in Multiple Sclerosis. New York, NY: National Multiple Sclerosis Society.
Ravaglia, G., Forti, P., Maioli, F., Chiappelli, M., Montesi, F., Tumini, E., et al. (2007). Blood inflammatory markers and risk of dementia: the conselice study of brain aging. Neurobiol. Aging 28, 1810–1820. doi: 10.1016/j.neurobiolaging.2006.08.012
Reis, P. A., Alexandre, P. C., D’Avila, J. C., Siqueira, L. D., Antunes, B., Estato, V., et al. (2017). Statins prevent cognitive impairment after sepsis by reverting neuroinflammation and microcirculatory/endothelial dysfunction. Brain Behav. Immun. 60, 293–303. doi: 10.1016/j.bbi.2016.11.006
Renna, N. F., Lembo, C., Diez, E., and Miatello, R. M. (2013). Role of renin-angiotensin system and oxidative stress on vascular inflammation in insulin resistence model. Int. J. Hypertens. 2013:420979. doi: 10.1155/2013/420979
Riederer, P., Korczyn, A. D., Ali, S. S., Bajenaru, O., Choi, M. S., Chopp, M., et al. (2017). The diabetic brain and cognition. J. Neural Transm. 124, 1431–1454. doi: 10.1007/s00702-017-1763-2
Rizzo, F., Riboldi, G., Salani, S., Nizzardo, M., Simone, C., Corti, S., et al. (2014). Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 71, 999–1015. doi: 10.1007/s00018-013-1480-4
Rodriguez-Grande, B., Ichkova, A., Lemarchant, S., and Badaut, J. (2017). Early to long-term alterations of CNS barriers after traumatic brain injury: considerations for drug development. AAPS J. 19, 1615–1625. doi: 10.1208/s12248-017-0123-3
Rodríguez-Mañas, L., El-Assar, M., Vallejo, S., López-Dóriga, P., Solís, J., Petidier, R., et al. (2009). Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 8, 226–238. doi: 10.1111/j.1474-9726.2009.00466.x
Roselli, F., Tartaglione, B., Federico, F., Lepore, V., Defazio, G., and Livrea, P. (2009). Rate of MMSE score change in Alzheimer’s disease: influence of education and vascular risk factors. Clin. Neurol. Neurosurg. 111, 327–330. doi: 10.1016/j.clineuro.2008.10.006
Rosenberg, G. A. (2009). Inflammation and white matter damage in vascular cognitive impairment. Stroke 40, S20–S23. doi: 10.1161/strokeaha.108.533133
Rothenburg, L. S., Herrmann, N., Swardfager, W., Black, S. E., Tennen, G., Kiss, A., et al. (2010). The relationship between inflammatory markers and post stroke cognitive impairment. J. Geriatr. Psychiatry Neurol. 23, 199–205. doi: 10.1177/0891988710373598
Ruet, A., Deloire, M., Charré-Morin, J., Hamel, D., and Brochet, B. (2013). Cognitive impairment differs between primary progressive and relapsing-remitting MS. Neurology 80, 1501–1508. doi: 10.1212/wnl.0b013e31828cf82f
Rust, R., Hofer, A.-S., and Schwab, M. E. (2018). Stroke promotes systemic endothelial inflammation and atherosclerosis. Trends Mol. Med. 24, 593–595. doi: 10.1016/j.molmed.2018.04.008
Ryan, S. M., and Nolan, Y. M. (2016). Neuroinflammation negatively affects adult hippocampal neurogenesis and cognition: can exercise compensate? Neurosci. Biobehav. Rev. 61, 121–131. doi: 10.1016/j.neubiorev.2015.12.004
Saiyasit, N., Sripetchwandee, J., Chattipakorn, N., and Chattipakorn, S. C. (2018). Potential roles of neurotensin on cognition in conditions of obese-insulin resistance. Neuropeptides 72, 12–22. doi: 10.1016/j.npep.2018.09.002
Salama, R., Sadaie, M., Hoare, M., and Narita, M. (2014). Cellular senescence and its effector programs. Genes Dev. 28, 99–114. doi: 10.1101/gad.235184.113
