 Lisbell D. Estrada
Lisbell D. Estrada Pablo Ahumada
Pablo Ahumada Daniel Cabrera
Daniel Cabrera Juan P. Arab
Juan P. Arab- 1Bionanotechnology Laboratory, Integrative Center for Applied Biology and Chemistry (CIBQA), Department of Chemical & Biological Sciences, Universidad Bernardo O’Higgins, Santiago, Chile
- 2Laboratório de Hepatologia Experimental, Gastroenterology Department, Facultad de Medicina, Centro de Envejecimiento y Regeneración (CARE Chile-UC), P. Universidad Catolica de Chile, Santiago, Chile
Alzheimer’s disease (AD) afflicts an estimated 20 million people worldwide and is the fourth-leading cause of death in the developed world. The most common cause of dementia in older individuals, AD is characterized by neuropathologies including synaptic and neuronal degeneration, amyloid plaques, and neurofibrillary tangles (NTFs). Amyloid plaques are primarily composed of amyloid-beta peptide (Aβ), which accumulates in the brains of patients with AD. Further, small aggregates termed Aβ oligomers are implicated in the synaptic loss and neuronal degeneration underlying early cognitive impairments. Whether Aβ accumulates in part because of dysregulated clearance from the brain remains unclear. The flow of substances (e.g., nutrients, drugs, toxins) in and out of the brain is mediated by the blood-brain-barrier (BBB). The BBB exhibits impairment in AD patients and animal models. The effect of BBB impairment on Aβ, and whether BBB function is affected by non-neurological pathologies that impair peripheral clearance requires further investigation. In particular, impaired peripheral clearance is a feature of nonalcoholic fatty liver disease (NAFLD), a spectrum of liver disorders characterized by accumulation of fat in the liver accompanied by varying degrees of inflammation and hepatocyte injury. NAFLD has reached epidemic proportions, with an estimated prevalence between 20% and 30% of the general population. This chronic condition may influence AD pathogenesis. This review article summarizes the current state of the literature linking NAFLD and AD, highlighting the role of the major Aβ efflux and clearance protein, the LRP-1 receptor, which is abundantly expressed in liver, brain, and vasculature.
Amyloid Beta Role in Alzheimer’s Disease
Alzheimer’s disease (AD) belongs to a large group of neurodegenerative diseases characterized by the pathophysiological brain changes related to the accumulation of misfolded proteins. Specifically, extracellular peptide variants of the amyloid-β (Aβ) accumulate in the form of amyloid plaques or senile plaques, and the intracellular accumulation of neurofibrillary tangles (NTFs) composed by phosphorylated Tau protein (pTau; Bloom, 2014; Héraud et al., 2014; He et al., 2018).
Both are reported to underlie progressive synaptic dysfunction in the AD brain, loss of dendritic spines, and neuronal death (Serrano-Pozo et al., 2011; Busche et al., 2019). Although AD was first described 100 years ago, its precise etiology remains unknown. Efforts to better understand AD have resulted in multiple hypotheses to explain events in AD pathogenesis, for example, the amyloid cascade theory that describes the imbalance between Aβ production and clearance (Selkoe and Hardy, 2016). Here, we provide an overview of the etiology of AD, and the principal concepts that support the critical role of the brain-blood barrier (BBB) and liver in AD development and progression.
In neurons under physiological conditions, Aβ is secreted to maintain normal synaptic function, morphology, and plasticity (Wang et al., 2012; Gouras et al., 2015; Klevanski et al., 2015). Aβ is a by-product generated from cleavage of the amyloid protein precursor (APP). APP plays an important physiological role in regulating γ-aminobutyric acid type B receptor (GABABR) and modulating synaptic transmission and plasticity (Chen et al., 2017; Doshina et al., 2017; Rice et al., 2019). In primary cortical neurons, APP modulates frequency and amplitude of calcium oscillations essential for synaptic transmission (Octave et al., 2013). A mouse model deficient for APP demonstrated that APP is necessary for the synapsis and maintenance of dendritic integrity in the hippocampus (Tyan et al., 2012). Likewise, hippocampal neurons in culture derived from APP knockout mice showed APP is critical for synaptogenesis and dendritic and axonal growth process and regulates substrate adhesion (Southam et al., 2019).
