 Qinqin Wang
Qinqin Wang Chunmei Wang
Chunmei Wang Bingyuan Ji1
Bingyuan Ji1 Jiawei Zhou
Jiawei Zhou Chunqing Yang
Chunqing Yang Jing Chen
Jing Chen- 1Neurobiology Key Laboratory, Jining Medical University, Jining, China
- 2State Key Laboratory of Neuroscience, Institute of Neuroscience, Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, China
- 3Division of Biomedical Sciences, Warwick Medical School, University of Warwick, Coventry, United Kingdom
Hyaluronan and proteoglycan link protein 2 (Hapln2) is important for the binding of chondroitin sulfate proteoglycans to hyaluronan. Hapln2 deficiency leads to the abnormal expression of extracellular matrix (ECM) proteins and dysfunctional neuronal conductivity, demonstrating the vital role of Hapln2 in these processes. Studies have revealed that Hapln2 promotes the aggregation of α-synuclein, thereby contributing to neurodegeneration in Parkinson’s disease (PD), and it was recently suggested to be in intracellular neurofibrillary tangles (NFTs). Additionally, the expression levels of Hapln2 showed lower in the anterior temporal lobes of individuals with schizophrenia than those of healthy subjects. Together, these studies implicate the involvement of Hapln2 in the pathological processes of neurological diseases. A better understanding of the function of Hapln2 in the central nervous system (CNS) will provide new insights into the molecular mechanisms of these diseases and help to establish promising therapeutic strategies. Herein, we review the recent progress in defining the role of Hapln2 in brain physiology and pathology.
Introduction
Parkinson’s disease (PD), Alzheimer’s disease (AD) and schizophrenia are neurological diseases characterized by the dysfunction of certain types of neurons (Hardy and Higgins, 1992; Korth, 2012; Ghosh et al., 2016). However, the molecular mechanisms underlying the pathological processes of these brain disorders remain elusive. Accumulating evidence suggests that the pathogenesis of these diseases involve abnormal protein aggregates (Hardy and Higgins, 1992; Korth, 2012; Ghosh et al., 2016). For example, in PD, the formation of Lewy bodies comprising α-synuclein aggregates leads to the degeneration of dopaminergic neurons in the substantia nigra (SN; Ghosh et al., 2016; Wang et al., 2016). The deposition of amyloid beta protein induces neuronal cell death in the development of AD (Hardy and Higgins, 1992). Recently, schizophrenia has been linked with the abnormal deposition of disrupted in schizophrenia 1 (DISC1) aggregates (Atkin and Kittler, 2012; Korth, 2012). Moreover, all of these three diseases have been linked with the dysfunction of the ubiquitin-proteasome pathway (UPP; Lam et al., 2000; Bousman et al., 2010; Shen et al., 2013), which balances protein synthesis with degradation (Tsukamoto and Yokosawa, 2006). Substrates for degradation within this pathway are specified by E3 ubiquitin ligases (Ardley and Robinson, 2005), and we recently demonstrated that the E3 ubiquitin ligases Hrd1, Gp78 and Parkin colocalized with hyaluronan and proteoglycan link protein 2 (Hapln2; Wang et al., 2016).
Studies have shown that Hapln2, also known as brain-derived link protein1 (Bral1), is vital for neuronal conductivity and the formation of the extracellular matrix (ECM), and besides its potential role in the UPP, it has been identified as a contributor to the pathological processes in several neurological disorders. For example, Martins-de-Souza et al. (2009) showed that the expression levels of Hapln2 in the anterior temporal lobe are lower in patients with schizophrenia than those of the control subjects, and Minjarez et al. (2013) suggested that Hapln2 was probably in the neurofibrillary tangles (NFTs) from AD brain. Our studies recently revealed that Hapln2 expression levels were dramatically increased in the SN of PD patients and in the 6-hydroxydopamine-induced rat PD model (Liu et al., 2015; Wang et al., 2016). This review article summarizes recent progress focusing on the roles of Hapln2 in the central nervous system (CNS) under physiological and pathological conditions, highlighting the therapeutic potential of Hapln2 in neurological diseases.
Hapln2 in CNS Physiology
Hapln2 Protein Structure
The structure and roles of Hapln2 in the CNS have been studied since around the turn of this century, when an analysis of the gene now known as HAPLN2 revealed seven exons encoding a polypeptide with an estimated molecular mass of 38 kDa (Hirakawa et al., 2000). The Hapln2 protein comprises three modules, namely, a proteoglycan tandem repeat 1 (PTR1) domain, a PTR2 domain, and an immunoglobulin-like fold (Hirakawa et al., 2000; Spicer et al., 2003). Western blotting results from Hapln2-EGFP transfected cell lysates have shown that an anti-Hapln2 antibody recognizes a band of around 55 kDa, which is in accordance with the predicted molecular mass (Wang et al., 2016). However, a band for Hapln2 of ~48 kDa is detected from SN tissue lysates of adult rats, revealing an inconsistency between the lysates from transfected cells and those from adult brain tissues (Wang et al., 2016). Given that Hapln2 was a kind of link protein, we attributed this discrepancy to hyaluronic acid modifications.
Hapln2 Expression
The hyaluronan and proteoglycan link family of proteins consists of four members: Hapln1, Hapln2, Hapln3, and Hapln4 (Spicer et al., 2003). An amino acid sequence alignment demonstrated that these four proteins have similarities of ~52%–62% (Spicer et al., 2003), with a high level of conservation among vertebrate species. Northern analyses and EST database searches revealed that Hapln2 and Hapln4 are specifically expressed in the brain, with the levels of Hapln2 significantly higher than that of Hapln4 (Spicer et al., 2003). These data indicate that the four-link proteins may have different roles.
