 Weixiang Chen
Weixiang Chen Chao Guo1,2,3,4†
Chao Guo1,2,3,4† Hua Feng
Hua Feng Yujie Chen
Yujie Chen- 1Department of Neurosurgery, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
- 2State Key Laboratory of Trauma, Burn and Combined Injury, Third Military Medical University (Army Medical University), Chongqing, China
- 3Chongqing Key Laboratory of Precision Neuromedicine and Neuroregenaration, Third Military Medical University (Army Medical University), Chongqing, China
- 4Collaborative Innovation Center for Brain Science, Third Military Medical University (Army Medical University), Chongqing, China
Intracerebral hemorrhage (ICH) is a destructive form of stroke that often results in death or disability. However, the survivors usually experience sequelae of neurological impairments and psychiatric disorders, which affect their daily functionality and working capacity. The recent MISTIE III and STICH II trials have confirmed that early surgical clearance of hematomas does not improve the prognosis of survivors of ICH, so it is vital to find the intervention target of secondary brain injury (SBI) after ICH. Mitochondrial dysfunction, which may be induced by oxidative stress, neuroinflammation, and autophagy, among others, is considered to be a novel pathological mechanism of ICH. Moreover, mitochondria play an important role in promoting neuronal survival and improving neurological function after a hemorrhagic stroke. This review summarizes the mitochondrial mechanism involved in cell death, reactive oxygen species (ROS) production, inflammatory activation, blood–brain barrier (BBB) disruption, and brain edema underlying ICH. We emphasize the potential of mitochondrial protection as a potential therapeutic target for SBI after stroke and provide valuable insight into clinical strategies.
Introduction
Hemorrhagic stroke, which is less common but far more likely to be fatal, makes up about 13% of stroke cases. Indeed, two-thirds of stroke survivors will experience moderate or severe disability (Balami and Buchan, 2012). The high incidence and mortality of intracerebral hemorrhage (ICH) are due to both primary brain injury (mainly caused by a mass effect) and secondary brain injury (SBI; mainly caused by hemoglobin degradation products), including neuronal death, oxidative stress injury, and cerebral edema (Wang et al., 2002; Cordonnier et al., 2018). Recently, the MISTIE III and STICH II trials confirmed that early surgical clearance of hematomas does not improve the prognosis of patients with ICH (Mendelow et al., 2013; Hanley et al., 2019), and the lack of evidence-based surgical treatment strategies has prompted researchers to seek effective intervention targets and therapies for secondary injuries after ICH.
The prevalence of ICH is ~120 per 100,000; 58% of patients die within 1 year, with two-thirds of the survivors being moderately or severely disabled (Balami and Buchan, 2012; Kumar et al., 2016). Furthermore, the 30-days mortality rate is ~30–55% (Feigin et al., 2015). ICH is associated with high morbidity and mortality from severe SBI, including white matter damage, neuronal death (apoptosis and necrosis), and inflammatory changes (Bobinger et al., 2018).
As the powerhouse of cells, mitochondria play a crucial role in cell energy homeostasis; thus, they are inevitably associated with the pathophysiology of ICH (Georgieva et al., 2017; Chen et al., 2020b). Mitochondrial injury in patients with ICH was first reported in 2006, suggesting that the damage is not caused by hypoxia and ischemia but rather by mitochondrial dysfunction (Kim-Han et al., 2006). Based on the knowledge gained by the greater attention paid to the development of methods for mitochondrial research, it is currently believed that the function of mitochondria involves both supplying energy for the cell and regulating cell death and inflammation (Dawson and Dawson, 2017; Gong et al., 2018). Metformin, a mitochondrion-related treatment drug, has been shown to reduce apoptosis, inflammation, oxidative stress, DNA damage, and mitochondrial damage after cerebral hemorrhage (Wang et al., 2018). Additionally, mitoquinone (MitoQ), a mitochondrial reactive oxygen-scavenging agent, improves mitochondrial function and motor function after ICH, reduces the hematoma volume, and alleviates cerebral edema (Chen et al., 2020a,b).
In this review, we discuss the latest perspectives on the role of mitochondria in cell death and survival and highlight the pathogenesis and treatment of mitochondrion-associated ICH.
Pathophysiology of ICH
Intracerebral hemorrhage is a subtype of stroke that is associated with high rates of mortality and disability (Feigin et al., 2015). There are two million cases of ICH worldwide each year, and those who survive often have severe neurological deficits (Kumar et al., 2016). At present, the mainstream view is that nerve injury after ICH can be divided into primary brain injury and SBI (Figure 1; Aronowski and Zhao, 2011). The former is mainly caused by mechanical disruption after initial bleeding, whereas the latter is caused by a series of mechanisms including oxidative stress, inflammation, mitochondrial dysfunction, and neuronal death (including apoptosis and necrosis; Yu et al., 2019).
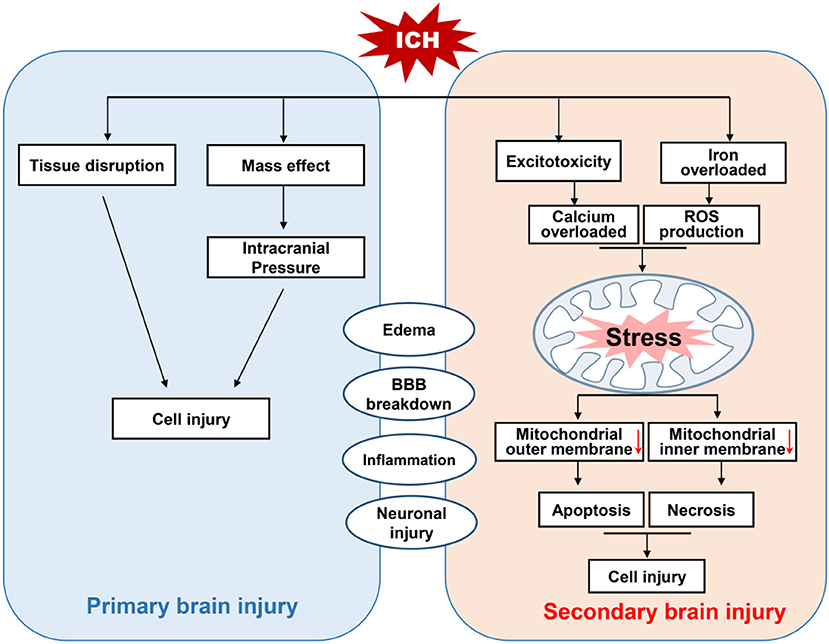
Figure 1. Schematic diagram of brain injury after intracerebral hemorrhage (ICH). Brain injury after ICH can be divided into primary and secondary brain injury (SBI). The primary brain injury is caused by tissue disruption and mass effect after initial bleeding. Whereas, SBI is induced by oxidative stress, the activation of TRP channels and excitotoxicity due to the accumulation of reactive oxygen species (ROS) triggered by iron overload and calcium. This excessive ROS and calcium cause mitochondrial dysfunction, and the activation of apoptotic factors leads to apoptotic and necrotic cell death.