On the other hand, in the amyloidogenic (i.e., disease-causing) pathway, APP is cleaved by β- and γ-secretase to generate Aβ, which accumulates as senile plaques (Hardy and Selkoe, 2002; Konietzko, 2011). AD-related plaques are associated with high levels of soluble oligomeric forms of Aβ (AβOs; Esparza et al., 2013). AβOs comprise soluble dimers and trimers of low molecular weight and soluble oligomeric forms of 12–14 monomers (Mroczko et al., 2018). In addition, these oligomers have been identified as the toxic conformers of Aβ plaques in AD (Jin et al., 2011; Verma et al., 2015). AβOs can diffuse across synaptic membranes (Hong et al., 2014) and trigger a cascade of injurious events in neurons, causing synaptic failure and memory loss (Morris et al., 2014; Brito-Moreira et al., 2017). Moreover, AβOs are associated with dystrophic neurites, reactive astrocytes, and aberrant activation of glutamatergic neurotransmission; the consequence of these changes is neuronal death by excessive neuronal influx of sodium and calcium (Ziegler-Waldkirch and Meyer-Luehmann, 2018). Postsynaptic protein disruption (Lésne et al., 2013) and hippocampal synaptic plasticity impairment by AβOs contributes to memory loss (Müller-Schiffmann et al., 2016). Intracellular AβOs are detectable in cholinergic neurons, suggesting that they play a critical role in cholinergic deficiency (Baker-Nigh et al., 2015). These devastating events not only lead to memory loss and learning impairment in AD patients, but also affect the capacities of reasoning, abstraction, and language (Duyckaerts et al., 2009).
Blood-Brain Barrier Breakdown and Role of LRP-1 in Alzheimer’s Disease
The blood-brain barrier (BBB) is a specialized structure that supports brain function. This structure supports the brain by regulating electrolyte flux, cerebral blood flow (CBF) and efficient oxygen and metabolite delivery, and restricting entry of potentially toxic and even some therapeutic agents into the brain (Provias and Jeynes, 2014; Andreone et al., 2015; Di Marco et al., 2015). BBB function is mediated by neurovascular units (NVU) comprising neurons, glial cells, pericytes, and brain endothelial cells, which maintain homeostasis of the cerebral microenvironment (Armulik et al., 2011). Brain endothelial cells are an important component mediating the flow between brain and blood by cell-to-cell communications called tight junctions and adherent junctions; these junctions connect cell networks (Deli et al., 2005; Van de Haar et al., 2015) and regulate the paracellular permeability of substances across the BBB (Bowman and Quinn, 2008; Viggars et al., 2011; Kook et al., 2013; Chow and Gu, 2015; Ulrich et al., 2015). Tight junctions proteins ZO-1, Occludin and CLN-5 are key to maintaining BBB integrity (Jiao et al., 2011). ZO-1 joins tight junctions with the actin cytoskeleton, working as accessory proteins (Xiao et al., 2017). Occludin and CLN-5 are transmembrane tight junction proteins involved in signal transduction of cytokines (Haseloff et al., 2015). The high expression of these proteins on brain endothelial cells regulates the transport of essential molecules through the BBB, such as the free and rapid diffusion of oxygen and carbon dioxide (Lin et al., 2015; Pardridge, 2015). Hydrophobic molecules permeate the BBB faster and more easily than hydrophilic molecules, while molecules that are larger than 180 KDa or water-soluble do not penetrate the BBB (Kroll and Neuwelt, 1998; Zlokovic, 2005; Masserini, 2013). For example, the BBB restricts the passage of albumin and immunoglobulins, high-molecular-weight proteins from the peripheral blood circulation (Xiao and Gan, 2013).
Another important component of brain endothelial cells is a complex and specific transport-receptor protein system that also contributes to BBB permeability (Zlokovic, 2011). The luminal side of the BBB contains transporters for specific classes of nutrients, such as glucose and vitamins, and receptors for peptides, proteins, and hormones. These mediators facilitate transport across the BBB from circulating blood into the brain (Deane and Zlokovic, 2007; Simpson et al., 2007). In contrast, the transport system of the abluminal side of the BBB eliminates neurotoxic molecules and metabolic waste (Begley and Brightman, 2003).
Dysfunction of the BBB, therefore, could result in altered permeability. Indeed, age-dependent BBB breakdown at the hippocampus is associated with mild cognitive impairment and correlates with pericytes injury. This finding suggests that the cerebrovascular integrity loss that begins at the hippocampus may contribute to early stages of dementia associated with AD (Montagne et al., 2015). Similarly, early cognitive dysfunction has been associated with capillary damage and BBB breakdown in older adults (Nation et al., 2019).
This breakdown of BBB function may be related to alterations in specific components of the BBB structure. Low-density lipoprotein receptor-related protein 1 (LRP-1) is a membrane receptor that mediates the cellular internalization of multiple ligands. Further, LRP-1 regulates several tight junction proteins in endothelial cells of the BBB (Zhao et al., 2016). Functional LRP-1 is expressed in liver sinusoidal endothelial cells (LECs), highly specialized scavenger cells, and LRP-1 expression contributes to the rapid removal of its blood ligands (Øie et al., 2011). Cell surface LRP-1 and circulating sLRP-1 are needed for brain and systemic clearance of Aβ; however, in AD, both cell surface LRP-1 and circulating sLRP-1 concentrations are dramatically reduced (Sagare et al., 2012). Importantly, these alterations may begin as early as two decades before the manifestation of cognitive impairment symptoms (Beason-Held et al., 2013; Jack et al., 2013; De Strooper, 2014). Clearance of Aβ may also be affected by other pathologies, however.