Salat, D. H., Buckner, R. L., Snyder, A. Z., Greve, D. N., Desikan, R. S., Busa, E., et al. (2004). Thinning of the cerebral cortex in aging. Cereb. Cortex 14, 721–730. doi: 10.1093/cercor/bhh032
Salat, D., Tuch, D., Hevelone, N., Fischl, B., Corkin, S., Rosas, H., et al. (2005). Age-related changes in prefrontal white matter measured by diffusion tensor imaging. Ann. N Y Acad. Sci. 1064, 37–49. doi: 10.1196/annals.1340.009
Salthouse, T. A. (1996). The processing-speed theory of adult age differences in cognition. Psychol. Rev. 103, 403–428. doi: 10.1037//0033-295x.103.3.403
Salvioli, S., Monti, D., Lanzarini, C., Conte, M., Pirazzini, C., Giulia Bacalini, M., et al. (2013). Immune system, cell senescence, aging and longevity-inflamm-aging reappraised. Curr. Pharm. Des. 19, 1675–1679. doi: 10.2174/138161213805219531
Samanez-Larkin, G. R., and Knutson, B. (2015). Decision making in the ageing brain: changes in affective and motivational circuits. Nat. Rev. Neurosci. 16, 278–289. doi: 10.1038/nrn3917
Sanchez, A., Tripathy, D., Luo, J., Yin, X., Martinez, J., and Grammas, P. (2013). Neurovascular unit and the effects of dosage in VEGF toxicity: role for oxidative stress and thrombin. J. Alzheimers Dis. 34, 281–291. doi: 10.3233/jad-121636
Sanchez-Marin, L., Pavon, F. J., Decara, J., Suarez, J., Gavito, A., Castilla-Ortega, E., et al. (2017). Effects of intermittent alcohol exposure on emotion and cognition: a potential role for the endogenous cannabinoid system and neuroinflammation. Front. Behav. Neurosci. 11:15. doi: 10.3389/fnbeh.2017.00015
Sa-Nguanmoo, P., Tanajak, P., Kerdphoo, S., Jaiwongkam, T., Wang, X., Liang, G., et al. (2018). FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. Biomed. Pharmacother. 97, 1663–1672. doi: 10.1016/j.biopha.2017.12.021
Savoia, C., and Schiffrin, E. L. (2006). Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 15, 152–158. doi: 10.1097/01.mnh.0000203189.57513.76
Schmidt, R., Schmidt, H., Curb, J. D., Masaki, K., White, L. R., and Launer, L. J. (2002). Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia aging study. Ann. Neurol. 52, 168–174. doi: 10.1002/ana.10265
Schram, M. T., Euser, S. M., De Craen, A. J., Witteman, J. C., Frölich, M., Hofman, A., et al. (2007). Systemic markers of inflammation and cognitive decline in old age. J. Am. Geriatr. Soc. 55, 708–716. doi: 10.1111/j.1532-5415.2007.01159.x
Schuitemaker, A., Kropholler, M. A., Boellaard, R., van der Flier, W. M., Kloet, R. W., van der Doef, T. F., et al. (2013). Microglial activation in Alzheimer’s disease: an (R)-[11C] PK11195 positron emission tomography study. Neurobiol. Aging 34, 128–136. doi: 10.1016/j.neurobiolaging.2012.04.021
Schuitemaker, A., van der Doef, T. F., Boellaard, R., van der Flier, W. M., Yaqub, M., Windhorst, A. D., et al. (2012). Microglial activation in healthy aging. Neurobiol. Aging 33, 1067–1072. doi: 10.1016/j.neurobiolaging.2010.09.016
Scuteri, A., Orru, M., Morrell, C., Piras, M. G., Taub, D., Schlessinger, D., et al. (2011). Independent and additive effects of cytokine patterns and the metabolic syndrome on arterial aging in the SardiNIA Study. Atherosclerosis 215, 459–464. doi: 10.1016/j.atherosclerosis.2010.12.023
Scuteri, A., Tesauro, M., Rizza, S., Iantorno, M., Federici, M., Lauro, D., et al. (2008). Endothelial function and arterial stiffness in normotensive normoglycemic first-degree relatives of diabetic patients are independent of the metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 18, 349–356. doi: 10.1016/j.numecd.2007.03.008
