 Bérénice Hatat1,2†
Bérénice Hatat1,2† Samir Yahiaoui1†
Samir Yahiaoui1† Cédric Lecoutey1
Cédric Lecoutey1 Audrey Davis1
Audrey Davis1 Thomas Freret3
Thomas Freret3 Michel Boulouard3
Michel Boulouard3 Sylvie Claeysen2
Sylvie Claeysen2 Christophe Rochais1*
Christophe Rochais1* Patrick Dallemagne1*
Patrick Dallemagne1*- 1Normandie Université, UNICAEN, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), Caen, France
- 2IGF, University of Montpellier, CNRS, INSERM, Montpellier, France
- 3Normandie Université, UNICAEN, INSERM, U1075, GIP CYCERON, COMETE, Caen, France
This work describes the conception, synthesis, in vitro and in vivo biological evaluation of novel Multi-Target Directed Ligands (MTDL) able to both activate 5-HT4 receptors, block 5-HT6 receptors and inhibit acetylcholinesterase activity (AChE), in order to exert a synergistic anti-amnesic effect, potentially useful in the treatment of Alzheimer’s disease (AD). Indeed, both activation of 5-HT4 and blockage of 5-HT6 receptors led to an enhanced acetylcholine release, suggesting it could lead to efficiently restoring the cholinergic neurotransmission deficit observed in AD. Furthermore, 5-HT4 receptor agonists are able to promote the non-amyloidogenic cleavage of the amyloid precursor protein (APP) and to favor the production of the neurotrophic protein sAPPα. Finally, we identified a pleiotropic compound, [1-(4-amino-5-chloro-2-methoxyphenyl)-3-(1-(3-methylbenzyl)piperidin-4-yl)propan-1-one fumaric acid salt (10)], which displayed in vivo an anti-amnesic effect in a model of scopolamine-induced deficit of working memory at a dose of 0.3 mg/kg.
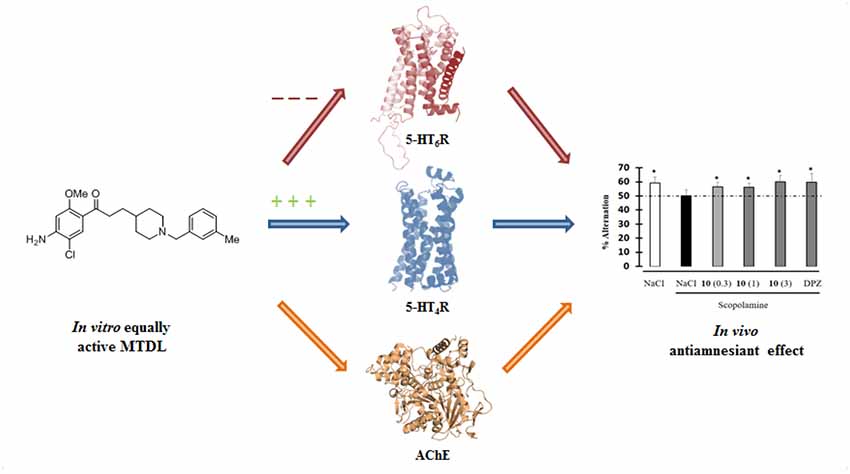
Graphical Abstract. Design of a MTDL able in vitro to both inactivate 5-HT6R and AChE and to activate 5-HT4R and finally displaying in vivo antiamnesiant effect.
Introduction
The pathogenesis of Alzheimer’s Disease (AD) is complex and related to the abnormality and dysfunction of multi-systems. Thus, to be potentially more effective, a treatment might consider more than a single target such as acetylcholinesterase (AChE) the main focus of marketed AD drugs.
Within this framework, the design of some pleiotropic ligands known as Multi-Target Directed Ligands (MTDL; Cavalli et al., 2008) appears as a promising approach to tackle the complex origin of the disease as demonstrated recently for several G Protein-Coupled Receptors (GPCRs) and enzymes (Dolles and Decker, 2017; Dolles et al., 2018). We recently described compound 1 (donecopride), which is currently a novel preclinical drug candidate exhibiting both an in vitro dual-binding site AChE inhibitory activity and a serotonergic subtype 4 receptor (5-HT4R) agonist effect leading to in vivo procognitive and anti-amnesic effects in mice (Figure 1; Lecoutey et al., 2014; Rochais et al., 2015). Indeed, 5-HT4R agonists are able to promote the “non-amyloidogenic” cleavage of the amyloid precursor protein (APP) by α-secretase, inducing the decrease in amyloid-β peptide (Aβ) production in primary neurons (Lezoualc’h, 2007; Russo et al., 2009), the release of soluble and neuroprotective sAPPα protein (Cho and Hu, 2007), and the in vivo improvement of memory in rodents (Lelong et al., 2003; Nirogi et al., 2018).
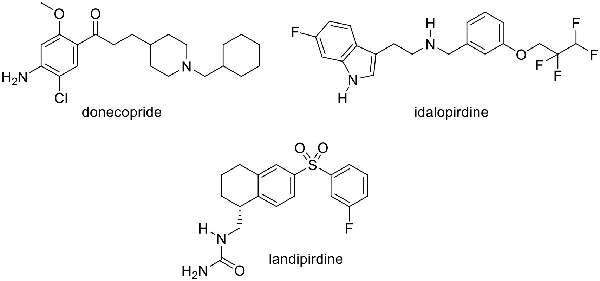
Figure 1. Structure of donecopride, idalopirdine and landipirdine.
5-HT4R is a GPCR. Interestingly, another serotonin GPCR, the serotonergic subtype 6 receptor (5-HT6R) appears as a valuable target to treat cognitive impairments in the field of neurodegenerative disorders, notably AD (Karila et al., 2015). In fact, its blockade confers to 5-HT6R antagonists procognitive effects (Benhamú et al., 2014). Among these 5-HT6R antagonists, 2 (idalopirdine) was studied in phase 3 of clinical trials (Wilkinson et al., 2014), and 3 (landipirdine), a dual antagonist of the 5-HT6 and 5-HT2A receptors is currently under investigation in the field of Parkinson Disease (Figure 1; Ellis and Fell, 2017).
The procognitive activity of 5-HT6R antagonists is probably mediated by modulation of neurotransmitters’ release. Indeed, 5-HT4R activation enhances the liberation of acetylcholine (Kilbinger and Wolf, 1992; Consolo et al., 1994), dopamine (Steward et al., 1996; Lucas et al., 2001) and serotonin (Ge and Barnes, 1996), while blockade of 5-HT6R enhances the liberation of acetylcholine (Shirazi-Southall et al., 2002; Riemer et al., 2003; Hirst et al., 2006; Marcos et al., 2006; Zhang et al., 2007) and glutamate (Dawson et al., 2000, 2001). Consequently, 5-HT6R antagonists can improve cognition since they limit the activation of the mTOR pathway (de Bruin and Kruse, 2015). The acute administration of 5-HT4R agonists or 5-HT6R antagonists leads to procognitive effects. However, recently we demonstrated that chronic 5-HT4R activation or chronic 5-HT6R blockade, also improved memory performances in object recognition test in mice (Quiedeville et al., 2015). These ligands are active at lower doses than those needed for an acute effect and furthermore, seem to be devoid of side effects in these conditions. We thus considered that co-modulation of these two receptors could represent a valuable strategy against memory deficits in AD (Claeysen et al., 2015; Lalut et al., 2017). Therefore, we recently reported the synthesis and the in vivo procognitive effect displayed by the dual compound 4 with in vitro 5-HT4R agonist and 5-HT6R antagonist effects (Figure 2; Yahiaoui et al., 2016). Based on the evidence that 5-HT4R, 5-HT6R and AChE are all valuable therapeutic targets in AD treatment, the present work aimed at designing, starting from our dual compound 4 and the benzyl analog of donecopride 5a (Rochais et al., 2015), novel MTDLs associating 5-HT4R agonist, 5-HT6R antagonist and AChE inhibitory balanced activities (Figure 2).
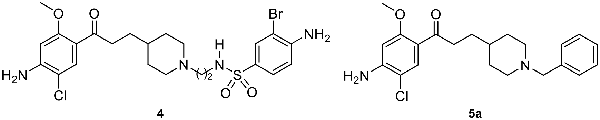
Figure 2. Structure of compounds 4 and 5a.
Experimental Section
Chemistry
General Materials and Methods
All chemical reagents and solvents were purchased from commercial sources and used without further purification. Melting points were determined on a STUART SMP50 melting point apparatus. 1H, 13C and 19F NMR spectra were recorded on a BRUKER AVANCE III 400MHz with chemical shifts expressed in parts per million (in chloroform-d, methanol-d4 or DMSO-d6) downfield from TMS as an internal standard and coupling in Hertz. IR spectra were recorded on a Perkin-Elmer BX FT-IR apparatus using KBr pellets. High resolution mass spectra (HRMS) were obtained by electrospray on a BrukermaXis. The purities of all tested compounds were analyzed by LC−MS, with the purity all being higher than 95%. Analyses were performed using a Waters Alliance 2695 as separating module (column XBridge C18 2.5 μM/4.6 × 50 mM) using the following gradients: A (95%)/B (5%) to A (5%)/B (95%) in 4.00 min. This ratio was held during 1.50 min before return to initial conditions in 0.50 min. Initial conditions were then maintained for 2.00 min (A = H2O, B = CH3CN; each containing HCOOH: 0.1%). MS were obtained on a SQ detector by positive ESI.
Procedures for the Deprotection and N-alkylation of tert-butyl 4-[3-(4-amino-5-chloro-2-methoxyphenyl)-3-oxo-propyl]piperidine-1-carboxylate (8)
Procedure A: to a solution of compound 8 in DCM (5 mL) was added TFA (1 mL) dropwise. The reaction mixture was stirred at room temperature for 1 h and then concentrated under reduced pressure. Evaporation of the solvent provided a light-yellow oil, which was thereafter dissolved in 1,4-dioxane (30 mL). To the resulting solution, 20 equivalent of K2CO3 were added and thereafter 1.2 equivalent of commercially available benzylbromide derivative was added. The reaction mixture was then stirred at reflux until the full consumption of the starting material. The mixture was then concentrated in vacuo, diluted with water and extracted twice with EtOAc. The combined organic phases were washed with brine, dried over MgSO4, filtrated and concentrated under pressure. The crude product was thereafter purified by silica column flash chromatography using the appropriate eluting system.
