 Mattéa J. Finelli
Mattéa J. Finelli- School of Medicine, Biodiscovery Institute, University of Nottingham, Nottingham, United Kingdom
Reactive oxygen species and reactive nitrogen species (RONS) are by-products of aerobic metabolism. RONS trigger a signaling cascade that can be transduced through oxidation-reduction (redox)-based post-translational modifications (redox PTMs) of protein thiols. This redox signaling is essential for normal cellular physiology and coordinately regulates the function of redox-sensitive proteins. It plays a particularly important role in the brain, which is a major producer of RONS. Aberrant redox PTMs of protein thiols can impair protein function and are associated with several diseases. This mini review article aims to evaluate the role of redox PTMs of protein thiols, in particular S-nitrosation, in brain aging, and in neurodegenerative diseases. It also discusses the potential of using redox-based therapeutic approaches for neurodegenerative conditions.
Introduction
Reactive oxygen species and reactive nitrogen species (RONS) are essential signaling molecules produced by the aerobic metabolism (Friedman, 2010a; Finkel, 2011; Sbodio et al., 2019). One essential route to transduce RONS signaling is through oxidation-reduction (redox)-based post-translational modifications (PTMs) of proteins (Stadtman, 1988; Stadtman and Levine, 2003; Finkel, 2011; Santos and Lindner, 2017).
The mammalian brain is metabolically very active and is a major producer of RONS (Colton and Gilbert, 2002; Friedman, 2010a,b). Therefore, RONS-dependent redox signaling is particularly important in the normal physiology of the brain (Colton and Gilbert, 2002; Beckhauser et al., 2016). Under pathological conditions, RONS can reach excessive levels, generating oxidative and nitrosative (O/N) stresses, which can damage DNA, lipid, and proteins and be detrimental to cell function (Jones, 2006; Ray et al., 2012). O/N stresses are active contributing factors to the pathophysiological mechanisms underpinning neurodegenerative conditions including Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS; Friedman, 2010a,b; Kim et al., 2015; Liu et al., 2017; Sbodio et al., 2019).
In aerobic organisms, RONS are constantly produced by the mitochondrial oxidative phosphorylation (OXPHOS) system and by several enzymes [e.g., NADPH oxidases (NOXs) and nitric oxide synthases (NOSs); Ignarro, 1990; Raha and Robinson, 2000; Forman et al., 2002; Bian and Murad, 2003; Murphy, 2009; Lambeth and Neish, 2014; Nayernia et al., 2014; Di Meo et al., 2016; Ma et al., 2017; Moldogazieva et al., 2018; Barua et al., 2019; Sbodio et al., 2019; Gantner et al., 2020]. Some of the major RONS include hydrogen peroxide (H2O2), hydroxyl radical (•OH), superoxide anion (O2•−), peroxyl radical (ROO•), singlet oxygen (1O2), nitic oxide or nitrogen monoxide (•NO), nitrogen dioxide (•NO2), and peroxynitrite (ONOO−; Commoner et al., 1954; McCord, 2000; Forman et al., 2002; Pham-Huy et al., 2008; Di Meo et al., 2016; Sbodio et al., 2019). A tightly regulated machinery of small molecules [e.g., cysteine, ascorbate, glutathione (GSH)] and proteins [e.g., superoxide dismutases (SODs), catalases (CATs), glutathione peroxidases (GPXs), peroxiredoxins (PRDXs)] reduce RONS to control their levels in cells (Forman et al., 2002; Birben et al., 2012; Moldogazieva et al., 2018; Paul et al., 2018; Sbodio et al., 2019). Under normal physiological (eustress) conditions, RONS production and reduction are balanced; however, any imbalance can generate O/N stresses (Jones, 2006; Moldogazieva et al., 2018; Sies, 2019). Crosstalk between ROS and RNS, their specific sources and scavengers regulate the distribution of the various RONS present in cells (RONS composition), their relative abundance and absolute levels (Forman et al., 2002; Daiber, 2010; Dikalov, 2011; Schulz et al., 2014; Daiber et al., 2017). This complex network of RONS triggers a downstream cascade of events, termed redox signaling (Forman et al., 2002, 2014; Jones, 2006; Ray et al., 2012; Moldogazieva et al., 2018).
This mini review article focuses on the signaling triggered by RONS and transduced through redox PTMs of protein thiols and highlights key features of this signaling in normal cell physiology. It also assesses the role played by this redox signaling in brain aging and neurodegenerative conditions. Lastly, it aims to argue for greater considerations of the complexities of this essential signaling when designing new redox-based therapeutic strategies for neurodegenerative diseases.
Redox Post-translational Modifications (PTMs) of Protein Thiols and Their Role as a Redox Switch for Protein Function
Proteins are the main targets of RONS in cells as a result of their high rate constants for oxidation reactions and their abundance (Stadtman and Levine, 2003; Davies, 2005, 2016; Wall et al., 2012; Moldogazieva et al., 2018). Redox PTMs can occur nearly on any amino acid side chains (Stadtman and Levine, 2003; Dickinson and Chang, 2011; Wall et al., 2012; Corcoran and Cotter, 2013; Go and Jones, 2013). Nitration can modify tyrosine residues, and carbonylation can modify lysine, arginine, proline and threonine residues; however, these modifications are generally stable or irreversible and associated with O/N stresses (Dickinson and Chang, 2011; Wall et al., 2012; Corcoran and Cotter, 2013; Houée-Lévin et al., 2015; Weng et al., 2017; Gonos et al., 2018). Redox PTMs of thiol-containing cysteine (Cys) residues are considered to be one of the main drivers of redox signaling in cells because these residues can be easily oxidized and their oxidation is mostly reversible, with rates of oxidation and reduction compatible with signaling initiation and transduction (Miseta and Csutora, 2000; Davies, 2005, 2016; Bindoli et al., 2008; Schöneich, 2011; Wall et al., 2012; Wani et al., 2014; Go et al., 2015). The electronic structure of the thiol group (–SH) of Cys residues enables multiple oxidation states from −2 to +6 leading to a range of reversible and irreversible redox PTMs (Reddie and Carroll, 2008; Wang et al., 2012b; Chung et al., 2013; Cremers and Jakob, 2013; Wani et al., 2014; Figure 1A). Suggesting a potential cross-regulation between different PTMs, Cys residues can be targeted by a number of redox PTMs; for instance, the active Cys sites of protein disulfide isomerase (PDI) can be S-nitrosated or S-glutathionylated (Halloran et al., 2013). Besides, an interconversion between redox PTMs can also occur, with sulfenic acid (–SOH) representing a potential precursor for other reversible redox PTMs, such as S-glutathionylation, or intramolecular disulfide bond formation, possibly through sequential thiol modifications (Gallogly and Mieyal, 2007; Mieyal et al., 2008; Poole and Nelson, 2008; Sabens Liedhegner et al., 2012). The precise mechanisms underlying cross-regulation and interconversion between different redox PTMs of protein thiols remain unclear and future studies investigating multiple redox PTMs in parallel and over appropriate time-course will be required to fully explore these important aspects of redox PTMs regulation.
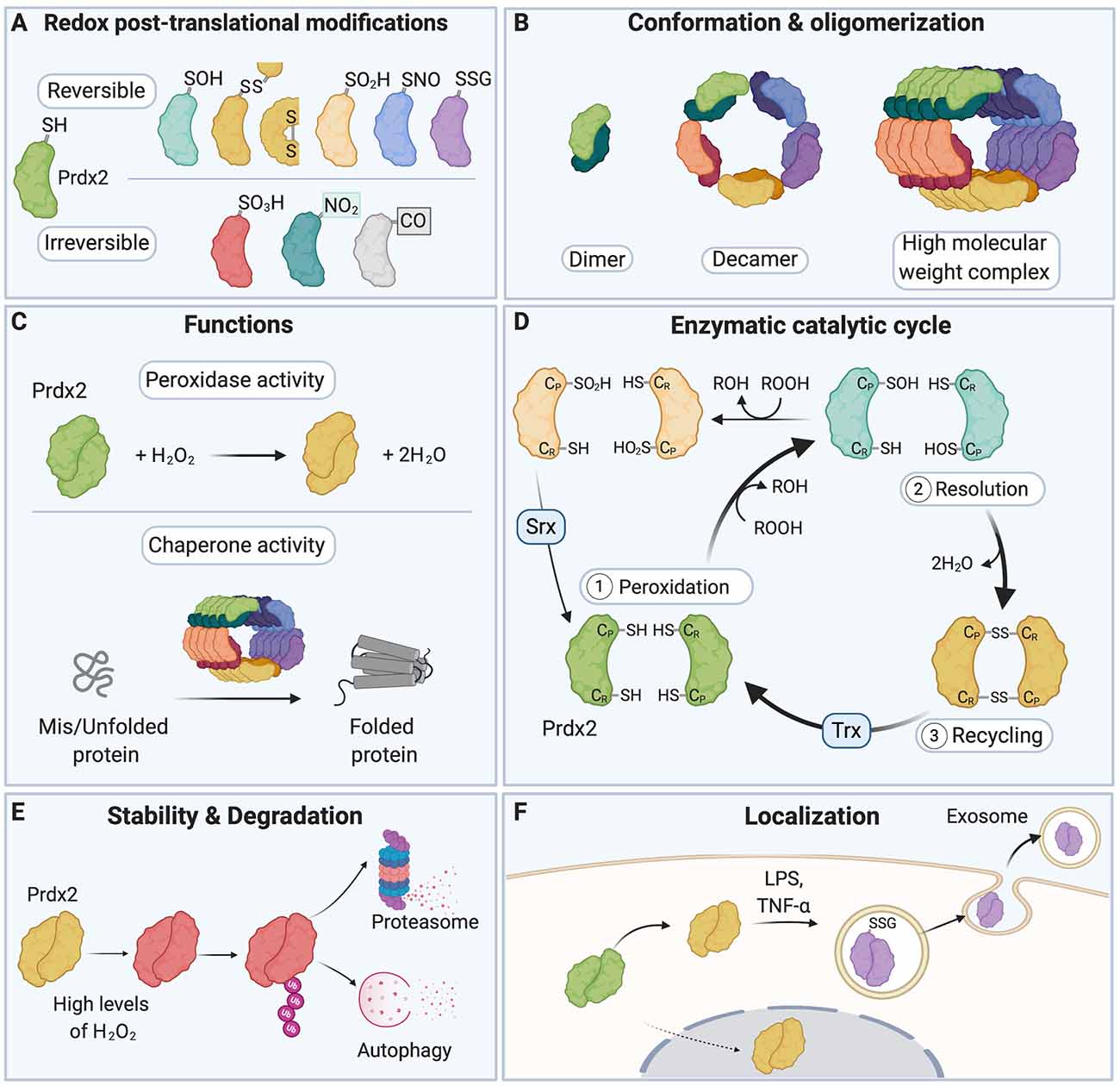
Figure 1. A redox switch modulates the multiple functions of Peroxiredoxin 2 (Prdx2). (A) The various residues of Prdx2 can be modified by S-sulfenylation (–SOH), intermolecular and intramolecular disulfide bonds (–SS–), S-sulfinylation (–SO2H), S-nitrosation (–SNO), S-glutathionylation (–SSG), S-sulfonylation (–SO3H), nitration (–NO2), and carbonylation (–CO: carbonyl groups). Only a subset of all the post-translational modifications (PTMs) that have been described for Prdx2 is shown here for clarity. (B) The redox state of the Cys residues of Prdx2 regulates its conformation as a dimer, decamer, or high molecular weight complex: the reduced and overoxidized dimers strongly tend to form decamers or high molecular weight complex, while oxidized forms are preferentially present as dimers. (C) Depending on its redox PTMs and conformation, Prdx2 can act as an antioxidant enzyme or as a chaperone. Multiple redox PTMs can coordinately regulate Prdx2 functions. (D) As part of Prdx2 catalytic cycle, its conserved peroxidatic cysteine (CP) first reduces H2O2 or ROOH (peroxidation step). Then a second free thiol (CR or resolving cysteine) forms a disulfide bond with its CP residue (resolution step). The catalytic cycle is completed when the disulfide bond is recycled, typically by a thioredoxin-like molecule (Trx), regenerating the free thiol forms of the CP and the CR residues (recycling step). CP-SOH Prdx2 can react with a second ROOH molecule before it can react with CR-SH, becoming overoxidized (CP-SO2H); this in turn inactivates Prdx2 peroxidase activity. CP-SO2H can be reduced by sulfiredoxin (Srx). (E) Oxidation of Prdx2 active site (Cys51) triggers conformational changes that bring to the protein surface of Prdx2 a flexible C-terminus that is subsequently polyubiquitinated; this leads to degradation of oxidized Prdx2 by the proteasome and autophagy. (F) Redox PTMs of Prdx2 can also regulate its secretion in the extracellular space. Upon exposure to inflammatory stimuli (e.g., LPS or TNF-α), Prdx2 Cys51 and Cys172 residues are oxidized forming disulfide-linked homodimers and mixed disulfides with glutathione, which triggers its secretion in exosomes into the extracellular milieu. Prdx2, peroxiredoxin 2; CP, peroxidatic Cys residue; CR, resolving Cys residue; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α; Trx, Thioredoxin; Srx, Sulfiredoxin.
Mass spectrometry-based redox proteomic studies that have mapped the redox-modified Cys residues across the proteome (redox Cys-proteome) have shown that, for a given condition, only a very specific subset of Cys residues is redox-modified, demonstrating that RONS modify Cys residues in a very selective manner (Doulias et al., 2010; Marino and Gladyshev, 2010, 2012; McDonagh et al., 2014; Gould et al., 2015; Raju et al., 2015; Araki et al., 2016; Sun et al., 2016; Topf et al., 2018; van der Reest et al., 2018; Mnatsakanyan et al., 2019; Xiao et al., 2020). This selectivity is modulated by several factors including the pKa of the –SH moiety of the Cys residue, its accessibility, the other PTMs present in the target protein, and the charges of the neighboring amino acids and local electrostatic effects (Hess et al., 2001; Davies, 2005; Doulias et al., 2010; Marino and Gladyshev, 2010, 2012; Chung et al., 2013; Roos et al., 2013; Gould et al., 2015; Sun et al., 2016; Mnatsakanyan et al., 2019; Xiao et al., 2020). Many potential consensus motifs have been identified and proposed as a chemical rationale that could underlie the redox sensitivity of specific Cys residues in a given protein (Stamler et al., 1997; Doulias et al., 2010; Marino and Gladyshev, 2010, 2012; Chen et al., 2014, 2015; Gould et al., 2015; Sun et al., 2016; Topf et al., 2018; Mnatsakanyan et al., 2019; Xiao et al., 2020). The variety of consensus motifs proposed so far may partly reflect the wide range of experimental and computational approaches employed to identify the protein thiols that are modified by redox PTMs. Indeed, several studies have focused on endogenous redox PTMs while others have studied redox PTMs induced by exogenously applied RONS (e.g., H2O2, S-Nitroso-l-cysteine, S-Nitrosoglutathione, etc.), and an array of proteomic approaches with varying degrees of sensitivity and selectivity has been used across studies. Thus, finding unifying motifs and rules overarching the specificity of redox PTMs on a proteome-wide scale remains an outstanding challenge. It has been proposed that under eustress, for a given RONS composition, RONS absolute levels, and relative abundance, only a subset of redox-sensitive Cys residues within a protein is potentially susceptible to several redox-PTMs. In contrast, O/N stresses can lead to aberrant redox PTMs including redox PTMs on even lowly-reactive Cys residues, and/or addition of excessive, persistent or irreversible redox PTMs (Chung et al., 2013). This continuum of reactivity of Cys residues in redox-sensitive proteins may provide a redox-sensing mechanism for cells to monitor RONS composition and RONS levels in the cellular environment and to coordinate an appropriate downstream response.
Redox PTMs on proteins act as a “redox switch” that dynamically regulates protein function (Go and Jones, 2013; Go et al., 2015; Fra et al., 2017). Redox PTMs can lead to structural changes in the target protein through disulfide bond formation, including intramolecular disulfide bonds, Cys-dependent metal cofactor interactions, and through alteration of the topography of the protein (Davies, 2005; Khoo and Norton, 2011; Wani et al., 2014). The way redox PTMs affect protein conformation is partly dictated by the position of the modified Cys residue in the target protein, e.g., on the protein surface or buried, or on the protein backbone or side-chains (Dean et al., 1997; Davies, 2005). It remains difficult to predict the precise effect of redox PTMs on protein structure and function, and thus computational and experimental approaches need to be combined to fully uncover the functional consequences of redox PTMs on a given protein (Raimondi et al., 2014; Wani et al., 2014). Redox PTMs can also modulate the stability and degradation of target proteins (Pajares et al., 2015, 2018). It has been suggested that depending on the degree of oxidation of a protein (esp. whether its redox PTMs are reversible or irreversible), degradation can occur through either ATP- and polyubiquitination-dependent or independent mechanisms (Pajares et al., 2015; Song et al., 2016). Redox PTMs can induce conformational changes that expose hydrophobic structures normally buried in the natively folded protein; these structures become recognition and binding surfaces for the 20S core subunit of the proteasome, which subsequently degrades the oxidized target protein (Grune et al., 1997; Jung and Grune, 2008; Jung et al., 2014). The redox state of protein thiols can also regulate the localization of target proteins (Wani et al., 2014). Importantly, redox PTMs can ultimately tune the activity of redox-sensitive enzymes and regulate their enzymatic cycle and recycling (Klomsiri et al., 2011). For instance, the function of the redox-sensitive protein, Prdx2, is regulated by a redox switch (Rhee and Kil, 2017). Depending on the cellular RONS composition and levels, the various residues of Prdx2 can be decorated by an array of different redox PTMs (Fang et al., 2007; Park et al., 2011; Engelman et al., 2013; Peskin et al., 2013, 2016; Wong et al., 2013; Randall et al., 2014, 2019; Salzano et al., 2014; Svistunova et al., 2019; Figure 1A). Redox PTMs of Prdx2 regulate its conformation (Wood et al., 2003; Barranco-Medina et al., 2009; Hall et al., 2009, 2011), catalytic cycle (Woo et al., 2005; Hall et al., 2009; Karplus, 2015; Rhee, 2016), degradation (Song et al., 2016) and secretion (Salzano et al., 2014; Mullen et al., 2015; Figures 1B–F). Crucially, redox PTMs can coordinately regulate Prdx2 multiple functions as an antioxidant enzyme and a chaperone. For example, overoxidation (Prdx2-SO2/3) or S-nitrosation (Prdx2-SNO) of Prdx2 can inactivate its antioxidant activity, while Prdx2-SO2/3 stabilizes Prdx2 high-molecular weight chaperone complexes (Wood et al., 2003; Jang et al., 2004; Fang et al., 2007; Hall et al., 2009, 2011; Rhee and Woo, 2011; Saccoccia et al., 2012; Rhee and Kil, 2017; Svistunova et al., 2019).
