 Georgia R. Frost
Georgia R. Frost Lauren A. Jonas
Lauren A. Jonas Yue-Ming Li
Yue-Ming Li- 1Chemical Biology Program, Memorial Sloan Kettering Cancer Center, Manhattan, NY, United States
- 2Pharmacology Program, Weill Cornell Graduate School of Medical Sciences, Cornell University, Ithaca, NY, United States
Alzheimer’s disease (AD) is marked by the presence of amyloid beta (Aβ) plaques, neurofibrillary tangles (NFT), neuronal death and synaptic loss, and inflammation in the brain. AD research has, in large part, been dedicated to the understanding of Aβ and NFT deposition as well as to the pharmacological reduction of these hallmarks. However, recent GWAS data indicates neuroinflammation plays a critical role in AD development, thereby redirecting research efforts toward unveiling the complexities of AD-associated neuroinflammation. It is clear that the innate immune system is intimately associated with AD progression, however, the specific roles of glia and neuroinflammation in AD pathology remain to be described. Moreover, inflammatory processes have largely been painted as detrimental to AD pathology, when in fact, many immune mechanisms such as phagocytosis aid in the reduction of AD pathologies. In this review, we aim to outline the delicate balance between the beneficial and detrimental aspects of immune activation in AD as a more thorough understanding of these processes is critical to development of effective therapeutics for AD.
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and the 6th leading cause of death in the United States, next to heart disease and cancer (James et al., 2014; Alzheimer’s Association, 2015). Currently, no treatment for AD exists, making the development of therapeutics essential to public health (Katsurabayashi et al., 2016). There are two well-established hallmarks of AD: amyloid beta (Aβ) plaques and neurofibrillary tangles (NFT) (Lue et al., 1996; DeTure and Dickson, 2019). In large part, therapeutic efforts have focused on targeting and reducing the accumulation of Aβ and NFT (Panza et al., 2009). However, recent genome-wide association studies (GWAS) indicate that a large percentage of AD risk genes are associated with innate immunity and inflammation, suggesting that the immune system plays a critical, and previously unappreciated, role in AD pathology (Bis et al., 2018; Pimenova et al., 2018).
Clinical observations and bioinformatics have demonstrated that inflammatory cytokines, chemokines and other mediators are upregulated in AD patients, which further emphasizes the involvement of the immune system and neuroinflammation in AD (Tarkowski et al., 2003; Cribbs et al., 2012; Zhang et al., 2013; Brosseron et al., 2014). Neuroinflammation is a broad term used to describe the innate immune response to CNS tissue damage. Neuroinflammation occurs when injury, infection, or disease (in the case of AD, misfolded proteins, namely Aβ and NFT) stimulates resident cells to produce inflammatory mediators such as cytokines and chemokines, but also, prostaglandins, free radicals, complement factors, and adhesion molecules which recruit and activate additional immune cells (Calsolaro and Edison, 2016; Schain and Kreisl, 2017; Shabab et al., 2017). Neuroinflammation also involves the proliferation and activation of glial cells, astrocytes and microglia, as well as the presence of the aforementioned inflammatory mediators in the brain (Shabab et al., 2017). Many different cells within the CNS, including microglia and astrocytes, but also, neurons and endothelial cells, are capable of initiating an inflammatory response (Schwartz et al., 2013).
The CNS innate immune response is poorly understood and research suggests there are many differences in comparison to the peripheral immune system (although, alterations in the peripheral immune system have also been implicated in AD progression) (Cao and Zheng, 2018). Notably, immune associated clearance and repair pathways face greater challenges within the CNS environment, such as the presence of the blood brain barrier and ineffective clearance, as well as the abundance of post-mitotic cells (i.e., neurons) and the inability for growth and repair (Herrup and Yang, 2007; Muldoon et al., 2013). Therefore, it is critical to develop a better understanding of the brain’s immune response in neurodegenerative disease and in AD.
While there is consensus that the immune system is intimately involved in AD, there is considerable debate over which aspects of inflammation are harmful and contribute to degeneration, and which are protective and may prevent cognitive decline. Furthermore, it has yet to be established which components of the immune system actively play a role in pathology and which are just a consequence of disease. As mentioned, gliosis, or increased numbers of activated astrocytes and microglia are a hallmark feature of neuroinflammation. However, past descriptions of this phenomenon, namely just “reactive” or “increased gliosis” are vastly oversimplified (Sofroniew and Vinters, 2010). Recent evidence highlights altered glia-specific pathways in post-mortem AD tissue and in mouse models of AD, suggesting that glial responses are much more heterogeneous and complex than previously thought (Thome et al., 2018). Overall, the broad spectrum of glial changes and functional consequences of gliosis in AD are not yet understood.
While neuroinflammation can promote efficient clearance of Aβ and neuronal debris it can also accelerate disease by causing neuronal and glial cell death (Qin et al., 2002; Block et al., 2007). This inflammatory balance is highly orchestrated and understanding how to regulate these responses is key to developing effective therapeutics for AD. The initiation of an immunological reaction can be beneficial and critical, allowing for a burst of glial activity to protect and repair the site of damage, and to clear toxic species or dysfunctional synapses (Wang M. M. et al., 2018). For example, in response to adverse conditions, microglia will undergo morphological changes, accompanied by the release of a storm of molecular mediators that increases clearances of Aβ (Sole-Domenech et al., 2016; Zhao et al., 2017). Furthermore, various types of non-neuronal cells are recruited to the site to assist in repairing the damage and consolidating excessive inflammation (Sofroniew, 2015). These reparative processes are beneficial, yet may also have harmful consequences such as sustained cytokine release which can become toxic to neuronal cells (Heneka et al., 2015). Therefore, understanding the specific cellular roles and inflammatory reactions in AD is of vital importance.
While studying familial Alzheimer’s disease (FAD) mutations has provided insight into disease etiology, FAD accounts for only a small proportion of all AD cases (St George-Hyslop et al., 1989). Sporadic Alzheimer’s disease (SAD) is a late onset, multi-factorial disease for which the biggest risk factor is age (Dorszewska et al., 2016). However, genetic predisposition still plays a considerable role in risk of developing SAD with a heritability estimate of 60–80% (Gatz et al., 2006). Linkage studies have highlighted genes involved in AD pathogenesis beyond those directly involved in Aβ production identified in FAD studies (Bertram et al., 2008). FAD mutations are associated with Mendelian patterns of inheritance and age-dependent penetrance. These genes cause AD but are extremely rare in the general population (Tanzi, 2012). In contrast, genes identified in SAD linkage studies have higher prevalence in the general population but confer a much smaller risk of developing AD (Figure 1; Pimenova et al., 2018; Naj et al., 2011). Understanding the role that these AD associated genes play will give further insight into AD pathology and possibly result in the identification or novel targets for therapeutic development.
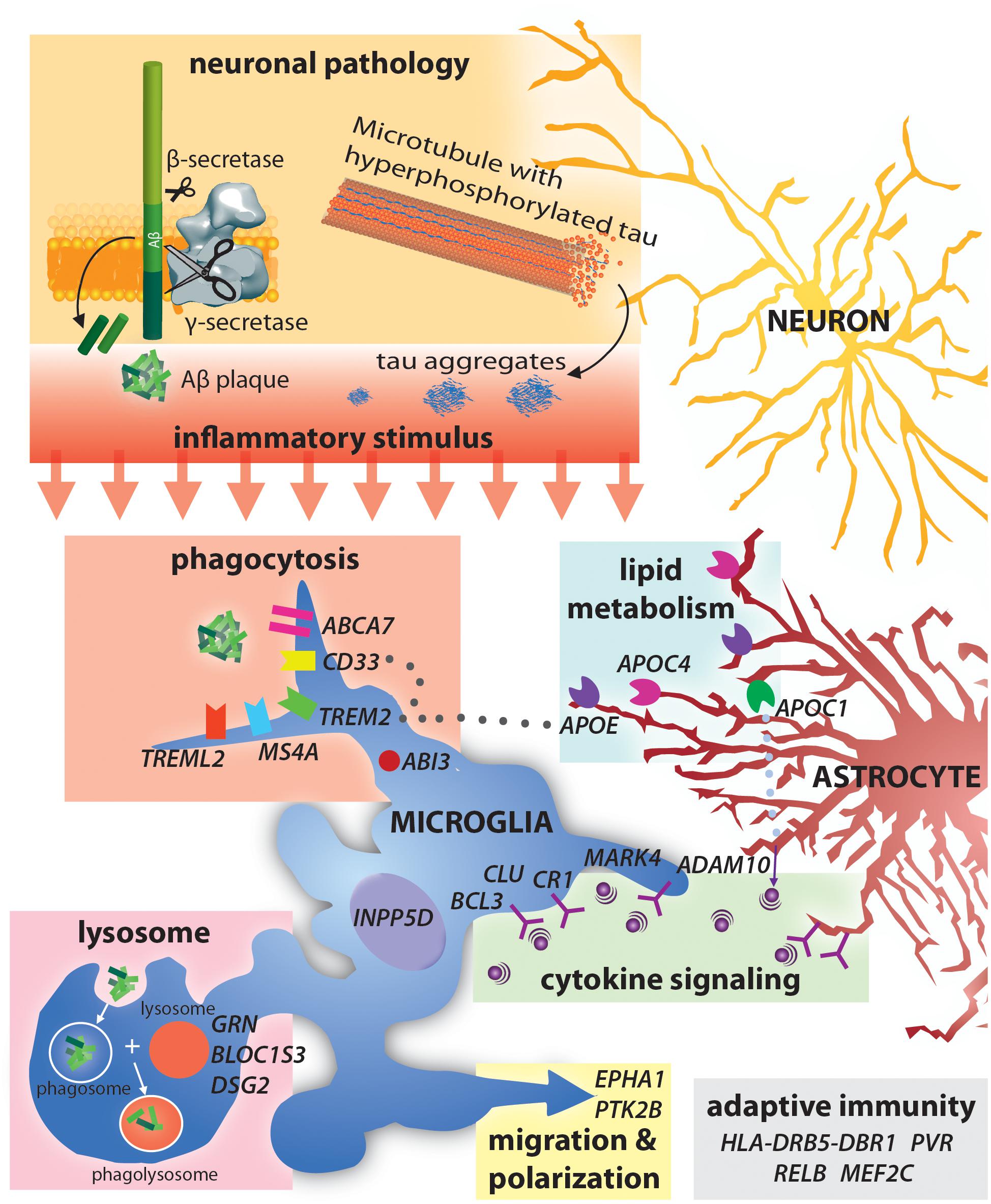
Figure 1. Alzheimer’s disease risk-loci are involved in a variety of immune functions including phagocytosis and lysocytic activity, cytokine signaling and adaptive immunity. Neuronal production of Aβ and tau aggregates may be the initial trigger for this immune activity in AD.
The identification of SAD associated genes has already provided novel insight into pathways involved in AD pathology. AD associated genes can be loosely grouped into those involved in Aβ accumulation, NFT formation and spread, cholesterol homeostasis, innate immunity and inflammation, endocytosis, the cytoskeleton, and epigenetics (Karch and Goate, 2015; Van Acker et al., 2019). Interestingly, many AD associated genes are involved in neuroinflammation and immune activation (Figure 1). Currently, AD associated inflammatory genes such as Trem2 and CD33 are perhaps the most widely discussed aspects of AD pathology. While the specific mechanisms as to how variants in these genes contribute to AD is still up for debate, an established feature of all three is activation of immune function. Notably, AD risk variants in triggering receptor expressed on myeloid cells 2 (TREM2) (Guerreiro et al., 2013; Jonsson et al., 2013) and myeloid cell surface antigen CD33 (Bradshaw et al., 2013) particularly highlight the importance of immune activation in clearing Aβ from the brain (Efthymiou and Goate, 2017).
Innate Immunity in AD
The original amyloid cascade hypothesis notes that immune system activation occurs after Aβ deposition. However, clinical studies suggest that inflammatory changes occur much earlier in disease than originally thought (Tarkowski et al., 2003; Brosseron et al., 2014). In fact, systemic immune challenge in mice with Polyriboinosinic-polyribocytidylic acid (polyI:C), a dsRNA analog, resulted in an AD phenotype including Aβ plaques, NFT and gliosis, suggesting immune challenge can trigger an AD phenotype (Krstic et al., 2012). Furthermore, a plethora of studies have demonstrated that modulating immune system specific proteins impacts the development of AD pathology in various experimental systems, suggesting that the immune system directly impacts pathology. Both systemic inflammatory conditions, such as obesity and diabetes, and activation of the immune system in the CNS, for example, traumatic brain injury, have been associated with the development of AD (Whitmer et al., 2008; Sivanandam and Thakur, 2012; Qizilbash et al., 2015).
The innate immune system is activated when cells expressing specialized pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) on invading pathogens (Akira et al., 2006; Suresh and Mosser, 2013). Host derived PRR ligands, known as danger-associated molecular patterns (DAMPs) will also trigger an immune response. In the case of AD, misfolded or aggregated protein, such as Aβ plaques or NFT are DAMPs that trigger neuroinflammation. Activation of PRR and DAMPs provide the cell with differential signaling outcomes, most likely due to the need to differentiate between pathogen-induced toll-like receptor (TLR) activation, which requires immune participation, and tissue damage-induced TLR activation, which requires a balance between immune intervention and tissue damage repair (Miyake, 2007). PRRs such as TLRs, Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing (NLRP)s, CD36 and RAGE mediate cellular activation by DAMP’s (Cribbs et al., 2012). This activation leads to transcriptional transduction of genes promoting NFkB dependent proinflammatory gene transcription and NLRP3 inflammasome assembly, ultimately leading to the release of proinflammatory cytokines (Latz et al., 2013; Liu et al., 2017).
AD Associated Proinflammatory Cytokines
Cytokines are a heterogeneous group of multifunctional proteins that can by synthesized and secreted by almost all cells (Feghali and Wright, 1997). They generally act locally in either a paracrine or autocrine manner (Zhang and An, 2007). Many cytokines are classified as interleukins (ILs) as they are secreted by, and act on, leukocytes. Other types of cytokines are tumor necrosis factors (TNFs), transforming growth factors (TGFs), interferons (IFNs), and chemokines (Feghali and Wright, 1997). Cytokine signaling is involved in a variety of cellular processes including: cell proliferation, gliogenesis and neurogenesis, cell migration, apoptosis, and neurotransmitter release (Boulanger, 2009; Borsini et al., 2015). Chemokines, while considered cytokines, function differently than ILs, TNFs, TGFs, and IFNs, by attracting both adaptive and innate immune cells to the site of insult.
Cytokines are often categorized as either pro inflammatory or anti-inflammatory. The balance between pro- and anti-inflammatory cytokines acts to successfully eliminate a pathogen, while also protecting surrounding tissue from excessive damage (Szelényi, 2001). Imbalance between pro- and anti-inflammatory cytokines could contribute to the development of AD. Cytokines function by binding to their receptors expressed on the surface of a variety of cell types. Expression of these receptors is tightly regulated, both spatially and temporally, as a means of controlling an inflammatory reaction (Szelényi, 2001). Research into the prevalence and function of cytokines in the AD is inconsistent, however, at least 23 cytokine polymorphisms in 13 different cytokines have been associated with risk of developing AD (Zheng C. et al., 2016; Table 1). Of these, studies have demonstrated that IL-1β, IL-6, IL-10, IL-18, IFN-γ, and TNF-α have altered expression in AD (Griffin et al., 1989; van der Wal et al., 1993; Hull et al., 1996). Furthermore, polymorphisms in IL-4, IL-12, and IL-23 and have been associated with AD but there is no consensus on whether their expression is changed with disease. Even though IL-10, IL-1ra and TGF-β are not genetically linked with AD, they have been found at higher levels in AD patients (Zheng C. et al., 2016).
