 Li Zhong1*
Li Zhong1* Xiao-Fen Chen1,2*
Xiao-Fen Chen1,2*- 1Fujian Provincial Key Laboratory of Neurodegenerative Disease and Aging Research, Institute of Neuroscience, Medical College, Xiamen University, Xiamen, China
- 2Shenzhen Research Institute of Xiamen University, Shenzhen, China
Alzheimer’s disease (AD) is the most common form of dementia characterized by the deposition of extracellular amyloid-β (Aβ)-containing plaques, the formation of intraneuronal neurofibrillary tangles as well as neuroinflammatory changes. As the key player in the brain innate immune system, microglia has now taken a center stage in AD research. A large number of AD risk loci identified by genome-wide association studies are located in or near the genes highly expressed in microglia. Among them, the triggering receptor expressed on myeloid cells 2 (TREM2) has drawn much attention. A rare variant in TREM2 increases AD risk with an odds ratio comparable to the strongest genetic risk factor apolipoprotein ε4 allele. In the past 6 years, extensive studies have dissected the mechanisms by which TREM2 and its variants modulate microglial functions impacting amyloid and tau pathologies in both animal models and human studies. In addition to the full-length TREM2, research on the soluble form of TREM2 (sTREM2) has facilitated the translation of preclinical findings on TREM2. In this review, we summarize our current understanding of the biology and pathobiology of sTREM2 including its origin, its emergence as a disease biomarker, and its potential neuroprotective functions. These aspects are important for understanding the involvement of sTREM2 in AD pathogenesis and may provide novel insights into applying sTREM2 for AD diagnosis and therapy.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly, typically presenting with a slow but progressive behavioral and cognitive impairment. Key histopathological hallmarks of AD include the deposition of extracellular neurotoxic plaques primarily composed of Aβ, and intracellular neurofibrillary tangles resulting from the aggregation of hyperphosphorylated tau (Holtzman et al., 2011). In addition to protein aggregation, neuroinflammatory changes have emerged as the third core feature of AD pathologies (Heneka et al., 2015; Karch and Goate, 2015). In the central nervous system (CNS), microglia are the resident immune cells that play central roles in neuroinflammation. Recent genome-wide association studies (GWAS) have uncovered a large number of mutations in microglial genes that are highly associated with increased AD risk (Gjoneska et al., 2015; Malik et al., 2015; Efthymiou and Goate, 2017). Hence, it has been proposed that microglia play a much larger role in AD pathogenesis than previously thought. Studies on the new genetic risk factors have aided the rapid progress towards unraveling the multiple facets of microglia under both healthy and diseased conditions (Colonna and Butovsky, 2017; Butovsky and Weiner, 2018; Hammond et al., 2018; Hickman et al., 2018; Heneka, 2019).
During development, microglia are actively involved in eliminating the apoptotic neurons via phagocytosis and the refinement of neural circuits by synaptic pruning (Wakselman et al., 2008; Paolicelli et al., 2011; Schafer et al., 2012; Cunningham et al., 2013). In the healthy adult brain, microglial processes are highly motile and constantly survey the surrounding environment in the parenchyma to maintain tissue homeostasis (Davalos et al., 2005; Nimmerjahn et al., 2005). In response to harmful stimuli such as Aβ aggregation, microglia rapidly transform from ramified to amoeboid morphology, facilitating the phagocytosis and clearance of Aβ aggregates (Itagaki et al., 1989; Bolmont et al., 2008). They also proliferate and migrate to the vicinity of plaques, forming a protective barrier around amyloid deposits to reduce the neurotoxicity of amyloid fibrils (Condello et al., 2015; Zhao et al., 2017). However, there is also abundant evidence that microglia have harmful actions in AD. Once activated, microglia can mediate the engulfment of neuronal synapses likely via a complement-dependent mechanism. They can also exacerbate tau pathology and secrete detrimental inflammatory factors that can directly or indirectly injure neurons (Hansen et al., 2018).
Hence, microglia may act as a double-edged sword being either protective or detrimental depending on the disease stage. Future efforts in profiling the microglial transcriptome particularly at the single-cell level and correlating such changes with disease progression are necessary to help us better understand the role of microglia in AD pathology (Keren-Shaul et al., 2017; Rangaraju et al., 2018; Hammond et al., 2019). Furthermore, expanding the studies from mouse models to human patients by using human microglia isolated from fresh postmortem brain tissues or human microglia-like cells differentiated from human induced pluripotent stem cells will significantly and greatly increase the success in translational research (Abud et al., 2017; Mizee et al., 2017; McQuade et al., 2018).
