 Domenico Sergi
Domenico Sergi Justine Renaud3†
Justine Renaud3† Nicola Simola
Nicola Simola Maria-Grazia Martinoli
Maria-Grazia Martinoli- 1Nutrition and Health Substantiation Group, Nutrition and Health Program, Health and Biosecurity, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Adelaide, SA, Australia
- 2Adelaide Medical School, University of Adelaide, Adelaide, SA, Australia
- 3Cellular Neurobiology, Department of Medical Biology, Université du Québec, Trois-Rivières, QC, Canada
- 4Department of Biomedical Sciences, University of Cagliari, Cagliari, Italy
- 5National Institute for Neuroscience (INN), University of Cagliari, Cagliari, Italy
- 6Department of Psychiatry and Neuroscience, Université Laval and CHU Research Center, Québec, QC, Canada
Diabetes mellitus (DM), a group of diseases characterized by defective glucose metabolism, is the most widespread metabolic disorder affecting over 400 million adults worldwide. This pathological condition has been implicated in the pathogenesis of a number of central encephalopathies and peripheral neuropathies. In further support of this notion, recent epidemiological evidence suggests a link between DM and Parkinson’s disease (PD), with hyperglycemia emerging as one of the culprits in neurodegeneration involving the nigrostriatal pathway, the neuroanatomical substrate of the motor symptoms affecting parkinsonian patients. Indeed, dopaminergic neurons located in the mesencephalic substantia nigra appear to be particularly vulnerable to oxidative stress and degeneration, likely because of their intrinsic susceptibility to mitochondrial dysfunction, which may represent a direct consequence of hyperglycemia and hyperglycemia-induced oxidative stress. Other pathological pathways induced by increased intracellular glucose levels, including the polyol and the hexosamine pathway as well as the formation of advanced glycation end-products, may all play a pivotal role in mediating the detrimental effects of hyperglycemia on nigral dopaminergic neurons. In this review article, we will examine the epidemiological as well as the molecular and cellular clues supporting the potential susceptibility of nigrostriatal dopaminergic neurons to hyperglycemia.
Introduction
Since the publication of the pioneering report of Dr. James Parkinson, “An Essay on the Shaking Palsy,” where he described six subjects suffering from a pathological state that he termed paralysis agitans, the field of Parkinson’s disease (PD) has phenomenally progressed. In this age of effervescent neuroscientific developments, clinical profiles are more accurately drawn, long-term patient management is improving, and the pursuit of etiological and pathophysiological explanations is starting to bear fruit. The preferential degeneration of the dopaminergic nigrostriatal pathway is well appreciated today, and additional pieces of the puzzle are continuously falling into place. More importantly, the apparent link between diabetes (DM) and PD is rising momentum, with hyperglycemia representing the metabolic culprit linking DM with PD. Even though a handful of medical conditions may cause hyperglycemia, we deliberately hold our focus on DM as it offers a tangible collection of experimental and clinical evidence interlaced with PD pathophysiology.
The present review will lay out the epidemiological evidence linking DM and PD as well as the molecular mechanisms underlying the exacerbated susceptibility of nigrostriatal dopaminergic neurons to hyperglycemia, by illustrating glucose transport and metabolism in the brain and the role of glucotoxicity in promoting neuronal damage and degeneration. The transport kinetics and metabolism of glucose are highly complex, especially in the central nervous system (CNS), and bear significant implications in the initiation of oxidative stress, induction of non-enzymatic glycation and activation of biochemical pathways mediating the effect of glucotoxicity in neurons.
Parkinson’s Disease and the Vulnerability of the Nigrostriatal Pathway
PD is the prototype of movement disorders. In fact, the most prominent feature of PD is the presence of motor symptoms expressed in a majority of patients as a classical triad of resting tremor, bradykinesia, and rigidity (Obeso et al., 2000). Non-dopaminergic (extranigral) motor features may also arise and result in falls, freezing of gait, speech impairment and difficulty in swallowing. Accompanying motor symptoms are equally disabling non-motor manifestations, such as psychiatric disturbances, dementia and autonomic failure (Schapira et al., 2017).
The neuropathological hallmark of PD is the progressive and relentless degeneration of dopaminergic neurons located in the substantia nigra pars compacta. Idiopathic PD still composes approximately 90–95% of diagnoses that are not always faithfully represented by genetic forms in terms of symptomatology and pathophysiology (Houlden and Singleton, 2012).
In the mid-1900s, the role of the substantia nigra pars compacta in PD was strengthened by findings enlisting dopamine (DA) as a key contributor to movement performance (Carlsson et al., 1958; Ehringer and Hornykiewicz, 1960) and demonstrating the projection of nigral neurons to the dorsal striatum (Hornykiewicz, 1966). Another significant leap forward in the field of PD was the identification of the first gene mutation, explicitly a single-point mutation in the SNCA gene coding for α-synuclein, found to cause a dominant familial form of early-onset PD (Polymeropoulos et al., 1997). This finding triggered a sequence of discoveries concerning genetic forms of the pathology. Disease-causing duplications (Chartier-Harlin et al., 2004) and triplications (Singleton et al., 2003) of the SNCA gene were identified, and other genes were added in subsequent years (Obeso et al., 2017).
The discovery of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) as a mitochondria-targeting agent of parkinsonism, broke new ground for the exploration of other chemicals, such as pesticides (Langston et al., 1983; Priyadarshi et al., 2000; Greenamyre et al., 2001; Furlong et al., 2015), solvents (Goldman et al., 2012; Pezzoli and Cereda, 2013), metals (Gorell et al., 1997; Finkelstein and Jerrett, 2007) and organochlorines (Steenland et al., 2006; Weisskopf et al., 2012), subsequently shown to constitute risk factors acting via bioenergetic disruption and oxidative mechanisms. Elements of infection were also suggested long ago to promote PD (Charcot, 1877; Gowers, 1886), substantiated by the occurrence of postencephalitic parkinsonism that succeeded the great pandemic of encephalitis lethargica in the early 1900s (Schwab et al., 1956; Vilensky et al., 2010). Later, studies found certain of these exogenous elements to represent a greater hazard in individuals carrying a specific genetic profile. Indeed, a host of genetic variants is known today to synergistically increase the odds of developing PD, in gene-environment interactions but also as standalone risk factors. For example, p.G2385R in the LRRK2 gene is common among Chinese and Japanese populations and approximately doubles the risk for PD (Bonifati, 2007), while the REP1 microsatellite marker of the SNCA promoter region was consistently associated with a 1.4-fold increased risk of PD (for a review, see Warner and Schapira, 2003; Polito et al., 2016).
Aside from these genetic and environmental factors specific to certain individuals, the substantia nigra pars compacta appears to be relatively susceptible to aging outside of a pathological context (Rudow et al., 2008; Buchman et al., 2012; Reeve et al., 2014), as spontaneous cell loss occurs at an estimated rate of 4.7–9.8% per decade in humans (Fearnley and Lees, 1991; Ma et al., 1999). Moreover, iron contents (Bilgic et al., 2012), levels of iron-binding neuromelanin (Double et al., 2003), neuroinflammation (Calabrese et al., 2018), oxidative stress-induced mitochondrial DNA deletions (Reeve et al., 2014), and malfunction of the ubiquitin-proteasome system and autophagy clearance pathways (Li and Li, 2011; Rubinsztein et al., 2011; Jana, 2012) were all shown to increase drastically with age, especially so in the substantia nigra pars compacta (Dexter et al., 1989; Corral-Debrinski et al., 1992; Soong et al., 1992; Fedorow et al., 2006; Daugherty and Raz, 2013). Coherently, in the setting of PD, nigrostriatal neurons that contain more neuromelanin are at higher risk of degenerating (Marsden, 1983; Hirsch et al., 1988; Gibb and Lees, 1991; Kastner et al., 1992; Zecca et al., 2001), microgliosis is relatively more pronounced in the substantia nigra pars compacta (McGeer et al., 1988; Lawson et al., 1990; Kim et al., 2000), nigral mitochondrial DNA deletions accumulate (Bender et al., 2006, 2008) and cellular clearance pathways are impaired (Cuervo et al., 2010; Dehay et al., 2010).
Despite their apparent disparity, multiple mechanisms converge to cause the degeneration of nigrostriatal neurons and possibly contribute to oxidative stress to which these neurons may be more vulnerable (Surmeier, 2007; Brichta and Greengard, 2014; Haddad and Nakamura, 2015; Oliveira et al., 2017; Surmeier et al., 2017). This concept dates back to the 1980s (Lenzi et al., 1979; Youdim et al., 1989; Götz et al., 1990; Jenner, 1991), but its popularity has waxed and waned at the rhythm of discoveries; nevertheless, the idea that metabolic oxidative insults may underlie the specific vulnerability of nigrostriatal neurons has lately gained momentum (Obeso et al., 2017).
It is critical that nigrostriatal neurons distinguish themselves from other dopaminergic neurons, especially their mesocorticolimbic analogs, as they all share the liability of employing dopamine as a neurotransmitter, recognized to generate reactive oxygen species (ROS) upon auto-oxidation (Segura-Aguilar et al., 2014) and to require ROS-producing monoamine oxidases for its degradation (Graham, 1984; Andersen et al., 1994). In this respect, we argue that nigrostriatal dopaminergic neurons display the following phenotypic idiosyncrasies that may underlie their vulnerability to oxidative stress instigated by genetic mutations, environmental toxins, aging and combinations of these factors:
1. They are embedded in a particular environment, the substantia nigra pars compacta, which stores considerable amounts of iron ions (Hirsch and Faucheux, 1998; Chinta and Andersen, 2008) accumulating with age (Daugherty and Raz, 2013) and known to participate in deleterious Fenton reactions with hydrogen peroxide to produce the very reactive hydroxyl radical (Youdim et al., 1989).
2. They express lower levels of vesicular monoamine transporter 2 (VMAT2) than most other catecholaminergic neurons, implying the protracted presence of dopamine in the cytosol leading to the inevitable formation of toxic oxidative by-products and, ultimately, potentially toxic neuromelanin (Liang et al., 2004).
3. Despite constituting a very small cluster of cells (German et al., 1988; Percheron et al., 1989), they are required to innervate a very large surface, the dorsal striatum, which entails tremendous branching of axons harboring numerous vesicular release sites ridden with potentially pathogenic α-synuclein (Parent and Parent, 2006; Matsuda et al., 2009);
4. To enable corticostriatal circuits, they constitute the greatest striatal input of uninterrupted tonic dopamine relying on autonomous pacemaking activity at the source of high baseline levels of calcium ions, which enhance oxidative phosphorylation, mitochondrial membrane hyperpolarization and superoxide anion production (Guzman et al., 2010; Pacelli et al., 2015).
5. To maintain their neurotransmission activities and drawn-out axonal harbors, nigral neurons keep an army of mitochondria to fulfill their exorbitant energetic needs, also implying elevated basal rates of oxidative phosphorylation and greater production of electron transport chain superoxide anion by-products (Pacelli et al., 2015).
6. Last, they are endowed with a limited calcium-buffering capacity (Foehring et al., 2009) and scanty endogenous antioxidative defenses (Bharath et al., 2002), consequent of their low expression of calbindin (Iacopino and Christakos, 1990; Yamada et al., 1990; Liang et al., 1996; Dopeso-Reyes et al., 2014) and glutathione (Kang et al., 1999), respectively.
These features unite to augment the basal oxidative burden to which nigrostriatal neurons are subjected. Upon further exposure to environmental or genetic insults, endogenous antioxidative defenses are eventually overwhelmed: this turning point typified by the failure of neurons to cope with the added oxidative load constitutes the origin of oxidative stress. Sustained in time and enhanced by aging processes, oxidative stress may eventually lead to the death of nigrostriatal neurons featuring PD. Following this premise, studies have employed primary neuron cultures (Pacelli et al., 2015), rat brain slices (Carbone et al., 2017), rodents (Vidyadhara et al., 2016) and primate (Dopeso-Reyes et al., 2014) models to demonstrate that nigrostriatal dopaminergic neurons are more vulnerable to neurotoxins that induce PD-like phenotypes in comparison to other neuronal populations, exemplarily dopaminergic neurons lodged in the neighboring ventral tegmental area.
Other sources of oxidative stress that are not specific to PD may yield similar results and shore up the idea of the selective vulnerability of nigrostriatal dopaminergic neurons. Indications of this are provided by epidemiological evidence showing a higher occurrence of PD in patients suffering from other pathologies with a significant component of oxidative stress, such as DM (Cereda et al., 2012; Sun et al., 2012; Santiago and Potashkin, 2013), ischemic stroke (Huang et al., 2013) and sleep apnea (Snyder and Cunningham, 2018). Interestingly, PD is significantly less frequent in patients with the hyperuricemic illness gout wherein the antioxidative effects of uric acid may, in fact, protect nigrostriatal neurons (Alonso and Sovell, 2010; Huang et al., 2017). Recently, our group has provided robust evidence for the selective demise of nigrostriatal neurons compared to mesocorticolimbic neurons over the course of 6 months of sustained hyperglycemia, attended by alterations in glial profiles that indirectly supported a role for oxidative stress as a key conspirator in neuronal death (Renaud et al., 2018), further evinced by in vitro groundwork (Bournival et al., 2012; Renaud et al., 2014). Notably, despite modest neurodegeneration, hyperglycemia evoked motor deficits strikingly reminiscent of parkinsonian motor symptoms.
Epidemiological Evidence: Parkinson’s Disease in Diabetic Patients
The first indications of a possible association between DM and PD date back to almost 60 years ago. Studies conducted in parallel showed that DM exacerbates the progression of motor and cognitive deficits in PD (Schwab, 1960), and that non-diabetic parkinsonian patients present impaired glucose tolerance (Barbeau et al., 1961) and hyperglycemia (Boyd et al., 1971). Since then, these findings have been reiterated in parkinsonian patients medicated with dopamine-replacing therapy (Lipman et al., 1974; Sandyk, 1993; Cereda et al., 2012), but also in drug-naïve parkinsonian patients who still displayed higher-than-normal levels of fasting blood glucose, within the prediabetic diagnostic range (Santiago and Potashkin, 2015). In addition, dietary habits featuring an elevated intake of high glycemic indexed carbohydrates have more recently been associated with greater odds of developing PD (Cheng et al., 2009; Murakami et al., 2010; Yang and Cheng, 2010; Okubo et al., 2012; Stafstrom and Rho, 2012).
In light of accumulating reports showing a possible association between DM and PD, numerous prospective cohort studies were elaborated to unveil their temporal and cause-effect relationship (Wirdefeldt et al., 2011; Santiago and Potashkin, 2013), with most of these studies reporting pre-existing DM as a risk factor for the development of PD (Arvanitakis et al., 2007; Hu et al., 2007; Becker et al., 2008; Driver et al., 2008; Cereda et al., 2011; Xu et al., 2011; Cereda et al., 2012). To dispel any doubt, one group addressed the question by quantifying comorbidity of DM in almost 40,000 single clinical measurements and robustly confirmed the relationship between DM and increased risk for the development of PD later in life (Klimek et al., 2015). Moreover, other studies provided evidence supporting a possible link between DM and the severity of PD-related symptoms (Schwab, 1960; Cereda et al., 2012). Likewise, in older individuals without PD who displayed parkinsonian-like signs, like gait disturbances, the severity of these signs was associated with existing DM (Arvanitakis et al., 2007).