Clearance of Aβ at The Periphery: Role of The Liver
Peripheral organs, including the kidney and the liver, play an essential role in the clearance of circulating Aβ. Elimination of Aβ from the circulation may contribute to AD progression, by helping to displace the dynamic equilibrium from Aβ deposited in the senile plaques toward soluble Aβ. This hypothesis is supported by evidence that peritoneal dialysis reduces the circulating levels of Aβ in humans and diminishes AD features in an animal model (Jin et al., 2017). Insufficient clearance of brain Aβ also contributes to the progression of sporadic AD (Wang et al., 2006). As brain Aβ equilibrates with Aβ in plasma, peripheral clearance of Aβ provides a potential approach to facilitate efflux of Aβ from the brain (Liu et al., 2015). Peripheral organs and tissues are key in clearing brain-derived Aβ under physiological conditions (Xiang et al., 2015).
The liver has many functions, one of which is metabolic detoxification. When the liver is under constant injury, as is found in metabolic diseases, it exhibits decreased detoxification capacity. Indeed, the expression of metabolic enzymes decreases in conditions such as obesity, diabetes, and cirrhosis (Rolle et al., 2018). Hepatocytes can act directly on circulating Aβ, promoting its clearance by degradation or through bile excretion. Further, Aβ uptake from circulation can be mediated through LRP-1, which is highly expressed in hepatocytes (Kanekiyo and Bu, 2014). Interestingly, liver dysfunction is accompanied by low LRP-1 hepatic expression and high levels of circulating Aβ. This correlation suggests that Aβ clearance decreases due to low hepatic LRP-1 activity (Wang et al., 2017; see Figure 1).
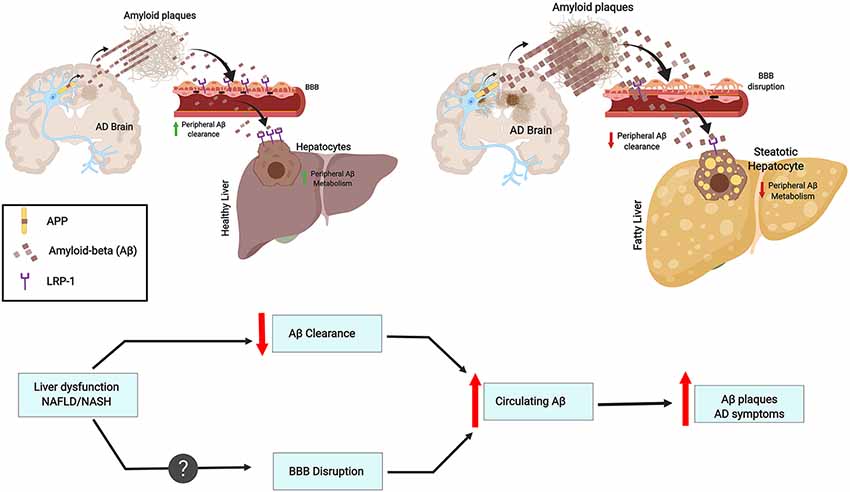
Figure 1. Chronic liver diseases may increase amyloid burden and Alzheimer’s pathology. This contribution results from an imbalance in peripheral amyloid-β (Aβ) clearance as a result of decreased LRP1 levels, general liver dysfunction, and chronic inflammation. These features may worsen blood-brain-barrier (BBB) impairment and contribute to a vicious cycle. As an example, the figure depicts fatty liver disease as a chronic liver condition.
AD pathophysiology has not been evaluated from a hepatic point of view; yet, the evidence points to a critical role for liver in AD pathogenesis. Aβ levels found in liver samples from AD patients are lower when compared to neurologically healthy controls, raising the possibility that the liver is not properly eliminating circulating Aβ (Roher et al., 2009). This observation is supported by studies where insulin promotes LRP-1 translocation to the cell membrane in hepatocytes, favoring Aβ clearance (Tamaki et al., 2007). The stimulation of LRP-1-mediated liver uptake improves cognitive impairment and decreases Aβ aggregation in the brain in AD transgenic mice (Sehgal et al., 2012).
NAFLD/NASH Affects Aβ Clearance
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of liver disorders characterized by excessive fat deposition in hepatocytes from individuals who drink little or no alcohol. NAFLD is an umbrella term for several subtypes ranging from isolated hepatic steatosis, or fatty liver, to nonalcoholic steatohepatitis (NASH). NASH is defined by the presence of fatty changes with inflammation and several degrees of hepatocellular injury or fibrosis. Thus, NASH is the aggressive form of NAFLD and can progress to advanced fibrosis and cirrhosis.
