 Amalie Clement
Amalie Clement Ove Wiborg
Ove Wiborg Ayodeji A. Asuni
Ayodeji A. Asuni- 1Laboratory of Neurobiology, Department of Health, Science and Technology, Aalborg University, Aalborg, Denmark
- 2Department of Physiology and Symptoms, H. Lundbeck A/S, Copenhagen, Denmark
Alzheimer’s disease (AD) is the most common form of dementia worldwide. It is mostly known for its devastating effect on memory and learning but behavioral alterations commonly known as neuropsychiatric disturbances (NPDs) are also characteristics of the disease. These include apathy, depression-like behavior, and sleep disturbances, and they all contribute to an increased caregiver burden and earlier institutionalization. The interaction between NPDs and AD pathology is not well understood, but the consensus is that they contribute to disease progression and faster decline. Consequently, recognizing and treating NPDs might improve AD pathology and increase the quality of life for both patients and caregivers. In this review article, we examine previous and current literature on apathy, depressive symptoms, and sleep disturbances in AD patients and preclinical AD mechanistic models. We hypothesize that tau accumulation, beta-amyloid (Aβ) aggregation, neuroinflammation, mitochondrial damage, and loss of the locus coeruleus (LC)-norepinephrine (NE) system all collectively impact the development of NPDs and contribute synergistically to AD pathology. Targeting more than one of these processes might provide the most optimal strategy for treating NPDs and AD. The development of such clinical approaches would be preceded by preclinical studies, for which robust and reliable mechanistic models of NPD-like behavior are needed. Thus, developing effective preclinical research models represents an important step towards a better understanding of NPDs in AD.
Introduction
Alzheimer’s disease (AD) remains a major cause of morbidity and mortality worldwide and is substantially burdensome to affected persons and their caregivers. It has been estimated that 47 million people worldwide were living with AD in 2015 (Alzheimer’s Disease International, 2015) and by 2060 15 million people will be diagnosed with AD in the US alone (Brookmeyer et al., 2018); thus, AD is a large social and financial burden to society.
The characteristic hallmarks of AD include β-amyloid (Aβ) plaques, neurofibrillary tangles (NFT), and neuroinflammation, and as the disease progresses almost all brain regions become affected with cell death and neuronal degeneration in the terminal stages eventually leading to substantial brain shrinkages and death. In addition to cognitive impairment, 80–97% of AD patients experience at least one non-mnemonic symptom at least once during the course of the disease (Gauthier et al., 2010; Van Dam et al., 2016; Tiel et al., 2019). These are denoted neuropsychiatric disturbances (NPDs) and include symptoms like apathy, depression-like behavior, sleep disturbances, aggression, and anxiety (Zhao et al., 2016). We have focused the discussion in this review on the three most common NPDs: apathy, depression-like behavior, and sleep disturbances. The factors that mediate the transition from prodromal AD to full-blown AD are not well characterized, but it is suggested that the onset, tempo, and rate of pathogenesis is impacted by significant contributions from NPDs (Geda et al., 2013; Zhao et al., 2016). Additionally, NPDs can appear before the prodromal phase of AD (Bartolini et al., 2004; Bidzan and Bidzan, 2014; Peters et al., 2015) and thus increase and prolong the caregiver burden; eventually, leading to earlier institutionalization (Steffens et al., 2005; Rea et al., 2014), which in turn increases the financial burden to society.
The diagnosing of AD is based on general health status, cognitive tests, cerebrospinal fluid (CSF) based biomarker assessment, magnetic resonance imaging (MRI), and positron emission tomography (PET) imaging. A CSF biomarker profile for AD includes low levels of Aβ42, reflecting Aβ plaque deposits, and high levels of total tau (T-tau) and hyperphosphorylated tau (P-tau), reflecting increased axonal damage and tau pathology, but other biomarkers have also been investigated to aid in early diagnosis, prognosis, and progression of AD (Blennow et al., 2010; Snyder et al., 2014; Tan et al., 2014). As NPDs have been shown to precede cognitive symptoms (Masters et al., 2015), they may represent additional biomarkers of early-stage AD and could enhance and accelerate the diagnosis of AD. However, few commonly accepted diagnostic tools for NPDs in AD exists. The Neuropsychiatric Inventory (NPI; Cummings et al., 1994; Cummings, 1997), and modifications of this (de Medeiros et al., 2010; Guercio et al., 2015; Morganti et al., 2018), is widely used to assess NPDs but it is not specific to AD and thus makes it difficult to use NPDs as biomarkers for early AD. The early and effective intervention of AD will improve quality of life for both patients and caregivers; therefore, recognizing and treating NPDs that may also contribute to AD pathophysiology (Geda et al., 2013; Zhao et al., 2016) is crucial. For this reason, we need to understand the underlying pathological mechanisms of NPDs and establish molecular biomarkers that will help in identifying and discriminating NPDs in early AD to support the development of better diagnostic tools and novel therapies.
The Unmet Need for Addressing NPDs
The Alzheimer’s Association Research Roundtable gathered in 2010 and 2016 to address the issue of NPDs in AD and improve the understanding of the underlining mechanisms (Lyketsos et al., 2011; Lanctôt et al., 2017). NPDs decrease quality of life (Robert et al., 2010), increase caregiver burden (D’Onofrio et al., 2015), and are associated with faster decline and earlier institutionalization (Steffens et al., 2005; Rea et al., 2014). Thus, the conclusion of the gathering was that better treatments of NPDs in AD patients are of the highest importance and that adequate research is necessary to support such developments. In spite of this, clinical trials for treating NPDs report conflicting results, no superiority of drug over placebo, and/or accompanying higher risk of adverse events (Banerjee et al., 2011; Brandt and Pythtila, 2013; Wang et al., 2015; Leyhe et al., 2017). The use of antipsychotics in the elderly can additionally increase the risk of mortality; moreover, dementia accelerates this risk which has led to an FDA black-box warning (Maust et al., 2015; Schmedt et al., 2016). AD patients, in particular, experience psychotropic-related adverse events when compared to age-matched non-AD patients (Sepassi and Watanabe, 2019). This suggests that different molecular circuits are involved in the development of non-mnemonic symptoms in AD patients when compared to younger patients experience similar symptoms. It further underlines the urgency for better understandings of the etiology and pathology of NPDs in AD.
Model organisms have been important tools in the studies of AD (Hall and Roberson, 2012) but unlike memory and learning, which are reasonably well modeled in AD transgenic rodents (Webster et al., 2014), NPD-like behavioral changes are less characterized in such models. Thus, valid preclinical models for NPDs need to be developed to support drug discovery research and allow us to expand the knowledge of NPD pathology.
Apathy and Depression-Like Behavior in AD
Apathy and depression can be difficult to distinguish and have overlapping symptomatology, like retarded executive function, and will be explored in parallel in this review. Apathy is the most common NPD in AD patients (Van Dam et al., 2016; Zhao et al., 2016; Lanctôt et al., 2017) with an overall estimated prevalence of 49%, while depressive behavior has an estimated prevalence of 42% (Zhao et al., 2016). Despite their similarities, symptoms like guilt, sadness or hopelessness are only associated with depressive behavior (Nobis and Husain, 2018).
Apathy is classified as a neurocognitive disturbance and defined as a reduced motivation for at least 4 weeks complemented by two of the following behaviors: reduced goal-directed behavior, reduced goal-directed cognitive activity, and emotional flattening (Van Dam et al., 2016). Meanwhile, apathy correlates with the severity of AD (Tschanz et al., 2011) and has been shown to persist if left untreated with a stronger association with mortality (van der Linde et al., 2017). On the other hand, depressive symptoms are more closely associated with reduced activity of daily living (ADL) and more serious aggression and wandering in AD patients compared to AD patients without depressive symptoms (Lyketsos et al., 1997). Finally, depressive symptoms accelerate cognitive decline in mild cognitive impairment (MCI; Brendel et al., 2015), while apathy predicts the transition from cognitively normal to MCI to AD (Guercio et al., 2015). In combination, patients with both apathy and depressive behavior are less independent and have lower ADL compared to AD patients with only apathy, depressive behavior, or none of the symptoms (Benoit et al., 2012). Thus, recognizing, discriminating, and treating apathy and depressive behavior are important but in AD pharmacological treatment of apathy (Rea et al., 2014) and depressive symptoms (Orgeta et al., 2017) have proven difficult; most likely, explained by the lack of knowledge about circuits involved in the symptom development.
