 Daniela Nunes-Costa1,2
Daniela Nunes-Costa1,2 João Duarte Magalhães1,2
João Duarte Magalhães1,2 Maria G-Fernandes1
Maria G-Fernandes1 Sandra Morais Cardoso1,3*
Sandra Morais Cardoso1,3* Nuno Empadinhas1,4*
Nuno Empadinhas1,4*- 1CNC–Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 2Ph.D. Programme in Biomedicine and Experimental Biology (PDBEB), Institute for Interdisciplinary Research, University of Coimbra, Coimbra, Portugal
- 3Institute of Cellular and Molecular Biology, Faculty of Medicine, University of Coimbra, Coimbra, Portugal
- 4Institute for Interdisciplinary Research (IIIUC), University of Coimbra, Coimbra, Portugal
The neurotoxin β-N-methylamino-L-alanine (BMAA) is a natural non-proteinogenic diamino acid produced by several species of both prokaryotic (cyanobacteria) and eukaryotic (diatoms and dinoflagellates) microorganisms. BMAA has been shown to biomagnify through the food chain in some ecosystems, accumulating for example in seafood such as shellfish and fish, common dietary sources of BMAA whose ingestion may have possible neuronal consequences. In addition to its excitotoxic potential, BMAA has been implicated in protein misfolding and aggregation, inhibition of specific enzymes and neuroinflammation, all hallmark features of neurodegenerative diseases. However, the exact molecular mechanisms of neurotoxicity remain to be elucidated in detail. Although BMAA is commonly detected in its free form, complex BMAA-containing molecules have also been identified such as the paenilamicins, produced by an insect gut bacterial pathogen. On the other hand, production of BMAA or BMAA-containing molecules by members of the human gut microbiota, for example by non-photosynthetic cyanobacteria, the Melainabacteria, remains only hypothetical. In any case, should BMAA reach the gut it may interact with cells of the mucosal immune system and neurons of the enteric nervous system (ENS) and possibly target the mitochondria. Here, we review the available evidence and hint on possible mechanisms by which chronic exposure to dietary sources of this microbial neurotoxin may drive protein misfolding and mitochondrial dysfunction with concomitant activation of innate immune responses, chronic low-grade gut inflammation, and ultimately the neurodegenerative features observed across the gut-brain axis in Parkinson’s disease (PD).
Chronic Exposure to BMAA
Microbial metabolism is an endless source of compounds with diverse biological activities ranging from vitamins to toxins, with a tremendous impact on human health and disease. The neurotoxin β-N-methylamino-L-alanine (BMAA) is an example of a microbial compound to which humans are differently exposed but whose health impacts are still not fully understood, even though current evidence points to an association between BMAA exposure and susceptibility to neurogenerative diseases.
Crucial to understanding how humans may be chronically exposed to BMAA and thus to evaluate how they may be more susceptible to neurodegenerative diseases is identifying its natural producers and how this neurotoxin moves through the food chain. On the island of Guam, symbiotic Nostoc cyanobacteria associated with the roots of cycad plants have historically been considered the main source of BMAA in the ecosystem. The hypothesis, in its most recent formulation, states that BMAA is produced by cycad-associated cyanobacteria, accumulates in cycad seeds consumed by fruit bats or flying foxes that were part of the local population’s diet, or directly used to make flour, thus biomagnifying in the food chain. According to this hypothesis, chronic exposure to BMAA through the ingestion of contaminated plants and animals would, therefore, be a contributing factor in the development of the ALS-PDC pathology in Guam residents (Bradley and Mash, 2009). High-incidence clusters of this condition have also been described in populations from the Kii peninsula of Japan and from western New Guinea, both genetically distinct from each other and from the Guam population but also with documented use of cycad-derived products, supporting an environmental etiology (Ince and Codd, 2005). Elsewhere, contaminated fish and shellfish have been proposed as a significant source of human exposure through biomagnification of BMAA produced not only by marine cyanobacteria but also by dinoflagellates and diatoms (Brand et al., 2010; Jonasson et al., 2010; Banack et al., 2014; Jiang et al., 2014; Lage et al., 2014). A few studies have investigated if crops irrigated with BMAA-contaminated water can bioaccumulate BMAA, and although this seems to be the case under laboratory-controlled conditions, the only study that conducted a field experiment with BMAA-containing water from a cyanobacterial bloom found no BMAA accumulation in the irrigated vegetables (Contardo-Jara et al., 2014, 2018; Esterhuizen-Londt and Pflugmacher, 2019). Thus, the BMAA exposure risk associated with the consumption of vegetables irrigated with cyanobacterial bloom-contaminated water remains to be demonstrated.
Cyanobacteria are widespread producers of BMAA, as shown by several studies employing highly selective analytical methods that confirmed the presence of BMAA in lab-maintained cyanobacterial cultures of different genera belonging to sections I (Chroococcus, Merismopedia, Mycrocystis, Synechocystis), II (Myxosarcina), III (Leptolyngbya, Lyngbya, Oscillatoria) and IV (Anabaena, Nostoc; Banack et al., 2007; Li et al., 2010; Spáčil et al., 2010; Downing et al., 2011; Berntzon et al., 2013; Combes et al., 2013; Jiang et al., 2013; Lage et al., 2016a; Monteiro et al., 2017; Metcalf et al., 2017; Violi et al., 2019). Although cyanobacteria are photoautotrophic bacteria, a nonphotosynthetic clade of divergent cyanobacteria named Melainabacteria was identified in groundwater, tap water, wastewater treatment plants and also as members of the human gut microbiota (Di Rienzi et al., 2013; Soo et al., 2014). The production of BMAA by members of the gut microbiota would be another possible route of chronic exposure but this hypothesis has not been investigated thus far (Brenner, 2013). However, the fact that the BMAA biosynthetic genes have not been identified in any of its natural producers prevents further speculation on whether Melainabacteria share the BMAA biosynthetic machinery with their photosynthetic counterparts. Indeed, and despite the ever-increasing interest in BMAA, the biosynthetic origins of this nonproteinogenic amino acid remain elusive and the proposed metabolic routes are still hypothetical (Brenner et al., 2003; Downing and Downing, 2016; Nunn and Codd, 2017). Recently, it was argued that cyanobacterial BMAA may originate from the hydrolysis of a methylated peptide (Nunn and Codd, 2017) and indeed, BMAA occurs in nature in a bound form in at least one family of secondary metabolites, the paenilamicins (Müller et al., 2014). These peptides are produced by the honey bee intestinal pathogen Paenibacillus larvae by a hybrid nonribosomal peptide/polyketide synthase (NRPS/PKS) and the genetic basis for the synthesis of the two BMAA moieties has been elucidated (Müller et al., 2014). Notably, homologs of the NRPS modules responsible for the BMAA moieties could be detected in some bacterial genera, none belonging to the phylum Cyanobacteria. Still, the possibility that free BMAA can originate in some organisms through the breakdown of more complex molecules such as the paenilamicins deserves further investigation.