Seals, D. R., Moreau, K. L., Gates, P. E., and Eskurza, I. (2006). Modulatory influences on ageing of the vasculature in healthy humans. Exp. Gerontol. 41, 501–507. doi: 10.1016/j.exger.2006.01.001
Seo, E.-J., Fischer, N., and Efferth, T. (2018). Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol. Res. 129, 262–273. doi: 10.1016/j.phrs.2017.11.030
Seth, P., and Koul, N. (2008). Astrocyte, the star avatar: redefined. J. Biosci. 33, 405–421. doi: 10.1007/s12038-008-0060-5
Sharp, S. I., Aarsland, D., Day, S., Sønnesyn, H., and Ballard, C. (2011). Hypertension is a potential risk factor for vascular dementia: systematic review. Int. J. Geriatr. Psychiatry 26, 661–669. doi: 10.1002/gps.2572
Shaw, A. C., Joshi, S., Greenwood, H., Panda, A., and Lord, J. M. (2010). Aging of the innate immune system. Curr. Opin. Immunol. 22, 507–513. doi: 10.1016/j.coi.2010.05.003
Shih, A. Y., Erb, H., Sun, X., Toda, S., Kalivas, P. W., and Murphy, T. H. (2006). Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 26, 10514–10523. doi: 10.1523/jneurosci.3178-06.2006
Shock, N. W., Greulich, R. C., Aremberg, D., Costa, P. T., Lakatta, E. G., and Tobin, J. D. (1984). Normal Human Aging: The Baltimore Longitudinal Study of Aging. Washington, DC: National Institutes of Health.
Sierra, A., Gottfried-Blackmore, A. C., McEwen, B. S., and Bulloch, K. (2007). Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55, 412–424. doi: 10.1002/glia.20468
Simen, A. A., Bordner, K. A., Martin, M. P., Moy, L. A., and Barry, L. C. (2011). Cognitive dysfunction with aging and the role of inflammation. Ther. Adv. Chronic Dis. 2, 175–195. doi: 10.1177/2040622311399145
Simpson, J. E., Ince, P. G., Matthews, F. E., Shaw, P. J., Heath, P. R., Brayne, C., et al. (2015). A neuronal DNA damage response is detected at the earliest stages of Alzheimer’s neuropathology and correlates with cognitive impairment in the Medical Research Council’s Cognitive Function and Ageing Study ageing brain cohort. Neuropathol. Appl. Neurobiol. 41, 483–496. doi: 10.1111/nan.12202
Singh-Manoux, A., Kivimaki, M., Glymour, M. M., Elbaz, A., Berr, C., Ebmeier, K. P., et al. (2012). Timing of onset of cognitive decline: results from Whitehall II prospective cohort study. BMJ 344:d7622. doi: 10.1136/bmj.d7622
Skaper, S. D., Facci, L., and Giusti, P. (2014). Neuroinflammation, microglia and mast cells in the pathophysiology of neurocognitive disorders: a review. CNS Neurol. Disord. Drug Targets 13, 1654–1666. doi: 10.2174/1871527313666141130224206
Skvarc, D. R., Berk, M., Byrne, L. K., Dean, O. M., Dodd, S., Lewis, M., et al. (2018). Post-operative cognitive dysfunction: an exploration of the inflammatory hypothesis and novel therapies. Neurosci. Biobehav. Rev. 84, 116–133. doi: 10.1016/j.neubiorev.2017.11.011
Solas, M., Milagro, F. I., Ramirez, M. J., and Martínez, J. A. (2017). Inflammation and gut-brain axis link obesity to cognitive dysfunction: plausible pharmacological interventions. Curr. Opin. Pharmacol. 37, 87–92. doi: 10.1016/j.coph.2017.10.005
Sparks, D., Sabbagh, M., Connor, D., Soares, H., Lopez, J., Stankovic, G., et al. (2006). Statin therapy in Alzheimer’s disease. Acta Neurol. Scand. 114, 78–86. doi: 10.1111/j.1600-0404.2006.00689.x
Stampanoni Bassi, M., Mori, F., Buttari, F., Marfia, G. A., Sancesario, A., Centonze, D., et al. (2017). Neurophysiology of synaptic functioning in multiple sclerosis. Clin. Neurophysiol. 128, 1148–1157. doi: 10.1016/j.clinph.2017.04.006