Among the AD risk-associated microglial genes, a special interest has been directed at the triggering receptor expressed on myeloid cells 2 (TREM2) since the rare R47H variant of TREM2 increases AD risk almost three-fold (Guerreiro et al., 2013; Jonsson et al., 2013). Thus, the effect size of TREM2 R47H is comparable to that for the ε4 allele of the gene encoding apolipoprotein E (apoE), the strongest genetic risk factor for sporadic AD identified 30 years earlier. As a receptor expressed on microglial cell surface, the ectodomain of TREM2 binds to an array of molecules that are important for AD, including the anionic and zwitterionic lipids, lipoproteins and apolipoproteins, oligomeric Aβ and galectin-3 as reported recently (Atagi et al., 2015; Bailey et al., 2015; Wang et al., 2015; Yeh et al., 2016; Lessard et al., 2018; Zhao et al., 2018; Zhong et al., 2018; Boza-Serrano et al., 2019). While the identities of these ligands remain uncertain, several functions of TREM2 have been well characterized in microglia. Recent studies have suggested that TREM2 impacts a multitude of microglial functions including activation, inflammation, phagocytosis, proliferation, survival and metabolism (Kleinberger et al., 2014, 2017; Cantoni et al., 2015; Wang et al., 2015; Zhong et al., 2015; Yeh et al., 2016; Ulland et al., 2017; Zheng et al., 2017). In the context of AD, TREM2 regulates the recruitment of microglia to the vicinity of amyloid plaque and limits amyloid or plaque tau seeding (Yuan et al., 2016; Cheng-Hathaway et al., 2018; Leyns et al., 2019; Parhizkar et al., 2019). TREM2 is also essential for microglia to eliminate supernumerary synapses in the developing brain (Filipello et al., 2018). Other studies have evaluated the impacts of Trem2 deficiency, TREM2 mutation, or human TREM2 expression in amyloid or tau mouse models (Ulrich et al., 2014; Jay et al., 2015; Wang et al., 2015; Bemiller et al., 2017; Leyns et al., 2017, 2019; Cheng-Hathaway et al., 2018; Lee et al., 2018; Sayed et al., 2018; Song et al., 2018). Surprisingly, these studies have produced conflicting results, with reports that AD-related pathology could be either increased or decreased in the presence of TREM2. It was proposed later that TREM2 modulates AD pathology dependent on disease progression (Jay et al., 2017; Shi and Holtzman, 2018). Future studies in comprehensive transgenic and knockout animal models are necessary to unravel the complex pathogenic roles for TREM2.
In addition to the full-length TREM2, research on the soluble form of TREM2 (sTREM2) has facilitated the translation of preclinical findings on TREM2. sTREM2 was produced from either the proteolytic cleavage of the membrane-anchored TREM2 receptor or the alternative splicing of TREM2 lacking the transmembrane domain (Schmid et al., 2002; Wunderlich et al., 2013). The levels of sTREM2 in the cerebrospinal fluid (CSF) have been quantified and found to be elevated in AD (Heslegrave et al., 2016; Piccio et al., 2016; Suárez-Calvet et al., 2016a). In cross-sectional studies, the levels of sTREM2 change dynamically during the progression of AD, peaking at the early symptomatic stages of the disease (Suárez-Calvet et al., 2016b). Importantly, the CSF concentrations of sTREM2 positively correlate with the levels of total tau and phosphorylated-tau (p-tau) in the CSF. These observations suggest that sTREM2 may play an important role in the development of AD pathology and neurodegeneration.
The presence of sTREM2 was once regarded as the loss-of-function consequences of full-length TREM2 on the cell surface. It could be either an inactive by-product from full-length TREM2 shedding or a decoy receptor opposing full-length TREM2 signaling. Nevertheless, emerging evidence from our study and others suggests that sTREM2 is actively involved in regulating microglial dynamics and AD-associated pathology. In this review article, we summarize the exciting research progress in the knowledge of sTREM2 including its origin, its biological and pathobiological functions. We further discuss the potential application of sTREM2 in AD diagnosis and therapy.
The Origin of Soluble TREM2
The existence of sTREM2 at the protein level was first reported in 2008 by immunoblotting of the precipitates from either human CSF or cultured dendritic cells supernatant fluid (Piccio et al., 2008). The sTREM2 protein was found to carry different levels of glycosylation and its size was reduced to 20 kDa after O- and N-deglycosylation. Interestingly, a TREM2 transcript that lacks the transmembrane domain was described in human and mouse myeloid cells (Figure 1), leading to the hypothesis that sTREM2 originates from alternative splicing of TREM2 (Schmid et al., 2002; Begum et al., 2004; Melchior et al., 2006). A recent study using bulk RNA-Seq data in the human brain tissue further demonstrates that around 25% of the sTREM2 protein originates from the TREM2 transcript that lacks the transmembrane domain (Deming et al., 2019). A strong and significant correlation of the sTREM2 band detected by Western-blots was found with the transcript that codifies for the soluble form of TREM2 (R2 = 0.42, p < 0.05). However, the predicted molecular weight of this transcript is ~27 kDa, which is larger than the 20 kDa deglycosylated sTREM2 protein detected in the CSF. Therefore, alternative splicing may not be the major source of sTREM2 protein.
