 Maria Pia Adorni
Maria Pia Adorni Massimiliano Ruscica
Massimiliano Ruscica Nicola Ferri
Nicola Ferri Franco Bernini1
Franco Bernini1 Francesca Zimetti
Francesca Zimetti- 1Dipartimento di Scienze degli Alimenti e del Farmaco, Università di Parma, Parma, Italy
- 2Dipartimento di Scienze Farmacologiche e Biomolecolari, Università degli Studi di Milano, Milan, Italy
- 3Dipartimento di Scienze del Farmaco, Università degli Studi di Padova, Padova, Italy
Alzheimer’s disease (AD) has been associated with dysregulation of brain cholesterol homeostasis. Proprotein convertase subtilisin/kexin type 9 (PCSK9), beyond the known role in the regulation of plasma low-density lipoprotein cholesterol, was first identified in the brain with a potential involvement in brain development and apoptosis. However, its role in the central nervous system (CNS) and in AD pathogenesis is still far from being understood. While in vitro and in vivo evidence led to controversial results, genetic studies apparently did not find an association between PCSK9 loss of function mutations and AD risk or prevalence. In addition, a potential impairment of cognitive performances by the treatment with the PCSK9 inhibitors, alirocumab and evolocumab, have been excluded, although ongoing studies with longer follow-up will provide further insights. PCSK9 is able to affect the expression of neuronal receptors involved in cholesterol homeostasis and neuroinflammation, and higher PCSK9 concentrations have been found in the cerebrospinal fluid (CSF) of AD patients. In this review article, we critically examined the science of PCSK9 with respect to its modulatory role of the mechanisms underlying the pathogenesis of AD. In addition, based on literature data, we made the hypothesis to consider brain PCSK9 as a negative modulator of brain cholesterol homeostasis and neuroinflammation and a potential pharmacological target for treatment.
Introduction
The Proprotein convertase subtilisin/kexin type 9 (PCSK9), acts as one of the major regulators of cholesterol homeostasis, by mediating the degradation of hepatic low density lipoprotein receptors (LDLr) (Macchi et al., 2019). Interestingly, PCSK9 was firstly identified in the brain where its expression in primary embryonic telencephalon cells was maximal between embryonic days 13–15, a gestational period characterized by intense neurogenesis (Seidah et al., 2003). In zebrafish, but not in mice, specific knockdown of PCSK9 mRNA led to a general disorganization of cerebellar neurons and loss of hindbrain-midbrain boundaries, with an end result of embryonic death (Poirier et al., 2006).
In this review article, we will focus on the role of PCSK9 at the cerebral level with particular attention on its potential involvement in neuronal functions and Alzheimer’s disease (AD) pathogenesis. We also critically examined the possibility to consider PCSK9 as a modulator of brain cholesterol homeostasis and inflammation and a potential pharmacological target for neurodegenerative disorders.
Brain Cholesterol Homeostasis
Cholesterol is one of the most important molecules in brain physiology (Chang et al., 2017): it is an important component of myelin, it is involved in neuronal development, synaptogenesis, outgrowth of neuritis, maintenance and repair of damaged membranes (Dietschy, 2009). Due to the presence of the blood-brain barrier (BBB), the brain relies on in situ local cholesterol synthesis (Björkhem and Meaney, 2004). In fact, cholesterol cannot cross the BBB, unlike its side-chain oxidized metabolites, 24S-hydroxycholesterol and 27-hydroxycholesterol (Björkhem et al., 2019). Central nervous system (CNS) cells are able to synthesize cholesterol but adult neurons progressively loose this capacity and become dependent on cholesterol provided from astrocytes (Dietschy and Turley, 2004; Saito et al., 2009). Depletion of neuronal cholesterol leads to excess tau phosphorylation, changes in β-amyloid (Aβ) peptides metabolism, neural oxidative stress reactions, ultimately resulting in neurodegeneration, as demonstrated in ex vivo rat hippocampus slices (Koudinov and Koudinova, 2005). The transport of cholesterol from astrocytes to neurons is warranted by peculiar molecules and receptors that cooperate in a coordinated manner. Cholesterol produced from astrocytes undergoes cholesterol efflux to Apolipoprotein E (ApoE)-containing particles through the activity of transporters, such as the ATP binding cassette transporters A1 (ABCA1), G1 (ABCG1) and G4 (ABCG4) (Chen et al., 2013). Subsequently, cholesterol transported by such particles, that resemble plasma HDL in composition and size, is finally incorporated into neurons by the particles binding to specific receptors, such as the LDL receptor (LDLr), the LDL receptor-related protein 1 (LRP1), the VLDL receptor (VLDLr) and the ApoE receptor 2 (ApoEr2) (Bu, 2009). Concerning the latter two, PCSK9 increases their degradation (Poirier et al., 2008; Canuel et al., 2013), implying PCSK9 in cerebral cholesterol homeostasis. This hypothesis is strengthened by in vivo findings showing that LDLr expression is reduced by PCSK9 during brain development and after transient ischemic stroke (Rousselet et al., 2011). It is therefore conceivable that the degrading activity of PCSK9 on lipoprotein receptors listed above may translate in a reduced cholesterol uptake by neurons, with potential deleterious consequences (Koudinov and Koudinova, 2005). However, not all data are consistent with this hypothesis. Liu et al. (2010) found that PCSK9 did not affect the expression of LDLR, VLDLR and apoEr2 in the mouse brain (Liu et al., 2010). These discrepancies highlight the need for further studies to dissect out the involvement of PCSK9 on brain cholesterol homeostasis.
Pcsk9 and Alzheimer’S Disease Pathogenesis
Alterations of CNS cholesterol homeostasis are associated with various neurodegenerative disorders, including AD (Sato and Morishita, 2015; Arenas et al., 2017). Genomic-wide association (GWAS) studies have identified several loci involved in lipid metabolism among AD susceptible genes (Lambert et al., 2013; Dong et al., 2017). A striking example of this association is the ε4 allele of the APOE gene encoding ApoE, the main apolipoprotein mediating the transport of cholesterol in the CNS (Mahoney-Sanchez et al., 2016). The E4 isoform is undoubtedly one of the most predictive factors for AD onset (Liu et al., 2013). However, recent studies have identified other genes involved in lipid metabolism, such as BIN1, CLU, PICALM, ABCA7, ABCA1, ABCG1 and SORL1 (Dong et al., 2017; Picard et al., 2018). From a molecular point of view, the cerebral cholesterol accumulates in lipid rafts, membrane microdomains where the processing of the amyloid precursor protein (APP; Picard et al., 2018) occurs, leading to deposition of insoluble fragments of Aβ in brain parenchyma. At this regards, it has been found that cholesterol promotes amyloidogenesis by providing structural stability to membrane-adjacent lipid rafts (Vetrivel and Thinakaran, 2010). Consequently, modulation of cholesterol content in lipid rafts is able to affect deposition of Aβ.