NAFLD/NASH is the leading cause of chronic liver disease worldwide and has reached epidemic proportions. Interestingly, most of the deaths in NAFLD patients are not restricted to liver-related morbidity or mortality; rather, cardiovascular disease (CVD) and cancer predominate (Armstrong et al., 2014). Therefore, the presence of fatty liver is not a benign pathology as was historically considered by most clinicians. Indeed, extensive evidence in recent years shows that NAFLD also increases the risk of end-stage liver disease, hepatocellular carcinoma (HCC), liver-related mortality, and all-cause mortality. These observations prompted the idea that NAFLD/NASH, either independently or concomitantly with other metabolic risk factors, determines or even drives extra-hepatic diseases such as CVD, chronic kidney disease, colorectal cancer, endocrine disorders like type 2 diabetes mellitus, osteoporosis, and, indeed, AD. Recent studies have linked insulin-resistance (the key pathophysiological feature of NAFLD) to several of the neurodegenerative mechanisms of AD including oxidative stress, mitochondrial dysfunction, and inflammation, via dysregulated insulin/IGF-1 signaling with attendant impairments in signal transduction and gene expression (de la Monte and Tong, 2014; de la Monte, 2017; Kim et al., 2016).
A network clustering analysis conducted by Karbalaei et al. (2018) indicated that there are 189 genes shared between NAFLD and AD. Further, three main groups of pathways are candidates for contributing to both AD and NAFLD: carbohydrate metabolism, long fatty acid metabolism, and IL-17 signaling pathways (Karbalaei et al., 2018). This suggests that diabetes and obesity might be considered as a risk factor for AD and NAFLD.
One study showed that NAFLD promotes AD in mice (Kim et al., 2016). This study evaluated whether NAFLD induction, through a dietary approach (high-fat diet), promotes the development of AD signs. Brains of HFD-fed mice showed increased levels of neuro-inflammation, characterized by higher levels of cytokines, toll-like receptors, and microgliosis. These features were accompanied by increased plaque formation in a transgenic mouse model of AD. In addition, intense and frequent signs of cerebral amyloid angiopathy (CAA)—a condition characterized by the Aβ deposition in the media and adventitia of small and mid-sized arteries—were observed in mice fed with HFD.
An abnormal lipid metabolism is linked with increased risk for AD development, and the liver plays a crucial role since is the main peripheral organ responsible for lipid metabolism (Fukumoto et al., 2002; Hooijmans and Kiliaan, 2008). Aβ is able to bind Apolipoprotein E (ApoE) and can be cleared from the brain together with cholesterol (Mahley, 1988). Interestingly, ApoE is a ligand of LRP-1 and both are genetically associated with AD and plasma Aβ levels (Kang et al., 2000). This link is intriguing since LRP-1 is suggested to facilitate Aβ clearance from the brain across the BBB (Deane et al., 2004; Sagare et al., 2012; see Figure 1).
Liver Inflammation and Aβ Levels
Hepatitis B is a liver infection that can become chronic and severe. Interestingly, Hepatitis B Virus (HBV) carriers have significantly higher plasma Aβ levels than non-carriers. Moreover, HBV carrier status is associated with plasma Aβ levels (Jin et al., 2017). Overall infectious burden including cytomegalovirus (CMV), herpes simplex virus type 1 (HSV-1), Borrelia burgdorferi, Chlamydophila pneumoniae and Helicobacter pylori was found to significantly contribute to AD pathogenesis (Bu et al., 2015). However, currently, no epidemiological study has been designed to understand the association between HBV infection and the risk for AD. The effect of chronic inflammation on Aβ clearance is lesser than the effects of HBV infection or liver dysfunction (Liu et al., 2013). Further, although plasma concentrations of cytokines IL-1β and IL-6 are significantly increased in cirrhosis patients and plasma IL-6 levels are correlated with Aβ40 levels (a 40 amino acid proteolytic product of APP cleavage that has gained attention as a biomarker correlating with AD), no association is observed by linear regression between IL-6 and Aβ40 levels. On the other hand, the ratio of AST/ALT, which is an indicator of liver functional impairment (Giannini et al., 1999), is significantly associated with circulating Aβ40 levels (Wang et al., 2017). Furthermore, hepatic dysfunction may lead to a plethora of systemic changes. Approximately 95% of Aβ in the blood is bound to serum albumin (Stanyon and Viles, 2012). The serum albumin pool represents an important reservoir for peripheral clearance of Aβ. Thus, a diminution in blood albumin in cirrhotic patients might contribute to the increase in plasma Aβ levels (see Figure 1).
Concluding Remarks
AD is a degenerative condition that will afflict an increasing number of people as the global population ages. Unfortunately, current treatments have only transient or modest effects. This article reviews evidence that supports the involvement of liver diseases, a growing health concern, in AD pathogenesis. The liver is the major player in the clearance of Aβ at the periphery, and an impairment of this clearance may shift the delicate Aβ equilibrium toward brain accumulation.
As to the possible role that the liver plays in brain-derived Aβ clearance, the impaired clearance of serum Aβ might contribute to the high Aβ levels in NAFLD patients. This effect is likely due to an intensification of the BBB disruption and drop in LRP-1 levels, the major receptor for Aβ efflux and important effector of clearance.
It is possible that hepatic malfunction contributes to AD in a plethora of non-excluding pathways, including: (i) the failure to maintain Aβ homeostasis at the periphery; (ii) acting as a source of pro-inflammatory cytokines when chronic inflammation follows different types of injury (like virus infection, drug-induced injury, and metabolic diseases); and (iii) through metabolic impairment.