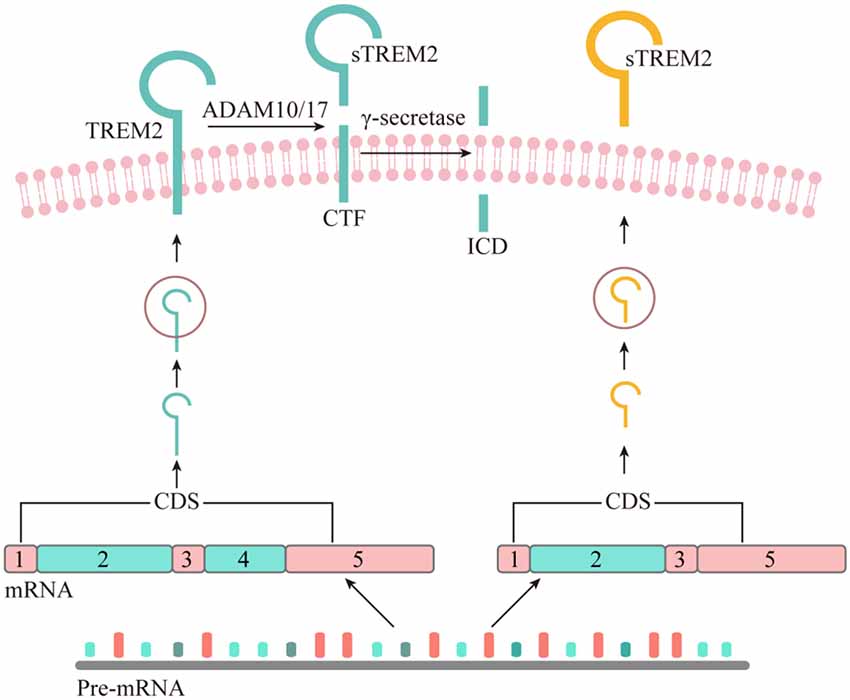
Figure 1. The origin of soluble form of TREM2. Although all the potential physiological sources of sTREM2 remain to be determined, recent studies suggest that sTREM2 can be generated by either alternative splicing or proteolytic cleavage of the full-length TREM2 protein. The TREM2 gene has three different alternative transcripts. The canonical and also the longest TREM2 transcript consists of five exons, with exon 4 encoding a transmembrane domain. This isoform is anchored to the cell membrane and shed by ADAM10 and/or ADAM17, leading to the production of a soluble TREM2 and a C-terminal fragment (CTF). The TREM2-CTF is further cleaved by γ-secretase to generate a TREM2 intracellular domain (ICD). It remains to be determined whether the second-longest transcript of TREM2, which lacks exon 5, can generate sTREM2 via a sequential proteolytic processing. The shortest TREM2 transcript encodes a soluble form of TREM2 due to the lack of exon 4 which encodes the transmembrane domain of the receptor.
As a single-pass type I transmembrane protein, TREM2 undergoes sequential proteolytic processing similar to the well-characterized amyloid precursor protein (APP; Wunderlich et al., 2013). The initial cleavage of TREM2 by a shedding enzyme results in the liberation of sTREM2 into the extracellular space. It was later reported that a disintegrin and metalloproteinase domain-containing protein (ADAM) family, including ADAM10 and ADAM17, are the proteases involved in the shedding of TREM2 ectodomain (Kleinberger et al., 2014; Feuerbach et al., 2017; Schlepckow et al., 2017; Thornton et al., 2017). However, which enzyme plays a dominant role remains controversial. In 2017, two independent groups reported simultaneously that sTREM2 is generated through the ADAM10/ADAM17 cleavage at the H157-S158 peptide bond (Schlepckow et al., 2017; Thornton et al., 2017). Interestingly, a late-onset AD-associated H157Y mutation significantly increases the shedding of full-length TREM2 in HEK293 cells, resulting in elevated levels of sTREM2 in the conditioned media.
The Genetic Modifiers of Soluble TREM2 Levels
The levels of sTREM2 in the CSF were quantified and found to be elevated in patients with AD and other neurological disorders with inflammatory etiologies (Piccio et al., 2008; Heslegrave et al., 2016). However, very little is known with regards to the mechanisms underlying sTREM2 generation under physiological and pathological conditions. Given that TREM2 mutations can affect protein production, maturation and cleavage, the amounts of sTREM2 in the CSF are likely influenced by TREM2 genetic status. Several studies have measured the CSF levels of sTREM2 in individuals carrying different TREM2 genetic variants and found these variants brought varying effects on sTREM2 levels in the CSF (Piccio et al., 2016; Deming et al., 2019). Carriers of the NHD-associated variants Q33X and T66M presented significantly lower CSF sTREM2 levels than controls. Similarly, the T96K/L211P/W191X carriers also had lower sTREM2 levels compared to controls. In contrast, the CSF sTREM2 levels were significantly higher in R47H carriers compared to non-carriers. Therefore, these variants may alter the sTREM2 level and increase the disease risk through distinct pathogenic mechanisms.