In consideration of the close relationship between DM and PD pathogenesis, anti-diabetic drugs are emerging as strong candidates for the development of novel promising therapeutic approaches for PD. A sharp interest in these substances surfaced following reports evoking a decreased risk of developing PD in diabetic patients employing anti-hyperglycemic drugs. Specifically, the incidence of PD was found to be lower in diabetic patients treated with metformin or thiazolidinediones, which are widely used glucose-lowering medications (Wahlqvist et al., 2012; Brauer et al., 2015). With this regard, current clinical trials are investigating the anti-diabetic and glucose-lowering effect of exenatide, a glucagon-like peptide 1 (GLP-1) receptor agonist acting as an incretin mimetic and increasing insulin secretion from the pancreatic beta cells of the islets of Langerhans. Moreover, activation of the GLP-1 receptor induces the activation of the PI3K/AKT and RAS-extracellular signal-related kinase (ERK) pathways which, as described below, are known to mediate the biological effects of insulin (Athauda and Foltynie, 2018). Furthermore, the stimulation of the GLP-1 receptor is able to improve insulin resistance and restore the insulin-mediated functions that become impaired in PD thereby acting as a neuroprotective factor, inhibiting apoptosis, inflammation and oxidative stress (Athauda and Foltynie, 2016a,b). An initial open-label trial carried out in 44 parkinsonian patients receiving dopamine-replacing therapy showed that twice-daily injections of exenatide for 12 months improved overnight off-medication motor symptoms and dementia, rated according to the unified PD rating scale and the Mattis dementia rating scale, respectively (Aviles-Olmos et al., 2013). This trial employed a washout design wherein exenatide treatments were ceased at 12 months and patients were re-evaluated at later time points. Remarkably, the benefits observed at 12 months still held at 14 (Aviles-Olmos et al., 2013) and 24 months (Aviles-Olmos et al., 2014). This encouraged the elaboration of subsequent double-blinded, placebo-controlled trials once again designed to include a washout period and to measure symptoms in off-medication patients. The benefits of exenatide were reiterated and supplementary DaTscan neuroimaging results, which conveyed information on the amount of presynaptic dopamine transporter (DAT) left in the striatum of patients, confirmed a significant positive difference between treated and control parkinsonian patients (Athauda et al., 2017). The link between DM and PD is further strengthened by the putative therapeutic potential of metformin in PD. Indeed, besides its well-established role in the treatment of type II DM, metformin is emerging for its ability to counteract neurodegenerative diseases by modulating intracellular pathways impinging on neuronal longevity (Rotermund et al., 2018). Particularly, metformin has been shown to exert beneficial effects on neuronal mitochondria (Fitzgerald et al., 2017) whose dysfunction has been described as a pivotal defect in the pathogenesis of PD (Haddad and Nakamura, 2015). Furthermore, metformin is able to modulate neuronal energy metabolism by promoting a mild inhibition of mitochondrial complex 1 and leading to a decrease in oxidative stress and the activation of AMP-activated protein kinase (AMPK; Rotermund et al., 2018) which, beside acting as a central intracellular energy sensor, may exert neuroprotective effects (Ng et al., 2012; Bobela et al., 2017). Despite from a mechanistic point of view metformin may represent a valuable disease-modifying tool, its therapeutic potential in PD remains to be fully investigated. Indeed, animal studies aimed at investigating the effect of metformin on PD remain controversial, with some (Patil et al., 2014; Bayliss et al., 2016; Lu et al., 2016; Katila et al., 2017) but not all reporting a neuroprotective effect of metformin against MPTP-induced dopaminergic neuron damage (Ismaiel et al., 2016). Importantly both metformin and MPTP target the mitochondrial complex 1 and their potential interaction at this level may explain the discrepancies among different studies. In regard to human studies, clinical data on the effect of metformin of PD are lacking with the majority of the studies conducted so far comparing metformin against or using it in combination with other anti-hyperglycemic drugs, making it difficult to identify a clear-cut effect of metformin on PD progression (Rotermund et al., 2018). Furthermore, there is also evidence that metformin may increase the risk of developing PD (Kuan et al., 2017), but this remains to be fully elucidated and further studies are warranted to confirm this relationship.
Despite the recent medical advances in teasing out the cause-effect relationship linking DM and PD, it is still unclear which of the multifarious metabolic changes that occur in DM trigger the nigrostriatal degeneration underlying PD. The two predominant lines of thought grant importance to the dimensions of defective insulin signaling and oxidative stress.
Diabetes, a Current Epidemic
DM is the most widespread chronic metabolic disorder, estimated to affect over 400 million adults worldwide, with the prevalence of type II DM having doubled since 1980 on the likely account of the marked rise in overweight and obese phenotypes in the population (World Health Organization, 2016). Today, DM has joined the infamous list of leading causes of death, responsible for 8.4% of global mortality in adulthood (International Diabetes Federation, 2013). Both types I and II DM occur from the progressive failure of the body to manage circulating blood glucose. However, the etiopathogenesis of type I DM (5–10% of diabetic patients) is specifically rooted in the autoimmune destruction of insulin-producing pancreatic β cells and usually, but not always, develops before adulthood (Yoon and Jun, 2001). On the other hand, type II DM ensues from peripheral resistance to and deficient production of insulin, alongside other secondary metabolic elements. Type II DM is more frequently diagnosed in adults although currently, youth-onset forms are dramatically on the rise (Correia et al., 2008; Nadeau et al., 2016; Sergi et al., 2019). In light of its relative or absolute absence, insulin fails to promote glucose transporter 4 (GLUT4)-dependent glucose uptake in skeletal muscle and adipose tissue, to inhibit hepatic glucose production and to regulate peripheral glucose homeostasis by acting directly on the brain which, in turn, relies on insulin signaling to control peripheral glucose metabolism (Tups et al., 2017; Sergi et al., 2019). The defective insulin signaling culminates in chronically sustained hyperglycemia and impaired postprandial glucose disposal, synonymous of glucose intolerance (for a detailed review, see Zaccardi et al., 2016).
Granted the multiple aspects of health it impinges on, DM nurtures the development of a myriad of diverse comorbidities, all having hyperglycemia as a common denominator. Indeed, DM has been associated with a variety of microvascular and macrovascular complications including retinopathies, nephropathies, coronary heart disease (Eppens et al., 2006; Pambianco et al., 2006; Gregg et al., 2016). However, for the purpose of this review only data relative to the effect of diabetic hyperglycemia on the nervous system will be examined. Indeed, the nervous system is not exempt from the deleterious effect of DM with neuropathies remaining the most prevalent neuronal affections in this metabolic disease. Peripheral nerves are more susceptible to the detrimental effects of hyperglycemia as they are devoid of blood-brain barrier protection, which may limit exposure to high circulating glucose levels (Bansal et al., 2006; Rajabally et al., 2017). Nonetheless, the protective effect of the blood-brain barrier against hyperglycemia has been challenged (Jacob et al., 2002). In agreement with this paradigm, the complications of DM also affect the CNS (Biessels et al., 2002) with hyperglycemia standing as a risk factor for PD (Pagano et al., 2018).
Glucotoxicity: Physiological Considerations
The brain accounts for approximately 2% of total body weight, nevertheless it requires up to 20% of total oxygen and energy consumed by the body (Raichle and Gusnard, 2002). Although neurons can use different energetic substrates, including lactate, pyruvate, ketone bodies, glutamate, glutamine, and aspartate (van Hall et al., 2009; Amaral, 2013; Lutas and Yellen, 2013), glucose represents their preferred metabolic substrate (Simpson et al., 2007, 2008). Unlike other energy-greedy cells endowed with glycogen stores, like striated muscle cells, neurons cannot call upon glycogenolysis for sustenance, as glycogen is almost exclusively confined to astrocytes in the brain (Watanabe and Passonneau, 1973; Sagar et al., 1987; Dienel and Cruz, 2006, 2015). Intraneuronal glucose is thus mainly provided from the extracellular environment, which itself is supplied by the circulation. This implies the need for glucose to cross several barriers whose transport kinetics assuredly influence its metabolism in neurons.
Glucose transporters (GLUTs) ubiquitously present at the surface of cells accomplish the function of taking up glucose from the extracellular environment. In peripheral tissues, such as the skeletal muscle and adipose tissue, glucose uptake is mediated by GLUT4, an insulin-dependent glucose transporter. On the contrary, in the vast majority of the CNS glucose transport is mediated by insulin-independent GLUTs (James et al., 1988; Huang and Czech, 2007). GLUT1, one of the insulin-independent GLUT, is particularly abundant in the brain microvasculature, distributed with a 1:4 ratio between the luminal and abluminal surfaces of the endothelium, and plays a pivotal role in mediating glucose transport across the blood-brain barrier (Maher et al., 1994; Simpson et al., 2001, 2008). GLUT1 is also the main transporter employed by glial cells, especially astrocytes (Dick et al., 1984; Gerhart et al., 1989; Maher et al., 1994; Simpson et al., 2001). Neurons, on the contrary, overwhelmingly employ the insulin-independent GLUT3 isoform densely located on the cell surface but more intensely so at the neuropil (Simpson et al., 2008). Thus, neuronal glucose uptake can be defined as an insulin-independent two-step process. Circulating glucose must first penetrate the endothelium and astrocytic endfeet of the blood-brain barrier via GLUT1 and subsequently be taken up by neurons in a GLUT3-dependent fashion.
Considering that GLUTs are facilitative, bidirectional, energy-independent transporters, they do not allow for glucose accumulation but for its equilibration between compartments in a manner that smoothens concentration gradients. Thus, the characteristic kinetics of brain GLUTs, rather than insulin, is the driver of glucose uptake into brain cells. Brain GLUTs possess variable degrees of affinity for glucose, with the highest being reported for neuronal GLUT3 (Km ~1.5 mM) which allows this transporter to work efficiently even at glucose concentrations surrounding the neurons in the order of 1–2 mM (Colville et al., 1993; Maher et al., 1996). In fact, GLUT3 also presents the greatest turnover for glucose, defined as the number of transport cycles per transporter per second (Rumsey et al., 1997; Manolescu et al., 2007) and considering its abundance in neurons provides them with preferential access to glucose (Simpson et al., 2008). Thus, in light of the kinetics of this transporter, glycemia must drop significantly before neuronal glucopenia manifests (Rao et al., 2006). However, under these circumstances, the brain can call upon different metabolic pathways and harvest energy from substrates other than glucose, as already discussed (van Hall et al., 2009; Amaral, 2013; Lutas and Yellen, 2013).
According to computational studies performed by Simpson et al. (2007), neurons and astrocytes only contain ~0.9 mM of glucose in normoglycemic steady-state conditions. Similarly, brain extracellular concentrations of glucose merely reach ~1.4 mM. Even endothelial cells that express GLUT1 and are in direct contact with blood-borne solutes display concentrations of ~3.8 mM. This discrepancy between expected compartment equilibration and markedly lower levels of glucose in brain parenchymal and endothelial cells may be explained by transport inhibition at the level of the blood-brain barrier. Indeed, endothelial GLUT1 undergoes allosteric inactivation by adenosine triphosphate (ATP) indicating that its activity is a direct function of energy availability (Lowe and Walmsley, 1986; Carruthers and Helgerson, 1989; Levine et al., 1998). Consequently, when glucose levels suffice brain energy demand its uptake by endothelial cells is inhibited and glucose supply to brain parenchyma decreases. Conversely, when glycolytic demands increase and ATP levels become depleted, GLUT1 activity is uninhibited and glucose may efficiently be transferred to surrounding tissues (Carruthers, 1986; Cloherty et al., 1996).
In opposition to the hypoglycemia-induced compensatory upregulation of brain GLUTs and metabolic switch to alternative substrates to meet brain energy requirements, hyperglycemia, as demonstrated in rodent models, does not trigger protective mechanisms aimed at limiting excess glucose entering the brain (Nagamatsu et al., 1994; Simpson et al., 1999; Santiago et al., 2006; Anitha et al., 2012). In support of this, in rodent models of sustained DM-induced hyperglycemia, intracerebral glucose concentrations are also augmented (Jacob et al., 2002; de Vries et al., 2003; Gomez and Barros, 2003; McCrimmon et al., 2003), which argues against the protective role of the blood-brain barrier against hyperglycemia and reinforces the susceptibility of the brain to chronically elevated circulating glucose levels. However, these experiments were performed on whole brain tissues and reliable intraneuronal measurements are still lacking. Nevertheless, computational renderings taking into account GLUT kinetics and their distribution predict greater glucose permeation in all brain regions under hyperglycemia (Simpson et al., 2007). In further support of this notion, our group has recently demonstrated that intracellular as well as extracellular glucose concentrations are increased in midbrain and striatum during long-term hyperglycemia in rats (Renaud et al., 2018). Thus, we can faithfully support the idea that hyperglycemia causes an upwelling of intraneuronal glucose likely to hold pathological implications for cellular fitness. Moreover, in humans, brain glucose uptake was reported to be inversely correlated with insulin sensitivity and positively associated with body mass index (BMI) as well as signs of inflammation in the hypothalamus (Rebelos et al., 2018). Thus, hypothalamic inflammation, may also occur in humans and induce insulin resistance leading to dysregulation of glucose homeostasis (Tups et al., 2017). This provides further evidence for the physiological relevance of the relationship between impaired glucose metabolism, insulin resistance, DM and enhanced brain glucose uptake.
Insulin and Neuronal Homeostasis
Although insulin occupies an increasingly acknowledged role in the human CNS, by activating insulin-responsive neurons responsible for controlling appetite, peripheral glucose homeostasis, reward, cognition and memory (Craft et al., 1996; Benedict et al., 2004, 2007; Anthony et al., 2006; Hallschmid et al., 2008; Reger et al., 2008; Khanh et al., 2014; Tups et al., 2017), this peptide hormone is not required for glucose transport in the brain parenchyma or in most brain resident cells (Tups et al., 2017). Nevertheless, besides the aforementioned biological effects in the CNS, insulin is also known to regulate physiological functions independent from and beyond the regulation of glucose homeostasis, including neuronal development, differentiation, plasticity and survival (Bassil et al., 2014). In physiological conditions, insulin concentrations are highest in the pons, medulla and hypothalamus, and lowest in the occipital cortex and thalamus (Banks and Kastin, 1998), with the midbrain, affected in PD, falling within the average. In agreement with the widespread effect of insulin in the brain, its receptor is also widely expressed throughout the CNS (Havrankova et al., 1978a; Hill et al., 1986; Gralle, 2017), and possesses similar kinetic properties to its peripheral counterpart (LeRoith et al., 1988; Zahniser et al., 1984). Insulin signal transduction pathway in neurons, similarly to peripheral cells, relies on the activation of PI3K/AKT and RAS-ERK pathways which are responsible for the stimulation of synaptic plasticity, inhibition of neuronal apoptosis and gene transcription (Athauda and Foltynie, 2016a). In light of its ability to activate pathways that promote survival, neurogenesis, and inhibition of apoptosis, insulin is generally accepted to act as a neurotrophic factor. In support of this, insulin has been shown to counteract the neurotoxic effect exerted by excitotoxicity or oxidative stress in vitro, confirming its neuroprotective role (Kim and Han, 2005; Duarte et al., 2006; Sun et al., 2010; Ribeiro et al., 2014). In vivo, near-direct application of insulin on the CNS, via intranasal or intracerebroventricular treatments, are highly effective in promoting various aspects of neuronal health, as demonstrated in a rat model of PD (Haas et al., 2016; Pang et al., 2016).