Procedure B: to a solution of compound 8 in DCM (5 mL/mmol) was added TFA (1 mL/mmol) dropwise. The reaction mixture was stirred at room temperature for 15 min and then concentrated under reduced pressure. Evaporation of the solvent provided a light-yellow oil, which was thereafter dissolved in DMF (10 mL/mmol). To the resulting solution, 10 equivalent of K2CO3 were added and thereafter 1.2 equivalent of commercially available benzylbromide derivative was added. The reaction mixture was then stirred at 110°C until the full consumption of the starting material. The mixture was then concentrated in vacuo, diluted with EtOAc and washed twice with brine. The organic layer was dried over MgSO4, filtrated and concentrated under pressure. The crude product was thereafter purified by silica column flash chromatography using the appropriate eluting system.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-benzyl)piperidin-4-yl)propan-1-one (5a; Rochais et al., 2015)
Following procedure A. Pale yellow oil (23%); 1H NMR (400 MHz, CDCl3) δ 7.71 (s, 1H), 7.19 (m, 5H), 6.18 (s, 1H), 4.39 (s, 2H), 3.76 (s, 3H), 3.45 (s, 2H), 2.81 (m, 4H), 1.90 (br t, J = 10.2 Hz, 2H), 1.61 (br d, J = 9.2 Hz, 2H), 1.52 (q, J = 6.9 Hz, 2H), 1.22 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.2, 159.5, 147.7, 137.7, 132.3, 129.5 (2C), 128.2 (2C), 127.1, 118.9, 111.3, 97.5, 63.3, 55.6, 53.7 (2C), 40.8, 35.4, 31.9 (2C), 31.4; HRMS (m/z) calcd for C22H28ClN2O2 [M + H]+ 387.183382, found 387.183120.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-chlorobenzyl)piperidin-4-yl)propan-1-one (5b)
Following procedure B. Pale yellow solid (33%); mp 125.4°C; 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.48 (dd, J = 7.7, 1.6 Hz, 1H), 7.32 (dd, J = 7.8, 1.4 Hz, 1H), 7.22 (dt, J = 7.5, 1.4 Hz, 1H), 7.16 (dt, J = 7.7, 1.8 Hz, 1H), 6.25 (s, 1H), 4.47 (s, 2H), 3.83 (s, 3H), 3.59 (s, 2H), 2.92–2.88 (m, 4H), 2.08–2.03 (m, 2H), 1.69–1.67 (m, 2H), 1.62–1.57 (m, 2H), 1.30–1.23 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.3, 159.6, 147.8, 136.5, 134.3, 132.3, 130.8, 129.4, 128.0, 126.7, 119.1, 111.4, 97.7, 59.7, 55.7, 54.1 (2C), 41.1, 35.7, 32.5 (2C), 31.5; MS (m/z) [M + H]+ 421.61–423.63; HRMS (m/z) calcd for C22H27Cl2N2O2 [M + H]+ 421.1450, found 421.1462.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-bromobenzyl)piperidin-4-yl)propan-1-one (5c)
Following procedure B. Pale brown solid (43%); mp 118.1°C; 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.51 (dd, J = 7.9, 1.1 Hz, 1H), 7.48 (dd, J = 7.3, 1.5 Hz, 1H), 7.26 (dt, J = 7.5, 1.1 Hz, 1H), 7.08 (dt, J = 7.8, 1.7 Hz, 1H), 6.25 (s, 1H), 4.49 (s, 2H), 3.82 (s, 3H), 3.56 (s, 2H), 2.92–2.88 (m, 4H), 2.09–2.04 (m, 2H), 1.69–1.67 (m, 2H), 1.62–1.57 (m, 2H), 1.28–1.25 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.3, 159.6, 147.8, 138.2, 132.7, 132.3, 130.8, 128.3, 127.3, 124.7, 119.0, 111.3, 97.6, 62.2, 55.7, 54.1 (2C), 41.1, 35.7, 32.5 (2C), 31.4; MS (m/z) [M + H]+ 465.59–467.59–469.59; HRMS (m/z) calcd for C22H27BrClN2O2 [M + H]+ 465.0944, found 465.0945.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-fluorobenzyl)piperidin-4-yl)propan-1-one (5d)
Following procedure B. Pale yellow solid (56%); mp 115°C; 1H NMR (399.75 MHz, CDCl3) δ 7.77 (s, 1H), 7.36 (td, J = 7.6, 1.8 Hz, 1H), 7.22 (m, 1H), 7.10 (td, J = 7.6, 1.2 Hz, 1H), 7.01 (m, 1H), 6.24 (s, 1H), 4.47 (s, 2H), 3.82 (s, 3H), 3.56 (s, 2H), 2.88 (m, 4H), 2.00 (br t, J = 10.7 Hz, 2H), 1.67 (br d, J = 9.3 Hz, 2H), 1.58 (m, 2H), 1.27 (m, 3H); 13C NMR (100.53 MHz, CDCl3) δ 199.1, 161.2 (d, J = 244.5 Hz), 159.5, 147.7, 132.2, 131.7 (d, J = 4.6 Hz), 128.6 (d, J = 8.0 Hz), 125.0 (d, J = 14.7 Hz), 123.8 (d, J = 3.4 Hz), 118.9, 115.2 (d, J = 22.5 Hz), 111.2, 97.5, 63.3, 55.6 (2C), 53.6 (2C), 40.9, 35.5, 32.3, 31.3, 29.7; 19F NMR (376.10 MHz, CDCl3) δ −117.75 (s); MS (m/z) [M + H]+ 405.56–407.55; HRMS (m/z) calcd for C22H27ClFN2O2 [M + H]+ 405.1745, found 405.1747.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-iodobenzyl)piperidin-4-yl)propan-1-one (5e)
Following procedure B. Pale yellow solid (44%); mp 106°C;1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J = 7.8, 1.1 Hz, 1H), 7.78 (s, 1H), 7.44 (d, J = 7.4 Hz, 1H), 7.30 (dt, J = 7.4, 1.1 Hz, 1H), 6.92 (dt, J = 7.6, 1.7 Hz, 1H), 6.25 (s, 1H), 4.48 (s, 2H), 3.83 (s, 3H), 3.50 (s, 2H), 2.92–2.88 (m, 4H), 2.11–2.06 (m, 2H), 1.69–1.67 (m, 2H), 1.62–1.57 (m, 2H), 1.31–1.25 (m, 3H) 13C NMR (100 MHz, CDCl3) δ 199.2, 159.6, 147.8, 141.0, 139.4, 132.3, 130.4, 128.7, 128.1, 119.0, 111.3, 100.7, 97.7, 67.0, 55.8, 54.0 (2C), 41.1, 35.7, 32.4 (2C), 31.4; MS (m/z) [M + H]+ 513.59–515.61; HRMS (m/z) calcd for C22H27ClIN2O2 [M + H]+ 513.0806, found 513.0824.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-methylbenzyl)piperidin-4-yl)propan-1-one (5f)
Following procedure B. Yellow-pale solid (45%); mp 119°C; 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.37 (m, 1H), 7.13 (m, 1H), 6.25 (s, 1H), 4.44 (br s, 2H), 3.84 (s, 3H), 3.41 (s, 2H), 2.91–2.83 (m, 4H), 2.35 (s, 3H), 1.95 (br t, J = 11.6 Hz, 2H), 1.67–1.53 (m, 4H), 1.28–1.18 (m, 3H);13C NMR (100 MHz, CDCl3) δ 199.3, 159.5, 137.4, 137.1, 132.2, 130.1, 129.7, 126.8, 125.4, 119.0, 111.2, 97.6, 61.1, 55.6, 54.1, 41.0, 35.8, 32.5, 31.4, 19.3; MS (m/z) [M + H]+ 401.65–403.64; HRMS (m/z) calcd for C23H30ClN2O2 [M + H]+ 401.1996, found 401.1994.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-benzyloxybenzyl)piperidin-4-yl)propan-1-one (5g)
Following procedure B. Yellow oil (16%); 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.46–7.44 (m, 2H), 7.40–7.37 (m, 3H), 7.32 (m, 1H), 7.21 (dt, J = 7.9, 1.6 Hz, 1H), 6.96–6.91 (m, 2H), 6.24 (s, 1H), 5.08 (s, 2H), 4.47 (s, 2H), 3.82 (s, 3H), 3.62 (s, 2H), 2.95–2.87 (m, 4H), 2.05–2.00 (m, 2H), 1.68–1.66 (m, 2H), 1.61–1.56 (m, 2H), 1.31–1.26 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.3, 159.6, 157.1, 147.8, 137.5, 132.3, 130.9, 128.6 (2C), 128.0, 127.8, 127.3 (3C), 120.7, 119.1, 112.0, 111.3, 97.7, 70.2, 56.7, 55.7, 54.0 (2C), 41.1, 35.7, 32.5 (2C), 31.5; MS (m/z) [M + H]+ 493.65–495.66; HRMS (m/z) calcd for C29H34ClN2O3 [M + H]+ 493.2258, found 493.2254.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-hydroxybenzyl)piperidin-4-yl)propan-1-one (5h)