To identify more genetic modifiers of CSF sTREM2 levels, Deming et al. (2019) performed a GWAS and found the membrane-spanning 4-domains superfamily A (MS4A) gene cluster as a key modulator of sTREM2 concentration in the CSF. Variants in the MS4A gene region have been previously linked to the risk of developing LOAD (Hollingworth et al., 2011; Naj et al., 2011; Lambert et al., 2013). Among them, the rs1582763 which increases the expression of both MS4A4A and MS4A6A genes in the brain is associated with elevated sTREM2 levels in the CSF, reduced AD risk and delayed age at onset. In contrast, the rs6591561 which creates a missense variant within the MS4A4A gene (M159V) is associated with reduced CSF sTREM2 levels, increased AD risk and accelerated age at onset. The study further provided functional evidence that CSF sTREM2 levels can be modified via modulating MS4A4A expression or targeting MS4A4A with a specific antibody. However, the study only focused on the common variants with a minor allele frequency larger than 0.02. Therefore, additional studies in a larger sample size will be necessary to fully identify the genetic modifiers of CSF sTREM2 levels. Moreover, future studies are necessary to uncover the molecular mechanisms by which the MS4A gene cluster modulates sTREM2 level. The MS4A transmembrane proteins are thought to play a role in intracellular protein trafficking (Cruse et al., 2015). It would be interesting to investigate whether the MS4A family regulates the trafficking of key proteins involved in sTREM2 generation, including the full-length TREM2 and ADAM10/17.
Soluble TREM2 as a Potential Disease Biomarker
The first attempt to quantify the levels of sTREM2 in the CSF was carried in subjects with multiple sclerosis (MS) and other CNS inflammatory diseases (Piccio et al., 2008). The amounts of sTREM2 were found to be significantly elevated in these subjects as compared to non-inflammatory conditions. In accordance with the finding, another study also observed higher CSF levels of sTREM2 in MS subjects than healthy controls, regardless of their clinical stages (Öhrfelt et al., 2016). Interestingly, after treatment with an anti-inflammatory drug natalizumab, there was a clinical improvement and simultaneously the levels of sTREM2 were decreased to levels comparable to the controls. Therefore, the CSF sTREM2 levels might serve as a biomarker for inflammatory diseases including MS, and it could be used to monitor the effects of anti-inflammatory drugs or the efficacy of therapeutics in the clinic.
Since TREM2 genetic variants are linked to AD, the CSF sTREM2 levels in AD cohorts have been extensively measured and correlated to the pathological changes in AD. By using either mass spectrometry or ELISA assay to quantify the amounts of sTREM2, two independent studies both reported significantly higher levels of sTREM2 in the CSF of mild AD subjects than healthy controls (Heslegrave et al., 2016; Piccio et al., 2016). Importantly, they showed that the CSF sTREM2 levels were positively correlated with that of total tau and p-tau. However, no significant correlation between sTREM2 and Aβ42 levels in the CSF was observed, suggesting that sTREM2 might play functions in the pathological processes occurring after Aβ accumulation. In a cross-sectional study, the CSF levels of sTREM2 were traced during the clinical course of AD, including the preclinical AD, mild cognitive impairment (MCI) due to AD (MCI-AD), and AD dementia (Suárez-Calvet et al., 2016b). Interestingly, the CSF levels of sTREM2 increase in a disease stage-dependent manner, reaching a peak in the early symptomatic phase of AD. The same group further investigated the temporal sequence of changes in CSF sTREM2 levels and markers for amyloid deposition and neurodegeneration as well as cognitive performance (Suárez-Calvet et al., 2016a). They found that the CSF sTREM2 levels were increased before the expected onset of symptoms but after the appearance of amyloidosis and neuronal injury. Thus, the CSF levels of sTREM2 have emerged as a valuable yet dynamic biomarker of the disease progression in AD.
The CSF AD biomarker tests are highly invasive, making them unsuitable for use in the primary care setting. Hence, the sTREM2 levels were also analyzed in the peripheral blood from AD patients as well as healthy subjects. A study has reported higher levels of sTREM2 in the CSF of AD patients, but no significant difference in the plasma levels of sTREM2 between AD cases and healthy controls (Piccio et al., 2016). Similarly, a recent meta-analysis of case-control and AD cohort studies revealed a prominent elevation in the CSF sTREM2 levels in AD groups compared with controls, but no significant difference in the plasma sTREM2 levels between the two groups (Liu et al., 2018). Based on these observations, it appears that only CSF but not plasma levels of sTREM2 could be a promising AD biomarker.