Northern blot analyses by Hirakawa et al. (2000) showed that HAPLN2 mRNA is expressed at high levels in the human hippocampus, medulla oblongata, putamen, SN, thalamus, and spinal cord but at lower levels in the cerebellum, cerebral cortex, frontal lobe, and nucleus accumbens. By in situ hybridization, we detected high expression levels of Hapln2 mRNA in the SN, olfactory bulb, red nucleus, cerebellum, brain stem, and hippocampus but relatively weak signals in the cerebral cortex of adult rat brain (Wang et al., 2016). The differential expression of Hapln2 among the various brain regions suggests that the protein has different roles in areas of high expression (SN and hippocampus) than in areas of low expression (cerebral cortex). The discrepancy between these studies regarding the expression in the cerebellum may reflect species specificity and/or the different experimental approaches used.
Hapln2 in ECM Formation and Neuronal Conductivity
Lecticans such as brevican and aggrecan mainly bind to the hyaluronans, which are the major components of the ECM in the brain (Yamaguchi, 2000; Theocharis et al., 2010). The proposed function of the link proteins is to stabilize these binding interactions (Oohashi et al., 2002; Bekku et al., 2003). Northern blot analyses revealed that the expression of Hapln2 largely coincides with that of brevican, a brain-specific lectican (Hirakawa et al., 2000). Previous studies have shown that aggrecan aggregates are denser in the presence of link protein (Mörgelin et al., 1988, 1992; Oohashi et al., 2002). Moreover, immunohistochemical analyses showed that the levels of ECM proteins, such as versican V2, brevican, hyaluronan, and tenascin-R, are much lower in the brain of mice with Hapln2 deficiency (Bekku et al., 2010). These findings not only reveal the proximity of Hapln2 and lecticans in the brain but also indicate the potential role of Hapln2 in stabilizing the binding between brain lecticans and hyaluronan (Hirakawa et al., 2000; Figure 1).

Figure 1. Physiological functions of hyaluronan and proteoglycan link protein 2 (Hapln2) in the central nervous system (CNS).
Northern blot analyses have shown that Hapln2 expression in the mouse brain begins at 20 days after birth and increases with age (Hirakawa et al., 2000). Immunohistochemical analyses revealed that the patterns of Hapln2 and versican V2 expression are similar in the late developmental stage of cerebellar development and in the adult mouse brain (Oohashi et al., 2002). Moreover, Hapln2 colocalizes with versican at myelinated retinal ganglion cell axons (Oohashi et al., 2002). The Ranvier nodes could be marked by the voltage-gated Na+ channels antibody (Oohashi et al., 2002). Interestingly, Hapln2 preferentially localizes with versican at nodes of Ranvier in adult mouse brain, as observed by labeling with antibodies against Hapln2, voltage-gated Na+ channels and the contactin-associated protein (Rasband et al., 1999; Oohashi et al., 2002).
As Na+ channel clustering coincides with myelination (Bekku et al., 2010), it was suggested that Hapln2 may also affect the clustering of the Na+ channels. Of note, the clustering of Na+ and K+ channels is critical for saltatory conduction (Oohashi et al., 2002; Bekku et al., 2010). Although immunohistochemical analyses showed that Hapln2 deletion did not affect the clustering of Na+ and K+ channels, flash visual evoked potentials had a longer latency and smaller amplitude in recordings from Hapln2 knockout mice compared with those from wild-type mice (Bekku et al., 2010). Besides, consistent with the previous results mentioned above, immunostaining analysis demonstrated that there were no signals of ECM proteins including brevican, neurocan and versican at the CNS nodes in adult mice with Hapln2 deletion (Susuki et al., 2013), suggesting the essential role of Hapln2 in the formation of nodal ECM. Additionally, Susuki et al. (2013) showed that single disruption of ECM or paranodal barriers or axonal cytoskeletal scaffolds (CS) had mild effects on the development of nodes, which was consistent with the previous results to some degree (Bekku et al., 2010). However, disruption of the paranodal barriers and ECM, or paranodal barriers and CS, or ECM and CS resulted in juvenile lethality and the robust decrease of clustering of Na+ channels (Susuki et al., 2013), which indicates the complex and complementary roles of the three elements in the formation of nodes. Early studies showed that clustering of nodal proteins such as neurofascin-186 (NF186) was responsible for action potential (AP) propagation (Salzer, 2003; Susuki et al., 2013). Further experiments suggested that there was a specific interaction between Hapln2 and NF186 using pull-down method (Susuki et al., 2013), suggesting the important role of Hapln2 in AP propagation. All of these data indicated the important roles of Hapln2 in clustering of Na+ channel, nodes formation and AP propagation. However, more studies should be performed to clarify the details about the relationship between Hapln2 and the paranodal barriers or the CS in these processes during the development.
As mentioned above, Hapln2 deficiency decreased the expression of ECM-associated proteins (Bekku et al., 2010). Moreover, diffusion-weighted magnetic resonance imaging and real-time iontophoretic assays showed an increase in the diffusion signals from the ECM in white matter in animals with Hapln2 depletion (Bekku et al., 2010). What was more, Hapln2 was essential for nodal ECM formation and interacted with nodal protein NF186 directly (Susuki et al., 2013). Altogether, these results suggest that Hapln2 alters neuronal conductivity by affecting the extracellular diffusion barriers at the nodes of Ranvier rather than by directly impacting saltatory conduction (Figure 1).