Pharmacologic Interventions
Methylphenidate, which increases dopamine, norepinephrine (NE), and other catecholamines in the brain, ameliorates apathy in both mild (Padala et al., 2018) and moderate (Herrmann et al., 2008; Rosenberg et al., 2013) stages of AD compared to placebo. Although the studies differed on apathy rating scales and length of treatment, it suggests that dopamine and/or NE are relevant for the molecular circuits involved in apathy in AD. Supporting this, several studies have mapped apathy to specific brain regions with abnormalities including the anterior cingulate cortex, the prefrontal cortex, and the basal ganglia (Stella et al., 2014; Theleritis et al., 2014; Le Heron et al., 2017), all of which are innervated by dopaminergic pathways. 11C-PiB-PET imaging revealed correlations between Aβ deposits and apathy in the right anterior cingulate cortex and the bilateral frontal cortex in apathetic AD patients (Mori et al., 2014), suggesting a direct link between Aβ pathology and apathy which might be further linked to loss of homeostasis in dopamine and/or NE. Lastly, Padala et al. (2018) reported cognitive and emotional improvements after methylphenidate treatment, which highlights the need to treat NPDs in AD. Cumulatively, these findings indicate that apathy is closely linked to the worsening of AD parameters and that ameliorating apathy by targeting the dopaminergic and/or norepinephrinergic circuits might improve core AD pathology.
Depressive symptoms in AD associate with cortical thinning which has been specified to the temporal and parietal regions (Lebedeva et al., 2014) and lower gray matter volume of the left middle frontal cortex (Hu et al., 2015). Reduced cerebral blood flow in the dorsolateral prefrontal area (middle frontal gyrus) has also been documented in AD patients with depressive symptoms (Akiyama et al., 2008; Levy-Cooperman et al., 2008; Terada et al., 2014). Most of these regions are innervated by projections of serotonergic neurons from the raphe nuclei (Charnay and Léger, 2010), and considering the role of serotonin in mood (Yohn et al., 2017) these structural damages to brain areas sensitive to serotonin might explain depressive symptoms. However, using selective serotonin reuptake inhibitors (SSRIs) to treat depressive symptoms in AD has had limited success. A recent meta-analysis of randomized controlled trials (RCT) for the use of antidepressants in AD patients indicated that antidepressants have no effect over placebo (Orgeta et al., 2017). This indicates that dysregulation of serotonin alone cannot explain the development of depressive symptoms in AD.
The structural brain changes observed in AD patients with depressive symptoms might also be explained by mitochondrial dysfunction. Studies using a rodent model of depression have shown impairment of oxidative phosphorylation (OXPHOS) possibly caused by changes in mitochondrial membrane integrity (Rezin et al., 2008; Gong et al., 2011) which ultimately can lead to apoptosis (Wang, 2001) and neuronal cell death and thereby structural brain changes. Mitochondrial damage has been reported in both AD (Swerdlow, 2018) and depression (Bansal and Kuhad, 2016) and both patient groups show reduced glucose metabolism using fluorodeoxyglucose (FDG) PET (Hunt et al., 2007; Wei et al., 2016; Rice and Bisdas, 2017; Fu et al., 2018). Supporting this, a Ginkgo biloba extract (GBE) that has free radical scavenging properties, enhances mitochondrial membrane potential, and increase ATP production (Lejri et al., 2019) revealed a significant effect on apathy and other NPDs in AD patients (Scripnikov et al., 2007). On the contrary, a recent RCT found no effect on NPDs when patients were treated with resveratrol which acts on several proteins important for mitochondrial function (Zhu et al., 2018). This indicates that specific mitochondrial pathways may, at least partly, drive depressive and apathetic symptoms in AD but more studies are needed to unravel these specific pathways. The positive effect of GBE might be driven by the free radical scavenging properties because reactive oxygen species (ROS) produced during electron transport chain and OXPHOS increase during mitochondrial damage and can induce neuroinflammation via NF-κB signaling pathways which in turn increases AD pathology (Kaur et al., 2015).
Lebedeva et al. (2014) found a negative correlation between cortical thickness and levels of CSF T-tau and P-tau in AD patients with depressive symptoms which were not observed for CSF Aβ42 and suggests that only tau pathology is linked to depressive symptoms in AD. Although studies on tau pathology and depressive symptoms in AD are limited, a recent study reported that Braak stage I/II scores (NFT in entorhinal cortex and hippocampus) in post-mortem AD patients was significantly associated with depressive behavior along with other NPDs (Ehrenberg et al., 2018). On the other hand, MCI patients with depressive symptoms had higher amyloid pathology in frontotemporal and insular cortices compared to MCI patients without depressive symptoms which further correlated to a faster cognitive decline (Brendel et al., 2015). Altogether, these studies indicate that depressive symptoms in AD might be unrelated to the serotonergic system and that AD-related pathology causes damage to specific brain regions resulting in the development of depressive symptoms. Mapping how such pathological damage is mediated in relation to depressive symptoms represents an important task in the development of novel treatment options for depression in AD.
Sleep Disturbances in AD
Thirty-nine percent of AD patients experience sleep disturbances (Zhao et al., 2016) and these cover a broad range of altered sleep-wake patterns including fragmented sleep, excessive daytime sleepiness, trouble falling asleep or maintaining sleep, and early morning awakening (Suzuki et al., 2017). Although it is unclear what drives sleep disturbances in AD, substantial evidence suggest that they significantly contribute to early pathological development (Spira et al., 2014; Kabeshita et al., 2017) and progression of disease (Mander et al., 2016; Musiek and Holtzman, 2016) and for this reason sleep disturbances have been investigated as a possible target for AD interventions (Mander et al., 2016).
Sleep disturbances can occur years before clinical AD symptoms (Spira et al., 2014; Kabeshita et al., 2017). Recently, a large systemic meta-analysis on sleep disturbances and risk of dementia showed that people with sleep disturbances at baseline have a 1.49 fold higher risk of developing AD compared to subjects without sleep disturbances (Shi et al., 2018). Alterations in sleep duration were also associated with an increased risk of cognitive decline (Chen et al., 2016) and ultimately dying from dementia (Benito-León et al., 2014). Similarly, Musiek et al. (2018) found that altered sleep patterns were associated with positive PiB-PET scanning in non-demented participants, underlining the link between sleep disturbances and AD pathology. However, age was also associated with circadian disruption and thus both age and AD pathology independently contributed to the association with sleep disturbances (Musiek et al., 2018). Supporting the link between age, sleep disturbances, and AD, Benedict et al. (2015) showed that 70-year old men with sleep disturbances have a 3-fold higher risk of developing AD compared to 70-year old men without sleep disturbances, while the risk of developing AD in 50-year old men was independent on sleep disturbances. Most AD patients are elderly and sleep disturbances are common in cognitively normal elder people too (Li et al., 2018); therefore, sleep disturbances in AD might be driven by age-dependent factors. The “mitochondrial cascade hypothesis” of sporadic AD postulate that mitochondrial dysfunction is the major cause of AD pathology and that Aβ accumulation is a secondary event (Swerdlow and Khan, 2004; Swerdlow et al., 2014; Swerdlow, 2018). Mitochondria are sensitive to aging due to lack of DNA repair mechanisms and thus mutations in mitochondrial DNA (mtDNA) accumulate over time (Grimm and Eckert, 2017). Consequently, mitochondrial dysfunction will increase with age and may represent one explanation for sleep disturbances in the elderly. Supportive of this view, Adler et al. (2020) found that aging disrupts the circadian rhythm in mice shown by loss of rhythmicity in proteins involved in circadian function. These proteins were linked to pathologies like AD and Parkinson’s Disease but also glycolysis and TCA cycle pathways (Adler et al., 2020) which are central to the electron transport chain and OXPHOS to produce ATP. Altogether, the reason for sleep disturbances to be so common in AD patients may be caused by age-dependent changes in mitochondrial function which might be even more severely compromised with AD pathology.
On the contrary, cognitively normal middle-aged people revealed that lower CSF Aβ42 was associated with worse objective sleep quality (Ju et al., 2013) and in 40-65-year old cognitively normal people self-reported worse sleep adequacy was associated with lower CSF Aβ42/Aβ40, higher CSF T-tau/Aβ42, and higher CSF P-tau/Aβ42 (Sprecher et al., 2017). Additionally, experimentally sleep-deprived healthy adults increased their Aβ production overnight with 25–30% compared to normal sleeping controls (Lucey et al., 2018) and the Aβ burden increased in the hippocampus and thalamus after one night of sleep deprivation in healthy individuals (Shokri-Kojori et al., 2018). These studies indicate that sleep disturbances can affect the levels of Aβ in the brain which might be due to increased production (Lucey et al., 2018) or decreased clearance of Aβ (Iliff et al., 2012), as described in the glymphatic system pathway (Plog and Nedergaard, 2018; Benveniste et al., 2019), or a combination of both. The default mode network (DMN) is active during awake non-task specific activities and inactive during sleep (Spreng et al., 2010). When activated it has a high neuronal activity which results in increased production of Aβ (Bero et al., 2011). Lastly, melatonin (the endogenous sleep-promoting hormone) acts on the DMN in an inhibitory manner (Zisapel, 2018) and AD patients have very low levels of melatonin which correlate with disease progression (Zhou et al., 2003). Thus, one explanation for the increased levels of Aβ with sleep deprivation could be increased activation of the DMN. Moreover, melatonin has several protective properties (for a more detailed description see Vincent, 2018) including the promotion of anti-inflammatory pathways and inhibition of pro-inflammatory pathways (Deng et al., 2006; Hardeland, 2018). The decrease of melatonin in AD patients could therefore both cause sleep disturbances and induce neuroinflammation both of which contribute to AD pathology.