By acting as a redox switch for protein function, redox PTMs allow for proteins to carry out different functions in different subcellular compartments. Indeed, the nature of the redox PTMs on protein thiols is greatly determined by the RONS composition and pH of the environment surrounding the target protein (Pace and Weerapana, 2013; Roos et al., 2013). Therefore, when a protein shuttles to a different compartment with a different RONS composition and pH, the redox PTMs of the protein as well as its activity will change accordingly (Go and Jones, 2008; Jones and Go, 2010). Together, redox PTMs of protein thiols constitute a powerful mechanism that allows for a very dynamic and compartment-specific regulation of protein functions in cells.
Role of Redox PTMs of Protein Thiols in Cell Physiology and Brain Aging
Advances in redox proteomics have allowed to explore further the functional role of redox PTMs of protein thiols in cell and organ physiology. In particular, a recent study has mapped the oxidized Cys residues landscape in ten different tissues from wild-type mice using a highly sensitive and specific approach (a Cys-reactive phosphate tag method) and has shown that oxidized Cys residues were detected in almost half of all proteins in all tissues tested suggesting that redox PTM of protein thiols is a widely used signaling mechanism in normal cellular physiology (Go et al., 2011; Xiao et al., 2020). Analyses of the redox Cys-proteome across subcellular compartments also demonstrated that redox PTM of protein thiols occurs in all subcellular compartments, from nucleus, cytoplasm, Golgi, endoplasmic reticulum (ER) to mitochondria (Go et al., 2011; Chung et al., 2013; Doulias et al., 2013; Xiao et al., 2020).
Redox PTMs have been shown to regulate the activity of proteins implicated in a range of major cellular functions, and cell survival/death (Trachootham et al., 2008; Brigelius-Flohé and Flohé, 2011; Handy and Loscalzo, 2012; Wall et al., 2012; Mailloux et al., 2014; Niforou et al., 2014; Go et al., 2015; Jones and Sies, 2015; Pajares et al., 2015; Nakamura and Lipton, 2017; Figure 2A). In particular, redox PTMs play an essential role in the regulation of energy metabolism in cells through S-nitrosation, S-glutathionylation, or S-sulfenylation of proteins of the mitochondrial respiratory complexes I, II, IV, and V, and ATP synthase (Hurd et al., 2008; Garcia et al., 2010; Handy and Loscalzo, 2012; Wang et al., 2013; Mailloux et al., 2014; Nakamura and Lipton, 2017; van der Reest et al., 2018; Xiao et al., 2020), enzymes of the tricarboxylic acid cycle (e.g., Alpha-ketoglutarate dehydrogenase, Isocitrate dehydrogenase, Aconitase; Kil and Park, 2005; McLain et al., 2013; Yan et al., 2013; Bulteau et al., 2017; Nakamura and Lipton, 2017, 2020; Xiao et al., 2020), enzymes of glycolysis [e.g., Hexokinase, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH); Riederer et al., 2009; Mailloux et al., 2014; McDonagh et al., 2014; Araki et al., 2016; van der Reest et al., 2018; Xiao et al., 2020] and of fatty acid metabolism (e.g., Very long chain acyl-coenzyme A dehydrogenase; Doulias et al., 2013). Similarly, for proteostasis, redox PTMs have been reported on chaperones (e.g., Heat shock protein 70, PDI; Wang C. et al., 2012; Grunwald et al., 2014), subunits of the proteasome (Aiken et al., 2011; Jung et al., 2014; Kors et al., 2019), and proteins involved in autophagy (e.g., Autophagy Related 3, 4 and 7; Frudd et al., 2018; Pajares et al., 2018; Scherz-Shouval et al., 2019), which ultimately contribute to the regulation of protein folding and degradation (Niforou et al., 2014; Pajares et al., 2015). Redox PTMs can also regulate the activity of several transcription factors (Brigelius-Flohé and Flohé, 2011). For instance, oxidation, S-nitrosation or nitration of p53, activating protein 1, myocyte enhancer factor 2 or nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) modulate the DNA binding and activity of these important transcription factors (Abate et al., 1990; Schreck et al., 1991; Rainwater et al., 1995; Klatt et al., 1999; Kabe et al., 2005; Yakovlev et al., 2007; Jung et al., 2008; Ljubuncic et al., 2008; Okamoto et al., 2014; Caviedes et al., 2020). Redox PTMs can also affect the activity of transcription factors through indirect mechanisms. For example, redox PTMs on Kelch-like ECH-associated protein 1 (Keap1) trigger the dissociation of the cytoplasmic complex formed by Keap1 and nuclear factor erythroid 2-related factor 2 (Nrf2), which allows Nrf2 to translocate to the nucleus and to bind to the antioxidant response element of its target genes (Dinkova-Kostova et al., 2002, 2017; Rachakonda et al., 2008).
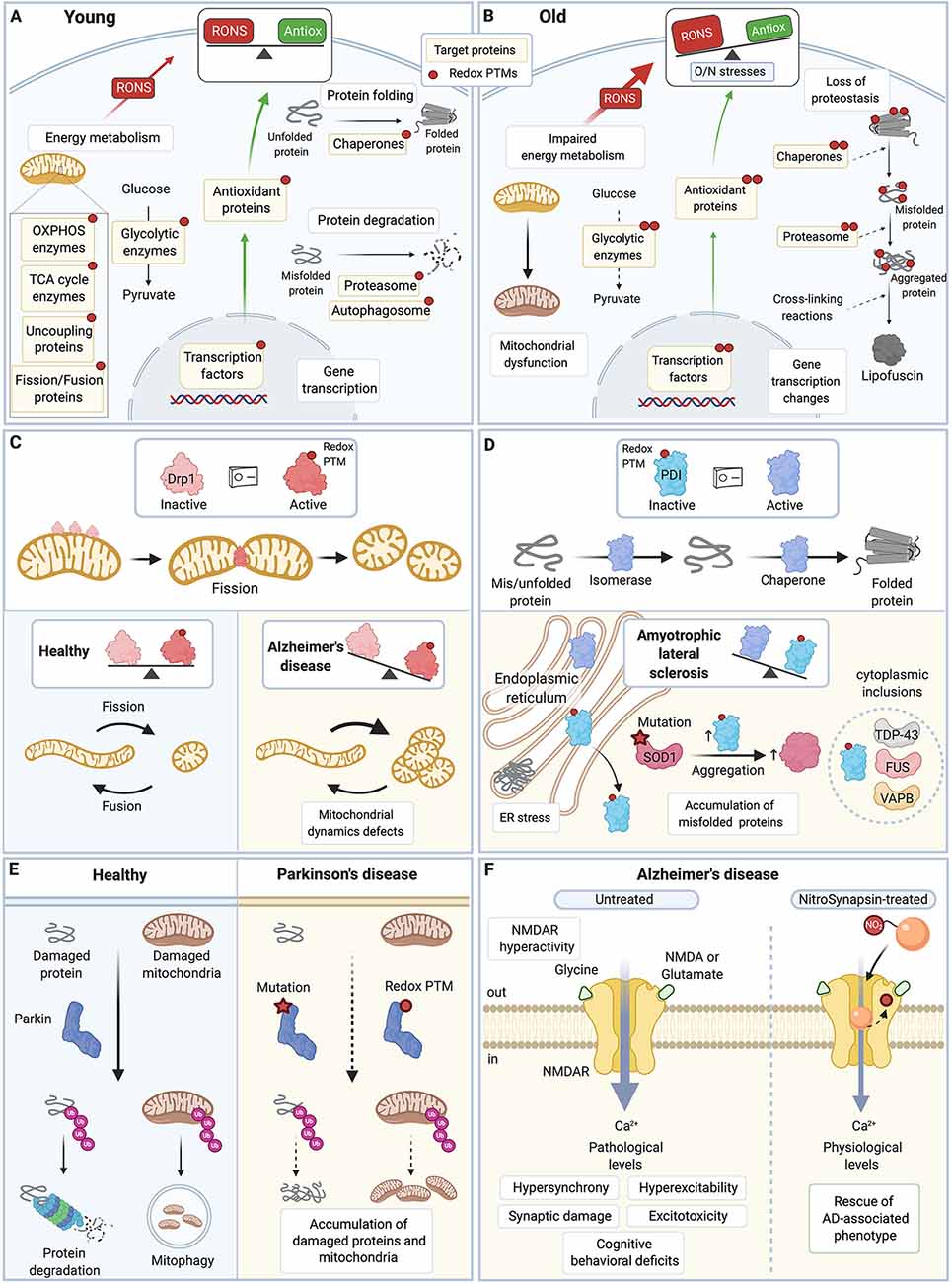
Figure 2. Redox PTMs of protein thiols are essential for normal cell physiology and contribute to aging and neurodegenerative diseases. (A) In young healthy cells, redox PTMs are an integral part of the normal cell signaling: they can coordinately modulate the activity of multiple proteins depending on the local RONS composition and levels. Redox PTMs regulate the function of proteins involved in essential pathways such as energy metabolism, protein folding and degradation, and gene transcription. Redox PTMs are represented by a red dot on the target proteins. (B) During aging, imbalance between RONS production and the antioxidant capacity of the cell leads to O/N stresses, and proteins may become differentially and/or aberrantly modified by redox PTMs, as represented by multiple red dots on the target proteins. These redox PTMs can affect the function of target proteins, ultimately altering key pathways that contribute to the aging process. (C) Drp1 is a GTPase protein involved in mitochondrial fission; when activated, especially by S-nitrosation, Drp1 forms oligomers that wrap around the mitochondrial outer membrane and scission it. Excessive levels of S-nitrosated Drp1, as seen in AD, disrupt the physiological equilibrium between mitochondrial fission and fusion,leading to increased mitochondrial fission, and synaptic and neuronal damage. (D) PDI is an ER protein with a dual isomerase and chaperone activity. S-nitrosation of PDI inhibits its functions. Excessive S-nitrosation of PDI, as observed in AD, PD, and ALS, leads to the accumulation of misfolded proteins in the ER and consequently, to increase in ER stress and neuronal cell death. In ALS, PDI is recruited to cytoplasmic inclusions containing ALS-associated proteins such as SOD1, TDP-43, FUS, or VABP. S-nitrosation of PDI also increases insoluble aggregates of ALS-associated mutant SOD1. (E) Parkin is an E3 ubiquitin ligase that targets damaged proteins and mitochondria to the ubiquitin-proteasome system and mitophagy. Loss-of-function mutations in Parkin are associated with familial PD. Importantly, excessive and persistent S-nitrosation of non-mutated Parkin, as detected in postmortem brains of patients with sporadic PD, also inhibits Parkin activity, which impairs ubiquitination of its substrate proteins and contributes to the accumulation of damaged proteins and mitochondria and ultimately to neuronal cell injury and death. (F) Hyperactivity of the extrasynaptic NMDAR leads to excessive intracellular calcium, hypersynchrony and hyperexcitability of neurons, and synaptic damage and excitotoxicity in AD. A new drug, NitroSynapsin, is constituted of a memantine moiety and an added nitro group (NO2). It can dampen the hyperactivity of NMDAR while preserving its physiological function by binding inside the excessively opened channel and by acting as an NO group donor, which triggers NMDAR inactivation through S-nitrosation of its redox-sensitive Cys residues. NitroSynapsin treatment significatively improves the pathological features associated with AD. TCA, tricarboxylic acid cycle; OXPHOS, oxidative phosphorylation; RONS, reactive oxygen species and reactive nitrogen species; Antiox, antioxidant capacity; Drp1, Dynamin related protein 1; PDI, protein disulfide isomerase; ER, endoplasmic reticulum; SOD1, superoxide dismutase 1; TDP-43, transactive response DNA binding protein 43 kDa; FUS, Fused in sarcoma; VAPB, vesicle-associated membrane protein; Ub, ubiquitin; NMDAR, N-methyl-D-aspartate receptor; AD, Alzheimer’s disease; PD, Parkinson’s disease; ALS, amyotrophic lateral sclerosis. All figures were created with BioRender.com.
More globally, comprehensive functional analyses on the redox Cys-proteomes of various cell types and tissues have demonstrated that redox PTMs can regulate whole networks of proteins that share similar biological activities (Doulias et al., 2010; Go et al., 2011, 2015; Xiao et al., 2020). Importantly, this coordinated redox-based regulation of functional protein networks underpins, at least in part, the unique physiological and metabolic states that characterize the different cell types and tissues present in organisms. Indeed, redox PTMs of protein thiols can tune the activity of ubiquitously expressed proteins in a tissue-specific manner and can also regulate functional networks specifically in different tissues to control both tissue-ubiquitous functions (e.g., tRNA aminoacylation) and tissue-specific functions (e.g., catabolism of glycogen and glucose; Doulias et al., 2013; Xiao et al., 2020). Therefore, redox PTMs of protein thiols play an essential part in normal cell physiology and coordinately regulate functional networks in a tissue- and cell type-specific manner.
Aging is characterized by molecular and cellular hallmarks including, but not limited to, altered mitochondrial function, impaired energy metabolism, increased O/N stresses, and loss of proteostasis (López-Otín et al., 2013; Figure 2B). Many aspects of the fundamental molecular mechanisms of brain aging remain unknown (Mattson and Arumugam, 2018). The “free radical theory of aging” postulates that aging is driven by an accumulation of harmful and irreversible RONS-induced damage to DNA, lipids, and proteins (Harman, 1956, 2006). In accordance with this theory, an increase in modified proteins (e.g., S-nitrosated Cys residues, and nitrated and carbonylated amino acids) has been observed in the brain of aging rodents and humans, especially in the hippocampus, substantia nigra and frontal cortex, brain regions particularly vulnerable to aging-associated diseases (Smith et al., 1991; Cini and Moretti, 1995; Farr et al., 2003; Dremina et al., 2005; Siqueira et al., 2005; Poon et al., 2006b; Gokulrangan et al., 2007; Prokai et al., 2007; Riederer et al., 2009; Grimm et al., 2011; Venkateshappa et al., 2012; Ortuño-Sahagún et al., 2014; Perluigi et al., 2014; Cabré et al., 2017; Gonos et al., 2018). In addition, in line with an age-dependent increase in O/N stresses, accumulation of redox PTMs (e.g., S-sulfonylation and excessive disulfide bonds) on key antioxidant proteins such as PRDXs and SODs has been described and shown to impair the enzymatic activity of these proteins (Kurokawa et al., 2001; Navarro and Boveris, 2004; Poon et al., 2006a,b; Musicco et al., 2009; Pérez et al., 2010; Gottfredsen et al., 2013; McDonagh et al., 2014; Perluigi et al., 2014).
Irreversible redox PTMs can participate in the aging process by triggering unfolding and aggregation of proteins through hydrophobic and electrostatic interactions between target proteins (Höhn et al., 2017). With time, these aggregates can be covalently stabilized, rendering protein aggregates insoluble and non-degradable (Pacifici et al., 1993; Giulivi et al., 1994; Johnston et al., 1998; Calderwood et al., 2009; Koga et al., 2011; Höhn et al., 2017). Existing protein aggregates can undergo further cross-linking reactions with molecules such as aldehydes and malondialdehyde, products of lipid peroxidation, resulting in the formation of highly cross-linked autofluorescent material referred to as lipofuscin, an important hallmark of aging (Stadtman and Levine, 2003; Terman and Brunk, 2004; Jung et al., 2007). O/N stresses also impair the activity of the proteasome through irreversible redox PTMs such as carbonylation of its subunits, leading to an age-dependent reduction of protein degradation and further accumulation of protein aggregates (Friguet, 2006; Jung et al., 2014; Höhn and Grune, 2014; Lefaki et al., 2017). These protein aggregates may in turn interfere with cell homeostasis.
Also, aberrant redox PTMs of proteins on Cys and other residues may be implicated in the impaired energy metabolism observed in aging (Yap et al., 2009; Barzilai et al., 2012; López-Otín et al., 2013; Chistiakov et al., 2014; Jové et al., 2014; Bouzier-Sore and Bolaños, 2015; Goyal et al., 2017; Mattson and Arumugam, 2018; Rizza et al., 2018; Hipkiss, 2019; Ravera et al., 2019). For instance, 3-nitrotyrosine and carbonylation have been detected on energy-related enzymes, proteins of the mitochondrial respiratory complex and ATP-synthase in aged rats, in the senescence-accelerated (SAMP8) mouse model of aging, and in human postmortem tissue from aged control patients (Smith et al., 1991; Navarro and Boveris, 2004; Poon et al., 2004, 2006a,b; Gokulrangan et al., 2007; Lam et al., 2009; Perluigi et al., 2010; Perluigi et al., 2014; Pérez et al., 2010). Aberrant redox PTMs of proteins may also contribute to the age-associated reduced synaptic transmission and impaired induction of long-term potentiation (Foster and Norris, 1997; Foster and Kumar, 2002; Foster, 2006, 2007, 2012; Kumar et al., 2009, 2018; Bodhinathan et al., 2010a,b; Oh et al., 2010; Bradley and Steinert, 2016; Hidalgo and Arias-Cavieres, 2016; Muñoz et al., 2020). For example, the redox-sensitive ryanodine receptor 3 (RyR3) essential for synaptic transmission and plasticity, is nitrated in the cerebellum of aged rats (Gokulrangan et al., 2007; Lanner et al., 2010; Abu-Omar et al., 2018; Arias-Cavieres et al., 2018; Muñoz et al., 2020). Interestingly, hyper-S-nitrosation of RyRs shifts these receptors from a finely regulated state to unregulated Ca2+ leak channels (Bull et al., 2008; Bellinger et al., 2009; Bodhinathan et al., 2010b; Liu et al., 2012; Lacampagne et al., 2017; Muñoz et al., 2020). S-glutathionylation of RyRs also compromises their function (Bull et al., 2003; Hidalgo, 2005; Aracena-Parks et al., 2006). Together, this suggests that redox PTMs of RyRs may contribute to the progressive age-dependent dysregulation of synaptic transmission.