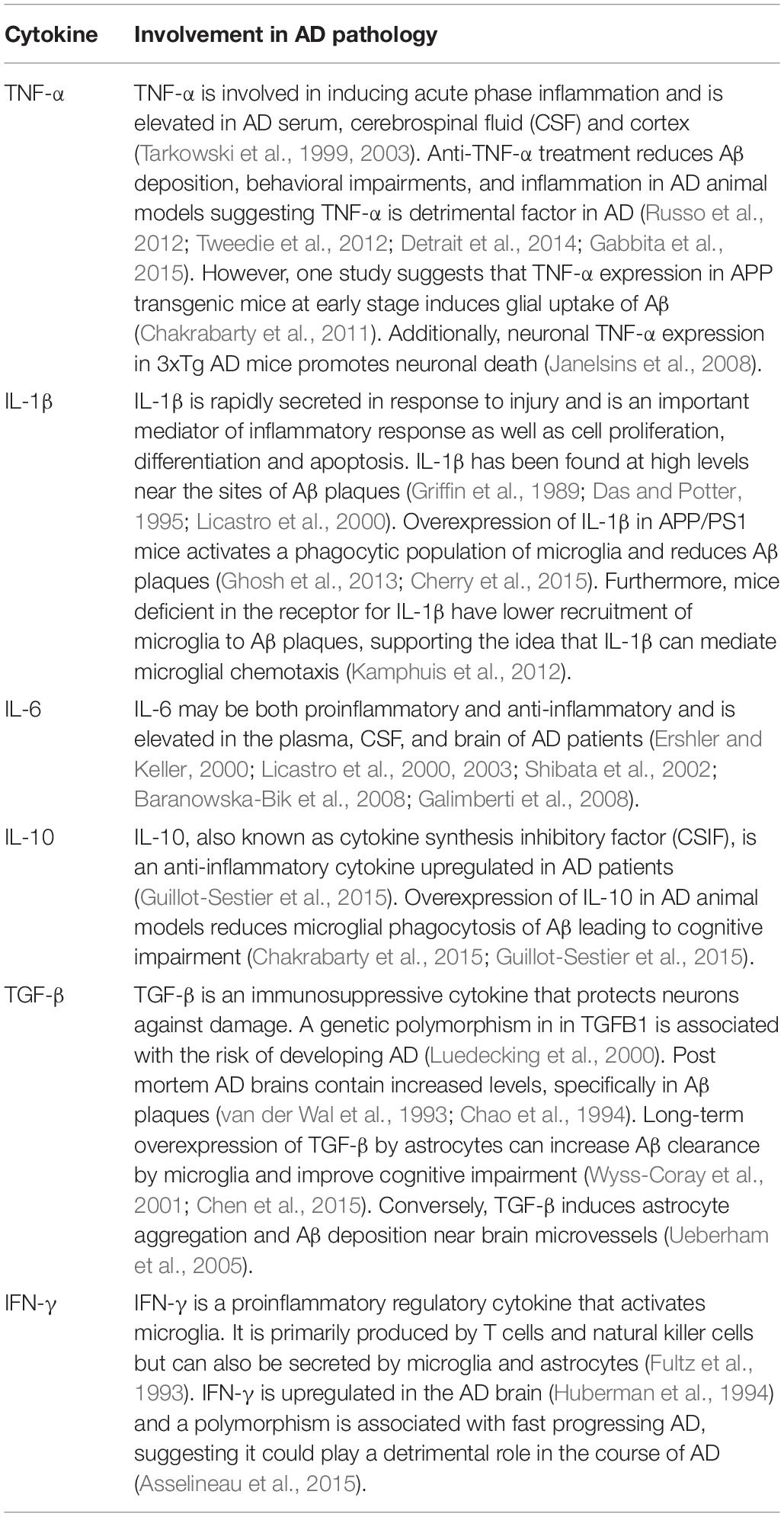
Table 1. Summary of the involvement of cytokines that play well established roles in AD pathology.
Interferons (IFNs) in AD
Termed for their ability to interfere with viral replication, IFNs are a family of pleiotropic cytokines with broad immunomodulatory functions (Lindenmann, 1982; de Weerd et al., 2013). IFNs are classified based on their cognate receptor: type-I IFN; 14 subtypes, notably IFNα and IFNβ, type-II IFN; IFN-γ, and type-III IFNs; contains three IFNλ subtypes (de Weerd et al., 2013). The three subtypes share pro-inflammatory properties and activate multiple signaling pathways. The most critical of these pathways is the janus-associated kinase (JAK) and signal transducer and activator of transcription (STAT) pathway (JAK-STAT) (Platanias, 2005).
Type-I IFNs signal through their cognate receptor: IFNα/β receptor (IFNAR) which is composed of two subunits, IFNAR1 and IFNAR2 (de Weerd et al., 2013). Upon binding the two subunits recruit JAKs (Tyk-2 for IFNAR1 and Jak1 for IFNAR2) which act as docking sites for the phosphorylation of STATs. Upon phosphorylation, STATs translocate to the nucleus and induce transcription of interferon-regulated genes (IRGs) (Platanias, 2005). Additionally, IFN-regulated factors (IRFs) may bind to STATs prior to nuclear translocation and further modify the IFN response (Honda et al., 2005). Type-I IFN signaling alone may result in regulation of over 2000 genes known collectively as interferon-stimulated genes (ISGs) (Schreiber and Piehler, 2015) – much is still unknown about the downstream outcomes of type-I IFN signaling (Ng et al., 2016).
The functional consequence of IFN signaling within the CNS is particularly diverse and is important for processes such as cell survival and immunomodulation (Gough et al., 2012). IFNα and IFNβ are used therapeutically in multiple sclerosis to suppress autoimmune attacks on myelination (Wingerchuk and Carter, 2014). Furthermore, IFNβ is a driver of microglial phagocytosis of myelin in a murine autoimmune encephalitis model (Kocur et al., 2015). However, an epidemiological study suggests IFNβ treated MS patients develop Parkinson’s disease (PD)-like symptoms, such as impaired motor function and cognitive deficits, with long term type-I IFN therapy (Manouchehrinia and Constantinescu, 2012). While IFN signaling may be beneficial, particularly in the periphery, within the CNS, IFNs may also be detrimental and contribute to the development of many neurodegenerative disorders. To highlight this contradiction, IFNβ-/- mice develop Lewy body and PD like symptoms (Ejlerskov et al., 2015), yet, genetic and pharmacological inhibition of type-I IFN signaling is neuroprotective in the MPTP model of PD (Main et al., 2017). The outcome of IFN signaling within the CNS is highly specific to environmental context and requires further study. Interestingly, there is a body of evidence suggesting that IFN signaling contributes to cognitive impairments in a variety of disease states such as human immunodeficiency virus (HIV) encephalopathy (Rho et al., 1995), West Nile virus (Cunningham et al., 2007), neonatal Aicardi–Goutières syndrome (Crow et al., 2006a, b), and even normal aging (Baruch et al., 2014; Zheng et al., 2014).
The staggering diversity of cytokines and chemokines implicated in the progression of AD pathology serves to highlight the complexity of immune activation in disease. Due to the influence of so many different proinflammatory and anti-inflammatory (or both) mediators in AD, it seems unlikely that targeting any specific cytokine, without fully understanding the downstream effect on the immune system as a whole, will be an effective treatment for AD. Furthermore, cytokines may elicit different effects depending on the cell type and cellular context so the relationship between different cell types in AD needs to be explored.
Glia
There are many different glial cell types: microglia, astrocytes, oligodendrocytes, ependymal and satellite cells. Until recently, glia were categorized as support cells for neuronal activity, however, accumulating evidence suggests glia have roles that expand far beyond neuronal support. Microglia, the most abundant mononuclear phagocyte in the CNS, are most studied for their role in immune surveillance, phagocytosis, neurodevelopment and synaptic plasticity, while astrocytes provide protection, support, and nutrients to neurons and to the vasculature (Allen and Eroglu, 2017; Li and Barres, 2018). Oligodendrocytes form myelin that coat nerve cell axons, aiding in the propagation of action potentials, while Schwann cells in the PNS act analogously (Lassmann, 1998). Ependymal cells line CNS ventricles and the central canal of the spinal cord and are responsible for the regulation of CSF (Johanson et al., 2008). It is well-established that neurons, astrocytes and microglia all play vital roles in the tripartite synapse and therefore are all necessary for synaptic transmission, learning and memory (Araque et al., 1999). A plethora of evidence highlights the roles of glia in immune activity in AD including phagocytosis of cellular debris and cytokine release (Leyns and Holtzman, 2017).
Microglia
The CNS contains microglia, resident immune cells which migrate to the brain during embryonic days 8–9 (Hanisch and Kettenmann, 2007; Quan and Banks, 2007; Ginhoux and Prinz, 2015; Louveau et al., 2015; Matcovitch-Natan et al., 2016). Microglia are found throughout the CNS and their immune activity within the brain is well defined (Lawson et al., 1990; Hanisch and Kettenmann, 2007; Ransohoff and Cardona, 2010; Kierdorf and Prinz, 2013; Jay et al., 2015; Lauro et al., 2015; Matcovitch-Natan et al., 2016). Microglia play a central role in the immune system and have receptors for neurotransmitters and hormones, cytokines and chemokines, and PRRs and therefore respond to a variety of inflammatory mediators that are implicated in AD, including Aβ (Liu et al., 2005; Smith et al., 2012; Dewapriya et al., 2013; Rangaraju et al., 2018a, b). Microglial activation is associated with AD progression: however, whether this is harmful, protective, or, both, remains to be established. Microglia may prevent AD pathology by reducing Aβ accumulation – Aβ can be directly removed from central nervous system circulation by activated microglia (Michaud et al., 2013). Microglia have been shown to recognize and phagocytose dysfunctional synapses (Wake et al., 2009; Tremblay et al., 2010) and stimulate synapse formation by secreting brain-derived neurotrophic factor (BDNF) (Parkhurst et al., 2013). On the other hand, microglial cytokine production has also been associated with neurotoxic effects that contribute to cognitive decline (Kettenmann et al., 2011; Heneka et al., 2015; Heppner et al., 2015; Calsolaro and Edison, 2016).
Microglia have a spectrum of phenotypes ranging from ameboid to ramified (Stence et al., 2001; Fontainhas et al., 2011) which are influenced by environmental factors such as the presence of cytokines and chemokines (Ginhoux et al., 2013). On one side of the spectrum, ramified microglia have many processes that facilitate the interaction with nearby neurons, astrocytes and blood vessels. These surveying interactions are necessary for CNS tissue maintenance and neuronal function (Wake et al., 2009; Paolicelli et al., 2011; Schafer et al., 2012; Parkhurst et al., 2013). On the other hand, amoeboid microglia have retracted processes and therefore, the highest level of motility and greatest capacity for phagocytosis (Karperien et al., 2013).
The importance of microglial phagocytosis in neuroprotection and maintaining homeostasis is well appreciated, however, the contribution of dysregulated phagocytosis to disease state has only recently emerged. In fact, impaired microglial clearance of Aβ is thought to play a more significant role in the development of SAD than aberrant Aβ production (Mawuenyega et al., 2010). A decrease in Aβ clearance has been demonstrated in AD patients compared to age-matched controls suggesting impaired microglia or astrocyte clearance may be a causative factor in AD (Mawuenyega et al., 2010). Phagocytic activity can differ with disease and environmental context, resulting in a wide spectrum of phagocytic responses. The phagocytotic pathway can be described in three steps: 1. find-me, 2. eat-me, and 3. digest-me (Sierra et al., 2013; Wolf et al., 2017). The first step is initiated when specific molecules bind to target-recognizing receptors on the microglia. These receptors can be specific toward signaling molecules on the surface of items to be phagocytosed, such as pathogens, dystrophic neurites or protein aggregates. In fact, different receptors trigger different signaling pathways that stimulate phagosome formation (Arcuri et al., 2017). In the case of AD, targets such as Aβ or NFTs are recognized by TLRs resulting in a proinflammatory cytokine storm associated with release of TNF, IL-1 and NO, while recognition of cell debris or dystrophic neurites by microglial TREM2 receptors is associated with a phagocytic response along with an increase in TGFB and IL10 signaling (Neumann and Takahashi, 2007; Takahashi et al., 2007; Neumann et al., 2009). During the second step, eat-me, the plasma membrane extends and encloses around the target forming a vesicular phagosome. This nascent phagosome subsequently fuses with lysosomes forming a phagolysosome. The third and final step, digest-me, occurs within the phagolysosome as the target is degraded. Following this, byproducts must be either stored or recycled by the phagocytic cell (Flannagan et al., 2012).
Specifically, Aβ triggers an inflammatory response which stimulates microglia to clear it from the brain and this prevents the formation of plaques (Colton et al., 2006; Jekabsone et al., 2006; Morgan, 2006; Zhao et al., 2017). After Aβ is engulfed it is targeted to the autophagic pathway for eventual degradation by the fusion of the autophagosome with a lysosome. Alternatively, reactive astrocytes can uptake Aβ and degrade it via the autophagic/lysosomal pathway. Additionally, Aβ may be cleared from the brain by proteolytic degradation. Aβ can be cleaved by the metalloprotease neprilysin (Takaki et al., 2000), therefore, mice treated with neprilysin inhibitors have increased Aβ load (Marr et al., 2003). Insulin degrading enzyme has also been shown to degrade monomeric Aβ (Vekrellis et al., 2000).
Beyond phagocytosis and the clearance of proteins from the brain, microglia are also essential to synaptic function and make direct contact with synapses and dendritic spines during neuronal activity. It is well-established that microglia play a vital role in synaptic pruning during development, but evidence suggests they contribute to synaptic plasticity in the developed brain by a similar mechanism (Hong et al., 2016; Sominsky et al., 2018). Microglia phagocytosis of synapsis and dendritic spines appears most common in the hippocampus (Paolicelli et al., 2011). In particular, microglial expression of complement proteins and receptors seems to play a critical role in recognizing which synapses to phagocytosis (Schafer et al., 2012). Microglia also remove damaged synapses in a neuroprotective process known as synaptic stripping (Trapp et al., 2007; Yamada et al., 2008). On the other hand, when microglia phagocytose Aβ it activates the NLRP3 inflammasome, leading to caspase-1 activation and IL-1β maturation and release which can be harmful to surrounding tissue (Halle et al., 2008). When NLRP3 and caspase-1 are genetically deleted there is reduced Aβ deposition, decreased IL-1β release and improved cognitive performance in APPPS1 mice (Heneka et al., 2013). Additionally, complement binding may stimulate microglia to engulf synaptic contents in the presence of Aβ (Hong et al., 2016). Therefore, microglial phagocytosis of Aβ may be both beneficial and harmful.
Activated microglia can secrete toxic proinflammatory cytokines (Colonna and Butovsky, 2017; Kinney et al., 2018) and also release mediators that induce astrocytes to secrete a neurotoxic substance (Liddelow et al., 2017) contributing to neurodegeneration. On the other hand, many of the previously discussed cytokines released by microglia, also have physiological roles in synaptic plasticity and neurogenesis (Albensi and Mattson, 2000; Bernardino et al., 2008; Morris et al., 2013). For example, IL-1β, TNF, IL-6. and complement cascade proteins are all involved in memory consolidation (Goshen et al., 2007; Boulanger, 2009; Yirmiya and Goshen, 2011). In conclusion, microglial activity and cytokine release are vital to normal cognitive processes, however, in other contexts these processes can be neurotoxic.