Although most researchers believe that these mechanisms are related to SBI after ICH, effective interventions are still lacking (Cordonnier et al., 2018). Therefore, it is important to explore ways of promoting the recovery of nerve function by reducing secondary injury in the treatment of ICH.
Structure and Function of Mitochondria
Mitochondria are ovoid or rod-shaped organelles with double-membrane structures comprising four distinct compartments: the outer membrane, the intermembrane space, the inner membrane, and the matrix (Figure 2; Giacomello et al., 2020). These organelles perform a variety of key functions in different cellular processes (Cunnane et al., 2020). The human nervous system consumes a great amount of energy, and by providing ATP through oxidative phosphorylation, mitochondria are the main source of energy for normal neuronal homeostasis and function (Area-Gomez et al., 2019).
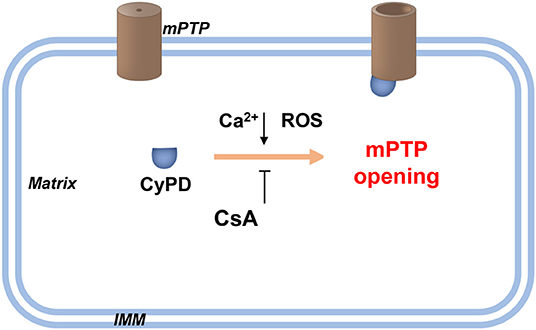
Figure 2. Schematic diagram of the PTP opening hypothesis. As illustrated in the figure above, an increasing level of Ca2+ and/or ROS in the mitochondrial matrix promotes the opening of high conductance of the PTP. This process is achieved by combining CyPD with PTP. Therefore, drugs targeting CypD, such as CsA, or genetic interventions for CyPD reduction can be applied without affecting PTP opening. Recently, Ca2+-induced PTP opening via small conductance PTP has been reported, and this process inhibits CsA. However, the same protein with different conformations may form pores of increasing size. CsA, cyclosporin A; CyPD, cyclophylin D; IMM, inner mitochondrial membrane; ROS, reactive oxygen species.
Indeed, mitochondria are pivotal regulators of cell survival, and their crucial role in cell survival is mainly reflected in three aspects (Bock and Tait, 2020). Mitochondria are the hubs of cellular calcium signaling, and they provide an important mechanism for regulating calcium concentration during signal transduction, which is particularly important for excitable cells, such as neurons (Giorgi et al., 2018). The enzyme complexes located in the mitochondrial inner membrane are essential for maintaining cellular energy requirements and metabolic homeostasis (Fricker et al., 2018). The survival of neuronal cells depends largely on the integrity and function of mitochondria. Therefore, mitochondrial defects have severe destructive effects on the central nervous system (CNS; Zsurka and Kunz, 2015).
Mitochondrial Quality Control Systems and ICH
Mitochondria control almost every aspect of cellular function, including managing REDOX status, modulating Ca2+ homeostasis, generating ATP, and regulating responses to cellular and environmental stresses (Liu et al., 2018). The function of mitochondria is also important for the health of neurons, and the activities of axons and dendritic neuron fibers are highly dependent on mitochondria, which are highly dynamic and exhibit an activity-induced interval distribution (Eisner et al., 2018; Bock and Tait, 2020). For example, mitochondrial transport to the remote synapse provides sufficient energy for local synaptic activity in the maintenance of ion channels, transporters, and synaptic transmission (Licznerski et al., 2020).
Moreover, abundant evidence supports the importance of maintaining normal clearance of damaged mitochondria for neuron survival after ICH (Huang and Jiang, 2019; Li et al., 2020). Mitochondrial dysfunction in SBI after ICH has been well-described, and mitochondrial quality and transport have a key function in the protection of neuronal injury. In fact, maintaining mitochondrial integrity and removing damaged mitochondria are important ways to prevent extensive mitochondrial dysfunction, oxidative stress, and cell death caused by hemorrhagic injury (Huang and Jiang, 2019). Therefore, mitochondrial dynamics and mitophagy are vital for maintaining cellular homeostasis and function after ICH. Here, we systematically review the critical roles of mitochondrial integrity and function in neuronal survival after ICH.
Mitochondrial ROS and ICH
Reactive oxygen species (ROS) are the causes of SBI after ICH (Duan et al., 2016; Qu et al., 2016). Excessive accumulation of ROS can lead to macromolecular damage, cellular signal transduction disorder, cell death, and tissue damage (Forrester et al., 2018).
After ICH, hematoma and perihematomal regions are rich with RBC lysis products, especially hemin. In addition, intracellular hemin is degraded into Fe2+. Fenton reaction caused by Fe2+ can generate a hydroxyl radical, which is the most reactive of all oxygen radicals, and leads to oxidative stress (Zille et al., 2017; Bertero and Maack, 2018). The initial bleed leads to an influx of glutamate from the bloodstream, and excessive glutamate is one of the most important damaging factors in the nervous system (Cheng et al., 2016). Excessive glutamate in the brain parenchyma can induce Ca2+ overload, which leads to membrane depolarization and ROS release (Joshi et al., 2015). Activation of inflammatory cells also contributes to the pathogenesis of brain injury in ICH, and granulocytes can be a source of ROS after ICH. They can cause the release of ROS via nicotine adenine dinucleotide phosphate (NADPH) oxidase and myeloperoxidase (Zia et al., 2009).