There is a growing body of evidence however that conflicts with the idea that RONS and associated redox PTMs of proteins in aging are solely detrimental to cell function. The “redox theory of aging” indeed presents the redox proteome as a system that allows organisms to best adapt to a changing environment and varied exposures (exposome) throughout their lifespan, and which contributes to building progressive resilience (Go and Jones, 2013; Jones, 2015; Jones and Sies, 2015). It also states that the process of aging could be a gradual breakdown of the redox proteome and redox metabolome, which act together as adaptive interfaces supporting the interaction between the genome and the exposome (Jones, 2015; Go and Jones, 2017). Other studies have also demonstrated that mitochondrial RONS can initiate a positive adaptive homeostasis cascade following a non-linear response (termed mitohormesis); both the dose and timing of RONS exposure determining whether RONS have either a beneficial or detrimental effect on cell function (Kawagishi and Finkel, 2014; Ristow, 2014; Ristow and Schmeisser, 2014; Yun and Finkel, 2014). Mild O/N stresses can indeed trigger a protective response that makes organisms more resistant to future, greater, oxidative insults and therefore increasing levels of RONS (and potentially of redox PTMs of protein thiols) can be beneficial at the cellular or organismal levels and can even promote healthspan and lifespan (Kawagishi and Finkel, 2014; Ristow, 2014; Ristow and Schmeisser, 2014; Yun and Finkel, 2014). It has been hypothesized that aging results in a generalized decline in this adaptive homeostasis, in particular through an age-dependent attenuation of proteolytic activity and capacity (Pomatto and Davies, 2018).
Besides, also contrary to the concept that RONS induce a bulk accumulation of non-specific redox PTMs of protein thiols in aging, a recent study that compared the redox Cys-proteome in tissues of young (16-week-old) and old (80-week-old) mice has shown that the overall levels of reversibly oxidized Cys residues are very similar in young and aged tissues (Xiao et al., 2020), which was consistent with previous observations made in D. melanogaster (Menger et al., 2015). Interestingly, it also found that young and aged tissues have largely distinct redox proteomic signatures with different highly modified Cys residues (Xiao et al., 2020). This demonstrates that a profound and very specific remodeling of the redox signaling landscape occurs during aging, which in turn induces tissue-specific functional shifts. It remains to explore however whether the redox PTMs that have been detected in aged tissue so far contribute to either accelerating or slowing down the the aging process. It is often difficult to ascertain whether redox PTMs of protein thiols have beneficial or detrimental effects on protein and cellular function, as exemplified by Prdxs, which are “gerontogenes” that can prevent the age-related decline in cognitive function and can increase healthspan and lifespan (Neumann et al., 2003; Oláhová et al., 2008; Timmermann et al., 2010; Kim et al., 2011; Nyström et al., 2012). Indeed, because Prdxs are overoxidized with age (which progressively inactivates their antioxidant activity), redox PTM of Prdxs was initially considered as a detrimental factor contributing to aging (Hall et al., 2009; Musicco et al., 2009; Molin et al., 2011; Nyström et al., 2012). However, because overoxidation of Prdxs also favors their chaperone activity, it was later suggested that redox PTM of Prdxs may constitute a positive adaption that helps counteract the detrimental protein aggregation associated with aging (Nyström et al., 2012). Somewhat related, redox-dependent dimerization of Prdx2 drives a Prdx2-dependent increase in overall life expectancy in C. elegans (De Haes et al., 2014), suggesting that redox PTM of Prdx2 plays a role in counteracting the aging process. Therefore, further studies are required to fully grasp the role played by redox PTMs of protein thiols in brain aging and to explore whether redox PTMs may have different effects (i.e., beneficial or detrimental) at different stages of the aging process.
In the past few decades, additional factors have been implicated in aging including low-grade chronic inflammation (“inflammaging”; Franceschi et al., 2007; von Bernhardi et al., 2015). Which one of the various factors contributing to the aging process is the main initiator of brain aging remains unclear. There are nevertheless many lines of evidence that suggest that these various contributing factors exacerbate each other, and that cellular redox dysregulation may be a common denominator (De la Fuente and Miquel, 2009; Ortuño-Sahagún et al., 2014; Yin et al., 2016). As a possible underlying molecular mechanism, it has been suggested that redox PTM of NF-kB, a pivotal transcription factor regulating inflammation, modulates NF-kB activity and subsequently the inflammatory response during aging (Ljubuncic et al., 2010; Zhang et al., 2013). Indeed, under sustained O/N stresses (as seen in aging), tyrosine residues of IκBα (one of NF-kB subunits) are nitrated, which leads to a prolonged and excessive NF-kB activation and signaling (Adler et al., 2007; Yakovlev et al., 2007). More integrated studies of the complex process of aging would be required to understand the interconnection and crosstalk between redox PTMs of protein and the various factors contributing to brain aging.
Role of Redox PTMs of Proteins in Neurodegenerative Conditions
In age-related neurodegenerative conditions such as AD, PD, and ALS, an increase in O/N stresses and associated redox PTMs has consistently been described; S-nitrosated, S-sulfonylated, nitrated and carbonylated proteins are amongst the redox PTMs detected in the cerebrospinal fluid, plasma and postmortem brains and spinal cords of patients with AD, PD or ALS (Markesbery, 1997; Jenner, 2003; Mitsumoto et al., 2008; Riederer et al., 2009; Di Domenico et al., 2011; Sultana et al., 2011a,b; Barone et al., 2012; Sultana and Butterfield, 2013; Zahid et al., 2014). Crucially, these O/N stresses-induced PTMs are also observed at the presymptomatic stages of neurodegenerative diseases (Andrus et al., 1998; Aluise et al., 2010, 2011; Butterfield et al., 2010, 2012; Hartl et al., 2012; Granold et al., 2015; Shen et al., 2015). For example, redox proteomics on postmortem brains from patients with preclinical AD (presymptomatic stage) or with AD with mild cognitive impairment (symptomatic stage) has shown that protein carbonylation increases at the transition between the two clinical stages and correlates with clinical features, pathology and biochemistry of AD (Butterfield et al., 2010, 2012; Sultana et al., 2010; Aluise et al., 2011). Together, this suggests that O/N stresses-induced PTMs are not simply a late consequence of the disease progression but that they play an active role in the pathophysiology of neurodegenerative conditions.
Redox PTMs contribute to neurodegenerative conditions by facilitating the aggregation of disease-associated proteins. For instance, redox PTMs such as oxidation, sumoylation, and nitration of amyloid β (Aβ) or α-synuclein (associated with AD and PD, respectively) inhibit their degradation by the ubiquitin/proteasome system and autophagy, which leads to further accumulation and aggregation of their toxic products (Kuo et al., 1998; Przedborski et al., 2001; Boutte et al., 2006; Butterfield et al., 2010; Stefanis, 2012; Chavarría and Souza, 2013; Barrett and Timothy Greenamyre, 2015; Roher et al., 2017; Savyon and Engelender, 2020).
Also, aberrant redox PTMs of proteins playing key roles in cell physiology are consistently observed in neurodegenerative conditions (Nakamura et al., 2013; Akhtar et al., 2016; Di Domenico et al., 2017; Tramutola et al., 2017; Valle and Carrì, 2017; Dyer et al., 2019; Sbodio et al., 2019; Tegeder, 2019; Nakamura and Lipton, 2020; Figures 2C–E). For example, the redox-sensitive dynamin-related protein 1 (Drp1), a GTPase involved in mitochondrial fission, is activated by S-nitrosation (Drp1-SNO; Cho et al., 2009; Willems et al., 2015; Figure 2C). Drp1 is aberrantly S-nitrosated in neurodegenerative disease as high levels of Drp1-SNO and hyperactive Drp1 are detected in postmortem brains and peripheral blood lymphocytes of AD patients (Cho et al., 2009; Wang et al., 2009; Manczak and Reddy, 2012; Wang S. et al., 2012). Crucially, exposure of primary neuron cultures to Aβ leads to increase in Drp1-SNO, excessive mitochondrial fission, impaired energy metabolism, and neuronal cell loss, mimicking what is seen in AD, while the use of a non-nitrosylable Drp1 mutant prevents these detrimental effects (Cho et al., 2009; Wang et al., 2009; Trushina et al., 2012; DuBoff et al., 2013; Joshi et al., 2017; Pérez et al., 2017; Flannery and Trushina, 2019; Oliver and Reddy, 2019). Therefore, S-nitrosation of Drp1 plays a key role in the mitochondrial dynamic defects and neurodegeneration observed in AD (Wang et al., 2009; Cho et al., 2010, 2013; Nakamura and Lipton, 2020). The multifunctional protein PDI is also aberrantly S-nitrosated in neurodegenerative conditions as shown in postmortem spinal cords of ALS patients and transgenic SOD1G93A mouse model of ALS as well as in postmortem brains of AD and PD patients (Uehara et al., 2006; Walker et al., 2009; Chen et al., 2013; Jeon et al., 2014; Conway and Harris, 2015; Figure 2D). PDI is an ER protein, but in disease conditions, triggered by redox PTMs in particular, it can translocate to the cytoplasm where it localizes to inclusions containing ALS-associated proteins (Turano et al., 2002; Honjo et al., 2011; Walker and Atkin, 2011; Farg et al., 2012; Jeon et al., 2014; Valle and Carrì, 2017; Matsusaki et al., 2020; Parakh et al., 2020). PDI, as an oxidoreductase chaperone, catalyzes the maturation of disulfide bond-containing proteins through oxidation and isomerization functions (Gonzalez et al., 2010). However, S-nitrosation of PDI Cys residues in its two thioredoxin-like domains inactivates both its chaperone and isomerase activities, which leads to the accumulation of misfolded proteins, in particular ALS-associated proteins like SOD1, and to a persistent ER stress that ultimately triggers neuronal injury and death (Uehara et al., 2006; Walker et al., 2009; Chen et al., 2012, 2013; Jeon et al., 2014; Medinas et al., 2018; Matsusaki et al., 2020). Similarly, excessive levels of S-nitrosation of another multifunctional protein, Prdx2, are observed in dopaminergic neurons differentiated from induced pluripotent stem cells from PD patients as well as in PQ/MB-exposed PD mouse model and postmortem brains of patients with PD (Fang et al., 2007; Sunico et al., 2016). Importantly, S-nitrosation of Prdx2 breaks the normal redox cycle that regenerates Prdx2, inhibits its peroxidase activity, and reduces its overoxidation (which is essential for stabilization of Prdx2 chaperone complexes; Jang et al., 2004; Fang et al., 2007; Engelman et al., 2013; Zhang et al., 2019). Together, this suggests that aberrant redox PTM of Prdx2 Cys residues impairs its antioxidant and chaperone functions, leading to accumulation of misfolded proteins and O/N stresses in neurodegenerative conditions. Neuroinflammation is another essential contributing factor to neurodegeneration (Hsieh and Yang, 2013; Guzman-Martinez et al., 2019). Crucially, redox PTMs of protein thiols have been implicated in inflammation regulation (Gloire and Piette, 2009; Nakamura et al., 2013; Ryan et al., 2014; Gorelenkova Miller and Mieyal, 2015). For instance, S-glutathionylation of various proteins of the NF-kB pathway can modulate the inflammatory response in different cell types, and it has been suggested that S-glutathionylation of protein thiols plays a key role in neuroinflammation in neurodegenerative conditions (Reynaert et al., 2006; Sabens Liedhegner et al., 2012; Gorelenkova Miller and Mieyal, 2015; Cha et al., 2017). Thus, redox PTMs of protein thiols can detrimentally impair the function of target proteins, which in turn can dysregulate many important pathways such as mitochondrial function, proteostasis, ER stress, neuroinflammation, and contribute to the neurodegenerative process (Sabens Liedhegner et al., 2012; Nakamura et al., 2013; Gorelenkova Miller and Mieyal, 2015; Nakato et al., 2015; Valle and Carrì, 2017; Nakamura and Lipton, 2020).
Aberrant redox PTMs have been described in several proteins that can be mutated in the familial forms of neurodegenerative conditions. Importantly, redox PTMs of non-mutated disease-associated proteins can phenocopy the effect of rare genetic mutations (Nakamura et al., 2013). For example, mutations in Parkin in patients with early-onset autosomal recessive juvenile PD impairs its E3 ubiquitin ligase activity, which leads to an abnormal accumulation of protein aggregates and cell death (Pankratz and Foroud, 2007; Nakamura et al., 2013; Arkinson and Walden, 2018; Figure 2E). Similar detrimental effects on Parkin function are observed in non-mutated Parkin modified by S-nitrosation (Chung et al., 2004; Yao et al., 2004; Sunico et al., 2013). Levels of S-nitrosated Parkin are increased in postmortem tissues of patients with sporadic PD, which implicates S-nitrosation of Parkin in the pathophysiology of these forms of PD (Chung et al., 2004; Meng et al., 2011; Sunico et al., 2013). Therefore, O/N stresses-induced PTM of protein thiols has been proposed to play an important role in the pathophysiology of the most common forms of neurodegenerative conditions (Sabens Liedhegner et al., 2012; Nakamura et al., 2013; Valle and Carrì, 2017; Dyer et al., 2019).
The Therapeutic Potential of Redox PTMs of Proteins
Given the contribution of O/N stresses in neurodegenerative conditions, therapeutic strategies aiming at reducing O/N stresses using common free radical scavengers such as coenzyme Q10 or Vitamin E have been tested in clinical trials for these conditions, but without much success (Feng and Wang, 2012; Filograna et al., 2016). Mainly due to a lack of appropriate biomarkers, there is still little evidence however that these interventions effectively reduce O/N stresses levels in the patients’ central nervous system. Also, the antioxidants tested target specific RONS but it is not known whether the targeted RONS are implicated in neurodegenerative disease. Indeed, not all RONS are equivalent (Murphy et al., 2011) and only a subset of specific RONS is likely implicated in a given disease state. Physiological levels of RONS are essential for normal cell function and RONS can be beneficial for healthspan and lifespan (Kawagishi and Finkel, 2014; Ristow and Schmeisser, 2014; Ristow, 2014; Yun and Finkel, 2014), therefore globally reducing RONS levels may not only fail to provide therapeutic benefits but may also trigger detrimental side-effects (Kawagishi and Finkel, 2014; Nakamura and Lipton, 2016). These represent few of the factors that may have contributed to the limited success of antioxidant-based therapeutic approaches for the treatment of neurodegenerative conditions.
As an alternative redox-based therapeutic approach, it has been proposed that modulation of redox PTMs of proteins playing part in the neurodegenerative process using small molecules could help prevent disease-associated dysfunction of these proteins and restore their normal activity (Lipton et al., 2002; Wani et al., 2014; Nakamura and Lipton, 2016). This approach has been shown to provide significant therapeutic benefits in several rodent models of neurological conditions. For example, deprenyl/selegiline and its derivatives (CGP3466B, TCH346), used in early PD (Moore and Saadabadi, 2020), has been shown to bind GAPDH (Kragten et al., 1998) and prevent S-nitrosation of its catalytic site (Hara et al., 2006b). S-nitrosation is essential for GAPDH function as it increases its binding to E3 ubiquitin-protein ligase Siah1, which leads to GAPDH translocation to the nucleus and its activity as a transcriptional activator of the apoptotic cascade (Sawa et al., 1997; Kragten et al., 1998; Hara et al., 2005, 2006a,b; Sen et al., 2008). CGP3466B, by preventing GAPDH S-nitrosation, blocks the apoptotic cascade induced by S-nitrosated GAPDH. Accordingly, CGP3466B has been shown to reduce neuronal cell death in animal models of PD (Andringa and Cools, 2000; Andringa et al., 2000; Waldmeier et al., 2000; Hara et al., 2006b; Naoi et al., 2020), ALS (Sagot et al., 2000; Waldmeier et al., 2000), AD (Sen et al., 2018), muscular dystrophy (Erb et al., 2009; Yu et al., 2013), cocaine addiction (Xu et al., 2013; Harraz and Snyder, 2015), traumatic brain injury (Liang et al., 2017), and ischemia (Simon et al., 2001; Ahmari et al., 2020). Together, these studies highlight the therapeutic potential of targeting redox PTMs of specific proteins to modulate protein function and to counteract the neurodegenerative process.