Astrocytes
Astrocytes are another major glial cell type within the CNS. Unlike microglia, which are known to be highly motile, astrocytes operate within territorial domains, either supporting the vasculature or enveloping neuronal synapses, through processes terminating in the endfoot (Oberheim et al., 2006). This enables astrocytes to exert control over various CNS functions including blood brain barrier regulation, nutrient delivery, ion and metabolite balance, and innate immune regulation (Attwell Buchan et al., 2010; Bazargani and Attwell, 2016). Astrocytic end-feet express Ca2+ ion channels and glutamate receptors, allowing for the propagation of calcium currents and the release of gliotransmitters. A pertinent member of the tripartite synapse, astrocytes enable communication with and between neurons (Lind et al., 2018). Specifically, astrocytes can uptake and release neurotransmitters such as glutamate, GABA and ATP, neuromodulators; d-serine and kynurenic acid, as well as growth factors and inflammatory mediators (Malarkey and Parpura, 2008; Choi et al., 2014; Martineau et al., 2014). Further, astrocytes offer protection against CNS injury and are critical for repair of nervous tissue. After injury, astrocytes propagate and become reactive, a process known as astrogliosis. Astrogliosis is an evolutionarily conserved and neuroprotective process that contributes to the isolation of damaged tissue through formation of a glial scars and consolidation.
Astrocytes are key regulators of the brains inflammatory response and are capable of releasing, and responding to, a spectrum of immune mediators (Farina et al., 2007). Astrocytes secrete many of the cytokines known to be upregulated in human AD brain samples and in transgenic mouse models of AD (Benzing et al., 1999; Apelt and Schliebs, 2001; Abbas et al., 2002; Tan et al., 2002), notably: IFNγ, IL-1β, TNFα, IL-6, and TGFβ (Constam et al., 1992; McGeer and McGeer, 1995; Hu et al., 1998; Johnstone et al., 1999) and are capable of secreting Aβ (Frost and Li, 2017). Furthermore, Aβ can stimulate the production and secretion of these cytokines from astrocytes (Craft et al., 2004; White et al., 2005). Similar to microglia, astrocytes undergo substantial changes in response to specific stimulus and may be resting or activated depending on the cellular environment (Mrak, 2012). The number of astrocytes is thought to remain constant throughout AD, however, some cells, particularly those in proximity to Aβ plaques, become reactive, while conversely, large numbers of astrocytes atrophy, a process known as astrodegeneration (Rodríguez et al., 2014).
Depending on the exact immune stimulus, astrocytes undergo a range of molecular and morphological changes (Zamanian et al., 2012). Characteristic of “astrogliosis,” astrocytes exhibit hypertrophy of processes, marked by increased expression of intermediate filament, most notably glial fibrillary acidic protein (GFAP) (Wilhelmsson et al., 2006; Kamphuis et al., 2012). Astrogliosis results in differential gene expression of structural proteins, synaptic modulators, and transcriptional, inflammatory and vascular regulators. Further, specific signaling pathways are associated with regulation of astrogliosis. JAK-STAT3 and NFkB signaling induces astrocyte reactivity and proinflammatory cytokine release (Ceyzeriat et al., 2016), while B1-itegrin-mediated signaling permits procurement of mature, non-reactive astrocytes (Robel et al., 2009) indicating the existance of feedback pathways for regulation of gliosis. Increasing evidence has shown the beneifts of astrogliosis, including excitotoxic glutamate uptake, inflammatory cell containment, and neuroprotection against ocidative stress via glutathione production (Sofroniew and Vinters, 2010).
In addition to their immune function, astrocytes are involved in learning and memory in several critical ways: (1) glutamate reuptake, synthesis and metabolism, (2) GABA recycling, (3) regulation of intracellular calcium, (4) regulation of extracellular K+ concentration, (5) release of gliotransmitters, (6) control of blood flow and therefore, glucose supply, and (7) regulation of lactate levels (Halassa et al., 2009; Suzuki et al., 2011; Han et al., 2012; Navarrete et al., 2012; Moraga-Amaro et al., 2014; Sahlender et al., 2014). Furthermore, astrocytes respond to neurotransmitters and can activate hundreds of synapses at once, enabling the synchronization of complete neuronal networks (Ben Haim and Rowitch, 2017). As with microglia, the contribution of astrocytes to normal cognition is undeniable, but further work needs to be done to further understand these mechanisms and how they can contribute to disease.
Oligodendrocytes
Alzheimer’s disease research has often been restricted to gray matter, however, it’s important to note that abnormalities in white matter have been identified in AD patients. In fact, white matter hyperintensities volume is found to be elevated in FAD patients 20 years prior to the onset of symptoms and can be used to predict incidence of AD and rate of cognitive decline in SAD cases as well (Lee et al., 2018; Nasrabady et al., 2018). White matter tracts of the brain are important for learning and memory and are the site of pronounced neuroinflammation and microgliosis (Raj et al., 2017). Oligodendrocytes are glia responsible for producing the myelin in the white matter regions. In addition to this, oligodendrocytes also produce neurotrophic factors which impact neurite growth and neuronal connectivity (Schwab, 1990), and can respond to immune activity in the brain. In fact, complement reactive oligodendrocytes in association with activated microglia can be found in the white matter of aged brains (Duce et al., 2006). Furthermore, microglia have diverse phenotypes depending of the region of the brain and may be more activated in white matter (Olah et al., 2011).
Glia Barriers
Damage to the CNS, in the form of injury or disease, results in a multifaceted immune response. The formation of glial barriers, also known as glial scars, is one type of response to injury and is formed by both glial and non-glial cells that surround the site of damage (Adams and Gallo, 2018). This phenomenon is most widely studied in the context of traumatic brain injury. Glial scarring has also been found after incidence of ischemic stroke and in neurodegenerative diseases such as multiple sclerosis (Woodcock and Morganti-Kossmann, 2013).
While many have reported the heterogeneity of glial scar formation across brain region and disease-type, the development of the glial barrier often begins with the proliferation of reactive astrocytes near the damaged tissue, and the subsequent orchestration of astrocytes, oligodendrocyte precursor cells and microglia to build a rigid wall surrounding damage (Sofroniew, 2009; Wanner et al., 2013). Ultimately a barrier is formed, culminating in the existence of two distinct regions: the lesion core, consisting of oligodendrocyte precursor, fibroblasts and macrophages, and the penumbra, containing reactive astrocytes and microglia (Anderson et al., 2014). The induction of glial scar formation is specific and is stimulated by a range of signaling molecules including Il-1, IL-6, TGF-B1 EGF, BMPs and LIF, from damaged tissue (Wang H. et al., 2018). These signaling molecules aid in the activation of transcription factors that regulate the astrocytic response to tissue damage. Specifically, signaling for the activation of transcription factors STAT3, SP1, OLIG2, Smad3, and NFkB pathways initiate glial scarring mechanisms including astrocytic hypertrophy, migration, proliferation, gliogenesis, and inflammation (Justicia et al., 2000; Hamby et al., 2012).
A defining feature of senile plaques is the presence of surrounding astrocytes and microglia (Itagaki et al., 1989), in fact, the presence of reactive astrocytes follows amyloid deposition throughout the course of AD (Vehmas et al., 2003). Glial barriers surrounding plaques are neuroprotective in two ways: (1) glia, particularly microglia, can break down and phagocytose amyloid plaques, (2) astrocytes can prevent the spread of toxic material and therefore, limit collateral damage (Faulkner et al., 2004) (Anderson et al., 2016). Studies have shown that a deficiency in the ability to form glial barriers worsens AD pathology. When GFAP was knocked out in a mouse model of AD there was a marked increase in plaque-associated dystrophic neurites (Kraft et al., 2013). Further, research shows that glial barriers can aid in axonal regrowth and repair in brain regions where barrier stiffness impacts neuronal growth are more widely affected (Yu and Bellamkonda, 2001; Bollmann et al., 2015).
Glial barriers surrounding amyloid plaques and NFT-associated damage may initially aid in the breakdown and consolidation of toxic debris from the CNS yet subsequently limit the brain’s ability to reform neuronal connections. Moreover, neurons are post mitotic and are unable to divide, making recovery from glial scarring even more difficult (Herrup and Yang, 2007). Thus, immune therapies aimed at regulating the formation of glial scars in AD could help to promote the beneficial regrowth of neuronal connections in the CNS.
AD Associated Genes
The discovery of immune related AD associated genes from AD linkage studies has provided substantial insight into the involvement of immunity in AD pathology. Numerous variants in immune related genes have been identified in whole-genome sequencing and GWAS analyses, including APOE, BIN1, CD33, CLU, CR1, INPP5D, PICALM, PLCG2, MS4A6D, and TREM2, these genes are involved in a wide range of immune related functions (Table 2; Figure 1; Efthymiou and Goate, 2017; Long and Holtzman, 2019; McQuade and Blurton-Jones, 2019; van der Lee et al., 2019). While most of the identified loci confer only a small contribution to AD development, they serve to highlight the involvement of various immune functions in the development of AD. A notable example is the Apoliprotein E (APOE) gene. APOE has three alleles (e2, e3, e4) accounting for proteins that differ by only one or two amino acids (Ward A. et al., 2012; Najm et al., 2019). A single copy of APOE4 increases risk of developing AD by 4-fold, and individuals homozygous for APOE4 have an approximately 12-fold increased risk of AD (Bu, 2009). The APOE4 allele is more common than FAD mutations and also has a substantial effect on risk of developing AD (Corder et al., 1993). Conversely, the e2 allele may protect against developing AD (Berlau et al., 2009). Post mortem analysis revealed that both AD patients and healthy controls with the APOEe2e3 genotype have reduced amyloid load (Lippa et al., 1997). A possible mechanism behind this phenomenon is that APOE allele can affect the efficiency of Aβ clearance (Kim et al., 2009) and also impact the formation of and morphology of NFT (Shi et al., 2017). The C→A substitution at position 158 allows for differential lipidation and higher APOE2 stability and resistance to thermal and chemical denaturation (Morrow et al., 2000; Ma et al., 2018). Thus, differential physical interactions and binding between APOE and soluble Aβ may be the cause for this change in clearance efficiency. Since presence of the e4 allele is thought to reduce Aβ clearance, this underscores the importance of immune clearance of toxic proteins from the brain in preventing AD symptoms. In fact, APOE alleles have been found to impact the efficacy of passive anti-Aβ immunization suggesting that the different alleles effect microglia’s phagocytic ability for Aβ (Pankiewicz et al., 2017). Many other SAD associated genes have also been shown to play vital roles in immune reactivity in the brain.
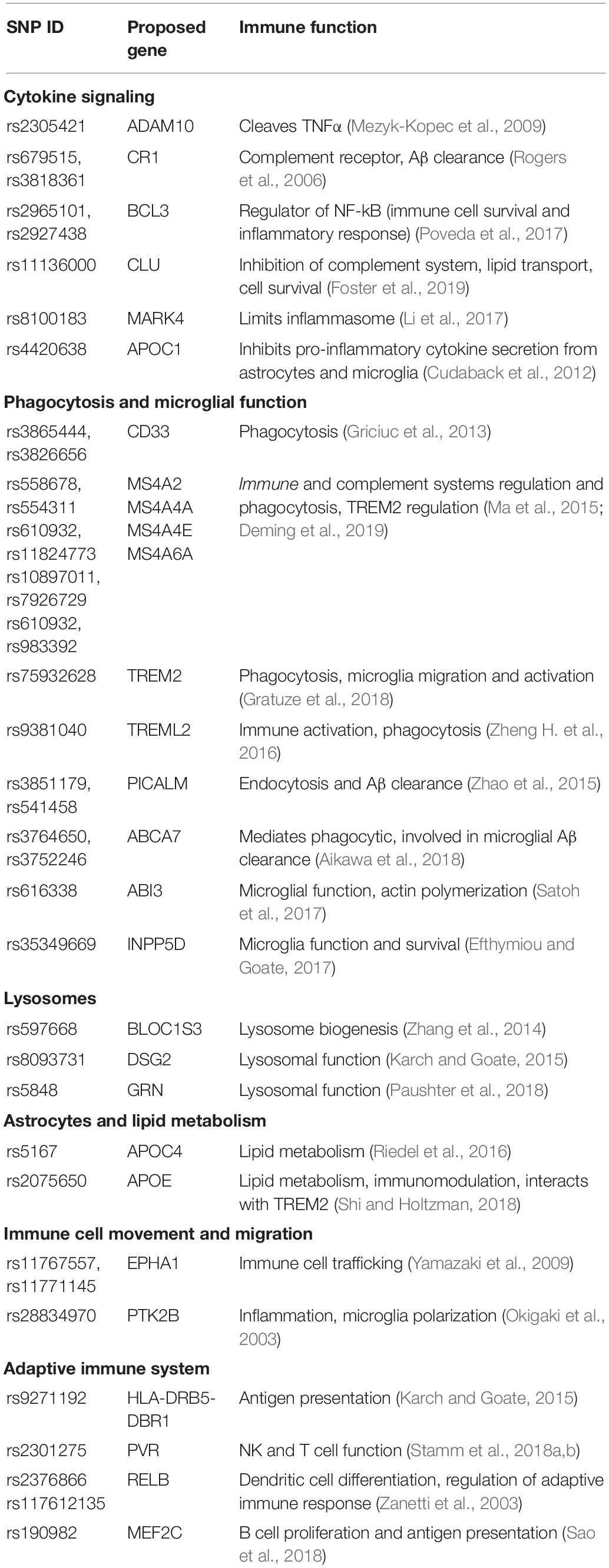
Table 2. Alzheimer’s disease risk-loci with proposed immune functions.
Rare variants in TREM2, which are thought to be loss of function, and increase the risk of developing AD by approximately 2- to 4-fold, were identified by whole-genome sequencing (Sierra et al., 2013; Wolf et al., 2017). A SNP resulting in an Arg-to-His change at amino acid 47 (R47H) is the best established of these variants (Guerreiro et al., 2013; Jonsson et al., 2013). TREM2 binds to the aforementioned AD associated gene APOE and other apolipoproteins, including APOA1, APOB, and APOJ (Yeh et al., 2016). TREM2 associates with DAP12, upon ligand binding, DAP12 is phosphorylated which leads to the recruitment of spleen tyrosine kinase (Syk). Subsequently, Syk signals through activation of phosphatidylinositol 3-kinase (PI3K), and mitogen-activated protein kinases (MAPKs) and the elevation of intracellular Ca2+ through release of IP3-gated Ca2+ stores (Colonna and Wang, 2016). A number of cellular functions have been attributed to this signaling, including: inhibition of inflammatory signaling, phagocytosis and cell survival (Colonna, 2003). Furthermore, Aβ has been demonstrated as a TREM2 ligand capable of triggering TREM2 signaling (Zhao et al., 2018; Zhong et al., 2018).
TREM2 mediated phagocytosis is critical for Aβ and neuronal debris clearance in AD (Kleinberger et al., 2014; Xiang et al., 2016; Yeh et al., 2016). Specifically, TREM2 expression is important for microglia to physically associate with Aβ plaques (Ulrich et al., 2014; Wang et al., 2016; Yuan et al., 2016; Jay et al., 2015, 2017a,b). In fact, high-resolution confocal microscopy revealed that microglial processes that are in contact with Aβ have enhanced expression of TREM2 and DAP12, possibly suggesting an enrichment of activated DAP12 signaling. These findings suggest that TREM2 is necessary for sustaining or initiating microgliosis in AD (Yuan et al., 2016). Presence of the Trem2 R47H allele in an AD mouse model results in reduced TREM2 expression around plaques, a decrease in microglia associated with plaques and an increase in neuritic dystrophy near plaques (Cheng-Hathaway et al., 2018). Additionally, TREM2 is important for microglia survival as TREM2−/− mice have increased apoptosis in plaque-associated microglia (Wang et al., 2016). Furthermore, soluble TREM2 (sTREM2) levels in the CSF are correlated with AD progression (Suarez-Calvet et al., 2019).