The blood–brain barrier (BBB) is a dynamic interface between the peripheral circulation and the CNS that prevents toxic substances from the CNS and contributes to the maintenance of brain homeostasis (Tschoe et al., 2020). Disruption of the BBB can cause brain edema, which is also an important secondary injury after ICH (Figure 3). Many researchers have demonstrated that mitochondrial membrane potential (MMP)-9 are upregulated after ICH, which is associated with oxidative stress and BBB disruption (Katsu et al., 2010). Inhibition of oxidative stress can decrease MMP-9 levels (Katsu et al., 2010). Therefore, ROS can trigger the activation of MMPs, leading to BBB disruption.
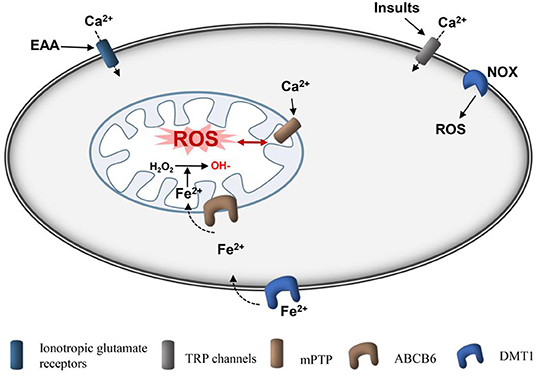
Figure 3. Schematic diagram of production of mitochondrial ROS after ICH. Ionotropic glutamate receptors, such as NMDA receptor and APMA receptor, are activated by glutamate, causing cellular Ca2+ overload in cells and mitochondria, respectively. Insults, such as ROS and pressure can induce cellular Ca2+ overload directly through TRP channels. Ferrous iron can be transported into cells by DMT1 and then loaded into mitochondria by ABCB6. Ferrous is used to convert H2O2 to hydroxyl radicals, which are the active radicals causing severe oxidative damage. mPTPs are activated by Ca2+ and ROS to open, and ROS are subsequently released. In addition, NOX can produce ROS. ROS, reactive oxygen species; Fe2+, ferrous iron; AMPA, amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor; NMDA, N-methyl-D-aspartic acid receptor; fer-1, ferrostatin-1; DMT1, divalent metal transporter 1; mPTP, mitochondrial permeability transition pore; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, nicotinamide adenine dinucleotide phosphate oxidase.
As mitochondria are the main sites of ROS production, mitochondrial accumulation and high oxygen consumption in the CNS render nerve tissue vulnerable to oxidative stress (Mariani et al., 2005; Sidlauskaite et al., 2018; Zheng et al., 2018).
Oligodendrocytes are rich in sphingomyelins, which are prone to injury from oxidative stress, leading to white matter damage (Zhuo et al., 2016). Some non-selective antioxidants, such as edaravone, have been shown to be effective in animal studies but have failed in clinical trials for use in a patient of ICH (Nakamura et al., 2008; Yang et al., 2011, 2015). Recent studies report that selective mitochondrial ROS (mROS) scavengers are superior to non-selective ROS scavengers in the treatment of many REDOX diseases involving mitochondrial dysfunction (Hu et al., 2016; Georgieva et al., 2017; Dey et al., 2018). Our group found that the MitoQ, a mitochondrial reactive oxygen-scavenging agent, can improve mitochondrial function and outcome of mice after ICH (Chen et al., 2020a,b).
Adaptive and non-adaptive responses to REDOX stress may involve mitochondrial channels, such as mitochondrial permeability transition pore (mPTP) and intimal anion channels (IMACs), and activation of these channels leads to an imbalance of REDOX homeostasis in cells and mitochondria, inducing ROS release (Augustynek et al., 2014; Bernardi et al., 2015). The regeneration cycle of the formation and release of mROS is termed as ROS-induced ROS release (Zorov et al., 2014). The mPTP is a multiprotein complex consisting of cyclophilin D (CyPD), mitochondrial peptide proline trans isomerase, voltage-dependent anion channel (VDAC), adenine nucleotide transporter (ANT), and other molecules that form channels in the mitochondrial inner membrane (Li et al., 2014; Bernardi et al., 2015; Chinopoulos, 2018). Regular mPTP opening plays a key physiological role in maintaining a healthy internal mitochondrial environment (Chinopoulos, 2018). At high ROS levels, continuous mPTP opening will trigger ROS release, leading to mitochondrial dysfunction; moreover, the transmission of ROS from damaged mitochondria to neighboring mitochondria results in uncontrollable damage (Zandalinas and Mittler, 2018). In addition, activation of the mPTP, a pore channel in the mitochondrial membrane, may also be a potential mechanism for necrosis and apoptosis (Porter and Beutner, 2018). Overall, mitochondria are the main sites of ROS production and participate in the amplification of ROS (Sies et al., 2017). The increase in mROS that occurs after ICH can be partially reversed by the VDAC inhibitor TRO-19622 or the mROS-specific-scavenging agent Mito-tempo (Ma et al., 2014).
In general, it is vital to explore the protective effect of selective mROS scavengers for treating ICH-induced SBI.
Mitochondria-Related Neuronal Apoptosis Pathway After ICH
Mitochondria are associated with a variety of apoptotic mechanisms. Apoptosis is a process by which early tissue injury around hematoma occurs after ICH (Lu et al., 2015; Bobinger et al., 2018). There are many factors influencing apoptosis after ICH, such as free-radical cascade reactions, inflammation, cytokines, thrombin, and erythrocyte lysis (Salihu et al., 2016). Apoptosis-related genes are involved in neuronal apoptosis after ICH, including BCL-2 associated x protein (Bax), which promotes apoptosis, and Bcl-2, which inhibits apoptosis (Song et al., 2019; Tang et al., 2020). Bcl-2 and Bax are located in the outer mitochondrial membrane, and the Bcl-2/Bax ratio can reflect the tendency of cells to undergo apoptosis or to survive after stimulation: When the Bcl-2 level increases and the Bax decreases, the ratio increases and cells tended to survive (Chen et al., 2015; Luo et al., 2020). The Bcl-2/Bax ratio in cerebral tissue decreases after ICH, which can be inhibited by melatonin (Wang et al., 2018).