Hapln2 in CNS Pathology
Hapln2 in PD
PD is characterized by the formation of Lewy bodies and selective loss of dopamine neurons in the SN (Liu et al., 2015; Wang et al., 2016). Despite the identification of a variety of risk factors including genetic elements, impairments in mitochondrial function and inflammatory responses, the pathogenesis of PD still remains unclear (Shao et al., 2013; Cruces-Sande et al., 2017; Billingsley et al., 2018; Segura-Aguilar and Huenchuguala, 2018). However, clinical analyses and animal experiments implicate the involvement of Hapln2 in PD. In a quantitative proteomics study, we demonstrated that the protein levels of Hapln2 were increased with the highest fold among all the upregulated proteins in the SN region of PD patients compared with the control subjects (Liu et al., 2015). This increase in the SN was confirmed both at the mRNA and protein levels by quantitative PCR and Western blotting analyses in a rat model of PD 2 weeks after 6-hydroxydopamine treatment as well as 4 weeks postlesion (Wang et al., 2016).
Flow cytometry and immunostaining analyses showed that the overexpression of Hapln2 increased the death of MES23.5 and primary neuronal cells (Wang et al., 2016). Confocal imaging of these cells demonstrated that the overexpressed Hapln2 formed aggregates, which were found to colocalize with E3 ubiquitin ligases Hrd1, Gp78, and Parkin (Wang et al., 2016). It is known that E3 ubiquitin ligases involve in specifying and catalyzing the transfer of substrates in the UPP (Uchida and Kitagawa, 2016). Moreover, the treatment of MES23.5 cells with the proteasome inhibitor MG132 increased Hapln2 levels, indicating that the UPP pathway regulates Hapln2 degradation (Wang et al., 2016). Of note, previous research showed that UPP dysfunction via proteasome inhibition resulted in the degeneration of dopaminergic neurons in vitro and in vivo (Kikuchi et al., 2003; Sun et al., 2006). These and the more recent results suggest that the increase in Hapln2 as a result of UPP dysfunction contributes to PD pathology.
Immunostaining analysis of MES23.5 cells overexpressing Hapln2 also showed an accumulation of α-synuclein, aggregates of which were found to colocalize with Hapln2 (Wang et al., 2016). Thus, Hapln2 may be another component of Lewy bodies in PD, which primarily comprise α-synuclein (Castro-Sánchez et al., 2018). The accumulation of α-synuclein not only promotes the death of dopaminergic neurons but also activates glial cells (La Vitola et al., 2018; Martinez et al., 2018). Mice overexpressing wild-type α-synuclein exhibit marked microglial activation (Su et al., 2008). Pathological α-synuclein aggregates activate astrocytes (Yu et al., 2018). Moreover, knockout of Hapln2 significantly reduces the fraction of α-synuclein that is insoluble in extracts from SN and cerebellum of 6-month-old mouse brain (Wang et al., 2016). These findings suggest that Hapln2 may contribute to neurodegeneration by regulating the activation of glial cells in the brain (Figure 2).
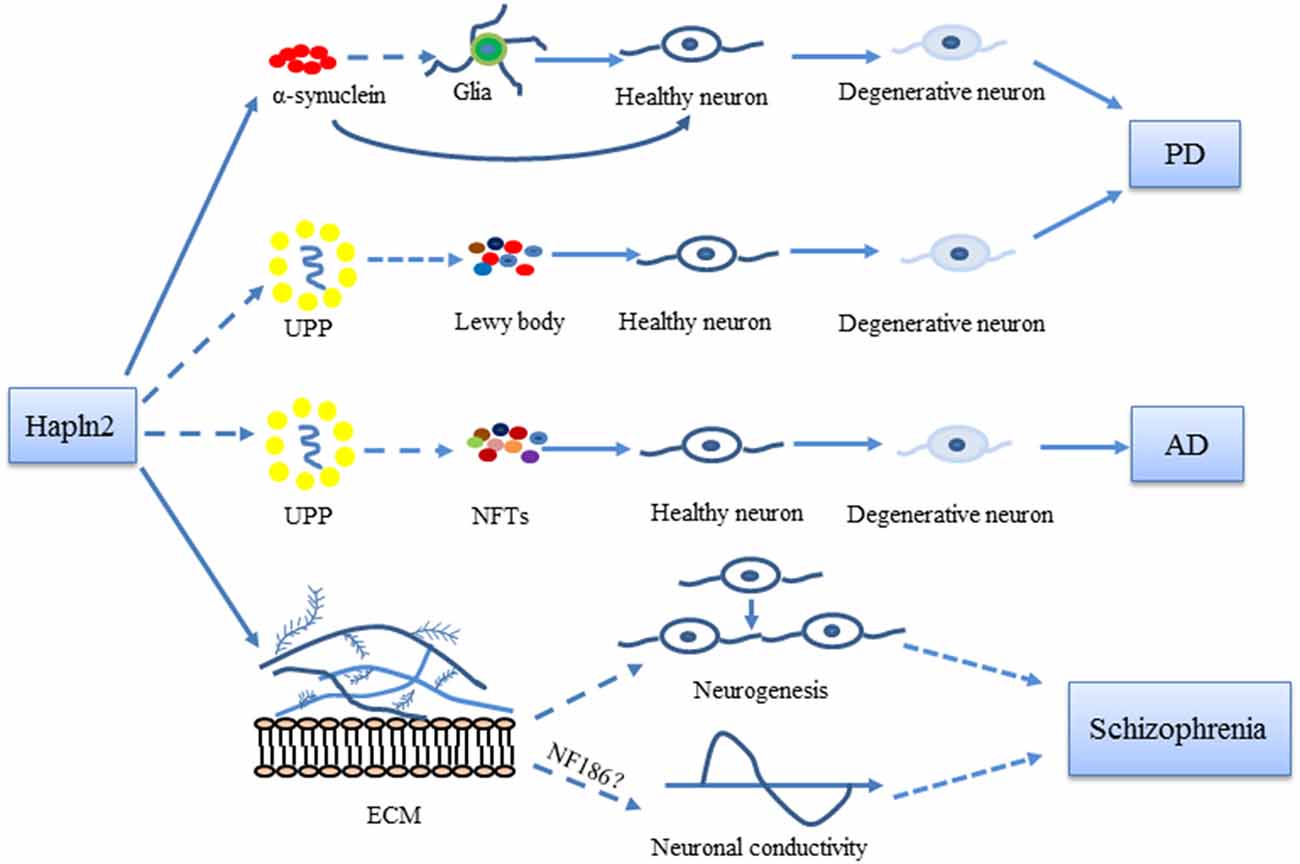
Figure 2. Possible roles of Hapln2 in Parkinson’s disease (PD), Alzheimer’s disease (AD), and schizophrenia. An upregulation of Hapln2 is observed in the substantia nigra (SN) of PD brain. The high expression levels of Hapln2 promote the aggregation of α-synuclein, which contributes to the degeneration of dopamine neurons and the pathophysiology of PD. Hapln2 has also been suggested in neurofibrillary tangles (NFTs). There is evidence showing the vital role of extracellular matrix (ECM) proteins during neurogenesis and neuronal conductivity, which become dysfunctional in schizophrenia. As Hapln2 helps maintain the ECM, it may also play a role in schizophrenia. Hapln2 colocalizes with several E3 ligases of the ubiquitin-proteasome pathway (UPP). The influence of Hapln2 on the ECM and UPP may contribute to PD, AD, and schizophrenia, neurological diseases in which ECM and UPP dysfunction have been observed.
Hapln2 in AD