The Gut-Brain Axis
Humans are constantly exposed to a wide variety of microbial metabolites including BMAA and the gastrointestinal (GI) tract represents the main interface, not only because it is in direct contact with microbial metabolites present in food and drinking water, but also because it is the most heavily colonized surface housing metabolically-active microorganisms at least as numerous as human cells. This enormous amount and diversity of microbes contributes with a giant genomic blueprint far superior to that of the human genome, and its products, together with dietary metabolites, have a pronounced impact on human biochemistry and physiology (Ley et al., 2006; Sekirov et al., 2010; Shibata et al., 2017). Intestinal microbes are known to be essential for the development and functionality of the host’s immune responses, for the regulation of gut motility, for maintenance of the intestinal barrier integrity and nutrient absorption, for the metabolism of therapeutic drugs, and prevention over inadvertent environmental exposure to toxic compounds (Sekirov et al., 2010; Cryan and Dinan, 2012). An imbalance in the normal microbiota elicited by diet, GI infection, stress or antibiotics, can significantly alter microbiome homeostasis with negative impacts on health (Stecher and Hardt, 2008; Claesson et al., 2012; Collins et al., 2012; Cryan and Dinan, 2012; Lee and Hase, 2014). In this article, we focus on the microbial-produced neurotoxin BMAA discussing how chronic exposure can affect the GI and nervous systems and lead to disease.
The human gut has its own nervous system, the enteric nervous system (ENS), a vast and complex network of neurons and glial cells within the bowel, grouped into ganglia located in the myenteric and the submucosal plexuses (Gershon, 1999; Lebouvier et al., 2009). The ENS depends extensively on the microbiota to develop neuronal electrophysiological properties, such as membrane action potential and sensory neuron excitability (McVey Neufeld et al., 2013), and it normally communicates with the central nervous system (CNS) via a bi-directional homeostatic route designated the gut-brain axis, involving neural, metabolic, hormonal and immunological signaling (Mayer, 2011; Collins et al., 2012; Cryan and Dinan, 2012). The vagus nerve is the main bidirectional pathway connecting the viscera to the brain (Forsythe et al., 2014). Microbial metabolites, namely neurotransmitters, can reach and affect brain development and functions through the gut-brain axis (Sekirov et al., 2010; Collins et al., 2012; Cryan and Dinan, 2012). Gut microbiota composition and dysbiosis also play a role in conditions of the CNS, including Parkinson’s disease (PD; Finegold et al., 2002; Parracho et al., 2005; Berer et al., 2011; Sampson et al., 2016).
Gastrointestinal Disorders and Neurodegeneration: The Case of PD
PD is usually defined as a movement disorder characterized by motor symptoms, such as tremor, postural imbalance, bradykinesia and rigidity (Obeso et al., 2010). The histopathological postmortem hallmark of PD is the presence of α-synuclein-containing (ASYN) insoluble fibrous aggregates, termed Lewy bodies (LBs), accompanied by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) in the CNS. Major causative gene mutations in familial PD are linked to proteins involved in mitochondrial metabolism and dynamics (Cardoso et al., 2005), which highlights a key role of mitochondria in the PD neurodegenerative process.
Sporadic PD has a long prodromal period during which several other features develop, namely olfactory impairment, sleep disturbances and depression (Reichmann et al., 2009). Additionally, GI dysfunction that includes dysphagia, gastroparesis and severe constipation (Wingate, 1999; Pfeiffer, 2003; Rao and Gershon, 2016) may precede the onset of motor symptoms by many years (Savica et al., 2009; Cersosimo and Benarroch, 2012). LBs were found in both the myenteric plexus and submucosal plexus neurons of postmortem PD patients (Wakabayashi et al., 1988; Braak et al., 2006) and even in presymptomatic cases (Braak et al., 2006). Indeed, accumulating evidence obtained from biopsy studies confirmed the presence of LBs in organs innervated by the vagus nerve such as the salivary glands, stomach, duodenum, colon and in the rectum (Cersosimo, 2015), indicating that the vagus nerve is the obvious route through which the disease spreads between the gut and the brain, leading to the hypothesis that a toxin or pathogen can trigger the disease (Hawkes et al., 2007; Brenner, 2013; Alam et al., 2017). Braak et al. (2003) showed that the progression of LBs, the PD histopathological marks, is in agreement with the hypothesis that a pathogen in the GI tract may trigger abnormal ASYN processing and then spread via retrograde axonal vagus transport from the ENS to the CNS and that the GI symptoms observed in the vast majority of PD patients are pre-motor manifestations of PD. Notably, ASYN fibrils injected in mice duodenum were able to propagate to the brain months after injection (Van Den Berge et al., 2019) with propagation occurring through the vagus since it could be inhibited by vagotomy (Kim et al., 2019). Indeed, individuals that underwent full truncal vagotomy had a decreased risk for subsequent PD, strongly implicating the vagus nerve in PD pathogenesis and again corroborating the involvement of an enteric pathogen or toxin (Svensson et al., 2015).
Amino Acid Misincorporation, Protein Folding and Neurodegeneration
Neurodegenerative disorders share similar histopathological hallmarks of protein misfolding and aggregation, even though the protein component and affected brain region may differ (Ross and Poirier, 2004). These neurodegenerative disorders are mostly sporadic without a known etiological factor, with aging being the most relevant risk factor. Although most proteins encoded in the human genome can be removed by cellular quality systems when damaged, some polypeptides are prone to misfold and aggregate into toxic oligomers. Interestingly, these protein aggregates can further downregulate the activities of proteolytic pathways since they are protease-resistant. Thus, accumulation of protein aggregates in different neurodegenerative diseases and disruption of translational fidelity in terminally differentiated post-mitotic neurons are potential pathways leading to cell death (Lee et al., 2006).
Hundreds of amino acids are known to occur in nature although only 22 are used in protein synthesis. Some of those non-proteinogenic amino acids can be mistakenly charged onto tRNA and misincorporated into proteins, potentially causing loss of function and/or misfolding (Bullwinkle et al., 2014). Primary metabolites such as ornithine, homoserine, and homocysteine are substrates for aminoacyl-tRNA synthetases, but cellular quality control mechanisms can detect their mischarging onto tRNA and prevent their inclusion into proteins. Other non-proteinogenic amino acids, though, are secondary metabolites produced by certain microbial species and can elude quality control pathways of nonproducing organisms, behaving as toxins (Bullwinkle et al., 2014). It has been speculated that these dietary or microbial-produced toxic amino acids could, in addition to host genetic factors, represent an environmental trigger for the onset of neurodegeneration (Rodgers, 2014).
Accumulating evidence suggests that this is the case for BMAA, which has been reported to be misincorporated into human proteins in place of serine, resulting in protein misfolding and aggregation, ER stress and apoptosis (Dunlop et al., 2013; Glover et al., 2014; Main et al., 2016). In light of these observations, BMAA misincorporation into ASYN could be a possible mechanism of BMAA-induced neurodegeneration, since serine phosphorylation is heavily correlated with ASYN aggregation. It is conceived that the vast majority of ASYN present in Lewy body aggregates is phosphorylated at serine 129 (S129; Oueslati, 2016), however, the modulation of ASYN aggregation by S129 phosphorylation is still a matter of debate, since in vivo studies demonstrate that this association is far more complex than what may appear (McFarland et al., 2009). Nonetheless, BMAA misincorporation in this position could provoke an imbalance in the extension of S129 phosphorylation, thereby promoting ASYN aggregation. Interestingly, S87 phosphorylation was found to inhibit ASYN aggregation and the substitution of serine by BMAA in this position could hypothetically reshape the sequence milieu and therefore reduce phosphorylated S87, increasing the propensity of ASYN to aggregate (Oueslati et al., 2012).