Mitochondria directly participate in the pre-apoptotic signaling pathway by releasing cytochrome C (CytoC) and apoptosis induction factor (AIF), which play a central role in the survival or death of neurons (Sabirzhanov et al., 2016). The latter induces apoptosis by activating caspases, whereas AIF is not related to caspase-mediated apoptosis (Hangen et al., 2010). Changes in mitochondrial outer membrane permeability lead to mitochondrial metabolic failure and the release of caspase-activating molecules as well as caspase-dependent death effectors, inducing mitochondrial swelling and neuron death (Augustynek et al., 2014). Previous studies have reported reduced mitochondrial respiratory function and CytoC release around the hematoma after ICH, suggesting that mitochondrial dysfunction is associated with brain injury (Lu et al., 2015; Ding et al., 2017). Moreover, based on double-labeled immunofluorescence, CytoC mainly colocalizes with CNPase, suggesting that the apoptosis of oligodendrocytes after intracapsular hemorrhage may be mediated by the mitochondrial pathway (Zhuo et al., 2016). CytoC levels reflect whether apoptosis in oligodendrocytes induced by ICH is mediated by the mitochondrial pathway (You et al., 2016). Recent studies have found that the expression of CytoC is upregulated in rats with ICH, which is consistent with the apoptosis trend of oligodendrocytes (Lu et al., 2015). Cell death after ICH can be induced by brain edema, inflammatory reaction, and BBB disruption, which are the results of secondary injury.
Mitochondrial Dynamics and ICH
Abnormal regulation of mitochondrial dynamics, which shifts the balance of fusion and fission to fission, is involved in the pathological process of hemorrhagic SBI (Wu et al., 2020a). Mitochondria are highly dynamic organelles that constantly undergo fission and fusion to maintain normal morphology and function (Fenton et al., 2020), as characterized by highly coordinated fission (the separation of a single mitochondria into two or more daughter organelles), fusion (an opposing reaction), and transport to a specific location in the cell (Giacomello et al., 2020) (Figure 4). These dynamic processes are accompanied by changes in mitochondrion size and shape (Pekkurnaz et al., 2014).
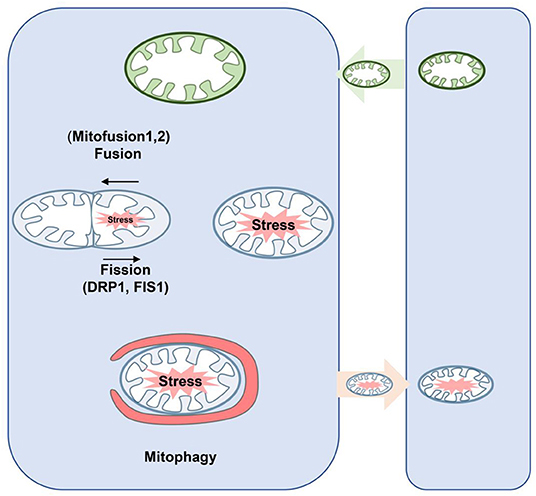
Figure 4. Schematic diagram of mitochondrial dynamics and mitophagy. Fusion is regulated by Mfn1/2 and Opa1, which mediate fusion of the outer and inner mitochondrial membrane, respectively. In the process of fission, Drp1 is recruited from the cytoplasm to the outer membrane of mitochondria and interacts with its receptor protein and Fis1 to form the fission complex in the outer membrane. Thus, damaged mitochondria can be repaired through fusion with healthy mitochondria; mitochondrial fission allows damaged mitochondria to be separated and eliminated by mitophagy. Neurons may secrete mitochondria, and these mitochondria may be unhealthy but may accept healthy mitochondria at the same time to maintain normal neuronal function. Drp, dynamin-related proteins; Mfn1/2, mitofusins; Opa1, optic atrophy 1.
Under physiological conditions, mitochondrial fission is crucial for removing damaged mitochondria to maintain mitochondrial stability (Weir et al., 2017). However, excessive fission damages mitochondrial structure, leading to impaired respiratory function, increased mROS production, ATP deficiency, and apoptotic pathway activation (Nakamura and Lipton, 2011; Zhou et al., 2019; Rong et al., 2020).
In turn, mitochondrial division is mainly regulated by dynamic protein-related protein 1 (Drp1), a membrane protein, which is recruited into the mitochondrial outer membrane and triggers mitochondrial division (Montessuit et al., 2010). Phosphorylation is a key step in regulating Drp1 recruitment under different conditions (Kashatus et al., 2011). Phosphorylation at Ser616 (S616) is generally believed to accelerate its recruitment to the mitochondrial membrane, whereas phosphorylation at S637 inhibits this process (Xu et al., 2016; Ma et al., 2020). Mitochondrial quality control mediated by Drp1 and mfn1/2 has been shown to play a crucial role in preventing ICH injury, such as neural apoptosis, brain edema, and inflammatory response (Wu et al., 2020a,b).
Previous studies have shown that the Drp1 inhibitor mdivi-1 can attenuate oxidative stress and neuronal apoptosis after subarachnoid hemorrhage (Fan et al., 2017). Furthermore, recent studies have shown that Drp1 inhibitors exert neuroprotective effects in the ICH models (Wu et al., 2020a,b), suggesting that excessive Drp1 activity may be an important risk factor for ICH-induced brain injury. In fact, mitochondrial injury induced by acrolein around the hematoma after ICH is associated with increased Drp1 translocation and excessive mitochondrial fission, and acrolein-scavenging agents can significantly inhibit Drp1-mediated nuclear fission after ICH and reduce mitochondrial morphological damage (Wu et al., 2020a). Most importantly, inhibition of mitochondrial fission significantly alleviates neuronal apoptosis, cerebral edema, and neurological deficits after ICH (Wu et al., 2020c).