Author Contributions
LE wrote and edited the manuscript. PA participated in manuscript writing. DC wrote the manuscript and designed figures. JA participated in manuscript writing and editing.
Funding
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) Initiation into Research Grant N° 11130561 to LE, and by FONDECYT Initiation into Research Grant N° 11171001, 11130561 and CARE-ChileUC to DC, Gastroenterology Department, Facultad de Medicina, P. Universidad Catolica de Chile to JA and Magister en Ciencias Quimico-Biologicas, Universidad Bernardo O Higgins.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the professional help of Mrs. Mariela Freire and Mr. Jonathan Leon for critical revision of the manuscript.
References
Andreone, B. J., Lacoste, B., and Gu, C. (2015). Neuronal and vascular interactions. Annu. Rev. Neurosci. 38, 25–46. doi: 10.1146/annurev-neuro-071714-033835
Armstrong, M. J., Adams, L. A., Canbay, A., and Syn, W.-K. (2014). Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 59, 1174–1197. doi: 10.1002/hep.26717
Armulik, A., Genové, G., and Betsholtz, C. (2011). Pericytes: developmental, physiological and pathological perspectives, problems and promises. Dev. Cell 21, 193–215. doi: 10.1016/j.devcel.2011.07.001
Baker-Nigh, A., Vahedi, S., Davis, E.-G., Weintraub, S., Bigio, E. H., Klein, W. L., et al. (2015). Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 138, 1722–1737. doi: 10.1093/brain/awv024
Beason-Held, L., Goh, J. O., An, Y., Kraut, M. A., O’Brien, R. J., Ferrucci, L., et al. (2013). Changes in brain function occurs years before the onset of cognitive impairment. J. Neurosci. 33, 18008–18014. doi: 10.1523/JNEUROSCI.1402-13.2013
Begley, D. J., and Brightman, M. W. (2003). Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 61, 39–78. doi: 10.1007/978-3-0348-8049-7_2
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Bowman, G. L., and Quinn, J. F. (2008). Alzheimer’s disease and the blood-brain barrier: past, present and future. Aging Health 4, 47–55. doi: 10.2217/1745509X.4.1.47
Brito-Moreira, J., Lourenco, M. V., Oliveira, M. M., Ribeiro, F. C., Ledo, J. H., Diniz, L. P., et al. (2017). Interaction of amyloid-β (Aβ) oligomers with neurexin 2α and neuroligin 1 mediates synapse damage and memory loss in mice. J. Biol. Chem. 292, 7327–7337. doi: 10.1074/jbc.M116.761189
Bu, X.-L., Yao, X.-Q., Jiao, S.-S., Zeng, F., Liu, Y.-H., Xiang, Y., et al. (2015). A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 22, 1519–1525. doi: 10.1111/ene.12477
Busche, M. A., Wegmann, S., Dujardin, S., Commins, C., Schiantarelli, J., Klickstein, N., et al. (2019). Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 22, 57–64. doi: 10.1038/s41593-018-0289-8
Chen, M., Wang, J., Jiang, J., Zheng, X., Justice, N. J., Wang, K., et al. (2017). APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. eLife 6:e20142. doi: 10.7554/eLife.20142
Chow, B. W., and Gu, C. (2015). The molecular constituents of the blood-brain barrier. Trends Neurosci. 38, 598–608. doi: 10.1016/j.tins.2015.08.003
Deane, R., Wu, Z., and Zlokovic, B. V. (2004). RAGE (Yin) versus LRP (Yang) balance regulates Alzheimer amyloid-peptide clearance through transport across the blood-brain barrier. Stroke 35, 2628–2631. doi: 10.1161/01.str.0000143452.85382.d1
Deane, R., and Zlokovic, B. V. (2007). Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 4, 191–197. doi: 10.2174/156720507780362245
de la Monte, S. M. (2017). Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 77, 47–65. doi: 10.1007/s40265-016-0674-0
de la Monte, S. M., and Tong, M. (2014). Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 88, 548–559. doi: 10.1016/j.bcp.2013.12.012
Deli, M. A., Abrahám, C. S., Kataoka, Y., and Niwa, M. (2005). Permeability studies on in vitro blood-brain barrier models: physiology, pathology and pharmacology. Cell. Mol. Neurobiol. 25, 59–127. doi: 10.1007/s10571-004-1377-8
De Strooper, B. (2014). Lessons from a failed γ-secretase Alzheimer trial. Cell 159, 721–726. doi: 10.1016/j.cell.2014.10.016
Di Marco, L. Y., Venneri, A., Farkas, E., Evans, P. C., Marzo, A., and Frangi, A. F. (2015). Vascular dysfunction in the pathogenesis of Alzheimer’s disease—a review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 82, 593–606. doi: 10.1016/j.nbd.2015.08.014