The Biological Functions of Soluble TREM2
Initially, sTREM2 was postulated to block the function of full-length TREM2 by competing with the ligand binding, in a way similar to the soluble version of another TREM family member, TREM1. Nevertheless, accumulating evidence suggests that sTREM2 possesses important biological and pathobiological functions rather than acting as a decoy receptor opposing full-length TREM2 signaling. One study presented the first evidence that sTREM2 efficiently prevents macrophage apoptosis when cultured under low concentrations of colony stimulating factor 1 (Wu et al., 2015). The study further suggested that sTREM2 promotes cell survival by acting as an intracellular messenger to trigger the activation of extracellular signal-regulated kinases 1/2 and mitogen-activated protein kinase 14. Our studies in microglia also confirmed a protective role of sTREM2 against cellular apoptosis (Zhong et al., 2017). However, sTREM2 forestalled apoptosis via activation of the AKT–glycogen synthase kinase 3β–β-catenin pathway in microglia, which is distinct from that in macrophage. Furthermore, sTREM2 induced inflammatory cytokine production in microglia by activating the nuclear factor-κB, accompanied by changes in morphology indicative of microglial activation. These functions of sTREM2 were recapitulated in Trem2-deficient microglial cells or mice model, suggesting that sTREM2 acts through a mechanism that is independent of the endogenous, full-length TREM2. Interestingly, sTREM2 derived from the AD-associated variants, R47H and R62H (sTREM2-R47H and sTREM2-R62H) are less potent in both suppressing apoptosis and triggering inflammatory responses in microglia. Thus, our results may constitute previously unknown molecular mechanisms linking TREM2 variants with increased risk of AD.
The Pathobiological Functions of Soluble TREM2
Since the CSF levels of sTREM2 are intimately associated with AD progression, it has been proposed that sTREM2 might modulate AD pathology. Accumulating evidence from the association analysis between CSF sTREM2 levels and regional brain volumes in AD cohorts suggests that sTREM2 functions are likely neuroprotective. One study reported that higher CSF sTREM2 levels were correlated with increased gray matter volume in MCI patients, when controlled for age, sex, and p-tau-associated brain atrophy (Gispert et al., 2016). A recent study also found that subjects with higher concentrations of CSF sTREM2 showed slower rate of decline in hippocampal volume in early-stage AD (Ewers et al., 2019). Intriguingly, an association between higher CSF sTREM2 concentrations and slower memory decline was observed in individuals with MCI or AD dementia. Furthermore, a higher ratio of sTREM2 to p-tau181 levels in the CSF predicted slower rate of clinical progression. These findings suggest that higher CSF sTREM2 levels are associated with attenuated cognitive and clinical decline in AD.
It was reported that sTREM2 bound to plaques and neurons when the common variant of human TREM2 was expressed on a bacterial artificial chromosome in Trem2 deficient 5xFAD mice, while no such findings were observed in mice expressing the R47H variant (Song et al., 2018). Since the R47H mutation constitutes a strong genetic risk factor for AD, the sTREM2 binding to neuron or plaque might have beneficial functions in AD brain. Our recent study in transgenic mouse model of AD provided direct insights into the pathobiological roles of sTREM2 (Zhong et al., 2019). We found that sTREM2 enhances a multitude of microglial functions, including survival, proliferation, migration, clustering in the vicinity of amyloid plaques, and the uptake and degradation of Aβ. Importantly, sTREM2 reduces amyloid plaque load and rescues the functional deficits of spatial memory and long-term potentiation (Figure 2). Depletion of microglia with an inhibitor of colony stimulating factor-1 receptor, PLX3397, abolishes the neuroprotective effects of sTREM2, supporting an indispensable role of microglia in mediating sTREM2 function. Hence, our study demonstrates a protective role of sTREM2 against amyloid pathology and related toxicity likely through modulating microglial activity and raises the interesting possibility that increasing sTREM2 can be explored for AD therapy.
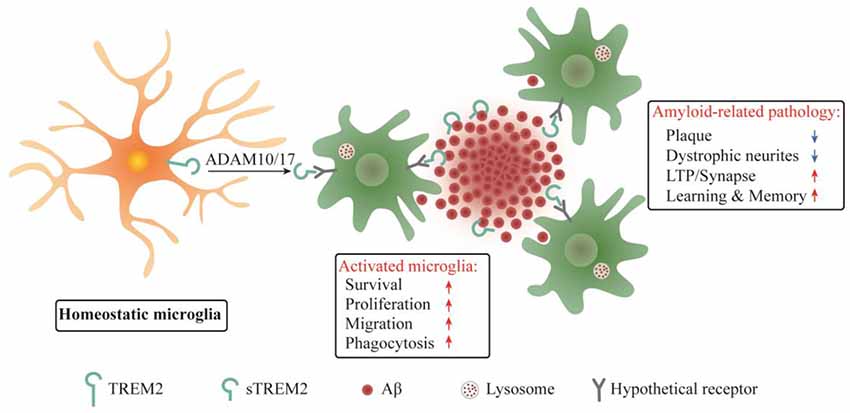
Figure 2. Schematic summary of sTREM2 functions in microglia and amyloid-related pathology. sTREM2 is released into the extracellular space via ADAM10 and/or ADAM17 shedding of the membrane-bound TREM2. Upon binding to a hypothetical microglial receptor, sTREM2 converts microglia from a homeostatic to an activated state. It enhances an array of microglial functions, including survival, proliferation, migration, clustering in the vicinity of amyloid plaques, and the uptake and degradation of Aβ. The consequences are the amelioration of amyloid-related pathologies.