The few and controversial data on PCSK9 and AD are summarized in Table 1. Concerning neuronal apoptosis, a pro-apoptotic activity of PCSK9 may occur through the upregulation of caspases or the reduction of the ApoEr2 levels (Wu et al., 2014). In APOE(−/−) mice fed with a high-fat diet, the hippocampal neuronal apoptosis was associated with an increase of PCSK9 expression (Zhao et al., 2017). Consistently, silencing of PCSK9 attenuates the neuronal apoptosis induced by cerebral ischemia reducing brain damage in mice (Wang et al., 2018). Conversely, a preventive action of PCSK9 on neuronal apoptosis may occur through the decrease in Aβ generation (Wu et al., 2014). In addition, the direct effect of PCSK9 on Aβ processing is still unresolved. In its absence, mice show increased expression of the β-site amyloid precursor protein-cleaving enzyme 1 (BACE1), the protease producing toxic Aβ that accumulates in neuritic plaques of AD brains. This effect translates in an increased total Aβ brain deposition: PCSK9 overexpression in mice reduced BACE1 levels (Jonas et al., 2008). On the other hand, in brain-damaged rats, the administration of a small molecule inhibiting PCSK9 prevented dendritic spine loss by attenuating the aggregation of Aβ and neuroinflammation (Apaijai et al., 2019). No evidence that PCSK9 regulates BACE1 levels or APP processing in the brain of mice has been reported by other authors (Liu et al., 2010; Fu et al., 2017). To the best of our knowledge, no data are available on the potential influence of PCSK9 on tau phosphorylation, another peculiar hallmark of AD pathogenesis.
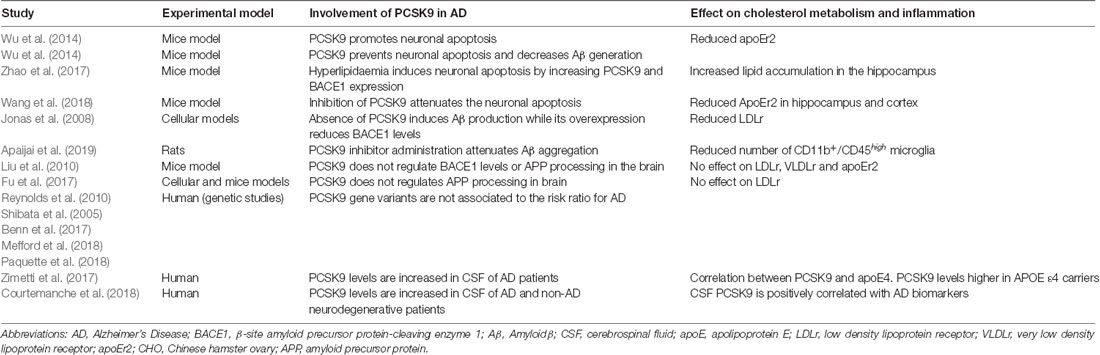
Table 1. Summary of studies investigating the involvement of PCSK9 in AD pathogenesis.
The impact of PCSK9 on neurocognitive performances in pre-clinical models has been indirectly suggested by the observation that deletion of the LRP1, which is sensitive to the degrading action of PCSK9 (Canuel et al., 2013), leads to a reduced Aβ clearance and to cognitive deficits in mice (Storck et al., 2016). Consistently, mice lacking the LDLr show signs of impaired learning and memory (Mulder et al., 2004), reduced hippocampal cell proliferation and synapses formation (Mulder et al., 2007).
Lipoprotein Receptors Target of PCSK9 and Their Role in Alzheimer’s Disease
Besides APOE, several cholesterol homeostasis related genes have been investigated for their association with AD (Wollmer, 2010) and some of them are targeted by PCSK9 (Figure 1). LRP1 is an ApoE receptor, and both positive and negative results on its genetic association with AD have been reported (Kang et al., 1997). LRP1 expression is reduced in total brain and brain capillaries with increasing age and even more reduced in AD (Kang et al., 2000; Shibata et al., 2000; Silverberg et al., 2010). Highly validated genetic risk factors for AD, like the APOE allele, may be linked to a reduced clearance of Aβ via LRP1 (Bell et al., 2007; Deane et al., 2008). LRP1 mediates Aβ clearance from the brain into the circulation at the BBB (Deane et al., 2009) and brain endothelial-specific LRP1 deletion elevates soluble brain Aβ, leading to aggravated spatial learning and memory deficits (Storck et al., 2016). Thus, LRP1 plays a pivotal role in the metabolism of the ApoE-Aβ complex and ApoE may compete with Aβ for the interaction with LRP1, resulting in impaired Aβ clearance (Verghese et al., 2013). LRP1 is also involved in the hepatic clearance of Aβ (Sagare et al., 2007). Because LRP1 is expressed in different cell types, including neurons, astrocytes and vascular cells in the brain, its levels may be altered differently in AD (Donahue et al., 2006; Ruzali et al., 2012). Indeed, LRP1 levels are decreased in neurons but increased in vascular cells or astrocytes that are proximate to amyloid plaques in AD brains (Arélin et al., 2002; Donahue et al., 2006; Ruzali et al., 2012). PCSK9 may induce the degradation of LRP1 in different cell types, including hepatocytes (Canuel et al., 2013) and vascular cells (Ferri et al., 2012; 2016b). Since PCSK9 is expressed in neurons and in vascular cells, it may influence the LRP1 levels in these cell types (Poirier et al., 2009). Thus, the observed increased PCSK9 cerebrospinal fluid (CSF) concentrations in AD (Zimetti et al., 2017) may determine a higher turnover of LRP1 on different cell types, thus affecting the Aβ elimination via the BBB.