Studies attempting to demonstrate that neuronal death directly ensues from hypoinsulinemia employed peripheral insulin administration to reverse cellular or structural damage induced by streptozotocin treatments in the CNS of rodents (Ramanathan et al., 1999; Moreira et al., 2005, 2006; Hung et al., 2014). However, they do not take into account the various peripheral benefits exerted by insulin replacement therapy, and specifically improved glycemic control. Such strategies exploiting organism-wide paradigms to establish causality between hypoinsulinemia and CNS neuronal death are based on the misconception that intracerebral insulin is solely provided from the circulation, itself supplied by the pancreas. As a matter of fact, insulin concentrations are considerably greater in the brain parenchyma than in the plasma, and are unexpectedly upregulated in hypoinsulinemic, hyperglycemic streptozotocin-treated rats (Havrankova et al., 1978a, b, 1979; Gupta et al., 1992; Banks et al., 1997b). In support of this, the insulin receptor is downregulated in peripheral tissues but remains unaffected in the brain of streptozotocin models (Pacold and Blackard, 1979; Pezzino et al., 1996). These preliminary findings led to the discovery that, besides being taken up through the blood-brain barrier (Margolis and Altszuler, 1967; Woods and Porte, 1977; Banks et al., 1997a), insulin is synthesized by neurons within the mammalian brain (Dorn et al., 1983; Birch et al., 1984; Young, 1986; Deltour et al., 1993; Devaskar et al., 1994; Schechter et al., 1994; Frölich et al., 1998; Mehran et al., 2012; Molnár et al., 2014). Importantly, one group demonstrated that hyperglycemia enhanced insulin production by cortical neurons (Molnár et al., 2014), which may be a mechanism to maintain appropriate intracerebral concentrations of insulin when pancreatic supplies plunge (Havrankova et al., 1979; Gupta et al., 1992; Banks et al., 1997b). Thus, hypoinsulinemia does not seem to be the metabolic culprit linking type I DM and PD, while hyperglycemia may represent a more plausible mediator bridging the gap between DM and PD. Nonetheless, we cannot overlook the fact that insulin resistance is the main feature of type II DM and that insulin resistance also occurs in the brain (Athauda and Foltynie, 2016a) with defective insulin signaling being identified as an important player in the pathogenesis of neurodegenerative disorders and particularly Alzheimer’s disease (AD; Steen et al., 2005; Talbot et al., 2012; Freiherr et al., 2013; Arnold et al., 2018). Besides its well-established role in AD, emerging evidence suggests insulin resistance also occurring in the brain of PD patients pointing to impaired brain insulin signaling as a potential contributing factor to the pathological defects of PD (Athauda and Foltynie, 2016a). In support of this, a downregulation of the insulin receptor in the substantia nigra pars compacta and increased insulin resistance has been described in patients affected by PD (Moroo et al., 1994; Takahashi et al., 1996; Duarte et al., 2012), a phenomenon which may occur before the death of dopaminergic neurons (Moroo et al., 1994). Particularly, AKT/protein kinase β appears to be a critical node in mediating the physiological effect of insulin, and impaired AKT signaling, as described in insulin-resistant individuals, may be involved in the pathogenesis of PD, further supporting its association with insulin resistance (Greene et al., 2011). In agreement with this, AKT phosphorylation has been reported to be decreased in the brain of PD patients following post mortem analysis (Malagelada et al., 2008). Moreover, inhibition of AKT signaling promotes dopaminergic cell death (Xu et al., 2014) and loss of dopaminergic phenotype in axons (Kim et al., 2011), providing a further mechanistic link between impaired insulin signaling and PD (Canal et al., 2014). Thus, the compromised neurotrophic action of insulin, due to defective insulin signaling, appears to be at the interphase between insulin resistance and PD pathogenesis. Nonetheless, other putative mechanisms may link insulin resistance and PD, including α-synuclein aggregation and toxicity, defects in mitochondrial function and neuroinflammation (Athauda and Foltynie, 2016a). Indeed, the activation of insulin signaling can modulate the degradation of alpha-synuclein and inhibit alpha-synuclein fibril formation by activating the insulin-degrading enzyme (Sharma et al., 2015) which is supported by the fact that reversing insulin resistance prevented alpha-synuclein-induced toxicity (Kao, 2009). In regard to the role of insulin in modulating mitochondrial function, activation of insulin signal transduction pathway and particularly PI3K/AKT plays a key role in maintain NAD+/NADH ratio which, in turn, regulates mitochondrial biogenesis and function by modulating the sirtuin 1/proliferator-activated receptor-gamma coactivator 1-α (SIRT/PGC-1α) axis (Cheng et al., 2010). Not surprisingly, animal models of insulin resistance display a decrease in mitochondrial proteins in the substantia nigra (Khang et al., 2015). Despite it is tempting to blame insulin resistance as the driver on neuronal mitochondrial disfunction, this remains elusive also in consideration of the fact that mitochondrial dysfunction itself may cause, and therefore precede, the onset of insulin resistance. Finally, insulin signaling may directly interfere with inflammation by upregulating IκBα and therefore inhibiting kappa-light-chain-enhancer of activated B cells (NF-κB) which is pivotal in regulating the expression of pro-inflammatory genes and has been implicated in PD neuro-inflammation (Kirkley et al., 2019).
Cellular Evidence: Neuronal Oxidative Stress in Hyperglycemia
The clinical significance of hyperglycemia emerged upon its formal identification as the prime culprit in comorbid diabetic complications (Nathan et al., 1993; UK Prospective Diabetes Study (UKPDS) Group, 1998; Gaede et al., 2008; Giacco and Brownlee, 2010). The most affected collateral targets in DM are the kidneys, the cardiovascular system and the nervous system (Nathan, 1993). Despite their many functional and phenotypic differences, the cells of these systems share the distinct liability of taking up glucose in an extracellular concentration-dependent but insulin-independent manner (Kaiser et al., 1993; Heilig et al., 1995). Indeed, mesangial and endothelial cells predominantly express the insulin-independent glucose transporter GLUT1 (Sone et al., 2000; Brosius and Heilig, 2005; Mueckler and Thorens, 2013); likewise, brain cells express the insulin-independent GLUTs, GLUT1 and GLUT3 with the latter being particularly abundant in neurons (Mantych et al., 1992; Nagamatsu et al., 1993; Simpson et al., 2008). Although it is clear that glucose concentrations rise in these cells under conditions of hyperglycemia, how exactly glucotoxicity-mediated cellular damage occurs is not as forthright. The current consensus is that oxidative stress may represent the principal mediator of hyperglycemia-induced diabetic complications (Giugliano et al., 1996; Ceriello, 2003; Brownlee, 2005; Araki and Nishikawa, 2010).
Owing to their steep demands in ATP, neurons possess a high basal oxidative metabolism compared to other types of cells in the brain or elsewhere and are thus plausibly more vulnerable to additional sources of stress. Although the literature does not provide an adequate account of the mechanisms underlying hyperglycemia-induced oxidative stress in the CNS, and most importantly in nigrostriatal dopaminergic neurons, we can speculate on the events that contribute to this critical coping threshold based on experimental data and our knowledge of neuronal glucose metabolism.
Mitochondrial Mechanisms
Following an upsurge in intracerebral and intraneuronal glucose, a plausible event that may occur is an amplified flux of electron donors toward oxidative phosphorylation. In a first scenario, high intraneuronal glucose concentrations may provide the glycolytic pathway and the tricarboxylic acid cycle (TCA) with more fuel, culminating in increased production of electron donors which are funneled toward the electron transport chain to fuel oxidative phosphorylation. In a second scenario, taking into account the possibility of an astrocyte-neuron lactate shuttle in hyperglycemia, lactate derived from glycolysis in astrocytes may enter the neurons and be converted to pyruvate to power the TCA. The end result is electron transport chain overload and subsequent increase in the proton gradient across the mitochondrial inner membrane which, in turn, impairs the transport of electrons across the mitochondrial complexes favoring the leakage of electrons and the formation of ROS (Figure 1).
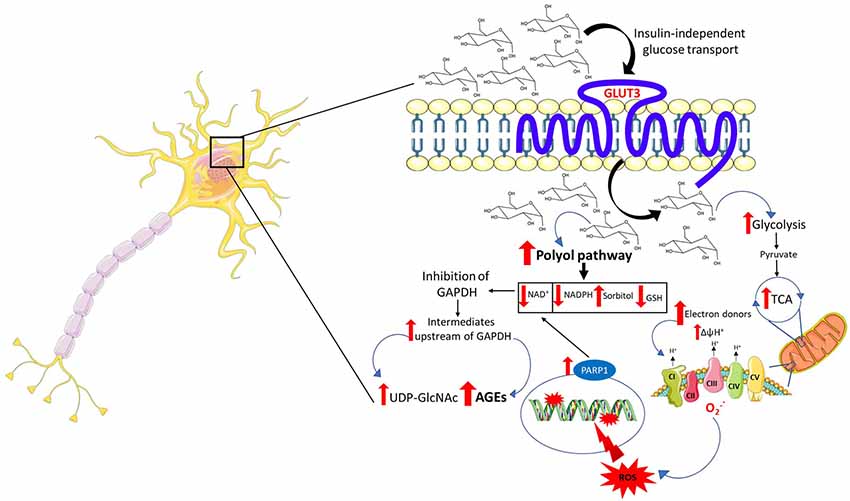
Figure 1. Mechanisms of neuronal glucotoxicity. In the early stages of hyperglycemia, there is an upregulation of glucose flux through glycolysis and the tricarboxylic acid cycle (TCA). This results in enhanced electron donor supply to the mitochondrial electron transport chain, leading to an increase in the proton gradient across the mitochondrial inner membrane which promotes the formation of reactive oxygen species (ROS). ROS-induced DNA damage triggers the activation of the NAD+-dependent DNA repair enzyme, poly (adenosine diphosphate-ribose) polymerase 1 (PARP1), responsible for depleting neuronal NAD+. The increase in intracellular glucose activates the polyol pathway which, in concert with PARP1, reduces intracellular NAD+ inhibiting glyceraldehyde triphosphate dehydrogenase (GAPDH) and therefore glycolysis. The inhibition of GAPDH provokes the accumulation of upstream triose phosphates then converted to glycating agents, thereby promoting the formation of advanced glycation end products (AGEs). The accumulation of fructose, another metabolite upstream GAPDH, triggers the activation of the hexosamine pathway and the subsequent build-up of UDP-N-acetylglucosamine (UDP-GlcNAc). Furthermore, the activation of the polyol pathway depletes NADPH and, alongside mitochondrial ROS, contributes to oxidative stress in light of its role as a cofactor for glutathione reductase, which, in turn, is crucial in maintaining the intracellular pool of reduced glutathione.
ROS induce cellular damage by targeting macromolecules, namely protein, lipids, and DNA. Particularly, oxidative stress-induced DNA damage results in strand breaks leading to the activation of the NAD+-dependent DNA repair enzyme, poly (adenosine diphosphate-ribose) polymerase (PARP1), which, in turn, by depleting intracellular NAD+, in concert with the polyol pathway, inhibits glyceraldehyde triphosphate dehydrogenase (GAPDH; Du et al., 2003; Figure 1). The inhibition of GAPDH represents a critical node in promoting glucotoxicity. Indeed, its inhibition disrupts intracellular glucose metabolism, promotes the buildup of metabolites acting as precursors of the advanced glycation end products (AGEs) and is responsible for the activation of the hexosamine pathway. Generation of ROS contributes to a self-perpetuating cycle of DNA damage and consequent PARP1 activation, which in concert with the polyol pathway inhibits GAPDH, leading to a permanent glycolytic impasse and sustained oxidative stress (Brownlee, 2005).
It is important to note that the key results in support of Brownlee’s theory were obtained in bovine aortic endothelial cells treated with 30 mM of glucose for 7 days (Nishikawa et al., 2000; Du et al., 2001). Consequently, these conditions may not reflect the pathophysiological mechanisms taking place in the brain of animals exposed to high levels of glucose. Indeed, a small body of literature investigating these mechanisms in the CNS and the periphery of rodents does not clearly validate this model in neurons (Thurston et al., 1995; Kaur and Bhardwaj, 1998; Thakran and Baquer, 2003; Price et al., 2006; Chowdhury et al., 2010; Akude et al., 2011; Hinder et al., 2013; Zheng et al., 2017). Despite, mitochondria appearing to be involved in perpetuating the effect of hyperglycemia by producing ROS, the impairment of mitochondrial oxidative metabolism may also play a pivotal role in the pathogenesis of PD as demonstrated by the downregulation of genes responsive to peroxisome PGC-1α (Zheng et al., 2010) which, in turn, is the master regulator of mitochondrial biogenesis and function (Sergi et al., 2019). In further support of this, most studies revealed a reduced activity or downregulation of electron transport chain proteins in the brain and in peripheral sensory neurons (Kaur and Bhardwaj, 1998; Chowdhury et al., 2010; Akude et al., 2011; Stančić et al., 2013; Aghanoori et al., 2017). The relationship between hyperglycemia and neuronal mitochondrial dysfunction is rather complex. In the early stages of hyperglycemia, an upregulation of glycolysis and tricarboxylic acid cycling occurs, most likely due to an increase in glucose supply (Thurston et al., 1995; Kaur and Bhardwaj, 1998; Thakran and Baquer, 2003; Price et al., 2006). Nevertheless, these catabolic pathways became impeded as hyperglycemia progresses (Chowdhury et al., 2010; Akude et al., 2011; Hinder et al., 2013; Aghanoori et al., 2017; Zheng et al., 2017). Remarkably, oxidative phosphorylation is impaired at all stages of hyperglycemia and is independent of the up- or down-regulation of glycolysis and Krebs cycle, indicating that mitochondrial dysfunction precedes glycolysis and TCA hindrance (Kaur and Bhardwaj, 1998; Chowdhury et al., 2010; Akude et al., 2011; Stančić et al., 2013; Aghanoori et al., 2017). This supports the theory that hyperglycemia exerts a metabolic insult to mitochondria leading to an impairment in oxidative phosphorylation (Nishikawa et al., 2000; Du et al., 2001; Brownlee, 2005; Tomlinson and Gardiner, 2008; Giacco and Brownlee, 2010). Thus, despite the initial increase in glycolysis and tricarboxylic acid cycling, this is not supported by a parallel increase in mitochondrial oxidative phosphorylation resulting in oxidative stress and lower energy efficiency. Apart from inducing oxidative stress and ROS, themselves contributing to mitochondrial DNA damage and mitochondrial dysfunction, defective mitochondrial oxidative metabolism also compromises ATP synthesis capacity. Neurons heavily rely on oxidative metabolism to efficiently obtain energy for their physiological functions with mitochondria playing a central role in this process. In light of this, mitochondrial dysfunction compromises neuronal energy availability. Particularly, nigrostriatal neurons have a high energy demand which may explain, albeit in part, their increased susceptibility to hyperglycemia (Renaud et al., 2018). Although hyperglycemia represents the more plausible primary driver of mitochondrial dysfunction, the potential involvement of insulin resistance should not be overlooked in consideration of the role of insulin in inducing mitochondrial biogenesis by promoting the SIRT1/PGC1α pathway, as discussed above (Cheng et al., 2010).
Rerouting Mechanisms: Polyol Pathway and Macromolecules Glycation
Aside from ROS generated through mitochondrial failure, hyperglycemia may trigger the onset of oxidative stress via pathways that dwell upstream the glycolytic enzyme GAPDH (Figure 1).
As previously discussed, free glucose is present in the cytosol of neurons and its concentrations rise during hyperglycemia leading to glucotoxicity. Under normal circumstances, hexokinase is mandated to transform available glucose into glucose 6-phosphate. However, due to substrate-mediated inhibition, in hyperglycemic conditions, it can only funnel a fraction of cytosolic glucose toward the glycolytic pathway before becoming saturated. Excess glucose is therefore consumed by other pathways and is constantly replenished by high extracellular levels. The principal compensatory outlets are the polyol pathway and macromolecule glycation which are two key pathways through which glucotoxicity manifests (Tups et al., 2017).