Doshina, A., Gourgue, F., Onizuka, M., Opsomer, R., Wang, P., Ando, K., et al. (2017). Cortical cells reveal APP as a new player in the regulation of GABAergic neurotransmission. Sci. Rep. 7:370. doi: 10.1038/s41598-017-00325-2
Duyckaerts, C., Delatour, B., and Potier, M. C. (2009). Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36. doi: 10.1007/s00401-009-0532-1
Esparza, T. J., Zhao, H., Cirrito, J. R., Cairns, N. J., Bateman, R. J., Holtzman, D. M., et al. (2013). Amyloid-β oligomerization in Alzheimer dementia versus highpathology controls. Ann. Neurol. 73, 104–119. doi: 10.1002/ana.23748
Fukumoto, H., Deng, A., Irizarry, M. C., Fitzgerald, M. L., and Rebeck, G. W. (2002). Induction of the cholesterol transporter ABCA1 in central nervous system cells by liver X receptor agonists increases secreted Aβ levels. J. Biol. Chem. 277, 48508–48513. doi: 10.1074/jbc.M209085200
Giannini, E., Botta, F., Fasoli, A., Ceppa, P., Risso, D., Lantieri, P. B., et al. (1999). Progressive liver functional impairment is associated with an increase in AST/ALT ratio. Dig. Dis. Sci. 44, 1249–1253. doi: 10.1023/A:1026609231094
Gouras, G. K., Olsson, T. T., and Hansson, O. (2015). β-amyloid peptide and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 12, 3–11. doi: 10.1007/s13311-014-0313-y
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Haseloff, R. F., Dithmer, S., Winkler, L., Wolburg, H., and Blasig, I. E. (2015). Transmembrane proteins of the tight junctions at the blood-brain barrier: structural and functional aspects. Semin. Cell Dev. Biol. 38, 16–25. doi: 10.1016/j.semcdb.2014.11.004
He, Z., Guo, J. L., McBride, J. D., Narashimhan, S., Kim, H., Changolkar, L., et al. (2018). Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque and aggregation. Nat. Med. 24, 29–38. doi: 10.1038/nm.4443
Héraud, C., Goufak, D., Ando, K., Leroy, K., Suain, V., Yilmaz, Z., et al. (2014). Increased misfolding and truncation of tau in APP/PS1/tau transgenic mice compared to mutant tau mice. Neurobiol. Dis. 62, 100–112. doi: 10.1016/j.nbd.2013.09.010
Hong, S., Ostaszewski, B. L., Yang, T., O’Malley, T. T., Jin, M., Yanagisawa, K., et al. (2014). Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319. doi: 10.1016/j.neuron.2014.02.027
Hooijmans, C. R., and Kiliaan, A. J. (2008). Fatty acids, lipid metabolism and Alzheimer pathology. Eur. J. Pharmacol. 585, 176–196. doi: 10.1016/j.ejphar.2007.11.081
Jack, C. R., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jiao, H., Wang, Y. Z., Liu, P., and Wang, Y. X. (2011). Specific role of tight junction proteins claudin-5, occludin and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J. Mol. Neurosci. 44, 130–139. doi: 10.1007/s12031-011-9496-4
Jin, W. S., Shen, L. L., Bu, X. L., Zhang, W. W., Chen, S. H., Huang, Z. L., et al. (2017). Peritoneal dialysis reduces amyloid-β plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 134, 207–220. doi: 10.1007/s00401-017-1721-y
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U S A 108, 5819–5824. doi: 10.1073/pnas.1017033108
Kanekiyo, T., and Bu, G. (2014). The low-density lipoprotein receptor-related protein 1 and amyloid-Î2 clearance in Alzheimer’s disease. Front Aging Neurosci. 6:93. doi: 10.3389/fnagi.2014.00093
Kang, D. E., Pietrzik, C. U., Baum, L., Chevallier, N., Merriam, D. E., Kounnas, M. Z., et al. (2000). Modulation of amyloid β-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Invest. 106, 1159–1166. doi: 10.1172/JCI11013
Karbalaei, R., Allahyari, M., Rezaei-Tavirani, M., Asadzadeh-Aghdaei, H., and Zali, M. R. (2018). Protein-protein interaction analysis of Alzheimer’s disease and NAFLD based on systems biology methods unhide common ancestor pathways. Gastroenterol. Hepatol. Bed Bench 1, 27–33. doi: 10.22037/ghfbb.v0i0.1327
Kim, D. G., Krenz, A., Toussaint, L. E., Maurer, K. J., Robinson, S. A., Yan, A., et al. (2016). Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflammation 13:1. doi: 10.1186/s12974-015-0467-5
Klevanski, M., Herrmann, U., Weyer, S. W., Fol, R., Cartier, N., Wolfer, D. P., et al. (2015). The APP intracellular domain is required for normal synaptic morphology, plasticity and hippocampus-dependent behavior. J. Neurosci. 35, 16018–16033. doi: 10.1523/JNEUROSCI.2009-15.2015
Konietzko, U. (2011). AICD nuclear signaling and its possible contribution to Alzheimer’s disease. Curr. Alzheimer Res. 9, 200–216. doi: 10.2174/156720512799361673