Stellos, K., Katsiki, N., Tatsidou, P., Bigalke, B., and Laske, C. (2014). Association of platelet activation with vascular cognitive impairment: implications in dementia development? Curr. Vasc. Pharmacol. 12, 152–154. doi: 10.2174/157016111201140327164641
Stellos, K., Panagiota, V., Kögel, A., Leyhe, T., Gawaz, M., and Laske, C. (2010). Predictive value of platelet activation for the rate of cognitive decline in Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 30, 1817–1820. doi: 10.1038/jcbfm.2010.140
Stranahan, A. M., Hao, S., Dey, A., Yu, X., and Baban, B. (2016). Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J. Cereb. Blood Flow Metab. 36, 2108–2121. doi: 10.1177/0271678x16642233
Sudduth, T. L., Schmitt, F. A., Nelson, P. T., and Wilcock, D. M. (2013). Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 34, 1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012
Sun, J., Zhang, S., Zhang, X., Zhang, X., Dong, H., and Qian, Y. (2015). IL-17A is implicated in lipopolysaccharide-induced neuroinflammation and cognitive impairment in aged rats via microglial activation. J. Neuroinflammation 12:165. doi: 10.1186/s12974-015-0394-5
Surendranathan, A., Su, L., Mak, E., Passamonti, L., Hong, Y. T., Arnold, R., et al. (2018). Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 141, 3415–3427. doi: 10.1093/brain/awy265
Suridjan, I., Pollock, B., Verhoeff, N., Voineskos, A., Chow, T., Rusjan, P., et al. (2015). In vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: a positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol. Psychiatry 20, 1579–1587. doi: 10.1038/mp.2015.1
Swardfager, W., Hennebelle, M., Yu, D., Hammock, B., Levitt, A., Hashimoto, K., et al. (2018). Metabolic/inflammatory/vascular comorbidity in psychiatric disorders; soluble epoxide hydrolase (sEH) as a possible new target. Neurosci. Biobehav. Rev. 87, 56–66. doi: 10.1016/j.neubiorev.2018.01.010
Takeda, S., Sato, N., Uchio-Yamada, K., Sawada, K., Kunieda, T., Takeuchi, D., et al. (2010). Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. U S A 107, 7036–7041. doi: 10.1073/pnas.1000645107
Tan, H., Bi, J., Wang, Y., Zhang, J., and Zuo, Z. (2015). Transfusion of old red blood cells induces neuroinflammation and cognitive impairment. Crit. Care Med. 43, e276–e286. doi: 10.1097/ccm.0000000000001023
Tarantini, S., Valcarcel-Ares, M. N., Yabluchanskiy, A., Tucsek, Z., Hertelendy, P., Kiss, T., et al. (2017). Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression and cognitive decline in mice, mimicking the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 73, 853–863. doi: 10.1093/gerona/glx177
Tatchum-Talom, R., and Martin, D. (2004). Tempol improves vascular function in the mesenteric vascular bed of senescent rats. Can. J. Physiol. Pharmacol. 82, 200–207. doi: 10.1139/y04-010
Terrando, N., Eriksson, L. I., Kyu Ryu, J., Yang, T., Monaco, C., Feldmann, M., et al. (2011). Resolving postoperative neuroinflammation and cognitive decline. Ann. Neurol. 70, 986–995. doi: 10.1002/ana.22664
Thirumangalakudi, L., Samany, P. G., Owoso, A., Wiskar, B., and Grammas, P. (2006). Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimers Dis. 10, 111–118. doi: 10.3233/jad-2006-10114
Tian, A., Ma, H., Cao, X., Zhang, R., Wang, X., and Wu, B. (2015). Vitamin D improves cognitive function and modulates T h 17/T reg cell balance after hepatectomy in mice. Inflammation 38, 500–509. doi: 10.1007/s10753-014-9956-4