The polyol pathway is driven by aldose reductase whose affinity for glucose is lower compared to hexokinase (Gabbay et al., 1966). Under physiological glucose concentrations, aldose reductase channels only a minimal amount of glucose toward the polyol pathway, with the aldose reductase-dependent conversion of glucose into sorbitol only significantly taking place when intraneuronal glucose concentrations are elevated. Increased processing of glucose through the polyol pathway drains pools of cofactors required for the activity of a plethora of enzymes, including glutathione reductase, GAPDH and others in the glycolytic pathway and TCA (Vander Jagt et al., 1995). The depletion of NADPH contributes to oxidative stress in light of its role as a cofactor for glutathione reductase which is crucial in maintaining the intracellular pool of reduced glutathione, an aspect of paramount importance for neurons considering their susceptibility to oxidative stress (Barnett et al., 1986). More importantly, the depletion of NAD+ operated by sorbitol dehydrogenase represents a key pathological mechanism exploited by the polyol pathway. The reduction in the intracellular availability of NAD+ leads to an inhibition of GAPDH with the consequent accumulation of the metabolic intermediates upstream of this enzyme, including triose phosphates. The buildup of triose phosphates leads to the synthesis of methylglyoxal a precursor of the AGEs (Figure 1), described below (Tups et al., 2017). The buildup of fructose, both upstream GAPDH as well as a product of the polyol pathway, may represent a further mediator of the neuronal damage induced by hyperglycemia. In keeping with this, fructose 6-phosphate represents the precursor of UDP-N-acetylglucosamine, the end-product of the hexosamine pathway (Figure 1; Brownlee, 2005). Excess UDP-N-acetylglucosamine induces pathological posttranslational modification by promoting aberrant protein glycosylation which compromises proteins’ function and, as a consequence, cellular homeostasis (Brownlee, 2005; Tomlinson and Gardiner, 2008). Nonetheless, despite the increase in intraneuronal glucose levels providing a mechanistic ground for the activation of these pathways, their role in mediating the effect of hyperglycemia in the pathogenesis of PD remains to be fully elucidated.
The second outlet of excess glucose is via macromolecule glycation, which may indirectly contribute to oxidative stress (Vicente Miranda et al., 2016). Glycation consists of the slow non-enzymatic, energy-independent, covalent addition of a monosaccharide to macromolecules, usually lipids or proteins; glycated macromolecules are termed AGEs. Although glucose is a poor glycating agent, it can react with free amino groups leading to the formation of a Schiff base which can then undergo an irreversible rearrangement to generate an Amadori product which accumulates in the tissues initiating the process of advanced glycation (Ulrich and Cerami, 2001). Intracellular AGEs are harmful to cells because they induce the loss or alteration of cellular normal protein or lipid functions, which occur despite the cells are empowered with mechanisms aimed at ensuring the prophylactic clearance of glycating agents (Ahmed et al., 2003). Nonetheless, in the setting of hyperglycemia, excess glycating agents may overwhelm these pathways leading to the formation of AGEs, evidenced by the proportional rise in CNS glycation as a function of glycemia, which can reach an exorbitant 34-fold increase (Uchiki et al., 2012). Most importantly, however, glyoxalase employs glutathione to deactivate the AGEs precursor methylglyoxal, while we know that aldose reductase depletes pools of NADPH (Thornalley, 1988; Schieber and Chandel, 2014). Therefore, methylglyoxal and other glycating agents derived from glucose or its metabolites indirectly worsen the oxidative status of neurons by monopolizing endogenous antioxidative defenses or the cofactors necessary for their restoration. Furthermore, AGEs may exacerbate oxidative stress and oxidative stress itself fosters the detrimental effects of AGEs by upregulating the receptor for AGEs (RAGE) via NF-κB activation thereby creating a vicious cycle (Nowotny et al., 2015).
AGEs may also occur extracellularly or being directly secreted from the cells in which they are generated to induce oxidative stress and inflammation in a variety of cells (Gao et al., 2017). AGEs trigger oxidative stress by binding their cognate membrane receptor, the RAGE (Stern et al., 2002) and activating downstream mediators know to induce oxidative stress, such as the inducible nitric oxide synthase (iNOS; Nowotny et al., 2015). Nevertheless, despite AGEs being described for their ability to promote oxidative stress in peripheral tissues (Nowotny et al., 2015), their role in oxidative stress in PD remains to be elucidated.
As described for the induction of oxidative stress, the activation of the RAGE by AGEs triggers deleterious inflammatory pathways (Singh et al., 2001; Jakus and Rietbrock, 2004). Binding of RAGEs by AGEs or other endogenous ligands induced by hyperglycemia is a known mechanism of systemic inflammation in DM and can trigger intracellular signaling cascades that lead to the activation of NF-κB (Bierhaus et al., 2005; Rong et al., 2005; Haslbeck et al., 2007). Although in the brain parenchyma neuroinflammation is primarily mediated by glial cells, it can also be triggered by a direct insult of nutrient-overload to neurons (Gao et al., 2017; Sergi et al., 2018a,b), which can exert additional extraneuronal oxidative injuries.
Gathering all the evidence together, hyperglycemia wields its detrimental effects on neurons by an assortment of conceivable mechanisms leading to oxidative stress:
1. The initial rise in glucose metabolism enhances electron donor supply to the mitochondrial electron transport chain, which intensifies the rate of superoxide anion formation. Superoxide anion damages mitochondrial DNA, which may lead to global mitochondrial failure in the long term. Furthermore, superoxide anion exacerbates ROS production that causes nuclear DNA strand breaks, thereby activating PARP1.
2. Depletion of NAD+ by PARP1 and the polyol pathway inhibit glycolysis, tricarboxylic acid cycling and oxidative enzymatic reactions operating via enzymes that require this cofactor. An intracellular decrease in NAD+ induces the inhibition of the glycolytic enzyme GAPDH, which provokes upstream accumulation of metabolites which are then diverted toward the synthesis of toxic intermediates.
3. Accrued levels of triose phosphates upstream of GAPDH are non-enzymatically converted into the glycating agent methylglyoxal whose decomposition by glyoxalase and aldose reductase expends antioxidative cofactors, such as glutathione and NADPH.
4. Excess glucose can be diverted into the deleterious polyol pathway, powered once again by aldose reductase that further depletes stocks of NADPH required for the renewal of the antioxidative cofactor glutathione and by sorbitol dehydrogenase which reduces intracellular NAD+.
What emerges here is a number of different pathways that collectively cause a shortage of antioxidant defenses layered with mitochondrial dysfunction, two pivotal factors likely responsible for pushing vulnerable cells beyond the critical coping threshold. The sum of these undesirable processes appears to be particularly relevant in neurons, in light of their relatively low glutathione levels and their reliance on the pentose phosphate pathway (PPP) for its rapid regeneration (Bolaños et al., 1996, 2008; Kang et al., 1999). In fact, insufficient provision of the PPP leads to poor regeneration of glutathione and neuronal apoptosis (Herrero-Mendez et al., 2009). Neurons may reach the coping threshold quicker than other brain cells, for instance, astrocytes that display comparatively greater PPP activity (García-Nogales et al., 2003; Herrero-Mendez et al., 2009).
Most importantly, the proposed molecular events leading to neuronal oxidative stress in hyperglycemia, explicitly via increased superoxide anion generation (Ziegler et al., 2015), mitochondrial dysfunction (Patti and Corvera, 2010), PARP1 activation (Obrosova et al., 2005), impairment of the PPP (Ziegler et al., 2017), glycation (Aubert et al., 2014) and glutathione system deficits (Kasznicki et al., 2012; Mendez et al., 2015), are all cogently established in diabetic patients with neuronal pathology. Remarkably, clinical trials employing pharmacological agents to target some of these pathways, including aldose reductase inhibitors to diminish polyol pathway influx (Greene et al., 1999; Obrosova et al., 2002; Kawai et al., 2010; Sekiguchi et al., 2019) or exogenous superoxide dismutase (SOD) to quench superoxide anion (Bertolotto and Massone, 2012), have been demonstrated to improve diabetic peripheral nerve damage and oxidative status, further solidifying the importance of oxidative stress in neuronal affections arising from hyperglycemia in humans. Unquestionably, though, glycemic control remains the most accessible means to preclude oxidative stress-induced diabetic complications in the brain and in peripheral nerves.
Conclusion
Today, little doubt remains as to the hazard that a diabetic prelude represents in the development of PD. Even if the etiopathogenesis of PD remains largely enigmatic, in part due to our lack of comprehension regarding the seemingly selective vulnerability of the nigrostriatal dopaminergic pathway to undergo degeneration, ascending hypotheses grant a preeminent role to the existence of a particular phenotypic liability that characterizes neurons of the substantia nigra pars compacta and that may render them more sensitive to oxidative stress.
Hyperglycemia represents an additional cause of superoxide anion production, mitochondrial dysfunction, antioxidative bankruptcy, ubiquitin-proteasome system failure and dopamine toxicity that may well favor the early demise of nigrostriatal dopaminergic neurons. Thus, considering the deleterious effects of neuronal glucotoxicity, improving glycemic control in diabetic patients should represent a valuable strategy to decrease the risk of developing PD.
Author Contributions
DS wrote and edited the manuscript and drew the figure. JR wrote the manuscript. NS revised the manuscript. M-GM designed the manuscript, drew the figure and revised the manuscript.
Funding
This research was funded by grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada no. 04321 to M-GM and by funds from the Autonomous Region of Sardinia (L.R. n 7/2007-2015) to NS. DS is the recipient of a Commonwealth Scientific and Industrial Research Organisation (CSIRO; Australia) Postdoctoral Research Fellowship. JR is the recipient of a Vanier Graduate Scholarship from the NSERC and a Doctoral Training Scholarship from the Fonds de Recherche en Santé du Québec.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aghanoori, M. R., Smith, D. R., Roy Chowdhury, S., Sabbir, M. G., Calcutt, N. A., and Fernyhough, P. (2017). Insulin prevents aberrant mitochondrial phenotype in sensory neurons of type 1 diabetic rats. Exp. Neurol. 297, 148–157. doi: 10.1016/j.expneurol.2017.08.005
Ahmed, N., Thornalley, P. J., Dawczynski, J., Franke, S., Strobel, J., Stein, G., et al. (2003). Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Invest. Ophthalmol. Vis. Sci. 44, 5287–5292. doi: 10.1167/iovs.03-0573
Akude, E., Zherebitskaya, E., Chowdhury, S. K., Smith, D. R., Dobrowsky, R. T., and Fernyhough, P. (2011). Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 60, 288–297. doi: 10.2337/db10-0818
Alonso, A., and Sovell, K. A. (2010). Gout, hyperuricemia, and Parkinson’s disease: a protective effect? Curr. Rheumatol. Rep. 12, 149–155. doi: 10.1007/s11926-010-0083-4
Amaral, A. I. (2013). Effects of hypoglycaemia on neuronal metabolism in the adult brain: role of alternative substrates to glucose. J. Inherit. Metab. Dis. 36, 621–634. doi: 10.1007/s10545-012-9553-3
Andersen, J. K., Frim, D. M., Isacson, O., Beal, M. F., and Breakefield, X. O. (1994). Elevation of neuronal MAO-B activity in a transgenic mouse model does not increase sensitivity to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Brain Res. 656, 108–114. doi: 10.1016/0006-8993(94)91371-4
Anitha, M., Abraham, P. M., and Paulose, C. S. (2012). Striatal dopamine receptors modulate the expression of insulin receptor, IGF-1 and GLUT-3 in diabetic rats: effect of pyridoxine treatment. Eur. J. Pharmacol. 696, 54–61. doi: 10.1016/j.ejphar.2012.09.006
Anthony, K., Reed, L. J., Dunn, J. T., Bingham, E., Hopkins, D., Marsden, P. K., et al. (2006). Attenuation of insulin-evoked responses in brain networks controlling appetite and reward in insulin resistance: the cerebral basis for impaired control of food intake in metabolic syndrome? Diabetes 55, 2986–2992. doi: 10.2337/db06-0376
Araki, E., and Nishikawa, T. (2010). Oxidative stress: a cause and therapeutic target of diabetic complications. J. Diabetes Investig. 1, 90–96. doi: 10.1111/j.2040-1124.2010.00013.x
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H. Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. doi: 10.1038/nrneurol.2017.185
Arvanitakis, Z., Wilson, R. S., Bienias, J. L., and Bennet, D. A. (2007). Diabetes and parkinsonian signs in older persons. Alzheimer Dis. Assoc. Disord. 21, 144–149. doi: 10.1097/wad.0b013e31805ba768
Athauda, D., and Foltynie, T. (2016a). Insulin resistance and Parkinson’s disease: a new target for disease modification? Prog. Neurobiol. 145–146, 98–120. doi: 10.1016/j.pneurobio.2016.10.001
Athauda, D., and Foltynie, T. (2016b). The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov. Today 21, 802–818. doi: 10.1016/j.drudis.2016.01.013
Athauda, D., and Foltynie, T. (2018). Protective effects of the GLP-1 mimetic exendin-4 in Parkinson’s disease. Neuropharmacology 136, 260–270. doi: 10.1016/j.neuropharm.2017.09.023
Athauda, D., Maclagan, K., Skene, S. S., Bajwa-Joseph, M., Letchford, D., Chowdhury, K., et al. (2017). Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675. doi: 10.1016/S0140-6736(17)31585-4
Aubert, C. E., Michel, P. L., Gillery, P., Jaisson, S., Fonfrede, M., Morel, F., et al. (2014). Association of peripheral neuropathy with circulating advanced glycation end products, soluble receptor for advanced glycation end products and other risk factors in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 30, 679–685. doi: 10.1002/dmrr.2529
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Ell, P., Soderlund, T., et al. (2013). Exanatide and the treatment of patients with Parkinson’s disease. J. Clin. Invest. 123, 2730–2736. doi: 10.1172/JCI68295
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Kahan, J., Fmedsci, P. E., et al. (2014). Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J. Parkinsons Dis. 4, 337–344. doi: 10.3233/jpd-140364
Banks, W. A., Jaspan, J. B., Huang, W., and Kastin, A. J. (1997a). Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides 18, 1423–1429. doi: 10.1016/s0196-9781(97)00231-3
Banks, W. A., Jaspan, J. B., and Kastin, A. J. (1997b). Effect of diabetes mellitus on the permeability of the blood-brain barrier to insulin. Peptides 18, 1577–1584. doi: 10.1016/s0196-9781(97)00238-6
Banks, W. A., and Kastin, A. J. (1998). Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19, 883–889. doi: 10.1016/s0196-9781(98)00018-7
Bansal, V., Kalita, J., and Misra, U. K. (2006). Diabetic neuropathy. Postgrad. Med. J. 82, 95–100. doi: 10.1136/pgmj.2005.036137
Barbeau, A., Giguere, R., and Hardy, J. (1961). Clinical experience with tolbutamide in Parkinson’s disease. Union Med. Can. 90, 147–151.