Kook, S. Y., Seok Hong, H., Moon, M., and Mook-Jung, I. (2013). Disruption of blood-brain barrier in Alzheimer disease pathogenesis. Tissue Barriers 1:e23993. doi: 10.4161/tisb.23993
Kroll, R. A., and Neuwelt, E. A. (1998). Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means. Neurosurgery 42, 1083–1099; discussion 1099–1100. doi: 10.1097/00006123-199805000-00082
Lésne, S. E., Sherman, M. A., Grant, M., Kuskowski, M., Schneider, J. A., Bannet, D. A., et al. (2013). Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 136, 1383–1398. doi: 10.1093/brain/awt062
Lin, L., Yee, S. W., Kim, R. B., and Giacomini, K. M. (2015). SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 14, 543–560. doi: 10.1038/nrd4626
Liu, Y. H., Wang, Y. R., Xiang, Y., Zhou, H. D., Giunta, B., Mañucat-Tan, N. B., et al. (2015). Clearance of amyloid-β in Alzheimer’s disease: shifting the action site from center to periphery. Mol. Neurobiol. 51, 1–7. doi: 10.1007/s12035-014-8694-9
Liu, Y. H., Zeng, F., Wang, Y. R., Zhou, H. D., Giunta, B., Tan, J., et al. (2013). Immunity and Alzheimer’s disease: immunological perspectives on the development of novel therapies. Drug Discov. Today 18, 1212–1220. doi: 10.1016/j.drudis.2013.07.020
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
Masserini, M. (2013). Nanoparticles for brain drug delivery. ISRN Biochem. 2013:238428. doi: 10.1155/2013/238428
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Morris, G. P., Clark, I. A., and Vissel, B. (2014). Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2:135. doi: 10.1186/s40478-014-0135-5
Mroczko, B., Groblewska, M., Litman-Zawadzka, A., Kornhuber, J., and Lewczuk, P. (2018). Cellular receptors of amyloid β oligomers (AβOs) in Alzheimer’s disease. Int. J. Mol. Sci. 19:1884. doi: 10.3390/ijms19071884
Müller-Schiffmann, A., Herring, A., Abdel-Hafiz, L., Chepkova, A. N., Schäble, S., Wedel, D., et al. (2016). Amyloid-β dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain 139, 509–525. doi: 10.1093/brain/awv355
Nation, D. A., Sweeney, M. D., Montagne, A., Sagare, A. P., D’ Orazio, L. M., Pachicano, M., et al. (2019). Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 25, 270–276. doi: 10.1038/s41591-018-0297-y
Octave, J. N., Pierrot, N., Ferao Santos, S., Nalivaeva, N. N., and Turner, A. (2013). From synaptic spines to nuclear signaling: nuclear and synaptic actions of the amyloid precursor protein. J. Neurochem. 126, 183–190. doi: 10.1111/jnc.12239
Øie, C. I., Appa, R. S., Hilden, I., Petersen, H. H., Gruhler, A., Smedsrød, B., et al. (2011). Rat liver sinusoidal endothelial cells (LSECs) express functional low density lipoprotein receptor-related protein-1 (LRP-1). J. Hepatol. 55, 1346–1352. doi: 10.1016/j.jhep.2011.03.013
Pardridge, W. M. (2015). Blood-brain barrier endogenous transporters as therapeutic targets: a new model for small molecule CNS drug discovery. Expert Opin. Ther. Targets 19, 1059–1072. doi: 10.1517/14728222.2015.1042364
Provias, J., and Jeynes, B. (2014). The role of the blood-brain barrier in the pathogenesis of senile plaques in Alzheimer’s disease. Int. J. Alzheimers Dis. 2014:191863. doi: 10.1155/2014/191863
Rice, H. C., de Malmazet, D., Schreurs, A., Frere, S., Van Molle, I., Volkov, A. N., et al. (2019). Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363:eaao4827. doi: 10.1126/science.aao4827
Roher, A. E., Esh, C. L., Kokjohn, T. A., Castaño, E. M., Van Vickle, G. D., Kalback, W. M., et al. (2009). Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 5, 18–29. doi: 10.1016/j.jalz.2008.10.004
Rolle, A., Paredes, S., Cortínez, L. I., Anderson, B.-J., Quezada, N., Solari, S., et al. (2018). Dexmedetomidine metabolic clearance is not affected by fat mass in obese patients. Br. J. Anaesth. 120, 969–977. doi: 10.1016/j.bja.2018.01.040
Sagare, A. P., Deane, R., and Zlokovic, B. V. (2012). Low-density lipoprotein receptor-related protein 1, A physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 136, 94–105. doi: 10.1016/j.pharmthera.2012.07.008
Sehgal, N., Gupta, A., Valli, R. K., Joshi, S. D., Mills, J. T., Hamel, E., et al. (2012). Withania somnifera reverses Alzheimer’s disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc. Natl. Acad. Sci. U S A 109, 3510–3515. doi: 10.1073/pnas.1112209109