Tremblay, M. È., Zettel, M. L., Ison, J. R., Allen, P. D., and Majewska, A. K. (2012). Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60, 541–558. doi: 10.1002/glia.22287
Triantafyllidi, H., Arvaniti, C., Lekakis, J., Ikonomidis, I., Siafakas, N., Tzortzis, S., et al. (2009). Cognitive impairment is related to increased arterial stiffness and microvascular damage in patients with never-treated essential hypertension. Am. J. Hypertens. 22, 525–530. doi: 10.1038/ajh.2009.35
Trollor, J. N., Smith, E., Baune, B. T., Kochan, N. A., Campbell, L., Samaras, K., et al. (2010). Systemic inflammation is associated with MCI and its subtypes: the sydney memory and aging study. Dement. Geriatr. Cogn. Disord. 30, 569–578. doi: 10.1159/000322092
Tumminia, A., Vinciguerra, F., Parisi, M., and Frittitta, L. (2018). Type 2 diabetes mellitus and Alzheimer’s disease: role of insulin signalling and therapeutic implications. Int. J. Mol. Sci. 19:E3306. doi: 10.3390/ijms19113306
Tzikas, S., Schlak, D., Sopova, K., Gatsiou, A., Stakos, D., Stamatelopoulos, K., et al. (2014). Increased myeloperoxidase plasma levels in patients with Alzheimer’s disease. J. Alzheimers Dis. 39, 557–564. doi: 10.3233/jad-131469
Ungvari, Z., Csiszar, A., and Kaley, G. (2004). Vascular inflammation in aging. Herz 29, 733–740. doi: 10.1007/s00059-004-2625-x
Ungvari, Z., Tarantini, S., Hertelendy, P., Valcarcel-Ares, M. N., Fülöp, G. A., Logan, S., et al. (2017). Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. Geroscience 39, 33–42. doi: 10.1007/s11357-017-9964-z
van Beek, A. H., Claassen, J. A., Rikkert, M. G. O., and Jansen, R. W. (2008). Cerebral autoregulation: an overview of current concepts and methodology with special focus on the elderly. J. Cereb. Blood Flow Metab. 28, 1071–1085. doi: 10.1038/jcbfm.2008.13
van Buchem, M. A., Biessels, G. J., Brunner la Rocca, H. P., de Craen, A. J., van der Flier, W. M., Ikram, M. A., et al. (2014). The heart-brain connection: a multidisciplinary approach targeting a missing link in the pathophysiology of vascular cognitive impairment. J. Alzheimers Dis. 42, S443–S451. doi: 10.3233/jad-141542
van Deursen, J. M. (2014). The role of senescent cells in ageing. Nature 509, 439–446. doi: 10.1038/nature13193
Varrone, A., and Nordberg, A. (2015). Molecular imaging of neuroinflammation in Alzheimer’s disease. Clin. Transl. Imaging. 3, 437–447. doi: 10.1007/s40336-015-0137-8
Vijayan, M., and Reddy, P. H. (2016). Stroke, vascular dementia and Alzheimer’s disease: molecular links. J. Alzheimers Dis. 54, 427–443. doi: 10.3233/JAD-160527
Von Bernhardi, R., Eugenín-von Bernhardi, L., and Eugenín, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Walters, H. E., Deneka-Hannemann, S., and Cox, L. S. (2016). Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 8, 231–244. doi: 10.18632/aging.100872
Wang, D. D., and Bordey, A. (2008). The astrocyte odyssey. Prog. Neurobiol. 86, 342–367. doi: 10.1016/j.pneurobio.2008.09.015
Wang, J., Li, L., Wang, Z., Cui, Y., Tan, X., Yuan, T., et al. (2018). Supplementation of lycopene attenuates lipopolysaccharide-induced amyloidogenesis and cognitive impairments via mediating neuroinflammation and oxidative stress. J. Nutr. Biochem. 56, 16–25. doi: 10.1016/j.jnutbio.2018.01.009