A variety of environmental and genetic factors have also been implicated in the pathology of AD (Chin-Chan et al., 2015; Kikis, 2017; Kocahan and Doğan, 2017). However, due to the complexity of this disease, the underlying molecular mechanism remains largely unknown. Although a major hallmark of AD is the accumulation of intracellular NFTs (Manczak et al., 2018), their insolubility impedes the identification of their integral components (Perry et al., 1991; Minjarez et al., 2013). Nevertheless, Benito and colleagues used different solubilization methods to identify lots of polypeptides, including Hapln2, in total homogenates of AD brain tissue containing NFTs by tandem mass spectrometry (Minjarez et al., 2013; Sugawara et al., 2018). However, there was no direct evidence for a colocalization of Hapln2 with tau, the main component of NFTs.
Notably, both AD and PD are age-associated neurodegenerative diseases with characteristic protein aggregates (Bridi and Hirth, 2018; Daniele et al., 2018; Theofilas et al., 2018). As mentioned above, the overexpression of Hapln2 promotes the aggregate formation and is involved with neuron death and the UPP in the pathology of PD (Wang et al., 2016). As Hapln2 was found to colocalize with some E3 ligases (Wang et al., 2016), it may also contribute to AD pathology via disruptions of the UPP. Postmortem tissue samples from the hippocampus, superior and middle temporal gyri, parahippocampal gyri, and the inferior parietal lobes of AD patients exhibit signs of reduced proteasome activity compared with those from control subjects (Keller et al., 2000). However, further animal experiments and clinical studies are needed to determine whether Hapln2 is a major constituent of the aggregates in these diseases.
Although high levels of Hapln2 in the hippocampus were measured by in situ hybridization (Wang et al., 2016), the function of Hapln2 in this brain region remains unknown. The hippocampus is involved in learning and memory (Portero-Tresserra et al., 2018; Wang D. et al., 2018), the dysfunction of which is a major symptom of AD (Axelrud et al., 2018). Behavioral tests in a mouse model of AD revealed a significant impairment in contextual conditioning and performance in pattern separation tests after neurogenesis in the hippocampus was inhibited (Hollands et al., 2017). Moreover, the performance of AD transgenic mice in spatial learning and memory tasks improved when hippocampal neurogenesis was enhanced by deep brain magnetic stimulation (Zhen et al., 2017). Of note, the fate determination of neural stem cells is guided by ECM components, such as the heparan sulfate proteoglycans glypican and perlecan (Yu et al., 2017). Given the vital role of Hapln2 in maintaining the ECM scaffold (Bekku et al., 2010), whether Hapln2 is involved in AD pathology by regulating hippocampal neurogenesis needs further investigation.
Hapln2 in Schizophrenia
Although the pathophysiology of schizophrenia has not been fully defined, genetic factors, epigenetic elements, and abnormal neurotransmission in the hippocampus are important contributors (Kim et al., 2018; Sugawara et al., 2018; Wang H. Y. et al., 2018). Previous studies of postmortem brain tissues from patients with schizophrenia revealed the presence of insoluble aggregates containing DISC1 (Atkin and Kittler, 2012; Korth, 2012). The overexpression of DISC in neuroblastoma cells leads to the formation of aggresomal deposits (Korth, 2012), which can invade cells and recruit other proteins that may contribute to the impairment of neuronal cells (Atkin and Kittler, 2012; Korth, 2012). Hapln2 represents another potential contributor, as a shotgun proteomic analysis of postmortem anterior temporal lobe tissues showed that Hapln2 protein levels are lower in schizophrenia patients than in control subjects (Martins-de-Souza et al., 2009). Additionally, a convergent pathway analysis indicated that there is dysregulation of the UPP in schizophrenia (Bousman et al., 2010). As mentioned above, the UPP is vital for protein catabolism via the degradation of particular substrate proteins (Tsukamoto and Yokosawa, 2006). The colocalization of Hapln2 with E3 ligases revealed in our previous work suggests a potential function of Hapln2 in the UPP (Wang et al., 2016), which suggesting that Hapln2 may also contribute to the pathophysiology of schizophrenia through dysfunction of UPP.