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Simpson, I. A., Carruthers, A., and Vannucci, S. J. (2007). Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow Metab. 27, 1766–1791. doi: 10.1038/sj.jcbfm.9600521
Southam, K. A., Stennard, F., Pavez, C., and Small, D. H. (2019). Knockout of amyloid-β protein precursor (APP) expression alters synaptogenesis, neurite branching and axonal morphology of hippocampal neurons. Neurochem. Res. 44, 1346–1355. doi: 10.1007/s11064-018-2512-0
Stanyon, H. F., and Viles, J. H. (2012). Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium. J. Biol. Chem. 287, 28163–28168. doi: 10.1074/jbc.C112.360800
Tamaki, C., Ohtsuki, S., and Terasaki, T. (2007). Insulin facilitates the hepatic clearance of plasma amyloid β-peptide (1–40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 72, 850–855. doi: 10.1124/mol.107.036913
Tyan, S. H., Shih, A., Walsh, J., Maruyama, H., Sarsoza, F., Ku, L., et al. (2012). Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 51, 43–52. doi: 10.1016/j.mcn.2012.07.009
Ulrich, J. D., Huynh, T. P., and Holtzman, D. M. (2015). Re-evaluation of the blood-brain barrier in the presence of Alzheimer’s disease pathology. Neuron 88, 237–239. doi: 10.1016/j.neuron.2015.10.008
Van de Haar, H. J., Burgmans, S., Hofman, P. A., Verhey, F. R., Jansen, J. F., and Backes, W. H. (2015). Blood-brain barrier impairment in dementia: current and future in vivo assessments. Neurosci. Biobehav. Rev. 49, 71–81. doi: 10.1016/j.neubiorev.2014.11.022
Verma, M., Vats, A., and Taneja, V. (2015). Toxic species in amyloid disorders: oligomers or mature fibrils. Ann. Indian Acad. Neurol. 18, 138–145. doi: 10.4103/0972-2327.144284
Viggars, A. P., Wharton, S. B., Simpson, J. E., Matthews, F. E., Brayne, C., Savva, G. M., et al. (2011). Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci. Lett. 505, 25–30. doi: 10.1016/j.neulet.2011.09.049
Wang, Y. R., Wang, Q. H., Zhang, T., Liu, Y. H., Yao, X. Q., Zeng, F., et al. (2017). Associations between hepatic functions and plasma amyloid-β levels—implications for the capacity of liver in peripheral amyloid-β clearance. Mol. Neurobiol. 54, 2338–2344. doi: 10.1007/s12035-016-9826-1
Wang, Z., Yang, L., and Zheng, H. (2012). Role of APP and Aβ in synaptic physiology. Curr. Alzheimer Res. 9, 217–226. doi: 10.2174/156720512799361691
Wang, Y. J., Zhou, H. D., and Zhou, X. F. (2006). Clearance of amyloid-beta in Alzheimer’s disease: progress, problems and perspectives. Drug Discov. Today 11, 931–938. doi: 10.1016/j.drudis.2006.08.004
Xiang, Y., Bu, X. L., Liu, Y. H., Zhu, C., Shen, L. L., Jiao, S. S., et al. (2015). Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 130, 487–499. doi: 10.1007/s00401-015-1477-1
Xiao, H., Deng, M., Yang, B., Tang, J., and Hu, Z. (2017). Role of glycogen synthase kinase 3 in ischemia-induced blood-brain barrier disruption in aged female rats. J. Neurochem. 142, 194–203. doi: 10.1111/jnc.14051
Xiao, G., and Gan, L. S. (2013). Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int. J. Cell Biol. 2013:703545. doi: 10.1155/2013/703545
Zhao, Y., Li, D., Zhao, J., Song, J., and Zhao, Y. (2016). The role of the low-density lipoprotein receptor-related protein 1 (LRP-1) in regulating blood-brain barrier integrity. Rev. Neurosci. 27, 623–634. doi: 10.1515/revneuro-2015-0069
Ziegler-Waldkirch, S., and Meyer-Luehmann, M. (2018). The role of glial cells and synapse loss in mouse models of Alzheimer’s disease. Front. Cell. Neurosci. 12:473. doi: 10.3389/fncel.2018.00473
Zlokovic, B. V. (2005). Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 28, 202–208. doi: 10.1016/j.tins.2005.02.001
Keywords: amyloid beta, NAFLD, LRP-1, BBB, Alzheimer’s
Citation: Estrada LD, Ahumada P, Cabrera D and Arab JP (2019) Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 11:174. doi: 10.3389/fnagi.2019.00174
Received: 31 March 2019; Accepted: 25 June 2019;
Published: 17 July 2019.
Edited by:
Ines Moreno-Gonzalez, University of Texas Health Science Center at Houston, United StatesReviewed by:
Xudong Huang, Massachusetts General Hospital, Harvard Medical School, United StatesEnrique Armijo, KBI Biopharma Inc., United States
Copyright © 2019 Estrada, Ahumada, Cabrera and Arab. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisbell D. Estrada, bGlzYmVsbC5lc3RyYWRhQHViby5jbA==