The locus coeruleus (LC) is implicated in controlling wakefulness and arousal by the release of NE with high levels of neural activity during wakefulness and low activity during sleep (Aston-Jones and Bloom, 1981; Mitchell and Weinshenker, 2010). The LC neurons project to a variety of brain areas and networks and have anti-inflammatory effects (Feinstein et al., 2016; Giorgi et al., 2019) but the neurons and thus NE release is compromised in MCI (Grudzien et al., 2007) and AD brains (Zarow et al., 2003; Braak et al., 2011; Mravec et al., 2014). Studies have shown that microglia respond to Aβ42 fibrils in a pro-inflammatory manner which is abolished in the presence of NE (Heneka et al., 2010), thus increased levels of Aβ in sleep disturbances may contribute to neuroinflammation which cannot be suppressed in MCI or AD brains due to loss of the LC-NE system. Furthermore, these studies found that depletion of NE by the degradation of LC neurons caused increased Aβ deposits in the hippocampus and increased levels of Aβ42 but not Aβ40 in APP transgenic mice, suggesting that NE depletion causes changes in clearance rather than the production of Aβ peptides (Heneka et al., 2010). Altogether, these studies provide evidence of a partial explanation of how sleep disturbances accelerate AD pathology via the neuroinflammatory response to depletion of NE.
Interestingly, tau pathology has been reported in LC neurons in both children and young adults (Mather and Harley, 2016) but become significantly more pathological in MCI and early AD cases (Grudzien et al., 2007). It is further postulated that tau pathology spreads from the LC to other brain areas (Iba et al., 2015) and that oligomeric tau induces mitochondrial membrane leakage and subsequently loss of OXPHOS and mitochondrial biogenesis (Camilleri et al., 2020). Lastly, dysregulation of the LC-NE system associate with depressive symptoms, apathy, and sleep disturbances in AD (Matthews et al., 2002). A recent study showed clear synergistic toxicity of tau and Aβ with both increased neurodegeneration and behavioral changes in C. elegance (Benbow et al., 2020). It is, therefore, reasonable to think that sleep disturbances, and other NPDs, are not driven by a single pathologic event; however, further studies are warranted to substantiate this hypothesis.
The Molecular Mechanisms of Sleep
The circadian system is the foundation of the sleep-wake cycle and in both humans and rodents, the system is regulated by the suprachiasmatic nucleus (SCN) of the hypothalamus (Johnston et al., 2016), which receives light/dark inputs via the intrinsically photosensitive retinal ganglion cells containing melanopsin (Paul et al., 2009). However, nearly all cells in the body contain a circadian clock where the circadian oscillations are generated by a negative feedback loop. This loop consists of the core transcriptional activators CLOCK and BMAL1 (also known as ARNTL), who control the transcription of PER and CRY genes among others. The SCN neurons project to different areas of the hypothalamus in a complex manner and these projections are responsible for the circuit activity of neurotransmitters and neuropeptides that regulate the sleep/wake cycle. These include melatonin, serotonin, NE, acetylcholine, glutamate, GABA, dopamine, orexin, neurotensin, vasopressin, and vasoactive intestinal peptide (VIP; Lim and Szymusiak, 2015; Van Erum et al., 2018). A detailed description of the circuits is beyond the scope of this review.
The circadian system network is compromised in AD (Van Erum et al., 2018). The ventrolateral preoptic nucleus of the hypothalamus is innervated by SCN GABAergic neuron projections (Chou et al., 2003) and is important for maintaining sleep (Lu et al., 2000). AD patients have fewer neurons in this area (Lim et al., 2014), suggesting a link between AD pathogenesis and dysregulated sleep. The ventrolateral preoptic nucleus produces orexins (also known as hypocretins) and regulates wakefulness (Sakurai et al., 1998; de Lecea et al., 1998) and its neurons project predominantly to the LC and raphe nuclei (Peyron et al., 1998; España and Scammell, 2011). Compared to non-demented subjects, patients with moderate to severe AD have significantly higher CSF orexin levels associating with impaired nighttime sleep (Liguori et al., 2014). These levels correlated positively with CSF T-tau and CSF P-tau, while the Mini-Mental State Examination (MMSE) score correlated positively with sleep efficiency (Liguori et al., 2014). Orexin has also been linked to higher levels of soluble Aβ in an AD transgenic mouse model (Kang et al., 2009). Lastly, Liguori et al. (2017) found a significant increase in CSF orexin in AD patients and that hypothalamic glucose consumption correlated negatively with the CSF T-tau/Aβ42 ratio suggesting a causative link between mitochondrial function, AD pathology, and orexin. Although hypothalamic glucose consumption did not correlate with CSF orexin levels, the authors argue that the hypothalamus is compromised by AD pathology and this may cause the sleep disturbances in AD patients (Liguori et al., 2017). Altogether, this is supportive of orexin dysfunction as a viable target for AD intervention. In this regard, a phase III clinical trial (NCT02750306) to treat insomnia in patients with AD using the orexin receptor antagonist (Suvorexant) was effective and generally well-tolerated (Herring et al., 2019).
Preclinical Research on NPD Related Behavior
Preclinical models are important tools for understanding pathological processes and developing novel treatment regimens; nevertheless, robust preclinical models for NPDs in AD are lacking. Although 189 AD transgenic rodent models exist today1, studies specifically characterizing NPD-like behavior in these models are limited.
Wister rats with oligomeric Aβ injected into the CA1 region of the hippocampus showed increased anxiety and memory impairment compared to vehicle injected rats without affecting hippocampal integrity (Salgado-Puga et al., 2015). This suggests, that Aβ oligomers trigger anxious behavior and memory impairment via cellular processes. In fact, others have shown that intracerebroventricular injected Aβ aggregates induce memory impairment and increased hippocampal levels of ROS and proinflammatory cytokines (Gupta et al., 2018). This indicates that Aβ oligomer injection to the brain can serve as a mechanistic model of NPD-like behavior. Double knockout PS1 and PS2 (DKO) mice experience age-dependent apathy shown by decreased nest building (Filali et al., 2009; Jirkof, 2014) activity compared to control littermates (Yan et al., 2013). Interestingly, this mouse model does not develop plaque or tangle pathology (Saura et al., 2004) but exhibit increased neuroinflammatory markers in the neocortex and hippocampus (Beglopoulos et al., 2004) and cortical neuron loss (Wines-Samuelson et al., 2010) at ages tested in the Yan-study. This suggests that neuroinflammation and/or neuronal loss can drive apathetic behavior unrelated to Aβ and tau toxicity. The 3xTg-AD mouse model shows anxiety and depressive behavior at ages where both Aβ and tau pathology is present. Interestingly, these behaviors could be ameliorated by melatonin treatment and protein changes on the protein levels were also normalized after treatment (Nie et al., 2017). Additionally, melatonin treatment significantly reduced the number of Aβ aggregates and neuroinflammation in 5xFAD mice (Jürgenson et al., 2019) and increased mitochondrial biogenesis together with a reduction in Aβ pathology in APPSwe/PS1dE9 mice (Song et al., 2018). Altogether, these studies indicate that the sleep promoting hormone, melatonin, is important for anxious and depressive behavior and that melatonin might act on both molecular and circuit pathways affected by Aβ pathology and neuroinflammation.
The APPSwe/PS1dE9 mouse model reported Aβ aggregation dependent disruption of the normal sleep-wake cycle with fewer sleep durations which could be rescued by preventing Aβ aggregation (Roh et al., 2012). These changes were accompanied by a significant reduction of Per1 and Per2 expression in the hippocampus and significant reduction of Per1, Per2, Cry1, and Cry2 expression in medulla-pons during the dark phase (Oyegbami et al., 2017). Similarly, APPxPS1 mice have decreased Per2 expression in the SCN at zeitgeber time (ZT) 2 and ZT10 (Duncan et al., 2012) while 5xFAD mice show significant changes in the mRNA expression patterns of both BMAL1 and Per2 in the SCN but these changes were less prominent on the protein level (Song et al., 2015). The McGill rats have increased expression levels of BMAL1 in the cortex and cerebellum but not in the hippocampus compared to age-matched control rats (Petrasek et al., 2018). Additionally, 3xTg-AD mice have reduced SCN neuron numbers and altered circadian rhythm (Sterniczuk et al., 2010) combined with lower numbers of norepinephrinergic neurons in the LC (Manaye et al., 2013).