While the observation that BMAA interacts very strongly with proteins causing their misfolding has not been disputed, some groups have failed to detect incorporation of BMAA into the primary structure of proteins thus calling into question the role of the misincorporation hypothesis in explaining BMAA’s observed neurotoxic effects (Beri et al., 2017; Onselen et al., 2017). Subsequently, alternative mechanisms have been investigated and BMAA has been found to strongly associate with melanin, to selectively inhibit the activity of certain enzymes and to interfere with and disrupt protein refolding in vitro by associating with proteins through electrostatic interactions strong enough to resist TCA precipitation and subsequent washing with SDS or DTT (van Onselen and Downing, 2018). In particular, the interaction of BMAA with melanin and neuromelanin, which seem to have an important role in neurotoxin sequestration and neurodegeneration, as well as its ability to inhibit human catalase, an enzyme directly involved in the detoxification of β-amyloid-linked cellular toxicity, have recently been proposed to also contribute to BMAA’s neurotoxicity (Delcourt et al., 2018; van Onselen and Downing, 2019). Whether BMAA causes proteins to misfold due to eventual misincorporation into their primary structure or through strong electrostatic interactions with nascent chains remains to be confirmed (Dunlop and Guillemin, 2019).
As indicated above, supporting evidence for BMAA’s putative role in triggering neurodegenerative diseases comes from the island of Guam, where endemic ALS-PDC has been linked to chronic exposure to BMAA in the diet of Guam residents (Bradley and Mash, 2009). Other factors have been proposed to be involved in the etiology of Guam ALS-PDC, namely secondary hyperparathyroidism due to low ingestion of calcium and magnesium together with high aluminum consumption, since deposits of these substances were observed in the brains of affected Guam residents (Garruto et al., 1984). Although we cannot fully discard this as a potential contributing factor, a study failed to prove altered levels of calcium, magnesium and heavy metal levels in patient blood, urine and hair. Parathyroid hormone and serum phosphorus levels—indicators of secondary hyperparathyroidism—were also reported to be within the normal range in Guam affected people (Ahlskog et al., 1995), rendering the heavy metal hypothesis unlikely to be the principal cause of endemic ALS-PDC. In addition, there is no report of secondary hyperparathyroidism treatment being successful in ALS-PDC patients. Nonetheless, BMAA was detected in brain proteins from PD, AD, and ALS patients but not in controls (Dunlop et al., 2013), and was also recently detected in the CNS of antemortem human individuals, including in one ALS patient where it was suggested to act as a disease-potentiating agent together with other environmental factors (Berntzon et al., 2015). Moreover, rats treated with BMAA display ALS-like neurological impairment (de Munck et al., 2013). Other studies have, however, failed to confirm such results casting doubt on the selectivity and reliability of the analytical methods employed by other groups (Meneely et al., 2016). On the other hand, a study with vervet monkeys found that dietary BMAA triggers the formation of neurofibrillary tangles and Aβ plaques characteristic of ALS-PDC and Alzheimer’s disease, whereas the co-administration of L-serine was shown to have a neuroprotective effect (Cox et al., 2016). The latter finding is consistent with BMAA being a substrate for seryl-tRNA synthetase and suggests that inhibition of the proposed BMAA misincorporation into brain proteins by L-serine may be linked with prevention of BMAA’s neurodegenerative effects (Main et al., 2016). Intriguingly, conversion of L-BMAA into D-BMAA has been detected in the liver and in the brain of mice, bringing additional complexity into the already puzzling field of BMAA research (Metcalf et al., 2017). More recently, an in vitro study revealed that human liver hepatocyte and intestinal epithelial cell cultures are incapable of metabolizing BMAA, suggesting the absence of detoxification pathways that should protect against dietary exposure to this toxin (Downing et al., 2019).
Mitochondrial-Driven Innate Immunity Activation: A Possible Link Between BMAA and Neurodegeneration
Neuronal loss is the ultimate consequence in the pathophysiology of neurodegenerative diseases, although the specificity of the neurons that degenerate is quite distinguishable. While in AD the entorhinal cortical and hippocampal neurons are the first to be affected, in PD and ALS the SNpc and the motor neurons are the first to perish, respectively (Martin, 1999). Despite these distinct features, neurodegenerative diseases share many common characteristics such as mitochondrial failure and marked proinflammatory profiles (Gan et al., 2018). It is also well established that in most neurodegenerative diseases, the activity of the electron transport chain (ETC) complexes are consistently impaired, along with aberrant mitochondrial dynamics, higher fragmentation, swelling with deformed cristae and increased reactive oxygen species (ROS) production (Johri and Beal, 2012; Hroudová et al., 2014; Muyderman and Chen, 2014). Furthermore, studies in post-mortem brain samples and analysis of cerebrospinal fluid (CSF) of AD, PD and ALS patients have demonstrated a sheer correlation between disease and up-regulation of several proinflammatory cytokines involved in the innate immune response (Stephenson et al., 2018). The cause of these striking neuroinflammatory events is very often intertwined with loss of neuronal mitochondria fitness. Upon a given damage, mitochondria expose danger-associated molecular patterns (DAMPs; Grazioli and Pugin, 2018), signals that are recognized by cellular mechanisms involving pattern recognition receptors (PRR) and which are divided into four families: toll-like receptors (TLR), Rig-1-like receptors (RLR), Nod-like receptors (NLR) and C-type lectin receptors (CLR). When activated by mitochondrial DAMPS, these PRR receptors can induce the upregulation of the NF-kB pathway, leading to the release of proinflammatory cytokines, such as TNFα and IL-1β, the latter through the activation of the NLRP3 inflammasome (Cardoso and Empadinhas, 2018). Moreover, mitochondrial ROS overproduction can also activate the inflammasome, thus amplifying the overall inflammatory response (Saint-Georges-Chaumet and Edeas, 2016).
Mitochondria are unique organelles with their origins tracing back to an ancient α-proteobacterial lineage (Hibbing et al., 2010) that grants them exclusive features such as their own DNA, an independent replication process, their own ribosomes, the mitoribosomes, and bacterial-type phospholipids such as cardiolipin (Cardoso and Empadinhas, 2018; Grazioli and Pugin, 2018). Bacterial populations compete for nutrients in many ways, one being the production of noxious substances that evolved to control or kill competitors. In fact, this is a common strategy used for the discovery of new potential antibiotics (Hibbing et al., 2010). Mitochondria’s resemblance with bacteria makes them natural targets of many microbial byproducts and, indeed, many examples of microbial substances targeting mitochondria have been identified. Streptococcus pneumoniae, for example, produces pneumolysin, a pore-forming substance that causes neuronal death by specifically targeting mitochondria (Braun et al., 2007). Some other toxins do not even require the actual presence of bacteria to promote mitochondrial failure. Although the molecular mechanisms are still to be explored, Staphylococcus aureus α-toxin and Helicobacter pylori vacuolating cytotoxin (VacA) can promote cytotoxicity through mitochondrial impairment (Arnoult et al., 2009). It should be noted here that BMAA may also have the ability to induce mitochondrial dysfunction (Beri et al., 2017). In vitro studies with NSC-34 cells, a cell line of motor neurons showed that BMAA elicited a pronounced decrease in oxidative phosphorylation, altered calcium homeostasis and exacerbated ROS production. In addition, treatment with BMAA dramatically decreased N2a neuronal cell line viability that was assessed by measuring the activity of the mitochondrial enzyme succinate dehydrogenase (SD; Takser et al., 2016). In brain samples from the population of the Kii peninsula of Japan, mitochondrial disruption has also been shown to occur leading to increased ROS production (Hata et al., 2018). Unfortunately, the mechanism of action remains elusive.