Moreover, it has been reported that ECM plays important roles in regulating neurogenesis, axonal outgrowth, synaptogenesis and cell migration (Bandtlow and Zimmermann, 2000; Faissner et al., 2010; Gundelfinger et al., 2010). The analysis of the schizophrenia risk genes showed that most of the genes were involved in the regulation of neuronal migration and cell adhesion (O’Dushlaine et al., 2011; Lips et al., 2012; Aberg et al., 2013). Additionally, the disruption of conduction velocity has been suggested in patients of schizophrenia (Thaker, 2008; Takahashi et al., 2011). The study of schizophrenia animal models indicated the reduction of conduction velocity, indicating the important role of nerve conduction in schizophrenia (Roy et al., 2007; Tanaka et al., 2009; Takahashi et al., 2011). As one of the important components of ECM, Hapln2 may be involved in the pathogenesis of schizophrenia through regulating the neuronal migration and velocity of nerve conduction. More studies should focus on the molecular mechanism of Hapln2 in the pathogenesis of schizophrenia using animal models and clinical analysis.
Conclusions
As a brain specific-hyaluronan and proteoglycan link protein, Hapln2 plays vital physiological roles in the formation and maintenance of the ECM scaffold. Studies in knockout mice have also revealed that Hapln2 influences neuronal conductivity. Several studies have revealed the involvement of Hapln2 in the pathogenesis of neurological diseases including PD, AD and schizophrenia, which provided new insights into the underlying molecular mechanisms of brain disorders (Figure 2).
Current research suggested that Hapln2 was involved in the formation of α-synuclein aggregates in PD pathology and study in AD suggested that Hapln2 might be the component of NFTs aggregates (Minjarez et al., 2013; Wang et al., 2016). It is noteworthy that, besides PD, AD and Schizophrenia, dysfunction of UPP system has also been implicated in some other neurological diseases including Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS; Zhao et al., 2016; Desai et al., 2018; Harding and Tong, 2018; Thibaudeau et al., 2018). Thus, it is likely that Hapln2 may also be involved in the pathological processes of these diseases through the UPP system. However, further studies of animal and clinical experiments are needed to clarify these mechanisms and the precise role that Hapln2 plays in these neurological disorders in the future. Nevertheless, the restriction of Hapln2 expression to the brain suggests that it is an important contributor.
Author Contributions
QW, CW, BJ, and CY wrote the manuscript. JC and JZ edited the manuscript. All the authors approved the final manuscript.
Funding
This work was funded by the Supporting Fund for Teachers’ research of Jining Medical University (JY2017KJ036), the Research Fund for the Doctoral Program of Jining Medical University, a Project of Shandong Province Higher Educational Science and Technology Program (J18KA124), Research Fund for Lin He’s Academician Workstation of New Medicine and Clinical Translation in Jining Medical University (JYHL2018MS03), the National Nature Science Foundation of China (81501018), the Natural Science Foundation of Shandong Province (ZR2018MC005) and Science and Technology plan project of Shandong higher education institutions (J15LE19).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aberg, K. A., Liu, Y., Bukszár, J., McClay, J. L., Khachane, A. N., Andreassen, O. A., et al. (2013). A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry 70, 573–581. doi: 10.1001/jamapsychiatry.2013.288
Ardley, H. C., and Robinson, P. A. (2005). E3 ubiquitin ligases. Essays Biochem. 41, 15–30. doi: 10.1042/bse0410015
Atkin, T., and Kittler, J. (2012). DISC1 and the aggresome: a disruption to cellular function? Autophagy 8, 851–852. doi: 10.4161/auto.19979
Axelrud, L. K., Santoro, M. L., Pine, D. S., Talarico, F., Gadelha, A., Manfro, G. G., et al. (2018). Polygenic risk score for Alzheimer’s disease: implications for memory performance and hippocampal volumes in early life. Am. J. Psychiatry 175, 555–563. doi: 10.1176/appi.ajp.2017.17050529
Bandtlow, C. E., and Zimmermann, D. R. (2000). Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol. Rev. 80, 1267–1290. doi: 10.1152/physrev.2000.80.4.1267
Bekku, Y., Su, W. D., Hirakawa, S., Fassler, R., Ohtsuka, A., Kang, J. S., et al. (2003). Molecular cloning of Bral2, a novel brain-specific link protein and immunohistochemical colocalization with brevican in perineuronal nets. Mol. Cell. Neurosci. 24, 148–159. doi: 10.1016/s1044-7431(03)00133-7
Bekku, Y., Vargova, L., Goto, Y., Vorisek, I., Dmytrenko, L., Narasaki, M., et al. (2010). Bral1: its role in diffusion barrier formation and conduction velocity in the CNS. J. Neurosci. 30, 3113–3123. doi: 10.1523/JNEUROSCI.5598-09.2010