Based on the fact that O/N stresses-induced PTM of proteins can affect the conformation of target proteins and their binding affinity to other proteins and potential drugs, pathologically-activated therapeutics have been developed to interact with “cryptic” sites of the target proteins that are exposed only under pathological conditions (Lipton, 2007). This approach aims to reduce the pathological activity of the target protein and to spare its normal activity in order to prevent any side-effects associated with drug treatment (Lipton, 2007). Such approach has been used to dampen the pathological hyperactivity of the N-methyl-D-aspartate receptor (NMDAR; Figure 2F). NMDAR is a glutamate-gated ion channel permeable to Ca2+ and is essential for neurotransmission, learning, and memory formation (Lipton, 2006). Hyperactivity of NMDAR is a common disease mechanism for many neurodegenerative conditions (Lipton and Rosenberg, 1994), and plays a critical role in excitotoxicity and synaptic damage in AD (Li et al., 2011; Talantova et al., 2013; Molokanova et al., 2014). To reduce NMDAR hyperactivity, the FDA-approved aminoadamantane compound, Memantine, an uncompetitive fast-off rate NMDAR antagonist that acts as an open-channel blocker was used initially (Chen et al., 1992; Chen and Lipton, 1997; Lipton, 2006). To improve the limited effect of Memantine on disease symptoms, and based on the observation that S-nitrosation of NMDAR reduces its activity (Lipton and Rosenberg, 1994; Choi et al., 2000; Lipton, 2006; Takahashi et al., 2007; Molokanova et al., 2014), NitroSynapsin (YQW-036, NitroMemantine), a derivative of Memantine that combines the aminoadamantane moiety and a nitro group was later synthesized and tested (Lipton, 2006, 2007; Wang et al., 2006; Talantova et al., 2013). NitroSynapsin utilizes the high-affinity Memantine binding site on NMDARs to target the nitro group for interaction with the S-nitrosation/inhibitory site of NMDAR (which is external to the Memantine-binding site); this increases NMDAR S-nitrosation and further reduces its excessive activity (Takahashi et al., 2015; Ghatak et al., 2020). Importantly, this new drug has been shown to reduce synaptic degeneration and improve the disease phenotype of a 3xTg AD mouse model (Talantova et al., 2013), a rat model of vascular dementia (Takahashi et al., 2015), a MEF2C haploinsufficiency mouse model of autism (Tu et al., 2017), a tuberous sclerosis mouse model (Okamoto et al., 2019) and an hAPP-J20 AD mouse model (Ghatak et al., 2020). Very recently, the effect of NitroSynapsin on neuronal activity has also been explored in neurons and cerebral organoids derived from human induced pluripotent stem cells (Ghatak et al., 2020). Interestingly, NitroSynapsin (but not Memantine) dampens the AD-associated increase in spontaneous action potentials and hypersynchronous network activity, suggesting that NitroSynapsin is more effective than Memantine as a human AD drug (Ghatak et al., 2020), which will be tested in future clinical trials.
The potential synergistic effect on drug efficacy of the combination of an existing drug and a nitro group has also been tested with the FDA-approved compound Clomethiazole (CMZ). CMZ is a well-established neuroprotective potentiator of the redox-sensitive gamma-aminobutyric acid receptor (GABAR; Castel and Vaudry, 2001; Wilby and Hutchinson, 2004; Gasulla et al., 2012; Dejanovic and Schwarz, 2014; Vandevrede et al., 2014; Calvo and Beltrán González, 2016). Crucially, NMZ/GT-1061, a CMZ compound analog possessing a nitro group, was shown in AD mouse models to significantly reduce Aβ deposition and cognitive decline, and to restore neuronal plasticity (Qin et al., 2012; Luo et al., 2015, 2016; Hollas et al., 2019); however, it remains unknown whether NO-mediated redox PTMs on GABAR play a part in the improved beneficial therapeutic effects provided by NMZ treatment.
Given the importance of O/N stresses and associated modifications in the onset and progression of many diseases, redox PTMs, especially irreversible redox PTMs, have been proposed as potential circulating biomarkers for a range of conditions including cardiovascular and pulmonary diseases (Di Domenico et al., 2011; Butterfield et al., 2014; Frijhoff et al., 2015; Mnatsakanyan et al., 2018; Tomin et al., 2019). Several studies have explored the proteins modified by irreversible redox PTMs (e.g., carbonylation) in cerebrospinal fluid and blood of AD and control patients and have detected differences in oxidation levels of a small number of proteins (Choi et al., 2002; Korolainen et al., 2007; Korolainen and Pirttilä, 2009; Cocciolo et al., 2012) suggesting that specific redox-modified proteins could be used as biomarkers for neurodegenerative conditions. Major technical challenges have however hindered so far the discovery of the full spectrum of proteins modified by redox PTMs in clinical samples (Delobel et al., 2016; Mnatsakanyan et al., 2018). Another major hurdle in the use of redox-modified proteins as biomarkers for neurodegenerative conditions is the limited passage of proteins or protein fragments through the blood-brain barrier (Di Domenico et al., 2011; Wang et al., 2012a; Hampel et al., 2018). So far, a handful of proteins present in biofluids of patients has been identified as potential biomarkers for neurodegenerative conditions, including Tau, Aβ, and neurofilament light (Hampel et al., 2018; Lewczuk et al., 2018; Robey and Panegyres, 2019; Ashton et al., 2020). Given that redox PTMs of these proteins contribute to the pathophysiology of neurodegenerative diseases (Horiguchi et al., 2003; Reynolds et al., 2006; Alkam et al., 2008; Kummer et al., 2011; Reyes et al., 2011), assessing the redox status of these proteins could provide crucial information, for example on the disease stage, which could potentially help follow disease progression or assess the efficacy of new drugs. It is therefore essential to further improve the sensitivity and robustness of current methodologies used to detect redox PTMs in clinical samples in order to discover redox-modified proteins that could discriminate pathological vs. healthy states and that could potentially be used as diagnostic or prognostic tools for neurodegenerative conditions.
Conclusion
Redox PTMs of protein thiols play an important role in normal cell physiology, brain aging, and in the pathophysiology of several neurodegenerative conditions, including AD, PD, and ALS. Aberrant redox PTMs can occur in response to O/N stresses and contribute to neurodegeneration by disrupting numerous pathways, from proteostasis to mitochondrial dynamics and function. Further developments in mass spectrometry-based redox proteomics will allow to explore the full redox proteome and to identify proteins and pathways modulated by specific redox PTMs in physiology and disease. A better understanding of drug targets’ redox regulations in disease conditions would also be required to design more effective and selective redox-based therapeutic approaches for neurodegenerative conditions. Approaches that would modulate redox PTMs on specific target proteins to elicit a selective cellular response could also be tested as an alternative redox-based therapeutic strategy for neurodegenerative disease.
Author Contributions
MF wrote and edited the entire manuscript.
Funding
This work was funded by an Anne McLaren Fellowship from the University of Nottingham awarded to MF.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abate, C., Patel, L., Rauscher, F. J. III., and Curran, T. (1990). Redox regulation of fos and jun DNA-binding activity in vitro. Science 249, 1157–1161. doi: 10.1126/science.2118682
Abu-Omar, N., Das, J., Szeto, V., and Feng, Z. P. (2018). Neuronal ryanodine receptors in development and aging. Mol. Neurobiol. 55, 1183–1192. doi: 10.1007/s12035-016-0375-4
Adler, A. S., Sinha, S., Kawahara, T. L., Zhang, J. Y., Segal, E., and Chang, H. Y. (2007). Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 21, 3244–3257. doi: 10.1101/gad.1588507
Ahmari, M., Sharafi, A., Mahmoudi, J., Jafari-Anarkoli, I., Gharbavi, M., and Hosseini, M. J. (2020). Selegiline (L-Deprenyl) mitigated oxidative stress, cognitive abnormalities and histopathological change in rats: alternative therapy in transient global ischemia. J. Mol. Neurosci. doi: 10.1007/s12031-020-01544-5 [Epub ahead of print].
Aiken, C. T., Kaake, R. M., Wang, X., and Huang, L. (2011). Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteomics 10:R110.006924. doi: 10.1074/mcp.M110.006924
Akhtar, M. W., Sanz-Blasco, S., Dolatabadi, N., Parker, J., Chon, K., Lee, M. S., et al. (2016). Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat. Commun. 7:10242. doi: 10.1038/ncomms10242
Alkam, T., Nitta, A., Mizoguchi, H., Itoh, A., Murai, R., Nagai, T., et al. (2008). The extensive nitration of neurofilament light chain in the hippocampus is associated with the cognitive impairment induced by amyloid β in mice. J. Pharmacol. Exp. Ther. 327, 137–147. doi: 10.1124/jpet.108.141309
Aluise, C. D., Robinson, R. A., Beckett, T. L., Murphy, M. P., Cai, J., Pierce, W. M., et al. (2010). Preclinical Alzheimer disease: brain oxidative stress, Aβ peptide and proteomics. Neurobiol. Dis. 39, 221–228. doi: 10.1016/j.nbd.2010.04.011
Aluise, C. D., Robinson, R. A. S., Cai, J., Pierce, W. M., Markesbery, W. R., and Butterfield, D. A. (2011). Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: insights into memory loss in mci. J. Alzheimers Dis. 23, 257–269. doi: 10.3233/jad-2010-101083
Andringa, G., and Cools, A. R. (2000). The neuroprotective effects of CGP 3466B in the best in vivo model of Parkinson’s disease, the bilaterally MPTP-treated rhesus monkey. J. Neural Transm. 60, 215–225. doi: 10.1007/978-3-7091-6301-6_14
Andringa, G., van Oosten, R. V., Unger, W., Hafmans, T. G., Veening, J., Stoof, J. C., et al. (2000). Systemic administration of the propargylamine CGP 3466B prevents behavioural and morphological deficits in rats with 6-hydroxydopamine-induced lesions in the substantia nigra. Eur. J. Neurosci. 12, 3033–3043. doi: 10.1046/j.1460-9568.2000.00181.x
Andrus, P. K., Fleck, T. J., Gurney, M. E., and Hall, E. D. (1998). Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 71, 2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x
Aracena-Parks, P., Goonasekera, S. A., Gilman, C. P., Dirksen, R. T., Hidalgo, C., and Hamilton, S. L. (2006). Identification of cysteines involved in S-nitrosylation, S-glutathionylation and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 281, 40354–40368. doi: 10.1074/jbc.m600876200
Araki, K., Kusano, H., Sasaki, N., Tanaka, R., Hatta, T., Fukui, K., et al. (2016). Redox sensitivities of global cellular cysteine residues under reductive and oxidative stress. J. Proteome Res. 15, 2548–2559. doi: 10.1021/acs.jproteome.6b00087
Arias-Cavieres, A., Barrientos, G. C., Sánchez, G., Elgueta, C., Muñoz, P., and Hidalgo, C. (2018). Ryanodine receptor-mediated calcium release has a key role in hippocampal ltd induction. Front. Cell. Neurosci. 12:403. doi: 10.3389/fncel.2018.00403
Arkinson, C., and Walden, H. (2018). Parkin function in Parkinson’s disease. Science 360, 267–268. doi: 10.1126/science.aar6606
Ashton, N. J., Hye, A., Rajkumar, A. P., Leuzy, A., Snowden, S., Suárez-Calvet, M., et al. (2020). An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat. Rev. Neurol. 16, 265–284. doi: 10.1038/s41582-020-0348-0
Barone, E., Di Domenico, F., Sultana, R., Coccia, R., Mancuso, C., Perluigi, M., et al. (2012). Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 52, 2292–2301. doi: 10.1016/j.freeradbiomed.2012.03.020
Barranco-Medina, S., Lázaro, J. J., and Dietz, K. J. (2009). The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 583, 1809–1816. doi: 10.1016/j.febslet.2009.05.029
Barrett, P. J., and Timothy Greenamyre, J. (2015). Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res. 1628, 247–253. doi: 10.1016/j.brainres.2015.06.002
Barua, S., Kim, J. Y., Yenari, M. A., and Lee, J. E. (2019). The role of NOX inhibitors in neurodegenerative diseases. IBRO Rep. 7, 59–69. doi: 10.1016/j.ibror.2019.07.1721
Barzilai, N., Huffman, D. M., Muzumdar, R. H., and Bartke, A. (2012). The critical role of metabolic pathways in aging. Diabetes 61, 1315–1322. doi: 10.2337/db11-1300
Beckhauser, T. F., Francis-Oliveira, J., and De Pasquale, R. (2016). Reactive oxygen species: physiological and physiopathological effects on synaptic plasticity. J. Exp. Neurosci. 10, 23–48. doi: 10.4137/jen.s39887
Bellinger, A. M., Reiken, S., Carlson, C., Mongillo, M., Liu, X., Rothman, L., et al. (2009). Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330. doi: 10.1038/nm.1916
Bian, K., and Murad, F. (2003). Nitric oxide (NO)—biogeneration, regulation, and relevance to human diseases. Front. Biosci. 8, d264–278. doi: 10.2741/997
Bindoli, A., Fukuto, J. M., and Forman, H. J. (2008). Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox Signal. 10, 1549–1564. doi: 10.1089/ars.2008.2063
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S., and Kalayci, O. (2012). Oxidative stress and antioxidant defense. World Allergy Organ. J. 5, 9–19. doi: 10.1097/WOX.0b013e3182439613
Bodhinathan, K., Kumar, A., and Foster, T. C. (2010a). Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 30, 1914–1924. doi: 10.1523/JNEUROSCI.5485-09.2010
Bodhinathan, K., Kumar, A., and Foster, T. C. (2010b). Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J. Neurophysiol. 104, 2586–2593. doi: 10.1152/jn.00577.2010
Boutte, A. M., Woltjer, R. L., Zimmerman, L. J., Stamer, S. L., Montine, K. S., Manno, M. V., et al. (2006). Selectively increased oxidative modifications mapped to detergent-insoluble forms of Aβ and β-III tubulin in Alzheimer’s disease. FASEB J. 20, 1473–1483. doi: 10.1096/fj.06-5920com
Bouzier-Sore, A.-K., and Bolaños, J. P. (2015). Uncertainties in pentose-phosphate pathway flux assessment underestimate its contribution to neuronal glucose consumption: relevance for neurodegeneration and aging. Front. Aging Neurosci. 7:89. doi: 10.3389/fnagi.2015.00089
Bradley, S. A., and Steinert, J. R. (2016). Nitric oxide-mediated posttranslational modifications: impacts at the synapse. Oxid. Med. Cell. Longev. 2016:5681036. doi: 10.1155/2016/5681036
Brigelius-Flohé, R., and Flohé, L. (2011). Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 15, 2335–2381. doi: 10.1089/ars.2010.3534
Bull, R., Finkelstein, J. P., Gálvez, J., Sánchez, G., Donoso, P., Behrens, M. I., et al. (2008). Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J. Neurosci. 28, 9463–9472. doi: 10.1523/JNEUROSCI.2286-08.2008
Bull, R., Marengo, J. J., Finkelstein, J. P., Behrens, M. I., and Alvarez, O. (2003). SH oxidation coordinates subunits of rat brain ryanodine receptor channels activated by calcium and ATP. Am. J. Physiol. Cell Physiol. 285, C119–128. doi: 10.1152/ajpcell.00296.2002
Bulteau, A. L., Mena, N. P., Auchère, F., Lee, I., Prigent, A., Lobsiger, C. S., et al. (2017). Dysfunction of mitochondrial Lon protease and identification of oxidized protein in mouse brain following exposure to MPTP: implications for Parkinson disease. Free Radic. Biol. Med. 108, 236–246. doi: 10.1016/j.freeradbiomed.2017.03.036
Butterfield, D. A., Galvan, V., Lange, M. B., Tang, H., Sowell, R. A., Spilman, P., et al. (2010). In vivo oxidative stress in brain of Alzheimer disease transgenic mice: requirement for methionine 35 in amyloid β-peptide of APP. Free Radic. Biol. Med. 48, 136–144. doi: 10.1016/j.freeradbiomed.2009.10.035
Butterfield, D. A., Gu, L., Domenico, F. D., and Robinson, R. A. S. (2014). Mass spectrometry and redox proteomics: applications in disease. Mass Spectrom. Rev. 33, 277–301. doi: 10.1002/mas.21374
Butterfield, D. A., Perluigi, M., Reed, T., Muharib, T., Hughes, C. P., Robinson, R. A. S., et al. (2012). Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid. Redox Signal. 17, 1610–1655. doi: 10.1089/ars.2011.4109
Cabré, R., Naudí, A., Dominguez-Gonzalez, M., Ayala, V., Jové, M., Mota-Martorell, N., et al. (2017). Sixty years old is the breakpoint of human frontal cortex aging. Free Radic. Biol. Med. 103, 14–22. doi: 10.1016/j.freeradbiomed.2016.12.010
Calderwood, S. K., Murshid, A., and Prince, T. (2009). The shock of aging: molecular chaperones and the heat shock response in longevity and aging—a mini-review. Gerontology 55, 550–558. doi: 10.1159/000225957
Calvo, D. J., and Beltrán González, A. N. (2016). Dynamic regulation of the GABAA receptor function by redox mechanisms. Mol. Pharmacol. 90, 326–333. doi: 10.1124/mol.116.105205
Castel, H., and Vaudry, H. (2001). Nitric oxide directly activates GABAA receptor function through a cGMP/protein kinase-independent pathway in frog pituitary melanotrophs. J. Neuroendocrinol. 13, 695–705. doi: 10.1046/j.1365-2826.2001.00683.x
Caviedes, A., Maturana, B., Corvalán, K., Engler, A., Gordillo, F., Varas-Godoy, M., et al. (2020). The eNOS-dependent S-nitrosylation of the NF-κB subunit p65 has neuroprotective consequences in excitotoxicity. bioRxiv [Preprint]. doi: 10.1101/2020.02.04.932772
Cha, S. J., Kim, H., Choi, H.-J., Lee, S., and Kim, K. (2017). Protein glutathionylation in the pathogenesis of neurodegenerative diseases. Oxid. Med. Cell. Longev. 2017:2818565. doi: 10.1155/2017/2818565
Chavarría, C., and Souza, J. M. (2013). Oxidation and nitration of α-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 533, 25–32. doi: 10.1016/j.abb.2013.02.009
Chen, X., Guan, T., Li, C., Shang, H., Cui, L., Li, X. M., et al. (2012). SOD1 aggregation in astrocytes following ischemia/reperfusion injury: a role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI). J. Neuroinflammation 9:237. doi: 10.1186/1742-2094-9-237
Chen, H. S., and Lipton, S. A. (1997). Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J. Physiol. 499, 27–46. doi: 10.1113/jphysiol.1997.sp021909
Chen, Y.-J., Lu, C.-T., Huang, K.-Y., Wu, H.-Y., Chen, Y.-J., and Lee, T.-Y. (2015). GSHSite: exploiting an iteratively statistical method to identify S-glutathionylation sites with substrate specificity. PLoS One 10:e0118752. doi: 10.1371/journal.pone.0118752
Chen, Y.-J., Lu, C.-T., Lee, T.-Y., and Chen, Y.-J. (2014). dbGSH: a database of S-glutathionylation. Bioinformatics 30, 2386–2388. doi: 10.1093/bioinformatics/btu301
Chen, H. S., Pellegrini, J. W., Aggarwal, S. K., Lei, S. Z., Warach, S., Jensen, F. E., et al. (1992). Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J. Neurosci. 12, 4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992
Chen, X., Zhang, X., Li, C., Guan, T., Shang, H., Cui, L., et al. (2013). S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J. Neurochem. 124, 45–58. doi: 10.1111/jnc.12046
Chistiakov, D. A., Sobenin, I. A., Revin, V. V., Orekhov, A. N., and Bobryshev, Y. V. (2014). Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res. Int. 2014:238463. doi: 10.1155/2014/238463
Cho, B., Choi, S. Y., Cho, H. M., Kim, H. J., and Sun, W. (2013). Physiological and pathological significance of dynamin-related protein 1 (drp1)-dependent mitochondrial fission in the nervous system. Exp. Neurobiol. 22, 149–157. doi: 10.5607/en.2013.22.3.149
Cho, D. H., Nakamura, T., and Lipton, S. A. (2010). Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol. Life Sci. 67, 3435–3447. doi: 10.1007/s00018-010-0435-2
Cho, D.-H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. doi: 10.1126/science.1171091
Choi, J., Malakowsky, C. A., Talent, J. M., Conrad, C. C., and Gracy, R. W. (2002). Identification of oxidized plasma proteins in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 293, 1566–1570. doi: 10.1016/S0006-291X(02)00420-5
Choi, Y. B., Tenneti, L., Le, D. A., Ortiz, J., Bai, G., Chen, H. S., et al. (2000). Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 3, 15–21. doi: 10.1038/71090
Chung, K. K. K., Thomas, B., Li, X., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2004). S-nitrosylation of parkin regulates ubiquitination and compromises Parkin’s protective function. Science 304, 1328–1331. doi: 10.1126/science.1093891
Chung, H. S., Wang, S.-B., Venkatraman, V., Murray, C. I., and Eyk, J. E. V. (2013). Cysteine oxidative posttranslational modifications. Circ. Res. 112, 382–392. doi: 10.1161/circresaha.112.268680
Cini, M., and Moretti, A. (1995). Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiol. Aging 16, 53–57. doi: 10.1016/0197-4580(95)80007-e
Cocciolo, A., Di Domenico, F., Coccia, R., Fiorini, A., Cai, J., Pierce, W. M., et al. (2012). Decreased expression and increased oxidation of plasma haptoglobin in Alzheimer disease: insights from redox proteomics. Free Radic. Biol. Med. 53, 1868–1876. doi: 10.1016/j.freeradbiomed.2012.08.596
Colton, C. A., and Gilbert, D. L. (2002). “Reactive oxygen species and neuronal function,” in Reactive Oxygen Species in Biological Systems: An Interdisciplinary Approach (Boston, MA: Springer), 569–589.