Wang, S., Moerman, E. J., Jones, R. A., Thweatt, R., and Goldstein, S. (1996). Characterization of IGFBP-3, PAI-1 and SPARC mRNA expression in senescent fibroblasts. Mech. Ageing Dev. 92, 121–132. doi: 10.1016/s0047-6374(96)01814-3
Wang, H., Zhang, K., Zhao, L., Tang, J., Gao, L., and Wei, Z. (2014). Anti-inflammatory effects of vinpocetine on the functional expression of nuclear factor-kappa B and tumor necrosis factor-α in a rat model of cerebral ischemia-reperfusion injury. Neurosci. Lett. 566, 247–251. doi: 10.1016/j.neulet.2014.02.045
Webster, S. J., Van Eldik, L. J., Watterson, D. M., and Bachstetter, A. D. (2015). Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J. Neurosci. 35, 6554–6569. doi: 10.1523/jneurosci.0291-15.2015
Wei, P., Liu, Q., Li, D., Zheng, Q., Zhou, J., and Li, J. (2015). Acute nicotine treatment attenuates lipopolysaccharide-induced cognitive dysfunction by increasing BDNF expression and inhibiting neuroinflammation in the rat hippocampus. Neurosci. Lett. 604, 161–166. doi: 10.1016/j.neulet.2015.08.008
Weiskopf, D., Weinberger, B., and Grubeck-Loebenstein, B. (2009). The aging of the immune system. Transpl. Int. 22, 1041–1050. doi: 10.1111/j.1432-2277.2009.00927.x
Weiss, S., Mori, F., Rossi, S., and Centonze, D. (2014). Disability in multiple sclerosis: when synaptic long-term potentiation fails. Neurosci. Biobehav. Rev. 43, 88–99. doi: 10.1016/j.neubiorev.2014.03.023
Wilson, C. J., Finch, C. E., and Cohen, H. J. (2002). Cytokines and cognition—the case for a head-to-toe inflammatory paradigm. J. Am. Geriatr. Soc. 50, 2041–2056. doi: 10.1046/j.1532-5415.2002.50619.x
Windham, B. G., Simpson, B. N., Lirette, S., Bridges, J., Bielak, L., Peyser, P. A., et al. (2014). Associations between inflammation and cognitive function in African Americans and European Americans. J. Am. Geriatr. Soc. 62, 2303–2310. doi: 10.1111/jgs.13165
Wolburg, H., Noell, S., Mack, A., Wolburg-Buchholz, K., and Fallier-Becker, P. (2009). Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 335, 75–96. doi: 10.1007/s00441-008-0658-9
Wolozin, B., Kellman, W., Ruosseau, P., Celesia, G. G., and Siegel, G. (2000). Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57, 1439–1443. doi: 10.1001/archneur.57.10.1439
Won, J.-S., Kim, J., Annamalai, B., Shunmugavel, A., Singh, I., and Singh, A. K. (2013). Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimers Dis. 34, 621–635. doi: 10.3233/JAD-121786
Wong, W. T. (2013). Microglial aging in the healthy CNS: phenotypes, drivers and rejuvenation. Front. Cell. Neurosci. 7:22. doi: 10.3389/fncel.2013.00022
Xie, G., Zhang, W., Chang, Y., and Chu, Q. (2009). Relationship between perioperative inflammatory response and postoperative cognitive dysfunction in the elderly. Med. Hypotheses 73, 402–403. doi: 10.1016/j.mehy.2009.01.056
Yaffe, K., Lindquist, K., Penninx, B., Simonsick, E., Pahor, M., Kritchevsky, S., et al. (2003). Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology 61, 76–80. doi: 10.1212/01.wnl.0000073620.42047.d7
Yano, Y., Matsuda, S., Hatakeyama, K., Sato, Y., Imamura, T., Shimada, K., et al. (2010). Plasma Pentraxin 3, but not high-sensitivity C-reactive protein, is a useful inflammatory biomarker for predicting cognitive impairment in elderly hypertensive patients. J. Gerontol. A Biol. Sci. Med. Sci. 65, 547–552. doi: 10.1093/gerona/glq030
Yasuno, F., Kosaka, J., Ota, M., Higuchi, M., Ito, H., Fujimura, Y., et al. (2012). Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 203, 67–74. doi: 10.1016/j.pscychresns.2011.08.013