Future Perspectives
The majority of AD therapies currently under investigation target Aβ accumulation by modulating the activity of key secretases involved in APP processing. Alternatively, antibodies were developed to prevent Aβ aggregation and/or to promote Aβ plaque clearance (Cao et al., 2018). Microglia and related neuroinflammation have been increasingly shown to play key roles in AD pathogenesis. In particular, studies on the functions of TREM2 and its variants in microglia and AD pathogenesis have prompted the design of therapeutic strategies targeting TREM2. While the full-length TREM2 has been suggested to play key roles in AD pathology, results have been conflicting regarding whether TREM2 is beneficial or harmful (Gratuze et al., 2018). In contrast, evidence accumulated from both preclinical and clinical studies suggests a protective effect of sTREM2 against AD pathology. However, the precise mechanisms mediating the functions of sTREM2 remain largely unknown. For example, the microglial surface receptor(s) for sTREM2 and the downstream signaling events remain to be determined. The molecular pathways underlying sTREM2 production also need to be fully elucidated. Since sTREM2 levels vary among different stages of AD, it is important to investigate the impacts of sTREM2 on amyloid and tau pathologies during AD progression. Further studies will be necessary to address these questions and to provide critical mechanistic insights to guide the application of sTREM2 for AD diagnosis and therapy.
Author Contributions
X-FC and LZ reviewed the literature and drafted the manuscript.
Funding
Research by the authors was supported by grants from the National Natural Science Foundation of China 81922021 (to X-FC), and 81701079 (to LZ), a grant from the Natural Science Foundation of Guangdong Province 2016A030306005 (to X-FC), a grant from the Natural Science Foundation of Fujian Province 2019J01014 (to X-FC), a grant from the Shenzhen Basic Research Program JCYJ20170818140904167 (to X-FC), a grant from the Fundamental Research Fund for the Central Universities 20720180055 (to X-FC), a grant from the Alzheimer’s Association AARG-18-56635 (to X-FC), and a grant from Open Research Fund of State Key Laboratory of Cellular Stress Biology, Xiamen University (to LZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abud, E. M., Ramirez, R. N., Martinez, E. S., Healy, L. M., Nguyen, C. H. H., Newman, S. A., et al. (2017). iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278.e279–293.e279. doi: 10.1016/j.neuron.2017.03.042
Atagi, Y., Liu, C. C., Painter, M. M., Chen, X. F., Verbeeck, C., Zheng, H., et al. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050. doi: 10.1074/jbc.M115.679043
Bailey, C. C., DeVaux, L. B., and Farzan, M. (2015). The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 290, 26033–26042. doi: 10.1074/jbc.m115.677286
Begum, N. A., Ishii, K., Kurita-Taniguchi, M., Tanabe, M., Kobayashi, M., Moriwaki, Y., et al. (2004). Mycobacterium bovis BCG cell wall-specific differentially expressed genes identified by differential display and cDNA subtraction in human macrophages. Infect. Immun. 72, 937–948. doi: 10.1128/iai.72.2.937-948.2004
Bemiller, S. M., McCray, T. J., Allan, K., Formica, S. V., Xu, G., Wilson, G., et al. (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 12:74. doi: 10.1186/s13024-017-0216-6
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C. A., Klunk, W. E., et al. (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292. doi: 10.1523/jneurosci.4814-07.2008
Boza-Serrano, A., Ruiz, R., Sanchez-Varo, R., Garcia-Revilla, J., Yang, Y., Jimenez-Ferrer, I., et al. (2019). Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 138, 251–273. doi: 10.1007/s00401-019-02013-z
Butovsky, O., and Weiner, H. L. (2018). Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 19, 622–635. doi: 10.1038/s41583-018-0057-5
Cantoni, C., Bollman, B., Licastro, D., Xie, M., Mikesell, R., Schmidt, R., et al. (2015). TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 129, 429–447. doi: 10.1007/s00401-015-1388-1
Cao, J., Hou, J., Ping, J., and Cai, D. (2018). Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol. Neurodegener. 13:64. doi: 10.1186/s13024-018-0299-8
Cheng-Hathaway, P. J., Reed-Geaghan, E. G., Jay, T. R., Casali, B. T., Bemiller, S. M., Puntambekar, S. S., et al. (2018). The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 13:29. doi: 10.1186/s13024-018-0262-8
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 6:6176. doi: 10.1038/ncomms7176
Cruse, G., Beaven, M. A., Music, S. C., Bradding, P., Gilfillan, A. M., and Metcalfe, D. D. (2015). The CD20 homologue MS4A4 directs trafficking of KIT toward clathrin-independent endocytosis pathways and thus regulates receptor signaling and recycling. Mol. Biol. Cell 26, 1711–1727. doi: 10.1091/mbc.e14-07-1221
Cunningham, C. L., Martínez-Cerdeño, V., and Noctor, S. C. (2013). Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 33, 4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Deming, Y., Filipello, F., Cignarella, F., Cantoni, C., Hsu, S., Mikesell, R., et al. (2019). The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 11:eaau2291. doi: 10.1126/scitranslmed.aau2291