Commoner, B., Townsend, J., and Pake, G. E. (1954). Free radicals in biological materials. Nature 174, 689–691. doi: 10.1038/174689a0
Conway, M. E., and Harris, M. (2015). S-nitrosylation of the thioredoxin-like domains of protein disulfide isomerase and its role in neurodegenerative conditions. Front. Chem. 3:27. doi: 10.3389/fchem.2015.00027
Corcoran, A., and Cotter, T. G. (2013). Redox regulation of protein kinases. FEBS J. 280, 1944–1965. doi: 10.1111/febs.12224
Cremers, C. M., and Jakob, U. (2013). Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 288, 26489–26496. doi: 10.1074/jbc.r113.462929
Daiber, A. (2010). Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797, 897–906. doi: 10.1016/j.bbabio.2010.01.032
Daiber, A., Di Lisa, F., Oelze, M., Kröller-Schön, S., Steven, S., Schulz, E., et al. (2017). Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 174, 1670–1689. doi: 10.1111/bph.13403
Davies, M. J. (2005). The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109. doi: 10.1016/j.bbapap.2004.08.007
Davies, M. J. (2016). Protein oxidation and peroxidation. Biochem. J. 473, 805–825. doi: 10.1042/bj20151227
De Haes, W., Frooninckx, L., Van Assche, R., Smolders, A., Depuydt, G., Billen, J., et al. (2014). Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. U S A 111, E2501–E2509. doi: 10.1073/pnas.1321776111
De la Fuente, M., and Miquel, J. (2009). An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 15, 3003–3026. doi: 10.2174/138161209789058110
Dean, R. T., Fu, S., Stocker, R., and Davies, M. J. (1997). Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 324, 1–18. doi: 10.1042/bj3240001
Dejanovic, B., and Schwarz, G. (2014). Neuronal nitric oxide synthase-dependent S-nitrosylation of gephyrin regulates gephyrin clustering at GABAergic synapses. J. Neurosci. 34, 7763–7768. doi: 10.1523/JNEUROSCI.0531-14.2014
Delobel, J., Prudent, M., Crettaz, D., ElHajj, Z., Riederer, B. M., Tissot, J. D., et al. (2016). Cysteine redox proteomics of the hemoglobin-depleted cytosolic fraction of stored red blood cells. Proteomics Clin. Appl. 10, 883–893. doi: 10.1002/prca.201500132
Di Domenico, F., Barone, E., Perluigi, M., and Butterfield, D. A. (2017). The triangle of death in Alzheimer’s disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling and protein homeostasis revealed by redox proteomics. Antioxid. Redox Signal. 26, 364–387. doi: 10.1089/ars.2016.6759
Di Domenico, F., Coccia, R., Butterfield, D. A., and Perluigi, M. (2011). Circulating biomarkers of protein oxidation for Alzheimer disease: expectations within limits. Biochim. Biophys. Acta 1814, 1785–1795. doi: 10.1016/j.bbapap.2011.10.001
Di Meo, S., Reed, T. T., Venditti, P., and Victor, V. M. (2016). Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016:1245049. doi: 10.1155/2016/1245049
Dickinson, B. C., and Chang, C. J. (2011). Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 7, 504–511. doi: 10.1038/nchembio.607
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole, R. N., Itoh, K., Wakabayashi, N., Katoh, Y., et al. (2002). Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U S A 99, 11908–11913. doi: 10.1073/pnas.172398899
Dinkova-Kostova, A. T., Kostov, R. V., and Canning, P. (2017). Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 617, 84–93. doi: 10.1016/j.abb.2016.08.005
Doulias, P.-T., Greene, J. L., Greco, T. M., Tenopoulou, M., Seeholzer, S. H., Dunbrack, R. L., et al. (2010). Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U S A 107, 16958–16963. doi: 10.1073/pnas.1008036107
Doulias, P.-T., Tenopoulou, M., Greene, J. L., Raju, K., and Ischiropoulos, H. (2013). Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 6:rs1. doi: 10.1126/scisignal.2003252
Dremina, E. S., Sharov, V. S., and Schöneich, C. (2005). Protein tyrosine nitration in rat brain is associated with raft proteins, flotillin-1 and α-tubulin: effect of biological aging. J. Neurochem. 93, 1262–1271. doi: 10.1111/j.1471-4159.2005.03115.x
DuBoff, B., Feany, M., and Götz, J. (2013). Why size matters—balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci. 36, 325–335. doi: 10.1016/j.tins.2013.03.002
Dyer, R. R., Ford, K. I., and Robinson, R. A. S. (2019). The roles of S-nitrosylation and S-glutathionylation in Alzheimer’s disease. Methods Enzymol. 626, 499–538. doi: 10.1016/bs.mie.2019.08.004
Engelman, R., Weisman-Shomer, P., Ziv, T., Xu, J., Arnér, E. S., and Benhar, M. (2013). Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation. J. Biol. Chem. 288, 11312–11324. doi: 10.1074/jbc.m112.433755
Erb, M., Meinen, S., Barzaghi, P., Sumanovski, L. T., Courdier-Früh, I., Rüegg, M. A., et al. (2009). Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-α2 deficiency. J. Pharmacol. Exp. Ther. 331, 787–795. doi: 10.1124/jpet.109.160754
Fang, J., Nakamura, T., Cho, D.-H., Gu, Z., and Lipton, S. A. (2007). S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 104, 18742–18747. doi: 10.1073/pnas.0705904104
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Horne, M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Farr, S. A., Poon, H. F., Dogrukol-Ak, D., Drake, J., Banks, W. A., Eyerman, E., et al. (2003). The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 84, 1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x
Feng, Y., and Wang, X. (2012). Antioxidant therapies for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012:472932. doi: 10.1155/2012/472932
Filograna, R., Beltramini, M., Bubacco, L., and Bisaglia, M. (2016). Anti-oxidants in Parkinson’s disease therapy: a critical point of view. Curr. Neuropharmacol. 14, 260–271. doi: 10.2174/1570159x13666151030102718
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Flannery, P. J., and Trushina, E. (2019). Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 98, 109–120. doi: 10.1016/j.mcn.2019.06.009
Forman, H. J., Torres, M., and Fukuto, J. (2002). “Redox signaling,” in Oxygen/Nitrogen Radicals: Cell Injury and Disease, eds V. Vallyathan, X. Shi and V. Castranova (Boston, MA: Springer US), 49–62.
Forman, H. J., Ursini, F., and Maiorino, M. (2014). An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 73, 2–9. doi: 10.1016/j.yjmcc.2014.01.018
Foster, T. C. (2006). Biological markers of age-related memory deficits: treatment of senescent physiology. CNS Drugs 20, 153–166. doi: 10.2165/00023210-200620020-00006
Foster, T. C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 6, 319–325. doi: 10.1111/j.1474-9726.2007.00283.x
Foster, T. C. (2012). Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Prog. Neurobiol. 96, 283–303. doi: 10.1016/j.pneurobio.2012.01.007
Foster, T. C., and Kumar, A. (2002). Calcium dysregulation in the aging brain. Neuroscientist 8, 297–301. doi: 10.1177/107385840200800404
Foster, T. C., and Norris, C. M. (1997). Age-associated changes in Ca2+-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus 7, 602–612. doi: 10.1002/(sici)1098-1063(1997)7:6<602::aid-hipo3>3.0.co;2-g
Fra, A., Yoboue, E. D., and Sitia, R. (2017). Cysteines as redox molecular switches and targets of disease. Front. Mol. Neurosci. 10:167. doi: 10.3389/fnmol.2017.00167
Franceschi, C., Capri, M., Monti, D., Giunta, S., Olivieri, F., Sevini, F., et al. (2007). Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 128, 92–105. doi: 10.1016/j.mad.2006.11.016
Friedman, J. (2010a). “The role of free radicals in the nervous system,” in Oxidative Stress and Free Radical Damage in Neurology. Oxidative Stress in Applied Basic Research and Clinical Practice, eds N. Gadoth and H. H. Göbel (Totowa, NJ: Humana Press), 1–17.
Friedman, J. (2010b). “Why is the nervous system vulnerable to oxidative stress?,” in Oxidative Stress in Applied Basic Research and Clinical Practice, eds N. Gadoth and H. H. Göbel (Totowa, NJ: Humana Press), 19–27.
Friguet, B. (2006). Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett. 580, 2910–2916. doi: 10.1016/j.febslet.2006.03.028
Frijhoff, J., Winyard, P. G., Zarkovic, N., Davies, S. S., Stocker, R., Cheng, D., et al. (2015). Clinical relevance of biomarkers of oxidative stress. Antioxid. Redox Signal. 23, 1144–1170. doi: 10.1089/ars.2015.6317
Frudd, K., Burgoyne, T., and Burgoyne, J. R. (2018). Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 9:95. doi: 10.1038/s41467-017-02352-z
Gallogly, M. M., and Mieyal, J. J. (2007). Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7, 381–391. doi: 10.1016/j.coph.2007.06.003
Gantner, B. N., LaFond, K. M., and Bonini, M. G. (2020). Nitric oxide in cellular adaptation and disease. Redox Biol. 34:101550. doi: 10.1016/j.redox.2020.101550
Garcia, J., Han, D., Sancheti, H., Yap, L. P., Kaplowitz, N., and Cadenas, E. (2010). Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654. doi: 10.1074/jbc.m110.164160
Gasulla, J., Beltrán González, A. N., and Calvo, D. J. (2012). Nitric oxide potentiation of the homomeric ρ1 GABA(C) receptor function. Br. J. Pharmacol. 167, 1369–1377. doi: 10.1111/j.1476-5381.2012.02087.x
Ghatak, S., Dolatabadi, N., Gao, R., Wu, Y., Scott, H., Trudler, D., et al. (2020). NitroSynapsin ameliorates hypersynchronous neural network activity in Alzheimer hiPSC models. Mol. Psychiatry doi: 10.1038/s41380-020-0776-7 [Epub ahead of print].