Yasuno, F., Ota, M., Kosaka, J., Ito, H., Higuchi, M., Doronbekov, T. K., et al. (2008). Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with [11C]DAA1106. Biol. Psychiatry 64, 835–841. doi: 10.1016/j.biopsych.2008.04.021
Ye, S.-M., and Johnson, R. W. (1999). Increased interleukin-6 expression by microglia from brain of aged mice. J. Neuroimmunol. 93, 139–148. doi: 10.1016/s0165-5728(98)00217-3
Yokokura, M., Mori, N., Yagi, S., Yoshikawa, E., Kikuchi, M., Yoshihara, Y., et al. (2011). In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 38, 343–351. doi: 10.1007/s00259-010-1612-0
Young, A. R., and Narita, M. (2009). SASP reflects senescence. EMBO Rep. 10, 228–230. doi: 10.1038/embor.2009.22
Younger, D. S. (2004). Vasculitis of the nervous system. Curr. Opin. Neurol. 17, 317–336. doi: 10.1097/00019052-200406000-00014
Yu, D., Corbett, B., Yan, Y., Zhang, G.-X., Reinhart, P., Cho, S. J., et al. (2012). Early cerebrovascular inflammation in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 2942–2947. doi: 10.1016/j.neurobiolaging.2012.02.023
Zanni, F., Vescovini, R., Biasini, C., Fagnoni, F., Zanlari, L., Telera, A., et al. (2003). Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: a contribution to understand the relationship between inflammation and immunosenescence. Exp. Gerontol. 38, 981–987. doi: 10.1016/s0531-5565(03)00160-8
Zhang, Y.-S., Li, J.-D., and Yan, C. (2017). An update on vinpocetine: new discoveries and clinical implications. Eur. J. Pharmacol. 819, 30–34. doi: 10.1016/j.ejphar.2017.11.041
Zhang, F., Yan, C., Wei, C., Yao, Y., Ma, X., Gong, Z., et al. (2018). Vinpocetine inhibits NF-κB-dependent inflammation in acute ischemic stroke patients. Transl. Stroke Res. 9, 174–184. doi: 10.1007/s12975-017-0549-z
Zhang, L., and Yang, L. (2014). Anti-inflammatory effects of vinpocetine in atherosclerosis and ischemic stroke: a review of the literature. Molecules 20, 335–347. doi: 10.3390/molecules20010335
Zhong, K. L., Chen, F., Hong, H., Ke, X., Lv, Y. G., Tang, S. S., et al. (2018). New views and possibilities of antidiabetic drugs in treating and/or preventing mild cognitive impairment and Alzheimer’s disease. Metab. Brain Dis. 33, 1009–1018. doi: 10.1007/s11011-018-0227-1
Ziv, Y., Ron, N., Butovsky, O., Landa, G., Sudai, E., Greenberg, N., et al. (2006). Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 9, 268–275. doi: 10.1038/nn1629
Zlokovic, B. V. (2005). Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 28, 202–208. doi: 10.1016/j.tins.2005.02.001
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. doi: 10.1038/nrn3114
Keywords: cognitive aging, cognitive decline, inflammation, inflamm-aging, vascular inflammation, neuroinflammation
Citation: Tangestani Fard M and Stough C (2019) A Review and Hypothesized Model of the Mechanisms That Underpin the Relationship Between Inflammation and Cognition in the Elderly. Front. Aging Neurosci. 11:56. doi: 10.3389/fnagi.2019.00056
Received: 05 October 2018; Accepted: 26 February 2019;
Published: 13 March 2019.
Edited by:
Craig Atwood, University of Wisconsin-Madison, United StatesReviewed by:
Murali Vijayan, Texas Tech University Health Sciences Center, United StatesXudong Huang, Massachusetts General Hospital, Harvard Medical School, United States
Copyright © 2019 Tangestani Fard and Stough. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Con Stough, Y3N0b3VnaEBzd2luLmVkdS5hdQ==