Taken together, the critical role of TREM2 in AD underscores the involvement of the immune system, particularly the aspect of microglial activation and phagocytosis, in pathology. Features of immune activation are thus beneficial, as microglia play a critical role in the surveillance and recognition of toxic species, in the initiation of an immune response and in the final clearance and degradation of pathogens.
CD33, a myeloid cell transmembrane receptor, is another top ranked AD associated gene identified by GWAS studies; two main variants, rs3865444 and rs12459419, confer risk of developing AD, and higher expression in the brain has been associated with advanced cognitive decline and AD (Carrasquillo et al., 2011; Hollingworth et al., 2011; Naj et al., 2011; Lambert et al., 2013; Malik et al., 2013; Li et al., 2015; Dos Santos et al., 2017). The rs9865444 variant is associated with increased CD33 expression and microglial activation with reduced AB42 internalization and increased brain amyloid load, while the rs12459419 variant has been identified as protective. CD33, a sialic-binding immunoglobulin-like lectin (Siglec-3), is most studied for its role in immunoreceptor tyrosine-based inhibitor motif (ITIM) signaling. CD33 signaling works counter to TREM2 signaling; Upon ligand binding to (i.e., sialylated glycolipids or amyloid plaques), CD33 signals for the phosphorylation of ITIM by SHP1/2, resulting in the inhibition of a variety of receptors containing the immunoreceptor-tyrosine-based activation motifs (ITAM). Inhibition of ITAM results in reduced cellular activity and suppression of proinflammatory cytokine release, oxidative burst and phagocytosis (Lajaunias et al., 2005; Jiang et al., 2014). The rs12459419 variant results in altered splicing of the CD33 mRNA and a loss of the sialic acid binding domain, thereby inhibiting ITIM inhibition and preserving the cell’s ability to phagocytose (Malik et al., 2013).
CD33 is elevated in the AD brain in microglia and infiltrating macrophages and is thought to modulate microglial activation and Aβ clearance (Griciuc et al., 2013; Walker et al., 2015). In fact, knock-out of CD33 in AD mouse models results in reduced Aβ plaque burden (Griciuc et al., 2013). Additionally, CD33 inhibits microglial cytokine release and immune cell proliferation (Tateno et al., 2007; Crocker et al., 2012) further supporting the involvement of inflammation and innate immunity in AD etiology. Further, the hypothesis that CD33 inhibition could increase phagocytosis of Aβ has fueled efforts to design immunotherapy agents and small molecule inhibitors for therapeutic use. However, strides to find specific pharmacological inhibitors of CD33 in the brain have been difficult due to the lack of an atomic resolution structure of the sialic binding domain.
Current Immune Related Therapies
Currently available treatments for AD are limited to cholinesterase inhibitors and memantine, both of which work by modifying transmission of the neurotransmitters: acetylcholine and glutamate, respectively. (Thomas and Grossberg, 2009; Ferreira-Vieira et al., 2016) While these drugs have proven effective for the slowing of symptoms in mild to moderate AD, no current therapies are effective in significantly slowing of AD progression (Burns et al., 1999; Reisberg et al., 2006). Epidemiological evidence has suggested non-steroidal anti-inflammatory drugs (NSAIDS) are beneficial in the slowing of AD progression – individuals taking NSAIDSs for chronic inflammatory conditions have a lower incidence of AD (de Craen et al., 2005). These studies led to a series of clinical trials testing the effects of naproxen or celecoxib on AD progression. Naproxen’s mechanism of action is through reversible inhibition of both COX-1 and COX-2 enzymes, resulting in the reduction of prostaglandin synthesis. Prostaglandins act as inflammatory inducers, and thus naproxen works as a non-selective inhibitor of an inflammatory response (Robinson, 1983). NSAIDS and naproxen, were originally thought to placate AD pathology by targeting the immune system. However, results from the INTREPAD study indicate that naproxen had no clinical benefits as adults that took naproxen for 2 years did not show slowed cognitive impairment or a decrease in AD-CSF markers (Pellicano, 2014).
It is possible that failure of these clinical trials was due to poor study design rather than a lack of efficacy. The majority of these trials took place early in disease progression, a time when inflammation is critical to initial clearance and consolidation. Therefore, while these studies deem NSAIDS not effective in preventing AD development, targeting inflammation remains a feasible goal. Many studies illustrate the ability to slow cognitive decline with a more selective reduction of detrimental inflammation in AD, while maintaining repair mechanisms. Thus, while studies for the use of NSAIDS in AD proved less than favorable, accumulated research supports the testing of immune modifying therapies.
Immunotherapy has emerged as one of the most promising strategies to tackle AD as it would enable specific clearance of the accumulated toxic proteins of Aβ and NFT without effective other functions in the brain (Valera and Masliah, 2013). Specifically, the development of active and passive vaccines against Aβ, has been utilized as a therapeutic approach (Folch et al., 2016). Active immunotherapy exploits the body’s own immune system and therefore is longer lasting, however, has a high risk of triggering an autoimmune reaction. Furthermore, active immunotherapy can be limited by epitope availability and specificity (Bard et al., 2003; Winblad et al., 2014). Alternatively, passive immunotherapy can be far more specific but has a short effect and therefore requires continuous antibody treatment (Ward M. et al., 2012).
One of the earliest Aβ immunotherapy trials utilized AN1792, a synthetic Aβ peptide, that induces the generation of antibodies against Aβ by active immunotherapy. Unfortunately, 6% of patients during phase 2 of clinical trials developed meningoencephalitis (Gilman et al., 2005; Cao et al., 2018). Furthermore, treatment did not provide a protective cognitive effect despite significantly reducing Aβ pathology. However, a follow-up study performed 4 years after immunization found improvements in cognition in 68% of patients. reduction in Aβ plaque load locally in the brain. Interestingly, a follow-up study, done 3.6–4.6 years after immunization with AN1792, found significant improvements in brain volume and cognitive decline in immunized vs. placebo group. Although, these improvements were only observed in 68% of antibody responders which was less than 20% of the total patients immunized (Vellas et al., 2009). Another active immunotherapy CAD106 has been found to be safe and well tolerated in clinical trials and is still in development (Farlow et al., 2015).
Two notable passive immunotherapeutic agents have entered clinical trials: bapineuzumab and solanezumab. Bapineuzumab that targets the N-terminus of Aβ and failed clinical trials due to a poor safety profile in mild to moderate AD patients (though some of these were APOE4 carriers) (Doody et al., 2014; Liu et al., 2015; Vandenberghe et al., 2016). Solanezumab is a humanized IgG1 monoclonal antibody targeting soluble monomeric Aβ that has passed Phases 1 and 2 of clinical trials with a favorable safety profile. Unfortunately, solanezumab did not show efficacy in phase 3 and was abandoned until it reentered clinical trials for FAD patients (Doody et al., 2014; Siemers et al., 2016).
Aducanumab is also a monoclonal antibody. In August 2016, very promising phase I results were published showing that Aducanumab decreased soluble and fibrillar forms of Aβ and slowed down cognitive decline 54 weeks after treatment in a dose-dependent manner (Sevigny et al., 2016). However, phase 3 trials for Aducanumab were canceled in March of 2019 after an independent data monitoring committee concluded that the trial would not meet its primary end points. Despite this, in October 2019, Biogen Inc., announced that they would resume clinical trials following further analysis of the phase 3 data (Biogen, 2019).
Infection Hypothesis
The antimicrobial peptide (AMP) hypothesis of AD suggests that Aβ is initially generated as a defense mechanism against a real or perceived infection signifying a direct link between AD and innate immunity (Moir et al., 2018). AMPs are evolutionarily conserved proteins that participate in the innate immune system’s first line of defense, with roles in microbial killing and immune regulation (Haney et al., 2017). AMPs require oligomerization for normal functioning, and soluble oligomers form fibrils that target viruses, fungi, bacteria, and some forms of cancerous host cells (Arnusch et al., 2007; Wiesner and Vilcinskas, 2010). Aβ has structural similarities to a family of host-encoded AMPs and possesses antimicrobial activity. In vitro studies show Aβ as having a greater potency compared to the archetypal human AMP LL-37 against microbes (Soscia et al., 2010). Further, in vivo and in vitro studies demonstrate how AMPs promote amyloid fibrilization for use in disruption of microbial cell membranes, neutralization of bacterial endotoxins and pathogen entrapment (Sood et al., 2008; Wang et al., 2014; Chai et al., 2017). Additionally, Aβ42-overexpressing cells are resistant to killing by yeast and bacteria (Eimer et al., 2018). The Aβ-yeast interaction caused Aβ fibril nucleation allowing for the entrapment of yeast, a mechanism that is comparable to the antimicrobial activity of many AMPs (Eimer et al., 2018). Moreover, a mouse model overexpressing Aβ42 displayed higher resistance to certain yeast and bacterial infections (Soscia et al., 2010). Further, recent reports have indicated higher levels of AMPs and pathogen derived epitopes in post mortem AD brain tissue (Williams et al., 2013; Alonso et al., 2014). Pathogen signatures (i.e., Chlamydia Pneumoniae, HSV-1 DNA) specifically colocalize with AD pathology (Balin et al., 1998; Wozniak et al., 2009). Further, HSV infection is significantly associated with development of AD and HSV-1 is shown to increase Aβ generation (Letenneur et al., 2008).
Amyloid beta was previously deemed physiologically irrelevant, however, the presence of antimicrobial activity supports a role in the innate immune response and thus could be an important finding for AD research. Knowledge of Aβ’s AMP capabilities may help put into perspective Aβ and the immune system’s causative role in Alzheimer’s disease. If the physiological role of APP is in pathogen/virus defense, then an overproduction of Aβ could be the result of an innate immune reaction. Indeed, the AMP hypothesis helps to shed a new light on aspects of immune involvement in AD pathogenesis. Understanding the initial benefits provided by amyloid’s AMP properties could prove beneficial to disease treatment.
Other Neurodegenerative Diseases
Recent genetic evidence has indicated a critical role for inflammation in many neurodegenerative diseases including Multiple Sclerosis (MS), Huntington’s disease (HD), Parkinson’s disease (PD), Ischemia, traumatic brain injury (TBI), and Parkinson’s disease (PD) (Dzamko et al., 2015; Baranzini and Oksenberg, 2017). A common link between these disorders is chronic activation of the innate immune system through specific activation of resident glial cells, release of inflammatory mediators, and the potential for recruitment of peripheral immune cells. While the specifics of immune activation vary between disorders, due to the pathogenic environment, it is clear that inflammation can play both a beneficial and harmful role in these other neurodegenerative diseases as well. Based on the initial trigger (i.e., alpha-synuclein in PD, ROS in Ischemia) chronic immune signaling can lead to neurotoxicity, increased oxidative stress and synaptic and neuronal death (Stephenson et al., 2018; Hassanzadeh and Rahimmi, 2019). However, immune activation can also be beneficial, resulting in the clearance of cell debris and aggregated/misfolded proteins, in the consolidation of inflammation, and in the mediation of cellular repair.
For example, MS is an autoimmune demyelinating disease of the CNS. While chronic immune activity is responsible for neurodegeneration in MS, evidence has also illustrated that inflammation is necessary for clearance of blood-borne inflammatory cells via emigration or in situ apoptosis. Proinflammatory cytokines, such as IFNγ and TNFa, promote in situ apoptosis of infiltrating T cells, as well as remyelination. Further, macrophages are critical in phagocytosing myelin debris in situ highlighting the beneficial immune activities in MS (Furlan et al., 2001).
Following brain injury, such as TBI and ischemic stroke, microglia actively promote injury−induced neurogenesis via production of insulin−like growth factor−1, which suppresses apoptosis and increases proliferation and differentiation of neural stem cells (Thored et al., 2009). Further, microglial activation aids in the consolidation of injured tissue and resultant inflammation, thereby limiting the spread of damage (Kitamura et al., 2009; Thored et al., 2009; Cunha et al., 2018). During PD, alpha-synuclein activates microglia, resulting in excessive release of pro-inflammatory mediators and an overactivation of the phagocytic pruning pathway on dopamine neurons (De Virgilio et al., 2016). Simultaneously, anti-inflammatory microglia are responsible for cytokine cleanup and phagocytosis of extracellular alpha-synuclein (Ferreira and Romero-Ramos, 2018). While it is clear the neuroinflammation can lead to disease progression, the beneficial aspects of inflammation remain critical to the underlying repair and clearance pathways in disorders of the CNS.
Conclusion
The genetic, cellular, and molecular changes associated with AD provide strong support for an innate immune contribution to disease pathology (Kim et al., 2018). However, which aspects of this contribution are positive or negative is still up for debate. In this review, we propose that there is a delicate balance involved in the innate immune response to AD, which can provide beneficial or detrimental effects. We suggest that some aspects of inflammation in AD are necessary and beneficial and are capable of restricting or preventing AD pathology. By harnessing the benefits provided by the AD immune reaction, we can work to find successful treatments for AD. For too long researchers have fixated on the negative consequences of microgliosis and astrogliosis, broadly categorizing glial cells as “reactive” or “activated.” However, recent genetic and cellular data has highlighted the extreme heterogeneity of glial cells, and the spectrum on which they function. Furthermore, while we have focused on the involvement of the innate immune system, substantial evidence suggests that the adaptive immune system also plays a role in pathology which is an area requiring further study (Sommer et al., 2017).
While it is clear that inflammation contributes to AD pathogenesis, it is too broad a word to describe the amalgam of innate immune mechanisms that arise during disease. Microgliosis involves phagocytosis of amyloid plaques and dysfunctional synapses, as well as the increased expression of complement proteins for neuronal pruning, and the release of trophic factors for cell plasticity and growth. Conversely, microgliosis increases quantities of cytokine and chemokines which can become toxic and harmful to neuronal cells. Astrogliosis can also be beneficial, resulting in propagation of calcium currents for enhanced signal transduction and enhanced repair and protection. On the other hand, excessive astrogliosis may result in the increase of toxic species. It is very clear that neuroinflammation is heavily involved in AD pathogenesis, but a more precise understanding of the AD-specific immune reaction is critical to our developing effective therapeutics for AD (Figure 2).