Barnett, P. A., González, R. G., Chylack, L. T. Jr., and Cheng, H. M. (1986). The effect of oxidation on sorbitol pathway kinetics. Diabetes 35, 426–432. doi: 10.2337/diab.35.4.426
Bassil, F., Fernagut, P. O., Bezard, E., and Meissner, W. G. (2014). Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog. Neurobiol. 118, 1–18. doi: 10.1016/j.pneurobio.2014.02.005
Bayliss, J. A., Lemus, M. B., Santos, V. V., Deo, M., Davies, J. S., Kemp, B. E., et al. (2016). Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS One 11:e0159381. doi: 10.1371/journal.pone.0159381
Becker, C., Brobert, G. P., Johansson, S., Jick, S. S., and Meier, C. R. (2008). Diabetes in patients with idiopathic Parkinson’s disease. Diabetes Care 31, 1808–1812. doi: 10.2337/dc08-0479
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bender, A., Schwarzkopf, R. M., McMillan, A., Krishnan, K. J., Rieder, G., Neumann, M., et al. (2008). Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J. Neurol. 255, 1231–1235. doi: 10.1007/s00415-008-0892-9
Benedict, C., Hallschmid, M., Hatke, A., Schultes, B., Fehm, H. L., Born, J., et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334. doi: 10.1016/j.psyneuen.2004.04.003
Benedict, C., Hallschmid, M., Schmitz, K., Schultes, B., Ratter, F., Fehm, H. L., et al. (2007). Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32, 239–243. doi: 10.1038/sj.npp.1301193
Bertolotto, F., and Massone, A. (2012). Combination of α lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs RD 12, 29–34. doi: 10.2165/11599200-000000000-00000
Bharath, S., Hsu, M., Kaur, D., Rajagopalan, S., and Andersen, J. K. (2002). Glutathione, iron and Parkinson’s disease. Biochem. Pharmacol. 64, 1037–1048. doi: 10.1016/s0006-2952(02)01174-7
Bierhaus, A., Humpert, P. M., Morcos, M., Wendt, T., Chavakis, T., Arnold, B., et al. (2005). Understanding RAGE, the receptor for advanced glycation end-products. J. Mol. Med. 83, 876–886. doi: 10.1007/s00109-005-0688-7
Biessels, G. J., van der Heide, L. P., Kamal, A., Bleys, R. L., and Gispen, W. H. (2002). Ageing and diabetes: implications for brain function. Eur. J. Pharmacol. 441, 1–14. doi: 10.1016/s0014-2999(02)01486-3
Bilgic, B., Pfefferbaum, A., Rohlfing, T., Sullivan, E. V., and Adalsteinsson, E. (2012). MRI estimates of brain iron concentration in normal aging using quantitative susceptibility mapping. Neuroimage 59, 2625–2635. doi: 10.1016/j.neuroimage.2011.08.077
Birch, N. P., Christie, D. L., and Renwick, A. G. C. (1984). Proinsulin-like material in mouse foetal brain cell cultures. FEBS Lett. 168, 299–302. doi: 10.1016/0014-5793(84)80266-5
Bobela, W., Nazeeruddin, S., Knott, G., Aebischer, P., and Schneider, B. L. (2017). Modulating the catalytic activity of AMPK has neuroprotective effects against α-synuclein toxicity. Mol. Neurodegener. 12:80. doi: 10.1186/s13024-017-0220-x
Bolaños, J. P., Delgado-Esteban, M., Herrero-Mendez, A., Fernandez-Fernandez, S., and Almeida, A. (2008). Regulation of glycolysis and pentose-phosphate pathway by nitric oxide: impact on neuronal survival. Biochim. Biophys. Acta 1777, 789–793. doi: 10.1016/j.bbabio.2008.04.011
Bolaños, J. P., Heales, S. J., Peuchen, S., Barker, J. E., Land, J. M., and Clark, J. B. (1996). Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic. Biol. Med. 21, 995–1001. doi: 10.1016/s0891-5849(96)00240-7
Bonifati, V. (2007). LRRK2 low-penetrance mutations (Gly2019Ser) and risk alleles (Gly2385Arg)-linking familial and sporadic Parkinson’s disease. Neurochem. Res. 32, 1700–1708. doi: 10.1007/s11064-007-9324-y
Bournival, J., Francoeur, M. A., Renaud, J., and Martinoli, M. G. (2012). Quercetin and sesamin protect neuronal PC12 cells from high-glucose-induced oxidation, nitrosative stress, and apoptosis. Rejuvenation Res. 15, 322–333. doi: 10.1089/rej.2011.1242
Boyd, A. E. III., Lebovitz, H. E., and Feldman, J. M. (1971). Endocrine function and glucose metabolism in patients with Parkinson’s disease and their alternation by L-dopa. J. Clin. Endocrinol. Metab. 33, 829–837. doi: 10.1210/jcem-33-5-829
Brauer, R., Bhaskaran, K., Chaturvedi, N., Dexter, D. T., Smeeth, L., and Douglas, I. (2015). Glitazone treatment and incidence of Parkinson’s disease among people with diabetes: a retrospective cohort study. PLoS Med. 12:e1001854. doi: 10.1371/journal.pmed.1001854
Brichta, L., and Greengard, P. (2014). Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: an update. Front. Neuroanat. 8:152. doi: 10.3389/fnana.2014.00152
Brosius, F. C., and Heilig, C. W. (2005). Glucose transporters in diabetic nephropathy. Pediatr. Nephrol. 20, 447–451. doi: 10.1007/s00467-004-1748-x
Brownlee, M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625. doi: 10.2337/diabetes.54.6.1615
Buchman, A. S., Shulman, J. M., Nag, S., Leurgans, S. E., Arnold, S. E., Morris, M. C., et al. (2012). Nigral pathology and Parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 71, 258–266. doi: 10.1002/ana.22588
Calabrese, V., Santoro, A., Monti, D., Crupi, R., Di Paola, R., Latteri, S., et al. (2018). Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 115, 80–91. doi: 10.1016/j.freeradbiomed.2017.10.379
Canal, M., Romaní-Aumedes, J., Martin-Flores, N., Pérez-Fernández, V., and Malagelada, C. (2014). RTP801/REDD1: a stress coping regulator that turns into a troublemaker in neurodegenerative disorders. Front. Cell. Neurosci. 8:313. doi: 10.3389/fncel.2014.00313
Carbone, C., Costa, A., Provensi, G., Mannaioni, G., and Masi, A. (2017). The hyperpolarization-activated current determines synaptic excitability, calcium activity and specific viability of substantia nigra dopaminergic neurons. Front. Cell. Neurosci. 11:187. doi: 10.3389/fncel.2017.00187
Carlsson, A., Lindqvist, M., Magnusson, T., and Waldeck, B. (1958). On the presence of 3- hydroxytyramine in brain. Science 127:471. doi: 10.1126/science.127.3296.471
Carruthers, A. (1986). ATP regulation of the human red cell sugar transporter. J. Biol. Chem. 261, 11028–11037.
Carruthers, A., and Helgerson, A. L. (1989). The human erythrocyte sugar transporter is also a nucleotide binding protein. Biochemistry 28, 8337–8346. doi: 10.1021/bi00447a011
Cereda, E., Barichella, M., Cassani, E., Caccialanza, R., and Pezzoli, G. (2012). Clinical features of Parkinson disease when onset of diabetes came first: a case-control study. Neurology 78, 1507–1511. doi: 10.1212/WNL.0b013e3182553cc9
Cereda, E., Barichella, M., Pedrolli, C., Klersy, C., Cassani, E., Caccialanza, R., et al. (2011). Diabetes and risk of Parkinson’s disease: a systematic review and meta-analysis. Diabetes Care 34, 2614–2623. doi: 10.2337/dc11-1584
Ceriello, A. (2003). New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care 26, 1589–1596. doi: 10.2337/diacare.26.5.1589
Charcot, J. M. (1877). Lectures on the Diseases of the Nervous System, Delivered at La Salpêtrière. London: The New Sydenham Society.
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. doi: 10.1016/S0140-6736(04)17103-1
Cheng, Z., Tseng, Y., and White, M. F. (2010). Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21, 589–598. doi: 10.1016/j.tem.2010.06.005
Cheng, B., Yang, X., An, L., Gao, B., Liu, X., and Liu, S. (2009). Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 1286, 25–31. doi: 10.1016/j.brainres.2009.06.060
Chinta, S. J., and Andersen, J. K. (2008). Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 1780, 1362–1367. doi: 10.1016/j.bbagen.2008.02.005
Chowdhury, S. K., Zherebitskaya, E., Smith, D. R., Akude, E., Chattopadhyay, S., Jolivalt, C. G., et al. (2010). Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59, 1082–1091. doi: 10.2337/db09-1299
Cloherty, E. K., Heard, K. S., and Carruthers, A. (1996). Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry 35, 10411–10421. doi: 10.1021/bi953077m
Colville, C. A., Seatter, M. J., Jess, T. J., Gould, G. W., and Thomas, H. M. (1993). Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem. J. 290, 701–706. doi: 10.1042/bj2900701
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Beal, M. F., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329. doi: 10.1038/ng1292-324
Correia, S., Carvalho, C., Santos, M. S., Seiça, R., Oliveira, C. R., and Moreira, P. I. (2008). Mechanisms of action of metformin in type 2 diabetes and associated complications: an overview. Mini Rev. Med. Chem. 8, 1343–1354. doi: 10.2174/138955708786369546
Craft, S., Newcomer, J., Kanne, S., Dagogo-Jack, S., Cryer, P., Sheline, Y., et al. (1996). Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 17, 123–130. doi: 10.1016/0197-4580(95)02002-0
Cuervo, A. M., Wong, E. S., and Martinez-Vicente, M. (2010). Protein degradation, aggregation, and misfolding. Mov. Disord. 25, 49–54. doi: 10.1002/mds.22718
Daugherty, A., and Raz, N. (2013). Age-related differences in iron content of subcortical nuclei observed in vivo: a meta-analysis. Neuroimage 70, 113–121. doi: 10.1016/j.neuroimage.2012.12.040
de Vries, M. G., Arseneau, L. M., Lawson, M. E., and Beverly, J. L. (2003). Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 52, 2767–2773. doi: 10.2337/diabetes.52.11.2767
Dehay, B., Bové, J., Rodríguez-Muela, N., Perier, C., Recasens, A., Boya, P., et al. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010
Deltour, L., Leduque, P., Blume, N., Madsen, O., Dubois, P., Jami, J., et al. (1993). Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc. Natl. Acad. Sci. U S A 90, 527–531. doi: 10.1073/pnas.90.2.527
Devaskar, S. U., Giddings, S. J., Rajakumar, P. A., Carnaghi, L. R., Menon, R. K., and Zahm, D. S. (1994). Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 269, 8445–8454.
Dexter, D. T., Wells, F. R., Lees, A. J., Agid, F., Agid, Y., Jenner, P., et al. (1989). Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 52, 1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x
Diabetes Control and Complications Trial Research Group, Nathan, D. M., Genuth, S., Lachin, J., Cleary, P., Crofford, O., et al. (1993). The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–986. doi: 10.1056/NEJM199309303291401
Dick, A. P., Harik, S. I., Klip, A., and Walker, D. M. (1984). Identification and characterization of the glucose transporter of the blood-brain barrier by cytochalasin B binding and immunological reactivity. Proc. Natl. Acad. Sci. U S A 81, 7233–7237. doi: 10.1073/pnas.81.22.7233
Dienel, G. A., and Cruz, N. F. (2006). Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem. Int. 48, 586–595. doi: 10.1016/j.neuint.2006.01.004
Dienel, G. A., and Cruz, N. F. (2015). Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 30, 281–298. doi: 10.1007/s11011-014-9493-8
Dopeso-Reyes, I. G., Rico, A. J., Roda, E., Sierra, S., Pignataro, D., Lanz, M., et al. (2014). Calbindin content and differential vulnerability of midbrain efferent dopaminergic neurons in macaques. Front. Neuroanat. 8:146. doi: 10.3389/fnana.2014.00146
Dorn, A., Bernstein, H. G., Rinne, A., Ziegler, M., Hahn, H. J., and Ansorge, S. (1983). Insulin and glucagonlike peptides in the brain. Anat. Rec. 207, 69–77. doi: 10.1002/ar.1092070108
Double, K. L., Gerlach, M., Schunemann, V., Trautwein, A. X., Zecca, L., Gallorini, M., et al. (2003). Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 66, 489–494. doi: 10.1016/s0006-2952(03)00293-4
Driver, J. A., Smith, A., Buring, J. E., Gaziano, J. M., Kurth, T., and Logroscino, G. (2008). Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 31, 2003–2005. doi: 10.2337/dc08-0688
Du, X. L., Edelstein, D., Dimmeler, S., Ju, Q., Sui, C., and Brownlee, M. (2001). Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Invest. 108, 1341–1348. doi: 10.1172/jci11235
Du, X., Matsumura, T., Edelstein, D., Rossetti, L., Zsengellér, Z., Szabo, C., et al. (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invest. 112, 1049–1057. doi: 10.1172/jci200318127
Duarte, A. I., Moreira, P. I., and Oliveira, C. R. (2012). Insulin in central nervous system: more than just a peripheral hormone. J. Aging Res. 2012:384017. doi: 10.1155/2012/384017
Duarte, A. I., Proenca, T., Oliveira, C. R., Santos, M. S., and Rego, A. C. (2006). Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes 55, 2863–2870. doi: 10.2337/db06-0030
Ehringer, H., and Hornykiewicz, O. (1960). Veteilung von noradrenalin und dopamine (3-hydroxytyramin) im gehirn des menschen und ihr verhalten bei erkankungen des extrapyramidalen systems. Klin. Wochenschr. 38, 1236–1239. doi: 10.1007/bf01485901
Eppens, M. C., Craig, M. E., Cusumano, J., Hing, S., Chan, A. K., Howard, N. J., et al. (2006). Prevalence of diabetes complications in adolescents with type 2 compared with type 1 diabetes. Diabetes Care 29, 1300–1306. doi: 10.2337/dc05-2470
Fearnley, J. M., and Lees, A. J. (1991). Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114, 2283–2301. doi: 10.1093/brain/114.5.2283
Fedorow, H., Halliday, G. M., Rickert, C. H., Gerlach, M., Riederer, P., and Double, K. L. (2006). Evidence for specific phases in the development of human neuromelanin. Neurobiol. Aging 27, 506–512. doi: 10.1016/j.neurobiolaging.2005.02.015
Finkelstein, M. M., and Jerrett, M. (2007). A study of the relationships between Parkinson’s disease and markers of traffic-derived and environmental manganese air pollution in two Canadian cities. Environ. Res. 104, 420–432. doi: 10.1016/j.envres.2007.03.002
Fitzgerald, J. C., Zimprich, A., Carvajal Berrio, D. A., Schindler, K. M., Maurer, B., Schulte, C., et al. (2017). Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson’s disease. Brain 140, 2444–2459. doi: 10.1093/brain/awx202
Foehring, R. C., Zhang, X. F., Lee, J. C., and Callaway, J. C. (2009). Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 102, 2326–2333. doi: 10.1152/jn.00038.2009
Freiherr, J., Hallschmid, M., Frey, W. H., Brunner, Y. F., Chapman, C. D., Holscher, C., et al. (2013). Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs 27, 505–514. doi: 10.1007/s40263-013-0076-8
Frölich, L., Blum-Degen, D., Bernstein, H. G., Engelsberger, S., Humrich, J., Laufer, S., et al. (1998). Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 105, 423–438. doi: 10.1007/s007020050068
Furlong, M., Tanner, C. M., Goldman, S. M., Bhudhikanok, G. S., Blair, A., Chade, A., et al. (2015). Protective glove use and hygiene habits modify the associations of specific pesticides with Parkinson’s disease. Environ. Int. 75, 144–150. doi: 10.1016/j.envint.2014.11.002
Gabbay, K. H., Merola, L. O., and Field, R. A. (1966). Sorbitol pathway: presence in nerve and cord with substrate accumulation in diabetes. Science 151, 209–210. doi: 10.1126/science.151.3707.209
Gaede, P., Lund-Andersen, H., Parving, H. H., and Pedersen, O. (2008). Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 358, 580–591. doi: 10.1056/NEJMoa0706245
Gao, Y., Bielohuby, M., Fleming, T., Grabner, G. F., Foppen, E., Bernhard, W., et al. (2017). Dietary sugars, not lipids, drive hypothalamic inflammation. Mol. Metab. 6, 897–908. doi: 10.1016/j.molmet.2017.06.008
García-Nogales, P., Almeida, A., and Bolaños, J. P. (2003). Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. J. Biol. Chem. 278, 864–874. doi: 10.1074/jbc.M206835200
Gerhart, D. Z., LeVasseur, R. J., Broderius, M. A., and Drewes, L. R. (1989). Glucose transporter localization in brain using light and electron immunocytochemistry. J. Neurosci. Res. 22, 464–472. doi: 10.1002/jnr.490220413
German, D. C., Dubach, M., Askari, S., Speciale, S. G., and Bowdon, D. M. (1988). 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonian syndrome in macaca fascicularis: which midbrain dopaminergic neurons are lost? Neuroscience 24, 161–174. doi: 10.1016/0306-4522(88)90320-x
Giacco, F., and Brownlee, M. (2010). Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070. doi: 10.1161/CIRCRESAHA.110.223545
Gibb, W. R., and Lees, A. J. (1991). Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra and differential cell death in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 54, 388–396. doi: 10.1136/jnnp.54.5.388
Giugliano, D., Ceriello, A., and Paolisso, G. (1996). Oxidative stress and diabetic vascular complications. Diabetes Care 19, 257–267. doi: 10.2337/diacare.19.3.257
Goldman, S. M., Quinlan, P. J., Ross, G. W., Marras, C., Meng, C., Bhudhikanok, G. S., et al. (2012). Solvent exposures and Parkinson disease risk in twins. Ann. Neurol. 71, 776–784. doi: 10.1002/ana.22629
Gomez, R., and Barros, H. M. (2003). Clonazepam increases in vivo striatal extracellular glucose in diabetic rats after glucose overload. Pharmacol. Biochem. Behav. 76, 443–450. doi: 10.1016/j.pbb.2003.08.018
Gorell, J. M., Johnson, C. C., Rybicki, B. A., Peterson, E. L., Kortsha, G. X., Brown, G. G., et al. (1997). Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 48, 650–658. doi: 10.1212/wnl.48.3.650
Götz, M. E., Freyberger, A., and Riederer, P. (1990). Oxidative stress: a role in the pathogenesis of Parkinson’s disease. J. Neural Transm. Suppl. 29, 241–249. doi: 10.1007/978-3-7091-9050-0_23
Graham, D. G. (1984). Catecholamine toxicity: a proposal for the molecular pathogenesis of manganese neurotoxicity and Parkinson’s disease. Neurotoxicology 5, 83–95.