Following procedure B. Yellow-brown oil (12%); 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.15 (dt, J = 8.0, 1.3 Hz, 1H), 6.95 (d, J = 7.3 Hz, 1H), 6.80 (d, J = 8.1 Hz, 1H), 6.76 (dt, J = 7.4, 1.1 Hz, 1H), 6.25 (s, 1H), 4.45 (s, 2H), 3.84 (s, 3H), 3.68 (s, 2H), 3.00–2.97 (m, 2H), 2.90 (t, J = 7.8 Hz, 2H), 2.10–2.07 (m, 2H), 1.76–1.73 (m, 2H), 1.63–1.58 (m, 2H), 1.33–1.25 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 198.9, 159.6, 158.3, 147.8, 132.4, 128.7, 128.6, 122.0, 119.1, 119.0, 116.2, 109.9, 97.7, 61.8, 55.8, 53.4 (2C), 40.8, 35.3, 32.2 (2C), 31.1; MS (m/z) [M + H]+ 403.60–405.58; HRMS (m/z) calcd for C22H28ClN2O3 [M + H]+ 403.1788, found 403.1788.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-bromobenzyl)piperidin-4-yl)propan-1-one (5i)
Following procedure A. Yellow-pale oil (34%); 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.47 (br t, J = 1.6 Hz, 1H), 7.36 (br dt, J = 1.6, 7.6 Hz, 1H), 7.23 (br dt, J = 1.6, 7.6 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 6.25 (s, 1H), 4.44 (s, 2H), 3.84 (s, 3H), 3.44 (s, 2H), 2.89 (br t, J = 7.8 Hz, 2H), 2.84 (br d, J = 10.8 Hz, 2H), 1.93 (br t, J = 11.0 Hz, 2H), 1.74–1.64 (m, 2H), 1.59 (br q, J = 6.9 Hz, 2H), 1.34–1.19 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.2, 159.6, 147.8, 141.3, 132.4, 132.1, 130.1, 129.8, 127.9, 122.5, 119.2, 111.4, 97.7, 63.0, 55.8, 54.0, 41.0, 35.7, 32.4, 31.4; HRMS (m/z) calcd for C22H27BrClN2O2 [M + H]+ 465.093894, found 465.093548.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-fluorobenzyl)piperidin-4-yl)propan-1-one (5j)
Following procedure B. White solid (48%); mp 95°C; 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.25 (m, 1H), 7.06 (m, 2H), 6.92 (m, 1H), 6.25 (s, 1H), 4.44 (s, 2H), 3.84 (s, 3H), 3.46 (s, 2H), 2.87 (m, 4H), 1.94 (br t, J = 11.1 Hz, 2H), 1.66 (m, 2H), 1.58 (m, 2H), 1.27 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.1, 162.9 (d, J = 243.7 Hz), 147.7, 141.5 (d, J = 6.7 Hz), 132.2, 129.5 (d, J = 8.1 Hz), 124.6 (d, J = 2.9 Hz), 118.9, 115.8 (d, J = 21.1 Hz), 113.7 (d, J = 20.8 Hz), 111.3, 97.6, 62.9, 55.6, 53.9, 40.9, 35.5, 32.3, 31.3; 19F NMR (376.10 MHz, CDCl3) δ −113.97 (s); MS (m/z) [M + H]+ 405.58–407.57; HRMS (m/z) calcd for C22H27ClFN2O2 [M + H]+ 405.1745, found 405.1745.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-methylbenzyl)piperidin-4-yl)propan-1-one (5k)
Following procedure A. Yellow-pale oil (41%); 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.19 (t, J = 7.4 Hz, 1H), 7.13 (br s, 1H), 7.09 (br d, J = 7.6 Hz, 1H), 7.06 (br d, J = 7.2 Hz, 1H), 6.25 (s, 1H), 4.44 (s, 2H), 3.83 (s, 3H), 3.45 (s, 2H), 2.96–2.80 (m, 4H), 2.34 (s, 3H), 1.93 (br t, J = 10.4 Hz, 2H), 1.73–1.52 (m, 4H), 1.36–1.17 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.3, 159.6, 147.8, 138.3, 137.9, 132.4, 130.2, 128.1, 127.8, 126.6, 119.2, 111.4, 97.7, 63.7, 55.8, 54.0, 41.0, 35.7, 32.3, 31.5, 21.5; HRMS (m/z) calcd for C23H30ClN2O2 [M + H]+ 401.1994, found 401.1992.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-methoxybenzyl)piperidin-4-yl)propan-1-one (5l)
Following procedure A. Colorless oil (27%); 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.22 (t, J = 8.0 Hz, 1H), 6.93–6.85 (m, 2H), 6.79 (ddd, J = 0.8, 2.4, 8.0 Hz, 1H), 6.25 (s, 1H), 4.44 (s, 2H), 3.83 (s, 3H), 3.81 (s, 3H), 3.47 (s, 2H), 2.95–2.81 (m, 4H), 1.94 (br t, J = 10.2 Hz, 2H), 1.75–1.52 (m, 4H), 1.36–1.17 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.3, 159.7, 159.6, 147.7, 140.2, 132.4, 129.2, 121.8, 119.2, 114.9, 112.5, 111.4, 97.7, 63.5, 55.8, 55.4, 54.0, 41.0, 35.7, 32.4, 31.5; HRMS (m/z) calcd for C23H30ClN2O3 [M + H]+ 417.193947, found 417.193907.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-aminobenzyl)piperidin-4-yl)propan-1-one (5m)
Following procedure A. Hygroscopic solid (36%); 1H NMR (400 MHz, CD3OD) δ 7.66 (s, 1H), 7.17 (t, J = 7.8 Hz, 1H), 6.87–6.70 (m, 3H), 6.69 (s, 2H), 6.44 (s, 1H), 4.11 (s, 2H), 3.85 (s, 3H), 3.43 (br d, J = 12.4 Hz, 2H), 3.05–2.76 (m, 4H), 1.95 (br d, J = 14.0 Hz, 2H), 1.73–1.28 (m, 5H).
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-nitrobenzyl)piperidin-4-yl)propan-1-one (5n)
Following procedure A. Yellow oil (80%); 1H NMR (400 MHz, CDCl3) δ 8.18 (br t, J = 1.6 Hz, 1H), 8.09 (br d, J = 8.0 Hz, 1H), 7.78 (s, 1H), 7.66 (br d, J = 7.6 Hz, 1H), 7.47 (t, J = 8.0 Hz, 1H), 6.26 (s, 1H), 4.45 (s, 2H), 3.84 (s, 3H), 3.55 (s, 2H), 2.90 (br t, J = 7.8 Hz, 2H), 2.83 (br d, J = 11.6 Hz, 2H), 1.99 (br t, J = 10.2 Hz, 2H), 1.78–1.64 (m, 2H), 1.60 (br q, J = 6.8 Hz, 2H), 1.35–1.19 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 199.2, 159.6, 148.4, 147.8, 141.4, 135.2, 132.4, 129.2, 123.9, 122.2, 119.1, 114.4, 97.7, 62.6, 55.8, 54.1, 41.0, 35.6, 32.4, 31.4; HRMS (m/z) calcd for C22H27ClN3O4 [M + H]+ 432.168460, found 432.168262.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(4-fluorobenzyl)piperidin-4-yl)propan-1-one (5o)
Following procedure B. Yellow pale solid (32%); mp 128°C; 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.26 (m, 2H), 7.06 (m, 2H), 6.98 (m, 2H), 6.25 (s, 1H), 4.48 (s, 2H), 3.83 (s, 3H), 3.43 (s, 2H), 2.86 (m, 4H), 1.91 (br t, J = 10.6 Hz, 2H), 1.66 (br d, J = 8.8 Hz, 2H), 1.58 (m, 2H), 1.25 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.1, 161.9 (d, J = 243.0 Hz), 159.5, 147.7, 134.3 (d, J = 3.0 Hz), 132.2, 130.7 (d, J = 7.9 Hz, 2C), 118.9, 114.9 (d, J = 21.1 Hz, 2C), 111.2, 97.5, 62.7, 55.6, 53.8, 40.9, 35.6, 32.3, 31.3 ; 19F NMR (376.1 MHz, CDCl3) δ −113.97 (s); MS (m/z) [M + H]+ 405.62–407.62; HRMS (m/z) calcd for C22H27ClFN2O2 [M + H]+ 405.1745, found 405.1740.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(4-hydroxybenzyl)piperidin-4-yl)propan-1-one (5p)
Following procedure B. Yellow oil (13%); 1H NMR (400 MHz, CD3OD) δ 7.64 (s, 1H), 7.14–7.12 (m, 2H), 6.74 (m, 2H), 6.43 (s, 1H), 3.84 (s, 3H), 3.44 (s, 2H), 2.93–2.87 (m, 4H), 2.04–1.99 (m, 2H), 1.72–1.70 (m, 2H), 1.57–1.51 (m, 2H), 1.32–1.22 (m, 4H); 13C NMR (100 MHz, CD3OD) δ 201.0, 161.6, 158.1, 151.7, 132.9, 132.4 (2C), 128.2, 117.9, 116.0 (2C), 111.6, 98.1, 63.7, 56.1, 54.4 (2C), 41.7, 36.6, 32.6 (3C); MS (m/z) [M + H]+ 403.65–405.66; HRMS (m/z) calcd for C22H28ClN2O3 [M + H]+ 403.1788, found 403.1786.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(4-methylbenzyl)piperidin-4-yl)propan-1-one (5q)
Following procedure B. White solid (20%); mp 97.0°C; 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.20 (d, J = 7.9 Hz, 2H), 7.11 (d, J = 7.9 Hz, 2H), 6.24 (s, 1H), 4.49 (br s, 2H), 3.81 (s, 3H), 3.47 (s, 2H), 2.90–2.86 (m, 4H), 2.33 (s, 3H), 1.96–1.91 (m, 2H), 1.68–1.65 (m, 2H), 1.60–1.55 (m, 2H), 1.33–1.23 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.2, 159.6, 147.8, 136.7, 135.0, 132.3, 129.5 (2C), 128.9 (2C), 118.9, 111.3, 97.6, 63.2, 55.7, 53.8, 53.5, 41.0, 35.6, 32.2 (2C), 31.4, 21.2; MS (m/z) [M + H]+ 401.66–403.65; HRMS (m/z) calcd for C23H30ClN2O2 [M + H]+ 401.1996, found 401.1996.