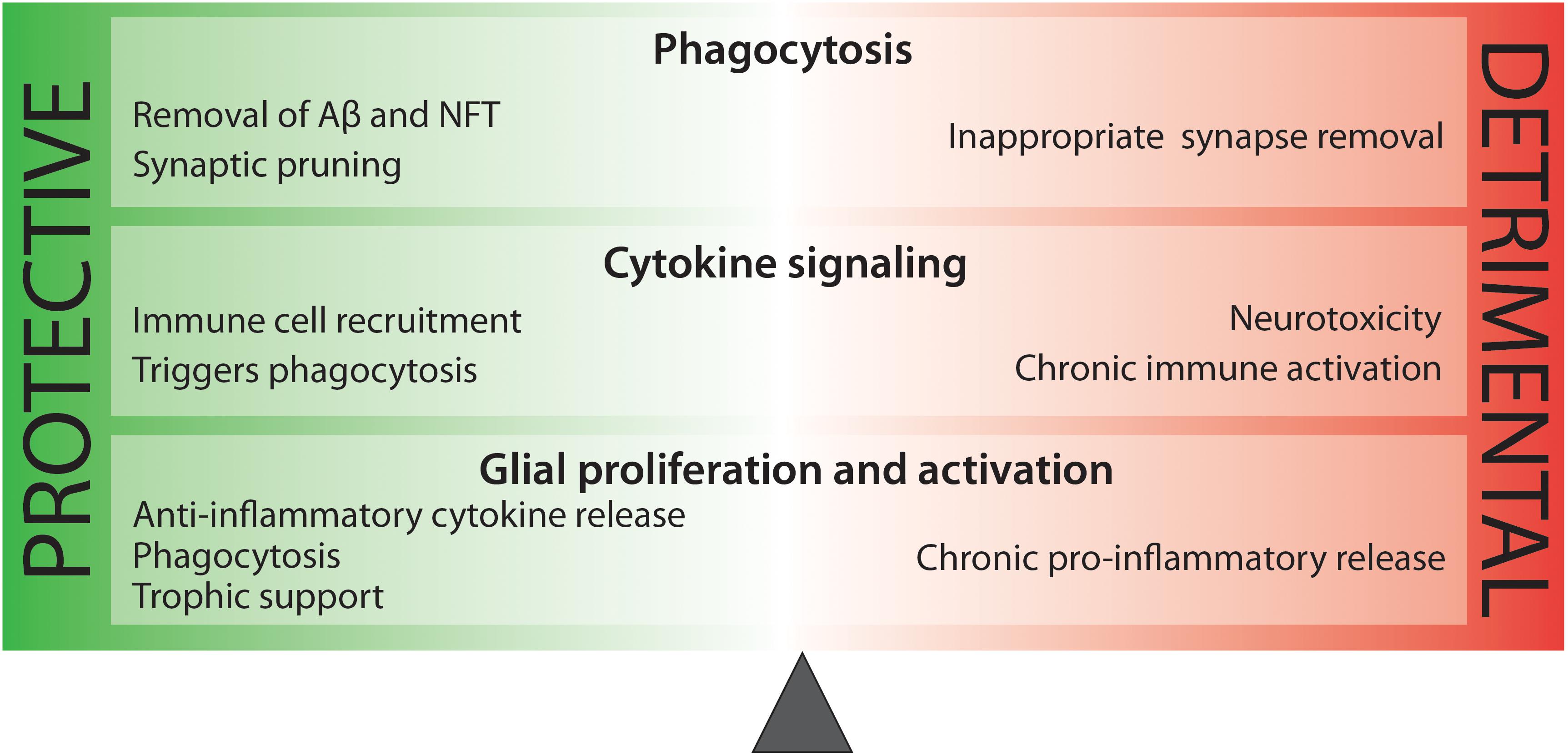
Figure 2. There is a delicate balance between beneficial and harmful immune activity in Alzheimer’s disease. While some aspects of inflammation are protective, such as the phagocytosis of toxic protein species, other processes, such as sustained pro-inflammatory cytokine release are neurotoxic.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH grant R01NS096275 (Y-ML), RF1AG057593 (Y-ML), the JPB Foundation (Y-ML), the MetLife Foundation (Y-ML), Cure Alzheimer’s Fund (Y-ML), The Edward and Della L. Thome Memorial Foundation (Y-ML), and Coins for the Alzheimer’s Research Trust (Y-ML). The Authors acknowledge the MSK Cancer Center Support Grant/Core Grant (Grant P30 CA008748), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbas, N., Bednar, I., Mix, E., Marie, S., Paterson, D., Ljungberg, A., et al. (2002). Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J. Neuroimmunol. 126, 50–57.
Adams, K. L., and Gallo, V. (2018). The diversity and disparity of the glial scar. Nat. Neurosci. 21, 9–15. doi: 10.1038/s41593-017-0033-9
Aikawa, T., Holm, M. L., and Kanekiyo, T. (2018). ABCA7 and pathogenic pathways of Alzheimer’s disease. Brain Sci. 8:E27. doi: 10.3390/brainsci8020027
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Albensi, B. C., and Mattson, M. P. (2000). Evidence for the involvement of TNF and NF-?B in hippocampal synaptic plasticity. Synapse 35, 151–159.
Allen, N. J., and Eroglu, C. (2017). Cell biology of Astrocyte-synapse interactions. Neuron 96, 697–708. doi: 10.1016/j.neuron.2017.09.056
Alonso, R., Pisa, D., Marina, A. I., Morato, E., Rábano, A., and Carrasco, L. (2014). Fungal infection in patients with Alzheimer’s disease. J. Alzheimers Dis. 41, 301–311. doi: 10.3233/JAD-132681
Alzheimer’s Association, (2015). 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384. doi: 10.1016/j.jalz.2015.02.003
Anderson, M. A., Ao, Y., and Sofroniew, M. V. (2014). Heterogeneity of reactive astrocytes. Neurosci. Lett. 565, 23–29. doi: 10.1016/j.neulet.2013.12.030
Anderson, M. A., Burda, J. E., Ren, Y., Ao, Y., O’Shea, T. M., Kawaguchi, R., et al. (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. doi: 10.1038/nature17623
Apelt, J., and Schliebs, R. (2001). Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 894, 21–30.
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215. doi: 10.1016/S0166-2236(98)01349-6
Arcuri, C., Mecca, C., Bianchi, R., Giambanco, I., and Donato, R. (2017). The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 10:191. doi: 10.3389/fnmol.2017.00191
Arnusch, C. J., Branderhorst, H., de Kruijff, B., Liskamp, R. M., Breukink, E., and Pieters, R. J. (2007). Enhanced membrane pore formation by multimeric/oligomeric antimicrobial peptides. Biochemistry 46, 13437–13442. doi: 10.1021/bi7015553
Asselineau, D., Benlhassan, K., Arosio, B., Mari, D., Ferri, E., Casati, M., et al. (2015). Interleukin-10 Production in response to amyloid-β differs between slow and fast decliners in patients with Alzheimer’s disease. J. Alzheimer Dis. 46, 837–842. doi: 10.3233/JAD-142832
Attwell Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Balin, B. J., Gerard, H. C., Arking, E. J., Appelt, D. M., Branigan, P. J., Abrams, J. T., et al. (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 187, 23–42.
Baranowska-Bik, A., Bik, W., Wolinska-Witort, E., Martynska, L., Chmielowska, M., and Barcikowska, M. (2008). Plasma beta amyloid and cytokine profile in women with Alzheimer’s disease. Neuro. Endocrinol. Lett. 29, 75–79.
Baranzini, E., and Oksenberg, J. R. (2017). The genetics of multiple sclerosis: from 0 to 200 in 50 years. Trends Gene. 33, 960–970. doi: 10.1016/j.tig.2017.09.004
Bard, F., Barbour, R., Cannon, C., Carretto, R., Fox, M., Games, D., et al. (2003). Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc. Natl. Acad. Sci. U.S.A. 100, 2023–2028. doi: 10.1073/pnas.0436286100
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Bazargani, N., and Attwell, D. (2016). Astrocyte calcium signaling: the third wave. Nat. Neurosci. 19, 182–189. doi: 10.1038/nn.4201
Ben Haim, L., and Rowitch, D. H. (2017). Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 18, 31–41. doi: 10.1038/nrn.2016.159
Benzing, W. C., Wujek, J. R., Ward, E. K., Shaffer, D., Ashe, K. H., Younkin, S. G., et al. (1999). Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 20, 581–589.
Berlau, D. J., Corrada, M. M., Head, E., and Kawas, C. H. (2009). APOE 2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology 72, 829–834. doi: 10.1212/01.wnl.0000343853.00346.a4
Bernardino, L., Agasse, F., Silva, B., Ferreira, R., Grade, S., and Malva, J. O. (2008). Tumor necrosis factor-α modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells 26, 2361–2371. doi: 10.1634/stemcells.2007-0914
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi: 10.1016/j.ajhg.2008.10.008
Bis, J. C., Jian, X., Kunkle, B. W., Chen, Y., Hamilton-Nelson, K. L., Bush, W. S., et al. (2018). Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 1–17. doi: 10.1038/s41380-018-0112-7
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Bollmann, L., Koser, D. E., Shahapure, R., Gautier, H. O., Holzapfel, G. A., Scarcelli, G., et al. (2015). Microglia mechanics: immune activation alters traction forces and durotaxis. Front. Cell Neurosci. 9:363. doi: 10.3389/fncel.2015.00363
Borsini, A., Zunszain, P. A., Thuret, S., and Pariante, C. M. (2015). The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 38, 145–157. doi: 10.1016/J.TINS.2014.12.006
Boulanger, L. M. (2009). Immune proteins in brain development and synaptic plasticity. Neuron 64, 93–109. doi: 10.1016/J.NEURON.2009.09.001
Bradshaw, E. M., Chibnik, L. B., Keenan, B. T., Ottoboni, L., Raj, T., Tang, A., et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. doi: 10.1038/nn.3435
Brosseron, F., Krauthausen, M., Kummer, M., and Heneka, M. T. (2014). Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol. Neurobiol. 50, 534–544. doi: 10.1007/s12035-014-8657-1
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Burns, A., Rossor, M., Hecker, J., Gauthier, S., Petit, H., Möller, H. J., et al. (1999). The effects of donepezil in Alzheimer’s disease - results from a multinational trial. Dement. Geriatr. Cogn. Disord. 10, 237–244. doi: 10.1159/000017126
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Cao, J., Hou, J., Ping, J., and Cai, D. (2018). Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol. Neurodegener. 13:64. doi: 10.1186/s13024-018-0299-8
Cao, W., and Zheng, H. (2018). Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 13:51. doi: 10.1186/s13024-018-0284-2
Carrasquillo, M. M., Belbin, O., Hunter, T. A., Ma, L., Bisceglio, G. D., Zou, F., et al. (2011). Replication of EPHA1 and CD33 associations with late-onset Alzheimer’s disease: a multi-centre case-control study. Mol. Neurodegener. 6:54. doi: 10.1186/1750-1326-6-54
Ceyzeriat, K., Abjean, L., Carrillo-de Sauvage, M. A., Ben Haim, L., and Escartin, C. (2016). The complex STATes of astrocyte reactivity: how are they controlled by the JAK-STAT3 pathway? Neuroscience 330, 205–218. doi: 10.1016/j.neuroscience.2016.05.043
Chai, Q., Jovasevic, V., Malikov, V., Sabo, Y., Morham, S., Walsh, D., et al. (2017). HIV-1 counteracts an innate restriction by amyloid precursor protein resulting in neurodegeneration. Nat. Commun. 8:1522. doi: 10.1038/s41467-017-01795-8
Chakrabarty, P., Herring, A., Ceballos-Diaz, C., Das, P., and Golde, T. E. (2011). Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 6:16. doi: 10.1186/1750-1326-6-16
Chakrabarty, P., Li, A., Ceballos-Diaz, C., Eddy, J. A., Funk, C. C., Moore, B., et al. (2015). IL-10 alters immunoproteostasis in APP Mice, increasing plaque burden and worsening cognitive behavior. Neuron 85, 519–533. doi: 10.1016/J.NEURON.2014.11.020
Chao, C. C., Hu, S., Frey, W. H., Ala, T. A., Tourtellotte, W. W., Peterson, P. K., et al. (1994). Transforming growth factor beta in Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1, 109–110.
Chen, J. H., Ke, K. F., Lu, J. H., Qiu, Y. H., and Peng, Y. P. (2015). Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1–42-Induced Alzheimer’s disease model rats. PLoS One 10:e0116549. doi: 10.1371/journal.pone.0116549
Cheng-Hathaway, J., Reed-Geaghan, E. G., Jay, T. R., Casali, B. T., Bemiller, S. M., Puntambekar, S. S., et al. (2018). The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 13:29. doi: 10.1186/s13024-018-0262-8
Cherry, D., Olschowka, J. A., and O’Banion, M. K. (2015). Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J. Neuroinflamm. 12:203. doi: 10.1186/s12974-015-0411-8
Choi, S. S., Lee, H. J., Lim, I., Satoh, J., and Kim, S. U. (2014). Human astrocytes: secretome profiles of cytokines and chemokines. PLoS One 9:e92325. doi: 10.1371/journal.pone.0092325
Colonna, M. (2003). TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453. doi: 10.1038/nri1106
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
Colonna, M., and Wang, Y. (2016). TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207. doi: 10.1038/nrn.2016.7
Colton, C. A., Mott, R. T., Sharpe, H., Xu, Q., Van Nostrand, W. E., and Vitek, M. P. (2006). Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J. Neuroinflamm. 3:27. doi: 10.1186/1742-2094-3-27
Constam, D. B., Philipp, J., Malipiero, U. V., ten Dijke, P., Schachner, M., and Fontana, A. (1992). Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 148, 1404–1410.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–323
Craft, J. M., Watterson, D. M., Frautschy, S. A., and Eldik, L. J. V. (2004). Aminopyridazines inhibit β-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol. Aging 25, 1283–1292. doi: 10.1016/j.neurobiolaging.2004.01.006
Cribbs, D. H., Berchtold, N. C., Perreau, V., Coleman, P. D., Rogers, J., Tenner, A. J., et al. (2012). Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J. Neuroinflamm. 9:643. doi: 10.1186/1742-2094-9-179
Crocker, R., McMillan, S. J., and Richards, H. E. (2012). CD33-related siglecs as potential modulators of inflammatory responses. Ann. N. Y. Acad. Sci. 1253, 102–111. doi: 10.1111/j.1749-6632.2011.06449.x
Crow, Y. J., Hayward, B. E., Parmar, R., Robins, P., Leitch, A., Ali, M., et al. (2006a). Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920. doi: 10.1038/ng1845
Crow, Y. J., Leitch, A., Hayward, B. E., Garner, A., Parmar, R., Griffith, E., et al. (2006b). Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 38, 910–916. doi: 10.1038/ng1842
Cudaback, E., Li, X., Yang, Y., Yoo, T., Montine, K. S., Craft, S., et al. (2012). Apolipoprotein C-I is an APOE genotype-dependent suppressor of glial activation. J. Neuroinflamm. 9:192. doi: 10.1186/1742-2094-9-192
Cunha, L. D., Yang, M., Carter, R., Guy, C., Harris, L., Crawford, J. C., et al. (2018). LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175:429-441.e16. doi: 10.1016/j.cell.2018.08.061
Cunningham, C., Campion, S., Teeling, J., Felton, L., and Perry, V. H. (2007). The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C). Brain Behav. Immun. 21, 490–502. doi: 10.1016/J.BBI.2006.12.007
Das, S., and Potter, H. (1995). Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron 14, 447–456.
de Craen, J., Gussekloo, J., Vrijsen, B., and Westendorp, R. G. (2005). Meta-analysis of nonsteroidal antiinflammatory drug use and risk of dementia. Am. J. Epidemiol. 161, 114–120. doi: 10.1093/aje/kwi029
De Virgilio, A., Greco, A., Fabbrini, G., Inghilleri, M., Rizzo, M. I., Gallo, A., et al. (2016). Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 15, 1005–1011. doi: 10.1016/j.autrev.2016.07.022
de Weerd, N. A., Vivian, J. P., Nguyen, T. K., Mangan, N. E., Gould, J. A., Braniff, S. J., et al. (2013). Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat. Immunol. 14, 901–907. doi: 10.1038/ni.2667
Deming, Y., Filipello, F., Cignarella, F., Cantoni, C., Hsu, S., Mikesell, R., et al. (2019). The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 11:eaau2291. doi: 10.1126/scitranslmed.aau2291
Detrait, E. R., Danis, B., Lamberty, Y., and Foerch, P. (2014). Peripheral administration of an anti-TNF-α receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-α levels and memory deficits in mice. Neurochem. Int. 72, 10–13. doi: 10.1016/J.NEUINT.2014.04.001
DeTure, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14:32. doi: 10.1186/s13024-019-0333-5
Dewapriya, P., Li, Y. X., Himaya, S. W. A., Pangestuti, R., and Kim, S. K. (2013). Neoechinulin A suppresses amyloid-β oligomer-induced microglia activation and thereby protects PC-12 cells from inflammation-mediated toxicity. Neuro. Toxicol. 35, 30–40. doi: 10.1016/j.neuro.2012.12.004
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
Dorszewska, J., Prendecki, M., Oczkowska, A., Dezor, M., and Kozubski, W. (2016). Molecular basis of familial and sporadic Alzheimer’s disease. Curr, Alzheimer Res. 13, 952–963.