Recent findings have confirmed that, in addition to the deleterious effects on mitochondria, BMAA exposure was also associated with the proinflammatory profile observed in neurodegenerative diseases. BMAA administration to rats was able to reproduce the neuronal phenotype of ALS, concomitantly with a strong expression of proinflammatory cytokines (Michaelson et al., 2017). Remarkably, BMAA was able to promote the upregulation of Glial Fibrillary Acidic Protein (GFAP) and, consequently, an evident astrogliosis in a rat model of ALS-PDC (Cai et al., 2018). Also, BMAA triggered cytotoxic effects in a RAW246.7 cell line and in BV-2 microglial cells (Takser et al., 2016), which confers potential immunomodulatory capacity to this neurotoxin.
Conclusion
Although considerable attention has been dedicated to BMAA research over the past decades, many aspects of this field remain uncertain. The fact that early analytical methods for BMAA detection were found to be unreliable hindered further progress (Faassen, 2014) and even with the implementation of more selective methods some conflicting results are still reported, particularly when it comes to assessing the BMAA biosynthetic potential of cyanobacterial strains. Such results increasingly suggest that BMAA production by cyanobacteria may be transient, inconsistent in laboratory cultures, and subject to fluctuations provoked by certain stimuli such as nitrogen availability (Downing et al., 2011; Monteiro et al., 2017). The fact is that the biological functions of this diamino acid in nature are still unknown. The few studies conducted on its natural producers have concluded that it interferes with nitrogen metabolism and have hypothesized an allelopathic role for BMAA, a common biological phenomenon by which metabolites produced by some organisms affect certain physiological traits of others (Lage et al., 2016b; Popova et al., 2018). In the case of the BMAA-containing paenilamicins, production of the compound was shown to impact competing bacterial populations in the larval gut thus confirming its allelopathic role (Müller et al., 2014). Although the ability of human gut microbiota to produce or be impacted by BMAA or BMAA-containing molecules is still unknown and warrants future investigation, there is little doubt that seafood and particularly marine bivalves can be a non-negligible source of dietary BMAA, which justifies further epidemiological studies to assess the real risk posed by their consumption (Lance et al., 2018).
Another topic warranting future investigation in this field is the identification of host genetic factors underlying individual predisposition to developing sporadic neurodegenerative diseases as a result of chronic BMAA exposure. Indeed, besides well-studied mutations that cause familial neurodegenerative diseases, some genetic polymorphisms have been associated with the susceptibility of their sporadic counterparts (Rocchi et al., 2003; Bras and Singleton, 2009). We propose that susceptible individuals with certain genetic polymorphisms, including in elements of the translational machinery (Bullwinkle et al., 2014), may more often misincorporate BMAA into aggregation-prone proteins whose accumulation in neurons may trigger the onset of neurodegeneration, initially in the ENS and ultimately in the CNS, owing to retrograde transport through the vagus nerve, or plasma, to the brain. Additionally, microbial metabolites can bypass the intestinal mucosa barrier due to increased permeabilization of the gut barrier, which is known to be increased in PD (Forsyth et al., 2011) and would facilitate BMAA toxicity. Aging accompanied with a decrease in the immune response (immunosenescence) often attributed to PD is greatly associated with a leaky gut (Nagpal et al., 2018). Also of note, a study performed by Dawson et al. (1998) demonstrated that male rats were more susceptible to BMAA-induced toxicity than females, a very interesting observation given that PD has a higher incidence in males than females.
In any case and as summarized in this review, BMAA effects in human cell lines and in animals are consistent with the hypothesis that chronic exposure to this neurotoxin can trigger features of sporadic neurodegenerative diseases such as PD (Figure 1). In this regard, it is reasonable to hypothesize that BMAA may promote protein misfolding, mitochondrial dysfunction and chronic innate immune activation in genetically susceptible individuals, initially in the ENS and later in the CNS by means of retrograde transport via the vagus nerve, and ultimately leading to brain neurodegeneration (Figure 1). The toxicological evidence from these studies is on its own sufficient to drive research towards the identification of the biosynthetic pathways and biological functions of this diamino acid in its natural producers, in-depth studies tracking BMAA through the food web in the various ecosystems, and epidemiological studies in populations exposed to BMAA-containing seafood. Only with this information, we will be able to accurately link the missing dots in the old but still controversial hypothesis of a BMAA-driven cause for sporadic neurodegenerative diseases in humans.
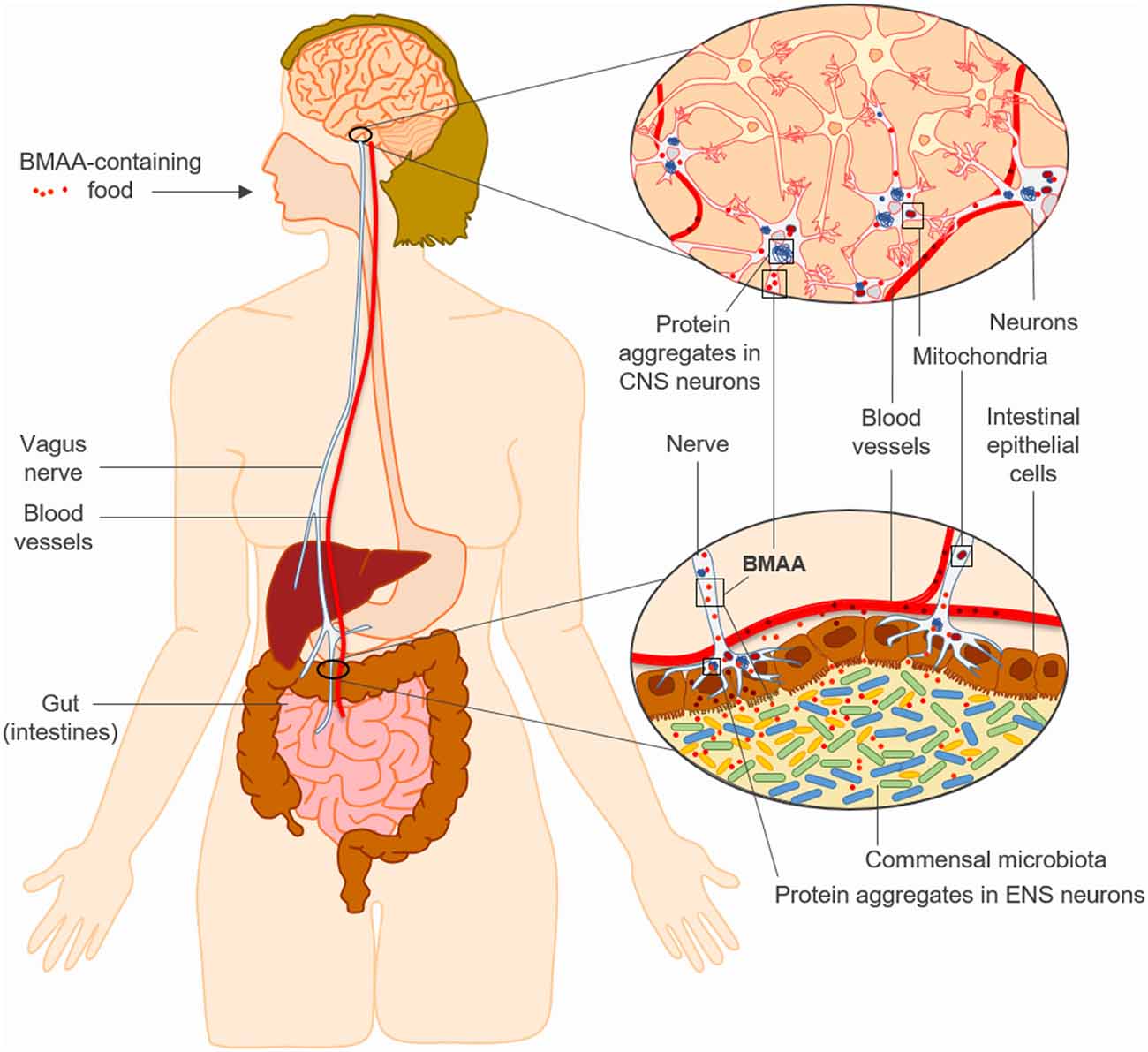
Figure 1. The “neurotoxin hypothesis” for sporadic neurodegenerative diseases such as Parkinson’s disease (PD). Chronic intestinal exposure to β-N-methylamino-L-alanine (BMAA) may trigger neurodegeneration by promoting protein misfolding, mitochondrial dysfunction and innate immune responses in genetically susceptible individuals, initially in the enteric nervous system (ENS) and later in the central nervous system (CNS) through retrograde transport via the vagus nerve.