N-(3-((4-(3-(4-Amino-5-chloro-2-methoxyphenyl)-3-oxopropyl)piperidin-1-yl)methyl)-4-bromophenyl) acetamide (5r)
Following procedure A. Yellow-pale oil (70%); 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.96 (br s, 1H), 7.52 (dd, J = 2.4, 8.8 Hz, 1H), 7.47–7.39 (m, 2H), 6.25 (s, 1H), 4.50 (s, 2H), 3.82 (s, 3H), 3.50 (s, 2H), 3.01–2.77 (m, 4H), 2.16 (s, 3H), 2.05 (br t, J = 10.8 Hz, 2H), 1.72–1.61 (m, 2H), 1.57 (br q, J = 6.8 Hz, 2H), 1.35–1.18 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 199.4, 168.7, 159.7, 147.9, 138.8, 137.5, 133.0, 132.3, 121.6, 120.0, 118.9, 118.8, 111.3, 97.7, 62.0, 55.8, 54.1, 41.1, 35.7, 32.5, 31.4, 24.7; HRMS (m/z) calcd for C24H30BrClN3O3 [M + H]+ 522.115358, found 522.114977.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(2-fluoro-5-nitrobenzyl)piperidin-4-yl)propan-1-one (5s)
Following procedure B. Yellow solid (35%); mp 133.4°C; 1H NMR (400 MHz, CDCl3) δ 8.36 (dd, J = 6.1 Hz, J = 2.8 Hz, 1H), 8.13 (ddd, J = 8.9, 4.3 Hz, J = 3.0 Hz, 1H), 7.78 (s, 1H), 7.15 (tapp, J = 8.8 Hz, 1H), 6.25 (s, 1H), 4.45 (s, 2H), 3.84 (s, 3H), 3.59 (s, 2H), 2.91–2.84 (m, 4H), 2.08–2.02 (m, 2H), 1.70–1.68 (m, 2H), 1.62–1.57 (m, 2H), 1.29–1.25 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 199.1, 164.8 (d, J = 256.4 Hz), 159.6, 147.8, 144.4 (d, J = 1.9 Hz), 132.4, 127.8 (d, J = 16.2 Hz), 127.2 (d, J = 6.9 Hz), 124.6 (d, J = 10.2 Hz), 119.1, 116.3 (d, J = 25.1 Hz), 111.4, 97.7, 55.8, 55.1, 53.9 (2C), 40.9, 35.4, 32.3 (2C), 31.3; MS (m/z) [M + H]+ 450.51–452.50; HRMS (m/z) calcd for C22H26ClFN3O4 [M + H]+ 450.1596, found 450.1594.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(5-amino-2-fluorobenzyl)piperidin-4-yl)propan-1-one (5t)
Following procedure B. Yellow-brown oil (11%); 1H NMR (400 MHz, CDCl3) δ 7.64 (s, 1H), 6.83 (dd, J = 9.5 Hz, J = 8.8 Hz, 1H), 6.72 (dd, J = 6.2, 2.8 Hz, 1H), 6.65 (ddd, J = 8.7, 4.1, 2.8 Hz, 1H), 6.44 (s, 1H), 3.85 (s, 3H), 3.55 (d, J = 1.2 Hz, 2H), 2.98–2.95 (m, 2H), 2.91–2.87 (m, 2H), 2.16–2.11 (m, 2H), 1.73–1.71 (m, 2H), 1.59–1.52 (m, 2H), 1.33–1.26 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 200.1, 161.7, 156.3 (d, J = 234.0 Hz), 151.7, 144.8, 132.9, 124.1 (d, J = 16.4 Hz), 119.7 (d, J = 3.3 Hz), 117.9, 117.3 (d, J = 7.7 Hz), 116.4 (d, J = 23.8 Hz), 111.6, 98.1, 56.3 (d, J = 1.3 Hz), 56.1, 54.4 (2C), 41.6, 36.4, 32.6 (3C); MS (m/z) [M + H]+ 420.60–422.60; HRMS (m/z) calcd for C22H28ClFN3O2 [M + H]+ 420.1854, found 420.1857.
1-(4-Amino-5-chloro-2-methoxyphenyl)-3-(1-(3-methylbenzyl)piperidin-4-yl)propan-1-one fumaric acid salt (10)
To a solution of 85 mg of compound 5k (0.212 mmol) in 3 mL of iPrOH was added 25 mg of fumaric acid (0.212 mmol, 1 equivalent). The solution was refluxed for 2 h. The mixture was then concentrated in vacuo. The residue is triturated in Et2O and then filtrated. Sixty milligram of desired compound were obtained as a beige solid (55%). Yellow solid; mp 185.4°C; 1H NMR (400 MHz, CD3OD) δ 7.56 (s, 1H), 7.28–7.16 (m, 4H), 6.60 (s, 2H), 6.35 (s, 1H), 4.11 (s, 2H), 3.75 (s, 3H), 3.32 (br d, J = 12.4 Hz, 2H), 2.88–2.81 (m, 4H), 2.28 (s, 3H), 1.86 (br d, J = 13.9 Hz, 2H), 1.55–1.52 (m, 3H), 1.38–1.32 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 198.7, 169.8 (2C), 160.3, 150.4, 139.0, 134.8 (2C), 131.5, 130.4, 129.3, 128.8, 127.9, 116.3, 110.3, 96.6, 60.2, 54.7 (2C), 52.1, 48.2, 39.8, 33.1, 30.1, 28.9, 20.0 (2C). HRMS (m/z) calcd for C23H30ClN2O2 [M + H]+ 401.1994, found 401.1996.
In vitro Biological Studies
Pharmacological Characterization of Drugs on Human 5-HT4R
For competition studies, 2.5 μg of proteins (5-HT4(b) membrane preparations, HTS110M, Eurofins. Eurofins’ 5-HT4(b) membrane preparations are crude membrane preparations made from their proprietary stable recombinant cell lines to ensure high-level of GPCR surface expression) were incubated in duplicate at 25°C for 60 min in the absence or the presence of 10−6 or 10−8M of each drug (9 was used as a reference standard) and 0.2 nM [3H]-GR113808 (NET 1152, Perkin Elmer) in 25 mM Tris buffer (pH 7.4, 25°C). At the end of the incubation, homogenates were filtered through Whatman GF/C filters (Alpha Biotech) presoaked with 0.5% polyethylenimine using a Brandel cell harvester. Filters were subsequently washed three times with 1 mL of ice-cold 25 mM Tris buffer (pH 7.4, 4°C). Non-specific binding was evaluated in parallel in the presence of 30 μM serotonin.
For some of these compounds, affinity constants were calculated from five-point inhibition curves using the GraphPad Prism 6 software and expressed as Ki ± SD.
Pharmacological Characterization of Drugs on Human 5-HT6R
Drugs were evaluated through their possibility to compete for the binding of [3H]-LSD on membranes of HEK-293 cells transiently expressing the human 5-HT6 receptors (ref. RBHS6M, Perkin Elmer). In brief, 4 μg of proteins were incubated at 37°C for 60 min in duplicate in the absence or the presence of 10−6 or 10−8M of each drug (2 was used as a reference standard) and 2.5 nM [3H]-LSD (ref. NET638250UC, Perkin Elmer), in 25 mM Tris-HCl buffer (pH 7.4) supplemented with 0.5 mM EDTA. At the end of the incubation, the homogenates were then filtered through Whatman GF/C filters and washed five times with ice-cold 25 mM Tris-HCl buffer. Non-specific binding was evaluated in the presence of 100 μM serotonin. Radioactivity associated to proteins was then quantified and expressed as the percentage of inhibition of the drugs under study.
For some of these compounds, affinity constants were calculated from five-point inhibition curves using the GraphPad Prism 6 software and expressed as Ki ± SD.
Determination of cAMP Production
COS-7 cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% dialyzed fetal calf serum (dFCS) and antibiotics. Cells were transiently transfected by electroporation with plasmids encoding HA-tagged 5-HT4R (100 ng/106 cells) or HA-tagged 5-HT6R (70 ng/106 cells or 300 ng/106 cells), then seeded in 96-well plates (16,000 cells/well).
5-HT4R-induced cAMP production: 24 h after transfection, cells were exposed to the indicated concentrations of 5-HT4R ligands in the presence of 0.1 mM of the phosphodiesterase inhibitor RO-20-1724, at 37°C in 100 μL of HBS (20 mM HEPES; 150 mM NaCl; 4.2 mM KCl; 0.9 mM CaCl2; 0.5 mM MgCl2; 0.1% glucose; 0.1% BSA). After 10 min, cells were then lysed by addition of the same volume of Triton-X100 (0.1%).
Blockade of the 5-HT6R-induced cAMP production: 24 h after transfection, cells were exposed to the indicated concentrations of 5-HT6R ligands at 37°C in 50 μL of HBS. After 7 min, 5-HT (5.10−7M final concentration) in the presence of RO-20-1724 (0.1 mM final concentration), in 50 μL HBS, was added to the wells. After 10 min at 37°C, cells were then lysed by addition of 100 μL of Triton-X100 (0.1%).
Reversion of the basal 5-HT6R-induced cAMP production: the 5-HT6R expression was augmented by transfection of higher quantities of cDNA (300 ng/106 cells) to increase the basal cAMP production linked to the constitutive activity of the receptor. Twenty-four hours after transfection, cells were exposed to 10−4M of 5-HT6R ligands in the presence of RO-20-1724 (0.1 mM final concentration), at 37°C in 100 μL of HBS. This experiment is performed in the absence of 5-HT. After 10 min at 37°C, cells were then lysed by addition of 100 μL of Triton-X100 (0.1%).
Quantification of cAMP production was performed by HTRF® by using the cAMP Dynamic kit (Cisbio Bioassays) according to the manufacturer’s instructions.
In vitro Tests of AChE Biological Activity
Inhibitory capacity of compounds on AChE biological activity was evaluated through the use of the spectrometric method of Ellman et al. (1961). Acetylthiocholine iodide and 5,5-dithiobis-(2-nitrobenzoic) acid (DTNB) were purchased from Sigma Aldrich. AChE from human erythrocytes (buffered aqueous solution, ≥500 units/mg protein (BCA), Sigma Aldrich) was diluted in 20 mM HEPES buffer pH 8, 0.1% Triton X-100 such as to have enzyme solution with 0.25 unit/mL enzyme activity. In the procedure, 100 μL of 0.3 mM DTNB dissolved in phosphate buffer pH 7.4 was added into the 96-well plate followed by 50 μL of test compound solution and 50 μL of enzyme (0.05 U final). After 5 min of preincubation at 25°C, the reaction was then initiated by the injection of 50 μL of 10 mM acetylthiocholine iodide solution. The hydrolysis of acetylthiocholine was monitored by the formation of yellow 5-thio-2-nitrobenzoate anion as the result of the reaction of DTNB with thiocholine, released by the enzymatic hydrolysis of acetylthiocholine, at a wavelength of 412 nm using a 96-well microplate plate reader (BioTek, Synergy 2). Test compounds were dissolved in analytical grade DMSO. Donepezil (DPZ) was used as a reference standard. The rate of absorbance increase at 412 nm was followed every minute for 10 min. Assays were performed with a blank containing all components except acetylthiocholine, in order to account for non-enzymatic reaction. The reaction slopes were compared and the percent inhibition due to the presence of test compounds was calculated by the following expression: 100 − (vi/v0 × 100) where vi is the rate calculated in the presence of inhibitor and v0 is the enzyme activity.