Gralle, M. (2017). The neuronal insulin receptor in its environment. J. Neurochem. 140, 359–367. doi: 10.1111/jnc.13909
Greenamyre, J. T., Sherer, T. B., Betarbet, R., and Panov, A. V. (2001). Complex I and Parkinson’s disease. IUBMB Life 52, 135–141. doi: 10.1080/15216540152845939
Greene, D. A., Arezzo, J. C., and Brown, M. B. (1999). Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Neurology 53, 580–591. doi: 10.1212/wnl.53.3.580
Greene, L. A., Levy, O., and Malagelada, C. (2011). Akt as a victim, villain and potential hero in Parkinson’s disease pathophysiology and treatment. Cell. Mol. Neurobiol. 31, 969–978. doi: 10.1007/s10571-011-9671-8
Gregg, E. W., Sattar, N., and Ali, M. K. (2016). The changing face of diabetes complications. Lancet Diabetes Endocrinol. 4, 537–547. doi: 10.1016/s2213-8587(16)30010-9
Gupta, G., Azam, M., and Baquer, N. Z. (1992). Effect of experimental diabetes on the catecholamine metabolism in rat brain. J. Neurochem. 58, 95–100. doi: 10.1111/j.1471-4159.1992.tb09282.x
Guzman, J. N., Sanchez-Padilla, J., Wokosin, D., Kondapalli, J., Ilijic, E., Schumacker, P. T., et al. (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468, 696–700. doi: 10.1038/nature09536
Haas, C. B., Kalinine, E., Zimmer, E. R., Hansel, G., Brochier, A. W., Oses, J. P., et al. (2016). Brain insulin administration triggers distinct cognitive and neurotrophic responses in young and aged rats. Mol. Neurobiol. 53, 5807–5817. doi: 10.1007/s12035-015-9494-6
Haddad, D., and Nakamura, K. (2015). Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett. 589, 3702–3713. doi: 10.1016/j.febslet.2015.10.021
Hallschmid, M., Benedict, C., Schultes, B., Born, J., and Kern, W. (2008). Obese men respond to cognitive but not to catabolic brain insulin signaling. Int. J. Obes. 32, 275–282. doi: 10.1038/sj.ijo.0803722
Haslbeck, K. M., Neundörfer, B., Schlötzer-Schrehardtt, U., Bierhaus, A., Schleicher, E., Pauli, E., et al. (2007). Activation of the RAGE pathway: a general mechanism in the pathogenesis of polyneuropathies? Neurol. Res. 29, 103–110. doi: 10.1179/174313206x152564
Havrankova, J., Roth, J., and Brownstein, M. (1978a). Insulin receptors are widely distributed in the central nervous system of the rat. Nature 272, 827–829. doi: 10.1038/272827a0
Havrankova, J., Schmechel, D., Roth, J., and Brownstein, M. (1978b). Identification of insulin in rat brain. Proc. Natl. Acad. Sci. U S A 75, 5737–5741. doi: 10.1073/pnas.75.11.5737
Havrankova, J., Roth, J., and Brownstein, M. J. (1979). Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels: studies of obese and streptozotocin-treated rodents. J. Clin. Invest. 64, 636–642. doi: 10.1172/jci109504
Heilig, C. W., Concepcion, L. A., Riser, B. L., Freytag, S. O., Zhu, M., and Cortes, P. (1995). Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J. Clin. Invest. 96, 1802–1814. doi: 10.1172/jci118226
Herrero-Mendez, A., Almeida, A., Fernandez, E., Maestre, C., Moncada, S., and Bolaños, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/ C-Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Hill, J., Lesniak, M., Pert, C., and Roth, J. (1986). Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience 17, 1127–1138. doi: 10.1016/0306-4522(86)90082-5
Hinder, L. M., Vivekanandan-Giri, A., McLean, L. L., Pennathur, S., and Feldman, E. L. (2013). Decreased glycolytic and tricarboxylic acid cycle intermediates coincide with peripheral nervous system oxidative stress in a murine model of type 2 diabetes. J. Endocrinol. 216, 1–11. doi: 10.1530/joe-12-0356
Hirsch, E. C., and Faucheux, B. A. (1998). Iron metabolism and Parkinson’s disease. Mov. Disord. 1, 39–45.
Hirsch, E., Graybiel, A. M., and Agid, Y. A. (1988). Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 334, 345–348. doi: 10.1038/334345a0
Hornykiewicz, O. (1966). “Metabolism of brain dopamine in human Parkinsonism: neurochemical and clinical aspects,” in Biochemistry and Pharmacology of the Basal Ganglia, ed. E. Costa (Hewlett, NY: Raven Press), 171–185.
Houlden, H., and Singleton, A. B. (2012). The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 124, 325–338. doi: 10.1007/s00401-012-1013-5
Hu, G., Jousilahti, P., Bidel, S., Antikainen, R., and Tuomilehto, J. (2007). Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 30, 842–847. doi: 10.2337/dc06-2011
Huang, Y. P., Chen, L. S., Yen, M. F., Fann, C. Y., Chiu, Y. H., Chen, H. H., et al. (2013). Parkinson’s disease is related to an increased risk of ischemic stroke-a population-based propensity score-matched follow-up study. PLoS One 8:e68314. doi: 10.1371/journal.pone.0068314
Huang, S., and Czech, M. P. (2007). The GLUT4 glucose transporter. Cell Metab. 5, 237–252. doi: 10.1016/j.cmet.2007.03.006
Huang, T. T., Hao, D. L., Wu, B. N., Mao, L. L., and Zhang, J. (2017). Uric acid demonstrates neuroprotective effect on Parkinson’s disease mice through Nrf2-ARE signaling pathway. Biochem. Biophys. Res. Commun. 493, 1443–1449. doi: 10.1016/j.bbrc.2017.10.004
Hung, L. M., Huang, J. P., Liao, J. M., Yang, M. H., Li, D. E., Day, Y. J., et al. (2014). Insulin renders diabetic rats resistant to acute ischemic stroke by arresting nitric oxide reaction with superoxide to form peroxynitrite. J. Biomed. Sci. 21:92. doi: 10.1186/s12929-014-0092-0
Iacopino, A. M., and Christakos, S. (1990). Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases. Proc. Natl. Acad. Sci. U S A 87, 4078–4082. doi: 10.1073/pnas.87.11.4078
International Diabetes Federation. (2013). Diabetes Atlas. 6th Edn. Brussels, Belgium: International Diabetes Federation.
Ismaiel, A. A., Espinosa-Oliva, A. M., Santiago, M., García-Quintanilla, A., Oliva-Martin, M. J., Herrera, A. J., et al. (2016). Metformin, besides exhibiting strong in vivo anti-inflammatory properties, increases mptp-induced damage to the nigrostriatal dopaminergic system. Toxicol. Appl. Pharmacol. 298, 19–30. doi: 10.1016/j.taap.2016.03.004
Jacob, R. J., Fan, X., Evans, M. L., Dziura, J., and Sherwin, R. S. (2002). Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: no evidence for protective adaptation by the blood brain barrier. Metab. Clin. Exp. 51, 1522–1524. doi: 10.1053/meta.2002.36347
Jakus, V., and Rietbrock, N. (2004). Advanced glycation end products and the progress of diabetic vascular complications. Physiol. Rev. 53, 131–142. doi: 10.4093/dmj.2017.0105
James, D. E., Brown, R., Navarro, J., and Pilch, P. F. (1988). Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 333, 183–185. doi: 10.1038/333183a0
Jana, N. R. (2012). Protein homeostasis and aging: role of ubiquitin protein ligases. Neurochem. Int. 60, 443–447. doi: 10.1016/j.neuint.2012.02.009
Jenner, P. (1991). Oxidative stress as a cause of Parkinson’s disease. Acta Neurol. Scand. Suppl. 136, 6–15. doi: 10.1111/j.1600-0404.1991.tb05013.x
Kaiser, N., Sasson, S., Feener, E. P., Boukobza-Vardi, N., Higashi, S., Moller, D. E., et al. (1993). Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 42, 80–89. doi: 10.2337/diabetes.42.1.80
Kang, Y., Viswanath, V., Jha, N., Qiao, X., Mo, J. Q., and Andersen, J. K. (1999). Brain γ-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J. Neurosci. Res. 58, 436–441. doi: 10.1002/(sici)1097-4547(19991101)58:3<436::aid-jnr9>3.3.co;2-2
Kao, S. Y. (2009). Rescue of α-synuclein cytotoxicity by insulin-like growth factors. Biochem. Biophys. Res. Commun. 385, 434–438. doi: 10.1016/j.bbrc.2009.05.089
Kastner, A., Hirsch, E. C., Lejeune, O., Javoy-Agid, F., Rascol, O., and Agid, Y. (1992). Is the vulnerability of neurons in the substantia nigra of patients with Parkinson’s disease related to their neuromelanin content? J. Neurochem. 59, 1080–1089. doi: 10.1111/j.1471-4159.1992.tb08350.x
Kasznicki, J., Kosmalski, M., Sliwinska, A., Mrowicka, M., Stanczyk, M., Majsterek, I., et al. (2012). Evaluation of oxidative stress markers in pathogenesis of diabetic neuropathy. Mol. Biol. Rep. 39, 8669–8678. doi: 10.1007/s11033-012-1722-9
Katila, N., Bhurtel, S., Shadfar, S., Srivastav, S., Neupane, S., Ojha, U., et al. (2017). Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 125, 396–407. doi: 10.1016/j.neuropharm.2017.08.015
Kaur, G., and Bhardwaj, S. K. (1998). The impact of diabetes on CNS. Role of bioenergetic defects. Mol. Chem. Neuropathol. 35, 119–131. doi: 10.1007/bf02815119
Kawai, T., Takei, I., Tokui, M., Funae, O., Miyamoto, K., Tabata, M., et al. (2010). Effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy in patients with type 2 diabetes, in relation to suppression of N(ε)-carboxymethyl lysine. J. Diabetes Complications 24, 424–432. doi: 10.1016/j.jdiacomp.2008.10.005
Khang, R., Park, C., and Shin, J. H. (2015). Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience 294, 182–192. doi: 10.1016/j.neuroscience.2015.03.017
Khanh, D. V., Choi, Y. H., Moh, S. H., Kinyua, A. W., and Kim, K. W. (2014). Leptin and insulin signaling in dopaminergic neurons: relationship between energy balance and reward system. Front. Psychol. 5:846. doi: 10.3389/fpsyg.2014.00846
Kim, S. J., and Han, Y. (2005). Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt). J. Neural Transm. 112, 179–191. doi: 10.1007/s00702-004-0163-6
Kim, W., Mohney, R. P., Wilson, B., Jeohn, G., Liu, B., and Hong, J. (2000). Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J. Neurosci. 20, 6309–6316. doi: 10.1523/jneurosci.20-16-06309.2000
Kim, S. R., Ries, V., Cheng, H. C., Kareva, T., Oo, T. F., Yu, W. H., et al. (2011). Age and α-synuclein expression interact to reveal a dependence of dopaminergic axons on endogenous Akt/PKB signaling. Neurobiol. Dis. 44, 215–222. doi: 10.1016/j.nbd.2011.07.003
Kirkley, K. S., Popichak, K. A., Hammond, S. L., Davies, C., Hunt, L., and Tjalkens, R. B. (2019). Genetic suppression of IKK2/NF-kappaB in astrocytes inhibits neuroinflammation and reduces neuronal loss in the MPTP-Probenecid model of Parkinson’s disease. Neurobiol. Dis. 127, 193–209. doi: 10.1016/j.nbd.2019.02.020
Klimek, P., Kautzky-Willer, A., Chmiel, A., Schiller-Frühwirth, I., and Thurner, S. (2015). Quantification of diabetes comorbidity risks across life using nation-wide big claims data. PLoS Comput. Biol. 11:e1004125. doi: 10.1371/journal.pcbi.1004125
Kuan, Y. C., Huang, K. W., Lin, C. L., Hu, C. J., and Kao, C. H. (2017). Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuropsychopharmacol. Biol. Psychiatry 79, 77–83. doi: 10.1016/j.pnpbp.2017.06.002
Langston, J. W., Ballard, P., Tetrud, J., and Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-w
Lenzi, G. L., Jones, T., Reid, J. L., and Moss, S. (1979). Regional impairment of cerebral oxidative metabolism in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 42, 59–62. doi: 10.1136/jnnp.42.1.59
LeRoith, D., Lowe, W. L. Jr., Shemer, J., Raizada, M. K., and Ota, A. (1988). Development of brain insulin receptors. Int. J. Biochem. 20, 225–230. doi: 10.1016/0020-711x(88)90345-x
Levine, K. B., Cloherty, E. K., Fidyk, N. J., and Carruthers, A. (1998). Structural and physiologic determinants of human erythrocyte sugar transport regulation by adenosine triphosphate. Biochemistry 37, 12221–12232. doi: 10.1021/bi980585y
Li, X. J., and Li, S. (2011). Proteasomal dysfunction in aging and Huntington disease. Neurobiol. Dis. 43, 4–8. doi: 10.1016/j.nbd.2010.11.018
Liang, C. L., Nelson, O., Yazdani, U., Pasbakhsh, P., and German, D. C. (2004). Inverse relationship between the contents of neuromelanin pigment and the vesicular monoamine transporter-2: human midbrain dopamine neurons. J. Comp. Neurol. 473, 97–106. doi: 10.1002/cne.20098
Liang, C. L., Sinton, C. M., Sonsalla, P. K., and German, D. C. (1996). Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration 5, 313–318. doi: 10.1006/neur.1996.0042
Lipman, I. J., Boykin, M. E., and Flora, R. E. (1974). Glucose intolerance in Parkinson’s disease. J. Chronic. Dis. 27, 573–579. doi: 10.1016/0021-9681(74)90031-9
Lowe, A. G., and Walmsley, A. R. (1986). The kinetics of glucose transport in human red blood cells. Biochim. Biophys. Acta 857, 146–154. doi: 10.1016/0005-2736(86)90342-1