Mitophagy and Its Role in ICH
Mitophagy was first described by Lemasters (2005), who determined that mitophagy is an autophagic response that is responsible for the specific removal of damaged mitochondria to maintain mitochondrial homeostasis and is an important mechanism for maintaining mitochondrial health (Lemasters, 2014) (Figure 4). Recently, it has been found that the inhibition of mitophagy is beneficial in the pathogenesis of various neurological diseases, such as subarachnoid hemorrhage (Li et al., 2014; Zhang et al., 2019). Mechanically, PINK1/Parkin-mediated fission is the most characteristic mitophagy pathway (Geisler et al., 2010). Moreover, other E3 ubiquitin-protein ligases, such as SMURF1, SIAH1, MUL1, Gp78, ARIH1, and HUWE1, are involved in the regulation of PINK1-Parkin independent mitophagy (Li et al., 2014).
In various pathological environments, “alternative” activation pathways of mitophagy are also involved in mitophagy (Ma et al., 2019). As an example, it has been demonstrated that FUNDC1, an OMM protein, is able to activate mitophagy. Indeed, FUNDC1 can directly recruit LC3 through its LIR domain [36]. Bcl-2-like 13 (BCL2 L13), another OMM protein and a homolog of Atg32, is a mitochondrial autophagy receptor in yeast that can also directly bind to LC3 and promote mitochondrial autophagy through its LIR motif (Jing et al., 2012). Recently, it has been found that mitophagy is involved in the pathogenesis of various neurological diseases, such as subarachnoid hemorrhage (Li et al., 2014; Zhang et al., 2019).
In general, it appears that the effect of mitophagy depends on its severity. Under physiological conditions, mitophagy prevents accelerated cellular senescence and programmed cell death (Kang, 2020; Zhou and Tan, 2020). However, excessive mitophagy plays a lethal role when overactivated by severe pathological stress like ischemia (Huang et al., 2020). NIX primarily regulates the basal level of mitophagy in physiological conditions, whereas BNIP3 exclusively activates excessive mitophagy, leading to cell death (Shi et al., 2014), although the role of mitophagy in ICH has not been fully appreciated to date. Further research is needed to understand whether mitophagy is beneficial after ICH.
Mitochondrial Membrane Potential
In cells, the MMP is typically maintained between 80 and 140 mV (Cloonan et al., 2016) and is regulated by mPTP. Although the structure and composition of mPTP remain controversial, CyPD has long been recognized as a pivotal regulator of the pore openness (Baines and Gutierrez-Aguilar, 2018; Porter and Beutner, 2018). The activity of CyPD is mainly regulated by post-translational modifications, including acetylation, S-glutathione, glycosylation, and S-nitrification (Hafner et al., 2010).
The mitochondrial injury induced by ICH is the result of mPTP opening, whereby various proteins, such as CytoC, are released into the cytoplasm, constituting important events that lead to apoptosis (Tomasello et al., 2009; Bock and Tait, 2020). Melatonin inhibits mPTP opening by upregulating antioxidants to reduce mitochondrial dysfunction therefore inhibiting SBI after ICH (Wang et al., 2018). It has been shown that melatonin treatment significantly inhibits apoptosis and mitochondrial damage, findings verified in vivo and in vitro, and some researchers have found that melatonin can significantly inhibit apoptosis and mitochondrial damage in vivo and in vitro (Wang et al., 2018). Our group found that selective mROS antioxidants MitoQ, but not the non-selective antioxidants, almost completely attenuated the iron-induced membrane potential decrease and cell death (Chen et al., 2020b). Additionally, Honokiol (HKL, a pharmacological agonist of sirt3) protects against hyperglycemic ICH-induced neuronal injury via the inhibition of mitochondrial depolarization (Zheng et al., 2018).
In addition, Miro1 is a glutamate receptor-dependent calcium sensor that targets the mitochondria of neurons, connecting mitochondria with motor proteins (Macaskill et al., 2009). The Miro1 protein is rapidly ubiquitinated and exhausted in damaged mitochondria to block microtubule-dependent transport of damaged mitochondria, thus promoting mitophagy in mitochondria with decreased membrane potential (Hsieh et al., 2019). Chen et al. reported that the upregulation of Miro1 significantly alleviated pathological symptoms on SBI in vivo and in vitro (Li et al., 2020). Miro1 may also be involved in a key mechanism for mitochondrial quality control after ICH.
Mitochondrial Transfer
Hayakawa et al. (2016) demonstrated for the first time in 2016 that astrocytes can transfer functional mitochondria to neighboring neurons and promote the survival of receptor neurons (Figure 4). These transferred mitochondria are involved in many important functions that benefit the recipient cells. In addition, these groups suggested that mitochondrial transfer improves functional neuron damage after stroke (Hayakawa et al., 2016).
Astrocytic release of extracellular mitochondria particles was mediated by a calcium-dependent mechanism involving CD38/cyclic ADP ribose signaling. Transient focal cerebral ischemia in mice induced astrocytic mitochondria entry to adjacent neurons that amplified cell survival signals (Hayakawa et al., 2016). In addition, damaged cells can produce phosphatidylserine, inducing the tunneling nanotubes (TNTs) formation promoting mitochondrial transfer (Liu et al., 2014). Similarly, TNT mitochondrial transfer from mesenchymal stem cells to cardiomyocytes improved survival and reduced cellular damage in an in vitro ischemia/reperfusion model (Han et al., 2016).
Studies have shown that the main function of transferred mitochondria is to enhance the capacity of recipient cells to metabolize energy (Picca et al., 2019). It has also been reported that mitochondria-derived humanin (HN), a cross-cellular signaling molecule, promotes neuron recovery after ICH: Mitochondrial transfer and the microglia phenotype regulate the function of “reparative” microglia after astrocytes are secreted as peptides or transferred among mitochondria (Chou et al., 2017). Additionally, neurons may secrete mitochondria, but these mitochondria may be unhealthy and may be considered cellular waste (Chou et al., 2017).
Perspectives and Conclusions
Mitochondrial dysfunction is an early initiating event in the pathophysiology of ICH. Bioenergy genetic defects, structural abnormalities and mitochondrial morphology, and aberrant mitochondrial dynamics play an important role in the activation of neuronal death signaling pathways. In preclinical studies, interventions aimed at mitochondrial quality control and mitochondrial dynamics have been shown to have neuroprotective effects (Chimeh et al., 2018).