SIRT1 is an NAD+-dependent deacetylase that is important for mitochondrial biogenesis (Wang and Chen, 2016) but also modulates the circadian cycle via its effect on the transcription of BMAL1 (Chang and Guarente, 2013). Serum levels of SIRT1 decrease with age and more dramatically with MCI and AD diagnosis (Kumar et al., 2013) and SIRT1 mRNA levels have been shown to decrease in an ApoE−/− mouse model of AD together with loss of circadian rhythmicity compared to normal (C57BL/6J) mice (Zhou et al., 2016). These ApoE−/− mice also had compromised mitochondria together with Aβ and tau pathology in the SCN (Zhou et al., 2016). Altogether, these studies provide evidence that dysfunctional mitochondria are implicated in the pathology of sleep disturbances in AD and that this implication might be triggered by obstruction of the normal function of SIRT1.
Altogether, these preclinical studies are consistent with the clinical data and provide evidence for a causal link between sleep disturbances, apathy, depressive behavior, and AD pathology. However, to substantiate these studies and our knowledge of NPD pathology we need to develop more robust preclinical models.
Conclusion
One notion put forward is that that NPDs appear because AD patients become unable to communicate or attend to their own needs and as a result feel misunderstood and become apathetic or depressive (Cohen-Mansfield et al., 2000). Although this hypothesis might explain some behavioral changes in AD patients, based on the literature reviewed here we propose a different hypothesis (summarized in Figure 1). Apathy, depressive behavior, and sleep disturbances are linked by pathophysiological events including mitochondrial damage, Aβ aggregations, tau accumulations, neuroinflammation, and loss of the LC-NE system, that together serve to drive and exacerbate AD progression. It is worth noting that separately, these pathophysiological processes are previous or current strategic targets for AD therapies, which have had limited success. Even though we still know little about these processes, targeting them in combination may prove to be the most optimal strategy and might pave the way for a better understanding of apathy, depressive symptoms, and sleep disturbances in AD; however, further investigations are needed to substantiate this hypothesis. Following this line of thinking, preclinical mechanistic models that allow us to dissect NPD processes are important. A strong commitment to building and studying such mechanistic models should be the next step towards developing and testing novel therapies in NPD research.
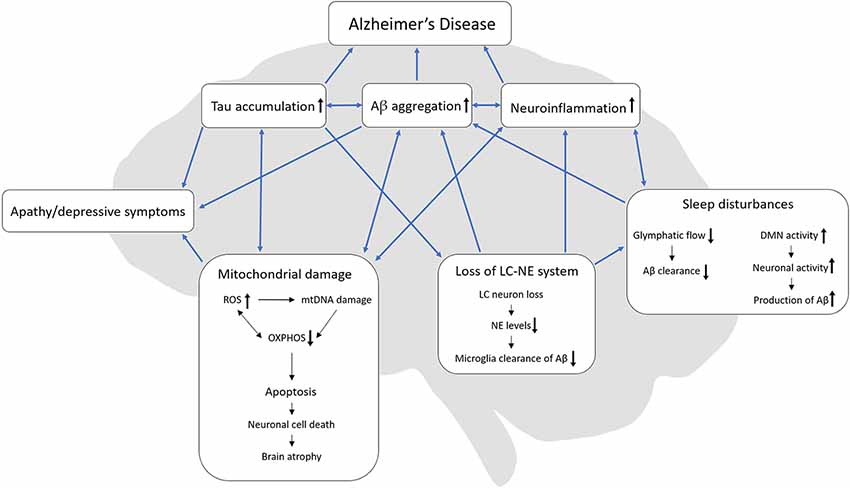
Figure 1. Interactions of AD-related pathologies, apathy, depressive symptoms, and sleep disturbances. We hypothesize, that loss of the LC-NE system might originate from tau accumulation in the LC which in turn can cause sleep disturbances. In turn, sleep disturbances can reduce the glymphatic flow and thereby decrease Aβ clearance. Also, sleep disturbances might increase the DMN activity due to the lack of sleep which then increases neuronal activity with a resulting increase in Aβ production. Both loss of the LC-NE system and sleep disturbances can increase neuroinflammation. During mitochondrial damage, ROS increases and can cause damage to both mtDNA and OXPHOS but ineffective OXPHOS can also increase the production of ROS. Mitochondrial damage can ultimately lead to apoptosis, neuronal cell death and lastly brain atrophy, which is present in late-stage AD. Both tau accumulation, Aβ aggregation and mitochondrial damage can lead to apathy/depressive behavior. AD, Alzheimer’s disease; Aβ, beta-amyloid; ROS, reactive oxygen species, OXPHOS, oxidative phosphorylation; mtDNA, mitochondrial DNA; LC, locus coeruleus; NE, norepinephrine, DMN, default mode network.
Author Contributions
All authors contributed equally to the conceptualization of the manuscript. AC wrote the manuscript. AC, OW, and AA reviewed the manuscript.
Funding
This work was partly supported by the Innovation Fund Denmark (Ref. no. 7038-00103B).
Conflict of Interest
AA is employed by the company H. Lundbeck A/S. AC is employed at both the company H. Lundbeck A/S and the University of Aalborg. OW declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to the department of health, science, and technology at Aalborg University and the department of physiology and symptoms and H. Lundbeck A/S for the academic discussion and inputs for this manuscript.
Footnotes
References
Adler, P., Chiang, C., Mayne, J., Ning, Z., Zhang, X., Xu, B., et al. (2020). Aging disrupts the circadian patterns of protein expression in the murine hippocampus. Front. Aging Neurosci. 11:368. doi: 10.3389/fnagi.2019.00368
Akiyama, H., Hashimoto, H., Kawabe, J., Higashiyama, S., Kai, T., Kataoka, K., et al. (2008). The relationship between depressive symptoms and prefrontal hypoperfusion demonstrated by eZIS in patients with DAT. Neurosci. Lett. 441, 328–331. doi: 10.1016/j.neulet.2008.06.053
Alzheimer’s Disease International. (2015). World Alzheimer Report: The Global Impact of Dementia. Available Online at: https://www.alz.co.uk/research/WorldAlzheimerReport2015-sheet.pdf. Accessed October 15, 2018.
Aston-Jones, G., and Bloom, F. E. (1981). Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1, 876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981
Banerjee, S., Hellier, J., Dewey, M., Romeo, R., Ballard, C., Baldwin, R., et al. (2011). Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet 378, 403–411. doi: 10.1016/S0140-6736(11)60830-1
Bansal, Y., and Kuhad, A. (2016). Mitochondrial dysfunction in depression. Curr. Neuropharmacol. 14, 610–618. doi: 10.2174/1570159x14666160229114755
Bartolini, M., Coccia, M., Luzzi, S., Provinciali, L., and Ceravolo, M. G. (2004). Motivational symptoms of depression mask preclinical Alzheimer’s disease in elderly subjects. Dement. Geriatr. Cogn. Disord. 19, 31–36. doi: 10.1159/000080968
Beglopoulos, V., Sun, X., Saura, C. A., Lemere, C. A., Kim, R. D., and Shen, J. (2004). Reduced β-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J. Biol. Chem. 279, 46907–46914. doi: 10.1074/jbc.M409544200
Benbow, S. J., Strovas, T. J., Darvas, M., Saxton, A., and Kraemer, B. C. (2020). Synergistic toxicity between tau and amyloid drives neuronal dysfunction and neurodegeneration in transgenic C. elegans. Hum. Mol. Genet. 29, 495–505. doi: 10.1093/hmg/ddz319
Benedict, C., Byberg, L., Cedernaes, J., Hogenkamp, P. S., Giedratis, V., Kilander, L., et al. (2015). Self-reported sleep disturbance is associated with Alzheimer’s disease risk in men. Alzheimers Dement. 11, 1090–1097. doi: 10.1016/j.jalz.2014.08.104
Benito-León, J., Louis, E. D., Villarejo-Galende, A., Romero, J. P., and Bermejo-Pareja, F. (2014). Long sleep duration in elders without dementia increases risk of dementia mortality (NEDICES). Neurology 83, 1530–1537. doi: 10.1212/wnl.0000000000000915
Benoit, M., Berrut, G., Doussaint, J., Bakchine, S., Bonin-Guillaume, S., Frémont, P., et al. (2012). Apathy and depression in mild Alzheimer’s disease: a cross-sectional study using diagnostic criteria. J. Alzheimers Dis. 31, 325–334. doi: 10.3233/JAD-2012-112003
Benveniste, H., Liu, X., Koundal, S., Sanggaard, S., Lee, H., and Wardlaw, J. (2019). The glymphatic system and waste clearance with brain aging. Gerontology 65, 106–119. doi: 10.1159/000490349
Bero, A. W., Yan, P., Roh, J. H., Cirrito, J. R., Stewart, F. R., Raichle, M. E., et al. (2011). Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 14, 750–756. doi: 10.1038/nn.2801
Bidzan, M., and Bidzan, L. (2014). Neurobehavioral manifestation in early period of Alzheimer disease and vascular dementia. Psychiatr. Pol. 48, 319–330.