Giulivi, C., Pacifici, R. E., and Davies, K. J. (1994). Exposure of hydrophobic moieties promotes the selective degradation of hydrogen peroxide-modified hemoglobin by the multicatalytic proteinase complex, proteasome. Arch. Biochem. Biophys. 311, 329–341. doi: 10.1006/abbi.1994.1245
Gloire, G., and Piette, J. (2009). Redox regulation of nuclear post-translational modifications during NF-κB activation. Antioxid. Redox Signal. 11, 2209–2222. doi: 10.1089/ars.2009.2463
Go, Y.-M., Chandler, J. D., and Jones, D. P. (2015). The cysteine proteome. Free Radic. Biol. Med. 84, 227–245. doi: 10.1016/j.freeradbiomed.2015.03.022
Go, Y.-M., Duong, D. M., Peng, J., and Jones, D. P. (2011). Protein cysteines map to functional networks according to steady-state level of oxidation. J. Proteomics Bioinform. 4, 196–209. doi: 10.4172/jpb.1000190
Go, Y.-M., and Jones, D. P. (2008). Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 1780, 1273–1290. doi: 10.1016/j.bbagen.2008.01.011
Go, Y.-M., and Jones, D. P. (2013). The redox proteome. J. Biol. Chem. 288, 26512–26520. doi: 10.1074/jbc.R113.464131
Go, Y.-M., and Jones, D. P. (2017). Redox theory of aging: implications for health and disease. Clin. Sci. 131, 1669–1688. doi: 10.1042/cs20160897
Gokulrangan, G., Zaidi, A., Michaelis, M. L., and Schöneich, C. (2007). Proteomic analysis of protein nitration in rat cerebellum: effect of biological aging. J. Neurochem. 100, 1494–1504. doi: 10.1111/j.1471-4159.2006.04334.x
Gonos, E. S., Kapetanou, M., Sereikaite, J., Bartosz, G., Naparło, K., Grzesik, M., et al. (2018). Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 10, 868–901. doi: 10.18632/aging.101450
Gonzalez, V., Pal, R., and Narayan, M. (2010). The oxidoreductase behavior of protein disulfide isomerase impedes fold maturation of endoplasmic reticulum-processed proteins in the pivotal structure-coupled step of oxidative folding: implications for subcellular protein trafficking. Biochemistry 49, 6282–6289. doi: 10.1021/bi100753s
Gorelenkova Miller, O., and Mieyal, J. J. (2015). Sulfhydryl-mediated redox signaling in inflammation: role in neurodegenerative diseases. Arch. Toxicol. 89, 1439–1467. doi: 10.1007/s00204-015-1496-7
Gottfredsen, R. H., Larsen, U. G., Enghild, J. J., and Petersen, S. V. (2013). Hydrogen peroxide induce modifications of human extracellular superoxide dismutase that results in enzyme inhibition. Redox Biol. 1, 24–31. doi: 10.1016/j.redox.2012.12.004
Gould, N. S., Evans, P., Martínez-Acedo, P., Marino, S. M., Gladyshev, V. N., Carroll, K. S., et al. (2015). Site-specific proteomic mapping identifies selectively modified regulatory cysteine residues in functionally distinct protein networks. Chem. Biol. 22, 965–975. doi: 10.1016/j.chembiol.2015.06.010
Goyal, M. S., Vlassenko, A. G., Blazey, T. M., Su, Y., Couture, L. E., Durbin, T. J., et al. (2017). Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26, 353.e3–360.e3. doi: 10.1016/j.cmet.2017.07.010
Granold, M., Moosmann, B., Staib-Lasarzik, I., Arendt, T., Del Rey, A., Engelhard, K., et al. (2015). High membrane protein oxidation in the human cerebral cortex. Redox Biol. 4, 200–207. doi: 10.1016/j.redox.2014.12.013
Grimm, S., Hoehn, A., Davies, K. J., and Grune, T. (2011). Protein oxidative modifications in the ageing brain: consequence for the onset of neurodegenerative disease. Free Radic. Res. 45, 73–88. doi: 10.3109/10715762.2010.512040
Grune, T., Reinheckel, T., and Davies, K. J. (1997). Degradation of oxidized proteins in mammalian cells. FASEB J. 11, 526–534. doi: 10.1096/fasebj.11.7.9212076
Grunwald, M. S., Pires, A. S., Zanotto-Filho, A., Gasparotto, J., Gelain, D. P., Demartini, D. R., et al. (2014). The oxidation of HSP70 is associated with functional impairment and lack of stimulatory capacity. Cell Stress Chaperones 19, 913–925. doi: 10.1007/s12192-014-0516-5
Guzman-Martinez, L., Maccioni, R. B., Andrade, V., Navarrete, L. P., Pastor, M. G., and Ramos-Escobar, N. (2019). Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 10:1008. doi: 10.3389/fphar.2019.01008
Hall, A., Karplus, P. A., and Poole, L. B. (2009). Typical 2-Cys peroxiredoxins—structures, mechanisms and functions. FEBS J. 276, 2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x
Hall, A., Nelson, K., Poole, L. B., and Karplus, P. A. (2011). Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 15, 795–815. doi: 10.1089/ars.2010.3624
Halloran, M., Parakh, S., and Atkin, J. D. (2013). The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013:797914. doi: 10.1155/2013/797914
Hampel, H., O’Bryant, S. E., Molinuevo, J. L., Zetterberg, H., Masters, C. L., Lista, S., et al. (2018). Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat. Rev. Neurol. 14, 639–652. doi: 10.1038/s41582-018-0079-7
Handy, D. E., and Loscalzo, J. (2012). Redox regulation of mitochondrial function. Antioxid. Redox Signal. 16, 1323–1367. doi: 10.1089/ars.2011.4123
Hara, M. R., Agrawal, N., Kim, S. F., Cascio, M. B., Fujimuro, M., Ozeki, Y., et al. (2005). S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674. doi: 10.1038/ncb1268
Hara, M. R., Cascio, M. B., and Sawa, A. (2006a). GAPDH as a sensor of NO stress. Biochim. Biophys. Acta 1762, 502–509. doi: 10.1016/j.bbadis.2006.01.012
Hara, M. R., Thomas, B., Cascio, M. B., Bae, B.-I., Hester, L. D., Dawson, V. L., et al. (2006b). Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. U S A 103, 3887–3889. doi: 10.1073/pnas.0511321103
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. doi: 10.1093/geronj/11.3.298
Harman, D. (2006). Free radical theory of aging: an update. Ann. N Y Acad. Sci. 1067, 10–21. doi: 10.1196/annals.1354.003
Harraz, M. M., and Snyder, S. H. (2015). Nitric oxide-GAPDH transcriptional signaling mediates behavioral actions of cocaine. CNS Neurol. Disord. Drug Targets 14, 757–763. doi: 10.2174/1871527314666150529150143
Hartl, D., Schuldt, V., Forler, S., Zabel, C., Klose, J., and Rohe, M. (2012). Presymptomatic alterations in energy metabolism and oxidative stress in the APP23 mouse model of Alzheimer disease. J. Proteome Res. 11, 3295–3304. doi: 10.1021/pr300021e
Hess, D. T., Matsumoto, A., Nudelman, R., and Stamler, J. S. (2001). S-nitrosylation: spectrum and specificity. Nat. Cell Biol. 3, E46–E48. doi: 10.1038/35055152
Hidalgo, C. (2005). Cross talk between Ca2+ and redox signalling cascades in muscle and neurons through the combined activation of ryanodine receptors/Ca2+ release channels. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 2237–2246. doi: 10.1098/rstb.2005.1759
Hidalgo, C., and Arias-Cavieres, A. (2016). Calcium, reactive oxygen species, and synaptic plasticity. Physiology 31, 201–215. doi: 10.1152/physiol.00038.2015
Hipkiss, A. R. (2019). Aging, Alzheimer’s disease and dysfunctional glycolysis; similar effects of too much and too little. Aging Dis. 10, 1328–1331. doi: 10.14336/ad.2019.0611
Höhn, T. J., and Grune, T. (2014). The proteasome and the degradation of oxidized proteins: part III-Redox regulation of the proteasomal system. Redox Biol. 2, 388–394. doi: 10.1016/j.redox.2013.12.029
Höhn, A., Weber, D., Jung, T., Ott, C., Hugo, M., Kochlik, B., et al. (2017). Happily (n)ever after: aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 11, 482–501. doi: 10.1016/j.redox.2016.12.001
Hollas, M. A., Ben Aissa, M., Lee, S. H., Gordon-Blake, J. M., and Thatcher, G. R. J. (2019). Pharmacological manipulation of cGMP and NO/cGMP in CNS drug discovery. Nitric Oxide 82, 59–74. doi: 10.1016/j.niox.2018.10.006
Honjo, Y., Kaneko, S., Ito, H., Horibe, T., Nagashima, M., Nakamura, M., et al. (2011). Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 444–450. doi: 10.3109/17482968.2011.594055
Horiguchi, T., Uryu, K., Giasson, B. I., Ischiropoulos, H., LightFoot, R., Bellmann, C., et al. (2003). Nitration of tau protein is linked to neurodegeneration in tauopathies. Am. J. Pathol. 163, 1021–1031. doi: 10.1016/s0002-9440(10)63462-1
Houée-Lévin, C., Bobrowski, K., Horakova, L., Karademir, B., Schöneich, C., Davies, M. J., et al. (2015). Exploring oxidative modifications of tyrosine: an update on mechanisms of formation, advances in analysis and biological consequences. Free Radic. Res. 49, 347–373. doi: 10.3109/10715762.2015.1007968
Hsieh, H.-L., and Yang, C.-M. (2013). Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res. Int. 2013:484613. doi: 10.1155/2013/484613
Hurd, T. R., Requejo, R., Filipovska, A., Brown, S., Prime, T. A., Robinson, A. J., et al. (2008). Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 283, 24801–24815. doi: 10.1074/jbc.m803432200
Ignarro, L. J. (1990). Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu. Rev. Pharmacol. Toxicol. 30, 535–560. doi: 10.1146/annurev.pa.30.040190.002535
Jang, H. H., Lee, K. O., Chi, Y. H., Jung, B. G., Park, S. K., Park, J. H., et al. (2004). Two enzymes in one: two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117, 625–635. doi: 10.1016/j.cell.2004.05.002
Jenner, P. (2003). Oxidative stress in Parkinson’s disease. Ann. Neurol. 53, S26–S38. doi: 10.1002/ana.10483
Jeon, G. S., Nakamura, T., Lee, J.-S., Choi, W.-J., Ahn, S.-W., Lee, K.-W., et al. (2014). Potential effect of S-nitrosylated protein disulfide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol. Neurobiol. 49, 796–807. doi: 10.1007/s12035-013-8562-z
Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. doi: 10.1083/jcb.143.7.1883
Jones, D. P. (2006). Redefining oxidative stress. Antioxid. Redox Signal. 8, 1865–1879. doi: 10.1089/ars.2006.8.1865
Jones, D. P., and Go, Y. M. (2010). Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 12, 116–125. doi: 10.1111/j.1463-1326.2010.01266.x
Jones, D. P., and Sies, H. (2015). The redox code. Antioxid. Redox Signal. 23, 734–746. doi: 10.1089/ars.2015.6247
Joshi, A. U., Saw, N. L., Shamloo, M., and Mochly-Rosen, D. (2017). Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 9, 6128–6143. doi: 10.18632/oncotarget.23640
Jové, M., Portero-Otín, M., Naudí, A., Ferrer, I., and Pamplona, R. (2014). Metabolomics of human brain aging and age-related neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 73, 640–657. doi: 10.1097/nen.0000000000000091
Jung, T., Bader, N., and Grune, T. (2007). Lipofuscin: formation, distribution, and metabolic consequences. Ann. N Y Acad. Sci. 1119, 97–111. doi: 10.1196/annals.1404.008
Jung, T., and Grune, T. (2008). The proteasome and its role in the degradation of oxidized proteins. IUBMB Life 60, 743–752. doi: 10.1002/iub.114
Jung, T., Höhn, A., and Grune, T. (2014). The proteasome and the degradation of oxidized proteins: part III—redox regulation of the proteasomal system. Redox Biol. 2, 388–394. doi: 10.1016/j.redox.2013.12.029
Jung, Y., Kim, H., Min, S. H., Rhee, S. G., and Jeong, W. (2008). Dynein light chain LC8 negatively regulates NF-κB through the redox-dependent interaction with IκBα. J. Biol. Chem. 283, 23863–23871. doi: 10.1074/jbc.M803072200
Kabe, Y., Ando, K., Hirao, S., Yoshida, M., and Handa, H. (2005). Redox regulation of NF-κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal. 7, 395–403. doi: 10.1089/ars.2005.7.395
Karplus, P. A. (2015). A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 80, 183–190. doi: 10.1016/j.freeradbiomed.2014.10.009
Kawagishi, H., and Finkel, T. (2014). Unraveling the truth about Antioxidants: ROS and disease: finding the right balance. Nat. Med. 20, 711–713. doi: 10.1038/nm.3625
Khoo, K. K., and Norton, R. S. (2011). “Role of disulfide bonds in peptide and protein conformation,” in Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Hughes (Weinheim: Wiley-VCH), 395–417.
Kil, I. S., and Park, J. W. (2005). Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 280, 10846–10854. doi: 10.1074/jbc.m411306200
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi: 10.5607/en.2015.24.4.325
Kim, S. U., Jin, M. H., Kim, Y. S., Lee, S. H., Cho, Y. S., Cho, K. J., et al. (2011). Peroxiredoxin II preserves cognitive function against age-linked hippocampal oxidative damage. Neurobiol. Aging 32, 1054–1068. doi: 10.1016/j.neurobiolaging.2009.05.017
Klatt, P., Molina, E. P., De Lacoba, M. G., Padilla, C. A., Martinez-Galesteo, E., Barcena, J. A., et al. (1999). Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 13, 1481–1490. doi: 10.1096/fasebj.13.12.1481
Klomsiri, C., Karplus, P. A., and Poole, L. B. (2011). Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 14, 1065–1077. doi: 10.1089/ars.2010.3376
Koga, H., Kaushik, S., and Cuervo, A. M. (2011). Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res. Rev. 10, 205–215. doi: 10.1016/j.arr.2010.02.001
Korolainen, M. A., and Pirttilä, T. (2009). Cerebrospinal fluid, serum and plasma protein oxidation in Alzheimer’s disease. Acta Neurol. Scand. 119, 32–38. doi: 10.1111/j.1600-0404.2008.01057.x
Korolainen, M. A., Nyman, T. A., Nyyssönen, P., Hartikainen, E. S., and Pirttilä, T. (2007). Multiplexed proteomic analysis of oxidation and concentrations of cerebrospinal fluid proteins in Alzheimer disease. Clin. Chem. 53, 657–665. doi: 10.1373/clinchem.2006.078014
Kors, S., Geijtenbeek, K., Reits, E., and Schipper-Krom, S. (2019). Regulation of proteasome activity by (Post-)transcriptional mechanisms. Front. Mol. Biosci. 6, 48–48. doi: 10.3389/fmolb.2019.00048
Kragten, E., Lalande, I., Zimmermann, K., Roggo, S., Schindler, P., Muller, D., et al. (1998). Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(−)-deprenyl. J. Biol. Chem. 273, 5821–5828. doi: 10.1074/jbc.273.10.5821
Kumar, A., Bodhinathan, K., and Foster, T. C. (2009). Susceptibility to calcium dysregulation during brain aging. Front. Aging Neurosci. 1:2. doi: 10.3389/neuro.24.002.2009
Kumar, A., Yegla, B., and Foster, T. C. (2018). Redox signaling in neurotransmission and cognition during aging. Antioxid. Redox Signal. 28, 1724–1745. doi: 10.1089/ars.2017.7111
Kummer, M. P., Hermes, M., Delekarte, A., Hammerschmidt, T., Kumar, S., Terwel, D., et al. (2011). Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 71, 833–844. doi: 10.1016/j.neuron.2011.07.001
Kuo, Y.-M., Webster, S., Emmerling, M. R., De Lima, N., and Roher, A. E. (1998). Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of Aβ peptides of Alzheimer’s disease. Biochim. Biophys. Acta 1406, 291–298. doi: 10.1016/s0925-4439(98)00014-3
Kurokawa, T., Asada, S., Nishitani, S., and Hazeki, O. (2001). Age-related changes in manganese superoxide dismutase activity in the cerebral cortex of senescence-accelerated prone and resistant mouse. Neurosci. Lett. 298, 135–138. doi: 10.1016/s0304-3940(00)01755-9
Lacampagne, A., Liu, X., Reiken, S., Bussiere, R., Meli, A. C., Lauritzen, I., et al. (2017). Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 134, 749–767. doi: 10.1007/s00401-017-1733-7
Lam, P. Y., Yin, F., Hamilton, R. T., Boveris, A., and Cadenas, E. (2009). Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radic. Res. 43, 431–439. doi: 10.1080/10715760902849813
Lambeth, J. D., and Neish, A. S. (2014). Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu. Rev. Pathol. 9, 119–145. doi: 10.1146/annurev-pathol-012513-104651
Lanner, J. T., Georgiou, D. K., Joshi, A. D., and Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2:a003996. doi: 10.1101/cshperspect.a003996
Lefaki, M., Papaevgeniou, N., and Chondrogianni, N. (2017). Redox regulation of proteasome function. Redox Biol. 13, 452–458. doi: 10.1016/j.redox.2017.07.005
Lewczuk, P., Riederer, P., O’Bryant, S. E., Verbeek, M. M., Dubois, B., Visser, P. J., et al. (2018). Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: an update of the consensus of the task force on biological markers in psychiatry of the world federation of societies of biological psychiatry. World J. Biol. Psychiatry 19, 244–328. doi: 10.1080/15622975.2017.1375556
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011). Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011
Liang, F., Shi, L., Zheng, J., Chen, S., Wang, Y., and Zhang, J. (2017). Neuroprotective effects of CGP3466B on apoptosis are modulated by protein-L-isoaspartate (D-aspartate) O-methyltransferase/Mst1 pathways after traumatic brain injury in rats. Sci. Rep. 7:9201. doi: 10.1038/s41598-017-08196-3
Lipton, S. A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discov. 5, 160–170. doi: 10.1038/nrd1958
Lipton, S. A. (2007). Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr. Drug Targets 8, 621–632. doi: 10.2174/138945007780618472
Lipton, S., Choi, Y.-B., Takahashi, H., Zhang, D., Li, W., Godzik, A., et al. (2002). Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci. 25, 474–480. doi: 10.1016/s0166-2236(02)02245-2
Lipton, S. A., and Rosenberg, P. A. (1994). Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 330, 613–622. doi: 10.1056/nejm199403033300907
Liu, X., Betzenhauser, M. J., Reiken, S., Meli, A. C., Xie, W., Chen, B. X., et al. (2012). Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell 150, 1055–1067. doi: 10.1016/j.cell.2012.06.052
Liu, Z., Zhou, T., Ziegler, A. C., Dimitrion, P., and Zuo, L. (2017). Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017:2525967. doi: 10.1155/2017/2525967
Ljubuncic, P. S., Bar-Shai, M., and Reznick, A. Z. (2008). “The role of reactive nitrogen species (RNS) in the activation of nuclear factor κ B (NFkB) and its implications for biological systems: the question of balance,” in Oxidants in Biology: A Question of Balance, eds G. Valacchi and P. A. Davis (Dordrecht: Springer), 67–109.
Ljubuncic, P., Gochman, E., and Reznick, A. Z. (2010). “Nitrosative stress in aging—its importance and biological implications in NF-κB signaling,” in Aging and Age-Related Disorders, eds S. Bondy and K. Maiese (Totowa, NJ: Humana Press), 27–54.
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Luo, J., Lee, S. H., VandeVrede, L., Qin, Z., Ben Aissa, M., Larson, J., et al. (2016). A multifunctional therapeutic approach to disease modification in multiple familial mouse models and a novel sporadic model of Alzheimer’s disease. Mol. Neurodegener. 11:35. doi: 10.1186/s13024-016-0103-6
Luo, J., Lee, S. H., VandeVrede, L., Qin, Z., Piyankarage, S., Tavassoli, E., et al. (2015). Re-engineering a neuroprotective, clinical drug as a procognitive agent with high in vivo potency and with GABAA potentiating activity for use in dementia. BMC Neurosci. 16:67. doi: 10.1186/s12868-015-0208-9
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., et al. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 12:7. doi: 10.1186/s13024-017-0150-7
Mailloux, R. J., Jin, X., and Willmore, W. G. (2014). Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2, 123–139. doi: 10.1016/j.redox.2013.12.011
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547. doi: 10.1093/hmg/dds072
Marino, S. M., and Gladyshev, V. N. (2010). Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 395, 844–859. doi: 10.1016/j.jmb.2009.10.042
Marino, S. M., and Gladyshev, V. N. (2012). Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 287, 4419–4425. doi: 10.1074/jbc.R111.275578
Markesbery, W. R. (1997). Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 23, 134–147. doi: 10.1016/s0891-5849(96)00629-6
Matsusaki, M., Kanemura, S., Kinoshita, M., Lee, Y. H., Inaba, K., and Okumura, M. (2020). The protein disulfide isomerase family: from proteostasis to pathogenesis. Biochim. Biophys. Acta Gen. Subj. 1864:129338. doi: 10.1016/j.bbagen.2019.04.003
Mattson, M. P., and Arumugam, T. V. (2018). Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 27, 1176–1199. doi: 10.1016/j.cmet.2018.05.011
McCord, J. M. (2000). The evolution of free radicals and oxidative stress. Am. J. Med. 108, 652–659. doi: 10.1016/s0002-9343(00)00412-5
McDonagh, B., Sakellariou, G. K., Smith, N. T., Brownridge, P., and Jackson, M. J. (2014). Differential cysteine labeling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J. Proteome Res. 13, 5008–5021. doi: 10.1021/pr5006394
McLain, A. L., Cormier, P. J., Kinter, M., and Szweda, L. I. (2013). Glutathionylation of α-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 61, 161–169. doi: 10.1016/j.freeradbiomed.2013.03.020
Medinas, D. B., Rozas, P., Martínez Traub, F., Woehlbier, U., Brown, R. H., Bosco, D. A., et al. (2018). Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A 115, 8209–8214. doi: 10.1073/pnas.1801109115
Meng, F., Yao, D., Shi, Y., Kabakoff, J., Wu, W., Reicher, J., et al. (2011). Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 6:34. doi: 10.1186/1750-1326-6-34
Menger, K. E., James, A. M., Cochemé, H. M., Harbour, M. E., Chouchani, E. T., Ding, S., et al. (2015). Fasting, but not aging, dramatically alters the redox status of cysteine residues on proteins in Drosophila melanogaster. Cell Rep. 11, 1856–1865. doi: 10.1016/j.celrep.2015.05.033
Mieyal, J. J., Gallogly, M. M., Qanungo, S., Sabens, E. A., and Shelton, M. D. (2008). Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 10, 1941–1988. doi: 10.1089/ars.2008.2089
Miseta, A., and Csutora, P. (2000). Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 17, 1232–1239. doi: 10.1093/oxfordjournals.molbev.a026406
Mitsumoto, H., Santella, R. M., Liu, X., Bogdanov, M., Zipprich, J., Wu, H.-C., et al. (2008). Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 9, 177–183. doi: 10.1080/17482960801933942
Mnatsakanyan, R., Markoutsa, S., Walbrunn, K., Roos, A., Verhelst, S. H. L., and Zahedi, R. P. (2019). Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat. Commun. 10:2195. doi: 10.1038/s41467-019-10182-4
Mnatsakanyan, R., Shema, G., Basik, M., Batist, G., Borchers, C. H., Sickmann, A., et al. (2018). Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev. Proteomics 15, 515–535. doi: 10.1080/14789450.2018.1483340
Moldogazieva, N. T., Mokhosoev, I. M., Feldman, N. B., and Lutsenko, S. V. (2018). ROS and RNS signalling: adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 52, 507–543. doi: 10.1080/10715762.2018.1457217
Molin, M., Yang, J., Hanzén, S., Toledano, M. B., Labarre, J., and Nyström, T. (2011). Life span extension and H2O2 resistance elicited by caloric restriction require the peroxiredoxin tsa1 in Saccharomyces cerevisiae. Mol. Cell 43, 823–833. doi: 10.1016/j.molcel.2011.07.027
Molokanova, E., Akhtar, M. W., Sanz-Blasco, S., Tu, S., Piña-Crespo, J. C., McKercher, S. R., et al. (2014). Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J. Neurosci. 34, 5023–5028. doi: 10.1523/JNEUROSCI.2907-13.2014
Moore, J. J., and Saadabadi, A. (2020). “Selegiline,” in StatPearls, (Treasure Island, FL: StatPearls Publishing).