Author Contributions
NE and SC: conceptualization. DN-C, JM, SC and NE: investigation. SC and NE: funding. DN-C, JM, MG-F, SC and NE: writing.
Funding
We acknowledge Fundação para a Ciência e a Tecnologia (FCT) and COMPETE 2020, Operational Programme for Competitiveness and Internationalization (POCI) through FEDER for projects UID/NEU/04539/2019 (POCI-01-0145-FEDER-007440), POCI-01-0145-FEDER-030712 and Ph.D. fellowships SFRH/BD/117777/2016 to DN-C and PD/BD/146409/2019 to JM. We also acknowledge the European Regional Development Fund Centro2020 Regional Operational Programme (CENTRO-01-0145-FEDER-000012-HealthyAging2020), the Faculty of Medicine, University of Coimbra (Project FMUC-PEPITA-2018) and Santa Casa da Misericórdia de Lisboa—Mantero Belard Neurosciences Prize 2016 (MB-40-2016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlskog, J. E., Waring, S. C., Kurland, L. T., Petersen, R. C., Moyer, T. P., Harmsen, W. S., et al. (1995). Guamanian neurodegenerative disease: investigation of the calcium metabolism/heavy metal hypothesis. Neurology 45, 1340–1344. doi: 10.1212/wnl.45.7.1340
Alam, M., Alam, Q., Kamal, M., Jiman-Fatani, A., Azhar, E., Khan, M., et al. (2017). Infectious agents and neurodegenerative diseases: exploring the links. Curr. Top. Med. Chem. 17, 1390–1399. doi: 10.2174/1568026617666170103164040
Arnoult, D., Carneiro, L., Tattoli, I., and Girardin, S. E. (2009). The role of mitochondria in cellular defense against microbial infection. Semin. Immunol. 21, 223–232. doi: 10.1016/j.smim.2009.05.009
Banack, S. A., Johnson, H. E., Cheng, R., and Cox, P. A. (2007). Production of the neurotoxin BMAA by a marine cyanobacterium. Mar. Drugs 5, 180–196. doi: 10.3390/md504180
Banack, S. A., Metcalf, J. S., Bradley, W. G., and Cox, P. A. (2014). Detection of cyanobacterial neurotoxin β-N-methylamino-L-alanine within shellfish in the diet of an ALS patient in Florida. Toxicon 90, 167–173. doi: 10.1016/j.toxicon.2014.07.018
Berer, K., Mues, M., Koutrolos, M., Rasbi, Z. A., Boziki, M., Johner, C., et al. (2011). Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541. doi: 10.1038/nature10554
Beri, J., Nash, T., Martin, R. M., and Bereman, M. S. (2017). Exposure to BMAA mirrors molecular processes linked to neurodegenerative disease. Proteomics 17:1700161. doi: 10.1002/pmic.201700161
Berntzon, L., Erasmie, S., Celepli, N., Eriksson, J., Rasmussen, U., and Bergman, B. (2013). BMAA inhibits nitrogen fixation in the cyanobacterium nostoc sp. PCC 7120. Mar. Drugs 11, 3091–3108. doi: 10.3390/md11083091
Berntzon, L., Ronnevi, L. O., Bergman, B., and Eriksson, J. (2015). Detection of BMAA in the human central nervous system. Neuroscience 292, 137–147. doi: 10.1016/j.neuroscience.2015.02.032
Braak, H., de Vos, R. A. I., Bohl, J., and Del Tredici, K. (2006). Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 396, 67–72. doi: 10.1016/j.neulet.2005.11.012
Braak, H., Rüb, U., Gai, W. P., and Del Tredici, K. (2003). Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 110, 517–536. doi: 10.1007/s00702-002-0808-2
Bradley, W. G., and Mash, D. C. (2009). Beyond Guam: the cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph. Lateral Scler. 10, 7–20. doi: 10.3109/17482960903286009
Brand, L. E., Pablo, J., Compton, A., Hammerschlag, N., and Mash, D. C. (2010). Cyanobacterial blooms and the occurrence of the neurotoxin, β-N-methylamino-L-alanine (BMAA), in South Florida aquatic food webs. Harmful Algae 9, 620–635. doi: 10.1016/j.hal.2010.05.002
Bras, J. M., and Singleton, A. (2009). Genetic susceptibility in Parkinson’s disease. Biochim. Biophys. Acta 1792, 597–603. doi: 10.1016/j.bbadis.2008.11.008
Braun, J. S., Hoffmann, O., Schickhaus, M., Freyer, D., Dagand, E., Bermpohl, D., et al. (2007). Pneumolysin causes neuronal cell death through mitochondrial damage. Infect. Immun. 75, 4245–4254. doi: 10.1128/iai.00031-07
Brenner, S. (2013). Blue-green algae or cyanobacteria in the intestinal micro-flora may produce neurotoxins such as β-N-Methylamino-L-Alanine (BMAA) which may be related to development of amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson-Dementia-Complex in humans and equine motor neuron disease in horses. Med. Hypotheses 80:103. doi: 10.1016/j.mehy.2012.10.010
Brenner, E. D., Stevenson, D. W., McCombie, R. W., Katari, M. S., Rudd, S. A., Mayer, K. F. X., et al. (2003). Expressed sequence tag analysis in Cycas, the most primitive living seed plant. Genome Biol. 4:R78. doi: 10.1186/gb-2003-4-12-r78
Bullwinkle, T., Lazazzera, B., and Ibba, M. (2014). Quality control and infiltration of translation by amino acids outside of the genetic code. Annu. Rev. Genet. 48, 149–166. doi: 10.1146/annurev-genet-120213-092101
Cai, H.-Y., Tian, K.-W., Zhang, Y.-Y., Jiang, H., and Han, S. (2018). Angiopoietin-1 and αnuβ3 integrin peptide promote the therapeutic effects of L-serine in an amyotrophic lateral sclerosis/Parkinsonism dementia complex model. Aging 10, 3507–3527. doi: 10.18632/aging.101661
Cardoso, S. M., and Empadinhas, N. (2018). The microbiome-mitochondria dance in prodromal Parkinson’s disease. Front. Physiol. 9:471. doi: 10.3389/fphys.2018.00471