Recent evidence of mitochondrial transfer has provided a new perspective for intercellular communication. The latest research suggests that mitochondria themselves can act as “help me” signals to respond to different extracellular stimuli and recruit adjacent cells to rescue those injured in stroke and those affected by aging, and disease (Heyck et al., 2019; Gollihue and Norris, 2020). Clearance of damaged mitochondria and replacement with healthy organelles is another promising treatment for CNS diseases, where mitochondria are abundant in distal axonal synapses and dendrites (He et al., 2020). The detailed mechanisms of mitochondrial release and receptor recognition in donor cells, the generalizability of beneficial results, and the ethical implications associated with the artificial transfer of mitochondria remain to be further studied.
Here, Table 1 lists the potential translational and clinical strategies targeting mitochondria in CNS disease. We have listed mitochondrial protectants that were used to protect neuronal injury from diseases occurring due to damage in the nervous system. Some of them, such as edaravone, are effective to improve prognosis in the studies of ischemic stroke (Shinohara et al., 2009). However, Cyclosporine was generally not effective in reducing infarct size after stroke (Nighoghossian et al., 2015). Regrettably, these drugs have not yet been tested in ICH.
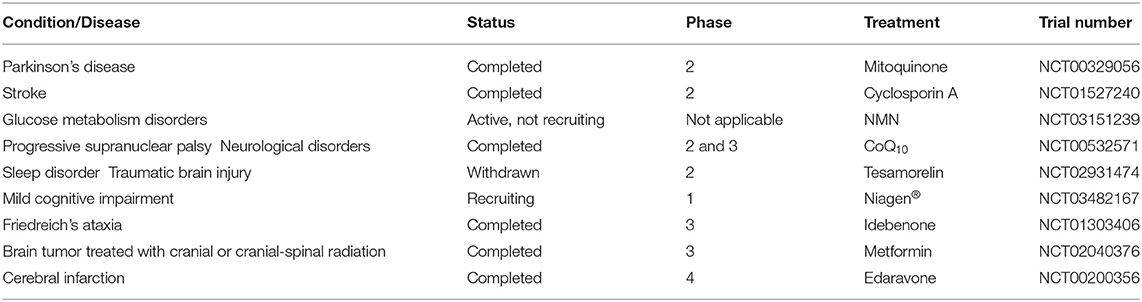
Table 1. Registered drugs targeting mitochondrial dysfunction on ClinicalTrials.gov.
The clinical trials using mitochondrial protectants or antioxidants have not been successful or fulfilled yet. ROS and mitochondrial damage are produced rapidly and have a cascade amplification effect (Qu et al., 2016). This time window of administration may be a problem for patients. In future clinical research, researchers should pay attention to the time window of antioxidant drugs to improve the therapeutic effect of ICH. In addition, a more comprehensive study of the mechanism of mitochondrial damage can also provide a more perfect strategy for the treatment of ICH.
Author Contributions
WC, CG, and YC drafted the manuscript and figures. HF and YC proofread and revised the manuscript. YC gave the final proof for this submission. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82001263 to WC), State Key Laboratory of Trauma, Burn and Combined Injury (SKLYQ202002 to YC), Southwest Hospital (SWH2018BJKJ-05 to YC), and Chongqing Talent Program (4139Z2391 to HF). The funders are not involved in the commissioning, conception, planning, design, conduct, or analysis of the work, the preparation or editing of the manuscript, or the decision to publish.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Area-Gomez, E., Guardia-Laguarta, C., Schon, E. A., and Przedborski, S. (2019). Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J. Clin. Invest. 129, 34–45. doi: 10.1172/JCI120848
Aronowski, J., and Zhao, X. (2011). Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42, 1781–1786. doi: 10.1161/STROKEAHA.110.596718
Augustynek, B., Kudin, A. P., Bednarczyk, P., Szewczyk, A., and Kunz, W. S. (2014). Hemin inhibits the large conductance potassium channel in brain mitochondria: a putative novel mechanism of neurodegeneration. Exp. Neurol. 257, 70–75. doi: 10.1016/j.expneurol.2014.04.022
Baines, C. P., and Gutierrez-Aguilar, M. (2018). The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 73, 121–130. doi: 10.1016/j.ceca.2018.05.003
Balami, J. S., and Buchan, A. M. (2012). Complications of intracerebral haemorrhage. Lancet Neurol. 11, 101–118. doi: 10.1016/S1474-4422(11)70264-2
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Bertero, E., and Maack, C. (2018). Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 122, 1460–1478. doi: 10.1161/CIRCRESAHA.118.310082
Bobinger, T., Burkardt, P., Huttner, B. H., and Manaenko, A. (2018). Programmed cell death after intracerebral hemorrhage. Curr. Neuropharmacol. 16, 1267–1281. doi: 10.2174/1570159X15666170602112851
Bock, F. J., and Tait, S. W.G. (2020). Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100. doi: 10.1038/s41580-019-0173-8
Chen, H. C., Kanai, M., Inoue-Yamauchi, A., Tu, H. C., Huang, Y., Ren, D., et al. (2015). An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat. Cell Biol. 17, 1270–1281. doi: 10.1038/ncb3236
Chen, W., Guo, C., Huang, S., Jia, Z., Wang, J., Zhong, J., et al. (2020a). MitoQ attenuates brain damage by polarizing microglia towards the M2 phenotype through inhibition of the NLRP3 inflammasome after ICH. Pharmacol. Res. 161:105122. doi: 10.1016/j.phrs.2020.105122
Chen, W., Guo, C., Jia, Z., Wang, J., Xia, M., Li, C., et al. (2020b). Inhibition of mitochondrial ROS by MitoQ alleviates white matter injury and improves outcomes after intracerebral haemorrhage in mice. Oxid. Med. Cell Longev. 2020:8285065. doi: 10.1155/2020/8285065
Cheng, A., Yang, Y., Zhou, Y., Maharana, C., Lu, D., Peng, W., et al. (2016). Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 23, 128–142. doi: 10.1016/j.cmet.2015.10.013
Chimeh, U., Zimmerman, M. A., Gilyazova, N., and Li, P. A. (2018). B355252, a novel small molecule, confers neuroprotection against cobalt chloride toxicity in mouse hippocampal cells through altering mitochondrial dynamics and limiting autophagy induction. Int. J. Med. Sci. 15, 1384–1396. doi: 10.7150/ijms.24702