Mullen, L., Hanschmann, E.-M., Lillig, C. H., Herzenberg, L. A., and Ghezzi, P. (2015). Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 21, 98–108. doi: 10.2119/molmed.2015.00033
Muñoz, P., Ardiles, Á. O., Pérez-Espinosa, B., Núñez-Espinosa, C., Paula-Lima, A., González-Billault, C., et al. (2020). Redox modifications in synaptic components as biomarkers of cognitive status, in brain aging and disease. Mech. Ageing Dev. 189:111250. doi: 10.1016/j.mad.2020.111250
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/bj20081386
Murphy, M. P., Holmgren, A., Larsson, N.-G., Halliwell, B., Chang, C. J., Kalyanaraman, B., et al. (2011). Unraveling the biological roles of reactive oxygen species. Cell Metab. 13, 361–366. doi: 10.1016/j.cmet.2011.03.010
Musicco, C., Capelli, V., Pesce, V., Timperio, A. M., Calvani, M., Mosconi, L., et al. (2009). Accumulation of overoxidized Peroxiredoxin III in aged rat liver mitochondria. Biochim. Biophys. Acta 1787, 890–896. doi: 10.1016/j.bbabio.2009.03.002
Nakamura, T., and Lipton, S. A. (2016). Protein S-nitrosylation as a therapeutic target for neurodegenerative diseases. Trends Pharmacol. Sci. 37, 73–84. doi: 10.1016/j.tips.2015.10.002
Nakamura, T., and Lipton, S. A. (2017). ‘SNO’-storms compromise protein activity and mitochondrial metabolism in neurodegenerative disorders. Trends Endocrinol. Metab. 28, 879–892. doi: 10.1016/j.tem.2017.10.004
Nakamura, T., and Lipton, S. A. (2020). Nitric oxide-dependent protein post-translational modifications impair mitochondrial function and metabolism to contribute to neurodegenerative diseases. Antioxid. Redox Signal. 32, 817–833. doi: 10.1089/ars.2019.7916
Nakamura, T., Tu, S., Akhtar, M. W., Sunico, C. R., Okamoto, S.-I., and Lipton, S. A. (2013). Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78, 596–614. doi: 10.1016/j.neuron.2013.05.005
Nakato, R., Ohkubo, Y., Konishi, A., Shibata, M., Kaneko, Y., Iwawaki, T., et al. (2015). Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 5:14812. doi: 10.1038/srep14812
Naoi, M., Maruyama, W., and Shamoto-Nagai, M. (2020). Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate α-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J. Neural Transm. 127, 131–147. doi: 10.1007/s00702-020-02150-w
Navarro, A., and Boveris, A. (2004). Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1244–R1249. doi: 10.1152/ajpregu.00226.2004
Nayernia, Z., Jaquet, V., and Krause, K. H. (2014). New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 20, 2815–2837. doi: 10.1089/ars.2013.5703
Neumann, C. A., Krause, D. S., Carman, C. V., Das, S., Dubey, D. P., Abraham, J. L., et al. (2003). Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424, 561–565. doi: 10.1038/nature01819
Niforou, K., Cheimonidou, C., and Trougakos, I. P. (2014). Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2, 323–332. doi: 10.1016/j.redox.2014.01.017
Nyström, T., Yang, J., and Molin, M. (2012). Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 26, 2001–2008. doi: 10.1101/gad.200006.112
Oh, M. M., Oliveira, F. A., and Disterhoft, J. F. (2010). Learning and aging related changes in intrinsic neuronal excitability. Front. Aging Neurosci. 2:2. doi: 10.3389/neuro.24.002.2010
Okamoto, S., Nakamura, T., Cieplak, P., Chan, S. F., Kalashnikova, E., Liao, L., et al. (2014). S-nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep. 8, 217–228. doi: 10.1016/j.celrep.2014.06.005
Okamoto, S. I., Prikhodko, O., Pina-Crespo, J., Adame, A., McKercher, S. R., Brill, L. M., et al. (2019). NitroSynapsin for the treatment of neurological manifestations of tuberous sclerosis complex in a rodent model. Neurobiol. Dis. 127, 390–397. doi: 10.1016/j.nbd.2019.03.029
Oláhová, M., Taylor, S. R., Khazaipoul, S., Wang, J., Morgan, B. A., Matsumoto, K., et al. (2008). A redox-sensitive peroxiredoxin that is important for longevity has tissue- and stress-specific roles in stress resistance. Proc. Natl. Acad. Sci. U S A 105, 19839–19844. doi: 10.1073/pnas.0805507105
Oliver, D. M. A., and Reddy, P. H. (2019). Molecular basis of Alzheimer’s disease: focus on mitochondria. J. Alzheimers Dis. 72, S95–S116. doi: 10.3233/JAD-190048
Ortuño-Sahagún, D., Pallàs, M., and Rojas-Mayorquín, A. E. (2014). Oxidative stress in aging: advances in proteomic approaches. Oxid. Med. Cell. Longev. 2014:573208. doi: 10.1155/2014/573208
Pace, N. J., and Weerapana, E. (2013). Diverse functional roles of reactive cysteines. ACS Chem. Biol. 8, 283–296. doi: 10.1021/cb3005269
Pacifici, R. E., Kono, Y., and Davies, K. J. (1993). Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. J. Biol. Chem. 268, 15405–15411.
Pajares, M., Cuadrado, A., Engedal, N., Jirsova, Z., and Cahova, M. (2018). The role of free radicals in autophagy regulation: implications for ageing. Oxid. Med. Cell. Longev. 2018:2450748. doi: 10.1155/2018/2450748
Pajares, M., Jiménez-Moreno, N., Dias, I. H. K., Debelec, B., Vucetic, M., Fladmark, K. E., et al. (2015). Redox control of protein degradation. Redox Biol. 6, 409–420. doi: 10.1016/j.redox.2015.07.003
Pankratz, N., and Foroud, T. (2007). Genetics of Parkinson disease. Genet. Med. 9, 801–811. doi: 10.1097/gim.0b013e31815bf97c
Parakh, S., Shadfar, S., Perri, E. R., Ragagnin, A. M. G., Piattoni, C. V., Fogolín, M. B., et al. (2020). The redox activity of protein disulfide isomerase inhibits ALS phenotypes in cellular and zebrafish models. iScience 23:101097. doi: 10.1016/j.isci.2020.101097
Park, J. W., Piszczek, G., Rhee, S. G., and Chock, P. B. (2011). Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 50, 3204–3210. doi: 10.1021/bi101373h
Paul, B. D., Sbodio, J. I., and Snyder, S. H. (2018). Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol. Sci. 39, 513–524. doi: 10.1016/j.tips.2018.02.007
Pérez, M. J., Ponce, D. P., Osorio-Fuentealba, C., Behrens, M. I., and Quintanilla, R. A. (2017). Mitochondrial bioenergetics is altered in fibroblasts from patients with sporadic Alzheimer’s disease. Front. Neurosci. 11:553. doi: 10.3389/fnins.2017.00553
Pérez, V. I., Pierce, A., de Waal, E. M., Ward, W. F., Bokov, A., Chaudhuri, A., et al. (2010). “Chapter 8—detection and quantification of protein disulfides in biological tissues: a fluorescence-based proteomic approach,” in Methods in Enzymology, eds E. Cadenas and L. Packer (Academic Press), 161–177.
Perluigi, M., Di Domenico, F., Giorgi, A., Schininà, M. E., Coccia, R., Cini, C., et al. (2010). Redox proteomics in aging rat brain: involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J. Neurosci. Res. 88, 3498–3507. doi: 10.1002/jnr.22500
Perluigi, M., Swomley, A. M., and Butterfield, D. A. (2014). Redox proteomics and the dynamic molecular landscape of the aging brain. Ageing Res. Rev. 13, 75–89. doi: 10.1016/j.arr.2013.12.005
Peskin, A. V., Dickerhof, N., Poynton, R. A., Paton, L. N., Pace, P. E., Hampton, M. B., et al. (2013). Hyperoxidation of Peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 288, 14170–14177. doi: 10.1074/jbc.m113.460881
Peskin, A. V., Pace, P. E., Behring, J. B., Paton, L. N., Soethoudt, M., Bachschmid, M. M., et al. (2016). Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J. Biol. Chem. 291, 3053–3062. doi: 10.1074/jbc.m115.692798
Pham-Huy, L. A., He, H., and Pham-Huy, C. (2008). Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 4, 89–96.
Pomatto, L. C. D., and Davies, K. J. A. (2018). Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 124, 420–430. doi: 10.1016/j.freeradbiomed.2018.06.016
Poole, L. B., and Nelson, K. J. (2008). Discovering mechanisms of signaling-mediated cysteine oxidation. Curr. Opin. Chem. Biol. 12, 18–24. doi: 10.1016/j.cbpa.2008.01.021
Poon, H. F., Calabrese, V., Calvani, M., and Butterfield, D. A. (2006a). Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid. Redox Signal. 8, 381–394. doi: 10.1089/ars.2006.8.381
Poon, H. F., Shepherd, H. M., Reed, T. T., Calabrese, V., Stella, A. M., Pennisi, G., et al. (2006b). Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiol. Aging 27, 1020–1034. doi: 10.1016/j.neurobiolaging.2005.05.014
Poon, H. F., Castegna, A., Farr, S. A., Thongboonkerd, V., Lynn, B. C., Banks, W. A., et al. (2004). Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience 126, 915–926. doi: 10.1016/j.neuroscience.2004.04.046
Prokai, L., Yan, L. J., Vera-Serrano, J. L., Stevens, S. M. Jr., and Forster, M. J. (2007). Mass spectrometry-based survey of age-associated protein carbonylation in rat brain mitochondria. J. Mass Spectrom. 42, 1583–1589. doi: 10.1002/jms.1345
Przedborski, S., Chen, Q., Vila, M., Giasson, B. I., Djaldatti, R., Vukosavic, S., et al. (2001). Oxidative post-translational modifications of α-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Neurochem. 76, 637–640. doi: 10.1046/j.1471-4159.2001.00174.x
Qin, Z., Luo, J., VandeVrede, L., Tavassoli, E., Fa, M., Teich, A. F., et al. (2012). Design and synthesis of neuroprotective methylthiazoles and modification as NO-chimeras for neurodegenerative therapy. J. Med. Chem. 55, 6784–6801. doi: 10.1021/jm300353r
Rachakonda, G., Xiong, Y., Sekhar, K. R., Stamer, S. L., Liebler, D. C., and Freeman, M. L. (2008). Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 21, 705–710. doi: 10.1021/tx700302s
Raha, S., and Robinson, B. H. (2000). Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 25, 502–508. doi: 10.1016/s0968-0004(00)01674-1
Raimondi, D., Orlando, G., and Vranken, W. F. (2014). Clustering-based model of cysteine co-evolution improves disulfide bond connectivity prediction and reduces homologous sequence requirements. Bioinformatics 31, 1219–1225. doi: 10.1093/bioinformatics/btu794
Rainwater, R., Parks, D., Anderson, M. E., Tegtmeyer, P., and Mann, K. (1995). Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 15, 3892–3903. doi: 10.1128/mcb.15.7.3892
Raju, K., Doulias, P.-T., Evans, P., Krizman, E. N., Jackson, J. G., Horyn, O., et al. (2015). Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal. 8:ra68. doi: 10.1126/scisignal.aaa4312
Randall, L. M., Dalla Rizza, J., Parsonage, D., Santos, J., Mehl, R. A., Lowther, W. T., et al. (2019). Unraveling the effects of peroxiredoxin 2 nitration; role of C-terminal tyrosine 193. Free Radic. Biol. Med. 141, 492–501. doi: 10.1016/j.freeradbiomed.2019.07.016
Randall, L. M., Manta, B., Hugo, M., Gil, M., Batthyàny, C., Trujillo, M., et al. (2014). Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J. Biol. Chem. 289, 15536–15543. doi: 10.1074/jbc.m113.539213
Ravera, S., Podestà, M., Sabatini, F., Dagnino, M., Cilloni, D., Fiorini, S., et al. (2019). Discrete changes in glucose metabolism define aging. Sci. Rep. 9:10347. doi: 10.1038/s41598-019-46749-w
Ray, P. D., Huang, B.-W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990. doi: 10.1016/j.cellsig.2012.01.008
Reddie, K. G., and Carroll, K. S. (2008). Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 12, 746–754. doi: 10.1016/j.cbpa.2008.07.028
Reyes, J. F., Fu, Y., Vana, L., Kanaan, N. M., and Binder, L. I. (2011). Tyrosine nitration within the proline-rich region of tau in Alzheimer’s disease. Am. J. Pathol. 178, 2275–2285. doi: 10.1016/j.ajpath.2011.01.030
Reynaert, N. L., van der Vliet, A., Guala, A. S., McGovern, T., Hristova, M., Pantano, C., et al. (2006). Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. U S A 103, 13086–13091. doi: 10.1073/pnas.0603290103
Reynolds, M. R., Reyes, J. F., Fu, Y., Bigio, E. H., Guillozet-Bongaarts, A. L., Berry, R. W., et al. (2006). Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer’s disease and other tauopathies. J. Neurosci. 26, 10636–10645. doi: 10.1523/jneurosci.2143-06.2006
Rhee, S. G., and Kil, I. S. (2017). Multiple functions and regulation of mammalian peroxiredoxins. Annu. Rev. Biochem. 86, 749–775. doi: 10.1146/annurev-biochem-060815-014431
Rhee, S. G., and Woo, H. A. (2011). Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2 and protein chaperones. Antioxid. Redox Signal. 15, 781–794. doi: 10.1089/ars.2010.3393
Riederer, I. M., Schiffrin, M., Kövari, E., Bouras, C., and Riederer, B. M. (2009). Ubiquitination and cysteine nitrosylation during aging and Alzheimer’s disease. Brain Res. Bull. 80, 233–241. doi: 10.1016/j.brainresbull.2009.04.018
Ristow, M. (2014). Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 20, 709–711. doi: 10.1038/nm.3624
Ristow, M., and Schmeisser, K. (2014). Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12, 288–341. doi: 10.2203/dose-response.13-035.ristow
Rizza, S., Cardaci, S., Montagna, C., Di Giacomo, G., De Zio, D., Bordi, M., et al. (2018). S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. U S A 115, E3388–E3397. doi: 10.1073/pnas.1722452115
Robey, T. T., and Panegyres, P. K. (2019). Cerebrospinal fluid biomarkers in neurodegenerative disorders. Future Neurol. 14:FNL6. doi: 10.2217/fnl-2018-0029
Roher, A. E., Kokjohn, T. A., Clarke, S. G., Sierks, M. R., Maarouf, C. L., Serrano, G. E., et al. (2017). APP/Aβ structural diversity and Alzheimer’s disease pathogenesis. Neurochem. Int. 110, 1–13. doi: 10.1016/j.neuint.2017.08.007
Roos, G., Foloppe, N., and Messens, J. (2013). Understanding the pKa of redox cysteines: the key role of hydrogen bonding. Antioxid. Redox Signal. 18, 94–127. doi: 10.1089/ars.2012.4521
Ryan, B. J., Nissim, A., and Winyard, P. G. (2014). Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases. Redox Biol. 2, 715–724. doi: 10.1016/j.redox.2014.05.004
Sabens Liedhegner, E. A., Gao, X. H., and Mieyal, J. J. (2012). Mechanisms of altered redox regulation in neurodegenerative diseases—focus on S—glutathionylation. Antioxid. Redox Signal. 16, 543–566. doi: 10.1089/ars.2011.4119
Saccoccia, F., Di Micco, P., Boumis, G., Brunori, M., Koutris, I., Miele, A. E., et al. (2012). Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure 20, 429–439. doi: 10.1016/j.str.2012.01.004
Sagot, Y., Toni, N., Perrelet, D., Lurot, S., King, B., Rixner, H., et al. (2000). An orally active anti-apoptotic molecule (CGP 3466B) preserves mitochondria and enhances survival in an animal model of motoneuron disease. Br. J. Pharmacol. 131, 721–728. doi: 10.1038/sj.bjp.0703633
Salzano, S., Checconi, P., Hanschmann, E.-M., Lillig, C. H., Bowler, L. D., Chan, P., et al. (2014). Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. U S A 111, 12157–12162. doi: 10.1073/pnas.1401712111
Santos, A. L., and Lindner, A. B. (2017). Protein posttranslational modifications: roles in aging and age-related disease. Oxid. Med. Cell. Longev. 2017:5716409. doi: 10.1155/2017/5716409
Savyon, M., and Engelender, S. (2020). SUMOylation in α-synuclein homeostasis and pathology. Front. Aging Neurosci. 12:167. doi: 10.3389/fnagi.2020.00167
Sawa, A., Khan, A. A., Hester, L. D., and Snyder, S. H. (1997). Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc. Natl. Acad. Sci. U S A 94, 11669–11674. doi: 10.1073/pnas.94.21.11669
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2019). Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 30, 1450–1499. doi: 10.1089/ars.2017.7321
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2019). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 38:e101812. doi: 10.15252/embj.2019101812
Schöneich, C. (2011). Cysteine residues as catalysts for covalent peptide and protein modification: a role for thiyl radicals? Biochem. Soc. Trans. 39, 1254–1259. doi: 10.1042/bst0391254
Schreck, R., Rieber, P., and Baeuerle, P. A. (1991). Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κ B transcription factor and HIV-1. EMBO J. 10, 2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x
Schulz, E., Wenzel, P., Münzel, T., and Daiber, A. (2014). Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 20, 308–324. doi: 10.1089/ars.2012.4609
Sen, N., Hara, M. R., Kornberg, M. D., Cascio, M. B., Bae, B. I., Shahani, N., et al. (2008). Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10, 866–873. doi: 10.1038/ncb1747
Sen, T., Saha, P., and Sen, N. (2018). Nitrosylation of GAPDH augments pathological tau acetylation upon exposure to amyloid-β. Sci. Signal. 11:eaao6765. doi: 10.1126/scisignal.aao6765
Shen, L., Chen, C., Yang, A., Chen, Y., Liu, Q., and Ni, J. (2015). Redox proteomics identification of specifically carbonylated proteins in the hippocampi of triple transgenic Alzheimer’s disease mice at its earliest pathological stage. J. Proteomics 123, 101–113. doi: 10.1016/j.jprot.2015.04.005
Sies, H. (2019). “Chapter 13—oxidative stress: eustress and distress in redox homeostasis,” in Stress: Physiology, Biochemistry and Pathology, ed. G. Fink (Cambridge, MA: Academic Press), 153–163.