Lu, M., Su, C., Qiao, C., Bian, Y., Ding, J., and Hu, G. (2016). Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int. J. Neuropsychopharmacol. 19:pyw047. doi: 10.1093/ijnp/pyw047
Lutas, A., and Yellen, G. (2013). The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40. doi: 10.1016/j.tins.2012.11.005
Ma, S. Y., Röytt, M., Collan, Y., and Rinne, J. O. (1999). Unbiased morphometrical measurements show loss of pigmented nigral neurones with ageing. Neuropathol. Appl. Neurobiol. 25, 394–399. doi: 10.1046/j.1365-2990.1999.00202.x
Maher, F., Davies-Hill, T. M., and Simpson, I. A. (1996). Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem. J. 315, 827–831. doi: 10.1042/bj3150827
Maher, F., Vannucci, S. J., and Simpson, I. A. (1994). Glucose transporter proteins in brain. FASEB J. 8, 1003–1011. doi: 10.1096/fasebj.8.13.7926364
Malagelada, C., Jin, Z. H., and Greene, L. A. (2008). RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 28, 14363–14371. doi: 10.1523/jneurosci.3928-08.2008
Manolescu, A. R., Augustin, R., Moley, K., and Cheeseman, C. (2007). A highly conserved hydrophobic motif in the exofacial vestibule of fructose transporting SLC2A proteins acts as a critical determinant of their substrate selectivity. Mol. Membr. Biol. 24, 455–463. doi: 10.1080/09687680701298143
Mantych, G. J., James, D. E., Chung, H. D., and Devaskar, S. U. (1992). Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology 131, 1270–1278. doi: 10.1210/endo.131.3.1505464
Margolis, R. U., and Altszuler, N. (1967). Insulin in the cerebrospinal fluid. Nature 215, 1375–1376. doi: 10.1038/2151375a0
Matsuda, W., Furuta, T., Nakamura, K. C., Hioki, H., Fujiyama, F., Arai, R., et al. (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453. doi: 10.1523/JNEUROSCI.4029-08.2009
McCrimmon, R. J., Jacob, R. J., Fan, X., McNay, E. C., and Sherwin, R. S. (2003). Effects of recurrent antecedent hypoglycaemia and chronic hyperglycaemia on brainstem extra-cellular glucose concentrations during acute hypoglycaemia in conscious diabetic BB rats. Diabetologia 46, 1658–1661. doi: 10.1007/s00125-003-1231-4
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. (1988). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/wnl.38.8.1285
Mehran, A. E., Templeman, N. M., Brigidi, G. S., Lim, G. E., Chu, K. Y., Hu, X., et al. (2012). Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 16, 723–737. doi: 10.1016/j.cmet.2012.10.019
Mendez, M. M., Folgado, J., Tormo, C., Artero, A., Martinez-Hervás, S., Chaves, J. F., et al. (2015). Altered glutathione system is associated with the presence of distal symmetric peripheral polyneuropathy in type 2 diabetic subjects. J. Diabetes Complications 29, 923–927. doi: 10.1016/j.jdiacomp.2015.05.023
Molnár, G., Faragó, N., Kocsis, Á. K., Rózsa, M., Lovas, S., Boldog, E., et al. (2014). GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J. Neurosci. 34, 1133–1137. doi: 10.1523/JNEUROSCI.4082-13.2014
Moreira, P. I., Rolo, A. P., Sena, C., Seiça, R., Oliveira, C. R., and Santos, M. S. (2006). Insulin attenuates diabetes-related mitochondrial alterations: a comparative study. Med. Chem. 2, 299–308. doi: 10.2174/157340606776930754
Moreira, P. I., Santos, M. S., Sena, C., Seiça, R., and Oliveira, C. R. (2005). Insulin protects against amyloid β-peptide toxicity in brain mitochondria of diabetic rats. Neurobiol. Dis. 18, 628–637. doi: 10.1016/j.nbd.2004.10.017
Moroo, I., Yamada, T., Makino, H., Tooyama, I., McGeer, P. L., McGeer, E. G., et al. (1994). Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson’s disease. Acta Neuropathol. 87, 343–348. doi: 10.1007/bf00313602
Mueckler, M., and Thorens, B. (2013). The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 34, 121–138. doi: 10.1016/j.mam.2012.07.001
Murakami, K., Miyake, Y., Sasaki, S., Tanaka, K., Fukushima, W., Kiyohara, C., et al. (2010). Dietary glycemic index is inversely associated with the risk of Parkinson’s disease: a case-control study in Japan. Nutrition 26, 515–521. doi: 10.1016/j.nut.2009.05.021
Nadeau, K. J., Anderson, B. J., Berg, E. G., Chiang, J. L., Chou, H., Copeland, K. C., et al. (2016). Youth-onset type 2 diabetes consensus report: current status, challenges and priorities. Diabetes Care 29, 1635–1642. doi: 10.2337/dc16-1066
Nagamatsu, S., Sawa, H., Inoue, N., Nakamichi, Y., Takeshima, H., and Hoshino, T. (1994). Gene expression of GLUT3 glucose transporter regulated by glucose in vivo in mouse brain and in vitro in neuronal cell cultures from rat embryos. Biochem. J. 300, 125–131. doi: 10.1042/bj3000125
Nagamatsu, S., Sawa, H., Kamada, K., Nakamichi, Y., Yoshimoto, K., and Hoshino, T. (1993). Neuron-specific glucose transporter (NSGT): CNS distribution of GLUT3 rat glucose transporter (RGT3) in rat central neurons. FEBS Lett. 334, 289–295. doi: 10.1016/0014-5793(93)80697-s
Nathan, D. M. (1993). Long-term complications of diabetes mellitus. N. Engl. J. Med. 328, 1676–1685. doi: 10.1056/NEJM199306103282306
Ng, C. H., Guan, M. S., Koh, C., Ouyang, X., Yu, F., Tan, E. K., et al. (2012). AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J. Neurosci. 32, 14311–14317. doi: 10.1523/jneurosci.0499-12.2012
Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., et al. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404, 787–790. doi: 10.1038/35008121
Nowotny, K., Jung, T., Hohn, A., Weber, D., and Grune, T. (2015). Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 5, 194–222. doi: 10.3390/biom5010194
Obeso, J. A., Rodríguez-Oroz, M. C., Rodriguez, M., Lanciego, J. L., Artieda, J., Gonzalo, N., et al. (2000). Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 23, S8–S19. doi: 10.1016/s1471-1931(00)00028-8
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, present and future of Parkinson’s disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 32, 1264–1310. doi: 10.1002/mds.27115
Obrosova, I. G., Drel, V. R., Pacher, P., Ilnytska, O., Wang, Z. Q., Stevens, M. J., et al. (2005). Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes 54, 3435–3441. doi: 10.2337/diabetes.54.12.3435
Obrosova, I. G., Van Huysen, C., Fathallah, L., Cao, X. C., Greene, D. A., and Stevens, M. J. (2002). An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism and antioxidative defense. FASEB J. 16, 123–125. doi: 10.1096/fj.01-0603fje
Okubo, H., Miyake, Y., Sasaki, S., Murakami, K., Tanaka, K., Fukushima, W., et al. (2012). Dietary patterns and risk of Parkinson’s disease: a case-control study in Japan. Eur. J. Neurol. 19, 681–688. doi: 10.1111/j.1468-1331.2011.03600.x
Oliveira, M. A. P., Balling, R., Smidt, M. P., and Fleming, R. M. T. (2017). Embryonic development of selectively vulnerable neurons in Parkinson’s disease. NPJ Parkinsons Dis. 3:21. doi: 10.1038/s41531-017-0022-4
Pacelli, C., Giguère, N., Bourque, M. J., Lévesque, M., Slack, R. S., and Trudeau, L. É. (2015). Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 25, 2349–2360. doi: 10.1016/j.cub.2015.07.050
Pacold, S. T., and Blackard, W. G. (1979). Central nervous system insulin receptors in normal and diabetic rats. Endocrinology 105, 1452–1457. doi: 10.1210/endo-105-6-1452
Pagano, G., Polychronis, S., Wilson, H., Giordano, B., Ferrara, N., Niccolini, F., et al. (2018). Diabetes mellitus and Parkinson disease. Neurology 90, e1654–e1662. doi: 10.1212/WNL.0000000000005475
Pambianco, G., Costacou, T., Ellis, D., Becker, D. J., Klein, R., and Orchard, T. J. (2006). The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes 55, 1463–1469. doi: 10.2337/db05-1423
Pang, Y., Lin, S., Wright, C., Shen, J., Carter, K., Bhatt, A., et al. (2016). Intranasal insulin protects against substantia nigra dopaminergic neuronal loss and alleviates motor deficits induced by 6-OHDA in rats. Neuroscience 318, 157–165. doi: 10.1016/j.neuroscience.2016.01.020
Parent, M., and Parent, A. (2006). Relationship between axonal collateralization and neuronal degeneration in basal ganglia. J. Neural Transm. Suppl. 2006, 85–88. doi: 10.1007/978-3-211-45295-0_14
Patil, S. P., Jain, P. D., Ghumatkar, P. J., Tambe, R., and Sathaye, S. (2014). Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 277, 747–754. doi: 10.1016/j.neuroscience.2014.07.046
Patti, M. E., and Corvera, S. (2010). The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 31, 364–395. doi: 10.1210/er.2009-0027
Percheron, G., François, C., Yelnik, J., and Fenelon, G. (1989). “The primate nigro-striato-pallido-nigral system. Not a mere loop,” in Neural Mechanisms in Disorders of Movement, eds A. R. Crossman and M. A. Sambrook (London: John Libbey), 103–109.
Pezzino, V., Costantino, A., Russo, P., Gullo, D., and Papa, V. (1996). Insulin receptor content in tissues of normal and diabetic rats measured by radioimmunoassay. J. Endocrinol. Invest. 19, 593–597. doi: 10.1007/BF03349023
Pezzoli, G., and Cereda, E. (2013). Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 80, 2035–2041. doi: 10.1212/WNL.0b013e318294b3c8
Polito, L., Greco, A., and Seripa, D. (2016). Genetic profile, environmental exposure, and their interaction in Parkinson’s disease. Parkinsons Dis. 2016:6465793. doi: 10.1155/2016/6465793
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Price, S. A., Gardiner, N. J., Duran-Jimenez, B., Zeef, L. A., Obrosova, I. G., and Tomlinson, D. R. (2006). Thioredoxin interacting protein is increased in sensory neurons in experimental diabetes. Brain Res. 1116, 206–214. doi: 10.1016/j.brainres.2006.07.109
Priyadarshi, A., Khuder, S. A., Schaub, E. A., and Shrivastava, S. (2000). A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology 21, 435–440.
Raichle, M. E., and Gusnard, D. A. (2002). Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. U S A 99, 10237–10239. doi: 10.1073/pnas.172399499
Rajabally, Y. A., Stettner, M., Kieseier, B. C., Hartung, H. P., and Malik, R. A. (2017). CIDP and other inflammatory neuropathies in diabetes—diagnosis and management. Nat. Rev. Neurol. 13, 599–611. doi: 10.1038/nrneurol.2017.123
Ramanathan, M., Jaiswal, A. K., and Bhattacharya, S. K. (1999). Superoxide dismutase, catalase and glutathione peroxidase activities in the brain of streptozotocin induced diabetic rats. Indian J. Exp. Biol. 37, 182–183.
Rao, J., Oz, G., and Seaquist, E. R. (2006). Regulation of cerebral glucose metabolism. Minerva Endocrinol. 31, 149–158.
Rebelos, E., Virtanen, K., Nuutila, P., Nummenmaa, L., Mari, A., Hyypia, A., et al. (2018). “The influence of brain metabolism in insulin secretion and action assessed with FDG-PET in humans,” in Proceedings of the 54th Annual Meeting of the European Association for the Study of Diabetes, Berlin.
Reeve, A., Simcox, E., and Turnbull, D. (2014). Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res. Rev. 14, 19–30. doi: 10.1016/j.arr.2014.01.004
Reger, M. A., Watson, G. S., Green, P. S., Wilkinson, C. W., Baker, L. D., Cholerton, B., et al. (2008). Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 70, 440–448. doi: 10.1212/01.WNL.0000265401.62434.36
Renaud, J., Bournival, J., Zottig, X., and Martinoli, M. G. (2014). Resveratrol protects DAergic PC12 cells from high glucose-induced oxidative stress and apoptosis: effect on p53 and GRP75 localization. Neurotox. Res. 25, 110–123. doi: 10.1007/s12640-013-9439-7
Renaud, J., Bassareo, V., Beaulieu, J., Pinna, A., Schlich, M., Lavoie, C., et al. (2018). Dopaminergic neurodegeneration in a rat model of long-term hyperglycemia: preferential degeneration of the nigrostriatal motor pathway. Neurobiol. Aging 69, 117–128. doi: 10.1016/j.neurobiolaging.2018.05.010
Ribeiro, M., Rosenstock, T. R., Oliveira, A. M., Oliveira, C. R., and Rego, A. C. (2014). Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 74, 129–144. doi: 10.1016/j.freeradbiomed.2014.06.023
Rong, L. L., Gooch, C., Szabolcs, M., Herold, K. C., Lalla, E., Hays, A. P., et al. (2005). RAGE: a journey from the complications of diabetes to disorders of the nervous system-striking a fine balance between injury and repair. Restor. Neurol. Neurosci. 23, 355–365.