Blennow, K., Hampel, H., Weiner, M., and Zetterberg, H. (2010). Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 6, 131–144. doi: 10.1038/nrneurol.2010.4
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/nen.0b013e318232a379
Brandt, N. J., and Pythtila, J. (2013). Psychopharmacological medication use among older adults with dementia in nursing homes. J. Gerontol. Nurs. 39, 8–14. doi: 10.3928/00989134-20130315-05
Brendel, M., Pogarell, O., Xiong, G., Delker, A., Bartenstein, P., Rominger, A., et al. (2015). Depressive symptoms accelerate cognitive decline in amyloid-positive MCI patients. Eur. J. Nucl. Med. Mol. Imaging 42, 716–724. doi: 10.1007/s00259-014-2975-4
Brookmeyer, R., Abdalla, N., Kawas, C. H., and Corrada, M. M. (2018). Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement. 14, 121–129. doi: 10.1016/j.jalz.2017.10.009
Camilleri, A., Ghio, S., Caruana, M., Weckbecker, D., Schmidt, F., Kamp, F., et al. (2020). Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 1862:183064. doi: 10.1016/j.bbamem.2019.183064
Chang, H.-C., and Guarente, L. (2013). SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153, 1448–1460. doi: 10.1016/j.cell.2013.05.027
Charnay, Y., and Léger, L. (2010). Brain serotonergic circuitries. Dialogues Clin. Neurosci. 12, 471–487.
Chen, J.-C., Espeland, M. A., Brunner, R. L., Lovato, L. C., Wallace, R. B., Leng, X., et al. (2016). Sleep duration, cognitive decline, and dementia risk in older women. Alzheimers Dement. 12, 21–33. doi: 10.1016/j.jalz.2015.03.004
Chou, T. C., Scammell, T. E., Gooley, J. J., Gaus, S. E., Saper, C. B., and Lu, J. (2003). Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J. Neurosci. 23, 10691–10702. doi: 10.1523/JNEUROSCI.23-33-10691.2003
Cohen-Mansfield, J., Golander, H., and Arnheim, G. (2000). Self-identity in older persons suffering from dementia: preliminary results. Soc. Sci. Med. 51, 381–394. doi: 10.1016/s0277-9536(99)00471-2
Cummings, J. L. (1997). The neuropsychiatric inventory: assessing psychopathology in dementia patients. Neurology 48, S10–S16. doi: 10.1212/wnl.48.5_suppl_6.10s
Cummings, J. L., Mega, M., Gray, K., Rosenberg-Thompson, S., Carusi, D. A., and Gornbein, J. (1994). The neuropsychiatric inventory: comprehensive assessment of psychopathology in dementia. Neurology 44, 2308–2314. doi: 10.1212/wnl.44.12.2308
D’Onofrio, G., Sancarlo, D., Addante, F., Ciccone, F., Cascavilla, L., Paris, F., et al. (2015). Caregiver burden characterization in patients with Alzheimer’s disease or vascular dementia. Int. J. Geriatr. Psychiatry 30, 891–899. doi: 10.1002/gps.4232
de Lecea, L., Kilduff, T. S., Peyron, C., Gao, X., Foye, P. E., Danielson, P. E., et al. (1998). The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U S A 95, 322–327. doi: 10.1073/pnas.95.1.322
de Medeiros, K., Robert, P., Gauthier, S., Stella, F., Politis, A., Leoutsakos, J., et al. (2010). The neuropsychiatric inventory-clinician rating scale (NPI-C): reliability and validity of a revised assessment of neuropsychiatric symptoms in dementia. Int. Psychogeriatr. 22, 984–994. doi: 10.1017/S1041610210000876
Deng, W.-G., Tang, S.-T., Tseng, H.-P., and Wu, K. K. (2006). Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108, 518–524. doi: 10.1182/blood-2005-09-3691
Duncan, M. J., Smith, J. T., Franklin, K. M., Beckett, T. L., Murphy, M. P., St Clair, D. K., et al. (2012). Effects of aging and genotype on circadian rhythms, sleep and clock gene expression in APPxPS1 knock-in mice, a model for Alzheimer’s disease. Exp. Neurol. 236, 249–258. doi: 10.1016/j.expneurol.2012.05.011
Ehrenberg, A. J., Suemoto, C. K., França Resende, E. P., Petersen, C., Leite, R. E. P., Rodriguez, R. D., et al. (2018). Neuropathologic correlates of psychiatric symptoms in Alzheimer’s disease. J. Alzheimers Dis. 66, 115–126. doi: 10.3233/JAD-180688
España, R. A., and Scammell, T. E. (2011). Sleep neurobiology from a clinical perspective. Sleep 34, 845–858. doi: 10.5665/sleep.1112
Feinstein, D. L., Kalinin, S., and Braun, D. (2016). Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J. Neurochem. 139, 154–178. doi: 10.1111/jnc.13447
Filali, M., Lalonde, R., and Rivest, S. (2009). Cognitive and non-cognitive behaviors in an APPswe/PS1 bigenic model of Alzheimer’s disease. Genes Brain Behav. 8, 143–148. doi: 10.1111/j.1601-183x.2008.00453.x
Fu, C., Zhang, H., Xuan, A., Gao, Y., Xu, J., and Shi, D. (2018). A combined study of 18F-FDG PET-CT and fMRI for assessing resting cerebral function in patients with major depressive disorder. Exp. Ther. Med. 16, 1873–1881. doi: 10.3892/etm.2018.6434
Gauthier, S., Cummings, J., Ballard, C., Brodaty, H., Grossberg, G., Robert, P., et al. (2010). Management of behavioral problems in Alzheimer’s disease. Int. Psychogeriatr. 22, 346–372. doi: 10.1017/S1041610209991505
Geda, Y. E., Schneider, L. S., Gitlin, L. N., Miller, D. S., Smith, G. S., Bell, J., et al. (2013). Neuropsychiatric symptoms in Alzheimer’s disease: past progress and anticipation of the future. Alzheimers Dement. 9, 602–608. doi: 10.1016/j.jalz.2012.12.001
Giorgi, F. S., Saccaro, L. F., Galgani, A., Busceti, C. L., Biagioni, F., Frati, A., et al. (2019). The role of locus coeruleus in neuroinflammation occurring in Alzheimer’s disease. Brain Res. Bull. 153, 47–58. doi: 10.1016/j.brainresbull.2019.08.007
Gong, Y., Chai, Y., Ding, J. H., Sun, X. L., and Hu, G. (2011). Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 488, 76–80. doi: 10.1016/j.neulet.2010.11.006
Grimm, A., and Eckert, A. (2017). Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 143, 418–431. doi: 10.1111/jnc.14037
Grudzien, A., Shaw, P., Weintraub, S., Bigio, E., Mash, D. C., and Mesulam, M. M. (2007). Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 28, 327–335. doi: 10.1016/j.neurobiolaging.2006.02.007
Guercio, B. J., Donovan, N. J., Munro, C. E., Aghjayan, S. L., Wigman, S. E., Locascio, J. J., et al. (2015). The apathy evaluation scale: a comparison of subject, informant, and clinician report in cognitively normal elderly and mild cognitive impairment. J. Alzheimers Dis. 47, 421–432. doi: 10.3233/jad-150146
Gupta, P., Sil, S., Ghosh, R., Ghosh, A., and Ghosh, T. (2018). Intracerebroventricular Aβ-induced neuroinflammation alters peripheral immune responses in rats. J. Mol. Neurosci. 66, 572–586. doi: 10.1007/s12031-018-1189-9
Hall, A. M., and Roberson, E. D. (2012). Mouse models of Alzheimer’s disease. Brain Res. Bull. 88, 3–12. doi: 10.1016/j.brainresbull.2011.11.017
Hardeland, R. (2018). Melatonin and inflammation-story of a double-edged blade. J. Pineal Res. 65:e12525. doi: 10.1111/jpi.12525
Heneka, M. T., Nadrigny, F., Regen, T., Martinez-Hernandez, A., Dumitrescu-Ozimek, L., Terwel, D., et al. (2010). Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U S A 107, 6058–6063. doi: 10.1073/pnas.0909586107
Herring, W. J., Ceesay, P., Snyder, E., Bliwise, D., Budd, K., Hutzelmann, J., et al. (2019). 0405 randomized controlled clinical polysomnography trial of suvorexant for treating insomnia in patients with Alzheimer’s disease. Sleep 42:A164. doi: 10.1093/sleep/zsz067.404
Herrmann, N., Rothenburg, L. S., Black, S. E., Ryan, M., Liu, B. A., Busto, U. E., et al. (2008). Methylphenidate for the treatment of apathy in Alzheimer disease. J. Clin. Psychopharmacol. 28, 296–301. doi: 10.1016/j.jalz.2008.05.333