Efthymiou, A. G., and Goate, A. M. (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Ewers, M., Franzmeier, N., Suárez-Calvet, M., Morenas-Rodriguez, E., Caballero, M. A. A., Kleinberger, G., et al. (2019). Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 11:eaav6221. doi: 10.1126/scitranslmed.aav6221
Feuerbach, D., Schindler, P., Barske, C., Joller, S., Beng-Louka, E., Worringer, K. A., et al. (2017). ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci. Lett. 660, 109–114. doi: 10.1016/j.neulet.2017.09.034
Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48, 979.e978–991.e978. doi: 10.1016/j.immuni.2018.04.016
Gispert, J. D., Suárez-Calvet, M., Monté, G. C., Tucholka, A., Falcon, C., Rojas, S., et al. (2016). Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement. 12, 1259–1272. doi: 10.1016/j.jalz.2016.06.005
Gjoneska, E., Pfenning, A. R., Mathys, H., Quon, G., Kundaje, A., Tsai, L. H., et al. (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. doi: 10.1038/nature14252
Gratuze, M., Leyns, C. E. G., and Holtzman, D. M. (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13:66. doi: 10.1186/s13024-018-0298-9
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Hammond, T. R., Dufort, C., Dissing-Olesen, L., Giera, S., Young, A., Wysoker, A., et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253.e256–271.e256. doi: 10.1016/j.immuni.2018.11.004
Hammond, T. R., Robinton, D., and Stevens, B. (2018). Microglia and the brain: complementary partners in development and disease. Annu. Rev. Cell Dev. Biol. 34, 523–544. doi: 10.1146/annurev-cellbio-100616-060509
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Heneka, M. T. (2019). Microglia take centre stage in neurodegenerative disease. Nat. Rev. Immunol. 19, 79–80. doi: 10.1038/s41577-018-0112-5
Heneka, M. T., Golenbock, D. T., and Latz, E. (2015). Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236. doi: 10.1038/ni.3102
Heslegrave, A., Heywood, W., Paterson, R., Magdalinou, N., Svensson, J., Johansson, P., et al. (2016). Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 11:3. doi: 10.1186/s13024-016-0071-x
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3:77sr71. doi: 10.1126/scitranslmed.3002369
Itagaki, S., McGeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182. doi: 10.1016/0165-5728(89)90115-x
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jay, T. R., von Saucken, V. E., and Landreth, G. E. (2017). TREM2 in neurodegenerative diseases. Mol. Neurodegener. 12:56. doi: 10.1186/s13024-017-0197-5
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Karch, C. M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e1217–1290.e1217. doi: 10.1016/j.cell.2017.05.018
Kleinberger, G., Brendel, M., Mracsko, E., Wefers, B., Groeneweg, L., Xiang, X., et al. (2017). The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion and glucose metabolism. EMBO J. 36, 1837–1853. doi: 10.15252/embj.201796516
Kleinberger, G., Yamanishi, Y., Suarez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6:243ra286. doi: 10.1126/scitranslmed.3009093
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L. L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032.e1035–1048.e1035. doi: 10.1016/j.neuron.2018.02.002
Lessard, C. B., Malnik, S. L., Zhou, Y., Ladd, T. B., Cruz, P. E., Ran, Y., et al. (2018). High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol. Med. 10:e9027. doi: 10.15252/emmm.201809027
Leyns, C. E. G., Gratuze, M., Narasimhan, S., Jain, N., Koscal, L. J., Jiang, H., et al. (2019). TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 22, 1217–1222. doi: 10.1038/s41593-019-0433-0
Leyns, C. E. G., Ulrich, J. D., Finn, M. B., Stewart, F. R., Koscal, L. J., Remolina Serrano, J., et al. (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. U S A 114, 11524–11529. doi: 10.1073/pnas.1710311114
Liu, D., Cao, B., Zhao, Y., Huang, H., McIntyre, R. S., Rosenblat, J. D., et al. (2018). Soluble TREM2 changes during the clinical course of Alzheimer’s disease: a meta-analysis. Neurosci. Lett. 686, 10–16. doi: 10.1016/j.neulet.2018.08.038
Malik, M., Parikh, I., Vasquez, J. B., Smith, C., Tai, L., Bu, G., et al. (2015). Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 10:52. doi: 10.1186/s13024-015-0048-1
McQuade, A., Coburn, M., Tu, C. H., Hasselmann, J., Davtyan, H., and Blurton-Jones, M. (2018). Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13:67. doi: 10.1186/s13024-018-0297-x
Melchior, B., Puntambekar, S. S., and Carson, M. J. (2006). Microglia and the control of autoreactive T cell responses. Neurochem. Int. 49, 145–153. doi: 10.1016/j.neuint.2006.04.002
Mizee, M. R., Miedema, S. S., van der Poel, M., Adelia, Schuurman, K. G., van Strien, M. E., et al (2017). Isolation of primary microglia from the human post-mortem brain: effects of ante- and post-mortem variables. Acta Neuropathol. Commun. 5:16. doi: 10.1186/s40478-017-0418-8