Rotermund, C., Machetanz, G., and Fitzgerald, J. C. (2018). The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 9:400. doi: 10.3389/fendo.2018.00400
Rubinsztein, D. C., Marino, G., and Kroemer, G. (2011). Autophagy and aging. Cell 146, 682–695. doi: 10.1016/j.cell.2011.07.030
Rudow, G., O’Brien, R., Savonenko, A. V., Resnick, S. M., Zonderman, A. B., Pletnikova, O., et al. (2008). Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol. 115, 461–470. doi: 10.1007/s00401-008-0352-8
Rumsey, S. C., Kwon, O., Xu, G. W., Burant, C. F., Simpson, I., and Levine, M. (1997). Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 272, 18982–18989. doi: 10.1074/jbc.272.30.18982
Sagar, S. M., Sharp, F. R., and Swanson, R. A. (1987). The regional distribution of glycogen in rat brain fixed by microwave irradiation. Brain Res. 417, 172–174. doi: 10.1016/0006-8993(87)90195-8
Sandyk, R. (1993). The relationship between diabetes mellitus and Parkinson’s disease. Int. J. Neurosci. 69, 125–130. doi: 10.3109/00207459309003322
Santiago, J. F., Carvalho, F. F., Perosa, S. R., Siliano, M. R., Cruz, J. W., Fernandes, M. J., et al. (2006). Effect of glycemic state in rats submitted to status epilepticus during development. Arq. Neuropsiquiatr. 64, 233–239. doi: 10.1590/s0004-282x2006000200012
Santiago, J. A., and Potashkin, J. A. (2013). Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 19, 176–186. doi: 10.1016/j.molmed.2013.01.002
Santiago, J. A., and Potashkin, J. A. (2015). Blood biomarkers associated with cognitive decline in early stage and drug-naive Parkinson’s disease patients. PLoS One 10:e0142582. doi: 10.1371/journal.pone.0142582
Schapira, A. H. V., Chaudhuri, K. R., and Jenner, P. (2017). Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18:509. doi: 10.1038/nrn.2017.91
Schechter, R., Whitmire, J., Wheet, G. S., Beju, D., Jackson, K. W., Harlow, R., et al. (1994). Immunohistochemical and in situ hybridization study of an insulin-like substance in fetal neuron cell cultures. Brain Res. 636, 9–27. doi: 10.1016/0006-8993(94)90170-8
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24, 453–462. doi: 10.1016/j.cub.2014.03.034
Schwab, R. S. (1960). Progression and prognosis in Parkinson’s disease. J. Nerv. Ment. Dis. 130, 556–566. doi: 10.1097/00005053-196006000-00017
Schwab, R. S., Doshay, L. J., Garland, H., Bradshaw, P., Garvey, E., and Crawford, B. (1956). Shift to older age distribution in parkinsonism: a report on 1,000 patients covering the past decade from three centers. Neurology 6, 783–790. doi: 10.1212/wnl.6.11.783
Segura-Aguilar, J., Paris, I., Muñoz, P., Ferrari, E., Zecca, L., and Zucca, F. A. (2014). Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 129, 898–915. doi: 10.1111/jnc.12686
Sekiguchi, K., Kohara, N., Baba, M., Komori, T., Naito, Y., Imai, T., et al. (2019). Aldose reductase inhibitor ranirestat significantly improves nerve conduction velocity in diabetic polyneuropathy: a randomized double-blind placebo-controlled study in Japan. J. Diabetes Investig. 10, 466–474. doi: 10.1111/jdi.12890
Sergi, D., Campbell, F. M., Grant, C., Morris, A. C., Bachmair, E.-M., Koch, C., et al. (2018a). SerpinA3N is a novel hypothalamic gene upregulated by a high-fat diet and leptin in mice. Genes Nutrit. 13:28. doi: 10.1186/s12263-018-0619-1
Sergi, D., Morris, A. C., Kahn, D. E., McLean, F. H., Hay, E. A., Kubitz, P., et al. (2018b). Palmitic acid triggers inflammatory responses in N42 cultured hypothalamic cells partially via ceramide synthesis but not via TLR4. Nutr. Neurosci. doi: 10.1080/1028415X.2018.1501533 [Epub ahead of print].
Sergi, D., Naumovski, N., Heilbronn, L. K., Abeywardena, M., O’Callaghan, N., Lionetti, L., et al. (2019). Mitochondrial (Dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front. Physiol. 10:532. doi: 10.3389/fphys.2019.00532
Sharma, S. K., Chorell, E., Steneberg, P., Vernersson-Lindahl, E., Edlund, H., and Wittung-Stafshede, P. (2015). Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 5:12531. doi: 10.1038/srep12531
Simpson, I. A., Appel, N. M., Hokari, M., Oki, J., Holman, G. D., Maher, F., et al. (1999). Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J. Neurochem. 72, 238–247. doi: 10.1046/j.1471-4159.1999.0720238.x
Simpson, I. A., Carruthers, A., and Vannucci, S. J. (2007). Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow Metab. 27, 1766–1791. doi: 10.1038/sj.jcbfm.9600521
Simpson, I. A., Dwyer, D., Malide, D., Moley, K. H., Travis, A., and Vannucci, S. J. (2008). The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 295, E242–E253. doi: 10.1152/ajpendo.90388.2008
Simpson, I. A., Vannucci, S. J., DeJoseph, M. R., and Hawkins, R. A. (2001). Glucose transporter asymmetries in the bovine blood-brain barrier. J. Biol. Chem. 276, 12725–12729. doi: 10.1074/jbc.M010897200
Singh, R., Barden, A., Mori, T., and Beilin, L. (2001). Advanced glycation end-products: a review. Diabetologia 44, 129–146. doi: 10.1007/s001250051591
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). α-synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Snyder, B., and Cunningham, R. L. (2018). Sex differences in sleep apnea and comorbid neurodegenerative diseases. Steroids 133, 28–33. doi: 10.1016/j.steroids.2017.12.006
Sone, H., Deo, B. K., and Kumagai, A. K. (2000). Enhancement of glucose transport by vascular endothelial growth factor in retinal endothelial cells. Invest. Ophthalmol. Vis. Sci. 41, 1876–1884.
Soong, N. W., Hinton, D. R., Cortopassi, G., and Arnheim, N. (1992). Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 2, 318–323. doi: 10.1038/ng1292-318
Stafstrom, C. E., and Rho, J. M. (2012). The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 3:59. doi: 10.3389/fphar.2012.00059
Stančić, A., Otašević, V., Janković, A., Vučetić, M., Ivanović-Burmazović, I., Filipović, M. R., et al. (2013). Molecular basis of hippocampal energy metabolism in diabetic rats: the effects of SOD mimic. Brain Res. Bull. 99, 27–33. doi: 10.1016/j.brainresbull.2013.09.009
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80. doi: 10.3233/jad-2005-7107
Steenland, K., Hein, M. J., Cassinelli, R. T. II., Prince, M. M., Nilsen, N. B., Whelan, E. A., et al. (2006). Polychlorinated biphenyls and neurodegenerative disease mortality in an occupational cohort. Epidemiology 17, 8–13. doi: 10.1097/01.ede.0000190707.51536.2b
Stern, D., Yan, S. D., Yan, S. F., and Schmidt, A. M. (2002). Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv. Drug Deliv. Rev. 54, 1615–1625. doi: 10.1016/s0169-409x(02)00160-6
Sun, Y., Chang, Y. H., Chen, H. F., Su, Y. H., Su, H. F., and Li, C. Y. (2012). Risk of Parkinson disease onset in patients with diabetes: a 9-year population-based cohort study with age and sex stratifications. Diabetes Care 35, 1047–1049. doi: 10.2337/dc11-1511
Sun, X., Yao, H., Douglas, R. M., Gu, X. Q., Wang, J., and Haddad, G. G. (2010). Insulin/PI3K signaling protects dentate neurons from oxygen-glucose deprivation in organotypic slice cultures. J. Neurochem. 112, 377–388. doi: 10.1111/j.1471-4159.2009.06450.x
Surmeier, D. J. (2007). Calcium, ageing and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 6, 933–938. doi: 10.1016/S1474-4422(07)70246-6
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Takahashi, M., Yamada, T., Tooyama, I., Moroo, I., Kimura, H., Yamamoto, T., et al. (1996). Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 204, 201–204. doi: 10.1016/0304-3940(96)12357-0
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903
Thakran, S., Salimuddin, and Baquer, N. Z. (2003). Oral administration of orthovanadate and Trigonella foenum graecum seed power restore the activities of mitochondrial enzymes in tissues of alloxan-induced diabetic rats. Mol. Cell. Biochem. 247, 45–53. doi: 10.1023/a:1024188600523
Thornalley, P. J. (1988). Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 254, 751–755. doi: 10.1042/bj2540751
Thurston, J. H., McDougal, D. B. Jr., Hauhart, R. E., and Schulz, D. W. (1995). Effects of acute, subacute and chronic diabetes on carbohydrate and energy metabolism in rat sciatic nerve. Diabetes 44, 190–195. doi: 10.2337/diab.44.2.190
Tomlinson, D. R., and Gardiner, N. J. (2008). Glucose neurotoxicity. Nat. Rev. Neurosci. 9, 36–45. doi: 10.1038/nrn2294
Tups, A., Benzler, J., Sergi, D., Ladyman, S. R., and Williams, L. M. (2017). Central regulation of glucose homeostasis. Compr. Physiol. 7, 741–764. doi: 10.1002/cphy.c160015
Uchiki, T., Weikel, K. A., Jiao, W., Shang, F., Caceres, A., Pawlak, D., et al. (2012). Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging and age-related disease (in nondiabetics). Aging Cell 11, 1–13. doi: 10.1111/j.1474-9726.2011.00752.x
UK Prospective Diabetes Study (UKPDS) Group. (1998). Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 352, 854–865. doi: 10.1016/s0140-6736(98)07037-8
Ulrich, P., and Cerami, A. (2001). Protein glycation, diabetes and aging. Recent Prog. Horm. Res. 56, 1–21. doi: 10.1210/rp.56.1.1
van Hall, G., Strømstad, M., Rasmussen, P., Jans, O., Zaar, M., Gam, C., et al. (2009). Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab. 29, 1121–1129. doi: 10.1038/jcbfm.2009.35
Vander Jagt, D. L., Kolb, N. S., Vander Jagt, T. J., Chino, J., Martinez, F. J., Hunsaker, L. A., et al. (1995). Substrate specificity of human aldose reductase: identification of 4-hydroxynonenal as an endogenous substrate. Biochim. Biophys. Acta 1249, 117–126. doi: 10.1016/0167-4838(95)00021-l
Vicente Miranda, H., El-Agnaf, O. M., and Outeiro, T. F. (2016). Glycation in Parkinson’s disease and Alzheimer’s disease. Mov. Disord. 31, 782–790. doi: 10.1002/mds.26566
Vidyadhara, D. J., Yarreiphang, H., Abhilash, P. L., Raju, T. R., and Alladi, P. A. (2016). Differential expression of calbindin in nigral dopaminergic neurons in two mice strains with differential susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Chem. Neuroanat. 76, 82–89. doi: 10.1016/j.jchemneu.2016.01.001
Vilensky, J. A., Gilman, S., and McCall, S. (2010). A historical analysis of the relationship between encephalitis lethargica and postencephalitic parkinsonism: a complex rather than a direct relationship. Mov. Disord. 25, 1116–1123. doi: 10.1002/mds.22908
Wahlqvist, M. L., Lee, M.-S., Hsu, C.-C., Chaung, S.-Y., Lee, J.-T., and Tsai, H.-N. (2012). Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 18, 753–758. doi: 10.1016/j.parkreldis.2012.03.010
Warner, T. T., and Schapira, A. H. (2003). Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 53, S16–S23; discussion S23–S25. doi: 10.1002/ana.10487
Watanabe, H., and Passonneau, J. V. (1973). Factors affecting the turnover of cerebral glycogen and limit dextrin in rive. J. Neurochem. 20, 1543–1554. doi: 10.1111/j.1471-4159.1973.tb00272.x
Weisskopf, M. G., Knekt, P., O’Reilly, E. J., Lyytinen, J., Reunanen, A., Laden, F., et al. (2012). Polychlorinated biphenyls in prospectively collected serum and Parkinson’s disease risk. Mov. Disord. 27, 1659–1665. doi: 10.1002/mds.25217
Wirdefeldt, K., Adami, H. O., Cole, P., Trichopoulos, D., and Mandel, J. (2011). Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur. J. Epidemiol. 26, S1–S58. doi: 10.1007/s10654-011-9581-6
Woods, S. C., and Porte, D. Jr. (1977). Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am. J. Physiol. 233, E331–E334. doi: 10.1152/ajpendo.1977.233.4.E331
World Health Organization. (2016). Global report on diabetes. Available online at: http://apps.who.int/iris/bitstream/10665/204871/1/9789241565257_eng.pdf?ua=1. Accessed October 27, 2019.
Xu, Y., Liu, C., Chen, S., Ye, Y., Guo, M., Ren, Q., et al. (2014). Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal. 26, 1680–1689. doi: 10.1016/j.cellsig.2014.04.009
Xu, Q., Park, Y., Huang, X., Hollenbeck, A., Blair, A., Schatzkin, A., et al. (2011). Diabetes and risk of Parkinson’s disease. Diabetes Care 34, 910–915. doi: 10.2337/dc10-1922
Yamada, T., McGeer, P. L., Baimbridge, K. G., and McGeer, E. G. (1990). Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 526, 303–307. doi: 10.1016/0006-8993(90)91236-a
Yang, X., and Cheng, B. (2010). Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 42, 145–153. doi: 10.1007/s12031-010-9336-y
Yoon, J. W., and Jun, H. S. (2001). Cellular and molecular pathogenic mechanisms of insulin-dependent diabetes mellitus. Ann. N Y Acad. Sci. 928, 200–211. doi: 10.1111/j.1749-6632.2001.tb05650.x
Youdim, M. B., Ben-Shachar, D., and Riederer, P. (1989). Is Parkinson’s disease a progressive siderosis of substantia nigra resulting in iron and melanin induced neurodegeneration? Acta Neurol. Scand. Suppl. 126, 47–54. doi: 10.1111/j.1600-0404.1989.tb01782.x
Young, W. S. III. (1986). Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides 8, 93–97. doi: 10.1016/0143-4179(86)90035-1
Zaccardi, F., Webb, D. R., Yates, T., and Davies, M. J. (2016). Pathophysiology of type 1 and type 2 diabetes mellitus: a 90-year perspective. Postgrad. Med. J. 92, 63–69. doi: 10.1136/postgradmedj-2015-133281
Zahniser, N. R., Goens, M. B., Hanaway, P. J., and Vinych, J. V. (1984). Characterization and regulation of insulin receptors in rat brain. J. Neurochem. 42, 1354–1362. doi: 10.1111/j.1471-4159.1984.tb02795.x
Zecca, L., Tampellini, D., Gerlach, M., Riederer, P., Fariello, R. G., and Sulzer, D. (2001). Substantia nigra neuromelanin: structure, synthesis and molecular behaviour. Mol. Pathol. 54, 414–418. doi: 10.1136/mp.54.6.414
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., et al. (2010). PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2:52ra73. doi: 10.1126/scitranslmed.3001059
Zheng, H., Zheng, Y., Zhao, L., Chen, M., Bai, G., Hu, Y., et al. (2017). Cognitive decline in type 2 diabetic db/db mice may be associated with brain region-specific metabolic disorders. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 266–273. doi: 10.1016/j.bbadis.2016.11.003
Ziegler, D., Buchholz, S., Sohr, C., Nourooz-Zadeh, J., and Roden, M. (2015). Oxidative stress predicts progression of peripheral and cardiac autonomic nerve dysfunction over 6 years in diabetic patients. Acta Diabetol. 52, 65–72. doi: 10.1007/s00592-014-0601-3
Keywords: dopamine, glucotoxicity, hyperglycemia, mitochondrial dysfunction, nigrostriatal pathway, oxidative stress
Citation: Sergi D, Renaud J, Simola N and Martinoli M-G (2019) Diabetes, a Contemporary Risk for Parkinson’s Disease: Epidemiological and Cellular Evidences. Front. Aging Neurosci. 11:302. doi: 10.3389/fnagi.2019.00302
Received: 10 September 2019; Accepted: 22 October 2019;
Published: 08 November 2019.
Edited by:
Ralf J. Braun, Danube Private University, AustriaReviewed by:
Christian Hölscher, Lancaster University, United KingdomMavis Akosua Asantewaa Tenkorang-Impraim, University of Nebraska Medical Center, United States
Rocío Martínez De Pablos, University of Seville, Spain
Copyright © 2019 Sergi, Renaud, Simola and Martinoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria-Grazia Martinoli, bWFyaWEtZ3JhemlhLm1hcnRpbm9saUB1cXRyLmNh
† These authors have contributed equally to this work