First screening of AChE activity was carried out at a 10−6M concentration of compounds under study. For the compounds with significant inhibition (≥50%), IC50 values were determined graphically by plotting the % inhibition vs. the logarithm of six inhibitor concentrations in the assay solution using the GraphPad Prism 6 software.
In vivo Biological Studies
Animals
Adult male NMRI mice (3 months old, weighing 35–40 g) from Janvier labs (Le Genest-Saint-Isle, France) were used to perform experiments. Mice were housed by ten in standard polycarbonate cages in standard controlled conditions (22 ± 2°C, 55 ± 10% humidity) with a reversed 12 h light/dark cycle (light on at 7 pm). Food and water were available ad libitum in the home cage. All experiments were conducted (between 9 am and 3 pm) during the active-dark-phase of the cycle, were approved by the ethics committee of Normandy (CENOMEXA, n°5161) and were in agreement with the European Directives and French law on animal experimentation (personal authorization n° 14–17 for MB and 14–60 for TF).
CNS-Activity and Acute Toxicity Test
Behavioral and neurological changes induced by graded doses (1, 10, 100 mg/kg) of the tested derivatives were evaluated in mice, in groups of four, by a standardized observation technique at different times (30 min, 3 and 24 h) after intraperitoneal administration (Morpugo, 1971). Major changes of behavioral data (for example, hypo- or hyperactivity, ataxia, tremors, convulsion, etc…) were noted in comparison to the control group. The approximate DL50 of the compounds were also calculated through the quantification of mortality after 24 h. Amphetamine (2 mg/kg), chlorpromazine (10 mg/kg) were used as a stimulant and depressive references, respectively.
Locomotor Activity
Locomotion of mice was measured using an actimeter (Imetronic®) through infrared detection. Eight individual removable polycarbonate cages (21 cm length, 7 cm wide and 12 cm high), where each mouse was placed, are disposed in the actimeter. Locomotor activity was measured by recording the number of interruption of beams of the red light over a period of 30 min through an attached recording system to the actimeter. Compound 10 was tested at 1, 3 and 10 mg/kg. Amphetamine (2 mg/kg), chlorpromazine (3 mg/kg) were used as a stimulant and depressive references, respectively (Freret et al., 2012).
Spatial Working Memory
Anti-amnesic activity of tested compounds was evaluated by reversal of scopolamine (0.5 mg/kg)-induced deficit on spontaneous alternation behavior in the Y maze test (Hooper et al., 1996). The Y maze made of gray plastic consisted of three equally spaced arms (21 cm long, 7 cm wide with walls 15-cm high). The mouse was placed at the end of one of the arms and allowed to move freely through the maze during a 5 min session while the sequence of arm entries was recorded by an observer. An arm entry was scored when all four feet crossed into the arm. An alternation was defined as entries into all three arms on a consecutive occasion. The number of possible alternation is thus the total number of arm entries minus two; the percentage of alternation was calculated as (actual alternation/possible alternation) × 100. Compound 10 was tested at 0.3, 1 and 3 mg/kg. DPZ (1 mg/kg) was tested as a clinical reference.
Pharmacological Treatments
Amphetamine, (+)-α-Methylphenethylamine hemisulfate, chlorpromazine hydrochloride, imipramine hydrochloride and scopolamine hydrobromide were purchased from Sigma (France). All those pharmacological compounds were dissolved in NaCl 0.9% as the vehicle were administered IP 30 min before tests, except scopolamine which was subcutaneously administered 20 min before spontaneous alternation test.
Statistical Analysis
Results were expressed as mean ± SD and were analyzed by one-way analysis of variance (ANOVA), with Statview® software. In case of significance, a SNK (Student-Newman-Keuls) post hoc test was realized. Additionally, for the spontaneous alternation test, the percentage of alternation was compared to a theoretical 50% value (random alternation) by an univariate t-test. Differences were considered as statistically significant if the p-value was strictly under 0.05.
Results
Chemistry
The targeted compounds 5a–t were obtained starting from 4-amino-5-chloro-2-methoxybenzoic acid (6) as reported for the synthesis of 5a (Scheme 1; Rochais et al., 2015). The synthesis of a β-ketoester (7) was achieved in 62% yield using the carbonyldiimidazole (CDI) activation of the carboxylic acid group of 6. A nucleophilic substitution, using N-Boc 4-(iodomethyl) piperidine at room temperature to avoid the risk of a double substitution, allowed the installation of the methylpiperidine moiety. The latter was immediately followed by a saponification–decarboxylation sequence using hydroalcoholic potassium hydroxide. The reaction yielded 8 in a 86% yield. Finally, the TFA-N-deprotection of 8, immediately followed by another nucleophilic substitution with various benzyl bromides yielded compounds 5a–t. The fumaric acid salt 10 was synthesized starting from 5k and using fumaric acid in iPrOH.
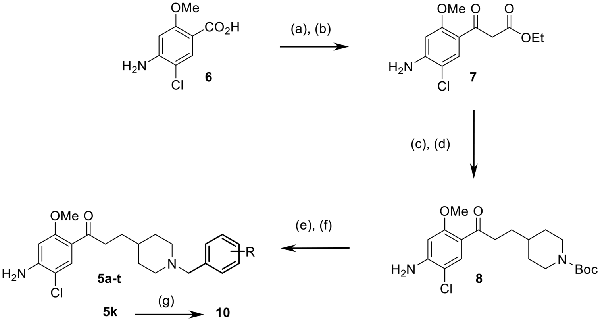
Scheme 1. Synthetic pathways for access to compounds 5a–t, 10. Conditions and reagents: (a) CDI, THF; (b) KO2CCH2CO2Et, MgCl2, THF; (c) N-Boc 4-iodomethylpiperidine, K2CO3, DMF; (d) KOH, EtOH/H2O; (e) TFA, 1,4-dioxan or DCM; (f) R-benzyl bromide, K2CO3, 1,4-dioxane or DMF; (g) fumaric acid, iPrOH.
In vitro Results
The biological evaluation of the synthesized compounds as potential inhibitors of human AChE was performed using the Ellman assay (Ellman et al., 1961), as well as their affinity for human 5-HT6R and 5-HT4R using a radioligand displacement assay (Table 1). In these tests, DPZ was used as a reference AChE inhibitor (AChEI), 2 as a 5-HT6R ligand and 9 (RS67333; Eglen et al., 1995) as a 5-HT4R ligand.
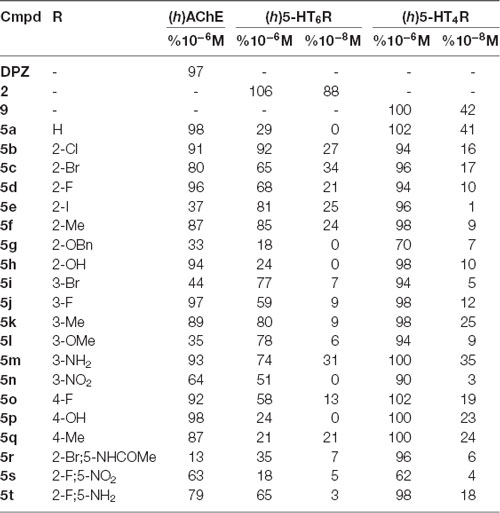
Table 1. (h)AChE inhibitory activity, (h)5-HT6R and (h)5-HT4R affinity for DPZ, 2, 9 and compounds 5a–t (% inhibition at 10−6M, 10−8M and 10−8 M, respectively).
The results of these in vitro evaluations are reported in a 3-fold-entry figure (Figure 3).
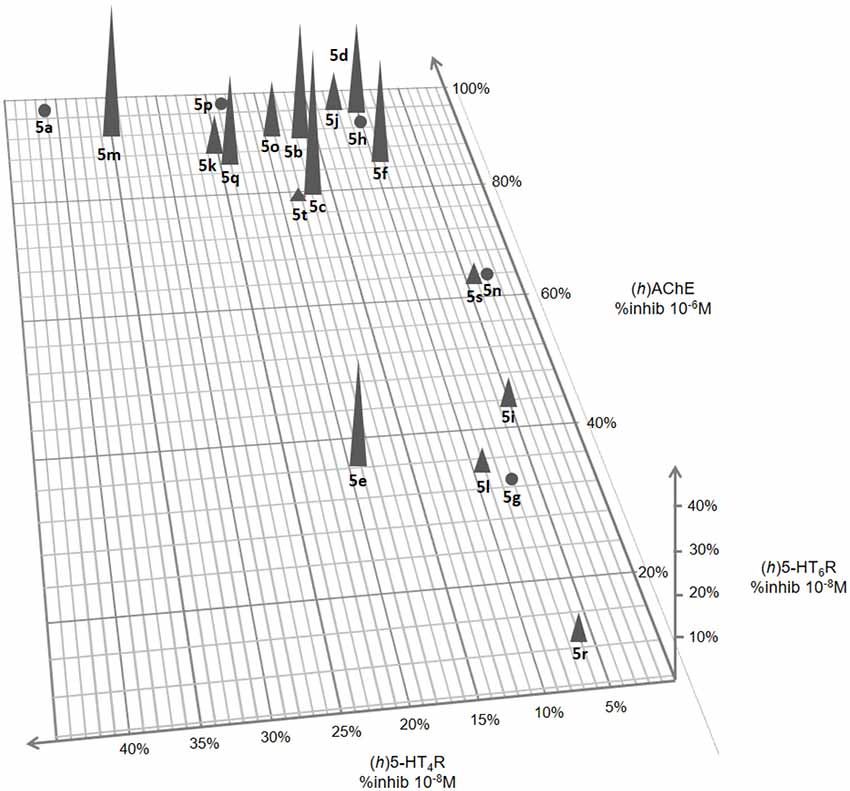
Figure 3. 3D representation of (h)5-HT6R and (h)5-HT4R affinity and (h)AChE inhibitory activity for compounds 5a–t.