Cardoso, S., Moreira, P., Agostinho, P., Pereira, C., and Oliveira, C. (2005). Neurodegenerative pathways in Parkinson’s disease: therapeutic strategies. Curr. Drug Targets CNS Neurol. Disord. 4, 405–419. doi: 10.2174/1568007054546072
Cersosimo, M. G. (2015). Gastrointestinal biopsies for the diagnosis of α-synuclein pathology in Parkinson’s disease. Gastroenterol. Res. Pract. 2015:476041. doi: 10.1155/2015/476041
Cersosimo, M. G., and Benarroch, E. E. (2012). Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 46, 559–564. doi: 10.1016/j.nbd.2011.10.014
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O’Connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. doi: 10.1038/nature11319
Collins, S. M., Surette, M., and Bercik, P. (2012). The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 10, 735–742. doi: 10.1038/nrmicro2876
Combes, A., El Abdellaoui, S., Sarazin, C., Vial, J., Mejean, A., Ploux, O., et al. (2013). Validation of the analytical procedure for the determination of the neurotoxin β-N-methylamino-L-alanine in complex environmental samples. Anal. Chim. Acta 771, 42–49. doi: 10.1016/j.aca.2013.02.016
Contardo-Jara, V., Schwanemann, T., Esterhuizen-Londt, M., and Pflugmacher, S. (2018). Protein association of β-N-methylamino-L-alanine in Triticum aestivum via irrigation irrigation. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 35, 732–740. doi: 10.1080/19440049.2018.1427283
Contardo-Jara, V., Schwanemann, T., and Pflugmacher, S. (2014). Uptake of a cyanotoxin, β-N-methylamino-L-alanine, by wheat (Triticum aestivum). Ecotoxicol. Environ. Saf. 104, 127–131. doi: 10.1016/j.ecoenv.2014.01.039
Cox, P. A., Davis, D. A., Mash, D. C., Metcalf, J. S., and Banack, S. A. (2016). Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc. R. Soc. 283:20152397. doi: 10.1098/rspb.2015.2397
Cryan, J. F., and Dinan, T. G. (2012). Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13, 701–712. doi: 10.1038/nrn3346
Dawson, R. Jr., Marschall, E. G., Chan, K. C., Millard, W. J., Eppler, B., and Patterson, T. A. (1998). Neurochemical and neurobehavioral effects of neonatal administration of β-N-methylamino-L-alanine and 3,3’-iminodipropionitrile. Neurotoxicol. Teratol. 20, 181–192. doi: 10.1016/s0892-0362(97)00078-0
de Munck, E., Muñoz-Sáez, E., Miguel, B. G., Solas, M. T., Ojeda, I., Martínez, A., et al. (2013). β-N-methylamino-L-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): the first step towards an experimental model for sporadic ALS. Environ. Toxicol. Pharmacol. 36, 243–255. doi: 10.1016/j.etap.2013.04.007
Delcourt, N., Claudepierre, T., Maignien, T., Arnich, N., and Mattei, C. (2018). Cellular and molecular aspects of the β-N-methylamino-L-alanine (BMAA) mode of action within the neurodegenerative pathway: facts and controversy. Toxins 10:E6. doi: 10.3390/toxins10010006
Di Rienzi, S. C., Sharon, I., Wrighton, K. C., Koren, O., Hug, L. A., Thomas, B. C., et al. (2013). The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife 2:e01102. doi: 10.7554/eLife.01102
Downing, S., Banack, S. A., Metcalf, J. S., Cox, P. A., and Downing, T. G. (2011). Nitrogen starvation of cyanobacteria results in the production of β-N-methylamino-L-alanine. Toxicon 58, 187–194. doi: 10.1016/j.toxicon.2011.05.017
Downing, S., and Downing, T. G. (2016). The metabolism of the non-proteinogenic amino acid β-N-methylamino-L-alanine (BMAA) in the cyanobacterium Synechocystis PCC6803. Toxicon 115, 41–48. doi: 10.1016/j.toxicon.2016.03.005
Downing, S., Van Onselen, R., Kemp, G., and Downing, T. G. (2019). Metabolism of the neurotoxic amino acid β-N-methylamino-L-alanine in human cell culture models. Toxicon 168, 131–139. doi: 10.1016/j.toxicon.2019.07.007
Dunlop, R. A., Cox, P. A., Banack, S. A., and Rodgers, K. J. (2013). The non-protein amino acid BMAA Is misincorporated into human proteins in place of L-serine causing protein misfolding and aggregation. PLoS One 8:e75376. doi: 10.1371/journal.pone.0075376
Dunlop, R. A., and Guillemin, G. J. (2019). The cyanotoxin and non-protein amino acid β-methylamino-L-alanine (L-BMAA) in the food chain: incorporation into proteins and its impact on human health. Neurotox. Res. 36, 602–611. doi: 10.1007/s12640-019-00089-9
Esterhuizen-Londt, M., and Pflugmacher, S. (2019). Vegetables cultivated with exposure to pure and naturally occurring β-N-methylamino-L-alanine (BMAA) via irrigation. Environ. Res. 169, 357–361. doi: 10.1016/j.envres.2018.11.030
Faassen, E. J. (2014). Presence of the neurotoxin BMAA in aquatic ecosystems: what do we really know? Toxins 6, 1109–1138. doi: 10.3390/toxins6031109
Finegold, S. M., Molitoris, D., Song, Y., Liu, C., Vaisanen, M., Bolte, E., et al. (2002). Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 35, S6–S16. doi: 10.1086/341914
Forsythe, P., Bienenstock, J., and Kunze, W. A. (2014). “Vagal pathways for microbiome-brain-gut axis communication,” in Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Advances in Experimental Medicine and Biology, vol 817, eds M. Lyte and J. Cryan (New York, NY: Springer), 115–133.