Hu, X., Meiberth, D., Newport, B., and Jessen, F. (2015). Anatomical correlates of the neuropsychiatric symptoms in Alzheimer’s disease. Curr. Alzheimer Res. 12, 266–277. doi: 10.2174/1567205012666150302154914
Hunt, A., Schönknecht, P., Henze, M., Seidl, U., Haberkorn, U., and Schröder, J. (2007). Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease. Psychiatry Res. 155, 147–154. doi: 10.1016/j.pscychresns.2006.12.003
Iba, M., McBride, J. D., Guo, J. L., Zhang, B., Trojanowski, J. Q., and Lee, V. M. Y. (2015). Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 130, 349–362. doi: 10.1007/s00401-015-1458-4
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Jirkof, P. (2014). Burrowing and nest building behavior as indicators of well-being in mice. J. Neurosci. Methods 234, 139–146. doi: 10.1016/j.jneumeth.2014.02.001
Johnston, J. D., Ordovas, J. M., Scheer, F. A., and Turek, F. W. (2016). Circadian rhythms, metabolism, and chrononutrition in rodents and humans. Adv. Nutr. 7, 399–406. doi: 10.3945/an.115.010777
Ju, Y.-E. S., McLeland, J. S., Toedebusch, C. D., Xiong, C., Fagan, A. M., Duntley, S. P., et al. (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 70, 587–593. doi: 10.1001/jamaneurol.2013.2334
Jürgenson, M., Zharkovskaja, T., Noortoots, A., Morozova, M., Beniashvili, A., Zapolski, M., et al. (2019). Effects of the drug combination memantine and melatonin on impaired memory and brain neuronal deficits in an amyloid-predominant mouse model of Alzheimer’s disease. J. Pharm. Pharmacol. 71, 1695–1705. doi: 10.1111/jphp.13165
Kabeshita, Y., Adachi, H., Matsushita, M., Kanemoto, H., Sato, S., Suzuki, Y., et al. (2017). Sleep disturbances are key symptoms of very early stage Alzheimer disease with behavioral and psychological symptoms: a Japan multi-center cross-sectional study (J-BIRD). Int. J. Geriatr. Psychiatry 32, 222–230. doi: 10.1002/gps.4470
Kang, J.-E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kaur, U., Banerjee, P., Bir, A., Sinha, M., Biswas, A., and Chakrabarti, S. (2015). Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: the NF-κB connection. Curr. Top. Med. Chem. 15, 446–457. doi: 10.2174/1568026615666150114160543
Kumar, R., Chaterjee, P., Sharma, P. K., Singh, A. K., Gupta, A., Gill, K., et al. (2013). Sirtuin1: a promising serum protein marker for early detection of Alzheimer’s disease. PLoS One 8:e61560. doi: 10.1371/journal.pone.0061560
Lanctôt, K. L., Amatniek, J., Ancoli-Israel, S., Arnold, S. E., Ballard, C., Cohen-Mansfield, J., et al. (2017). Neuropsychiatric signs and symptoms of Alzheimer’s disease: new treatment paradigms. Alzheimers Dement. 3, 440–449. doi: 10.1016/j.trci.2017.07.001
Lebedeva, A., Westman, E., Lebedev, A. V., Li, X., Winblad, B., Simmons, A., et al. (2014). Structural brain changes associated with depressive symptoms in the elderly with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 85, 928–933. doi: 10.1136/jnnp-2013-307110
Le Heron, C., Apps, M. A. J., and Husain, M. (2017). The anatomy of apathy: a neurocognitive framework for amotivated behaviour. Neuropsychologia 118, 54–67. doi: 10.1016/j.neuropsychologia.2017.07.003
Lejri, I., Agapouda, A., Grimm, A., and Eckert, A. (2019). Mitochondria- and oxidative stress-targeting substances in cognitive decline-related disorders- and molecular mechanisms to clinical evidence. Oxid. Med. Cell. Longev. 2019:9695412. doi: 10.1155/2019/9695412
Levy-Cooperman, N., Burhan, A. M., Rafi-Tari, S., Kusano, M., Ramirez, J., Caldwell, C., et al. (2008). Frontal lobe hypoperfusion and depressive symptoms in Alzheimer disease. J. Psychiatry Neurosci. 33, 218–226.
Leyhe, T., Reynolds, C. F. III., Melcher, T., Linnemann, C., Klöppel, S., Blennow, K., et al. (2017). A common challenge in older adults: classification, overlap, and therapy of depression and dementia. Alzheimers Dement. 13, 59–71. doi: 10.1016/j.jalz.2016.08.007
Li, J., Vitiello, M. V., and Gooneratne, N. S. (2018). Sleep in normal aging. Sleep Med. Clin. 13, 1–11. doi: 10.1016/j.jsmc.2017.09.001
Liguori, C., Chiaravalloti, A., Nuccetelli, M., Izzi, F., Sancesario, G., Cimini, A., et al. (2017). Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer’s disease. J. Neurol. 264, 2215–2223. doi: 10.1007/s00415-017-8613-x
Liguori, C., Romigi, A., Nuccetelli, M., Zannino, S., Sancesario, G., Martorana, A., et al. (2014). Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 71, 1498–1505. doi: 10.1001/jamaneurol.2014.2510
Lim, A. S. P., Ellison, B. A., Wang, J. L., Yu, L., Schneider, J. A., Buchman, A. S., et al. (2014). Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain 137, 2847–2861. doi: 10.1093/brain/awu222
Lim, M. M., and Szymusiak, R. (2015). Neurobiology of arousal and sleep: updates and insights into neurological disorders. Curr. Sleep Med. Reports 1, 91–100. doi: 10.1007/s40675-015-0013-0
Lu, J., Greco, M. A., Shiromani, P., and Saper, C. B. (2000). Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J. Neurosci. 20, 3830–3842. doi: 10.1523/JNEUROSCI.20-10-03830.2000
Lucey, B. P., Hicks, T. J., McLeland, J. S., Toedebusch, C. D., Boyd, J., Elbert, D. L., et al. (2018). Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 83, 197–204. doi: 10.1002/ana.25117
Lyketsos, C. G., Carrillo, M. C., Ryan, J. M., Khachaturian, A. S., Trzepacz, P., Amatniek, J., et al. (2011). Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 7, 532–539. doi: 10.1016/j.jalz.2011.05.2410
Lyketsos, C. G., Steele, C., Baker, L., Galik, E., Kopunek, S., Steinberg, M., et al. (1997). Major and minor depression in Alzheimer’s disease: prevalence and impact. J. Neuropsychiatry Clin. Neurosci. 9, 556–561. doi: 10.1176/jnp.9.4.556
Manaye, K. F., Mouton, P. R., Xu, G., Drew, A., Lei, D.-L., Sharma, Y., et al. (2013). Age-related loss of noradrenergic neurons in the brains of triple transgenic mice. Age 35, 139–147. doi: 10.1007/s11357-011-9343-0
Mander, B. A., Winer, J. R., Jagust, W. J., and Walker, M. P. (2016). Sleep: a novel mechanistic pathway, biomarker and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci. 39, 552–566. doi: 10.1016/j.tins.2016.05.002
Masters, M. C., Morris, J. C., and Roe, C. M. (2015). “Noncognitive” symptoms of early Alzheimer disease: a longitudinal analysis. Neurology 84, 617–622. doi: 10.1212/WNL.0000000000001238
Mather, M., and Harley, C. W. (2016). The locus coeruleus: essential for maintaining cognitive function and the aging brain. Trends Cogn. Sci. 20, 214–226. doi: 10.1016/j.tics.2016.01.001
Matthews, K. L., Chen, C. P. L. H., Esiri, M. M., Keene, J., Minger, S. L., and Francis, P. T. (2002). Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol. Psychiatry 51, 407–416. doi: 10.1016/s0006-3223(01)01235-5
Maust, D. T., Kim, H. M., Seyfried, L. S., Chiang, C., Kavanagh, J., Schneider, L. S., et al. (2015). Antipsychotics, other psychotropics, and the risk of death in patients with dementia: number needed to harm. JAMA Psychiatry 72, 438–445. doi: 10.1001/jamapsychiatry.2014.3018
Mitchell, H. A., and Weinshenker, D. (2010). Good night and good luck: norepinephrine in sleep pharmacology. Biochem. Pharmacol. 79, 801–809. doi: 10.1016/j.bcp.2009.10.004