Among the 20 tested compounds, 12 novel derivatives appear as moderate to potent AChEIs with %inhibition at 10−6M ≥ 80%. Their IC50 values ranged from 450 (5q) to 6 nM (5p; Table 2). Some of these AChEI showed an additional affinity towards 5-HT6R with %inhibition at 10−6M > 70% (5b, f, k, m) and Ki ranging from 1,150 to 110 nM. Three derivatives (5e, i, l) displayed affinity for 5-HT6R, while being devoid of inhibitory effect towards AChE. Finally, among the four dual compounds, 5k and 5m further exhibited an affinity towards 5-HT4R with %inhibition at 10−8M ≥ 25% and Ki of 168 and 43 nM respectively. Three other compounds (5o–q) displayed good affinity towards 5-HT4R but are devoid of such an affinity towards 5-HT6R.
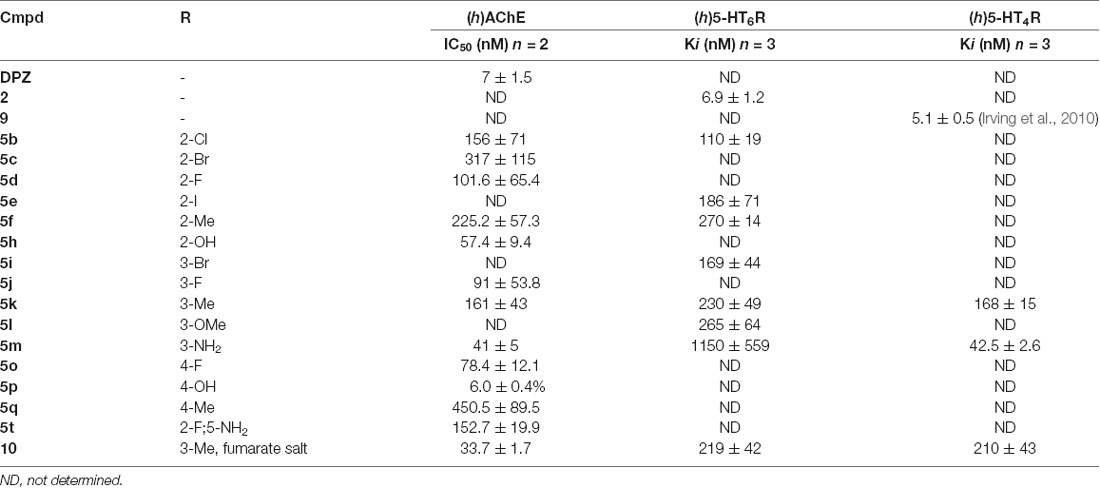
Table 2. (h)AChE inhibitory activity (IC50) and (h)5-HT6R and (h)5-HT4R affinity (Ki) for DPZ, 2, 9, 5b–f, h–m, o–q, t and the fumarate 10.
By virtue of its well-balanced in vitro activities towards the three targets, 5k was selected for further investigation. According to the concept of MTDL and the synergy theoretically displayed by the association of activities into a sole compound, 5k has been preferred to more potent compounds (e.g., 5m) but with unbalanced levels of activity towards the three targets. This derivative has then been used as its fumaric acid salt 10. The in vitro activities of 10 are depicted in Table 3.
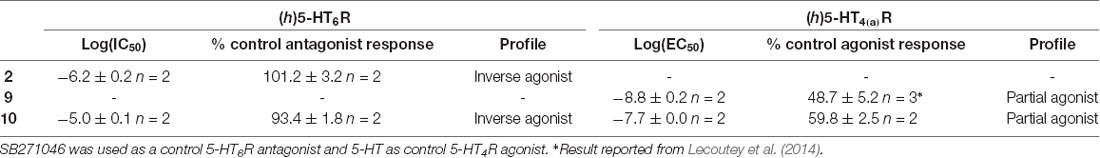
Table 3. (h)5-HT6R and (h)5-HT4(a)R pharmacological profile of compounds 2, 9 and 10.
The pharmacological profile of 10 was established towards (h)5-HT4R and (h)5-HT6R, respectively. It acts as a partial agonist towards (h)5-HT4R, in a similar manner as RS67333 (9), and as an inverse agonist towards (h)5-HT6R, in a similar manner as SB271046, used as a control 5-HT6R antagonist, and idalopirdine (2; Figure 4 and Table 3). Indeed, in our hands, these three compounds decreased cAMP production below the basal, in the absence of agonist (Figure 4C).
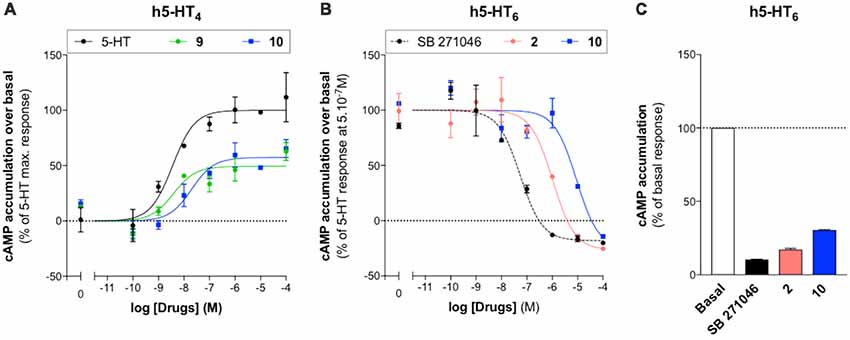
Figure 4. Pharmacological profile of compounds 2, 9 and 10. Representative experiments illustrating agonist activities of 9 and 10 towards (h)5-HT4(a)R (A) and antagonist activities of SB271046, 2 and 10 towards (h)5-HT6R stimulated with 5.10−7M of 5-HT [B; data are means ± standard error of the mean (SEM); n = 2]. (C) Inverse agonist activities of SB271046, 2 and 10 (10−4M) towards (h)5-HT6R in absence of 5-HT (means ± SEM of two independent experiments performed in duplicates).
In vivo Results
Whatever the dose tested (1, 10 and 100 mg/kg), preliminary toxicological and pharmacological screening of 10 (Table 4) did not show any deleterious signs, suggesting thus a LD50 quite higher than 100 mg/kg.
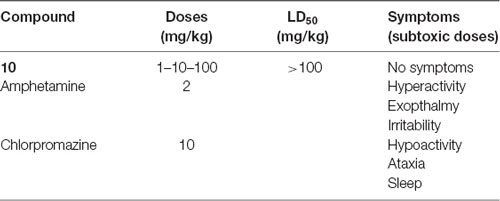
Table 4. Pharmacological and toxicological properties of 10.
None of the tested doses of 10 (1, 3 and 10 mg/kg) did neither modify spontaneous locomotor activity (Figure 5).
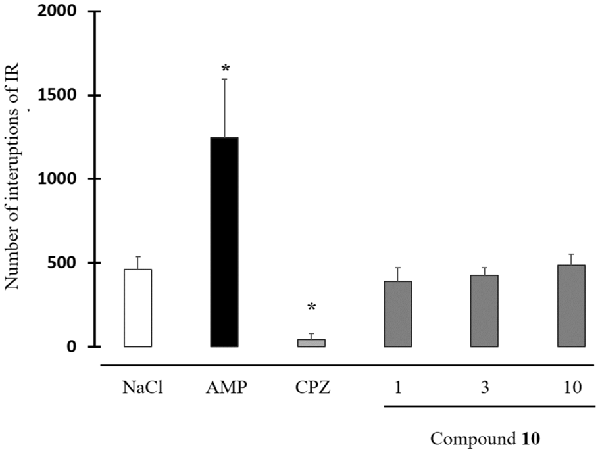
Figure 5. Effect of compound 10 on spontaneous locomotor activity. Data are expressed as the mean ± standard deviation (n = 8). Drugs were administered intraperitoneally (IP) 30 min before the behavioral test. Compound 10: 1–3–10 mg/kg; AMP: amphetamine 2 mg/kg; CPZ: chlorpromazine 3 mg/kg [*p < 0.05 NaCl, Student-Newman-Keuls (SNK) test].
Moreover, an anti-amnesic effect was observed in the scopolamine-induced spontaneous alternation deficit test, after IP administration of 10 at doses starting from 0.3 mg/kg (Figure 6). The degree of effect was similar to that of the dual-targeted compound donecopride in the same dose range (Rochais et al., 2015).
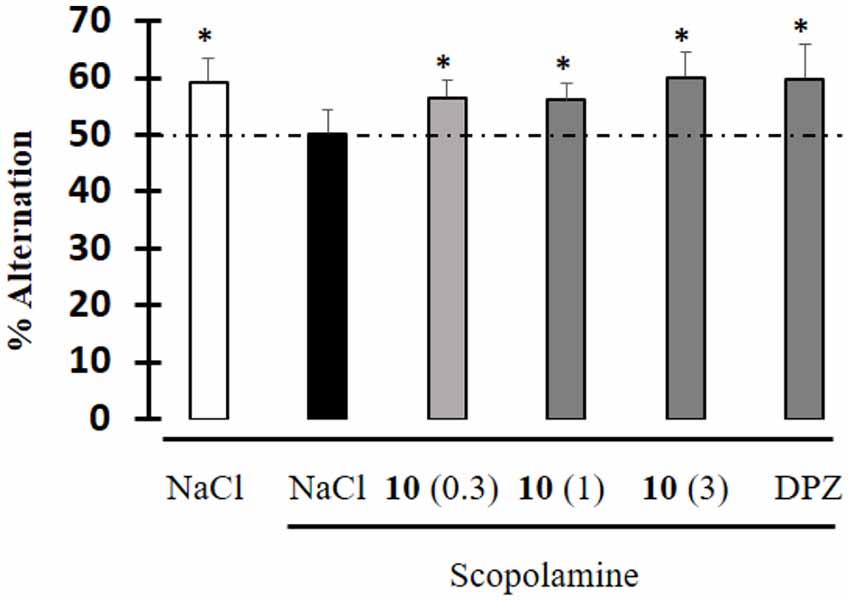
Figure 6. Effect of compound 10 on scopolamine-induced impairment during the spontaneous alternation test. Data are expressed as the mean ± standard deviation (n = 8). Drugs were administered IP 30 min before the test, and scopolamine was administered SC 20 min before the test. Compound 10: 0.3, 1, 3 mg/kg; DPZ: Donepezil; 1 mg/kg; Scopolamine: 0.5 mg/kg (*p < 0.05 vs. 50%; univariate t-test).