Dos Santos, L. R., Pimassoni, L. H. S., Sena, G. G. S., Camporez, D., Belcavello, L., Trancozo, M., et al. (2017). Validating GWAS Variants from microglial genes implicated in Alzheimer’s disease. J. Mol. Neurosci. 62, 215–221. doi: 10.1007/s12031-017-0928-7
Duce, J. A., Hollander, W., Jaffe, R., and Abraham, C. R. (2006). Activation of early components of complement targets myelin and oligodendrocytes in the aged rhesus monkey brain. Neurobiol. Aging 27, 633–44. doi: 10.1016/j.neurobiolaging.2005.03.027
Dzamko, N., Geczy, C. L., and Halliday, G. M. (2015). Inflammation is genetically implicated in Parkinson’s disease. Neuroscience 302, 89–102. doi: 10.1016/j.neuroscience.2014.10.028
Efthymiou, G., and Goate, A. M. (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Eimer, W. A., Vijaya Kumar, D. K., Navalpur Shanmugam, N. K., Rodriguez, A. S., Mitchell, T., Washicosky, K. J., et al. (2018). Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99:53-63.e3. doi: 10.1016/j.neuron.2018.06.030
Ejlerskov, P., Jeanette Hultberg, G., Wang, J., Carlsson, R., Ambjørn, M., Kuss, M., et al. (2015). Lack of neuronal IFN-β-IFNAR causes lewy body- and Parkinson’s disease-like dementia. Cell 163, 324–339. doi: 10.1016/J.CELL.2015.08.069
Ershler, W. B., and Keller, E. T. (2000). Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu. Rev. Med. 51, 245–270. doi: 10.1146/annurev.med.51.1.245
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145. doi: 10.1016/j.it.2007.01.005
Farlow, M. R., Andreasen, N., Riviere, M. E., Vostiar, I., Vitaliti, A., Sovago, J., et al. (2015). Long-term treatment with active Abeta immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res. Ther. 7:23. doi: 10.1186/s13195-015-0108-3
Faulkner, R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B., and Sofroniew, M. V. (2004). Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24, 2143–2155. doi: 10.1523/jneurosci.3547-03.2004
Feghali, C. A., and Wright, T. M. (1997). Cytokines in acute and chronic inflammation. Front. Biosci. 2:d12–d26. doi: 10.2741/a171
Ferreira, S. A., and Romero-Ramos, M. (2018). Microglia response during Parkinson’s disease: Alpha-Synuclein intervention. Front. Cell. Neurosci. 12:247–247. doi: 10.3389/fncel.2018.00247
Ferreira-Vieira, T. H., Guimaraes, I. M., Silva, F. R., and Ribeiro, F. M. (2016). Alzheimer’s disease: targeting the cholinergic system. Curr. Neuropharmacol. 14, 101–115.
Flannagan, R. S., Jaumouille, V., and Grinstein, S. (2012). The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98. doi: 10.1146/annurev-pathol-011811-132445
Folch, J., Petrov, D., Ettcheto, M., Abad, S., Sanchez-Lopez, E., Garcia, M. L., et al. (2016). Current research therapeutic strategies for Alzheimer’s disease treatment. Neural. Plast. 2016:8501693. doi: 10.1155/2016/8501693
Fontainhas, M., Wang, M., Liang, K. J., Chen, S., Mettu, P., Damani, M., et al. (2011). Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One 6:e15973. doi: 10.1371/journal.pone.0015973
Foster, E. M., Dangla-Valls, A., Lovestone, S., Ribe, E. M., and Buckley, N. J. (2019). Clusterin in Alzheimer’s disease: mechanisms, genetics, and lessons from other pathologies. Front. Neurosci. 13:164. doi: 10.3389/fnins.2019.00164
Frost, R., and Li, Y. M. (2017). The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 7:170228. doi: 10.1098/rsob.170228
Fultz, M. J., Barber, S. A., Dieffenbach, C. W., and Vogel, S. N. (1993). Induction of IFN-gamma in macrophages by lipopolysaccharide. Int. Immunol. 5, 1383–1392. doi: 10.1093/intimm/5.11.1383
Furlan, R., Brambilla, E., Ruffini, F., Poliani, P. L., Bergami, A., Marconi, P. C., et al. (2001). Intrathecal delivery of IFN-gamma protects C57BL/6 mice from chronic-progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system-infiltrating lymphocytes. J. Immunol. 167, 1821–1829. doi: 10.4049/jimmunol.167.3.1821
Gabbita, S. P., Johnson, M. F., Kobritz, N., Eslami, P., Poteshkina, A., Varadarajan, S., et al. (2015). Oral TNFα modulation alters neutrophil infiltration, improves cognition and diminishes tau and amyloid pathology in the 3xTgAD mouse model. PLoS One 10:e0137305. doi: 10.1371/journal.pone.0137305
Galimberti, D., Venturelli, E., Fenoglio, C., Guidi, I., Villa, C., Bergamaschini, L., et al. (2008). Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer’s disease and frontotemporal lobar degeneration. J. Neurol. 255, 539–544. doi: 10.1007/s00415-008-0737-6
Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., et al. (2006). Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. doi: 10.1001/archpsyc.63.2.168
Ghosh, S., Wu, M. D., Shaftel, S. S., Kyrkanides, S., LaFerla, F. M., Olschowka, J. A., et al. (2013). Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 33, 5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013
Gilman, S., Koller, M., Black, R. S., Jenkins, L., Griffith, S. G., Fox, N. C., et al. (2005). Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7:a020537. doi: 10.1101/cshperspect.a020537
Ginhoux, F., Lim, S., Hoeffel, G., Low, D., and Huber, T. (2013). Origin and differentiation of microglia. Front. Cell. Neurosci. 7:45. doi: 10.3389/fncel.2013.00045
Goshen, I., Kreisel, T., Ounallah-Saad, H., Renbaum, P., Zalzstein, Y., Ben-Hur, T., et al. (2007). A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 32, 1106–1115. doi: 10.1016/J.PSYNEUEN.2007.09.004
Gough, D. J., Messina, N. L., Clarke, C. J., Johnstone, R. W., and Levy, D. E. (2012). Constitutive Type I interferon modulates homeostatic balance through tonic signaling. Immunity 36, 166–174. doi: 10.1016/J.IMMUNI.2012.01.011
Gratuze, M., Leyns, C. E. G., and Holtzman, D. M. (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13:66. doi: 10.1186/s13024-018-0298-9
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., et al. (2013). Alzheimer’s disease risk Gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615. doi: 10.1073/PNAS.86.19.7611
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. New Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guillot-Sestier, M. V., Doty, K. R., Gate, D., Rodriguez, J. Jr., Leung, B. P., Rezai-Zadeh, K., et al. (2015). Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 85, 534–548. doi: 10.1016/J.NEURON.2014.12.068
Halassa, M., Florian, C., Fellin, T., Munoz, J. R., Lee, S. Y., Abel, T., et al. (2009). Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61, 213–219. doi: 10.1016/J.NEURON.2008.11.024
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hamby, M. E., Coppola, G., Ao, Y., Geschwind, D. H., Khakh, B. S., and Sofroniew, M. V. (2012). Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J. Neurosci. 32, 14489–14510. doi: 10.1523/JNEUROSCI.1256-12.2012
Han, J., Kesner, P., Metna-Laurent, M., Duan, T., Xu, L., Georges, F., et al. (2012). Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 148, 1039–1050. doi: 10.1016/J.CELL.2012.01.037
Haney, F., Mansour, S. C., and Hancock, R. E. (2017). Antimicrobial peptides: an introduction. Methods Mol. Biol. 1548, 3–22. doi: 10.1007/978-1-4939-6737-7-1
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hassanzadeh, K., and Rahimmi, A. (2019). Oxidative stress and neuroinflammation in the story of Parkinson’s disease: could targeting these pathways write a good ending? J. Cell. Physiol. 234, 23–32. doi: 10.1002/jcp.26865
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet. Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Herrup, K., and Yang, Y. (2007). Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378. doi: 10.1038/nrn2124
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Honda, K., Yanai, H., Negishi, H., Asagiri, M., Sato, M., Mizutani, T., et al. (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. doi: 10.1038/nature03464
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hu, J., Akama, K. T., Krafft, G. A., Chromy, B. A., and Van Eldik, L. J. (1998). Amyloid-β peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 785, 195–206. doi: 10.1016/S0006-8993(97)01318-8
Huberman, M., Shalit, F., Roth-Deri, I., Gutman, B., Brodie, C., Kott, E., et al. (1994). Correlation of cytokine secretion by mononuclear cells of Alzheimer patients and their disease stage. J. Neuroimmunol. 52, 147–152. doi: 10.1016/0165-5728(94)90108-2
Hull, M., Berger, M., Volk, B., and Bauer, J. (1996). Occurrence of interleukin-6 in cortical plaques of Alzheimer’s disease patients may precede transformation of diffuse into Neuritic Plaquesa. Ann. N. Y. Acad. Sci. 777, 205–212. doi: 10.1111/j.1749-6632.1996.tb34420.x
Itagaki, S., McGeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182
James, B. D., Leurgans, S. E., Hebert, L. E., Scherr, P. A., Yaffe, K., and Bennett, D. A. (2014). Contribution of Alzheimer disease to mortality in the United States. Neurology 82, 1045–1050. doi: 10.1212/wnl.0000000000000240
Janelsins, M. C., Mastrangelo, M. A., Park, K. M., Sudol, K. L., Narrow, W. C., Oddo, S., et al. (2008). Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD Mice. Am. J. Pathol. 173, 1768–1782. doi: 10.2353/AJPATH.2008.080528
Jay, T. R., Hirsch, A. M., Broihier, M. L., Miller, C. M., Neilson, L. E., Ransohoff, R. M., et al. (2017a). Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37, 637–647. doi: 10.1523/JNEUROSCI.2110-16.2016
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jay, T. R., von Saucken, V. E., and Landreth, G. E. (2017b). TREM2 in neurodegenerative diseases. Mol. Neurodegener. 12:56. doi: 10.1186/s13024-017-0197-5
Jekabsone, A., Mander, P. K., Tickler, A., Sharpe, M., and Brown, G. C. (2006). Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J. Neuroinflamm. 3:24. doi: 10.1186/1742-2094-3-24
Jiang, T., Yu, J. T., Hu, N., Tan, M. S., Zhu, X. C., and Tan, L. (2014). CD33 in Alzheimer’s disease. Mol. Neurobiol. 49, 529–535. doi: 10.1007/s12035-013-8536-1
Johanson, C. E., Duncan, J. A. III, Klinge, P. M., Brinker, T., Stopa, E. G., and Silverberg, G. D. (2008). Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res. 5:10. doi: 10.1186/1743-8454-5-10
Johnstone, M., Gearing, A. J., and Miller, K. M. (1999). A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 93, 182–193.
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. New Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Justicia, C., Gabriel, C., and Planas, A. M. (2000). Activation of the JAK/STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes. Glia 30, 253–270
Kamphuis, W., Mamber, C., Moeton, M., Kooijman, L., Sluijs, J. A., Jansen, A. H. P., et al. (2012). GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7:e42823. doi: 10.1371/journal.pone.0042823
Karch, M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Karperien, A., Ahammer, H., and Jelinek, H. F. (2013). Quantitating the subtleties of microglial morphology with fractal analysis. Front. Cell. Neurosci. 7:3. doi: 10.3389/fncel.2013.00003
Katsurabayashi, S., Kawano, H., Ii, M., Nakano, S., Tatsumi, C., Kubota, K., et al. (2016). Overexpression of Swedish mutant APP in aged astrocytes attenuates excitatory synaptic transmission. Physiol. Rep. 4:e12665. doi: 10.14814/phy2.12665
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kierdorf, K., and Prinz, M. (2013). Factors regulating microglia activation. Front. Cell. Neurosci. 7:44. doi: 10.3389/fncel.2013.00044
Kim, D. K., Park, J., Han, D., Yang, J., Kim, A., Woo, J., et al. (2018). Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 13:2. doi: 10.1186/s13024-017-0234-4
Kim, J., Basak, J. M., and Holtzman, D. M. (2009). The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303. doi: 10.1016/j.neuron.2009.06.026
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590. doi: 10.1016/j.trci.2018.06.014
Kitamura, Y., Yanagisawa, D., Takata, K., and Taniguchi, T. (2009). Neuroprotective function in brain microglia. Curr. Anaesth. Crit. Care 20, 142–147.
Kleinberger, G., Yamanishi, Y., Suárez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6:243ra86. doi: 10.1126/scitranslmed.3009093
Kocur, M., Schneider, R., Pulm, A. K., Bauer, J., Kropp, S., Gliem, M., et al. (2015). IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol. Commun. 3:20. doi: 10.1186/s40478-015-0192-4
Kraft, W., Hu, X., Yoon, H., Yan, P., Xiao, Q., Wang, Y., et al. (2013). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198. doi: 10.1096/fj.12-208660
Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., et al. (2012). Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflamm. 9:151. doi: 10.1186/1742-2094-9-151
Lajaunias, F., Dayer, J. M., and Chizzolini, C. (2005). Constitutive repressor activity of CD33 on human monocytes requires sialic acid recognition and phosphoinositide 3-kinase-mediated intracellular signaling. Eur. J. Immunol. 35, 243–251. doi: 10.1002/eji.200425273
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lauro, C., Catalano, M., Trettel, F., and Limatola, C. (2015). Fractalkine in the nervous system: neuroprotective or neurotoxic molecule? Ann. N. Y. Acad. Sci. 1351, 141–148. doi: 10.1111/nyas.12805
Lawson, J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-W
Lee, S., Zimmerman, M. E., Narkhede, A., Nasrabady, S. E., Tosto, G., Meier, I. B., et al. (2018). White matter hyperintensities and the mediating role of cerebral amyloid angiopathy in dominantly-inherited Alzheimer’s disease. PLoS One 13:e0195838. doi: 10.1371/journal.pone.0195838
Letenneur, L., Peres, K., Fleury, H., Garrigue, I., Barberger-Gateau, P., Helmer, C., et al. (2008). Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: a population-based cohort study. PLoS One 3:e3637. doi: 10.1371/journal.pone.0003637
Leyns, E. G., and Holtzman, D. M. (2017). Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 12:50. doi: 10.1186/s13024-017-0192-x
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. doi: 10.1038/nri.2017.125
Li, X., Shen, N., Zhang, S., Liu, J., Jiang, Q., Liao, M., et al. (2015). CD33 rs3865444 polymorphism contributes to Alzheimer’s disease susceptibility in Chinese, European, and North American Populations. Mol. Neurobiol. 52, 414–421. doi: 10.1007/s12035-014-8880-9
Li, X., Thome, S., Ma, X., Amrute-Nayak, M., Finigan, A., Kitt, L., et al. (2017). MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun. 8:15986. doi: 10.1038/ncomms15986
Licastro, F., Grimaldi, L. M. E., Bonafè, M., Martina, C., Olivieri, F., Cavallone, L., et al. (2003). Interleukin-6 gene alleles affect the risk of Alzheimer’s disease and levels of the cytokine in blood and brain. Neurobiol. Aging 24, 921–926. doi: 10.1016/S0197-4580(03)00013-7
Licastro, F., Pedrini, S., Caputo, L., Annoni, G., Davis, L. J., Ferri, C., et al. (2000). Increased plasma levels of interleukin-1, interleukin-6 and α-1-antichymotrypsin in patients with Alzheimer’s disease: peripheral inflammation or signals from the brain? J. Neuroimmunol. 103, 97–102. doi: 10.1016/S0165-5728(99)00226-X
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lind, B. L., Jessen, S. B., Lonstrup, M., Josephine, C., Bonvento, G., and Lauritzen, M. (2018). Fast Ca(2+) responses in astrocyte end-feet and neurovascular coupling in mice. Glia 66, 348–358. doi: 10.1002/glia.23246
Lindenmann, J. (1982). From interference to interferon: a brief historical introduction [and Discussion]. Philos. Trans. R. Soc. B Biol. Sci. 299, 3–6. doi: 10.1098/rstb.1982.0101
Lippa, C. F., Smith, T. W., Saunders, A. M., Hulette, C., Pulaski-Salo, D., and Roses, A. D. (1997). Apolipoprotein E-epsilon 2 and Alzheimer’s disease: genotype influences pathologic phenotype. Neurology 48, 515–519. doi: 10.1212/wnl.48.2.515
Liu, E., Schmidt, M. E., Margolin, R., Sperling, R., Koeppe, R., Mason, N. S., et al. (2015). Amyloid-beta 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology 85, 692–700. doi: 10.1212/WNL.0000000000001877
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-κB signaling in inflammation. Signal Trans. Target. Ther. 2:17023. doi: 10.1038/sigtrans.2017.23
Liu, Y., Walter, S., Stagi, M., Cherny, D., Letiembre, M., Schulz-Schaeffer, W., et al. (2005). LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 128, 1778–1789. doi: 10.1093/brain/awh531
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Louveau, A., Harris, T. H., and Kipnis, J. (2015). Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577. doi: 10.1016/J.IT.2015.08.006
Lue, L. F., Brachova, L., Civin, W. H., and Rogers, J. (1996). Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J. Neuropathol. Exp. Neurol. 55, 1083–1088.