Simon, L., Szilágyi, G., Bori, Z., Orbay, P., and Nagy, Z. (2001). (−)-d-Deprenyl attenuates apoptosis in experimental brain ischaemia. Eur. J. Pharmacol. 430, 235–241. doi: 10.1016/s0014-2999(01)01375-9
Siqueira, I. R., Fochesatto, C., de Andrade, A., Santos, M., Hagen, M., Bello-Klein, A., et al. (2005). Total antioxidant capacity is impaired in different structures from aged rat brain. Int. J. Dev. Neurosci. 23, 663–671. doi: 10.1016/j.ijdevneu.2005.03.001
Smith, C. D., Carney, J. M., Starke-Reed, P. E., Oliver, C. N., Stadtman, E. R., Floyd, R. A., et al. (1991). Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. U S A 88, 10540–10543. doi: 10.1073/pnas.88.23.10540
Song, I.-K., Lee, J.-J., Cho, J.-H., Jeong, J., Shin, D.-H., and Lee, K.-J. (2016). Degradation of redox-sensitive proteins including peroxiredoxins and dj-1 is promoted by oxidation-induced conformational changes and ubiquitination. Sci. Rep. 6:34432. doi: 10.1038/srep34432
Stadtman, E. R. (1988). Protein modification in aging. J. Gerontol. 43, B112–B120. doi: 10.1093/geronj/43.5.b112
Stadtman, E. R., and Levine, R. L. (2003). Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25, 207–218. doi: 10.1007/s00726-003-0011-2
Stamler, J. S., Toone, E. J., Lipton, S. A., and Sucher, N. J. (1997). (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18, 691–696. doi: 10.1016/s0896-6273(00)80310-4
Stefanis, L. (2012). α-Synuclein in Parkinson’s disease. Cold Spring Harb. perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Sultana, R., and Butterfield, D. A. (2013). Oxidative modification of brain proteins in Alzheimer’s disease: perspective on future studies based on results of redox proteomics studies. J. Alzheimers Dis. 33, S243–S251. doi: 10.3233/jad-2012-129018
Sultana, R., Mecocci, P., Mangialasche, F., Cecchetti, R., Baglioni, M., and Butterfield, D. A. (2011a). Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J. Alzheimers Dis. 24, 77–84. doi: 10.3233/jad-2011-101425
Sultana, R., Robinson, R. A. S., Di Domenico, F., Abdul, H. M., St Clair, D. K., Markesbery, W. R., et al. (2011b). Proteomic identification of specifically carbonylated brain proteins in APP(NLh)/APP(NLh) × PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice model of Alzheimer disease as a function of age. J. Proteomics 74, 2430–2440. doi: 10.1016/j.jprot.2011.06.015
Sultana, R., Perluigi, M., Newman, S. F., Pierce, W. M., Cini, C., Coccia, R., et al. (2010). Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease. Antioxid. Redox Signal. 12, 327–336. doi: 10.1089/ars.2009.2810
Sun, M.-a., Zhang, Q., Wang, Y., Ge, W., and Guo, D. (2016). Prediction of redox-sensitive cysteines using sequential distance and other sequence-based features. BMC Bioinformatics 17:316. doi: 10.1186/s12859-016-1185-4
Sunico, C. R., Nakamura, T., Rockenstein, E., Mante, M., Adame, A., Chan, S. F., et al. (2013). S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 8:29. doi: 10.1186/1750-1326-8-29
Sunico, C. R., Sultan, A., Nakamura, T., Dolatabadi, N., Parker, J., Shan, B., et al. (2016). Role of sulfiredoxin as a peroxiredoxin-2 denitrosylase in human iPSC-derived dopaminergic neurons. Proc. Natl. Acad. Sci. U S A 113, E7564–E7571. doi: 10.1073/pnas.1608784113
Svistunova, D. M., Simon, J. N., Rembeza, E., Crabtree, M., Yue, W. W., Oliver, P. L., et al. (2019). Oxidation resistance 1 regulates post-translational modifications of peroxiredoxin 2 in the cerebellum. Free Radic. Biol. Med. 130, 151–162. doi: 10.1016/j.freeradbiomed.2018.10.447
Takahashi, H., Shin, Y., Cho, S. J., Zago, W. M., Nakamura, T., Gu, Z., et al. (2007). Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron 53, 53–64. doi: 10.1016/j.neuron.2006.11.023
Takahashi, H., Xia, P., Cui, J., Talantova, M., Bodhinathan, K., Li, W., et al. (2015). Pharmacologically targeted NMDA receptor antagonism by NitroMemantine for cerebrovascular disease. Sci. Rep. 5:14781. doi: 10.1038/srep14781
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S., et al. (2013). Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation and synaptic loss. Proc. Natl. Acad. Sci. U S A 110, E2518–2527. doi: 10.1073/pnas.1306832110
Tegeder, I. (2019). Nitric oxide mediated redox regulation of protein homeostasis. Cell. Signal. 53, 348–356. doi: 10.1016/j.cellsig.2018.10.019
Terman, A., and Brunk, U. T. (2004). Lipofuscin. Int. J. Biochem. Cell Biol. 36, 1400–1404. doi: 10.1016/j.biocel.2003.08.009
Timmermann, B., Jarolim, S., Russmayer, H., Kerick, M., Michel, S., Krüger, A., et al. (2010). A new dominant peroxiredoxin allele identified by whole-genome re-sequencing of random mutagenized yeast causes oxidant-resistance and premature aging. Aging 2, 475–486. doi: 10.18632/aging.100187
Tomin, T., Schittmayer, M., Honeder, S., Heininger, C., and Birner-Gruenberger, R. (2019). Irreversible oxidative post-translational modifications in heart disease. Expert Rev. Proteomics 16, 681–693. doi: 10.1080/14789450.2019.1645602
Topf, U., Suppanz, I., Samluk, L., Wrobel, L., Böser, A., Sakowska, P., et al. (2018). Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 9:324. doi: 10.1038/s41467-017-02694-8
Trachootham, D., Lu, W., Ogasawara, M. A., Nilsa, R.-D. V., and Huang, P. (2008). Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374. doi: 10.1089/ars.2007.1957
Tramutola, A., Lanzillotta, C., Perluigi, M., and Butterfield, D. A. (2017). Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 133, 88–96. doi: 10.1016/j.brainresbull.2016.06.005
Trushina, E., Nemutlu, E., Zhang, S., Christensen, T., Camp, J., Mesa, J., et al. (2012). Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One 7:e32737. doi: 10.1371/journal.pone.0032737
Tu, S., Akhtar, M. W., Escorihuela, R. M., Amador-Arjona, A., Swarup, V., Parker, J., et al. (2017). NitroSynapsin therapy for a mouse MEF2C haploinsufficiency model of human autism. Nat. Commun. 8:1488. doi: 10.1038/s41467-017-01563-8
Turano, C., Coppari, S., Altieri, F., and Ferraro, A. (2002). Proteins of the PDI family: unpredicted non-ER locations and functions. J. Cell. Physiol. 193, 154–163. doi: 10.1002/jcp.10172
Uehara, T., Nakamura, T., Yao, D., Shi, Z.-Q., Gu, Z., Ma, Y., et al. (2006). S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517. doi: 10.1038/nature04782
Valle, C., and Carrì, M. T. (2017). Cysteine modifications in the pathogenesis of ALS. Front. Mol. Neurosci. 10:5. doi: 10.3389/fnmol.2017.00005
van der Reest, J., Lilla, S., Zheng, L., Zanivan, S., and Gottlieb, E. (2018). Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 9:1581. doi: 10.1038/s41467-018-04003-3
Vandevrede, L., Tavassoli, E., Luo, J., Qin, Z., Yue, L., Pepperberg, D. R., et al. (2014). Novel analogues of chlormethiazole are neuroprotective in four cellular models of neurodegeneration by a mechanism with variable dependence on GABAA receptor potentiation. Br. J. Pharmacol. 171, 389–402. doi: 10.1111/bph.12454
Venkateshappa, C., Harish, G., Mahadevan, A., Srinivas Bharath, M. M., and Shankar, S. K. (2012). Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer’s disease. Neurochem. Res. 37, 1601–1614. doi: 10.1007/s11064-012-0755-8
von Bernhardi, R., Eugenin-von Bernhardi, L., and Eugenin, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Waldmeier, P. C., Boulton, A. A., Cools, A. R., Kato, A. C., and Tatton, W. G. (2000). Neurorescuing effects of the GAPDH ligand CGP 3466B. J. Neural Transm. Suppl. 60, 197–214. doi: 10.1007/978-3-7091-6301-6_13
Walker, A. K., and Atkin, J. D. (2011). Mechanisms of neuroprotection by protein disulphide isomerase in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011:317340. doi: 10.1155/2011/317340
Walker, A. K., Farg, M. A., Bye, C. R., McLean, C. A., Horne, M. K., and Atkin, J. D. (2009). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
Wall, S. B., Oh, J.-Y., Diers, A. R., and Landar, A. (2012). Oxidative modification of proteins: an emerging mechanism of cell signaling. Front. Physiol. 3:369. doi: 10.3389/fphys.2012.00369
Wang, Y., Eu, J., Washburn, M., Gong, T., Chen, H. S., James, W. L., et al. (2006). The pharmacology of aminoadamantane nitrates. Curr. Alzheimer Res. 3, 201–204. doi: 10.2174/156720506777632808
Wang, S. B., Murray, C. I., Chung, H. S., and Van Eyk, J. E. (2013). Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med. 23, 14–18. doi: 10.1016/j.tcm.2012.08.005
Wang, S., Song, J., Tan, M., Albers, K. M., and Jia, J. (2012). Mitochondrial fission proteins in peripheral blood lymphocytes are potential biomarkers for Alzheimer’s disease. Eur. J. Neurol. 19, 1015–1022. doi: 10.1111/j.1468-1331.2012.03670.x
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wang, Y., Sørensen, M. G., Zheng, Q., Zhang, C., Karsdal, M. A., and Henriksen, K. (2012a). Will posttranslational modifications of brain proteins provide novel serological markers for dementias? Int. J. Alzheimers Dis. 2012:209409. doi: 10.1155/2012/209409
Wang, Y., Yang, J., and Yi, J. (2012b). Redox sensing by proteins: oxidative modifications on cysteines and the consequent events. Antioxid. Redox Signal. 16, 649–657. doi: 10.1089/ars.2011.4313
Wang, C., Yu, J., Huo, L., Wang, L., Feng, W., and Wang, C.-C. (2012). Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a’. J. Biol. Chem. 287, 1139–1149. doi: 10.1074/jbc.m111.303149
Wani, R., Nagata, A., and Murray, B. W. (2014). Protein redox chemistry: post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front. Pharmacol. 5:224. doi: 10.3389/fphar.2014.00224
Weng, S.-L., Huang, K.-Y., Kaunang, F. J., Huang, C.-H., Kao, H.-J., Chang, T.-H., et al. (2017). Investigation and identification of protein carbonylation sites based on position-specific amino acid composition and physicochemical features. BMC Bioinformatics 18:66. doi: 10.1186/s12859-017-1472-8
Wilby, M. J., and Hutchinson, P. J. (2004). The pharmacology of chlormethiazole: a potential neuroprotective agent? CNS Drug Rev. 10, 281–294. doi: 10.1111/j.1527-3458.2004.tb00028.x
Willems, P. H., Rossignol, R., Dieteren, C. E., Murphy, M. P., and Koopman, W. J. (2015). Redox homeostasis and mitochondrial dynamics. Cell Metab. 22, 207–218. doi: 10.1016/j.cmet.2015.06.006
Wong, C.-M., Marcocci, L., Das, D., Wang, X., Luo, H., Zungu-Edmondson, M., et al. (2013). Mechanism of protein decarbonylation. Free Radic. Biol. Med. 65, 1126–1133. doi: 10.1016/j.freeradbiomed.2013.09.005
Woo, H. A., Jeong, W., Chang, T.-S., Park, K. J., Park, S. J., Yang, J. S., et al. (2005). Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 280, 3125–3128. doi: 10.1074/jbc.C400496200
Wood, Z. A., Schröder, E., Robin Harris, J., and Poole, L. B. (2003). Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 28, 32–40. doi: 10.1016/s0968-0004(02)00003-8
Xiao, H., Jedrychowski, M. P., Schweppe, D. K., Huttlin, E. L., Yu, Q., Heppner, D. E., et al. (2020). A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 180, 968.e24–983.e24. doi: 10.1016/j.cell.2020.02.012
Xu, R., Serritella, A. V., Sen, T., Farook, J. M., Sedlak, T. W., Baraban, J., et al. (2013). Behavioral effects of cocaine mediated by nitric oxide-GAPDH transcriptional signaling. Neuron 78, 623–630. doi: 10.1016/j.neuron.2013.03.021
Yakovlev, V. A., Barani, I. J., Rabender, C. S., Black, S. M., Leach, J. K., Graves, P. R., et al. (2007). Tyrosine nitration of IκBα: a novel mechanism for NF-κB activation. Biochemistry 46, 11671–11683. doi: 10.1021/bi701107z
Yan, L. J., Sumien, N., Thangthaeng, N., and Forster, M. J. (2013). Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic. Res. 47, 123–133. doi: 10.3109/10715762.2012.752078
Yao, D., Gu, Z., Nakamura, T., Shi, Z.-Q., Ma, Y., Gaston, B., et al. (2004). Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U S A 101, 10810–10814. doi: 10.1073/pnas.0405661101
Yap, L.-P., Garcia, J. V., Han, D., and Cadenas, E. (2009). The energy-redox axis in aging and age-related neurodegeneration. Adv. Drug Deliv. Rev. 61, 1283–1298. doi: 10.1016/j.addr.2009.07.015
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 100, 108–122. doi: 10.1016/j.freeradbiomed.2016.04.200
Yu, Q., Sali, A., Van der Meulen, J., Creeden, B. K., Gordish-Dressman, H., Rutkowski, A., et al. (2013). Omigapil treatment decreases fibrosis and improves respiratory rate in dy(2J) mouse model of congenital muscular dystrophy. PLoS One 8:e65468. doi: 10.1371/journal.pone.0065468
Yun, J., and Finkel, T. (2014). Mitohormesis. Cell Metab. 19, 757–766. doi: 10.1016/j.cmet.2014.01.011
Zahid, S., Khan, R., Oellerich, M., Ahmed, N., and Asif, A. R. (2014). Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 256, 126–136. doi: 10.1016/j.neuroscience.2013.10.026
Zhang, G., Li, J., Purkayastha, S., Tang, Y., Zhang, H., Yin, Y., et al. (2013). Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 497, 211–216. doi: 10.1038/nature12143
Keywords: redox, post-translational modifications, cysteine residues, aging, neurodegenerative diseases, brain, S-nitrosation
Citation: Finelli MJ (2020) Redox Post-translational Modifications of Protein Thiols in Brain Aging and Neurodegenerative Conditions—Focus on S-Nitrosation. Front. Aging Neurosci. 12:254. doi: 10.3389/fnagi.2020.00254
Received: 30 April 2020; Accepted: 24 July 2020;
Published: 03 September 2020.
Edited by:
Bindu Diana Paul, Johns Hopkins University, United StatesReviewed by:
Ana Denicola, Universidad de la República, UruguayGuoyuan Qi, University of Arizona, United States
John J. Mieyal, Case Western Reserve University, United States
Copyright © 2020 Finelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mattéa J. Finelli, bWF0dGVhLmZpbmVsbGlAbm90dGluZ2hhbS5hYy51aw==