Chinopoulos, C. (2018). Mitochondrial permeability transition pore: back to the drawing board. Neurochem. Int. 117, 49–54. doi: 10.1016/j.neuint.2017.06.010
Chou, S. H., Lan, J., Esposito, E., Ning, M., Balaj, L., Ji, X., et al. (2017). Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke 48, 2231–2237. doi: 10.1161/STROKEAHA.117.017758
Cloonan, S. M., Glass, K., Laucho-Contreras, M. E., Bhashyam, A. R., Cervo, M., Pabon, M. A., et al. (2016). Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 22, 163–174. doi: 10.1038/nm.4021
Cordonnier, C., Demchuk, A., Ziai, W., and Anderson, C. S. (2018). Intracerebral haemorrhage: current approaches to acute management. Lancet 392, 1257–1268. doi: 10.1016/S0140-6736(18)31878-6
Cunnane, S. C., Trushina, E., Morland, C., Prigione, A., Casadesus, G., Andrews, Z. B., et al. (2020). Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 19, 609–633. doi: 10.1038/s41573-020-0072-x
Dawson, T. M., and Dawson, V. L. (2017). Mitochondrial mechanisms of neuronal cell death: potential therapeutics. Annu. Rev. Pharmacol. Toxicol. 57, 437–454. doi: 10.1146/annurev-pharmtox-010716-105001
Dey, S., Demazumder, D., Sidor, A., Foster, D. B., and O'rourke, B. (2018). Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ. Res. 123, 356–371. doi: 10.1161/CIRCRESAHA.118.312708
Ding, W., Chen, R., Wu, C., Chen, W., Zhang, H., Fan, X., et al. (2017). Increased expression of HERPUD1 involves in neuronal apoptosis after intracerebral hemorrhage. Brain Res. Bull. 128, 40–47. doi: 10.1016/j.brainresbull.2016.11.006
Duan, X., Wen, Z., Shen, H., Shen, M., and Chen, G. (2016). Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid. Med. Cell Longev. 2016:1203285. doi: 10.1155/2016/1203285
Eisner, V., Picard, M., and Hajnoczky, G. (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765. doi: 10.1038/s41556-018-0133-0
Fan, L. F., He, P. Y., Peng, Y. C., Du, Q. H., Ma, Y. J., Jin, J. X., et al. (2017). Mdivi-1 ameliorates early brain injury after subarachnoid hemorrhage via the suppression of inflammation-related blood-brain barrier disruption and endoplasmic reticulum stress-based apoptosis. Free Radic. Biol. Med. 112, 336–349. doi: 10.1016/j.freeradbiomed.2017.08.003
Feigin, V. L., Krishnamurthi, R. V., Parmar, P., Norrving, B., Mensah, G. A., Bennett, D. A., et al. (2015). Update on the global burden of ischemic and hemorrhagic stroke in 1990–2013: the GBD 2013 study. Neuroepidemiology 45, 161–176. doi: 10.1159/000441085
Fenton, A. R., Jongens, T. A., and Holzbaur, E. L.F. (2020). Mitochondrial dynamics: shaping and remodeling an organelle network. Curr. Opin. Cell Biol. 68, 28–36. doi: 10.1016/j.ceb.2020.08.014
Forrester, S. J., Kikuchi, D. S., Hernandes, M. S., Xu, Q., and Griendling, K. K. (2018). Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 122, 877–902. doi: 10.1161/CIRCRESAHA.117.311401
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Georgieva, E., Ivanova, D., Zhelev, Z., Bakalova, R., Gulubova, M., and Aoki, I. (2017). Mitochondrial dysfunction and redox imbalance as a diagnostic marker of “free radical diseases”. Anticancer Res. 37, 5373–5381. doi: 10.21873/anticanres.11963
Giacomello, M., Pyakurel, A., Glytsou, C., and Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. doi: 10.1038/s41580-020-0210-7
Giorgi, C., Marchi, S., and Pinton, P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730. doi: 10.1038/s41580-018-0052-8
Gollihue, J. L., and Norris, C. M. (2020). Astrocyte mitochondria: Central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res. Rev. 59:101039. doi: 10.1016/j.arr.2020.101039
Gong, Z., Pan, J., Shen, Q., Li, M., and Peng, Y. (2018). Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 15:242. doi: 10.1186/s12974-018-1282-6
Hafner, A. V., Dai, J., Gomes, A. P., Xiao, C. Y., Palmeira, C. M., Rosenzweig, A., et al. (2010). Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923. doi: 10.18632/aging.100252
Han, H., Hu, J., Yan, Q., Zhu, J., Zhu, Z., Chen, Y., et al. (2016). Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 13, 1517–1524. doi: 10.3892/mmr.2015.4726
Hangen, E., Blomgren, K., Benit, P., Kroemer, G., and Modjtahedi, N. (2010). Life with or without AIF. Trends Biochem. Sci. 35, 278–287. doi: 10.1016/j.tibs.2009.12.008
Hanley, D. F., Thompson, R. E., Rosenblum, M., Yenokyan, G., Lane, K., Mcbee, N., et al. (2019). Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): a randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet 393, 1021–1032. doi: 10.1016/S0140-6736(19)30195-3
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. doi: 10.1038/nature18928
He, Z., Ning, N., Zhou, Q., Khoshnam, S. E., and Farzaneh, M. (2020). Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med. 146, 45–58. doi: 10.1016/j.freeradbiomed.2019.11.005
Heyck, M., Bonsack, B., Zhang, H., Sadanandan, N., Cozene, B., Kingsbury, C., et al. (2019). The brain and eye: treating cerebral and retinal ischemia through mitochondrial transfer. Exp. Biol. Med. 244, 1485–1492. doi: 10.1177/1535370219881623
Hsieh, C. H., Li, L., Vanhauwaert, R., Nguyen, K. T., Davis, M. D., Bu, G., et al. (2019). Miro1 Marks Parkinson's disease subset and Miro1 reducer rescues neuron loss in Parkinson's models. Cell Metab. 30, 1131–1140 e1137. doi: 10.1016/j.cmet.2019.08.023