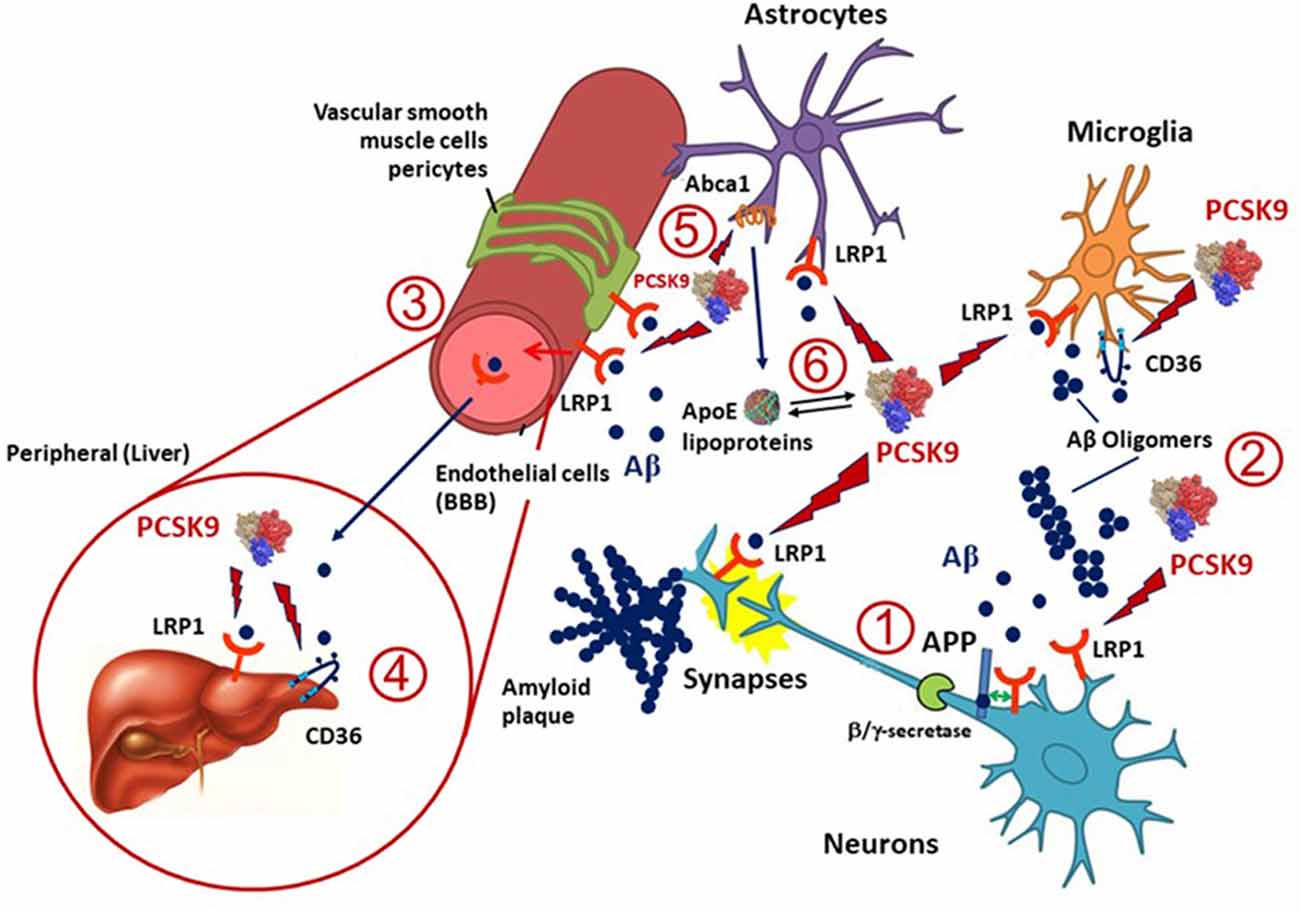
Figure 1. Potential implication of proprotein convertase subtilisin/kexin type 9 (PCSK9) in amyloid β (Aβ) clearance in Alzheimer’s disease (AD). LDL receptor-related protein 1 (LRP1), expressed in microglia, neurons, astrocytes and pericytes, and CD36, mainly present in microglia, are the two main lipoprotein receptors involved in Aβ clearance and are potentially targeted by PCSK9. (1) LRP1 may also influence the production of Aβ from amyloid precursor protein (APP) in neurons through a direct protein-protein interaction or competition with the α/β-secretase cleavage of APP. (2) Once Aβ is released into the extracellular space in the brain can form amyloid plaques or oligomers and LRP1 or CD36 can mediate its cellular uptake by neurons, microglia, astrocytes, vascular smooth muscle cells, pericytes and endothelial cells. (3) A portion of Aβ may be transported through LRP1 at the blood-brain barrier (BBB) and reversed into the blood, thus PCSK9 may also interfere with this process. (4) Both LRP1 and CD36 expressed in the liver might also help the clearance of Aβ from the blood, and PCSK9 may affect this pathway by reducing their expression levels in hepatocytes. (5) Apolipoprotein E (ApoE), which is mainly produced and secreted from astrocytes in the brain, is lipidated by ATP binding cassette transporters A1 (ABCA1) to supply cholesterol/lipids to neurons and other cells through LRP1 and CD36. PCSK9 has been shown to downregulate the expression of ABCA1, thus opening to a possible modulation of the release of ApoE containing lipoproteins and thus LRP1- or CD36-mediated Aβ metabolism. Indeed, ApoE isoforms may affect LRP1-mediated Aβ metabolism by directly interacting with Aβ or competing with Aβ for receptor binding. (6) ApoE lipoprotein may also interact with PCSK9 hence influencing its action on LRP1 and CD36.
The LDLr is a major ApoE receptor in the brain and genetic studies aiming at identifying its link to AD are controversial, although one large study found an association in men but not in women (Lendon et al., 1997; Zou et al., 2008). In a mouse model of AD, LDLr demonstrated a beneficial effect via enhancement of Aβ clearance (Kim et al., 2009), suggesting its promising association with the risk for AD and consequently the involvement of PCSK9.
VLDLr can be involved in the clearance of ApoE-Aβ complexes (Helbecque and Amouyel, 2000). However, a meta-analysis of genetic studies conducted on the VLDLr gene polymorphic triplet (CGG) repeat in the 5-UTR showed contradictory results, being a protective factor for AD in Caucasians and as a risk factor in Asian people (Llorca et al., 2008). In addition, genetic association with AD was described in one study but not confirmed in two replication studies (Taguchi et al., 2005). Thus, the direct association of VLDLr and AD still needs to be proven.
The scavenger receptor CD36 is involved in fibrillar Aβ-mediated microglial activation and consecutive activation of an innate immune response (Coraci et al., 2002; Moore et al., 2002; Bamberger et al., 2003). PCSK9 leads to increased CD36 expression in macrophages and microglial-like cells (Ding et al., 2018), suggesting that PCSK9 may regulate both the CD36-mediated clearance of Aβ and the innate host response to Aβ and oxidized-LDL (oxLDL) in brain cells. Indeed, CD36 acts as a co-receptor for the toll-like receptors (TLRs) heterodimerization, an essential step for the initiation of the inflammatory signals and microglia-dependent neurodegeneration (Stewart et al., 2010). Consistently, PCSK9 elicits a proinflammatory effect on macrophages (Ricci et al., 2018) and the administration of a PCSK9 inhibitor leads to a neuroinflammation attenuation in mice models (Apaijai et al., 2019).