Naj, A. C., Jun, G., Beecham, G. W., Wang, L. S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Öhrfelt, A., Axelsson, M., Malmestrom, C., Novakova, L., Heslegrave, A., Blennow, K., et al. (2016). Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult. Scler. 22, 1587–1595. doi: 10.1177/1352458515624558
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parhizkar, S., Arzberger, T., Brendel, M., Kleinberger, G., Deussing, M., Focke, C., et al. (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 22, 191–204. doi: 10.1038/s41593-018-0296-9
Piccio, L., Buonsanti, C., Cella, M., Tassi, I., Schmidt, R. E., Fenoglio, C., et al. (2008). Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131, 3081–3091. doi: 10.1093/brain/awn217
Piccio, L., Deming, Y., Del-Aguila, J. L., Ghezzi, L., Holtzman, D. M., Fagan, A. M., et al. (2016). Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 131, 925–933. doi: 10.1007/s00401-016-1533-5
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 13:24. doi: 10.1186/s13024-018-0254-8
Sayed, F. A., Telpoukhovskaia, M., Kodama, L., Li, Y., Zhou, Y., Le, D., et al. (2018). Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. U S A 115, 10172–10177. doi: 10.1073/pnas.1811411115
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schlepckow, K., Kleinberger, G., Fukumori, A., Feederle, R., Lichtenthaler, S. F., Steiner, H., et al. (2017). An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 9, 1356–1365. doi: 10.15252/emmm.201707672
Schmid, C. D., Sautkulis, L. N., Danielson, P. E., Cooper, J., Hasel, K. W., Hilbush, B. S., et al. (2002). Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83, 1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x
Shi, Y., and Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772. doi: 10.1038/s41577-018-0051-1
Song, W. M., Joshita, S., Zhou, Y., Ulland, T. K., Gilfillan, S., and Colonna, M. (2018). Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 215, 745–760. doi: 10.1084/jem.20171529
Suárez-Calvet, M., Araque Caballero, M. A., Kleinberger, G., Bateman, R. J., Fagan, A. M., Morris, J. C., et al. (2016a). Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 8:369ra178. doi: 10.1126/scitranslmed.aag1767
Suárez-Calvet, M., Kleinberger, G., Araque Caballero, M. A., Brendel, M., Rominger, A., Alcolea, D., et al. (2016b). sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 8, 466–476. doi: 10.15252/emmm.201506123
Thornton, P., Sevalle, J., Deery, M. J., Fraser, G., Zhou, Y., Stahl, S., et al. (2017). TREM2 shedding by cleavage at the H157–S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 9, 1366–1378. doi: 10.15252/emmm.201707673.
Ulland, T. K., Song, W. M., Huang, S. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649.e613–663.e613. doi: 10.1016/j.cell.2017.07.023
Ulrich, J. D., Finn, M. B., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener. 9:20. doi: 10.1186/1750-1326-9-20
Wakselman, S., Bechade, C., Roumier, A., Bernard, D., Triller, A., and Bessis, A. (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wu, K., Byers, D. E., Jin, X., Agapov, E., Alexander-Brett, J., Patel, A. C., et al. (2015). TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 212, 681–697. doi: 10.1084/jem.20141732
Wunderlich, P., Glebov, K., Kemmerling, N., Tien, N. T., Neumann, H., and Walter, J. (2013). Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036. doi: 10.1074/jbc.M113.517540
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Zhao, R., Hu, W., Tsai, J., Li, W., and Gan, W. B. (2017). Microglia limit the expansion of beta-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:47. doi: 10.1186/s13024-017-0188-6
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron 97, 1023.e1027–1031.e1027. doi: 10.1016/j.neuron.2018.01.031
Zheng, H., Jia, L., Liu, C. C., Rong, Z., Zhong, L., Yang, L., et al. (2017). TREM2 promotes microglial survival by activating Wnt/beta-catenin pathway. J. Neurosci. 37, 1772–1784. doi: 10.1523/JNEUROSCI.2459-16.2017
Zhong, L., Chen, X. F., Wang, T., Wang, Z., Liao, C., Wang, Z., et al. (2017). Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 214, 597–607. doi: 10.1084/jem.20160844
Zhong, L., Chen, X. F., Zhang, Z. L., Wang, Z., Shi, X. Z., Xu, K., et al. (2015). DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against lps-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877. doi: 10.1084/jem.20160844
Zhong, L., Wang, Z., Wang, D., Wang, Z., Martens, Y. A., Wu, L., et al. (2018). Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 13:15. doi: 10.1186/s13024-018-0247-7
Keywords: Alzheimer’s disease, microglia, soluble TREM2, biomarker, neuroinflammation
Citation: Zhong L and Chen X-F (2019) The Emerging Roles and Therapeutic Potential of Soluble TREM2 in Alzheimer’s Disease. Front. Aging Neurosci. 11:328. doi: 10.3389/fnagi.2019.00328
Received: 24 October 2019; Accepted: 12 November 2019;
Published: 26 November 2019.
Edited by:
Xiongwei Zhu, Case Western Reserve University, United StatesCopyright © 2019 Zhong and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhong, emhvbmdsaUB4bXUuZWR1LmNu; Xiao-Fen Chen, Y2hlbnhmQHhtdS5lZHUuY24=