Luedecking, E. K., DeKosky, S. T., Mehdi, H., Ganguli, M., and Kamboh, M. I. (2000). Analysis of genetic polymorphisms in the transforming growth factor-beta1 gene and the risk of Alzheimer’s disease. Hum. Genet. 106, 565–569.
Ma, J., Yu, J. T., and Tan, L. (2015). MS4A cluster in Alzheimer’s disease. Mol. Neurobiol. 51, 1240–1248. doi: 10.1007/s12035-014-8800-z
Ma, Q., Zhao, Z., Sagare, A. P., Wu, Y., Wang, M., Owens, N. C., et al. (2018). Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 13:57. doi: 10.1186/s13024-018-0286-0
Main, B. S., Zhang, M., Brody, K. M., Kirby, F. J., Crack, P. J., and Taylor, J. M. (2017). Type-I interferons mediate the neuroinflammatory response and neurotoxicity induced by rotenone. J. Neurochem. 141, 75–85. doi: 10.1111/jnc.13940
Malarkey, E. B., and Parpura, V. (2008). Mechanisms of glutamate release from astrocytes. Neurochem. Int. 52, 142–154. doi: 10.1016/j.neuint.2007.06.005
Malik, M., Simpson, J. F., Parikh, I., Wilfred, B. R., Fardo, D. W., Nelson, P. T., et al. (2013). CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and Exon 2 splicing. J. Neurosci. 33, 13320–13325. doi: 10.1523/JNEUROSCI.1224-13.2013
Manouchehrinia, A., and Constantinescu, C. S. (2012). Cost-effectiveness of disease-modifying therapies in multiple sclerosis. Curr. Neurol. Neurosci. Rep. 12, 592–600. doi: 10.1007/s11910-012-0291-6
Marr, R. A., Rockenstein, E., Mukherjee, A., Kindy, M. S., Hersh, L. B., Gage, F. H., et al. (2003). Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996.
Martineau, M., Parpura, V., and Mothet, J. P. (2014). Cell-type specific mechanisms of D-serine uptake and release in the brain. Front. Syn. Neurosci. 6:12. doi: 10.3389/fnsyn.2014.00012
Matcovitch-Natan, O., Winter, D. R., Giladi, A., Vargas Aguilar, S., Spinrad, A., Sarrazin, S., et al. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science 353:aad8670. doi: 10.1126/science.aad8670
Mawuenyega, G., Sigurdson, W., Ovod, V., Munsell, L., Kasten, T., Morris, J. C., et al. (2010). Decreased Clearance of CNS -amyloid in Alzheimer’s disease. Science 330, 1774–1774. doi: 10.1126/science.1197623
McGeer, L., and McGeer, E. G. (1995). The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 21, 195–218.
McQuade, A., and Blurton-Jones, M. (2019). Microglia in Alzheimer’s disease: exploring how genetics and phenotype influence risk. J. Mol. Biol. 431, 1805–1817. doi: 10.1016/j.jmb.2019.01.045
Mezyk-Kopec, R., Bzowska, M., Stalinska, K., Chelmicki, T., Podkalicki, M., Jucha, J., et al. (2009). Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts. Cytokine 46, 309–315. doi: 10.1016/j.cyto.2009.03.002
Michaud, J. P., Halle, M., Lampron, A., Theriault, P., Prefontaine, P., Filali, M., et al. (2013). Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. U.S.A. 110, 1941–1946. doi: 10.1073/pnas.1215165110
Miyake, K. (2007). Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 19, 3–10. doi: 10.1016/j.smim.2006.12.002
Moir, R. D., Lathe, R., and Tanzi, R. E. (2018). The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 14, 1602–1614.
Moraga-Amaro, R., Jerez-Baraona, J. M., Simon, F., and Stehberg, J. (2014). Role of astrocytes in memory and psychiatric disorders. J. Physiol. Paris 108, 240–251. doi: 10.1016/J.JPHYSPARIS.2014.08.005
Morgan, D. (2006). Modulation of microglial activation state following passive immunization in amyloid depositing transgenic mice. Neurochem. Int. 49, 190–194. doi: 10.1016/j.neuint.2006.03.017
Morris, G. P., Clark, I. A., Zinn, R., and Vissel, B. (2013). Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 105, 40–53. doi: 10.1016/J.NLM.2013.07.002
Morrow, J. A., Segall, M. L., Lund-Katz, S., Phillips, M. C., Knapp, M., Rupp, B., et al. (2000). Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry 39, 11657–11666. doi: 10.1021/bi000099m
Mrak, R. E. (2012). Microglia in Alzheimer Brain: a neuropathological perspective. Int. J. Alzheimers Dis. 2012, 1–6. doi: 10.1155/2012/165021
Muldoon, L. L., Alvarez, J. I., Begley, D. J., Boado, R. J., Del Zoppo, G. J., Doolittle, N. D., et al. (2013). Immunologic privilege in the central nervous system and the blood-brain barrier. J. Cereb. Blood Flow Metab. 33, 13–21. doi: 10.1038/jcbfm.2012.153
Naj, A. C., Jun, G., Beecham, G. W., Wang, L. S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Najm, R., Jones, E. A., and Huang, Y. (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol. Neurodegener. 14:24. doi: 10.1186/s13024-019-0324-6
Nasrabady, S. E., Rizvi, B., Goldman, J. E., and Brickman, A. M. (2018). White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6:22. doi: 10.1186/s40478-018-0515-3
Navarrete, M., Perea, G., Fernandez de Sevilla, D., Gómez-Gonzalo, M., Núñez, A., Martín, E. D., et al. (2012). Astrocytes mediate In Vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10:e1001259. doi: 10.1371/journal.pbio.1001259
Neumann, H., and Takahashi, K. (2007). Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J. Neuroimmunol. 184, 92–99. doi: 10.1016/j.jneuroim.2006.11.032
Neumann, H., Kotter, M. R., and Franklin, R. J. M. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132(Pt 2), 288–295. doi: 10.1093/brain/awn109
Ng, C. T., Mendoza, J. L., Garcia, K. C., and Oldstone, M. B. (2016). Alpha and Beta Type 1 interferon signaling: passage for diverse biologic outcomes. Cell 164, 349–352. doi: 10.1016/J.CELL.2015.12.027
Oberheim, N. A., Wang, X., Goldman, S., and Nedergaard, M. (2006). Astrocytic complexity distinguishes the human brain. Trends Neurosci. 29, 547–553. doi: 10.1016/j.tins.2006.08.004
Okigaki, M., Davis, C., Falasca, M., Harroch, S., Felsenfeld, D. P., Sheetz, M. P., et al. (2003). Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc. Natl. Acad. Sci. U.S.A. 100, 10740–10745. doi: 10.1073/pnas.1834348100
Olah, M., Biber, K., Vinet, J., and Boddeke, H. W. (2011). Microglia phenotype diversity. CNS Neurol. Disord. Drug Targets 10, 108–118. doi: 10.2174/187152711794488575
Pankiewicz, J. E., Baquero-Buitrago, J., Sanchez, S., Lopez-Contreras, J., Kim, J., Sullivan, P. M., et al. (2017). APOE genotype differentially modulates effects of Anti-Abeta, passive immunization in APP transgenic mice. Mol. Neurodegener. 12:12. doi: 10.1186/s13024-017-0156-1
Panza, F., Solfrizzi, V., Frisardi, V., Imbimbo, B. P., Capurso, C., D’Introno, A., et al. (2009). Beyond the neurotransmitter-focused approach in treating Alzheimer’s disease: drugs targeting beta-amyloid and tau protein. Aging Clin. Exp. Res. 21, 386–406.
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/J.CELL.2013.11.030
Paushter, D. H., Du, H., Feng, T., and Hu, F. (2018). The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 136, 1–17. doi: 10.1007/s00401-018-1861-8
Pellicano, R. (2014). Gastrointestinal damage by non-steroidal anti-inflammatory drugs: updated clinical considerations. Minerva Gastroenterol. Dietol. 60, 255–261.
Pimenova, A. A., Raj, T., and Goate, A. M. (2018). Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 83, 300–310. doi: 10.1016/j.biopsych.2017.05.014
Platanias, L. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Poveda, J., Sanz, A. B., Carrasco, S., Ruiz-Ortega, M., Cannata-Ortiz, P., Sanchez-Nino, M. D., et al. (2017). Bcl3: a regulator of NF-kappaB inducible by TWEAK in acute kidney injury with anti-inflammatory and antiapoptotic properties in tubular cells. Exp. Mol. Med. 49:e352. doi: 10.1038/emm.2017.89
Qin, L., Liu, Y., Cooper, C., Liu, B., Wilson, B., and Hong, J. S. (2002). Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 83, 973–983.
Qizilbash, N., Gregson, J., Johnson, M. E., Pearce, N., Douglas, I., Wing, K., et al. (2015). BMI and risk of dementia in two million people over two decades: a retrospective cohort study. Lancet Diabetes Endocrinol. 3, 431–436. doi: 10.1016/s2213-8587(15)00033-9
Quan, N., and Banks, W. A. (2007). Brain-immune communication pathways. Brain Behav. Immun. 21, 727–735. doi: 10.1016/J.BBI.2007.05.005
Raj, D., Yin, Z., Breur, M., Doorduin, J., Holtman, I. R., Olah, M., et al. (2017). Increased white matter inflammation in aging- and Alzheimer’s disease brain. Front. Mol. Neurosci. 10:206. doi: 10.3389/fnmol.2017.00206
Rangaraju, S., Dammer, E. B., Raza, S. A., Gao, T., Xiao, H., Betarbet, R., et al. (2018a). Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol. Neurodegener. 13:34. doi: 10.1186/s13024-018-0266-4
Rangaraju, S., Dammer, E. B., Raza, S. A., Rathakrishnan, P., Xiao, H., Gao, T., et al. (2018b). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 13:24. doi: 10.1186/s13024-018-0254-8
Ransohoff, R. M., and Cardona, A. E. (2010). The myeloid cells of the central nervous system parenchyma. Nature 468, 253–262. doi: 10.1038/nature09615
Reisberg, B., Doody, R., Stoffler, A., Schmitt, F., Ferris, S., and Mobius, H. J. (2006). A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease. Arch. Neurol. 63, 49–54. doi: 10.1001/archneur.63.1.49
Rho, M. B., Wesselingh, S., Glass, J. D., Mcarthur, J. C., Choi, S., Griffin, J., et al. (1995). A potential role for interferon-α in the pathogenesis of HIV-associated dementia. Brain Behav. Immun. 9, 366–377. doi: 10.1006/BRBI.1995.1034
Riedel, B. C., Thompson, P. M., and Brinton, R. D. (2016). Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 160, 134–147. doi: 10.1016/j.jsbmb.2016.03.012
Robel, S., Mori, T., Zoubaa, S., Schlegel, J., Sirko, S., Faissner, A., et al. (2009). Conditional deletion of beta1-integrin in astroglia causes partial reactive gliosis. Glia 57, 1630–1647. doi: 10.1002/glia.20876
Robinson, D. R. (1983). Prostaglandins and the mechanism of action of anti-inflammatory drugs. Am. J. Med. 75, 26–31. doi: 10.1016/0002-9343(83)90325-x
Rodríguez, J. J., Yeh, C. Y., Terzieva, S., Olabarria, M., Kulijewicz-Nawrot, M., and Verkhratsky, A. (2014). Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23. doi: 10.1016/j.neurobiolaging.2013.07.002
Rogers, J., Li, R., Mastroeni, D., Grover, A., Leonard, B., Ahern, G., et al. (2006). Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol. Aging 27, 1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043
Russo, I., Caracciolo, L., Tweedie, D., Choi, S. H., Greig, N. H., Barlati, S., et al. (2012). 3,6’-Dithiothalidomide, a new TNF-a synthesis inhibitor, attenuates the effect of Aß1-42 intracerebroventricular injection on hippocampal neurogenesis and memory deficit. J. Neurochem. 122, 1181–1192. doi: 10.1111/j.1471-4159.2012.07846.x
Sahlender, D. A., Savtchouk, I., and Volterra, A. (2014). What do we know about gliotransmitter release from astrocytes? Philos. Trans. R. Soc. B Biol. Sci. 369:20130592. doi: 10.1098/rstb.2013.0592
Sao, T., Yoshino, Y., Yamazaki, K., Ozaki, Y., Mori, Y., Ochi, S., et al. (2018). MEF2C mRNA expression and cognitive function in Japanese patients with Alzheimer’s disease. Psychiatr. Clin. Neurosci. 72, 160–167. doi: 10.1111/pcn.12618
Satoh, J. I., Kino, Y., Yanaizu, M., Tosaki, Y., Sakai, K., Ishida, T., et al. (2017). Microglia express ABI3 in the brains of Alzheimer’s disease and Nasu-Hakola disease. Intractable Rare Dis. Res. 6, 262–268. doi: 10.5582/irdr.2017.01073
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/J.NEURON.2012.03.026
Schain, M., and Kreisl, W. C. (2017). Neuroinflammation in neurodegenerative disorders-a review. Curr. Neurol. Neurosci. Rep. 17:25. doi: 10.1007/s11910-017-0733-2
Schreiber, G., and Piehler, J. (2015). The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol. 36, 139–149. doi: 10.1016/J.IT.2015.01.002
Schwab, M. E. (1990). Myelin-associated inhibitors of neurite growth and regeneration in the CNS. Trends Neurosci. 13, 452–456. doi: 10.1016/0166-2236(90)90098-u
Schwartz, M., Kipnis, J., Rivest, S., and Prat, A. (2013). How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 33, 17587–17596. doi: 10.1523/jneurosci.3241-13.2013
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56. doi: 10.1038/nature19323
Shabab, T., Khanabdali, R., Moghadamtousi, S. Z., Kadir, H. A., and Mohan, G. (2017). Neuroinflammation pathways: a general review. Int. J. Neurosci. 127, 624–633. doi: 10.1080/00207454.2016.1212854
Shi, Y., and Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772. doi: 10.1038/s41577-018-0051-1
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Shibata, N., Ohnuma, T., Takahashi, T., Baba, H., Ishizuka, T., Ohtsuka, M., et al. (2002). Effect of IL-6 polymorphism on risk of Alzheimer disease: Genotype-phenotype association study in Japanese cases. Am. J. Med. Genet. 114, 436–439. doi: 10.1002/ajmg.10417
Siemers, E. R., Sundell, K. L., Carlson, C., Case, M., Sethuraman, G., Liu-Seifert, H., et al. (2016). Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 12, 110–120. doi: 10.1016/j.jalz.2015.06.1893
Sierra, A., Abiega, O., Shahraz, A., and Neumann, H. (2013). Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell Neurosci. 7:6. doi: 10.3389/fncel.2013.00006
Sivanandam, T. M., and Thakur, M. K. (2012). Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 36, 1376–1381. doi: 10.1016/j.neubiorev.2012.02.013
Smith, J. A., Das, A., Ray, S. K., and Banik, N. L. (2012). Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. doi: 10.1016/j.brainresbull.2011.10.004
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263. doi: 10.1038/nrn3898
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sole-Domenech, S., Cruz, D. L., Capetillo-Zarate, E., and Maxfield, F. R. (2016). The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 32, 89–103. doi: 10.1016/j.arr.2016.07.002
Sominsky, L., De Luca, S., and Spencer, S. J. (2018). Microglia: key players in neurodevelopment and neuronal plasticity. Int. J. Biochem. Cell Biol. 94, 56–60. doi: 10.1016/j.biocel.2017.11.012
Sommer, A., Winner, B., and Prots, I. (2017). The trojan horse - neuroinflammatory impact of T cells in neurodegenerative diseases. Mol. Neurodegener. 12:78. doi: 10.1186/s13024-017-0222-8
Sood, R., Domanov, Y., Pietiäinen, M., Kontinen, V. P., and Kinnunen, P. K. J. (2008). Binding of LL-37 to model biomembranes: Insight into target vs host cell recognition. Biochim. Biophys. Acta Biomembr. 1778, 983–996. doi: 10.1016/j.bbamem.2007.11.016
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
St George-Hyslop, P. H., Myers, R. H., Haines, J. L., Farrer, L. A., Tanzi, R. E., Abe, K., et al. (1989). Familial Alzheimer’s disease: progress and problems. Neurobiol. Aging 10, 417–425.