Finally, our group reported that human recombinant PCSK9 inhibits the ABCA1-mediated cholesterol efflux in macrophages (Adorni et al., 2017). In this regard, data on the influence of ABCA1 in AD are conflicting as well. For example, carriers of the R219K SNP in the ABCA1 gene shown 33% lower total cholesterol in CSF compared to non-carriers. This allele is also associated with a delay of disease onset by 1.7 years on average (Wollmer et al., 2003). This suggests that a genetic variability of ABCA1 influences the development of AD, possibly by interfering with CNS cholesterol homeostasis. Nevertheless, an association of the same ABCA1 variant R219K with increased AD risk has been described (Rodríguez-Rodríguez et al., 2007). Similarly, additional evidence reported the association between genetic variants of ABCA1 gene with either reduced or increased AD risk (Katzov et al., 2004; Nordestgaard et al., 2015; Beecham et al., 2018). Specifically, a genetic study involving more than 90,000 subjects evidenced a significant association between ABCA1 loss-of-function mutation and 41% increased risk of AD (Nordestgaard et al., 2015). Nevertheless, also for ABCA1, several negative studies have been published, such as a meta-analysis where no association has been found between R219K, I883M and R1587K polymorphisms and risk of AD (Jiang et al., 2012). The involvement of ABCA1 in AD has been investigated in ABCA1 knock-out mice cross-bred with an amyloid pathology of AD. The absence of ABCA1 determined a higher amyloid load in the brains (Koldamova et al., 2005; Wahrle et al., 2005, 2008), although other similar experiments failed to show an effect of ABCA1 on amyloid pathology (Hirsch-Reinshagen et al., 2005, 2007). Since ABCA1 regulates ApoE levels and the transfer of cholesterol from the glial to the neuronal compartment (Wahrle et al., 2004), its role on AD may involve PCSK9, which affects ABCA1 expression (Adorni et al., 2017). Consistent with this hypothesis, a strong decrease in ApoE levels in both the cortex and CSF, together with an impairment of its lipidation, has been described in ABCA1 knock-out mice (Wahrle et al., 2004). Interestingly, CSF samples extracted from AD patients have lower ex-vivo capacity to promote ABCA1-mediated cholesterol efflux compared to controls (Yassine et al., 2016). Finally, a further level of complexity could be the possible binding between PCSK9 and apoE-containing lipoproteins as previously described for LDL (Tavori et al., 2013), Lipoprotein (a) (Tavori et al., 2016) and HDL (Ferri et al., 2016a; Ruscica et al., 2018).
PCSK9 and Alzheimer’s Disease in Humans
The genetic studies conducted so far in humans (Table 1) are not conclusive on the impact of PCSK9 mutations on AD. Although Wollmer (2010) first identified PCSK9 among the cholesterol-related genes that have been matched with AD genes listed in the AlzGene database, no association was found between PCSK9 polymorphism and the risk of AD onset, neither in a Japanese nor in a Swedish cohort study (Shibata et al., 2005; Reynolds et al., 2010). Consistently, in a recent Mendelian randomization analysis, PCSK9 loss-of-function mutations were not associated to a rise in the risk of AD [Hazard Ratio (HR) = 0.50; p = 0.37; Benn et al., 2017].
To a negative conclusion came also the results of genetic studies among African American REGARDS (Reasons for Geographic and Racial Differences in Stroke) participants with and without the PCSK9 loss-of-function variants C697X or Y142X. The presence of these variants did not affect the primary endpoint of the study, i.e., the neurocognitive performance (Mefford et al., 2018). In another study conducted in French Canadian subjects, carriers of the PCSK9 loss of function mutations, R46L and InsLEU, did not differ from non-carriers as either AD prevalence or age of disease onset (Paquette et al., 2018).
In humans, PCSK9 has been detected in CSF even though at a much lower concentration compared to plasma (Chen et al., 2014). CSF PCSK9 concentrations appeared to be constant throughout the day, thus not undergoing the typical diurnal pattern of plasma PCSK9 (Persson et al., 2010) and suggesting a differential mechanism of PCSK9 regulation in the peripheral and central body compartments (Chen et al., 2014).
In a previous work, we demonstrated increased levels of PCSK9 in the CSF of AD patients with the highest levels in APOE ε4 carriers (Zimetti et al., 2017). These data suggest an involvement of PCSK9 in the disease and a pathophysiological link with APOE4 that deserves further investigations. Our observation has been confirmed by Courtemanche et al. (2018), however, they show a trend for increased CSF PCSK9 levels also in non-AD neurodegenerative disease, confirming a link to the neurodegenerative process but not specifically to AD.
PCSK9 Inhibitors and Neurocognitive Disorders
Plasma cholesterol under the PCSK9 inhibitor (alirocumab, evolocumab or bococizumab) treatment reached very low levels (mean level 30 mg/dL in the FOURIER trial and 25 mg/dL in ODYSSEY LONG-TERM and SPIRE trials) (Robinson et al., 2015; Ridker et al., 2017; Sabatine et al., 2017). These observations raised some concerns about the potential side effects related to the clinical use of PCSK9 inhibitors. In particular, some clinical trials highlighted a potential association between treatment with PCSK9 monoclonal antibodies and cognitive adverse events (Robinson et al., 2015; Sabatine et al., 2015). However, such disorders were often self-reported and occurred in very few patients with pre-existing medical conditions or other confounders, as emerged from the results of an analysis made by the FDA (Food and Drug Administration Briefing Document, 2015a,b). In order to better clarify this issue, a recent study prospectively and objectively evaluated the effect of the PCSK9 inhibitor evolocumab on cognitive functions (Giugliano et al., 2017a). In this study, a total of 1,204 subjects with mean age of 65 years, that did not present neurological disorders on treatment with evolocumab or placebo, were followed for 1.6 years without evidencing any association with adverse cognitive effects (Giugliano et al., 2017b). This result was further confirmed by a recent meta-analysis (Bajaj et al., 2018; Harvey et al., 2018). However, considering the short follow-up period of the EBBINGHAUS, a 5-year extension of the FOURIER trial will provide further findings on neurocognitive functions1.
The lack of an evident effect by PCSK9 inhibitors on cognitive functions is very likely explained by the BBB presence, with the consequence that high or low levels of cholesterol in the circulation are not likely to have direct effects on lipid level in the brain (Olsson et al., 2017). With this respect, in the genetic study with carriers of PCSK9 loss-of-function variants, lifelong exposure to low levels of LDL-cholesterol (LDL-C) was not indeed associated with neurocognitive effects (Mefford et al., 2018; Paquette et al., 2018).
Moreover, BBB limits the access of both PCSK9 (Rousselet et al., 2011) and more so of monoclonal antibodies, such as alirocumab or evolocumab to the CNS. Under the conditions where the integrity of BBB is intact, the presence of tight junctions prevents the transcellular route for diffusion of antibodies across the capillary. Therefore, in general, the antibodies penetration into the brain has been estimated to be about 0.1%, both in humans and animals (Tabrizi et al., 2010). In some pathological conditions, such as diabetes, the BBB might be compromised (Rom et al., 2019). However, some indications ruling out the possibility that the antibodies cross the BBB in such conditions, come from the EBBINGHAUS study, which also involves diabetic subjects (37.2%), and in which no variation of cognitive functions was observed (Giugliano et al., 2017b).
Thus, it would be of great interest to evaluate the effect of small molecules PCSK9 inhibitors capable of crossing the BBB.
Conclusion
Although several extrahepatic effects of PCSK9 beyond LDL-C (Stoekenbroek et al., 2018) have been identified and well characterized, its role in the brain and the potential involvement in CNS diseases is still under investigation. In this regard, pathophysiological studies on pre-clinical models of AD led to controversial results, leaving open the question of the potential implication of PCSK9 in the disease pathogenesis. Furthermore, the few genetic studies available focused only on PCSK9 genetic variants leading to loss-of-function mutations and are not supportive of an association between PCSK9 and AD risk (Mefford et al., 2018; Paquette et al., 2018). Notably, in all of the studies, only plasma PCSK9 concentrations have been evaluated, while PCSK9 cannot cross the BBB and its regulation may be different in the central and periphery body compartments. Based on the observation made by ourselves and others regarding increased PCSK9 levels in the CSF of AD patients and considering that PCSK9 may interfere with CNS cholesterol transport by degrading the neuronal ApoE-receptors responsible for astrocyte-derived cholesterol uptake, it is conceivable to hypothesize a PCSK9-induced impairment of cholesterol supply to neurons occurring in AD. The consequences of this cholesterol-depletion would include a loss of neuronal physiological functions and ultimately neurodegeneration. In addition, PCSK9 may contribute to exacerbate neuroinflammation, possibly acting on the receptors CD36 and TLR4 (Stewart et al., 2010). These hypotheses, that are being tested by our research group, may set the basis for testing new pharmacological approaches, including existing small molecules potentially able to cross the BBB by either diffusion or transporter-mediated processes, differing from the anti- PCSK9 antibodies. These molecules, by restoring the physiological brain cholesterol transport from astrocytes to neurons in CNS and by attenuating the neuroinflammation through CD36-TRL4 pathway inhibition, might clear the path for a potential future innovative AD therapy.
Author Contributions
FZ and MA wrote the first draft of the review and prepared the table. NF, MR and FB wrote sections of the review article. NF made the figure. All authors critically revised the text and all approved the submitted version.
Funding
The open access publication of this review article is supported by a grant from Amgen (Amgen’s PSCK9 Competitive Grant Program 2018, recipient FB).
Conflict of Interest Statement
FB received a financial grant from Amgen (Amgen’s PSCK9 Competitive Grant Program 2018) with a project entitled: “EXplorIng the paThophysological role of PCSK9 in Alzheimer’s Disease: focus on inflammation and lipid metabolism (EXIT-AD)”.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Adorni, M. P., Cipollari, E., Favari, E., Zanotti, I., Zimetti, F., Corsini, A., et al. (2017). Inhibitory effect of PCSK9 on Abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis 256, 1–6. doi: 10.1016/j.atherosclerosis.2016.11.019
Apaijai, N., Moisescu, D. M., Palee, S., Mcsweeney, C. M., Saiyasit, N., Maneechote, C., et al. (2019). Pretreatment with PCSK9 inhibitor protects the brain against cardiac ischemia/reperfusion injury through a reduction of neuronal inflammation and amyloid β aggregation. J. Am. Heart Assoc. 8:e010838. doi: 10.1161/jaha.118.010838
Arélin, K., Kinoshita, A., Whelan, C. M., Irizarry, M. C., Rebeck, G. W., Strickland, D. K., et al. (2002). LRP and senile plaques in Alzheimer’s disease: colocalization with apolipoprotein E and with activated astrocytes. Mol. Brain Res. 104, 38–46. doi: 10.1016/s0169-328x(02)00203-6
Arenas, F., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2017). Intracellular cholesterol trafficking and impact in neurodegeneration. Front. Mol. Neurosci. 10:382. doi: 10.3389/fnmol.2017.00382
Bajaj, N. S., Patel, N., Kalra, R., Ahmad, A., Venkatraman, A., Arora, G., et al. (2018). Neurological effects of proprotein convertase subtilisin/kexin type 9 inhibitors: direct comparisons. Eur. Heart J. Qual. Care Clin. Outcomes 4, 132–141. doi: 10.1093/ehjqcco/qcx037
Bamberger, M. E., Harris, M. E., Mcdonald, D. R., Husemann, J., and Landreth, G. E. (2003). A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J. Neurosci. 23, 2665–2674. doi: 10.1523/jneurosci.23-07-02665.2003
Beecham, G. W., Vardarajan, B., Blue, E., Bush, W., Jaworski, J., Barral, S., et al. (2018). Rare genetic variation implicated in non-Hispanic white families with Alzheimer disease. Neurol. Genet. 4:e286. doi: 10.1212/nxg.0000000000000286
Bell, R. D., Sagare, A. P., Friedman, A. E., Bedi, G. S., Holtzman, D. M., Deane, R., et al. (2007). Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 27, 909–918. doi: 10.1038/sj.jcbfm.9600419
Benn, M., Nordestgaard, B. G., Frikke-Schmidt, R., and Tybjaerg-Hansen, A. (2017). Low LDL cholesterol, PCSK9 and HMGCR genetic variation and risk of Alzheimer’s disease and Parkinson’s disease: Mendelian randomisation study. BMJ 357:j1648. doi: 10.1136/bmj.j1648
Björkhem, I., Leoni, V., and Svenningsson, P. (2019). On the fluxes of side-chain oxidized oxysterols across blood-brain and blood-CSF barriers and origin of these steroids in CSF (Review). J. Steroid Biochem. Mol. Biol. 188, 86–89. doi: 10.1016/j.jsbmb.2018.12.009
Björkhem, I., and Meaney, S. (2004). Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 24, 806–815. doi: 10.1161/01.ATV.0000120374.59826.1b
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Canuel, M., Sun, X., Asselin, M. C., Paramithiotis, E., Prat, A., and Seidah, N. G. (2013). Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 8:e64145. doi: 10.1371/journal.pone.0064145
Chang, T. Y., Yamauchi, Y., Hasan, M. T., and Chang, C. (2017). Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid Res. 58, 2239–2254. doi: 10.1194/jlr.R075630
Chen, Y. Q., Troutt, J. S., and Konrad, R. J. (2014). PCSK9 is present in human cerebrospinal fluid and is maintained at remarkably constant concentrations throughout the course of the day. Lipids 49, 445–455. doi: 10.1007/s11745-014-3895-6
Chen, J., Zhang, X., Kusumo, H., Costa, L. G., and Guizzetti, M. (2013). Cholesterol efflux is differentially regulated in neurons and astrocytes: implications for brain cholesterol homeostasis. Biochim. Biophys. Acta 1831, 263–275. doi: 10.1016/j.bbalip.2012.09.007
Coraci, I. S., Husemann, J., Berman, J. W., Hulette, C., Dufour, J. H., Campanella, G. K., et al. (2002). CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to β-amyloid fibrils. Am. J. Pathol. 160, 101–112. doi: 10.1016/s0002-9440(10)64354-4
Courtemanche, H., Bigot, E., Pichelin, M., Guyomarch, B., Boutoleau-Bretonniere, C., Le May, C., et al. (2018). PCSK9 concentrations in cerebrospinal fluid are not specifically increased in Alzheimer’s disease. J. Alzheimers Dis. 62, 1519–1525. doi: 10.3233/jad-170993
Deane, R., Bell, R. D., Sagare, A., and Zlokovic, B. V. (2009). Clearance of amyloid-β peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 8, 16–30. doi: 10.2174/187152709787601867
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/jci36663
Dietschy, J. M. (2009). Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 390, 287–293. doi: 10.1515/bc.2009.035
Dietschy, J. M., and Turley, S. D. (2004). Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45, 1375–1397. doi: 10.1194/jlr.r400004-jlr200
Ding, Z., Liu, S., Wang, X., Theus, S., Deng, X., Fan, Y., et al. (2018). PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc. Res. 114, 1145–1153. doi: 10.1093/cvr/cvy079
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A. III., Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1 and amyloid-β protein in Alzheimer’s disease. Acta Neuropathol. 112, 405–415. doi: 10.1007/s00401-006-0115-3
Dong, H. K., Gim, J. A., Yeo, S. H., and Kim, H. S. (2017). Integrated late onset Alzheimer’s disease (LOAD) susceptibility genes: cholesterol metabolism and trafficking perspectives. Gene 597, 10–16. doi: 10.1016/j.gene.2016.10.022
Ferri, N., Corsini, A., Macchi, C., Magni, P., and Ruscica, M. (2016a). Proprotein convertase subtilisin kexin type 9 and high-density lipoprotein metabolism: experimental animal models and clinical evidence. Transl. Res. 173, 19–29. doi: 10.1016/j.trsl.2015.10.004
Ferri, N., Marchiano, S., Tibolla, G., Baetta, R., Dhyani, A., Ruscica, M., et al. (2016b). PCSK9 knock-out mice are protected from neointimal formation in response to perivascular carotid collar placement. Atherosclerosis 253, 214–224. doi: 10.1016/j.atherosclerosis.2016.07.910
Ferri, N., Tibolla, G., Pirillo, A., Cipollone, F., Mezzetti, A., Pacia, S., et al. (2012). Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis 220, 381–386. doi: 10.1016/j.atherosclerosis.2011.11.026
Food and Drug Administration Briefing Document. (2015a). The endocrinologic and metabolic drugs advisory committee meeting. Repatha (evolocumab) injections. Available online at: https://wayback.archive-it.org/7993/20170405215129/https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM450072.pdf. Accessed June 10, 2015.