Billingsley, K. J., Bandres-Ciga, S., Saez-Atienzar, S., and Singleton, A. B. (2018). Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 373, 9–20. doi: 10.1007/s00441-018-2817-y
Bousman, C. A., Chana, G., Glatt, S. J., Chandler, S. D., Lucero, G. R., Tatro, E., et al. (2010). Preliminary evidence of ubiquitin proteasome system dysregulation in schizophrenia and bipolar disorder: convergent pathway analysis findings from two independent samples. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 494–502. doi: 10.1002/ajmg.b.31006
Bridi, J. C., and Hirth, F. (2018). Mechanisms of α-synuclein induced synaptopathy in Parkinson’s disease. Front. Neurosci. 12:80. doi: 10.3389/fnins.2018.00080
Castro-Sánchez, S., García-Yagüe, Á. J., López-Royo, T., Casarejos, M., Lanciego, J. L., and Lastres-Becker, I. (2018). Cx3cr1-deficiency exacerbates α-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 66, 1752–1762. doi: 10.1002/glia.23338
Chin-Chan, M., Navarro-Yepes, J., and Quintanilla-Vega, B. (2015). Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 9:124. doi: 10.3389/fncel.2015.00124
Cruces-Sande, A., Méndez-Álvarez, E., and Soto-Otero, R. (2017). Copper increases the ability of 6-hydroxydopamine to generate oxidative stress and the ability of ascorbate and glutathione to potentiate this effect: potential implications in Parkinson’s disease. J. Neurochem. 141, 738–749. doi: 10.1111/jnc.14019
Daniele, S., Frosini, D., Pietrobono, D., Petrozzi, L., Lo Gerfo, A., Baldacci, F., et al. (2018). α-synuclein heterocomplexes with β-amyloid are increased in red blood cells of Parkinson’s disease patients and correlate with disease severity. Front. Mol. Neurosci. 11:53. doi: 10.3389/fnmol.2018.00053
Desai, S., Juncker, M., and Kim, C. (2018). Regulation of mitophagy by the ubiquitin pathway in neurodegenerative diseases. Exp. Biol. Med. 243, 554–562. doi: 10.1177/1535370217752351
Faissner, A., Pyka, M., Geissler, M., Sobik, T., Frischknecht, R., Gundelfinger, E. D., et al. (2010). Contributions of astrocytes to synapse formation and maturation—potential functions of the perisynaptic extracellular matrix. Brain Res. Rev. 63, 26–38. doi: 10.1016/j.brainresrev.2010.01.001
Ghosh, A., Tyson, T., George, S., Hildebrandt, E. N., Steiner, J. A., Madaj, Z., et al. (2016). Mitochondrial pyruvate carrier regulates autophagy, inflammation and neurodegeneration in experimental models of Parkinson’s disease. Sci. Transl. Med. 8:368ra174. doi: 10.1126/scitranslmed.aag2210
Gundelfinger, E. D., Frischknecht, R., Choquet, D., and Heine, M. (2010). Converting juvenile into adult plasticity: a role for the brain’s extracellular matrix. Eur. J. Neurosci. 31, 2156–2165. doi: 10.1111/j.1460-9568.2010.07253.x
Harding, R. J., and Tong, Y. F. (2018). Proteostasis in Huntington’s disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 39, 754–769. doi: 10.1038/aps.2018.11
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hirakawa, S., Oohashi, T., Su, W. D., Yoshioka, H., Murakami, T., Arata, J., et al. (2000). The brain link protein-1 (BRAL1): cDNA cloning, genomic structure, and characterization as a novel link protein expressed in adult brain. Biochem. Biophys. Res. Commun. 276, 982–989. doi: 10.1006/bbrc.2000.3583
Hollands, C., Tobin, M. K., Hsu, M., Musaraca, K., Yu, T. S., Mishra, R., et al. (2017). Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol. Neurodegener. 12:64. doi: 10.1186/s13024-017-0207-7
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 75, 436–439. doi: 10.1046/j.1471-4159.2000.0750436.x
Kikis, E. A. (2017). Nature versus nurture: does proteostasis imbalance underlie the genetic, environmental, and age-related risk factors for Alzheimer’s disease? Healthcare 5:E46. doi: 10.3390/healthcare5030046
Kikuchi, S., Shinpo, K., Tsuji, S., Takeuchi, M., Yamagishi, S., Makita, Z., et al. (2003). Effect of proteasome inhibitor on cultured mesencephalic dopaminergic neurons. Brain Res. 964, 228–236. doi: 10.1016/s0006-8993(02)04030-1
Kim, P., Scott, M. R., and Meador-Woodruff, J. H. (2018). Abnormal expression of ER quality control and ER associated degradation proteins in the dorsolateral prefrontal cortex in schizophrenia. Schizophr. Res. 197, 484–491. doi: 10.1016/j.schres.2018.02.010
Kocahan, S., and Doğan, Z. (2017). Mechanisms of Alzheimer’s disease pathogenesis and prevention: the brain, neural pathology, N-methyl-D-aspartate receptors, tau protein and other risk factors. Clin. Psychopharmacol. Neurosci. 15, 1–8. doi: 10.9758/cpn.2017.15.1.1
Korth, C. (2012). Aggregated proteins in schizophrenia and other chronic mental diseases: DISC1opathies. Prion 6, 134–141. doi: 10.4161/pri.18989
La Vitola, P., Balducci, C., Cerovic, M., Santamaria, G., Brandi, E., Grandi, F., et al. (2018). α-synuclein oligomers impair memory through glial cell activation and via Toll-like receptor 2. Brain Behav. Immun. 69, 591–602. doi: 10.1016/j.bbi.2018.02.012