Discussion
Trying to establish SAR for MTDL is always quite challenging taking into consideration that the structural requirements can be divergent between the expected interactions with the different targets. Considering the three present targets, the SAR appeared similar between AChE and 5-HT4R, but partially different from those observed for 5-HT6R.
Indeed, with regard to AChE and 5-HT4R activities, the best substituent of the piperidine moiety of compounds 5 is the unsubstituted benzyl group (5a). According to our previous work (Yahiaoui et al., 2016), the nature of the substitution of a phenyl group distant from the piperidine moiety could influence in a great manner the affinity of the ligands towards both 5-HT receptors. For the two targets, poly or bulky substituents (Br, I, OBn, OMe, NO2), appear detrimental to the activities (5c, g, i, l, n) while a smaller one (OH, NH2, F, Me) seems likely to relatively maintain the activity (5h, p, m, d, j, o), especially when present in para position (5p).
Conversely, the 5-HT6R affinity appears linked to the substitution of the benzyl group within the studied scaffold, since the unsubstituted derivative 5a was devoid of such an activity. Further, small substituents (OH, F) do not favor, this time, the affinity (5d, h, j, o, p), whatever their position on the phenyl ring, while bulkier ones (Cl, Br, I, Me, OMe) promote it, especially in ortho or meta positions (5b, c, e, f, i, k, l).
These contradictory requirements explain that 5k with a methyl group in meta position displayed balanced activities towards the three targets and was chosen for further investigation under its fumarate form (10). Indeed, according to the MTDL concept (Cavalli et al., 2008), pleiotropic ligand could present moderate in vitro activities toward their targets that could, however, lead to potent and promising in vivo activities explained by their synergistic effect (Prati et al., 2015).
Of interest, compound 10, idalopirdine and SB271046 were able to reverse the constitutive activity of the 5-HT6 receptor, thus demonstrating an inverse agonist profile. Such a property is of high interest regarding this receptor as it is known to possess a high constitutive activity in the brain, exerting a tonic activity (Duhr et al., 2014; Deraredj Nadim et al., 2016). Regarding the 5-HT4R, 10 demonstrated a partial agonist activity similar to the one of 9, which had proven its efficiency to prevent amyloid production in a mouse model of AD (Hashimoto et al., 2012; Giannoni et al., 2013; Baranger et al., 2017).
The main result of the behavioral study concerns the anti-amnesic effect of 10 in a model of scopolamine-induced deficit of working memory from the dose of 0.3 mg/kg.
Although no pharmacokinetic data are available for compound 10 yet, this result indicates that it reaches the general circulation and crosses the blood-brain barrier upon IP administration. The anti-amnesic effect of compound 10 suggests that the co-modulation of the three targets could be a therapeutic strategy of interest in the field of memory impairments. The anti-amnesic properties of the compound 10, together with the absence of any deleterious signs during the pharmacological screening (even when tested at high dose: 100 mg/kg), are arguing in favor of 10 as a new promising drug, with excellent general tolerance.
Conclusion
We have synthesized a variety of compounds possessing both in vitro and in vivo activities towards three therapeutic targets (5-HT4R/5-HT6R, AChE) in the potential treatment of AD. The use of these innovative MTDL which may exhibit a greater benefit than single-targeted drugs towards the neuropathological hallmarks of the disease and the associated behavioral deficits will require further studies in animal models and potential future clinical trials to demonstrate their efficacy.
Associated Content
Analytical data of final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Ethics Statement
All experiments were conducted in agreement with the European Directives and French law on animal experimentation (personal authorization n° 14-17 for MB and 14-60 for TF).
Author Contributions
The manuscript was written through contributions of SC, TF, MB, CR and PD. All authors have given approval to the final version of the manuscript.
Funding
This work was supported by funding from the Fondation Vaincre Alzheimer (#FR-15072) and the Fondation Plan Alzheimer (AAP2015 Project TRIAD 016). The authors gratefully acknowledge the Conseil Régional de Normandie, as well as the European Community (FEDER) for their contribution to the CERMN’s analytical platform. European COST action CA15135 (Multi-Target Paradigm for Innovative Ligand Identification in the Drug Discovery Process, MuTaLig) supports this article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Steve Dubel for final proofreading of the manuscript.
Abbreviations
AD, Alzheimer’s disease; ACh, acetylcholine; AChE, acetylcholinesterase; AChEI, acetylcholinesterase inhibitor; DPZ, donepezil; MTDL, multi-target directed ligands.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2019.00148/full#supplementary-material
References
Baranger, K., Giannoni, P., Girard, S. D., Girot, S., Gaven, F., Stephan, D., et al. (2017). Chronic treatments with a 5-HT4 receptor agonist decrease amyloid pathology in the entorhinal cortex and learning and memory deficits in the 5xFAD mouse model of Alzheimer’s disease. Neuropharmacology 126, 128–141. doi: 10.1016/j.neuropharm.2017.08.031
Benhamú, B., Martín-Fontecha, M., Vázquez-Villa, H., Pardo, L., and López-Rodríguez, M. L. (2014). Serotonin 5-HT6 receptor antagonists for the treatment of cognitive deficiency in Alzheimer’s disease. J. Med. Chem. 57, 7160–7181. doi: 10.1021/jm5003952
Cavalli, A., Bolognesi, M. L., Minarini, A., Rosini, M., Tumiatti, V., Recanatini, M., et al. (2008). Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 51, 347–372. doi: 10.1021/jm7009364
Cho, S., and Hu, Y. (2007). Activation of 5-HT4 receptors inhibits secretion of β-amyloid peptides and increases neuronal survival. Exp. Neurol. 203, 274–278. doi: 10.1016/j.expneurol.2006.07.021
Claeysen, S., Bockaert, J., and Giannoni, P. (2015). Serotonin: a new hope in Alzheimer’s disease? ACS Chem. Neurosci. 6, 940–943. doi: 10.1021/acschemneuro.5b00135
Consolo, S., Arnaboldi, S., Giorgi, S., Russi, G., and Ladinsky, H. (1994). 5-HT4 receptor stimulation facilitates acetylcholine release in rat frontal cortex. Neuroreport 5, 1230–1232. doi: 10.1097/00001756-199406020-00018
Dawson, L. A., Nguyen, H. Q., and Li, P. (2000). In vivo effects of the 5-HT6 antagonist SB-271046 on striatal and frontal cortex extracellular concentrations of noradrenaline, dopamine, 5-HT, glutamate and aspartate. Br. J. Pharmacol. 130, 23–26. doi: 10.1038/sj.bjp.0703288
Dawson, L., Nguyen, H., and Li, P. (2001). The 5-HT(6) receptor antagonist SB-271046 selectively enhances excitatory neurotransmission in the rat frontal cortex and hippocampus. Neuropsychopharmacology 25, 662–668. doi: 10.1016/s0893-133x(01)00265-2
de Bruin, N. M., and Kruse, C. G. (2015). 5-HT6 receptor antagonists: potential efficacy for the treatment of cognitive impairment in schizophrenia. Curr. Pharm. Des. 21, 3739–3759. doi: 10.2174/1381612821666150605112105
Deraredj Nadim, W., Chaumont-Dubel, S., Madouri, F., Cobret, L., De Tauzia, M. L., Zajdel, P., et al. (2016). Physical interaction between neurofibromin and serotonin 5-HT6 receptor promotes receptor constitutive activity. Proc. Natl. Acad. Sci. U S A 113, 12310–12315. doi: 10.1073/pnas.1600914113
Dolles, D., and Decker, M. (2017). “Dual-acting compounds acting as receptor ligands and enzyme inhibitors,” in Design of Hybrid Molecules for Drug Development, ed. M. Decker (Oxford: Elsevier), 137–165.
