 Eitan Wong
Eitan Wong Georgia R. Frost
Georgia R. Frost Yue-Ming Li
Yue-Ming Li- Chemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY, United States
Described as the “proteasome of the membrane” or the “scissors in the membrane,” γ-secretase has notoriously complicated biology, and even after decades of research, the full extent of its regulatory mechanism remains unclear. γ-Secretase is an intramembrane aspartyl protease complex composed of four obligatory subunits: Nicastrin (NCT), Presenilin (PS), Presenilin Enhancer-2 (Pen-2), and Anterior pharynx-defective-1 (Aph-1). γ-Secretase cleaves numerous type 1 transmembrane substrates, with no apparent homology, and plays major roles in broad biological pathways such as development, neurogenesis, and cancer. Notch and the amyloid precursor protein (APP) and are undoubtedly the best-studied γ-secretase substrates because of their role in cancer and Alzheimer’s disease (AD) and therefore became the focus of increasing studies as an attractive therapeutic target. The regulation of γ-secretase is intricate and involves the function of multiple cellular entities. Recently, γ-secretase modulatory proteins (GSMPs), which are non-essential subunits and yet modulate γ-secretase activity and specificity, have emerged as an important component in guiding γ-secretase. GSMPs are responsive to cellular and environmental changes and therefore, provide another layer of regulation of γ-secretase. This type of enzymatic regulation allows for a rapid and fine-tuning of γ-secretase activity when appropriate signals appear enabling a temporal level of regulation. In this review article, we discuss the latest developments on GSMPs and implications on the development of effective therapeutics for γ-secretase-associated diseases such as AD and cancer.
γ-Secretase and Alzheimer Disease (AD)
Alzheimer’s disease (AD) is the most common neurodegenerative disease manifested in dementia symptoms that gradually worsen with age. AD pathology is characterized by the presence of extracellular plaques and intracellular neurofibrillary tangles in the brain. According to the amyloid cascade hypothesis (Hardy and Higgins, 1992; Hardy and Selkoe, 2002), β-amyloid peptide (Aβ), the major constituent of plaques, is the causative mediator driving the deterioration and neuropathology of AD. The Aβ peptides are derived from amyloid precursor protein (APP) after processing by two proteases, first, β-secretase (BACE1) producing the β-C-terminal fragment (βCTF), and second, γ-secretase cleavage, creating an amyloid intracellular domain (AICD) and Aβ of different lengths (Crump et al., 2013).
AD can be classified into two major types: familial AD (FAD) and sporadic AD (SAD; Bateman et al., 2011). The pathological features and functional connectivity of both forms of AD are similar (Bateman et al., 2013; Thomas et al., 2014), suggesting that FAD can be an effective model to study the pathogenic mechanism of SAD. Mutations in one of three genes, APP, presenilin-1 (PSEN1), and presenilin-2 (PSEN2), or duplication of APP cause FAD (Goate et al., 1991; Levy-Lahad et al., 1995; Sherrington et al., 1995). Presenilin-1 (PS1) and presenilin-2 (PS2) are polytopic membrane proteins with nine transmembrane domains that are synthesized as a single polypeptide (Spasic et al., 2006). Both the PS1 and PS2 proteins are endoproteolyzed, whereby the N- and C-terminal cleavage products (NTF and CTF) remain associated in the heterodimeric form (Thinakaran et al., 1996).
The discovery of PS has provided critical insight into the identity of γ-secretase. (1) FAD mutations in PSEN1 and PSEN2 increase in the ratio of Aβ42 to Aβ40 in transfected cells and in transgenic mice (Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996; De Strooper, 2007; Wolfe, 2007). (2) Cultured neurons isolated from PS1-deficient mice produce significantly less Aβ and APP fragments that are not processed by γ-secretase is being accumulated (Naruse et al., 1998; De Strooper et al., 1998). (3) γ-Secretase activity is abolished in cells cultured from PS1 and PS2 deficient mice (Herreman et al., 2000; Zhang et al., 2000). (4) γ-Secretase activity is reduced by mutagenesis of two conserved aspartate residues in the transmembrane regions of PS1 (Wolfe et al., 1999). (5) γ-Secretase activity is connected to PS-containing macromolecular complexes (Li et al., 2000a). (6) Active-site directed γ-secretase inhibitors directly bind to PS1 or PS2 (Esler et al., 2000; Li et al., 2000b; Xu et al., 2002). (7) A recombinant PS1 variant alone in the purified reconstitution system has γ-secretase activity, offering the final proof that PS is γ-secretase (Ahn et al., 2010).
γ-Secretase Essential Subunits and Basic Regulation
A broad range of γ-secretase substrates has been identified (Haapasalo and Kovacs, 2011), indicating the diverse biological functions of this protease. The most notable substrate of γ-secretase is APP, the precursor for Aβ peptides, which aggregates to form the core of senile plaque in AD patients (Guo et al., 2020). Another critical substrate which has been extensively investigated is Notch, a cell surface protein, which participates in cell-cell contact signaling and is essential for propriate embryonic development (De Strooper et al., 1999). Notch plays an important role in different types of cancer (Lobry et al., 2011), therefore, γ-secretase is an appealing drug target in both AD and cancer. However, because of the wide spectrum of γ-secretase substrates, it is extremely difficult to develop selective inhibitors, as evidenced by the failure of γ-secretase inhibitors in clinical trials due to side effects from non-selective inhibition (Haapasalo and Kovacs, 2011).
It is known that γ-secretase is an intramembrane protease dependent on the assembly of four subunits: Nicastrin (NCT), PS, Presenilin Enhancer-2 (Pen-2), and Anterior pharynx-defective-1 (Aph-1; De Strooper et al., 2012; Crump et al., 2013). Structures of human γ-secretase complex alone or with substrates obtained by cryo-electron microscopy (cryo-EM), have offered novel insights into the structural basis of function and recognition as well as the flexibility and complexity of this enzyme (Lu et al., 2014; Sun et al., 2015; Yang et al., 2019; Zhou et al., 2019). Also, the endogenous γ-secretase complexes appear to range in size with different levels of activity (Gu et al., 2004; Evin et al., 2005), suggesting that different subunit stoichiometry and cofactors composition might contribute to activity and substrate specificity (Placanica et al., 2010). The mechanisms by which γ-secretase is being regulated at the cellular level have been the focus of extensive studies that uncover in part its intricate biology.
The regulation of γ-secretase starts with the synthesis and assembly of the complex. The correct assembly of the γ-secretase complex is tightly regulated and requires multiple cellular events to generate the mature and active form (Takasugi et al., 2003; Kim et al., 2004). While the basic functional γ-secretase complex requires at least one of each of its essential subunits (Sato et al., 2007), the existence of PS and Aph-1 isoforms (PS1/PS2 and Aph-1a/Aph-1b), as well as splice variants (Aph-1aS and Aph-1aL), introduces further permutations to the basic complex (Lai et al., 2003; Shirotani et al., 2004). Furthermore, different variations of the γ-secretase complex can exist in the same tissue and even in the same cells (Placanica et al., 2009a, b).
Compartmentalization between γ-secretase and its different substrates in distinct cellular domains likely plays a role in γ-secretase substrate availability. Rather than altering γ-secretase activity to favor one substrate over another, spatial separation offers a post-translational regulation mechanism. For example, APP is processed by intracellular γ-secretase, while notch, which functions in the plasma membrane, is processed at the cell surface (Tarassishin et al., 2004). Multiple lines of evidence also suggest that lipid composition and lipid rafts which simulate the different cellular compartments directly affect the cleavage and distribution of γ-secretase. Changes to lipid composition in the membrane also impact substrate processing (Osenkowski et al., 2008) and in particular cholesterol-rich membranes are the major site of Aβ production (Wahrle et al., 2002; Marquer et al., 2014). Interestingly, a recent report suggests that not only are APP and Notch processing modulated by membrane lateral organization but also that γ-secretase can actively recruit specific membrane components that create a lipid environment favorable for substrate recognition and activity (Barros et al., 2020).
The Context-Dependent γ-Secretase Modulatory Proteins (GSMPs)
The regulation of γ-secretase has been and continues to be, a major question in the field because of its numerous substrates that regulate many biological processes ranging from neuronal development, angiogenesis to tumorigenesis (Jurisch-Yaksi et al., 2013). While the expression of the essential γ-secretase components is ubiquitous in all tissues, only a small percentage of γ-secretase complexes are catalytically active (Lai et al., 2003). The existence of the inactive complex raises three critical questions: (1) Why does the inactive complex exist? (2) Can the inactive complex be activated? and (3) Does the activation play a role in AD and cancer? Furthermore, since the gene expression and the protein level of PS are not correlated with enzymatic activity in cells, it is challenging to assess the active γ-secretase complex (Lai et al., 2003; Placanica et al., 2009a). Therefore, the development of accurate γ-secretase activity assays (Shelton et al., 2009a, b), as well as photoaffinity labeling using a transition state inhibitor, are valuable tools to study γ-secretase (Crump et al., 2013; Nie et al., 2020).
The notion that an abundant protease like γ-secretase must respond to changes in the environment has led to the hypothesis that a fraction of the γ-secretase complexes exist in a partially dormant state and can be activated and attenuated by binding of γ-secretase modulatory proteins (GSMPs; Figure 1). Numerous proteins had been identified to interact with the core complex and impact γ-secretase activity (Chen et al., 2006; Vetrivel et al., 2007; Gertsik et al., 2014; Villa et al., 2014; Wong et al., 2019; Hur et al., 2020; Jung et al., 2020). In this review article, we will focus on the recent progress GSMPs and their regulation.
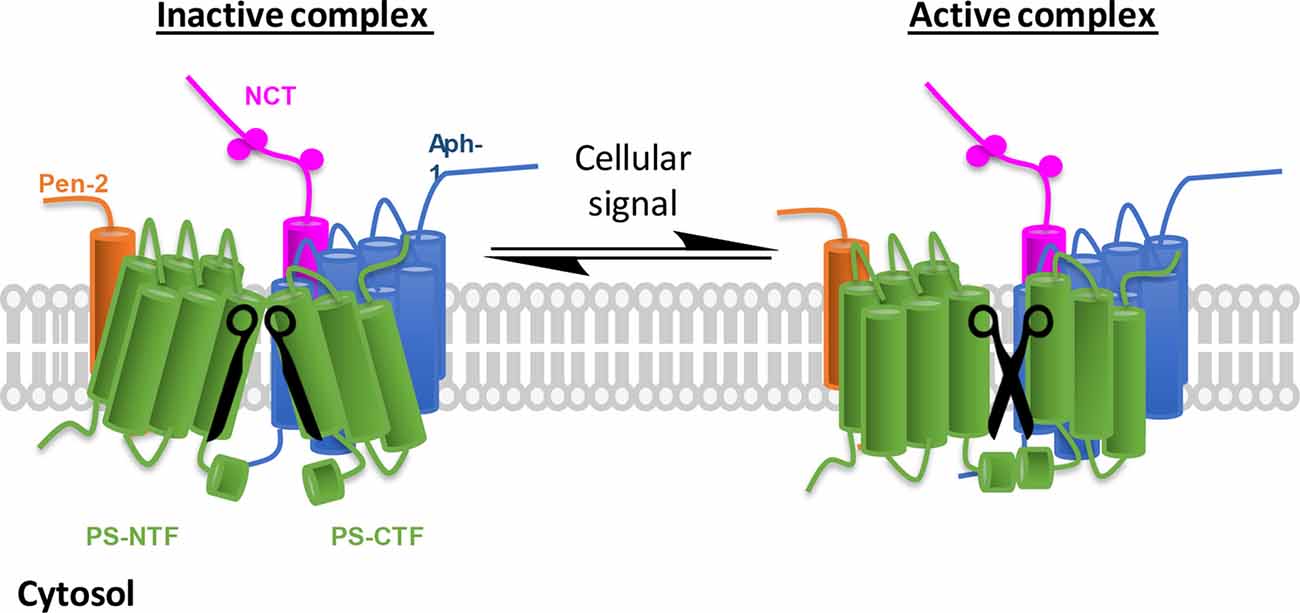
Figure 1. γ-Secretase complex activation in response to cellular environmental changes. Schematic representation of inactive/active γ-secretase equilibrium. γ-Secretase obligatory subunits are abundantly expressed in all tissues and cells, however, most of the γ-secretase complexes are not catalytically active (left, the two blades of scissors are not at correct position). To adapt cellular changes, γ-secretase can be activated by turning the inactive complexes to active ones (right, functional scissors) that selectively promote the cleavage of a specific substrate. The implication is 2-fold: first, inactive γ-secretase complexes are physiologically important, and second, γ-secretase can be temporally regulated.
The Missing Link Between Neuroinflammation and γ-Secretase in AD
Although genetic and animal studies support that Aβ peptides play a causative role in AD, the physiological role of Aβ is not clear. Recent studies suggest that Aβ peptides have antimicrobial and antiviral properties and function as part of the innate immune response of the brain (Vijaya Kumar et al., 2016; Eimer et al., 2018). Also, neuroinflammation has been established as a critical component of AD pathogenesis (Heneka et al., 2015), therefore, a connection between the two elements is under active investigation. Recently, a molecular link between innate immunity and AD was reported, interferon-induced transmembrane protein 3 (IFITM3) binds to γ-secretase and intensifies Aβ plaque formation (Hur et al., 2020).
IFITM3 is an innate immune response protein known to be involved in the cellular response to viral infections (Bailey et al., 2014) by inhibiting the entry of viruses to the host cells (Amini-Bavil-Olyaee et al., 2013). IFITM3 is highly upregulated by pro-inflammatory cytokines including both types I and II Interferon. Although IFITM3 was suggested to be a binding partner for γ-secretase, the underlying mechanism of interaction remained unclear (Wakabayashi et al., 2009). Recently, it was demonstrated that not only does IFITM3 directly bind to the γ-secretase complex, but it also upregulates γ-secretase activity for Aβ production. This means that IFITM3, in addition to its role in viral entry restriction, may trigger the production of anti-microbial peptide Aβ as an innate immune response by increasing γ-secretase activity. Interestingly, the binding of IFTIM3 to γ-secretase reduces Notch cleavage. This unprecedented property of IFITM3 to upregulated Aβ as an innate immune response may be a double-edged sword as it also contributes to the accumulation of Aβ in the brain. IFITM3 knockout in the AD mouse model (5XFAD) significantly reduces plaque deposition (Hur et al., 2020). This finding provides mechanistic evidence that immune activation contributes to the production of Aβ by increasing γ-secretase activity during infection and inflammation.
Risk Factors and Sporadic AD (SAD)
Though rare, FAD mutations have provided critical insights into the underlying mechanisms of AD through the involvement of APP and γ-secretase, however, the causes for SAD are poorly understood. SAD is a complex and heterogeneous neurodegenerative disease attributed to multiple factors including genetics, aging, lifestyle, and environment. Apolipoprotein E4 (APOE4) is a major risk factor for SAD in an isoform-dependent manner influencing susceptibility for ~50% of cases (Corder et al., 1993; Bu, 2009). A recent study shows that Apoe−/− transgenic mice recapitulate transcriptomic signatures of human SAD samples (Pandey et al., 2019). Gender impacts the APOE effect, as females have almost a 2-fold greater risk of AD compared to males (Chene et al., 2015), which might be associated with the gut microbiome (Maldonado Weng et al., 2019). It is thought that APOE influences AD risk through regulating the inflammatory response and Aβ aggregation and clearance (Bales et al., 2009; Keren-Shaul et al., 2017; Krasemann et al., 2017; Liu et al., 2017). However, recent studies have shown that peripheral APOE has no apparent effect on Aβ accumulation in APP/PS1 transgenic AD mouse model (Huynh et al., 2019). Furthermore, APOE isoforms affect the expression of APP and the production of Aβ in neuronal cells (Huang et al., 2017; Wang et al., 2018). Whether APOE regulates γ-secretase in neurons remains to be investigated. Also, it has been reported that APOE4 affects Tau-mediated neurodegeneration (Shi et al., 2017). APOE4 contributes to the pathogenesis of AD through multiple pathways and is an appareling target for drug development (Liu et al., 2013).
Genome-wide association studies and identification of rare variants associated with AD have highlighted genes that are potential risk factors for AD (Karch and Goate, 2015). Triggering receptor expressed on myeloid cells 2 (TREM2) and CD33 are both expressed in myeloid-linage cells including microglia are associated with AD (Bertram et al., 2008; Guerreiro et al., 2013; Jonsson et al., 2013). Both TREM2 and CD33 regulate microglia-mediated uptake and clearance of Aβ (Griciuc et al., 2013; Wang et al., 2015; Ulrich and Holtzman, 2016). TREM2 was shown to directly bind to Aβ oligomers, activating the TREM2-dependent signaling pathway, and promotes microglial migration in vitro and clustering in vivo (Zhao et al., 2018; Zhong et al., 2018). While a loss of function variant of TREM2 (R47H), which is linked with a high risk of SAD, exhibits impairment in microglia associated plaque in an AD mouse model with knock-in TREM2 (R47H; Cheng-Hathaway et al., 2018). The role of TREM2 which is strongly associated with the risk of developing AD, confirms the important part of microglia and innate immune system in AD pathogenesis (Gratuze et al., 2018).
The identification and profiling of Disease-Associated Microglia (DAM) have further deepened our understanding of the contribution of innate immunity to AD (Keren-Shaul et al., 2017). Markers of inflammation are upregulated in AD mouse models and AD patients (Patel et al., 2005; Taylor et al., 2014) and peripheral myeloid cells in patients of different AD stages have increased pro-inflammatory profile correlated with disease progression (Thome et al., 2018). Proteomic profiling of microglia from the AD mouse model (5×FAD) identified similar pro-inflammatory changes as have been observed in LPS-treated mice (Rangaraju et al., 2018). Collectively, this suggests that AD pathology presents characteristics of chronic inflammatory disease. Interestingly, aging, the biggest risk factor for AD, has been shown to induce type I IFNs that modulate brain function (Baruch et al., 2014) and IFITM3 was found to be increased in a subset of SAD patients postmortem samples and in aged mice (Hur et al., 2020). Additionally, the strong correlation between the amount IFITM3 associated with γ-secretase and enzymatic activity for Aβ cleavage indicates that IFITM3 protein could be a marker of γ-secretase activity in SAD (Hur et al., 2020). Moreover, TREM2 is a substrate of γ-secretase (Wunderlich et al., 2013) and how GMSPs, such as IFITM3, affect the processing of TREM2 and the function of microglia remains to be investigated.
The Modulation of γ-Secretase Under Hypoxic Condition
Hypoxia is a condition in which cells or tissues are deprived of adequate oxygen supply which triggers a cellular response orchestrated by the master transcriptional regulator Hypoxia-inducible factor 1-α (Hif-1α; Mukherjee et al., 2011). Hif-1α is continuously expressed but rapidly degrades in normal oxygen conditions (Jaakkola et al., 2001), whereas in hypoxic conditions Hif-1α is stabilized and translocates to the nucleus. In the nucleus, Hif-1α binds to and activates genes that are involved with vasomotor control, cell proliferation, angiogenesis, and cell metabolism (Maxwell et al., 1999). Surprisingly, a non-canonical role of Hif-1α was revealed as a GMSP. Under hypoxic conditions, Hif-1α can directly bind to the γ-secretase complex and upregulates Notch cleavage activity (Villa et al., 2014). It was shown that hypoxia increases γ-secretase cleavage of Notch, and this is dependent on Hif-1α. Interestingly, in a breast cancer model, the primary tumor grows into a proportion which generates hypoxic regions that stabilize Hif-1α. In turn, Hif-1α binds to and modulates γ-secretase activity which promotes migration and metastasis through activation of Notch downstream genes. It was suggested that Hif-1α can shift the equilibrium from inactive to active γ-secretase complexes, in response to low oxygen (Villa et al., 2014). Thus, increased γ-secretase activity and Notch signaling are essential for hypoxia-induced cell migration, invasion, and metastasis of breast cancer. This study provided the first evidence for temporal activation of γ-secretase complexes caused by changes in the cellular environment. This type of enzymatic regulation allows for a rapid switch from “off” state to “on” state when an appropriate signal appears enabling a temporal level of regulation.
γ-Secretase Activating Protein (GSAP)
Imatinib (Gleevec), a drug for cancer treatment, was found to be selectively inhibiting Aβ secretion with no effect on Notch processing (Netzer et al., 2003). To identify Imatinib target protein, a photoactivatable form was synthesized and was found to specifically label an orphan protein, namely γ-secretase activating protein (GSAP; He et al., 2010). Knockdown of GSAP in an AD model transgenic mice (APPsweXPS1ΔE9) reduces Aβ burden (He et al., 2010) and treatment with imatinib, which binds to GSAP, reduces GSAP expression, Aβ levels, and tau phosphorylation in another AD mouse model (3xTg; Chu et al., 2014). It was hypothesized that GSAP simultaneously interacts with APP βCTF and γ-secretase, increasing Aβ generation (He et al., 2010). However, the following study brought the mechanism of GSAP in γ-secretase regulation into question, reporting that while knockdown of GSAP reduces Aβ production in cells, it is not through the interaction with βCTF (Hussain et al., 2013). A later study revealed that GSAP modulates γ-secretase activity by altering the active site and subsequently enhancing APP processing while not affecting Notch processing (Wong et al., 2019). In the case of GSAP, the precise cellular cue that is responsible for its interaction with γ-secretase is unclear, but there is an SNP in GSAP that is associated with AD, which might associate GSAP as a disease-related risk factor (Zhu et al., 2014; Perez et al., 2017). This was the first study that demonstrated the extent to which GSMPs attenuate γ-secretase activity by interacting with the enzyme in the allosteric site and changing its active site conformation.
SERP1 Functions as the GSMP in Metabolic Stress Conditions
Diabetes is one of many risk factors associated with a greater risk of dementia diseases including AD (Ninomiya, 2014). High glucose levels, like in diabetes, induce endoplasmic reticulum (ER) stress and directly enhance Aβ production both in vitro and in vivo (Zhong et al., 2012; Nagai et al., 2016; Tharp et al., 2016). Stress-associated ER protein 1 (SERP1) is a small protein that interacts with and protects unfolded target proteins against degradation and is upregulated during ER stress. It was found that SERP1 stabilizes the APH1A/NCT subcomplex and promotes the localization of the γ-secretase complex in lipid raft (Jung et al., 2020). Subsequently, SERP1 facilitates the spatial distribution of γ-secretase resulting in increased APP processing in lipid rafts, the suggested site for APP cleavage (Wahrle et al., 2002; Osenkowski et al., 2008), while having virtually no impact on Notch cleavage. Postmortem AD samples have increased SERP1 expression and knocking down SERP1 in cells and mouse hippocampus decreases Aβ production. This study reveals the modulation of γ-secretase activity under ER stress enabled by the SERP1 acting as GSMP.
Conclusions
From the initial discoveries that mutations in PS1 and PS2 cause FAD and that PS1 and PS2 are the catalytic subunit of γ-secretase, extensive research on γ-secretase has revealed an overwhelming number of substrates of this protease and its involvement in broad aspects of biological processes. At first glance, γ-secretase may seem like a promiscuous enzyme earning its moniker as the “scissors in the membrane” its regulation is as complex and as variable as its involvement in multiple signaling pathways. The scientific community has come a long way in understanding these regulatory mechanisms from the assembly of essential subunits, complex formation to lipid composition. Yet, the discoveries of GMSPs present an interesting mode of regulation, adding another layer to the already complicated biology of γ-secretase. These nonessential subunits can fine-tune γ-secretase activity and specificity upon the appropriate cellular signal. It makes intuitive sense that an omnipresence protease like γ-secretase, which participates in numerous pathways, requires a quick attenuation when a signal appears to trigger the proper cellular response. Cells possess multiple γ-secretase complexes with different levels of activities, in which the majority are inactive and can serve as a pool by which a GSMP can bind and rapidly “activate”. Recent studies demonstrate that multiple GSMP can be recruited to modify γ-secretase activity and specificity under certain circumstances: IFITM3 induced by innate immune response and aging, Hif-1α stabilized by hypoxic condition, GSAP supposedly elevated in aging and AD and SERP1 under ER stress (Figure 2). Interestingly, each modulator has a different effect on γ-secretase substrates selectivity, indicating the wide range of impact on γ-secretase by GSMPs. The multiple γ-secretase-dependent signaling pathways suggest that more GMSPs might exist which modulates γ-secretase in response to other environmental changes. Thus, metaphorically, GMSPs may be the guiding hands behind “the running scissors” (Figure 3).
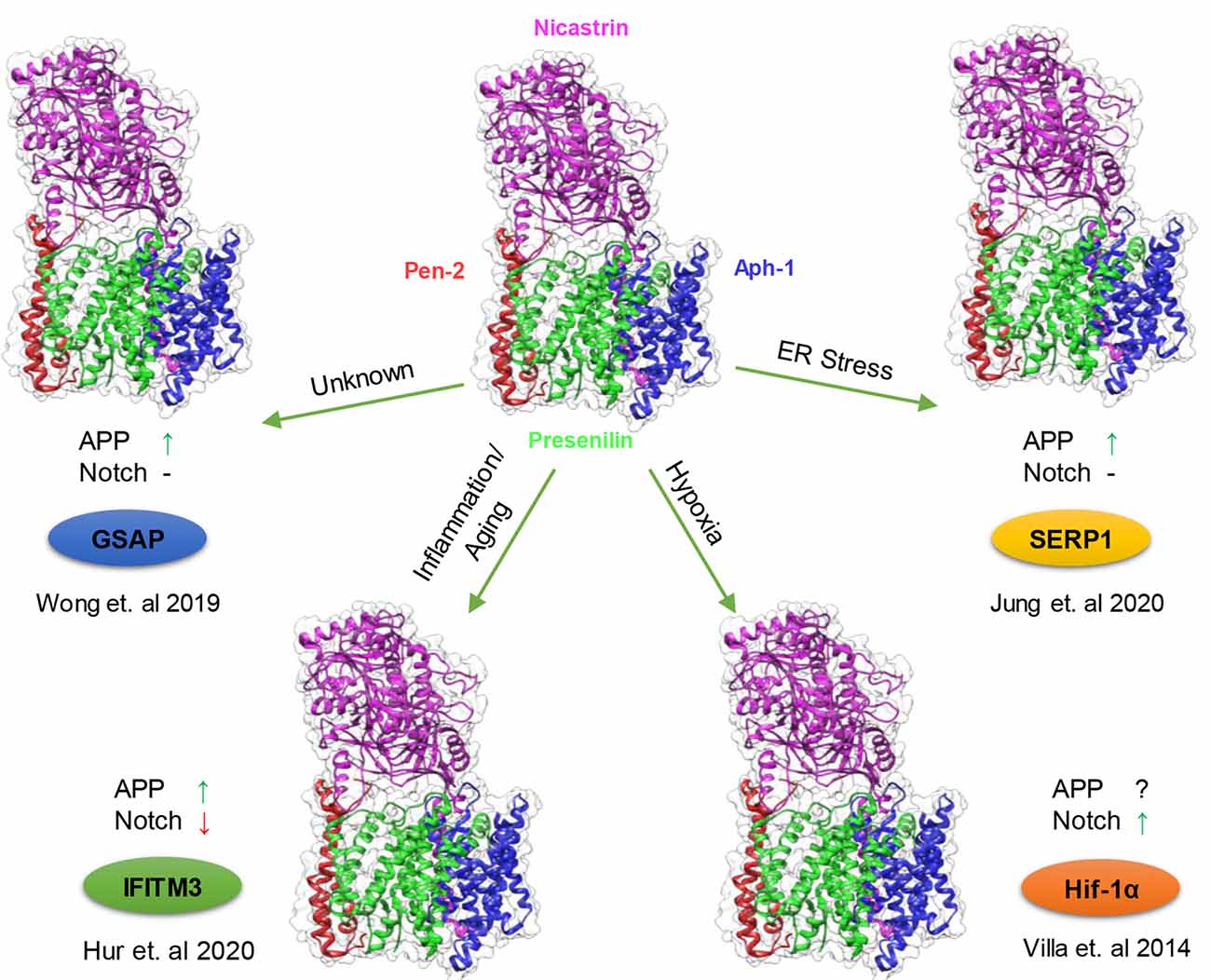
Figure 2. Modulation of γ-secretase by γ-secretase modulatory proteins (GSMPs). Schematic representation of the conditions in which GSMP triggers the attenuation of γ-secretase activity and selectivity. Cells possess a different type of γ-secretase complexes with variable levels of activities (PDB structure: 6IDF), which under certain cellular circumstances GSMP can bind and modify its activity and selectivity (clockwise): endoplasmic reticulum (ER) stress upregulate stress-associated ER protein 1 (SERP1) expression which binds and localize the γ-secretase complex to lipid rafts where amyloid precursor protein (APP) resides thus increasing APP cleavage but not Notch processing (Jung et al., 2020), hypoxic conditions stabilize Hif-1α which in turn binds to γ-secretase and increase its activity for Notch substrates (Villa et al., 2014), innate immune response and aging triggers the binding of interferon-induced transmembrane protein 3 (IFITM3) thus enhancing γ-secretase for APP but reducing Notch processing (Hur et al., 2020) and under unknown conditions GSAP attenuates γ-secretase activity solely towards APP but not Notch (Wong et al., 2019; Magenta—Nicastrin, Green—Presenilin, Blue—Aph-1 and Red—Pen-2).

Figure 3. GSMPs pull the strings of γ-secretase activity. GSMPs act as a guiding hand to modulate γ-secretase activity in response to cellular and environmental cues such as hypoxia, inflammation, ER stress, and aging.
The molecular understanding of this type of regulation can be harnessed for therapeutic intervention. Although small-molecule γ-secretase modulators are still being developed to selectively inhibit γ-secretase activity for APP, while sparing Notch, as potential treatment (Mekala et al., 2020), it is worthwhile to also explore this new avenue and target GSMPs. Developing therapeutics to prevent the engagement of transient and context-dependent interactions with GSMP, instead of directly targeting γ-secretase, could reduce potential side effects. Thus, this emerging level of γ-secretase regulation may enable the development of targeted therapies for AD and cancer.
Author Contributions
All authors prepared the manuscript and figures. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge the support of this research by National Institutes of Health Grants R01NS096275, RF1AG057593, and R01AG061350 (Y-ML), the JPB Foundation (Y-ML), The Cure Alzheimer’s Fund (Y-ML), and the BrightFocus Foundation (EW). We also acknowledge the MSK Cancer Center Support Grant/Core Grant (Grant P30 CA008748).
Conflict of Interest
YM-L is a co-inventor of the intellectual property (assay for gamma secretase activity and screening method for gamma secretase inhibitors) owned by MSKCC and licensed to Jiangsu Continental Medical Development.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, K., Shelton, C. C., Tian, Y., Zhang, X., Gilchrist, M. L., Sisodia, S. S., et al. (2010). Activation and intrinsic γ-secretase activity of presenilin 1. Proc. Natl. Acad. Sci. U S A 107, 21435–21440. doi: 10.3233/JAD-2011-101065
Amini-Bavil-Olyaee, S., Choi, Y. J., Lee, J. H., Shi, M., Huang, I. C., Farzan, M., et al. (2013). The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13, 452–464. doi: 10.1016/j.chom.2013.03.006
Bailey, C. C., Zhong, G., Huang, I. C., and Farzan, M. (2014). IFITM-family proteins: the cell’s first line of antiviral defense. Annu. Rev. Virol. 1, 261–283. doi: 10.1146/annurev-virology-031413-085537
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., DeLong, C., et al. (2009). Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009
Barros, M., Houlihan, W. J., Paresi, C. J., Brendel, M., Rynearson, K. D., Lee, C. W., et al. (2020). γ-Secretase partitioning into lipid bilayers remodels membrane microdomains after direct insertion. Langmuir 36, 6569–6579. doi: 10.1021/acs.langmuir.0c01178
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Bateman, R. J., Aisen, P. S., De Strooper, B., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2011). Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 3:1. doi: 10.1186/alzrt59
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi: 10.1016/j.ajhg.2008.10.008
Borchelt, D. R., Thinakaran, G., Eckman, C. B., Lee, M. K., Davenport, F., Ratovitsky, T., et al. (1996). Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013. doi: 10.1016/s0896-6273(00)80230-5
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Chen, F., Hasegawa, H., Schmitt-Ulms, G., Kawarai, T., Bohm, C., Katayama, T., et al. (2006). TMP21 is a presenilin complex component that modulates γ-secretase but not epsilon-secretase activity. Nature 440, 1208–1212. doi: 10.1038/nature04667
Chene, G., Beiser, A., Au, R., Preis, S. R., Wolf, P. A., Dufouil, C., et al. (2015). Gender and incidence of dementia in the Framingham Heart Study from mid-adult life. Alzheimers Dement. 11, 310–320. doi: 10.1016/j.jalz.2013.10.005
Cheng-Hathaway, P. J., Reed-Geaghan, E. G., Jay, T. R., Casali, B. T., Bemiller, S. M., Puntambekar, S. S., et al. (2018). The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 13:29. doi: 10.1186/s13024-018-0262-8
Chu, J., Lauretti, E., Craige, C. P., and Pratico, D. (2014). Pharmacological modulation of GSAP reduces amyloid-β levels and tau phosphorylation in a mouse model of Alzheimer’s disease with plaques and tangles. J. Alzheimers Dis. 41, 729–737. doi: 10.3233/JAD-140105
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Crump, C. J., Johnson, D. S., and Li, Y. M. (2013). Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry 52, 3197–3216. doi: 10.1021/bi400377p
Crump, C. J., Johnson, D. S., and Li, Y.-M. (2013). Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry 52, 3197–3216. doi: 10.1021/bi400377p
De Strooper, B. (2007). Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 141–146. doi: 10.1038/sj.embor.7400897
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., et al. (1999). A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. doi: 10.1038/19083
De Strooper, B., Iwatsubo, T., and Wolfe, M. S. (2012). Presenilins and γ-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006304. doi: 10.1101/cshperspect.a006304
De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., et al. (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390. doi: 10.1038/34910
Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C. M., Perez-tur, J., et al. (1996). Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. doi: 10.1038/383710a0
Eimer, W. A., Vijaya Kumar, D. K., Navalpur Shanmugam, N. K., Rodriguez, A. S., Mitchell, T., Washicosky, K. J., et al. (2018). Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 100, 1527–1532. doi: 10.1016/j.neuron.2018.11.043
Esler, W. P., Kimberly, W. T., Ostaszewski, B. L., Diehl, T. S., Moore, C. L., Tsai, J. Y., et al. (2000). Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol. 2, 428–434. doi: 10.1038/35017062
Evin, G., Canterford, L. D., Hoke, D. E., Sharples, R. A., Culvenor, J. G., and Masters, C. L. (2005). Transition-state analogue γ-secretase inhibitors stabilize a 900 kDa presenilin/nicastrin complex. Biochemistry 44, 4332–4341. doi: 10.1021/bi0481702
Gertsik, N., Chiu, D., and Li, Y.-M. (2014). Complex regulation of γ-secretase: from obligatory to modulatory subunits. Front. Aging Neurosci. 6:342. doi: 10.3389/fnagi.2014.00342
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Gratuze, M., Leyns, C. E. G., and Holtzman, D. M. (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13:66. doi: 10.1186/s13024-018-0298-9
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Gu, Y., Sanjo, N., Chen, F., Hasegawa, H., Petit, A., Ruan, X., et al. (2004). The presenilin proteins are components of multiple membrane-bound complexes that have different biological activities. J. Biol. Chem. 279, 31329–31336. doi: 10.1074/jbc.M401548200
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, T., Zhang, D., Zeng, Y., Huang, T. Y., Xu, H., and Zhao, Y. (2020). Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 15:40. doi: 10.1186/s13024-020-00391-7
Haapasalo, A., and Kovacs, D. M. (2011). The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28. doi: 10.3233/JAD-2011-101065
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
He, G., Luo, W., Li, P., Remmers, C., Netzer, W. J., Hendrick, J., et al. (2010). γ-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. doi: 10.1038/nature09325
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Herreman, A., Serneels, L., Annaert, W., Collen, D., Schoonjans, L., and De Strooper, B. (2000). Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2, 461–462. doi: 10.1038/35017105
Huang, Y.-A., Zhou, B., Wernig, M., and Südhof, T. C. (2017). ApoE2, ApoE3 and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168, 427.e21–441.e21. doi: 10.1016/j.cell.2016.12.044
Hur, J.-Y., Frost, G. R., Wu, X., Crump, C., Pan, S. J., Wong, E., et al. (2020). The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 586, 735–740. doi: 10.1038/s41586-020-2681-2
Hussain, I., Fabrègue, J., Anderes, L., Ousson, S., Borlat, F., Eligert, V., et al. (2013). The role of γ-secretase activating protein (GSAP) and imatinib in the regulation of γ-secretase activity and amyloid-β generation. J. Biol. Chem. 288, 2521–2531. doi: 10.1074/jbc.M112.370924
Huynh, T.-P. V., Wang, C., Tran, A. C., Tabor, G. T., Mahan, T. E., Francis, C. M., et al. (2019). Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model. Mol. Neurodegener. 14:37. doi: 10.1186/s13024-019-0337-1
Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., et al. (2001). Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. doi: 10.1126/science.1059796
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Jung, S., Hyun, J., Nah, J., Han, J., Kim, S. H., Park, J., et al. (2020). SERP1 is an assembly regulator of γ-secretase in metabolic stress conditions. Sci. Signal. 13:eaax8949. doi: 10.1126/scisignal.aax8949
Jurisch-Yaksi, N., Sannerud, R., and Annaert, W. (2013). A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta 1828, 2815–2827. doi: 10.1016/j.bbamem.2013.04.016
Karch, C. M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e17–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kim, S.-H., Yin, Y. I., Li, Y.-M., and Sisodia, S. S. (2004). Evidence that assembly of an active γ-secretase complex occurs in the early compartments of the secretory pathway. J. Biol. Chem. 279, 48615–48619. doi: 10.1074/jbc.C400396200
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566.e9–581.e9. doi: 10.1016/j.immuni.2017.08.008
Lai, M.-T., Chen, E., Crouthamel, M.-C., DiMuzio-Mower, J., Xu, M., Huang, Q., et al. (2003). Presenilin-1 and presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J. Biol. Chem. 278, 22475–22481. doi: 10.1074/jbc.M300974200
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. doi: 10.1126/science.7638622
Li, Y. M., Lai, M. T., Xu, M., Huang, Q., DiMuzio-Mower, J., Sardana, M. K., et al. (2000a). Presenilin 1 is linked with γ-secretase activity in the detergent solubilized state. Proc. Natl. Acad. Sci. U S A 97, 6138–6143. doi: 10.1073/pnas.110126897
Li, Y. M., Xu, M., Lai, M. T., Huang, Q., Castro, J. L., DiMuzio-Mower, J., et al. (2000b). Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. doi: 10.1038/35015085
Liu, C.-C., Liu, C.-C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, C.-C., Zhao, N., Fu, Y., Wang, N., Linares, C., Tsai, C. W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron 96, 1024.e3–1032.e3. doi: 10.1016/j.neuron.2017.11.013
Lobry, C., Oh, P., and Aifantis, I. (2011). Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J. Exp. Med. 208, 1931–1935. doi: 10.1084/jem.20111855
Lu, P., Bai, X.-C., Ma, D., Xie, T., Yan, C., Sun, L., et al. (2014). Three-dimensional structure of human γ-secretase. Nature 512, 166–170. doi: 10.1038/nature13567
Maldonado Weng, J., Parikh, I., Naqib, A., York, J., Green, S. J., Estus, S., et al. (2019). Synergistic effects of APOE and sex on the gut microbiome of young EFAD transgenic mice. Mol. Neurodegener. 14:47. doi: 10.1186/s13024-019-0352-2
Marquer, C., Laine, J., Dauphinot, L., Hanbouch, L., Lemercier-Neuillet, C., Pierrot, N., et al. (2014). Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol. Neurodegener. 9:60. doi: 10.1186/1750-1326-9-60
Maxwell, P. H., Wiesener, M. S., Chang, G. W., Clifford, S. C., Vaux, E. C., Cockman, M. E., et al. (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275. doi: 10.1038/20459
Mekala, S., Nelson, G., and Li, Y.-M. (2020). Recent developments of small molecule γ-secretase modulators for Alzheimer’s disease. RSC Med. Chem. 11, 1003–1022. doi: 10.1039/d0md00196a
Mukherjee, T., Kim, W. S., Mandal, L., and Banerjee, U. (2011). Interaction between Notch and Hif-α in development and survival of Drosophila blood cells. Science 332, 1210–1213. doi: 10.1126/science.1199643
Nagai, N., Ito, Y., and Sasaki, H. (2016). Hyperglycemia enhances the production of amyloid β1–42 in the lenses of otsuka long-evans tokushima fatty rats, a model of human type 2 diabetes. Invest. Ophthalmol. Vis. Sci. 57, 1408–1417. doi: 10.1167/iovs.15-19026
Naruse, S., Thinakaran, G., Luo, J. J., Kusiak, J. W., Tomita, T., Iwatsubo, T., et al. (1998). Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 21, 1213–1221. doi: 10.1016/s0896-6273(00)80637-6
Netzer, W. J., Dou, F., Cai, D., Veach, D., Jean, S., Li, Y., et al. (2003). Gleevec inhibits β-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. U S A 100, 12444–12449. doi: 10.1073/pnas.1534745100
Nie, P., Vartak, A., and Li, Y. M. (2020). γ-Secretase inhibitors and modulators: mechanistic insights into the function and regulation of γ-secretase. Semin. Cell Dev. Biol. 105, 43–53. doi: 10.1016/j.semcdb.2020.03.002
Ninomiya, T. (2014). Diabetes mellitus and dementia. Curr. Diab. Rep. 14:487. doi: 10.1007/s11892-014-0487-z
Osenkowski, P., Ye, W., Wang, R., Wolfe, M. S., and Selkoe, D. J. (2008). Direct and potent regulation of γ-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540. doi: 10.1074/jbc.M801925200
Pandey, R. S., Graham, L., Uyar, A., Preuss, C., Howell, G. R., and Carter, G. W. (2019). Genetic perturbations of disease risk genes in mice capture transcriptomic signatures of late-onset Alzheimer’s disease. Mol. Neurodegener. 14:50. doi: 10.1186/s13024-019-0351-3
Patel, N. S., Paris, D., Mathura, V., Quadros, A. N., Crawford, F. C., and Mullan, M. J. (2005). Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflammation 2:9. doi: 10.1186/1742-2094-2-9
Perez, S. E., Nadeem, M., Malek-Ahmadi, M. H., He, B., and Mufson, E. J. (2017). Frontal cortex and hippocampal γ-secretase activating protein levels in prodromal Alzheimer disease. Neurodegener. Dis. 17, 235–241. doi: 10.1159/000477937
Placanica, L., Chien, J. W., and Li, Y.-M. (2010). Characterization of an atypical γ-secretase complex from hematopoietic origin. Biochemistry 49, 2796–2804. doi: 10.1021/bi901388t
Placanica, L., Tarassishin, L., Yang, G., Peethumnongsin, E., Kim, S. H., Zheng, H., et al. (2009a). Pen2 and presenilin-1 modulate the dynamic equilibrium of presenilin-1 and presenilin-2 γ-secretase complexes. J. Biol. Chem. 284, 2967–2977. doi: 10.1074/jbc.M807269200
Placanica, L., Zhu, L., and Li, Y.-M. (2009b). Gender- and age-dependent γ-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One 4:e5088. doi: 10.1371/journal.pone.0005088
Rangaraju, S., Dammer, E. B., Raza, S. A., Gao, T., Xiao, H., Betarbet, R., et al. (2018). Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol. Neurodegener. 13:34. doi: 10.1186/s13024-018-0266-4
Sato, T., Diehl, T. S., Narayanan, S., Funamoto, S., Ihara, Y., De Strooper, B., et al. (2007). Active γ-secretase complexes contain only one of each component. J. Biol. Chem. 282, 33985–33993. doi: 10.1074/jbc.M705248200
Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., et al. (1996). Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 2, 864–870. doi: 10.1038/nm0896-864
Shelton, C. C., Tian, Y., Frattini, M. G., and Li, Y. M. (2009a). An exo-cell assay for examining real-time γ-secretase activity and inhibition. Mol. Neurodegener. 4:22. doi: 10.1186/1750-1326-4-22
Shelton, C. C., Tian, Y., Shum, D., Radu, C., Djaballah, H., and Li, Y. M. (2009b). A miniaturized 1536-well format γ-secretase assay. Assay Drug Dev. Technol. 7, 461–470. doi: 10.1089/adt.2009.0202
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Shirotani, K., Edbauer, D., Prokop, S., Haass, C., and Steiner, H. (2004). Identification of distinct γ-secretase complexes with different APH-1 variants. J. Biol. Chem. 279, 41340–41345. doi: 10.1074/jbc.M405768200
Spasic, D., Tolia, A., Dillen, K., Baert, V., De Strooper, B., Vrijens, S., et al. (2006). Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 281, 26569–26577. doi: 10.1074/jbc.M600592200
Sun, L., Zhao, L., Yang, G., Yan, C., Zhou, R., Zhou, X., et al. (2015). Structural basis of human γ-secretase assembly. Proc. Natl. Acad. Sci. U S A 112, 6003–6008. doi: 10.1073/pnas.1506242112
Takasugi, N., Tomita, T., Hayashi, I., Tsuruoka, M., Niimura, M., Takahashi, Y., et al. (2003). The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441. doi: 10.1038/nature01506
Tarassishin, L., Yin, Y. I., Bassit, L., and Li, Y. M. (2004). Processing of Notch and amyloid precursor protein by γ-secretase is spatially distinct. Proc. Natl. Acad. Sci. U S A 101, 17050–17055. doi: 10.1073/pnas.0408007101
Taylor, J. M., Minter, M. R., Newman, A. G., Zhang, M., Adlard, P. A., and Crack, P. J. (2014). Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol. Aging 35, 1012–1023. doi: 10.1016/j.neurobiolaging.2013.10.089
Tharp, W. G., Gupta, D., Smith, J., Jones, K. P., Jones, A. M., and Pratley, R. E. (2016). Effects of glucose and insulin on secretion of amyloid-β by human adipose tissue cells. Obesity 24, 1471–1479. doi: 10.1002/oby.21494
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. doi: 10.1016/s0896-6273(00)80291-3
Thomas, J. B., Brier, M. R., Bateman, R. J., Snyder, A. Z., Benzinger, T. L., Xiong, C., et al. (2014). Functional connectivity in autosomal dominant and late-onset Alzheimer disease. JAMA Neurol. 71, 1111–1122. doi: 10.1001/jamaneurol.2014.1654
Thome, A. D., Faridar, A., Beers, D. R., Thonhoff, J. R., Zhao, W., Wen, S., et al. (2018). Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol. Neurodegener. 13:61. doi: 10.1186/s13024-018-0293-1
Ulrich, J. D., and Holtzman, D. M. (2016). TREM2 function in Alzheimer’s disease and neurodegeneration. ACS Chem. Neurosci. 7, 420–427. doi: 10.1021/acschemneuro.5b00313
Vetrivel, K. S., Gong, P., Bowen, J. W., Cheng, H., Chen, Y., Carter, M., et al. (2007). Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol. Neurodegener. 2:4. doi: 10.1186/1750-1326-2-4
Vijaya Kumar, D. K., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
Villa, J. C., Chiu, D., Brandes, A. H., Escorcia, F. E., Villa, C. H., Maguire, W. F., et al. (2014). Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 8, 1077–1092. doi: 10.1016/j.celrep.2014.07.028
Wahrle, S., Das, P., Nyborg, A. C., McLendon, C., Shoji, M., Kawarabayashi, T., et al. (2002). Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23. doi: 10.1006/nbdi.2001.0470
Wakabayashi, T., Craessaerts, K., Bammens, L., Bentahir, M., Borgions, F., Herdewijn, P., et al. (2009). Analysis of the γ-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 11, 1340–1346. doi: 10.1038/ncb1978
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, C., Najm, R., Xu, Q., Jeong, D. E., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. doi: 10.1038/s41591-018-0004-z
Wolfe, M. S. (2007). When loss is gain: reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 136–140. doi: 10.1038/sj.embor.7400896
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517. doi: 10.1038/19077
Wong, E., Liao, G. P., Chang, J. C., Xu, P., Li, Y. M., and Greengard, P. (2019). GSAP modulates γ-secretase specificity by inducing conformational change in PS1. Proc. Natl. Acad. Sci. U S A 116, 6385–6390. doi: 10.1073/pnas.1820160116
Wunderlich, P., Glebov, K., Kemmerling, N., Tien, N. T., Neumann, H., and Walter, J. (2013). Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036. doi: 10.1074/jbc.M113.517540
Xu, M., Lai, M.-T., Huang, Q., DiMuzio-Mower, J., Castro, J. L., Harrison, T., et al. (2002). γ-Secretase: characterization and implication for Alzheimer disease therapy. Neurobiol. Aging 23, 1023–1030. doi: 10.1016/s0197-4580(02)00126-4
Yang, G., Zhou, R., Zhou, Q., Guo, X., Yan, C., Ke, M., et al. (2019). Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. doi: 10.1038/s41586-018-0813-8
Zhang, Z., Nadeau, P., Song, W., Donoviel, D., Yuan, M., Bernstein, A., et al. (2000). Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2, 463–465. doi: 10.1038/35017108
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023.e7–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zhong, Y., Li, J., Chen, Y., Wang, J. J., Ratan, R., and Zhang, S. X. (2012). Activation of endoplasmic reticulum stress by hyperglycemia is essential for Muller cell-derived inflammatory cytokine production in diabetes. Diabetes 61, 492–504. doi: 10.2337/db11-0315
Zhong, L., Wang, Z., Wang, D., Wang, Z., Martens, Y. A., Wu, L., et al. (2018). Amyloid-β modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 13:15. doi: 10.1186/s13024-018-0247-7
Zhou, R., Yang, G., Guo, X., Zhou, Q., Lei, J., and Shi, Y. (2019). Recognition of the amyloid precursor protein by human γ-secretase. Science 363:eaaw0930. doi: 10.1126/science.aaw0930
Keywords: IFITM, hypoxia, neuroinflammation, neurodegeneration, enzyme
Citation: Wong E, Frost GR and Li Y-M (2020) γ-Secretase Modulatory Proteins: The Guiding Hand Behind the Running Scissors. Front. Aging Neurosci. 12:614690. doi: 10.3389/fnagi.2020.614690
Received: 06 October 2020; Accepted: 10 November 2020;
Published: 02 December 2020.
Edited by:
Xiongwei Zhu, Case Western Reserve University, United StatesReviewed by:
Riqiang Yan, University of Connecticut, United StatesWeiming Xia, Boston University, United States
Copyright © 2020 Wong, Frost and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eitan Wang, d29uZ3cyQG1za2NjLm9yZw==; Yue-Ming Li, bGl5MkBtc2tjYy5vcmc=