Lam, Y. A., Pickart, C. M., Alban, A., Landon, M., Jamieson, C., Ramage, R., et al. (2000). Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 9902–9906. doi: 10.1073/pnas.170173897
Lips, E. S., Cornelisse, L. N., Toonen, R. F., Min, J. L., Hultman, C. M., Holmans, P. A., et al. (2012). Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol. Psychiatry 17, 996–1006. doi: 10.1038/mp.2011.117
Liu, Y., Zhou, Q., Tang, M., Fu, N., Shao, W., Zhang, S., et al. (2015). Upregulation of αB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol. Aging 36, 1686–1691. doi: 10.1016/j.neurobiolaging.2015.01.015
Manczak, M., Kandimalla, R., Yin, X., and Reddy, P. H. (2018). Hippocampal mutant app and amyloid β induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 1332–1342. doi: 10.1093/hmg/ddy042
Martinez, J. H., Alaimo, A., Gorojod, R. M., Porte Alcon, S., Fuentes, F., Coluccio Leskow, F., et al. (2018). Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing α-synuclein. Mol. Cell. Neurosci. 88, 107–117. doi: 10.1016/j.mcn.2018.01.004
Martins-de-Souza, D., Gattaz, W. F., Schmitt, A., Rewerts, C., Marangoni, S., Novello, J. C., et al. (2009). Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J. Neural Transm. 116, 275–289. doi: 10.1007/s00702-008-0156-y
Minjarez, B., Valero Rustarazo, M. L., Sanchez del Pino, M. M., González-Robles, A., Sosa-Melgarejo, J. A., Luna-Muñoz, J., et al. (2013). Identification of polypeptides in neurofibrillary tangles and total homogenates of brains with Alzheimer’s disease by tandem mass spectrometry. J. Alzheimers Dis. 34, 239–262. doi: 10.3233/jad-121480
Mörgelin, M., Engel, J., Heinegård, D., and Paulsson, M. (1992). Proteoglycans from the swarm rat chondrosarcoma. Structure of the aggregates extracted with associative and dissociative solvents as revealed by electron microscopy. J. Biol. Chem. 267, 14275–14284.
Mörgelin, M., Paulsson, M., Hardingham, T. E., Heinegård, D., and Engel, J. (1988). Cartilage proteoglycans. Assembly with hyaluronate and link protein as studied by electron microscopy. Biochem. J. 253, 175–185. doi: 10.1042/bj2530175
O’Dushlaine, C., Kenny, E., Heron, E., Donohoe, G., Gill, M., Morris, D., et al. (2011). Molecular pathways involved in neuronal cell adhesion and membrane scaffolding contribute to schizophrenia and bipolar disorder susceptibility. Mol. Psychiatry 16, 286–292. doi: 10.1038/mp.2010.7
Oohashi, T., Hirakawa, S., Bekku, Y., Rauch, U., Zimmermann, D. R., Su, W. D., et al. (2002). Bral1, a brain-specific link protein, colocalizing with the versican V2 isoform at the nodes of Ranvier in developing and adult mouse central nervous systems. Mol. Cell. Neurosci. 19, 43–57. doi: 10.1006/mcne.2001.1061
Perry, G., Siedlak, S. L., Richey, P., Kawai, M., Cras, P., Kalaria, R. N., et al. (1991). Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 11, 3679–3683. doi: 10.1523/jneurosci.11-11-03679.1991
Portero-Tresserra, M., Marti-Nicolovius, M., Tarres-Gatius, M., Candalija, A., Guillazo-Blanch, G., and Vale-Martinez, A. (2018). Intra-hippocampal D-cycloserine rescues decreased social memory, spatial learning reversal and synaptophysin levels in aged rats. Psychopharmacology 235, 1463–1477. doi: 10.1007/s00213-018-4858-z
Rasband, M. N., Peles, E., Trimmer, J. S., Levinson, S. R., Lux, S. E., and Shrager, P. (1999). Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J. Neurosci. 19, 7516–7528. doi: 10.1523/jneurosci.19-17-07516.1999
Roy, K., Murtie, J. C., El-Khodor, B. F., Edgar, N., Sardi, S. P., Hooks, B. M., et al. (2007). Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. U S A 104, 8131–8136. doi: 10.1073/pnas.0702157104
Salzer, J. L. (2003). Polarized domains of myelinated axons. Neuron 40, 297–318. doi: 10.1016/s0896-6273(03)00628-7
Segura-Aguilar, J., and Huenchuguala, S. (2018). Aminochrome induces irreversible mitochondrial dysfunction by inducing autophagy dysfunction in Parkinson’s disease. Front. Neurosci. 12:106. doi: 10.3389/fnins.2018.00106
Shao, W., Zhang, S. Z., Tang, M., Zhang, X. H., Zhou, Z., Yin, Y. Q., et al. (2013). Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature 494, 90–94. doi: 10.1038/nature11748
Shen, Y. F., Tang, Y., Zhang, X. J., Huang, K. X., and Le, W. D. (2013). Adaptive changes in autophagy after UPS impairment in Parkinson’s disease. Acta Pharmacol. Sin. 34, 667–673. doi: 10.1038/aps.2012.203
Spicer, A. P., Joo, A., and Bowling, R. A. Jr. (2003). A hyaluronan binding link protein gene family whose members are physically linked adjacent to chondroitin sulfate proteoglycan core protein genes: the missing links. J. Biol. Chem. 278, 21083–21091. doi: 10.1074/jbc.m213100200
Su, X., Maguire-Zeiss, K. A., Giuliano, R., Prifti, L., Venkatesh, K., and Federoff, H. J. (2008). Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 29, 1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006
Sugawara, H., Murata, Y., Ikegame, T., Sawamura, R., Shimanaga, S., Takeoka, Y., et al. (2018). DNA methylation analyses of the candidate genes identified by a methylome-wide association study revealed common epigenetic alterations in schizophrenia and bipolar disorder. Psychiatry Clin. Neurosci. 72, 245–254. doi: 10.1111/pcn.12645
Sun, F., Anantharam, V., Zhang, D., Latchoumycandane, C., Kanthasamy, A., and Kanthasamy, A. G. (2006). Proteasome inhibitor MG-132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology 27, 807–815. doi: 10.1016/j.neuro.2006.06.006
Susuki, K., Chang, K. J., Zollinger, D. R., Liu, Y., Ogawa, Y., Eshed-Eisenbach, Y., et al. (2013). Three mechanisms assemble central nervous system nodes of Ranvier. Neuron 78, 469–482. doi: 10.1016/j.neuron.2013.03.005
Takahashi, N., Sakurai, T., Davis, K. L., and Buxbaum, J. D. (2011). Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 93, 13–24. doi: 10.1016/j.pneurobio.2010.09.004
Tanaka, H., Ma, J., Tanaka, K. F., Takao, K., Komada, M., Tanda, K., et al. (2009). Mice with altered myelin proteolipid protein gene expression display cognitive deficits accompanied by abnormal neuron-glia interactions and decreased conduction velocities. J. Neurosci. 29, 8363–8371. doi: 10.1523/jneurosci.3216-08.2009
Thaker, G. K. (2008). Neurophysiological endophenotypes across bipolar and schizophrenia psychosis. Schizophr. Bull. 34, 760–773. doi: 10.1093/schbul/sbn049
Theocharis, A. D., Skandalis, S. S., Tzanakakis, G. N., and Karamanos, N. K. (2010). Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277, 3904–3923. doi: 10.1111/j.1742-4658.2010.07800.x
Theofilas, P., Ehrenberg, A. J., Nguy, A., Thackrey, J. M., Dunlop, S., Mejia, M. B., et al. (2018). Probing the correlation of neuronal loss, neurofibrillary tangles and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol. Aging 61, 1–12. doi: 10.1016/j.neurobiolaging.2017.09.007
Thibaudeau, T. A., Anderson, R. T., and Smith, D. M. (2018). A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 9:1097. doi: 10.1038/s41467-018-03509-0
Tsukamoto, S., and Yokosawa, H. (2006). Natural products inhibiting the ubiquitin-proteasome proteolytic pathway, a target for drug development. Curr. Med. Chem. 13, 745–754. doi: 10.2174/092986706776055571
Uchida, C., and Kitagawa, M. (2016). RING-, HECT-, and RBR-type E3 ubiquitin ligases: involvement in human cancer. Curr. Cancer Drug Targets 16, 157–174. doi: 10.2174/1568009616666151112122801
Wang, D., Wang, X., Liu, X., Jiang, L., Yang, G., Shi, X., et al. (2018). Inhibition of miR-219 alleviates arsenic-induced learning and memory impairments and synaptic damage through up-regulating camkii in the hippocampus. Neurochem. Res. 43, 948–958. doi: 10.1007/s11064-018-2500-4
Wang, H. Y., Liu, Y., Yan, J. W., Hu, X. L., Zhu, D. M., Xu, X. T., et al. (2018). Gene polymorphisms of DISC1 is associated with schizophrenia: evidence from a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 81, 64–73. doi: 10.1016/j.pnpbp.2017.10.008
Wang, Q., Zhou, Q., Zhang, S., Shao, W., Yin, Y., Li, Y., et al. (2016). Elevated Hapln2 expression contributes to protein aggregation and neurodegeneration in an animal model of Parkinson’s disease. Front. Aging Neurosci. 8:197. doi: 10.3389/fnagi.2016.00197
Yamaguchi, Y. (2000). Lecticans: organizers of the brain extracellular matrix. Cell. Mol. Life Sci. 57, 276–289. doi: 10.1007/pl00000690
Yu, W. W., Cao, S. N., Zang, C. X., Wang, L., Yang, H. Y., Bao, X. Q., et al. (2018). Heat shock protein 70 suppresses neuroinflammation induced by α-synuclein in astrocytes. Mol. Cell. Neurosci. 86, 58–64. doi: 10.1016/j.mcn.2017.11.013
Yu, C., Griffiths, L. R., and Haupt, L. M. (2017). Exploiting heparan sulfate proteoglycans in human neurogenesis-controlling lineage specification and fate. Front. Integr. Neurosci. 11:28. doi: 10.3389/fnint.2017.00028
Zhao, T., Hong, Y., Li, X. J., and Li, S. H. (2016). Subcellular clearance and accumulation of Huntington disease protein: a mini-review. Front. Mol. Neurosci. 9:27. doi: 10.3389/fnmol.2016.00027
Keywords: Hapln2, aggregates, Parkinson’s disease, Alzheimer’s disease, schizophrenia
Citation: Wang Q, Wang C, Ji B, Zhou J, Yang C and Chen J (2019) Hapln2 in Neurological Diseases and Its Potential as Therapeutic Target. Front. Aging Neurosci. 11:60. doi: 10.3389/fnagi.2019.00060
Received: 02 September 2018; Accepted: 01 March 2019;
Published: 21 March 2019.
Edited by:
Anne Eckert, University Psychiatric Clinic Basel, SwitzerlandReviewed by:
Toshitaka Oohashi, Okayama University, JapanJin Xu, Institute of Neuroscience, Shanghai Institutes for Biological Sciences (CAS), China
Copyright © 2019 Wang, Wang, Ji, Zhou, Yang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Chen, amluZy5jaGVuQHdhcndpY2suYWMudWs=