Forsyth, C. B., Shannon, K. M., Kordower, J. H., Voigt, R. M., Shaikh, M., Jaglin, J. A., et al. (2011). Increased intestinal permeability correlates with sigmoid mucosa α-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 6:e28032. doi: 10.1371/journal.pone.0028032
Gan, L., Cookson, M. R., Petrucelli, L., and La Spada, A. R. (2018). Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 21, 1300–1309. doi: 10.1038/s41593-018-0237-7
Garruto, R. M., Fukatsu, R., Yanagihara, R., Gajdusek, D. C., Hook, G., and Fiori, C. E. (1984). Imaging of calcium and aluminum in neurofibrillary tangle-bearing neurons in parkinsonism-dementia of Guam. Proc. Natl. Acad. Sci. U S A 81, 1875–1879. doi: 10.1073/pnas.81.6.1875
Gershon, M. D. (1999). The enteric nervous system: a second brain. Hosp. Pract. 34, 31–52. doi: 10.3810/hp.1999.07.153
Glover, W. B., Mash, D. C., and Murch, S. J. (2014). The natural non-protein amino acid N-β-methylamino-L-alanine (BMAA) is incorporated into protein during synthesis. Amino Acids 46, 2553–2559. doi: 10.1007/s00726-014-1812-1
Grazioli, S., and Pugin, J. (2018). Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front. Immunol. 9:832. doi: 10.3389/fimmu.2018.00832
Hata, Y., Ma, N., Yoneda, M., Morimoto, S., Okano, H., Murayama, S., et al. (2018). Nitrative stress and tau accumulation in amyotrophic lateral sclerosis/Parkinsonism-dementia complex (ALS/PDC) in the kii peninsula, japan. Front. Neurosci. 11:751. doi: 10.3389/fnins.2017.00751
Hawkes, C. H., Del Tredici, K., and Braak, H. (2007). Parkinson’s disease: a dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614. doi: 10.1111/j.1365-2990.2007.00874.x
Hibbing, M. E., Fuqua, C., Parsek, M. R., and Peterson, S. B. (2010). Bacterial competition: surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 8, 15–25. doi: 10.1038/nrmicro2259
Hroudová, J., Singh, N., and Fišar, Z. (2014). Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. Biomed. Res. Int. 2014:175062. doi: 10.1155/2014/175062
Ince, P. G., and Codd, G. A. (2005). Return of the cycad hypothesis—does the amyotrophic lateral sclerosis/parkinsonism dementia complex (ALS/PDC) of Guam have new implications for global health? Neuropathol. Appl. Neurobiol. 31, 345–353. doi: 10.1111/j.1365-2990.2005.00686.x
Jiang, L., Eriksson, J., Lage, S., Jonasson, S., Shams, S., Mehine, M., et al. (2014). Diatoms: a novel source for the neurotoxin BMAA in aquatic environments. PLoS One 9:e84578. doi: 10.1371/journal.pone.0084578
Jiang, L., Johnston, E., Åberg, K. M., Nilsson, U., and Ilag, L. L. (2013). Strategy for quantifying trace levels of BMAA in cyanobacteria by LC/MS/MS. Anal. Bioanal. Chem. 405, 1283–1292. doi: 10.1007/s00216-012-6550-1
Johri, A., and Beal, M. F. (2012). Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 342, 619–630. doi: 10.1124/jpet.112.192138
Jonasson, S., Eriksson, J., Berntzon, L., Spácil, Z., Ilag, L. L., Ronnevi, L.-O., et al. (2010). Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for human exposure. Proc. Natl. Acad. Sci. U S A 107, 9252–9257. doi: 10.1073/pnas.0914417107
Kim, S., Kwon, S.-H., Kam, T.-I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627.e7–641.e7. doi: 10.1016/j.neuron.2019.05.035
Lage, S., Burian, A., Rasmussen, U., Costa, P. R., Annadotter, H., Godhe, A., et al. (2016a). BMAA extraction of cyanobacteria samples: which method to choose? Environ. Sci. Pollut. Res. 23, 338–350. doi: 10.1007/s11356-015-5266-0
Lage, S., Ström, L., Godhe, A., and Rydberg, S. (2016b). The effect of exogenous β-N-methylamino-L-alanine (BMAA) on the diatoms Phaeodactylum tricornutum and Thalassiosira weissflogii. Harmful Algae 58, 85–92. doi: 10.1016/j.hal.2016.08.005
Lage, S., Costa, P. R., Moita, T., Eriksson, J., Rasmussen, U., and Rydberg, S. J. (2014). BMAA in shellfish from two Portuguese transitional water bodies suggests the marine dinoflagellate Gymnodinium catenatum as a potential BMAA source. Aquat. Toxicol. 152, 131–138. doi: 10.1016/j.aquatox.2014.03.029
Lance, E., Arnich, N., Maignien, T., and Biré, R. (2018). Occurrence of β-N-methylamino-L-alanine (BMAA) and isomers in aquatic environments and aquatic food sources for humans. Toxins 10:E191. doi: 10.3390/toxins10050191
Lebouvier, T., Chaumette, T., Paillusson, S., Duyckaerts, C., Bruley des Varannes, S., Neunlist, M., et al. (2009). The second brain and Parkinson’s disease. Eur. J. Neurosci. 30, 735–741. doi: 10.1111/j.1460-9568.2009.06873.x
Lee, J. W., Beebe, K., Nangle, L. A., Jang, J., Longo-Guess, C. M., Cook, S. A., et al. (2006). Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443, 50–55. doi: 10.1038/nature05096
Lee, W.-J., and Hase, K. (2014). Gut microbiota-generated metabolites in animal health and disease. Nat. Chem. Biol. 10, 416–424. doi: 10.1038/nchembio.1535
Ley, R. E., Peterson, D. A., and Gordon, J. I. (2006). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. doi: 10.1016/j.cell.2006.02.017
Li, A., Tian, Z., Li, J., Yu, R., Banack, S. A., and Wang, Z. (2010). Detection of the neurotoxin BMAA within cyanobacteria isolated from freshwater in China. Toxicon 55, 947–953. doi: 10.1016/j.toxicon.2009.09.023
Main, B. J., Dunlop, R. A., and Rodgers, K. J. (2016). The use of L-serine to prevent β-methylamino-L-alanine (BMAA)-induced proteotoxic stress in vitro. Toxicon 109, 7–12. doi: 10.1016/j.toxicon.2015.11.003
Martin, J. B. (1999). Molecular basis of the neurodegenerative disorders. N. Engl. J. Med. 340, 1970–1980. doi: 10.1056/NEJM199906243402507
Mayer, E. A. (2011). Gut feelings: the emerging biology of gut-brain communication. Nat. Rev. Neurosci. 12, 453–466. doi: 10.1038/nrn3071
McFarland, N. R., Fan, Z., Xu, K., Schwarzschild, M. A., Feany, M. B., Hyman, B. T., et al. (2009). α-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of parkinson disease. J. Neuropathol. Exp. Neurol. 68, 515–524. doi: 10.1097/NEN.0b013e3181a24b53
McVey Neufeld, K. A., Mao, Y. K., Bienenstock, J., Foster, J. A., and Kunze, W. A. (2013). The microbiome is essential for normal gut intrinsic primary afferent neuron excitability in the mouse. Neurogastroenterol. Motil. 25, 183–190. doi: 10.1111/nmo.12049
Meneely, J. P., Chevallier, O. P., Graham, S., Greer, B., Green, B. D., and Elliott, C. T. (2016). β-methylamino-L-alanine (BMAA) is not found in the brains of patients with confirmed Alzheimer’s disease. Sci. Rep. 6:36363. doi: 10.1038/srep36363
Metcalf, J. S., Lobner, D., Banack, S. A., Cox, G. A., Nunn, P. B., Wyatt, P. B., et al. (2017). Analysis of BMAA enantiomers in cycads, cyanobacteria, and mammals: in vivo formation and toxicity of D-BMAA. Amino Acids 49, 1427–1439. doi: 10.1007/s00726-017-2445-y
Michaelson, N., Facciponte, D., Bradley, W., and Stommel, E. (2017). Cytokine expression levels in ALS: a potential link between inflammation and BMAA-triggered protein misfolding. Cytokine Growth Factor Rev. 37, 81–88. doi: 10.1016/j.cytogfr.2017.05.001
Monteiro, M., Costa, M., Moreira, C., Vasconcelos, V. M., and Baptista, M. S. (2017). Screening of BMAA-producing cyanobacteria in cultured isolates and in in situ blooms. J. Appl. Phycol. 29, 879–888. doi: 10.1007/s10811-016-1003-4
Müller, S., Garcia-Gonzalez, E., Mainz, A., Hertlein, G., Heid, N. C., Mösker, E., et al. (2014). Paenilamicin: structure and biosynthesis of a hybrid nonribosomal peptide/polyketide antibiotic from the bee pathogen paenibacillus larvae. Angew. Chem. Int. Ed Engl. 53, 10821–10825. doi: 10.1002/anie.201404572
Muyderman, H., and Chen, T. (2014). Mitochondrial dysfunction in amyotrophic lateral sclerosis—a valid pharmacological target? Br. J. Pharmacol. 171, 2191–2205. doi: 10.1111/bph.12476
Nagpal, R., Mainali, R., Ahmadi, S., Wang, S., Singh, R., Kavanagh, K., et al. (2018). Gut microbiome and aging: physiological and mechanistic insights. Nutr. Healthy Aging 4, 267–285. doi: 10.3233/nha-170030
Nunn, P. B., and Codd, G. A. (2017). Metabolic solutions to the biosynthesis of some diaminomonocarboxylic acids in nature: formation in cyanobacteria of the neurotoxins 3-N-methyl-2,3-diaminopropanoic acid (BMAA) and 2,4-diaminobutanoic acid (2,4-DAB). Phytochemistry 144, 253–270. doi: 10.1016/j.phytochem.2017.09.015
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
Onselen, R. van., Downing, S., Kemp, G., and Downing, T. (2017). Investigating β-N-methylamino-L-alanine misincorporation in human cell cultures: a comparative study with known amino acid analogues. Toxins 9:E400. doi: 10.3390/toxins9120400
Oueslati, A. (2016). Implication of α-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J. Parkinsons Dis. 6, 39–51. doi: 10.3233/jpd-160779
Oueslati, A., Paleologou, K. E., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2012). Mimicking phosphorylation at serine 87 inhibits the aggregation of human α-synuclein and protects against its toxicity in a rat model of parkinson’s disease. J. Neurosci. 32, 1536–1544. doi: 10.1523/JNEUROSCI.3784-11.2012
Parracho, H. M. R. T., Bingham, M. O., Gibson, G. R., and McCartney, A. L. (2005). Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 54, 987–991. doi: 10.1099/jmm.0.46101-0
Pfeiffer, R. F. (2003). Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2, 107–116. doi: 10.1016/S1474-4422(03)00307-7
Popova, A. A., Semashko, T. A., Kostina, N. V., Rasmussen, U., Govorun, V. M., and Koksharova, O. A. (2018). The cyanotoxin BMAA induces heterocyst specific gene expression in Anabaena sp. PCC 7120 under repressive conditions. Toxins 10:E478. doi: 10.3390/toxins10110478
Rao, M., and Gershon, M. D. (2016). The bowel and beyond: the enteric nervous system in neurological disorders. Nat. Rev. Gastroenterol. Hepatol. 13, 517–528. doi: 10.1038/nrgastro.2016.107
Reichmann, H., Schneider, C., and Löhle, M. (2009). Non-motor features of Parkinson’s disease: depression and dementia. Parkinsonism Relat Disord. 15, S87–S92. doi: 10.1016/S1353-8020(09)70789-8
Rocchi, A., Pellegrini, S., Siciliano, G., and Murri, L. (2003). Causative and susceptibility genes for Alzheimer’s disease: a review. Brain Res. Bull. 61, 1–24. doi: 10.1016/s0361-9230(03)00067-4
Rodgers, K. J. (2014). Non-protein amino acids and neurodegeneration: the enemy within. Exp. Neurol. 253, 192–196. doi: 10.1016/j.expneurol.2013.12.010
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17. doi: 10.1038/nm1066
Saint-Georges-Chaumet, Y., and Edeas, M. (2016). Microbiota-mitochondria inter-talk: consequence for microbiota-host interaction. Pathog. Dis. 74:ftv096. doi: 10.1093/femspd/ftv096
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469.e12–1480.e12. doi: 10.1016/j.cell.2016.11.018
Savica, R., Carlin, J. M., Grossardt, B. R., Bower, J. H., Ahlskog, J. E., Maraganore, D. M., et al. (2009). Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology 73, 1752–1758. doi: 10.1212/WNL.0b013e3181c34af5
Sekirov, I., Russell, S., Antunes, L., and Finlay, B. B. (2010). Gut microbiota in health and disease. Physiol. Rev. 90, 859–904. doi: 10.1152/physrev.00045.2009
Shibata, N., Kunisawa, J., and Kiyono, H. (2017). Dietary and microbial metabolites in the regulation of host immunity. Front. Microbiol. 8:2171. doi: 10.3389/fmicb.2017.02171
Soo, R. M., Skennerton, C. T., Sekiguchi, Y., Imelfort, M., Paech, S. J., Dennis, P. G., et al. (2014). An expanded genomic representation of the phylum cyanobacteria. Genome Biol. Evol. 6, 1031–1045. doi: 10.1093/gbe/evu073
Spáčil, Z., Eriksson, J., Jonasson, S., Rasmussen, U., Ilag, L. L., and Bergman, B. (2010). Analytical protocol for identification of BMAA and DAB in biological samples. Analyst 135, 127–132. doi: 10.1039/b921048b
Stecher, B., and Hardt, W. D. (2008). The role of microbiota in infectious disease. Trends Microbiol. 16, 107–114. doi: 10.1016/j.tim.2007.12.008
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219. doi: 10.1111/imm.12922
Svensson, E., Horváth-Puhó, E., Thomsen, R. W., Djurhuus, J. C., Pedersen, L., Borghammer, P., et al. (2015). Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 78, 522–529. doi: 10.1002/ana.24448
Takser, L., Benachour, N., Husk, B., Cabana, H., and Gris, D. (2016). Cyanotoxins at low doses induce apoptosis and inflammatory effects in murine brain cells: potential implications for neurodegenerative diseases. Toxicol. Rep. 3, 180–189. doi: 10.1016/j.toxrep.2015.12.008
Van Den Berge, N., Ferreira, N., Gram, H., Mikkelsen, T. W., Alstrup, A. K. O., Casadei, N., et al. (2019). Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of α-synuclein in rats. Acta Neuropathol. 138, 535–550. doi: 10.1007/s00401-019-02040-w
van Onselen, R., and Downing, T. G. (2018). BMAA-protein interactions: a possible new mechanism of toxicity. Toxicon 143, 74–80. doi: 10.1016/j.toxicon.2018.01.011
van Onselen, R., and Downing, T. G. (2019). β-N-methylamino-L-alanine inhibits human catalase activity: possible implications for neurodegenerative disease development. Int. J. Toxicol. 38, 129–134. doi: 10.1177/1091581818821921
Violi, J. P., Mitrovic, S. M., Colville, A., Main, B. J., and Rodgers, K. J. (2019). Ecotoxicology and Environmental Safety Prevalence of β-methylamino-L-alanine (BMAA) and its isomers in freshwater cyanobacteria isolated from eastern Australia. Ecotoxicol. Environ. Saf. 172, 72–81. doi: 10.1016/j.ecoenv.2019.01.046
Wakabayashi, K., Takahashi, H., Takeda, S., Ohama, E., and Ikuta, F. (1988). Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 76, 217–221. doi: 10.1007/bf00687767
Keywords: Parkinson’s disease, mitochondrial dysfunction, innate immunity, neurodegeneration, microbial β-N-Methylamino-L-alanine (BMAA)
Citation: Nunes-Costa D, Magalhães JD, G-Fernandes M, Cardoso SM and Empadinhas N (2020) Microbial BMAA and the Pathway for Parkinson’s Disease Neurodegeneration. Front. Aging Neurosci. 12:26. doi: 10.3389/fnagi.2020.00026
Received: 14 November 2019; Accepted: 23 January 2020;
Published: 07 February 2020.
Edited by:
Nicola Simola, University of Cagliari, ItalyReviewed by:
Steven Robert Brenner, Saint Louis University, United StatesAgata Copani, University of Catania, Italy
Copyright © 2020 Nunes-Costa, Magalhães, G-Fernandes, Cardoso and Empadinhas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Morais Cardoso, Y2FyZG9zby5zYW5kcmEubUBnbWFpbC5jb20=; Nuno Empadinhas, bnVtZW5pdXNAY25jLnVjLnB0