Dolles, D., Hoffmann, M., Gunesch, S., Marinelli, O., Möller, J., Santoni, G., et al. (2018). Structure-activity relationships and computational investigations into the development of potent and balanced dual-acting butyrylcholinesterase inhibitors and human cannabinoid receptor 2 ligands with pro-cognitive in vivo profiles. J. Med. Chem. 61, 1646–1663. doi: 10.1021/acs.jmedchem.7b01760
Duhr, F., Déléris, P., Raynaud, F., Séveno, M., Morisset-Lopez, S., Mannoury la Cour, C., et al. (2014). Cdk5 induces constitutive activation of 5-HT6 receptors to promote neurite growth. Nat. Chem. Biol. 10, 590–597. doi: 10.1038/nchembio.1547
Eglen, R. M., Bonhaus, D. W., Johnson, L. G., Leung, E., and Clark, R. D. (1995). Pharmacological characterization of two novel and potent 5-HT4 receptor agonists, RS67333 and RS67506, in vitro and in vivo. Br. J. Pharmacol. 115, 1387–1392. doi: 10.1111/j.1476-5381.1995.tb16628.x
Ellis, J. M., and Fell, M. J. (2017). Current approaches to the treatment of Parkinson’s disease. Bioorg. Med. Chem. Lett. 27, 4247–4255. doi: 10.1016/j.bmcl.2017.07.075
Ellman, G. L., Courtney, K. D., Andres, V. Jr., and Featherstone, R. M. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95. doi: 10.1016/0006-2952(61)90145-9
Freret, T., Bouet, V., Quiedeville, A., Nee, G., Dallemagne, P., Rochais, C., et al. (2012). Synergistic effect of acetylcholinesterase inhibition (donepezil) and 5-HT(4) receptor activation (RS67333) on object recognition in mice. Behav. Brain Res. 230, 304–308. doi: 10.1016/j.bbr.2012.02.012
Ge, J., and Barnes, N. M. (1996). 5-HT4 receptor-mediated modulation of 5-HT release in the rat hippocampus in vivo. Br. J. Pharmacol. 117, 1475–1480. doi: 10.1111/j.1476-5381.1996.tb15309.x
Giannoni, P., Gaven, F., De Bundel, D., Baranger, K., Marchetti-Gauthier, E., Roman, F., et al. (2013). Early administration of RS 67333, a specific 5-HT4 receptor agonist, prevents amyloidogenesis and behavioral deficits in the 5XFAD mouse model of Alzheimer’s disease. Front. Aging Neurosci. 5:96. doi: 10.3389/fnagi.2013.00096
Hashimoto, G., Sakurai, M., Teich, A. F., Saeed, F., Aziz, F., and Arancio, O. (2012). 5-HT4 receptor stimulation leads to soluble AβPPα production through MMP-9 upregulation. J. Alzheimers Dis. 32, 437–445. doi: 10.3233/jad-2012-111235
Hirst, W. D., Stean, T. O., Rogers, D. C., Sunter, D., Pugh, P., Moss, S. F., et al. (2006). SB-399885 is a potent, selective 5-HT6 receptor antagonist with cognitive enhancing properties in aged rat water maze and novel object recognition models. Eur. J. Pharmacol. 553, 109–119. doi: 10.1016/j.ejphar.2006.09.049
Hooper, N., Fraser, C., and Stone, T. W. (1996). Effect of purines analogues on spontaneous alternation in mice. Psychopharmacology 123, 250–257. doi: 10.1007/bf02246579
Irving, H. R., Tochon-Danguy, N., Chinkwo, K. A., Li, J. G., Grabbe, C., Shapiro, M., et al. (2010). Investigations into the binding affinities of different human 5-HT4 receptor splice variants. Pharmacology 85, 224–233. doi: 10.1159/000280418
Karila, D., Freret, T., Bouet, V., Boulouard, M., Dallemagne, P., and Rochais, C. (2015). Therapeutic potential of 5-HT6 receptor agonists. J. Med. Chem. 58, 7901–7912. doi: 10.1021/acs.jmedchem.5b00179
Kilbinger, H., and Wolf, D. (1992). Effects of 5-HT4 receptor stimulation on basal and electrically evoked release of acetylcholine from guinea-pig myenteric plexus. Naunyn Schmiedebergs Arch. Pharmacol. 345, 270–275. doi: 10.1007/bf00168686
Lalut, J., Karila, D., Dallemagne, P., and Rochais, C. (2017). Modulating 5-HT4 and 5-HT6 receptors in Alzheimer’s disease treatment. Future Med. Chem. 9, 781–795. doi: 10.4155/fmc-2017-0031
Lecoutey, C., Hédou, D., Freret, T., Giannoni, P., Gaven, F., Since, M., et al. (2014). Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. U S A 111, E3825–E3830. doi: 10.1073/pnas.1410315111
Lelong, V., Lhonneur, L., Dauphin, F., and Boulouard, M. (2003). BIMU 1 and RS 67333, two 5-HT4 receptor agonists, modulate spontaneous alternation deficits induced by scopolamine in the mouse. Naunyn Schmiedebergs Arch. Pharmacol. 367, 621–628. doi: 10.1007/s00210-003-0743-2
Lezoualc’h, F. (2007). 5-HT4 receptor and Alzheimer’s disease: the amyloid connection. Exp. Neurol. 205, 325–329. doi: 10.1016/j.expneurol.2007.02.001
Lucas, G., Di Matteo, V., De Deurwaerdère, P., Porras, G., Martín-Ruiz, R., Artigas, F., et al. (2001). Neurochemical and electrophysiological evidence that 5-HT4 receptors exert a state-dependent facilitatory control in vivo on nigrostriatal, but not mesoaccumbal, dopaminergic function. Eur. J. Neurosci. 13, 889–898. doi: 10.1046/j.0953-816x.2000.01453.x
Marcos, B., Gil-Bea, F. J., Hirst, W. D., Garcia-Alloza, M., and Ramirez, M. J. (2006). Lack of localization of 5-HT6 receptors on cholinergic neurons: implication of multiple neurotransmitter systems in 5-HT6 receptor-mediated acetylcholine release. Eur. J. Neurosci. 24, 1299–1306. doi: 10.1111/j.1460-9568.2006.05003.x
Morpugo, C. (1971). A new design for the screening of CNS-active drugs in mice. Arzneimittelforschung 11, 1727–1734.
Nirogi, R., Mohammed, A. R., Shinde, A. K., Gagginapally, S. R., Kancharla, D. M., Middekadi, V. R., et al. (2018). Synthesis, structure-activity relationships, and preclinical evaluation of heteroaromatic amides and 1,3,4-oxadiazole derivatives as 5-HT4 receptor partial agonists. J. Med. Chem. 61, 4993–5008. doi: 10.1021/acs.jmedchem.8b00457
Prati, F., De Simone, A., Bisignano, P., Armirotti, A., Summa, M., Pizzirani, D., et al. (2015). Multitarget drug discovery for Alzheimer’s disease: triazinones as BACE-1 and GSK-3β inhibitors. Angew. Chem. Int. Ed Engl. 54, 1578–1582. doi: 10.1002/anie.201410456
Quiedeville, A., Boulouard, M., Hamidouche, K., Da Silva Costa-Aze, V., Nee, G., Rochais, C., et al. (2015). Chronic activation of 5-HT4 receptors or blockade of 5-HT6 receptors improve memory performances. Behav. Brain Res. 293, 10–17. doi: 10.1016/j.bbr.2015.07.020
Riemer, C., Borroni, E., Levet-Trafit, B., Martin, J. R., Poli, S., Porter, R. H. P., et al. (2003). Influence of the 5-HT6 receptor on acetylcholine release in the cortex: pharmacological characterization of 4–(2-bromo-6-pyrrolidin-1-ylpyridine-4-sulfonyl)phenylamine, a potent and selective 5-HT6 receptor antagonist. J. Med. Chem. 46, 1273–1276. doi: 10.1021/jm021085c
Rochais, C., Lecoutey, C., Gaven, F., Giannoni, P., Hamidouche, K., Hedou, D., et al. (2015). Multi target-directed ligands (MTDLs) with acetylcholinesterase (AChE) inhibitory and serotonergic subtype 4 receptor (5-HT4R) agonist activities as potential agents against Alzheimer’s disease: the design of donecopride. J. Med. Chem. 58, 3172–3187. doi: 10.1021/acs.jmedchem.5b00115
Russo, O., Cachard-chastel, M., Giner, M., Soulier, J., Berthouze, M., Richard, T., et al. (2009). Design, synthesis, and biological evaluation of new 5-HT4 receptor agonists: application as amyloid cascade modulators and potential therapeutic utility in Alzheimer’s disease design. J. Med. Chem. 52, 2214–2225. doi: 10.1021/jm801327q
Shirazi-Southall, S., Rodriguez, D. E., and Nomikos, G. G. (2002). Effects of typical and atypical antipsychotics and receptor selective compounds on acetylcholine efflux in the hippocampus of the rat. Neuropsychopharmacology 26, 583–594. doi: 10.1016/s0893-133x(01)00400-6
Steward, L. J., Ge, J., Stowe, R. L., Brown, D. C., Bruton, R. K., Stokes, P. R. A., et al. (1996). Ability of 5-HT4 receptor ligands to modulate rat striatal dopamine release in vitro and in vivo. Br. J. Pharmacol. 117, 55–62. doi: 10.1111/j.1476-5381.1996.tb15154.x
Wilkinson, D., Windfeld, K., and Colding-Jørgensen, E. (2014). Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 13, 1092–1099. doi: 10.1016/s1474-4422(14)70198-x
Yahiaoui, S., Hamidouche, K., Ballandonne, C., Davis, A., De Oliveira Santos, J. S., Freret, T., et al. (2016). Design, synthesis, and pharmacological evaluation of multitarget-directed ligands with both serotonergic subtype 4 receptor (5-HT4) partial agonist and 5-HT6 antagonist activities, as potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 121, 283–293. doi: 10.1016/j.ejmech.2016.05.048
Zhang, M. Y., Hughes, Z. A., Kerns, E. H., Lin, Q., and Beyer, C. E. (2007). Development of a liquid chromatography/tandem mass spectrometry method for the quantitation of acetylcholine and related neurotransmitters in brain microdialysis samples. J. Pharm. Biomed. Anal. 44, 586–593. doi: 10.1016/j.jpba.2007.02.024
Keywords: Alzheimer’s disease, acetylcholinesterase, 5-HT4 receptors, 5-HT6 receptors, MTDL
Citation: Hatat B, Yahiaoui S, Lecoutey C, Davis A, Freret T, Boulouard M, Claeysen S, Rochais C and Dallemagne P (2019) A Novel in vivo Anti-amnesic Agent, Specially Designed to Express Both Acetylcholinesterase (AChE) Inhibitory, Serotonergic Subtype 4 Receptor (5-HT4R) Agonist and Serotonergic Subtype 6 Receptor (5-HT6R) Inverse Agonist Activities, With a Potential Interest Against Alzheimer’s Disease. Front. Aging Neurosci. 11:148. doi: 10.3389/fnagi.2019.00148
Received: 11 April 2019; Accepted: 05 June 2019;
Published: 19 June 2019.
Edited by:
Rommy Von Bernhardi, Pontifical Catholic University of Chile, ChileReviewed by:
Romain Lefebvre, Ghent University, BelgiumJoachim Neumann, Institut für Pharmakologie und Toxikologie, Germany
Copyright © 2019 Hatat, Yahiaoui, Lecoutey, Davis, Freret, Boulouard, Claeysen, Rochais and Dallemagne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Rochais, Y2hyaXN0b3BoZS5yb2NoYWlzQHVuaWNhZW4uZnI=; Patrick Dallemagne, cGF0cmljay5kYWxsZW1hZ25lQHVuaWNhZW4uZnI=
† These authors have contributed equally to this work