Morganti, F., Soli, A., Savoldelli, P., and Belotti, G. (2018). The neuropsychiatric inventory-diary rating scale (NPI-DIARY): a method for improving stability in assessing neuropsychiatric symptoms in dementia. Dement. Geriatr. Cogn. Dis. Extra 8, 306–320. doi: 10.1159/000490380
Mori, T., Shimada, H., Shinotoh, H., Hirano, S., Eguchi, Y., Yamada, M., et al. (2014). Apathy correlates with prefrontal amyloid β deposition in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 85, 449–455. doi: 10.1136/jnnp-2013-306110
Mravec, B., Lejavova, K., and Cubinkova, V. (2014). Locus (coeruleus) minoris resistentiae in pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 11, 992–1001. doi: 10.2174/1567205011666141107130505
Musiek, E. S., Bhimasani, M., Zangrilli, M. A., Morris, J. C., Holtzman, D. M., and Ju, Y.-E. S. (2018). Circadian rest-activity pattern changes in aging and preclinical alzheimer disease. JAMA Neurol. 75, 582–590. doi: 10.1001/jamaneurol.2017.4719
Musiek, E. S., and Holtzman, D. M. (2016). Mechanisms linking circadian clocks, sleep and neurodegeneration. Science 354, 1004–1008. doi: 10.1126/science.aah4968
Nie, L., Wei, G., Peng, S., Qu, Z., Yang, Y., Yang, Q., et al. (2017). Melatonin ameliorates anxiety and depression-like behaviors and modulates proteomic changes in triple transgenic mice of Alzheimer’s disease. Biofactors 43, 593–611. doi: 10.1002/biof.1369
Nobis, L., and Husain, M. (2018). Apathy in Alzheimer’s disease. Curr. Opin. Behav. Sci. 22, 7–13. doi: 10.1016/j.cobeha.2017.12.007
Orgeta, V., Tabet, N., Nilforooshan, R., and Howard, R. (2017). Efficacy of antidepressants for depression in Alzheimer’s disease: systematic review and meta-analysis. J. Alzheimers Dis. 58, 725–733. doi: 10.3233/JAD-161247
Oyegbami, O., Collins, H. M., Pardon, M.-C., Ebling, F. J. P., Heery, D. M., and Moran, P. M. (2017). Abnormal clock gene expression and locomotor activity rhythms in two month-old female APPSwe/PS1dE9 mice. Curr. Alzheimer Res. 14, 850–860. doi: 10.2174/1567205014666170317113159
Padala, P. R., Padala, K. P., Lensing, S. Y., Ramirez, D., Monga, V., Bopp, M. M., et al. (2018). Methylphenidate for apathy in community-dwelling older veterans with mild Alzheimer’s disease: a double-blind, randomized, placebo-controlled trial. Am. J. Psychiatry 175, 159–168. doi: 10.1176/appi.ajp.2017.17030316
Paul, K. N., Saafir, T. B., and Tosini, G. (2009). The role of retinal photoreceptors in the regulation of circadian rhythms. Rev. Endocr. Metab. Disord. 10, 271–278. doi: 10.1007/s11154-009-9120-x
Peters, M. E., Schwartz, S., Han, D., Rabins, P. V., Steinberg, M., Tschanz, J. T., et al. (2015). Neuropsychiatric symptoms as predictors of progression to severe Alzheimer’s dementia and death: the cache county dementia progression study. Am. J. Psychiatry 172, 460–465. doi: 10.1176/appi.ajp.2014.14040480
Petrasek, T., Vojtechova, I., Lobellova, V., Popelikova, A., Janikova, M., Brozka, H., et al. (2018). The mcgill transgenic rat model of Alzheimer’s disease displays cognitive and motor impairments, changes in anxiety and social behavior, and altered circadian activity. Front. Aging Neurosci. 10:250. doi: 10.3389/fnagi.2018.00250
Peyron, C., Tighe, D. K., van den Pol, A. N., de Lecea, L., Heller, H. C., Sutcliffe, J. G., et al. (1998). Neurons containing hypocretin (orexin) project to multiple neuronal systems. J. Neurosci. 18, 9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998
Plog, B. A., and Nedergaard, M. (2018). The glymphatic system in central nervous system health and disease: past, present, and future. Annu. Rev. Pathol. Mech. Dis. 13, 379–394. doi: 10.1146/annurev-pathol-051217-111018
Rea, R., Carotenuto, A., Fasanaro, A. M., Traini, E., and Amenta, F. (2014). Apathy in Alzheimer’s disease: any effective treatment? ScientificWorldJournal 2014:421385. doi: 10.1155/2014/421385
Rezin, G. T., Cardoso, M. R., Gonçalves, C. L., Scaini, G., Fraga, D. B., Riegel, R. E., et al. (2008). Inhibition of mitochondrial respiratory chain in brain of rats subjected to an experimental model of depression. Neurochem. Int. 53, 395–400. doi: 10.1016/j.neuint.2008.09.012
Rice, L., and Bisdas, S. (2017). The diagnostic value of FDG and amyloid PET in Alzheimer’s disease—A systematic review. Eur. J. Radiol. 94, 16–24. doi: 10.1016/j.ejrad.2017.07.014
Robert, P. H., Mulin, E., Malléa, P., and David, R. (2010). Apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci. Ther. 16, 263–271. doi: 10.1111/j.1755-5949.2009.00132.x
Roh, J. H., Huang, Y., Bero, A. W., Kasten, T., Stewart, F. R., Bateman, R. J., et al. (2012). Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 4:150ra122. doi: 10.1126/scitranslmed.3004291
Rosenberg, P. B., Lanctôt, K. L., Drye, L. T., Herrmann, N., Scherer, R. W., Bachman, D. L., et al. (2013). Safety and efficacy of methylphenidate for apathy in Alzheimer’s disease: a randomized, placebo-controlled trial. J. Clin. Psychiatry 74, 810–816. doi: 10.4088/JCP.12m08099
Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., et al. (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585. doi: 10.1016/s0092-8674(00)80949-6
Salgado-Puga, K., Prado-Alcalá, R. A., and Peña-Ortega, F. (2015). Amyloid-β enhances typical rodent behavior while it impairs contextual memory consolidation. Behav. Neurol. 2015:526912. doi: 10.1155/2015/526912
Saura, C. A., Choi, S.-Y., Beglopoulos, V., Malkani, S., Zhang, D., Shankaranarayana Rao, B. S., et al. (2004). Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36. doi: 10.1016/s0896-6273(04)00182-5
Schmedt, N., Kollhorst, B., Enders, D., Jobski, K., Krappweis, J., Garbe, E., et al. (2016). Comparative risk of death in older adults treated with antipsychotics: a population-based cohort study. Eur. Neuropsychopharmacol. 26, 1390–1400. doi: 10.1016/j.euroneuro.2016.07.006
Scripnikov, A., Khomenko, A., and Napryeyenko, O. (2007). Effects of Ginkgo biloba extract EGb 761°ledR on neuropsychiatric symptoms of dementia: findings from a randomised controlled trial. Wien. Med. Wochenschr. 157, 295–300. doi: 10.1007/s10354-007-0427-5
Sepassi, A., and Watanabe, J. H. (2019). Emergency department visits for psychotropic-related adverse drug events in older adults with Alzheimer disease, 2013–2014. Ann. Pharmacother. 53, 1173–1183. doi: 10.1177/1060028019866927
Shi, L., Chen, S.-J., Ma, M.-Y., Bao, Y.-P., Han, Y., Wang, Y.-M., et al. (2018). Sleep disturbances increase the risk of dementia: a systematic review and meta-analysis. Sleep Med. Rev. 40, 4–16. doi: 10.1016/j.smrv.2017.06.010
Shokri-Kojori, E., Wang, G.-J., Wiers, C. E., Demiral, S. B., Guo, M., Kim, S. W., et al. (2018). β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. U S A 115, 4483–4488. doi: 10.1073/pnas.1721694115
Snyder, H. M., Carrillo, M. C., Grodstein, F., Henriksen, K., Jeromin, A., Lovestone, S., et al. (2014). Developing novel blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 10, 109–114. doi: 10.1016/j.jalz.2013.10.007
Song, C., Li, M., Xu, L., Shen, Y., Yang, H., Ding, M., et al. (2018). Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 29, 1517–1524. doi: 10.1097/wnr.0000000000001139
Song, H., Moon, M., Choe, H. K., Han, D.-H., Jang, C., Kim, A., et al. (2015). Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol. Neurodegener. 10:13. doi: 10.1186/s13024-015-0007-x