Hu, X., Tao, C., Gan, Q., Zheng, J., Li, H., and You, C. (2016). Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid. Med. Cell Longev. 2016:3215391. doi: 10.1155/2016/3215391
Huang, J., and Jiang, Q. (2019). Dexmedetomidine protects against neurological dysfunction in a mouse intracerebral hemorrhage model by inhibiting mitochondrial dysfunction-derived oxidative stress. J. Stroke Cerebrovasc. Dis. 28, 1281–1289. doi: 10.1016/j.jstrokecerebrovasdis.2019.01.016
Huang, Y., Gao, X., Zhou, X., Xie, B., Zhang, Y., Zhu, J., et al. (2020). Mitophagy in the hippocampus is excessive activated after cardiac arrest and cardiopulmonary resuscitation. Neurochem. Res. 45, 322–330. doi: 10.1007/s11064-019-02916-z
Jing, C. H., Wang, L., Liu, P. P., Wu, C., Ruan, D., and Chen, G. (2012). Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience 213, 144–153. doi: 10.1016/j.neuroscience.2012.03.055
Joshi, D. C., Tewari, B. P., Singh, M., Joshi, P. G., and Joshi, N. B. (2015). AMPA receptor activation causes preferential mitochondrial Ca(2)(+) load and oxidative stress in motor neurons. Brain Res. 1616, 1–9. doi: 10.1016/j.brainres.2015.04.042
Kang, T. C. (2020). Nuclear factor-erythroid 2-related factor 2 (Nrf2) and mitochondrial dynamics/mitophagy in neurological diseases. Antioxidants (Basel) 9:617. doi: 10.3390/antiox9070617
Kashatus, D. F., Lim, K. H., Brady, D. C., Pershing, N. L., Cox, A. D., and Counter, C. M. (2011). RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 13, 1108–1115. doi: 10.1038/ncb2310
Katsu, M., Niizuma, K., Yoshioka, H., Okami, N., Sakata, H., and Chan, P. H. (2010). Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J. Cereb. Blood Flow Metab. 30, 1939–1950. doi: 10.1038/jcbfm.2010.45
Kim-Han, J. S., Kopp, S. J., Dugan, L. L., and Diringer, M. N. (2006). Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke 37, 2457–2462. doi: 10.1161/01.STR.0000240674.99945.4e
Kumar, S., Selim, M., Marchina, S., and Caplan, L. R. (2016). Transient neurological symptoms in patients with intracerebral hemorrhage. JAMA Neurol. 73, 316–320. doi: 10.1001/jamaneurol.2015.4202
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuv. Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Lemasters, J. J. (2014). Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (Type 3). Redox. Biol. 2, 749–754. doi: 10.1016/j.redox.2014.06.004
Li, B., Zhang, Y., Li, H., Shen, H., Wang, Y., Li, X., et al. (2020). Miro1 regulates neuronal mitochondrial transport and distribution to alleviate neuronal damage in secondary brain injury after intracerebral hemorrhage in rats. Cell Mol. Neurobiol. 40. doi: 10.1007/s10571-020-00887-2. [Epub ahead of print].
Li, J., Lu, J., Mi, Y., Shi, Z., Chen, C., Riley, J., et al. (2014). Voltage-dependent anion channels (VDACs) promote mitophagy to protect neuron from death in an early brain injury following a subarachnoid hemorrhage in rats. Brain Res. 1573, 74–83. doi: 10.1016/j.brainres.2014.05.021
Licznerski, P., Park, H. A., Rolyan, H., Chen, R., Mnatsakanyan, N., Miranda, P., et al. (2020). ATP synthase c-subunit leak causes aberrant cellular metabolism in fragile X syndrome. Cell 182, 1170–1185 e1179. doi: 10.1016/j.cell.2020.07.008
Liu, F., Lu, J., Manaenko, A., Tang, J., and Hu, Q. (2018). Mitochondria in ischemic stroke: new insight and implications. Aging Dis. 9, 924–937. doi: 10.14336/AD.2017.1126
Liu, K., Ji, K., Guo, L., Wu, W., Lu, H., Shan, P., et al. (2014). Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 92, 10–18. doi: 10.1016/j.mvr.2014.01.008
Lu, H., Jiang, M., Lu, L., Zheng, G., and Dong, Q. (2015). Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of Huperzine A after acute intracerebral hemorrhage. Neuropsychiatr. Dis. Treat. 11, 2649–2657. doi: 10.2147/NDT.S92158
Luo, X., O'neill, K. L., and Huang, K. (2020). The third model of Bax/Bak activation: a Bcl-2 family feud finally resolved? F1000Research 9:F1000 Faculty Rev-935. doi: 10.12688/f1000research.25607.1
Ma, J., Ni, H., Rui, Q., Liu, H., Jiang, F., Gao, R., et al. (2019). Potential roles of NIX/BNIP3L pathway in rat traumatic brain injury. Cell Transplant. 28, 585–595. doi: 10.1177/0963689719840353
Ma, Q., Chen, S., Hu, Q., Feng, H., Zhang, J. H., and Tang, J. (2014). NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann. Neurol. 75, 209–219. doi: 10.1002/ana.24070
Ma, R., Ma, L., Weng, W., Wang, Y., Liu, H., Guo, R., et al. (2020). DUSP6 SUMOylation protects cells from oxidative damage via direct regulation of Drp1 dephosphorylation. Sci. Adv. 6:eaaz0361. doi: 10.1126/sciadv.aaz0361
Macaskill, A. F., Rinholm, J. E., Twelvetrees, A. E., Arancibia-Carcamo, I. L., Muir, J., Fransson, A., et al. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. doi: 10.1016/j.neuron.2009.01.030
Mariani, E., Polidori, M. C., Cherubini, A., and Mecocci, P. (2005). Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 827, 65–75. doi: 10.1016/j.jchromb.2005.04.023
Mendelow, A. D., Gregson, B. A., Rowan, E. N., Murray, G. D., Gholkar, A., Mitchell, P. M., et al. (2013). Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet 382, 397–408. doi: 10.1016/S0140-6736(13)60986-1
Montessuit, S., Somasekharan, S. P., Terrones, O., Lucken-Ardjomande, S., Herzig, S., Schwarzenbacher, R., et al. (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901. doi: 10.1016/j.cell.2010.08.017
Nakamura, T., Kuroda, Y., Yamashita, S., Zhang, X., Miyamoto, O., Tamiya, T., et al. (2008). