 Jennifer M. Hafycz
Jennifer M. Hafycz Nirinjini N. Naidoo*
Nirinjini N. Naidoo*- Center for Sleep and Circadian Neurobiology, Perelman School of Medicine, University of Pennsylvania, Pennsylvania, PA, United States
Many neurodegenerative diseases manifest in an overall aged population, the pathology of which is hallmarked by abnormal protein aggregation. It is known that across aging, sleep quality becomes less efficient and protein homeostatic regulatory mechanisms deteriorate. There is a known relationship between extended wakefulness and poorly consolidated sleep and an increase in cellular stress. In an aged population, when sleep is chronically poor, and proteostatic regulatory mechanisms are less efficient, the cell is inundated with misfolded proteins and suffers a collapse in homeostasis. In this review article, we explore the interplay between aging, sleep quality, and proteostasis and how these processes are implicated in the development and progression of neurodegenerative diseases like Alzheimer’s disease (AD). We also present data suggesting that reducing cellular stress and improving proteostasis and sleep quality could serve as potential therapeutic solutions for the prevention or delay in the progression of these diseases.
Introduction
Sleep is found in all animals that have been studied to date. It is a universal and crucial aspect of overall health. While the specific functions of sleep are still actively researched, some benefits of sleep, including memory consolidation (Stickgold and Walker, 2007; Diekelmann and Born, 2010) and management of cellular processes (Benington and Heller, 1995; Cirelli et al., 2004), have been demonstrated. However, it is known that sleep quality decreases with age (Manderville and Wetmore, 2017) and poor sleep is often a symptom of many aged-related neurodegenerative diseases, such as Alzheimer’s disease (AD; Ju et al., 2013). This begs the question to what extent is sleep causally related to the progression of aging and the symptoms of neurodegeneration. Perhaps the most effective way to examine this question is to investigate cellular health at the molecular level. Most neurodegenerative diseases display protein aggregation as a main feature of the disease, suggesting that proteostatic dysfunction could be causally related to the development and progression of these diseases. Proteostasis, or protein homeostasis, is maintained by quality control systems and signal transduction pathways such as the unfolded protein response (UPR; Braakman and Hebert, 2013). The UPR is initiated in the endoplasmic reticulum (ER), which is a major site of protein folding and processing. The ER is responsible for ensuring proper protein folding of all secretory and membrane proteins, through the use of other chaperone proteins. Protein misfolding due to changes in the ER environment such as redox, energy, pH and calcium perturbations activate the UPR (Kaufman, 2002; Ron, 2002). Activation of the UPR is an adaptive response and works in the short term to restore proteostasis (Koga et al., 2011). Importantly, as we age, the protein regulatory mechanisms of the cell deteriorate, as does sleep quality (Koga et al., 2011). This review will explore the interplay of sleep quality and protein homeostatic changes across aging, with a focus on the UPR, and the implications that these changes have for the development, progression, and treatment of neurodegenerative diseases. The goal of this is review is to present a cellular perspective on sleep and aging in terms of neurodegenerative diseases that can have broad impacts on the overall health of the individual across the normal aging process, and can serve as a potential therapeutic target for these diseases.
Sleep and Proteostasis
Sleep is a ubiquitous physiological process. The current theory for the physiological regulation of sleep is the two-process model of sleep (Borbély, 1982). This model suggests that there is a circadian component to regulate the timing of sleep across the 24-h day, as well as a homeostatic process that regulates sleep based on the homeostatic regulatory processes of the cell (Borbély, 1982). The circadian process is regulated by light across the 24-h day as well as the cycling of proteins such as melatonin (Cassone, 1990; Brown, 1994). For the purposes of this review, however, we will focus on the homeostatic process, which regulates sleep on a cellular/molecular level based on cellular demands. This hypothesis posits that there is a buildup of molecules that correlate with the duration of wakefulness. Extending wakefulness, therefore, results in an accumulation of molecules and proteins commensurate with the prior duration of wakefulness.
The waking cell is incredibly active transcribing DNA and translating proteins that are necessary to maintain wake function. As mentioned previously, the ER is the site of secretory and membrane protein synthesis in the cell, with the neuron being no exception. The ER of neurons extends through the entire axon and cell body of the neuron (Berridge, 2002; Ramírez and Couve, 2011). The vastness of this organelle, relative to the size of the neuron itself, indicates that protein synthesis is especially critical for neural function. Newly synthesized proteins are located in the ER lumen, which contains molecular chaperones that bind to the newly formed proteins in order to assist them in achieving their appropriate conformation. Once proteins are properly folded, they are transferred out of the ER to serve their cellular function. If the proteins cannot be properly folded, they are targeted to the proteasome for degradation to prevent aggregation (Ellgaard et al., 1999; Ellgaard and Helenius, 2001). Despite quality checkpoints and the presence of molecular chaperones, some proteins will be misfolded and will accumulate in the lumen of the ER.
There are instances when protein accumulation in the ER is too overwhelming for chaperone proteins alone. This state, once reached, is referred to as ER stress, which initiates a cascade of cellular signaling collectively referred to as the UPR, to cope with the aggregated proteins in the lumen of the ER (Berridge, 2002). This UPR has many downstream consequences for the cell and affects many important cellular mechanisms, including metabolic pathways (Bravo et al., 2013). The UPR activates three main types of protective responses: up-regulation of chaperone proteins such as BiP [also known as glucose regulated protein 78 (GRP78) or Hspa5], down-regulation of protein translation, and increasing degradation of misfolded proteins. When there is extended stress on the ER and these three mechanisms of the UPR are not sufficient to relieve that stress, apoptotic pathways are activated, ultimately leading to cell death (see reviews by Ron and Walter, 2007); and (Szegezdi et al., 2006). Interestingly, several microarray studies have shown that protein translation factors and ER resident factors such as BiP that are involved in these pathways of the UPR are increased with prolonged wakefulness, or sleep deprivation (Cirelli and Tononi, 2000; Cirelli et al., 2004; Naidoo et al., 2005; Mackiewicz et al., 2007; Naidoo et al., 2007). These observations suggest that protein homeostatic mechanisms could be linked to sleep/wake patterns.
There are three ER transmembrane proteins that serve as the “stress detectors” of the cell; PKR-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6; Zhang and Kaufman, 2004; Ron and Walter, 2007). BiP is bound to these three receptors under normal cellular conditions. When misfolded proteins are present, BiP dissociates from these receptors, thereby activating their signaling pathways, and binds to the unfolded proteins. Activation of IRE1 results in downstream signals that increase molecules involved in endoplasmic reticulum associated degradation (ERAD), targeting misfolded proteins for degradation. ATF6 activation results in the ultimate translation of more chaperone proteins to assist in protein folding. Finally, PERK activation leads to a temporary global reduction in protein synthesis by inhibiting translation (Harding et al., 2000; Hetz and Mollereau, 2014). Together, these processes allow the cell to cope with or manage accumulated misfolded proteins, and we know that these systems are activated in states of sleep deprivation. Specifically, several studies have shown that sleep deprivation leads to an increase in levels of the chaperone protein, BiP/GRP78, as well as activation of the PERK pathway of the UPR (Cirelli et al., 2004; Naidoo et al., 2005, 2007). Importantly, recovery sleep following sleep deprivation in Drosophila leads to a reduction in BiP levels (Naidoo et al., 2007).
Autophagy is another well-studied proteostasis pathway that is activated by the integrated stress response downstream of the UPR (see review by Kroemer et al., 2010). Autophagy leads to the degradation of aggregated proteins and defective organelles and is activated when protein homeostasis cannot be achieved by the cell, leading to apoptosis. It is known from studies in yeast that ER stress induces the autophagy response and that a downstream signaling molecule, Atg1, is necessary to induce autophagosome formation (Yorimitsu et al., 2006). In particular, it has been shown that the IRE1-JNK pathway is necessary for autophagy activation (Ogata et al., 2006). Further, ER stress molecules negatively regulate the AKT/mTOR pathway, which leads to an induction in autophagy (Qin et al., 2010). Recent data suggest that autophagy is regulated by the circadian clock with a direct role for Rev-erbα and transcription factor CEBPB linking these two processes (Huang et al., 2016). Interestingly, the cycling of autophagy proteins in hippocampal tissue is altered with sleep fragmentation (He et al., 2016). It is possible that sleep and the circadian clock could together influence how the cell is able to degrade aggregated proteins or dysfunctional organelles.
Fascinatingly, deep slow wave sleep that occurs during non-rapid eye movement (NREM) sleep, reflects the intensity of the previous waking period. The greater the previous bout of wakefulness, the longer the duration of slow wave sleep (Borbély et al., 1989). According to (Tononi and Cirelli, 2003) this slow-wave pattern could be involved in homeostatic regulation. They propose that wakefulness is associated with synaptic potentiation and slow-wave activity is associated with the corresponding synaptic downscaling, together resulting in the beneficial consequences of sleep on cognition. This hypothesis specifically addresses a role for sleep in the regulation of protein homeostasis in the brain in terms of the dynamic synaptic changes that occur with learning and daily neural activity.
Aging: Effects on Sleep and Protein Homeostatic Regulation
The quality of sleep changes across the healthy aging process. In general, as humans reach maturity, some studies suggest that sleep need decreases (Manderville and Wetmore, 2017). Across aging, there are key changes in sleep architecture. Some of these changes include increased sleep onset latency, shorter sleep duration, impaired sleep consolidation by increased awakening, increased daytime sleepiness, decreased melatonin levels, and reduced the amount of deep slow wave sleep (Welsh et al., 1986; Pandi-Perumal et al., 2002; Wolkove et al., 2007; Yaffe et al., 2014; Mander et al., 2017 and references therein). Importantly, these age-related changes in sleep characteristics are conserved across many animal species studied (Mendelson and Bergmann, 1999; Koh et al., 2006; Naidoo et al., 2008; Wimmer et al., 2013; Brown et al., 2014). While sleep need decreases with age, the fact that sleep remains necessary even in adulthood in many species underscores that idea that sleep has been preserved across evolution because it serves a critical function.
The age-related alterations in sleep characteristics could be related to, or even contribute to, cognitive impairment. Some work has shown that poor slow-wave oscillations and sleep spindles during NREM sleep in aged individuals is linked to poor memory and forgetting (Helfrich et al., 2018). Interestingly, other work has shown that sleep quality in middle-aged individuals could affect cognitive abilities later in life. In this middle-aged population, sleep quality was positively correlated with working memory and visual-spatial episodic memory (Rana et al., 2018). Deficits in sleep at this stage could be indicative of increased risk for cognitive decline at a later age, and improving sleep quality during mid-life, could reduce susceptibility to cognitive decline. Other studies have shown that there is a correlation between poor sleep quality and working memory, long term memory, verbal knowledge, and visuospatial reasoning (Schmutte et al., 2007; Nebes et al., 2009). Further, sleep fragmentation and increased daytime sleepiness are associated with increased cognitive impairment (Ohayon and Vecchierini, 2002; Naismith et al., 2010; Jaussent et al., 2012). Importantly, sleep disruptions such as those discussed here are also associated with neurodegenerative diseases, such as AD (Ju et al., 2013; Lim et al., 2013). While the biological mechanisms and pathways that could underlie this relationship between sleep and cognition are poorly understood, it is clear that there is a connection between them with respect to aging.
Furthermore, with aging, the protein quality control systems discussed above become dysfunctional. The ER stress response becomes less adaptive as we age, resulting in reduced efficiency of the refolding aspects of the UPR and increasing the prominence of the apoptotic pathways (Hussain and Ramaiah, 2007; Naidoo et al., 2008; Brown et al., 2014). Interestingly, protein aggregations as a result of maladaptive UPR function occur in almost all of the tissues of an aged organism (see review by Koga et al., 2011). Deficits in chaperone protein function have also been reported in age-related diseases (Macario and Conway de Macario, 2002; Nuss et al., 2008), as well as overall decreases in the levels of chaperone proteins in aged wildtype rodents (Naidoo et al., 2008). Conversely, increasing chaperone protein levels results in an increase in longevity in flies and worms (Tatar et al., 1997; Kang et al., 2002; Morley and Morimoto, 2004). In addition, improving protein homeostasis by treatment with a small chemical chaperone molecule, 4-phenyl butyrate (PBA), improves sleep in aged flies (Brown et al., 2014). Further, work done in C. elegans has demonstrated that some protein misfolding earlier in adulthood can lead to the eventual overall collapse of protein homeostasis in older organisms (Ben-Zvi et al., 2009). It is clear that in addition to sleep quality changing with age so does overall protein homeostasis. What remains unclear is how these phenomena are related and if improving one is sufficient to rescue the other, particularly in disease states.
Neurodegenerative Diseases: Intersection of Age-Related Changes in Sleep and Proteostasis
Protein misfolding and aggregation is a key feature of many neurodegenerative diseases, particularly AD (Cornejo and Hetz, 2013), Parkinson’s disease (PD; Breydo et al., 2012), Huntington’s disease (HD; Arrasate and Finkbeiner, 2012), fronto-temporal dementia (FTD; Ling et al., 2013), and amyotrophic lateral sclerosis (ALS; Lindberg et al., 2005). Most of those who suffer from neurodegenerative diseases are aged individuals, which is a population that, as discussed above, displays reduced sleep quality and a decreased ability to maintain protein homeostasis due to aged-related dysfunctions in cellular regulatory mechanisms [(Selkoe, 2003) and see the review by Hetz and Saxena (2017)]. Therefore, it is reasonable to consider that these aged-related changes in both sleep and protein homeostasis could increase the risk of neurodegeneration by ultimately leading to increases in levels of abnormal protein aggregates and an inability for individual cells to ameliorate this homeostatic imbalance.
The over activation of the UPR/maladaptive ER stress response could contribute to neurodegenerative disease. The expression of disease-associated proteins that aggregate and cause an imbalance in protein homeostasis have long-term consequences on the folding of other proteins. Part of the downstream signaling branches of the UPR reduces translation (Harding et al., 2000). This reduction could have negative consequences for synaptic health, memory formation, and basic cellular function, as none of these can occur without proper protein quantities. To this end, some work has shown that inhibiting PERK improves memory (see review by Halliday and Mallucci, 2014). This feature of protein regulation demonstrates that the cell does not have the capacity to deal with chronic protein misfolding, suggesting that the quality control mechanisms may be maladaptive to chronic ER stress in these disease states (Morimoto, 2008; Brown and Naidoo, 2012).
Several animal models of neurodegenerative diseases, such as ALS, FTD, AD, PD, and HD display an accumulation of misfolded proteins and an activation of the UPR (Reddy et al., 1999; Rao et al., 2002; Hoozemans et al., 2005); see review by (Hetz and Saxena, 2017). In AD, increased levels of β-amyloid precursor proteins in neurons sensitize these cells to ER stress (Chafekar et al., 2008), for review see Cornejo and Hetz (2013). Studies have shown that amyloid-β, which can accumulate in the ER lumen, can disrupt ER calcium homeostasis, leading to a proapoptotic stress response (Cornejo and Hetz, 2013). Phosphorylation of PERK and its downstream target eIF2α, a branch of the UPR, is observed in AD patients (Chang et al., 2002). Further, reduction of PERK rescues memory and cholinergic neurodegeneration by preventing increases in BACE1, which is involved in Aβ production (Devi and Ohno, 2014). Some evidence in PD has shown that ER stress in the form of increased phosphorylation of PERK and eIF2α occurs in the substantia nigra early in the disease (Hoozemans et al., 2012). In addition, another study showed that iPSCs derived from PD patients display early ER stress, which leads to a disruption of proteostasis (Chung et al., 2013). In HD, ER calcium homeostasis has been identified in patient-derived iPSCs, which could contribute to protein misfolding (Nekrasov et al., 2016), and increased levels of UPR effectors BiP and CHOP have been observed in HD patients (Carnemolla et al., 2009; Kalathur et al., 2015). Finally, all three UPR branches are expressed in ALS (Atkin et al., 2008; Hetz et al., 2009; Ito et al., 2009; Saxena et al., 2009; Kiskinis et al., 2014).
In addition, autophagy is a prominent pathological feature of many neurodegenerative diseases (see review Martinez-Vicente and Cuervo, 2007). In PD, α-synuclein can be degraded via autophagy (Webb et al., 2003), and it is known that the effectiveness of these degradation pathways decays with age (Martinez-Vicente et al., 2005). In AD, the lysosomal system that provides enzymes for degradation of toxic proteins in autophagosomes becomes inefficient (Yu et al., 2005). Further, in HD, macroautophagy is upregulated in response to Huntingtin aggregation (Kegel et al., 2000). It is possible that with age and poor sleep quality, increased levels of ER stress coupled with a maladaptive autophagic response all contribute to the development and progression of neurodegenerative disease.
Several studies using animal models of neurodegenerative diseases, specifically AD, have shown that increasing protein chaperone levels that assist in the folding of proteins, and thereby improve these protein quality control mechanisms, the progression of these diseases can be ameliorated (Ricobaraza et al., 2011; Wiley et al., 2011; Cuadrado-Tejedor et al., 2013; Hafycz et al., 2019). In models of ALS, treatment with eIF2α phosphatase inhibitors improved motor neuron performance and survival (Jiang et al., 2014; Wang et al., 2014; Das et al., 2015). In mouse models of FTD, PERK inhibition resulted in improved neuronal function (Radford et al., 2015). This data together suggests that reducing the activation of the UPR in these disease models can, to a degree, rescue progression of the diseases. Further evidence demonstrates that modulating ER stress signaling in an effort to make it more effective results in neuroprotection (Hughes and Mallucci, 2019). Together, these studies suggest that cellular stress underlies many neurodegenerative diseases and could serve as a therapeutic target for the treatment of patients suffering with these disorders.
As mentioned previously, most patients suffering from protein misfolding neurodegenerative diseases also report sleep disruptions (see Table 1). AD and PD in particular report a high percentage of patients suffering from sleep alterations such as increased sleep fragmentation, increased daytime sleepiness, and REM sleep disruptions (Factor et al., 1990; Iranzo, 2011; Ju et al., 2013). Additional studies examining ALS and FTD show that in a mutant animal model of these diseases expressing FUS protein demonstrate sleep and circadian disturbances prior to the cognitive deficits (Hetz and Saxena, 2017; Zhang et al., 2018). Some work has also investigated the effect of neurodegenerative disease, specifically AD, on the circadian clock, suggesting that once the neurodegeneration begins in sleep and circadian rhythm regulatory regions, the brain becomes even more susceptible to the effects of neurodegeneration (see review by Chauhan et al., 2017 and citations therein).
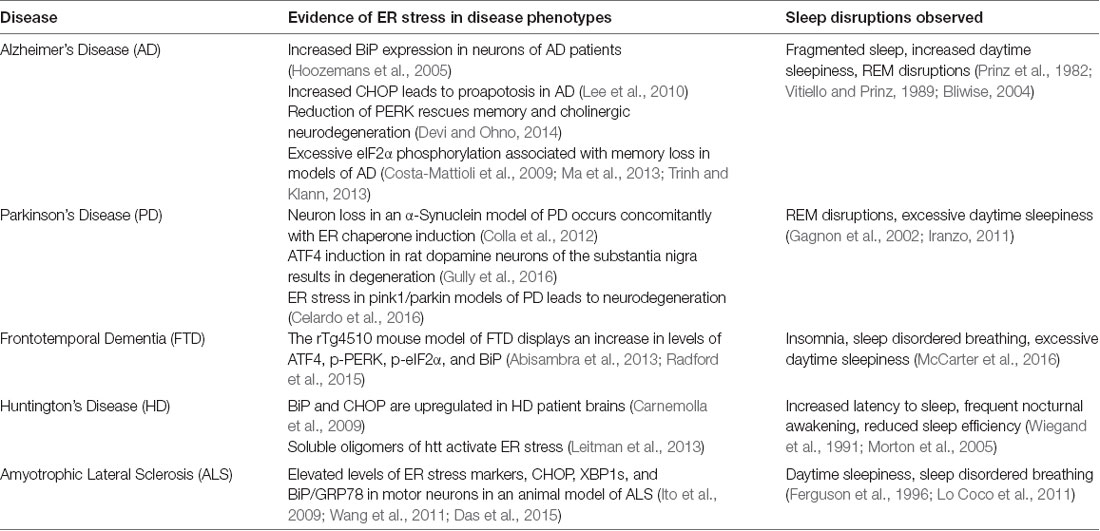
Table 1. Neurodegenerative diseases that display alterations in protein homeostatic regulation and sleep disruptions.
An ongoing investigation into these diseases is required to establish which comes first, protein homeostatic disruptions or sleep disturbances, and in particular, if sleep quality is improved, can the spread of these protein misfolding diseases be delayed or halted altogether?
Concluding Remarks
There is sufficient evidence that cellular health and sleep quality go hand-in-hand. Across the aging process, both sleep quality and protein homeostatic mechanisms decrease in efficiency. Many studies demonstrate that poor sleep quality is correlated with poor memory and cognition. While it is impossible to examine protein homeostasis and protein levels of neurons in humans, evidence from neurodegenerative diseases clearly shows increases in cellular stress and poor regulation of protein folding, leading to aggregations commonly seen in these diseases. What remains unclear is the link between sleep and protein homeostasis in these disease states. Some evidence suggests that it is the interplay between poor sleep and maladaptive protein regulation that contributes to age-related neurodegenerative diseases like AD. We suggest, since both sleep quality and the efficacy of protein homeostatic maintenance machinery decreases with aging, it seems plausible that these two key aspects of aging could contribute to the onset of neurodegenerative age-related diseases. In accordance with this perspective, improving sleep quality and relieving cellular stress could serve as critical target areas for therapeutic intervention of these devastating diseases.
Author Contributions
JH and NN wrote and edited the manuscript.
Funding
This work was supported by National Institutes of Health (NIH) AG17628.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O’Leary, J. C. III., Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 33, 9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Benington, J. H., and Heller, H. C. (1995). Restoration of brain energy metabolism as the function of sleep. Prog. Neurobiol. 45, 347–360. doi: 10.1016/0301-0082(94)00057-o
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. U S A 106, 14914–14919. doi: 10.1073/pnas.0902882106
Berridge, M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249. doi: 10.1016/s0143416002001823
Bliwise, D. L. (2004). Sleep disorders in Alzheimer’s disease and other dementias. Clin. Cornerstone 6, S16–S28. doi: 10.1016/S1098-3597(04)90014-2
Borbély, A. A., Achermann, P., Trachsel, L., and Tobler, I. (1989). Sleep initiation and initial sleep intensity: interactions of homeostatic and circadian mechanisms. J. Biol. Rhythms 4, 37–48. doi: 10.1177/074873048900400205
Braakman, I., and Hebert, D. N. (2013). Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5:a013201. doi: 10.1101/cshperspect.a013201
Bravo, R., Parra, V., Gatica, D., Rodriguez, A. E., Torrealba, N., Paredes, F., et al. (2013). Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 301, 215–290. doi: 10.1016/B978-0-12-407704-1.00005-1
Breydo, L., Wu, J. W., and Uversky, V. N. (2012). α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 1822, 261–285. doi: 10.1016/j.bbadis.2011.10.002
Brown, G. M. (1994). Light, melatonin and the sleep-wake cycle. J. Psychiatry Neurosci. 19, 345–353.
Brown, M. K., Chan, M. T., Zimmerman, J. E., Pack, A. I., Jackson, N. E., and Naidoo, N. (2014). Aging induced endoplasmic reticulum stress alters sleep and sleep homeostasis. Neurobiol. Aging 35, 1431–1441. doi: 10.1016/j.neurobiolaging.2013.12.005
Brown, M. K., and Naidoo, N. (2012). The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3:263. doi: 10.3389/fphys.2012.00263
Carnemolla, A., Fossale, E., Agostoni, E., Michelazzi, S., Calligaris, R., De Maso, L., et al. (2009). Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J. Biol. Chem. 284, 18167–18173. doi: 10.1074/jbc.m109.018325
Cassone, V. M. (1990). Effects of melatonin on vertebrate circadian systems. Trends Neurosci. 13, 457–464. doi: 10.1016/0166-2236(90)90099-v
Celardo, I., Costa, A. C., Lehmann, S., Jones, C., Wood, N., Mencacci, N. E., et al. (2016). Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 7:e2271. doi: 10.1038/cddis.2016.173
Chafekar, S. M., Zwart, R., Veerhuis, R., Vanderstichele, H., Baas, F., and Scheper, W. (2008). Increased Aβ1-42 production sensitizes neuroblastoma cells for ER stress toxicity. Curr. Alzheimer Res. 5, 469–474. doi: 10.2174/156720508785908883
Chang, R. C., Wong, A. K., Ng, H. K., and Hugon, J. (2002). Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13, 2429–2432. doi: 10.1097/01.wnr.0000048020.74602.bb
Chauhan, R., Chen, K. F., Kent, B. A., and Crowther, D. C. (2017). Central and peripheral circadian clocks and their role in Alzheimer’s disease. Dis. Model Mech. 10, 1187–1199. doi: 10.1242/dmm.030627
Chung, C. Y., Khurana, V., Auluck, P. K., Tardiff, D. F., Mazzulli, J. R., Soldner, F., et al. (2013). Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. doi: 10.1126/science.1245296
Cirelli, C., Gutierrez, C. M., and Tononi, G. (2004). Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41, 35–43. doi: 10.1016/s0896-6273(03)00814-6
Cirelli, C., and Tononi, G. (2000). Gene expression in the brain across the sleep-waking cycle. Brain Res. 885, 303–321. doi: 10.1016/s0006-8993(00)03008-0
Colla, E., Jensen, P. H., Pletnikova, O., Troncoso, J. C., Glabe, C., and Lee, M. K. (2012). Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 32, 3301–3305. doi: 10.1523/jneurosci.5368-11.2012
Cornejo, V. H., and Hetz, C. (2013). The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 35, 277–292. doi: 10.1007/s00281-013-0373-9
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26. doi: 10.1016/j.neuron.2008.10.055
Cuadrado-Tejedor, M., Ricobaraza, A. L., Torrijo, R., Franco, R., and Garcia-Osta, A. (2013). Phenylbutyrate is a multifaceted drug that exerts neuroprotective effects and reverses the Alzheimer’s disease-like phenotype of a commonly used mouse model. Curr. Pharm. Des. 19, 5076–5084. doi: 10.2174/1381612811319280006
Das, I., Krzyzosiak, A., Schneider, K., Wrabetz, L., D’Antonio, M., Barry, N., et al. (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242. doi: 10.1126/science.aaa4484
Devi, L., and Ohno, M. (2014). PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 35, 2272–2281. doi: 10.1016/j.neurobiolaging.2014.04.031
Diekelmann, S., and Born, J. (2010). The memory function of sleep. Nat. Rev. Neurosci. 11, 114–126. doi: 10.1038/nrn2762
Ellgaard, L., and Helenius, A. (2001). ER quality control: towards an understanding at the molecular level. Curr. Opin. Cell Biol. 13, 431–437. doi: 10.1016/s0955-0674(00)00233-7
Ellgaard, L., Molinari, M., and Helenius, A. (1999). Setting the standards: quality control in the secretory pathway. Science 286, 1882–1888. doi: 10.1126/science.286.5446.1882
Factor, S. A., McAlarney, T., Sanchez-Ramos, J. R., and Weiner, W. J. (1990). Sleep disorders and sleep effect in Parkinson’s disease. Mov. Disord. 5, 280–285. doi: 10.1002/mds.870050404
Ferguson, K. A., Strong, M. J., Ahmad, D., and George, C. F. (1996). Sleep-disordered breathing in amyotrophic lateral sclerosis. Chest 110, 664–669. doi: 10.1378/chest.110.3.664
Gagnon, J. F., Bédard, M. A., Fantini, M. L., Petit, D., Panisset, M., Rompre, S., et al. (2002). REM sleep behavior disorder and REM sleep without atonia in Parkinson’s disease. Neurology 59, 585–589. doi: 10.1212/wnl.59.4.585
Gully, J. C., Sergeyev, V. G., Bhootada, Y., Mendez-Gomez, H., Meyers, C. A., Zolotukhin, S., et al. (2016). Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 627, 36–41. doi: 10.1016/j.neulet.2016.05.039
Hafycz, J., Zhu, J., and Naidoo, N. (2019). A role for protein homeostasis in sleep: implications for alzheimer’s disease. Sleep 42:A121. doi: 10.1093/sleep/zsz067.296
Halliday, M., and Mallucci, G. R. (2014). Targeting the unfolded protein response in neurodegeneration: a new approach to therapy. Neuropharmacology 76, 169–174. doi: 10.1016/j.neuropharm.2013.08.034
Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904. doi: 10.1016/s1097-2765(00)80330-5
He, Y., Cornelissen-Guillaume, G. G., He, J., Kastin, A. J., Harrison, L. M., and Pan, W. (2016). Circadian rhythm of autophagy proteins in hippocampus is blunted by sleep fragmentation. Chronobiol. Int. 33, 553–560. doi: 10.3109/07420528.2015.1137581
Helfrich, R. F., Mander, B. A., Jagust, W. J., Knight, R. T., and Walker, M. P. (2018). Old brains come uncoupled in sleep: slow wave-spindle synchrony, brain atrophy, and forgetting. Neuron 97, 221.e4–230.e4. doi: 10.1016/j.neuron.2017.11.020
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Hoozemans, J. J., van Haastert, E. S., Nijholt, D. A., Rozemuller, A. J., and Scheper, W. (2012). Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 10, 212–215. doi: 10.1159/000334536
Hoozemans, J. J., Veerhuis, R., Van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., et al. (2005). The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 110, 165–172. doi: 10.1007/s00401-005-1038-0
Huang, G., Zhang, F., Ye, Q., and Wang, H. (2016). The circadian clock regulates autophagy directly through the nuclear hormone receptor Nr1d1/Rev-erbα and indirectly via Cebpb/(C/ebpβ) in zebrafish. Autophagy 12, 1292–1309. doi: 10.1080/15548627.2016.1183843
Hughes, D., and Mallucci, G. R. (2019). The unfolded protein response in neurodegenerative disorders—therapeutic modulation of the PERK pathway. FEBS J. 286, 342–355. doi: 10.1111/febs.14422
Hussain, S. G., and Ramaiah, K. V. (2007). Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 355, 365–370. doi: 10.1016/j.bbrc.2007.01.156
Iranzo, A. (2011). Sleep-wake changes in the premotor stage of Parkinson disease. J. Neurol. Sci. 310, 283–285. doi: 10.1016/j.jns.2011.07.049
Ito, Y., Yamada, M., Tanaka, H., Aida, K., Tsuruma, K., Shimazawa, M., et al. (2009). Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 36, 470–476. doi: 10.1016/j.nbd.2009.08.013
Jaussent, I., Bouyer, J., Ancelin, M. L., Berr, C., Foubert-Samier, A., Ritchie, K., et al. (2012). Excessive sleepiness is predictive of cognitive decline in the elderly. Sleep 35, 1201–1207. doi: 10.5665/sleep.2070
Jiang, H. Q., Ren, M., Jiang, H. Z., Wang, J., Zhang, J., Yin, X., et al. (2014). Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 277, 132–138. doi: 10.1016/j.neuroscience.2014.03.047
Ju, Y. E., McLeland, J. S., Toedebusch, C. D., Xiong, C., Fagan, A. M., Duntley, S. P., et al. (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 70, 587–593. doi: 10.1001/jamaneurol.2013.2334
Kalathur, R. K., Giner-Lamia, J., Machado, S., Barata, T., Ayasolla, K. R., and Futschik, M. E. (2015). The unfolded protein response and its potential role in Huntington’s disease elucidated by a systems biology approach. F1000Res. 4:103. doi: 10.12688/f1000research.6358.2
Kang, H. L., Benzer, S., and Min, K. T. (2002). Life extension in Drosophila by feeding a drug. Proc. Natl. Acad. Sci. U S A 99, 838–843. doi: 10.1073/pnas.022631999
Kaufman, R. J. (2002). Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 110, 1389–1398. doi: 10.1172/JCI16886
Kegel, K. B., Kim, M., Sapp, E., McIntyre, C., Castaño, J. G., Aronin, N., et al. (2000). Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20, 7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Koga, H., Kaushik, S., and Cuervo, A. M. (2011). Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215. doi: 10.1016/j.arr.2010.02.001
Koh, K., Evans, J. M., Hendricks, J. C., and Sehgal, A. (2006). A Drosophila model for age-associated changes in sleep:wake cycles. Proc. Natl. Acad. Sci. U S A 103, 13843–13847. doi: 10.1073/pnas.0605903103
Kroemer, G., Mariño, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. doi: 10.1016/j.molcel.2010.09.023
Lee, J. H., Won, S. M., Suh, J., Son, S. J., Moon, G. J., Park, U. J., et al. (2010). Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 42, 386–394. doi: 10.3858/emm.2010.42.5.040
Leitman, J., Ulrich Hartl, F., and Lederkremer, G. Z. (2013). Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 4:2753. doi: 10.1038/ncomms3753
Lim, A. S., Kowgier, M., Yu, L., Buchman, A. S., and Bennett, D. A. (2013). Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 36, 1027–1032. doi: 10.5665/sleep.2802
Lindberg, M. J., Byström, R., Boknäs, N., Andersen, P. M., and Oliveberg, M. (2005). Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc. Natl. Acad. Sci. U S A 102, 9754–9759. doi: 10.1073/pnas.0501957102
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Lo Coco, D., Mattaliano, P., Spataro, R., Mattaliano, A., and La Bella, V. (2011). Sleep-wake disturbances in patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 839–842. doi: 10.1136/jnnp.2010.228007
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Macario, A. J., and Conway de Macario, E. (2002). Sick chaperones and ageing: a perspective. Ageing Res. Rev. 1, 295–311. doi: 10.1016/s1568-1637(01)00005-8
Mackiewicz, M., Shockley, K. R., Romer, M. A., Galante, R. J., Zimmerman, J. E., Naidoo, N., et al. (2007). Macromolecule biosynthesis: a key function of sleep. Physiol. Genomics 31, 441–457. doi: 10.1152/physiolgenomics.00275.2006
Mander, B. A., Winer, J. R., and Walker, M. P. (2017). Sleep and human aging. Neuron 94, 19–36. doi: 10.1016/j.neuron.2017.02.004
Manderville, R. A., and Wetmore, S. D. (2017). Mutagenicity of ochratoxin a: role for a carbon-linked C8-deoxyguanosine adduct? J. Agric. Food Chem. 65, 7097–7105. doi: 10.1021/acs.jafc.6b03897
Martinez-Vicente, M., and Cuervo, A. M. (2007). Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 6, 352–361. doi: 10.1016/s1474-4422(07)70076-5
Martinez-Vicente, M., Sovak, G., and Cuervo, A. M. (2005). Protein degradation and aging. Exp. Gerontol. 40, 622–633. doi: 10.1016/j.exger.2005.07.005
McCarter, S. J., St Louis, E. K., and Boeve, B. F. (2016). Sleep disturbances in frontotemporal dementia. Curr. Neurol. Neurosci. Rep. 16:85. doi: 10.1007/s11910-016-0680-3
Mendelson, W. B., and Bergmann, B. M. (1999). Age-related changes in sleep in the rat. Sleep 22, 145–150. doi: 10.1093/sleep/22.2.145
Morimoto, R. I. (2008). Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438. doi: 10.1101/gad.1657108
Morley, J. F., and Morimoto, R. I. (2004). Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 15, 657–664. doi: 10.1091/mbc.e03-07-0532
Morton, A. J., Wood, N. I., Hastings, M. H., Hurelbrink, C., Barker, R. A., and Maywood, E. S. (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 25, 157–163. doi: 10.1523/JNEUROSCI.3842-04.2005
Naidoo, N., Casiano, V., Cater, J., Zimmerman, J., and Pack, A. I. (2007). A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep 30, 557–565. doi: 10.1093/sleep/30.5.557
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/JNEUROSCI.5685-07.2008
Naidoo, N., Giang, W., Galante, R. J., and Pack, A. I. (2005). Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J. Neurochem. 92, 1150–1157. doi: 10.1111/j.1471-4159.2004.02952.x
Naismith, S. L., Rogers, N. L., Mackenzie, J., Hickie, I. B., and Lewis, S. J. (2010). The relationship between actigraphically defined sleep disturbance and REM sleep behaviour disorder in Parkinson’s disease. Clin. Neurol. Neurosurg. 112, 420–423. doi: 10.1016/j.clineuro.2010.02.011
Nebes, R. D., Buysse, D. J., Halligan, E. M., Houck, P. R., and Monk, T. H. (2009). Self-reported sleep quality predicts poor cognitive performance in healthy older adults. J. Gerontol. B Psychol. Sci. Soc. Sci. 64, 180–187. doi: 10.1093/geronb/gbn037
Nekrasov, E. D., Vigont, V. A., Klyushnikov, S. A., Lebedeva, O. S., Vassina, E. M., Bogomazova, A. N., et al. (2016). Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 11:27. doi: 10.1186/s13024-016-0092-5
Nuss, J. E., Choksi, K. B., DeFord, J. H., and Papaconstantinou, J. (2008). Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 365, 355–361. doi: 10.1016/j.bbrc.2007.10.194
Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231. doi: 10.1128/MCB.01453-06
Ohayon, M. M., and Vecchierini, M. F. (2002). Daytime sleepiness and cognitive impairment in the elderly population. Arch. Intern. Med. 162, 201–208. doi: 10.1001/archinte.162.2.201
Pandi-Perumal, S. R., Seils, L. K., Kayumov, L., Ralph, M. R., Lowe, A., Moller, H., et al. (2002). Senescence, sleep, and circadian rhythms. Ageing Res. Rev. 1, 559–604. doi: 10.1016/S1568-1637(02)00014-4
Prinz, P. N., Vitaliano, P. P., Vitiello, M. V., Bokan, J., Raskind, M., Peskind, E., et al. (1982). Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol. Aging 3, 361–370. doi: 10.1016/0197-4580(82)90024-0
Qin, L., Wang, Z., Tao, L., and Wang, Y. (2010). ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6, 239–247. doi: 10.4161/auto.6.2.11062
Radford, R. A., Morsch, M., Rayner, S. L., Cole, N. J., Pountney, D. L., and Chung, R. S. (2015). The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci. 9:414. doi: 10.3389/fncel.2015.00414
Ramírez, O. A., and Couve, A. (2011). The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol. 21, 219–227. doi: 10.1016/j.tcb.2010.12.003
Rana, B. K., Panizzon, M. S., Franz, C. E., Spoon, K. M., Jacobson, K. C., Xian, H., et al. (2018). Association of sleep quality on memory-related executive functions in middle age. J. Int. Neuropsychol. Soc. 24, 67–76. doi: 10.1017/s1355617717000637
Rao, R. V., Peel, A., Logvinova, A., del Rio, G., Hermel, E., Yokota, T., et al. (2002). Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 514, 122–128. doi: 10.1016/s0014-5793(02)02289-5
Reddy, P. H., Williams, M., and Tagle, D. A. (1999). Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 22, 248–255. doi: 10.1016/s0166-2236(99)01415-0
Ricobaraza, A., Cuadrado-Tejedor, M., and Garcia-Osta, A. (2011). Long-term phenylbutyrate administration prevents memory deficits in Tg2576 mice by decreasing Aβ. Front. Biosci. 3, 1375–1384. doi: 10.2741/e340
Ron, D. (2002). Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388. doi: 10.1172/JCI16784
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schmutte, T., Harris, S., Levin, R., Zweig, R., Katz, M., and Lipton, R. (2007). The relation between cognitive functioning and self-reported sleep complaints in nondemented older adults: results from the Bronx aging study. Behav. Sleep Med. 5, 39–56. doi: 10.1207/s15402010bsm0501_3
Stickgold, R., and Walker, M. P. (2007). Sleep-dependent memory consolidation and reconsolidation. Sleep Med. 8, 331–343. doi: 10.1016/j.sleep.2007.03.011
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Tatar, M., Khazaeli, A. A., and Curtsinger, J. W. (1997). Chaperoning extended life. Nature 390:30. doi: 10.1038/36237
Tononi, G., and Cirelli, C. (2003). Sleep and synaptic homeostasis: a hypothesis. Brain Res. Bull. 62, 143–150. doi: 10.1016/j.brainresbull.2003.09.004
Trinh, M. A., and Klann, E. (2013). Translational control by eIF2α kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol. Learn. Mem. 105, 93–99. doi: 10.1016/j.nlm.2013.04.013
Vitiello, M. V., and Prinz, P. N. (1989). Alzheimer’s disease. Sleep and sleep/wake patterns. Clin. Geriatr. Med. 5, 289–299. doi: 10.1016/S0749-0690(18)30679-7
Wang, J. W., Brent, J. R., Tomlinson, A., Shneider, N. A., and McCabe, B. D. (2011). The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest. 121, 4118–4126. doi: 10.1172/jci57883
Wang, L., Popko, B., and Roos, R. P. (2014). An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 23, 2629–2638. doi: 10.1093/hmg/ddt658
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.M300227200
Welsh, D. K., Richardson, G. S., and Dement, W. C. (1986). Effect of age on the circadian pattern of sleep and wakefulness in the mouse. J. Gerontol. 41, 579–586. doi: 10.1093/geronj/41.5.579
Wiegand, M., Möller, A. A., Schreiber, W., Lauer, C., and Krieg, J. C. (1991). Brain morphology and sleep EEG in patients with Huntington’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 240, 148–152. doi: 10.1007/bf02190755
Wiley, J. C., Pettan-Brewer, C., and Ladiges, W. C. (2011). Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 10, 418–428. doi: 10.1111/j.1474-9726.2011.00680.x
Wimmer, M. E., Rising, J., Galante, R. J., Wyner, A., Pack, A. I., and Abel, T. (2013). Aging in mice reduces the ability to sustain sleep/wake states. PLoS One 8:e81880. doi: 10.1371/journal.pone.0081880
Wolkove, N., Elkholy, O., Baltzan, M., and Palayew, M. (2007). Sleep and aging: 2. Management of sleep disorders in older people. CMAJ 176, 1449–1454. doi: 10.1503/cmaj.070335
Yaffe, K., Falvey, C. M., and Hoang, T. (2014). Connections between sleep and cognition in older adults. Lancet Neurol. 13, 1017–1028. doi: 10.1016/s1474-4422(14)70172-3
Yorimitsu, T., Nair, U., Yang, Z., and Klionsky, D. J. (2006). Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299–30304. doi: 10.1074/jbc.M607007200
Yu, W. H., Cuervo, A. M., Kumar, A., Peterhoff, C. M., Schmidt, S. D., Lee, J. H., et al. (2005). Macroautophagy—a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 171, 87–98. doi: 10.1083/jcb.200505082
Zhang, T., Jiang, X., Xu, M., Wang, H., Sang, X., Qin, M., et al. (2018). Sleep and circadian abnormalities precede cognitive deficits in R521C FUS knockin rats. Neurobiol. Aging 72, 159–170. doi: 10.1016/j.neurobiolaging.2018.08.025
Keywords: sleep, aging, proteostasis cell stress and aging, neurodegenarative disease, unfolded protein response
Citation: Hafycz JM and Naidoo NN (2019) Sleep, Aging, and Cellular Health: Aged-Related Changes in Sleep and Protein Homeostasis Converge in Neurodegenerative Diseases. Front. Aging Neurosci. 11:140. doi: 10.3389/fnagi.2019.00140
Received: 16 April 2019; Accepted: 23 May 2019;
Published: 11 June 2019.
Edited by:
Paula I. Moreira, University of Coimbra, PortugalReviewed by:
Cláudia Pereira, Faculty of Medicine, University of Coimbra, PortugalRussell H. Swerdlow, University of Kansas, United States
Copyright © 2019 Hafycz and Naidoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nirinjini N. Naidoo, bmFpZG9vQHBlbm5tZWRpY2luZS51cGVubi5lZHU=