Food and Drug Administration Briefing Document. (2015b). The endocrinologic and metabolic drugs advisory committee meeting. Pralutent (alirocumab) injection. Available online at: https://wayback.archive-it.org/7993/20170405215212/https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM449865.pdf. Accessed June 9, 2015.
Fu, T., Guan, Y., Xu, J., and Wang, Y. (2017). APP, APLP2 and LRP1 interact with PCSK9 but are not required for PCSK9-mediated degradation of the LDLR in vivo. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 883–889. doi: 10.1016/j.bbalip.2017.05.002
Giugliano, R. P., Mach, F., Zavitz, K., Kurtz, C., Schneider, J., Wang, H., et al. (2017a). Design and rationale of the EBBINGHAUS trial: a phase 3, double-blind, placebo-controlled, multicenter study to assess the effect of evolocumab on cognitive function in patients with clinically evident cardiovascular disease and receiving statin background lipid-lowering therapy-A cognitive study of patients enrolled in the FOURIER trial. Clin. Cardiol. 40, 59–65. doi: 10.1002/clc.22678
Giugliano, R. P., Sabatine, M. S., and Ott, B. R. (2017b). Cognitive function in a randomized trial of evolocumab. N. Engl. J. Med. 377:1997. doi: 10.1056/nejmc1712102
Harvey, P. D., Sabbagh, M. N., Harrison, J. E., Ginsberg, H. N., Chapman, M. J., Manvelian, G., et al. (2018). No evidence of neurocognitive adverse events associated with alirocumab treatment in 3340 patients from 14 randomized Phase 2 and 3 controlled trials: a meta-analysis of individual patient data. Eur. Heart J. 39, 374–381. doi: 10.1093/eurheartj/ehx661
Helbecque, N., and Amouyel, P. (2000). Very low density lipoprotein receptor in Alzheimer disease. Microsc. Res. Tech. 50, 273–277. doi: 10.1002/1097-0029(20000815)50:4<273::aid-jemt4>3.0.co;2-0
Hirsch-Reinshagen, V., Chan, J. Y., Wilkinson, A., Tanaka, T., Fan, J., Ou, G., et al. (2007). Physiologically regulated transgenic ABCA1 does not reduce amyloid burden or amyloid-β peptide levels in vivo. J. Lipid Res. 48, 914–923. doi: 10.1194/jlr.m600543-jlr200
Hirsch-Reinshagen, V., Maia, L. F., Burgess, B. L., Blain, J. F., Naus, K. E., Mcisaac, S. A., et al. (2005). The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280, 43243–43256. doi: 10.1074/jbc.m508781200
Jiang, M., Lv, L., Wang, H., Yang, X., Ji, H., Zhou, F., et al. (2012). Meta-analysis on association between the ATP-binding cassette transporter A1 gene (ABCA1) and Alzheimer’s disease. Gene 510, 147–153. doi: 10.1016/j.gene.2012.09.009
Jonas, M. C., Costantini, C., and Puglielli, L. (2008). PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 9, 916–922. doi: 10.1038/embor.2008.132
Kang, D. E., Pietrzik, C. U., Baum, L., Chevallier, N., Merriam, D. E., Kounnas, M. Z., et al. (2000). Modulation of amyloid β-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Invest. 106, 1159–1166. doi: 10.1172/jci11013
Kang, D. E., Saitoh, T., Chen, X., Xia, Y., Masliah, E., Hansen, L. A., et al. (1997). Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer’s disease. Neurology 49, 56–61. doi: 10.1212/wnl.49.1.56
Katzov, H., Chalmers, K., Palmgren, J., Andreasen, N., Johansson, B., Cairns, N. J., et al. (2004). Genetic variants of ABCA1 modify Alzheimer disease risk and quantitative traits related to β-amyloid metabolism. Hum. Mutat. 23, 358–367. doi: 10.1002/humu.20012
Kim, J., Castellano, J. M., Jiang, H., Basak, J. M., Parsadanian, M., Pham, V., et al. (2009). Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron 64, 632–644. doi: 10.1016/j.neuron.2009.11.013
Koldamova, R., Staufenbiel, M., and Lefterov, I. (2005). Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235. doi: 10.1074/jbc.m504513200
Koudinov, A. R., and Koudinova, N. V. (2005). Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J. Neurol. Sci. 229–230, 233–240. doi: 10.1016/j.jns.2004.11.036
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lendon, C. L., Talbot, C. J., Craddock, N. J., Han, S. W., Wragg, M., Morris, J. C., et al. (1997). Genetic association studies between dementia of the Alzheimer’s type and three receptors for apolipoprotein E in a Caucasian population. Neurosci. Lett. 222, 187–190. doi: 10.1016/s0304-3940(97)13381-x
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, M., Wu, G., Baysarowich, J., Kavana, M., Addona, G. H., Bierilo, K. K., et al. (2010). PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J. Lipid Res. 51, 2611–2618. doi: 10.1194/jlr.m006635
Llorca, J., Rodriguez-Rodriguez, E., Dierssen-Sotos, T., Delgado-Rodriguez, M., Berciano, J., and Combarros, O. (2008). Meta-analysis of genetic variability in the β-amyloid production, aggregation and degradation metabolic pathways and the risk of Alzheimer’s disease. Acta Neurol. Scand. 117, 1–14. doi: 10.1111/j.1600-0404.2007.00899.x
Macchi, C., Banach, M., Corsini, A., Sirtori, C. R., Ferri, N., and Ruscica, M. (2019). Changes in circulating pro-protein convertase subtilisin/kexin type 9 levels—experimental and clinical approaches with lipid-lowering agents. Eur. J. Prev. Cardiol. doi: 10.1177/2047487319831500 [Epub ahead of print].
Mahoney-Sanchez, L., Belaidi, A. A., Bush, A. I., and Ayton, S. (2016). The complex role of apolipoprotein E in Alzheimer’s disease: an overview and update. J. Mol. Neurosci. 60, 325–335. doi: 10.1007/s12031-016-0839-z
Mefford, M. T., Rosenson, R. S., Cushman, M., Farkouh, M. E., Mcclure, L. A., Wadley, V. G., et al. (2018). PCSK9 variants, low-density lipoprotein cholesterol and neurocognitive impairment: reasons for geographic and racial differences in stroke study (REGARDS). Circulation 137, 1260–1269. doi: 10.1161/CIRCULATIONAHA.117.029785
Moore, K. J., El Khoury, J., Medeiros, L. A., Terada, K., Geula, C., Luster, A. D., et al. (2002). A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. J. Biol. Chem. 277, 47373–47379. doi: 10.1074/jbc.m208788200
Mulder, M., Jansen, P. J., Janssen, B. J., Van De Berg, W. D., Van Der Boom, H., Havekes, L. M., et al. (2004). Low-density lipoprotein receptor-knockout mice display impaired spatial memory associated with a decreased synaptic density in the hippocampus. Neurobiol. Dis. 16, 212–219. doi: 10.1016/j.nbd.2004.01.015
Mulder, M., Koopmans, G., Wassink, G., Al Mansouri, G., Simard, M. L., Havekes, L. M., et al. (2007). LDL receptor deficiency results in decreased cell proliferation and presynaptic bouton density in the murine hippocampus. Neurosci. Res. 59, 251–256. doi: 10.1016/j.neures.2007.07.004
Nordestgaard, L. T., Tybjaerg-Hansen, A., Nordestgaard, B. G., and Frikke-Schmidt, R. (2015). Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement. 11, 1430–1438. doi: 10.1016/j.jalz.2015.04.006
Olsson, A. G., Angelin, B., Assmann, G., Binder, C. J., Bjorkhem, I., Cedazo-Minguez, A., et al. (2017). Can LDL cholesterol be too low? Possible risks of extremely low levels. J. Intern. Med. 281, 534–553. doi: 10.1111/joim.12614
Paquette, M., Saavedra, Y. G. L., Poirier, J., Theroux, L., Dea, D., Baass, A., et al. (2018). Loss-of-function PCSK9 mutations are not associated with Alzheimer disease. J. Geriatr. Psychiatry Neurol. 31, 90–96. doi: 10.1177/0891988718764330
Persson, L., Cao, G., Ståhle, L., Sjöberg, B. G., Troutt, J. S., Konrad, R. J., et al. (2010). Circulating proprotein convertase subtilisin kexin type 9 has a diurnal rhythm synchronous with cholesterol synthesis and is reduced by fasting in humans. Arterioscler. Thromb. Vasc. Biol. 30, 2666–2672. doi: 10.1161/atvbaha.110.214130
Picard, C., Julien, C., Frappier, J., Miron, J., Theroux, L., Dea, D., et al. (2018). Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol. Aging 66, 180.e1–180.e9. doi: 10.1016/j.neurobiolaging.2018.01.018
Poirier, S., Mayer, G., Benjannet, S., Bergeron, E., Marcinkiewicz, J., Nassoury, N., et al. (2008). The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372. doi: 10.1074/jbc.m708098200
Poirier, S., Mayer, G., Poupon, V., Mcpherson, P. S., Desjardins, R., Ly, K., et al. (2009). Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864. doi: 10.1074/jbc.m109.037085
Poirier, S., Prat, A., Marcinkiewicz, E., Paquin, J., Chitramuthu, B. P., Baranowski, D., et al. (2006). Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J. Neurochem. 98, 838–850. doi: 10.1111/j.1471-4159.2006.03928.x
Reynolds, C. A., Hong, M. G., Eriksson, U. K., Blennow, K., Wiklund, F., Johansson, B., et al. (2010). Analysis of lipid pathway genes indicates association of sequence variation near SREBF1/TOM1L2/ATPAF2 with dementia risk. Hum. Mol. Genet. 19, 2068–2078. doi: 10.1093/hmg/ddq079
Ricci, C., Ruscica, M., Camera, M., Rossetti, L., Macchi, C., Colciago, A., et al. (2018). PCSK9 induces a pro-inflammatory response in macrophages. Sci. Rep. 8:2267. doi: 10.1038/s41598-018-20425-x
Ridker, P. M., Revkin, J., Amarenco, P., Brunell, R., Curto, M., Civeira, F., et al. (2017). Cardiovascular efficacy and safety of bococizumab in high-risk patients. N. Engl. J. Med. 376, 1527–1539. doi: 10.1056/NEJMoa1701488
Robinson, J. G., Farnier, M., Krempf, M., Bergeron, J., Luc, G., Averna, M., et al. (2015). Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1489–1499. doi: 10.1056/NEJMoa1501031
Rodríguez-Rodríguez, E., Mateo, I., Llorca, J., Sanchez-Quintana, C., Infante, J., Garcia-Gorostiaga, I., et al. (2007). Association of genetic variants of ABCA1 with Alzheimer’s disease risk. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 964–968. doi: 10.1002/ajmg.b.30552
Rom, S., Zuluaga-Ramirez, V., Gajghate, S., Seliga, A., Winfield, M., Heldt, N. A., et al. (2019). Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus (DM) type 1 and type 2 mouse models. Mol. Neurobiol. 56, 1883–1896. doi: 10.1007/s12035-018-1195-5
Rousselet, E., Marcinkiewicz, J., Kriz, J., Zhou, A., Hatten, M. E., Prat, A., et al. (2011). PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J. Lipid Res. 52, 1383–1391. doi: 10.1194/jlr.m014118
Ruscica, M., Simonelli, S., Botta, M., Ossoli, A., Lupo, M. G., Magni, P., et al. (2018). Plasma PCSK9 levels and lipoprotein distribution are preserved in carriers of genetic HDL disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 991–997. doi: 10.1016/j.bbalip.2018.05.015
Ruzali, W. A., Kehoe, P. G., and Love, S. (2012). LRP1 expression in cerebral cortex, choroid plexus and meningeal blood vessels: relationship to cerebral amyloid angiopathy and APOE status. Neurosci. Lett. 525, 123–128. doi: 10.1016/j.neulet.2012.07.065
Sabatine, M. S., Giugliano, R. P., Keech, A. C., Honarpour, N., Wiviott, S. D., Murphy, S. A., et al. (2017). Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 376, 1713–1722. doi: 10.1056/NEJMoa1615664
Sabatine, M. S., Giugliano, R. P., Wiviott, S. D., Raal, F. J., Blom, D. J., Robinson, J., et al. (2015). Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1500–1509. doi: 10.1056/NEJMoa1500858
Sagare, A., Deane, R., Bell, R. D., Johnson, B., Hamm, K., Pendu, R., et al. (2007). Clearance of amyloid-β by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031. doi: 10.1038/nm1635
Saito, K., Dubreuil, V., Arai, Y., Wilsch-Brauninger, M., Schwudke, D., Saher, G., et al. (2009). Ablation of cholesterol biosynthesis in neural stem cells increases their VEGF expression and angiogenesis but causes neuron apoptosis. Proc. Natl. Acad. Sci. U S A 106, 8350–8355. doi: 10.1073/pnas.0903541106
Sato, N., and Morishita, R. (2015). The roles of lipid and glucose metabolism in modulation of β-amyloid, tau and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 7:199. doi: 10.3389/fnagi.2015.00199
Seidah, N. G., Benjannet, S., Wickham, L., Marcinkiewicz, J., Jasmin, S. B., Stifani, S., et al. (2003). The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U S A 100, 928–933. doi: 10.1073/pnas.0335507100
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/JCI10498
Shibata, N., Ohnuma, T., Higashi, S., Higashi, M., Usui, C., Ohkubo, T., et al. (2005). No genetic association between PCSK9 polymorphisms and Alzheimer’s disease and plasma cholesterol level in Japanese patients. Psychiatr. Genet. 15:239. doi: 10.1097/00041444-200512000-00004
Silverberg, G. D., Messier, A. A., Miller, M. C., Machan, J. T., Majmudar, S. S., Stopa, E. G., et al. (2010). Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 69, 1034–1043. doi: 10.1097/nen.0b013e3181f46e25
Stewart, C. R., Stuart, L. M., Wilkinson, K., Van Gils, J. M., Deng, J., Halle, A., et al. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161. doi: 10.1038/ni.1836
Stoekenbroek, R. M., Lambert, G., Cariou, B., and Hovingh, G. K. (2018). Inhibiting PCSK9—biology beyond LDL control. Nat. Rev. Endocrinol. 15, 52–62. doi: 10.1038/s41574-018-0110-5
Storck, S. E., Meister, S., Nahrath, J., Meissner, J. N., Schubert, N., Di Spiezio, A., et al. (2016). Endothelial LRP1 transports amyloid-β(1–42) across the blood-brain barrier. J. Clin. Invest. 126, 123–136. doi: 10.1172/jci81108
Tabrizi, M., Bornstein, G. G., and Suria, H. (2010). Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 12, 33–43. doi: 10.1208/s12248-009-9157-5
Taguchi, K., Yamagata, H. D., Zhong, W., Kamino, K., Akatsu, H., Hata, R., et al. (2005). Identification of hippocampus-related candidate genes for Alzheimer’s disease. Ann. Neurol. 57, 585–588. doi: 10.1002/ana.20433
Tavori, H., Christian, D., Minnier, J., Plubell, D., Shapiro, M. D., Yeang, C., et al. (2016). PCSK9 association with lipoprotein(a). Circ. Res. 119, 29–35. doi: 10.1161/CIRCRESAHA.116.308811
Tavori, H., Giunzioni, I., Linton, M. F., and Fazio, S. (2013). Loss of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) after lipoprotein apheresis. Circ. Res. 113, 1290–1295. doi: 10.1161/circresaha.113.302655
Verghese, P. B., Castellano, J. M., Garai, K., Wang, Y., Jiang, H., Shah, A., et al. (2013). ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U S A 110, E1807–E1816. doi: 10.1073/pnas.1220484110
Vetrivel, K. S., and Thinakaran, G. (2010). Membrane rafts in Alzheimer’s disease β-amyloid production. Biochim. Biophys. Acta 1801, 860–867. doi: 10.1016/j.bbalip.2010.03.007
Wahrle, S. E., Jiang, H., Parsadanian, M., Hartman, R. E., Bales, K. R., Paul, S. M., et al. (2005). Deletion of Abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242. doi: 10.1074/jbc.m508780200
Wahrle, S. E., Jiang, H., Parsadanian, M., Kim, J., Li, A., Knoten, A., et al. (2008). Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682. doi: 10.1172/jci33622
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., et al. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993. doi: 10.1074/jbc.m407963200
Wang, L., Wang, Z., Shi, J., Jiang, Q., Wang, H., Li, X., et al. (2018). Inhibition of proprotein convertase subtilisin/kexin type 9 attenuates neuronal apoptosis following focal cerebral ischemia via apolipoprotein E receptor 2 downregulation in hyperlipidemic mice. Int. J. Mol. Med. 42, 2098–2106. doi: 10.3892/ijmm.2018.3797
Wollmer, M. A. (2010). Cholesterol-related genes in Alzheimer’s disease. Biochim. Biophys. Acta 1801, 762–773. doi: 10.1016/j.bbalip.2010.05.009
Wollmer, M. A., Streffer, J. R., Lutjohann, D., Tsolaki, M., Iakovidou, V., Hegi, T., et al. (2003). ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol. Aging 24, 421–426. doi: 10.1016/s0197-4580(02)00094-5
Wu, Q., Tang, Z. H., Peng, J., Liao, L., Pan, L. H., Wu, C. Y., et al. (2014). The dual behavior of PCSK9 in the regulation of apoptosis is crucial in Alzheimer’s disease progression (Review). Biomed. Rep. 2, 167–171. doi: 10.3892/br.2013.213
Yassine, H. N., Feng, Q., Chiang, J., Petrosspour, L. M., Fonteh, A. N., Chui, H. C., et al. (2016). ABCA1-mediated cholesterol efflux capacity to cerebrospinal fluid is reduced in patients with mild cognitive impairment and Alzheimer’s disease. J. Am. Heart Assoc. 5:e002886. doi: 10.1161/jaha.115.002886
Zhao, X. S., Wu, Q., Peng, J., Pan, L. H., Ren, Z., Liu, H. T., et al. (2017). Hyperlipidemia-induced apoptosis of hippocampal neurons in ApoE(−/−) mice may be associated with increased PCSK9 expression. Mol. Med. Rep. 15, 712–718. doi: 10.3892/mmr.2016.6055
Zimetti, F., Caffarra, P., Ronda, N., Favari, E., Adorni, M. P., Zanotti, I., et al. (2017). Increased PCSK9 cerebrospinal fluid concentrations in Alzheimer’s disease. J. Alzheimers Dis. 55, 315–320. doi: 10.3233/JAD-160411
Keywords: PCSK9 (proprotein convertase subtilisin/kexin type 9), Alzheimer, cholesterol, apolipoprotein E, neuron, brain, cognitive, apoE receptors
Citation: Adorni MP, Ruscica M, Ferri N, Bernini F and Zimetti F (2019) Proprotein Convertase Subtilisin/Kexin Type 9, Brain Cholesterol Homeostasis and Potential Implication for Alzheimer’s Disease. Front. Aging Neurosci. 11:120. doi: 10.3389/fnagi.2019.00120
Received: 22 February 2019; Accepted: 07 May 2019;
Published: 22 May 2019.
Edited by:
Patrizia Giannoni, University of Nîmes, FranceReviewed by:
Monique Mulder, Erasmus University Rotterdam, NetherlandsDaniel Gaudet, Université de Montréal, Canada
Copyright © 2019 Adorni, Ruscica, Ferri, Bernini and Zimetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Ferri, bmljb2xhLmZlcnJpQHVuaXBkLml0