Spira, A. P., Chen-Edinboro, L. P., Wu, M. N., and Yaffe, K. (2014). Impact of sleep on the risk of cognitive decline and dementia. Curr. Opin. Psychiatry 27, 478–483. doi: 10.1097/yco.0000000000000106
Sprecher, K. E., Koscik, R. L., Carlsson, C. M., Zetterberg, H., Blennow, K., Okonkwo, O. C., et al. (2017). Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89, 445–453. doi: 10.1212/wnl.0000000000004171
Spreng, R. N., Stevens, W. D., Chamberlain, J. P., Gilmore, A. W., and Schacter, D. L. (2010). Default network activity, coupled with the frontoparietal control network, supports goal-directed cognition. NeuroImage 53, 303–317. doi: 10.1016/j.neuroimage.2010.06.016
Steffens, D. C., Maytan, M., Helms, M. J., and Plassman, B. L. (2005). Prevalence and clinical correlates of neuropsychiatric symptoms in dementia. Am. J. Alzheimers Dis. Other Demen. 20, 367–373. doi: 10.1177/153331750502000611
Stella, F., Radanovic, M., Aprahamian, I., Canineu, P. R., de Andrade, L. P., and Forlenza, O. V. (2014). Neurobiological correlates of apathy in Alzheimer’s disease and mild cognitive impairment: a critical review. J. Alzheimers Dis. 39, 633–648. doi: 10.3233/JAD-131385
Sterniczuk, R., Dyck, R. H., LaFerla, F. M., and Antle, M. C. (2010). Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res. 1348, 139–148. doi: 10.1016/j.brainres.2010.05.013
Suzuki, K., Miyamoto, M., and Hirata, K. (2017). Sleep disorders in the elderly: diagnosis and management. J. Gen. Fam. Med. 18, 61–71. doi: 10.1002/jgf2.27
Swerdlow, R. H. (2018). Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 62, 1403–1416. doi: 10.3233/JAD-170585
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Swerdlow, R. H., and Khan, S. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 63, 8–20. doi: 10.1016/j.mehy.2003.12.045
Tan, C.-C., Yu, J.-T., and Tan, L. (2014). Biomarkers for preclinical Alzheimer’s disease. J. Alzheimers Dis. 42, 1051–1069. doi: 10.3233/JAD-140843
Terada, S., Oshima, E., Sato, S., Ikeda, C., Nagao, S., Hayashi, S., et al. (2014). Depressive symptoms and regional cerebral blood flow in Alzheimer’s disease. Psychiatry Res. 221, 86–91. doi: 10.1016/j.pscychresns.2013.11.002
Theleritis, C., Politis, A., Siarkos, K., and Lyketsos, C. G. (2014). A review of neuroimaging findings of apathy in Alzheimer’s disease. Int. Psychogeriatr. 26, 195–207. doi: 10.1017/S1041610213001725
Tiel, C., Sudo, F. K., and Calmon, A. B. (2019). Neuropsychiatric symptoms and executive function impairments in Alzheimer’s disease and vascular dementia: the role of subcortical circuits. Dement. Neuropsychol. 13, 293–298. doi: 10.1590/1980-57642018dn13-030005
Tschanz, J. T., Corcoran, C. D., Schwartz, S., Treiber, K., Green, R. C., Norton, M. C., et al. (2011). Progression of cognitive, functional and neuropsychiatric symptom domains in a population cohort with Alzheimer dementia: the cache county dementia progression study. Am. J. Geriatr. Psychiatry 19, 532–542. doi: 10.1097/jgp.0b013e3181faec23
Van Dam, D., Vermeiren, Y., Dekker, A. D., Naudé, P. J. W., and De Deyn, P. P. (2016). Neuropsychiatric disturbances in Alzheimer’s disease: what have we learned from neuropathological studies? Curr. Alzheimer Res. 13, 1145–1164. doi: 10.2174/1567205013666160502123607
van der Linde, R. M., Matthews, F. E., Dening, T., and Brayne, C. (2017). Patterns and persistence of behavioural and psychological symptoms in those with cognitive impairment: the importance of apathy. Int. J. Geriatr. Psychiatry 32, 306–315. doi: 10.1002/gps.4464
Van Erum, J., Van Dam, D., and De Deyn, P. P. (2018). Sleep and Alzheimer’s disease: a pivotal role for the suprachiasmatic nucleus. Sleep Med. Rev. 40, 17–27. doi: 10.1016/j.smrv.2017.07.005
Vincent, B. (2018). Protective roles of melatonin against the amyloid-dependent development of Alzheimer’s disease: a critical review. Pharmacol. Res. 134, 223–237. doi: 10.1016/j.phrs.2018.06.011
Wang, J., and Chen, G. J. (2016). Mitochondria as a therapeutic target in Alzheimer’s disease. Genes Dis. 3, 220–227. doi: 10.1016/j.gendis.2016.05.001
Wang, J., Yu, J.-T., Wang, H.-F., Meng, X.-F., Wang, C., Tan, C.-C., et al. (2015). Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 86, 101–109. doi: 10.1136/jnnp-2014-308112
Webster, S. J., Bachstetter, A. D., Nelson, P. T., Schmitt, F. A., and Van Eldik, L. J. (2014). Using mice to model Alzheimer’s dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 5:88. doi: 10.3389/fgene.2014.00088
Wei, K., Xue, H. L., Guan, Y. H., Zuo, C. T., Ge, J. J., Zhang, H. Y., et al. (2016). Analysis of glucose metabolism of 18F-FDG in major depression patients using PET imaging: Correlation of salivary cortisol and α-amylase. Neurosci. Lett. 629, 52–57. doi: 10.1016/j.neulet.2016.06.039
Wines-Samuelson, M., Schulte, E. C., Smith, M. J., Aoki, C., Liu, X., Kelleher, R. J., et al. (2010). Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS One 5:e10195. doi: 10.1371/journal.pone.0010195
Yan, L., Li, L., Han, W., Pan, B., Xue, X., and Mei, B. (2013). Age-related neuropsychiatric symptoms in presenilins conditional double knockout mice. Brain Res. Bull. 97, 104–111. doi: 10.1016/j.brainresbull.2013.06.002
Yohn, C. N., Gergues, M. M., and Samuels, B. A. (2017). The role of 5-HT receptors in depression. Mol. Brain 10:28. doi: 10.1186/s13041-017-0306-y
Zarow, C., Lyness, S. A., Mortimer, J. A., and Chui, H. C. (2003). Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 60, 337–341. doi: 10.1001/archneur.60.3.337
Zhao, Q.-F., Tan, L., Wang, H.-F., Jiang, T., Tan, M.-S., Tan, L., et al. (2016). The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: systematic review and meta-analysis. J. Affect. Disord. 190, 264–271. doi: 10.1016/j.jad.2015.09.069
Zhou, L., Gao, Q., Nie, M., Gu, J. L., Hao, W., Wang, L., et al. (2016). Degeneration and energy shortage in the suprachiasmatic nucleus underlies the circadian rhythm disturbance in ApoE−/− mice: implications for Alzheimer’s disease. Sci. Rep. 6:36335. doi: 10.1038/srep36335
Zhou, J.-N., Liu, R.-Y., Kamphorst, W., Hofman, M. A., and Swaab, D. F. (2003). Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J. Pineal Res. 35, 125–130. doi: 10.1034/j.1600-079x.2003.00065.x
Zhu, C. W., Grossman, H., Neugroschl, J., Parker, S., Burden, A., Luo, X., et al. (2018). A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: a pilot study. Alzheimers Dement. 4, 609–616. doi: 10.1016/j.trci.2018.09.009
Keywords: Alzheimer’s disease, sleep disturbance, apathy, depression, neuropsychiatric disturbances, preclinical animal models
Citation: Clement A, Wiborg O and Asuni AA (2020) Steps Towards Developing Effective Treatments for Neuropsychiatric Disturbances in Alzheimer’s Disease: Insights From Preclinical Models, Clinical Data, and Future Directions. Front. Aging Neurosci. 12:56. doi: 10.3389/fnagi.2020.00056
Received: 28 November 2019; Accepted: 18 February 2020;
Published: 06 March 2020.
Edited by:
Silvia Fossati, Temple University, United StatesReviewed by:
Yong Tang, Chongqing Medical University, ChinaRamesh Kandimalla, Texas Tech University Health Sciences Center, United States
Copyright © 2020 Clement, Wiborg and Asuni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amalie Clement, YW1jbEBsdW5kYmVjay5jb20=