Stamm, H., Klingler, F., Grossjohann, E. M., Muschhammer, J., Vettorazzi, E., Heuser, M., et al. (2018a). Immune checkpoints PVR and PVRL2 are prognostic markers in AML and their blockade represents a new therapeutic option. Oncogene 37, 5269–5280. doi: 10.1038/s41388-018-0288-y
Stamm, H., Wellbrock, J., and Fiedler, W. (2018b). Interaction of PVR/PVRL2 with TIGIT/DNAM-1 as a novel immune checkpoint axis and therapeutic target in cancer. Mamm. Genome 29, 694–702. doi: 10.1007/s00335-018-9770-7
Stence, N., Waite, M., and Dailey, M. E. (2001). Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266.
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219. doi: 10.1111/imm.12922
Suarez-Calvet, M., Morenas-Rodriguez, E., Kleinberger, G., Schlepckow, K., Araque Caballero, M. A., Franzmeier, N., et al. (2019). Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-beta pathology. Mol. Neurodegener. 14:1. doi: 10.1186/s13024-018-0301-5
Suresh, R., and Mosser, D. M. (2013). Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 37, 284–291. doi: 10.1152/advan.00058.2013
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/J.CELL.2011.02.018
Szelényi, J. (2001). Cytokines and the central nervous system. Brain Res. Bull. 54, 329–338. doi: 10.1016/S0361-9230(01)00428-2
Takahashi, K., Prinz, M., Stagi, M., Chechneva, O., and Neumann, H. (2007). TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 4:e124. doi: 10.1371/journal.pmed.0040124
Takaki, Y., Iwata, N., Tsubuki, S., Taniguchi, S., Toyoshima, S., Lu, B., et al. (2000). Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid beta peptide in the brain. J. Biochem. 128, 897–902.
Tan, J., Town, T., Crawford, F., Mori, T., DelleDonne, A., Crescentini, R., et al. (2002). Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat. Neurosci. 5, 1288–1293. doi: 10.1038/nn968
Tanzi, R. E. (2012). The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006296. doi: 10.1101/cshperspect.a006296
Tarkowski, E., Andreasen, N., Tarkowski, A., and Blennow, K. (2003). Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 74, 1200–1205. doi: 10.1136/JNNP.74.9.1200
Tarkowski, E., Blennow, K., Wallin, A., and Tarkowski, A. (1999). Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol. 19, 223–230.
Tateno, H., Li, H., Schur, M. J., Bovin, N., Crocker, P. R., Wakarchuk, W. W., et al. (2007). Distinct endocytic mechanisms of CD22 (Siglec-2) and Siglec-F reflect roles in cell signaling and innate immunity. Mol. Cell. Biol. 27, 5699–5710. doi: 10.1128/MCB.00383-07
Thomas, J., and Grossberg, G. T. (2009). Memantine: a review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clin. Interv. Aging 4, 367–377.
Thome, D., Faridar, A., Beers, D. R., Thonhoff, J. R., Zhao, W., Wen, S., et al. (2018). Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol. Neurodegener. 13:61. doi: 10.1186/s13024-018-0293-1
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., et al. (2009). Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57, 835–849. doi: 10.1002/glia.20810
Trapp, B. D., Wujek, J. R., Criste, G. A., Jalabi, W., Yin, X., Kidd, G. J., et al. (2007). Evidence for synaptic stripping by cortical microglia. Glia 55, 360–368. doi: 10.1002/glia.20462
Tremblay, M. È, Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Tweedie, D., Ferguson, R. A., Fishman, K., Frankola, K. A., Van Praag, H., Holloway, H. W., et al. (2012). Tumor necrosis factor-a synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 9:575. doi: 10.1186/1742-2094-9-106
Ueberham, U., Ueberham, E., Brückner, M. K., Seeger, G., Gärtner, U., Gruschka, H., et al. (2005). Inducible neuronal expression of transgenic TGF-β1 in vivo : dissection of short-term and long-term effects. Eur. J. Neurosci. 22, 50–64. doi: 10.1111/j.1460-9568.2005.04189.x
Ulrich, J. D., Finn, M., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 9:20. doi: 10.1186/1750-1326-9-20
Valera, E., and Masliah, E. (2013). Immunotherapy for neurodegenerative diseases: focus on alpha-synucleinopathies. Pharmacol. Ther. 138, 311–322. doi: 10.1016/j.pharmthera.2013.01.013
Van Acker, Z. P., Bretou, M., and Annaert, W. (2019). Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol. Neurodegener. 14:20. doi: 10.1186/s13024-019-0323-7
van der Lee, S. J., Knol, M. J., Chauhan, G., Satizabal, C. L., Smith, A. V., Hofer, E., et al. (2019). A genome-wide association study identifies genetic loci associated with specific lobar brain volumes. Commun Biol, 2:285. doi: 10.1038/s42003-019-0537-9
van der Wal, E. A., Gómez-Pinilla, F., and Cotman, C. W. (1993). Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 4, 69–72.
Vandenberghe, R., Rinne, J. O., Boada, M., Katayama, S., Scheltens, P., Vellas, B., et al. (2016). Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 8:18. doi: 10.1186/s13195-016-0189-7
Vehmas, K., Kawas, C. H., Stewart, W. F., and Troncoso, J. C. (2003). Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 24, 321–331
Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D., Hartley, D., Chesneau, V., et al. (2000). Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665.
Vellas, B., Black, R., Thal, L. J., Fox, N. C., Daniels, M., McLennan, G., et al. (2009). Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr. Alzheimer Res. 6, 144–151. doi: 10.2174/156720509787602852
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses In Vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
Walker, D. G., Whetzel, A. M., Serrano, G., Sue, L. I., Beach, T. G., and Lue, L. F. (2015). Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 36, 571–582. doi: 10.1016/j.neurobiolaging.2014.09.023
Wang, H., Song, G., Chuang, H., Chiu, C., Abdelmaksoud, A., Ye, Y., et al. (2018). Portrait of glial scar in neurological diseases. Int. J. Immunopathol. Pharmacol. 31:2058738418801406. doi: 10.1177/2058738418801406
Wang, J., Li, Y., Wang, X., Chen, W., Sun, H., and Wang, J. (2014). Lipopolysaccharide induces amyloid formation of antimicrobial peptide HAL-2. Biochim. Biophys. Acta 1838, 2910–2918. doi: 10.1016/j.bbamem.2014.07.028
Wang, M. M., Miao, D., Cao, X. P., Tan, L., and Tan, L. (2018). Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 6, 177–177. doi: 10.21037/atm.2018.04.20
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wanner, B., Anderson, M. A., Song, B., Levine, J., Fernandez, A., Gray-Thompson, Z., et al. (2013). Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 33, 12870–12886. doi: 10.1523/jneurosci.2121-13.2013
Ward, A., Crean, S., Mercaldi, C. J., Collins, J. M., Boyd, D., Cook, M. N., et al. (2012). Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology 38, 1–17. doi: 10.1159/000334607
Ward, M., Himmelstein, D. S., Lancia, J. K., and Binder, L. I. (2012). Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 40, 667–671. doi: 10.1042/BST20120134
White, J. A., Manelli, A. M., Holmberg, K. H., Van Eldik, L. J., and LaDu, M. J. (2005). Differential effects of oligomeric and fibrillar amyloid-β1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 18, 459–465. doi: 10.1016/j.nbd.2004.12.013
Whitmer, R. A., Gustafson, D. R., Barrett-Connor, E., Haan, M. N., Gunderson, E. P., and Yaffe, K. (2008). Central obesity and increased risk of dementia more than three decades later. Neurology 71, 1057–1064. doi: 10.1212/01.wnl.0000306313.89165.ef
Wiesner, J., and Vilcinskas, A. (2010). Antimicrobial peptides: the ancient arm of the human immune system. Virulence 1, 440–464. doi: 10.4161/viru.1.5.12983
Wilhelmsson, U., Bushong, E. A., Price, D. L., Smarr, B. L., Phung, V., Terada, M., et al. (2006). Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl. Acad. Sci. U.S.A. 103, 17513–17518. doi: 10.1073/pnas.0602841103
Williams, M., Torres, S., Siedlak, S. L., Castellani, R. J., Perry, G., Smith, M. A., et al. (2013). Antimicrobial peptide beta-defensin-1 expression is upregulated in Alzheimer’s brain. J. Neuroinflamm. 10:127. doi: 10.1186/1742-2094-10-127
Winblad, B., Graf, A., Riviere, M. E., Andreasen, N., and Ryan, J. M. (2014). Active immunotherapy options for Alzheimer’s disease. Alzheimers Res. Ther. 6:7. doi: 10.1186/alzrt237
Wingerchuk, D. M., and Carter, J. L. (2014). Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin. Proc. 89, 225–240. doi: 10.1016/J.MAYOCP.2013.11.002
Wolf, S. A., Boddeke, H. W., and Kettenmann, H. (2017). Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619–643. doi: 10.1146/annurev-physiol-022516-034406
Woodcock, T., and Morganti-Kossmann, M. C. (2013). The role of markers of inflammation in traumatic brain injury. Front. Neurol. 4:18 doi: 10.3389/fneur.2013.00018
Wozniak, M. A., Mee, A. P., and Itzhaki, R. F. (2009). Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 217, 131–138. doi: 10.1002/path.2449
Wyss-Coray, T., Lin, C., Yan, F., Yu, G. Q., Rohde, M., McConlogue, L., et al. (2001). TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat. Med. 7, 612–618. doi: 10.1038/87945
Xiang, X., Werner, G., Bohrmann, B., Liesz, A., Mazaheri, F., Capell, A., et al. (2016). TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol. Med. 8, 992–1004. doi: 10.15252/emmm.201606370
Yamada, J., Hayashi, Y., Jinno, S., Wu, Z., Inoue, K., Kohsaka, S., et al. (2008). Reduced synaptic activity precedes synaptic stripping in vagal motoneurons after axotomy. Glia 56, 1448–1462. doi: 10.1002/glia.20711
Yamazaki, T., Masuda, J., Omori, T., Usui, R., Akiyama, H., and Maru, Y. (2009). EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J. Cell Sci. 122(Pt 2), 243–255. doi: 10.1242/jcs.036467
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 91, 328–340. doi: 10.1016/J.NEURON.2016.06.015
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213. doi: 10.1016/J.BBI.2010.10.015
Yu, X., and Bellamkonda, R. V. (2001). Dorsal root ganglia neurite extension is inhibited by mechanical and chondroitin sulfate-rich interfaces. J. Neurosci. Res. 66, 303–310. doi: 10.1002/jnr.1225
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Thomas Bird, D., Steven Paul, M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/J.NEURON.2016.05.003
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/jneurosci.6221-11.2012
Zanetti, M., Castiglioni, P., Schoenberger, S., and Gerloni, M. (2003). The role of relB in regulating the adaptive immune response. Ann. N. Y. Acad. Sci. 987, 249–257. doi: 10.1111/j.1749-6632.2003.tb06056.x
Zhang, A., He, X., Zhang, L., Yang, L., Woodman, P., and Li, W. (2014). Biogenesis of lysosome-related organelles complex-1 subunit 1 (BLOS1) interacts with sorting nexin 2 and the endosomal sorting complex required for transport-I (ESCRT-I) component TSG101 to mediate the sorting of epidermal growth factor receptor into endosomal compartments. J. Biol. Chem. 289, 29180–29194. doi: 10.1074/jbc.M114.576561
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. doi: 10.1016/J.CELL.2013.03.030
Zhang, J. M., and An, J. (2007). Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45, 27–37. doi: 10.1097/AIA.0b013e318034194e
Zhao, R., Hu, W., Tsai, J., Li, W., and Gan, W. B. (2017). Microglia limit the expansion of beta-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:47. doi: 10.1186/s13024-017-0188-6
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 Is a receptor for beta-amyloid that mediates microglial function. Neuron 97:1023-e1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zhao, Z., Sagare, A. P., Ma, Q., Halliday, M. R., Kong, P., Kisler, K., et al. (2015). Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci. 18, 978–987. doi: 10.1038/nn.4025
Zheng, C., Zhou, X. W., and Wang, J. Z. (2016). The dual roles of cytokines in Alzheimer’s disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 5:7. doi: 10.1186/s40035-016-0054-4
Zheng, H., Liu, C. C., Atagi, Y., Chen, X. F., Jia, L., Yang, L., et al. (2016). Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 42, 132–141. doi: 10.1016/j.neurobiolaging.2016.03.004
Zheng, L. S., Hitoshi, S., Kaneko, N., Takao, K., Miyakawa, T., Tanaka, Y., et al. (2014). Mechanisms for Interferon-α-induced depression and neural stem cell dysfunction. Stem Cell Rep. 3, 73–84. doi: 10.1016/J.STEMCR.2014.05.015
Keywords: neuroinflammation and neurodegeneration, glia, cytokine, amyloid, tau
Citation: Frost GR, Jonas LA and Li Y-M (2019) Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front. Aging Neurosci. 11:337. doi: 10.3389/fnagi.2019.00337
Received: 31 October 2019; Accepted: 21 November 2019;
Published: 10 December 2019.
Edited by:
Xiongwei Zhu, Case Western Reserve University, United StatesReviewed by:
Riqiang Yan, University of Connecticut, United StatesJuan C. Piña-Crespo, Sanford Burnham Prebys Medical Discovery Institute, United States
Copyright © 2019 Frost, Jonas and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgia R. Frost, ZnJvc3RnQG1za2NjLm9yZw==
†These authors have contributed equally to this work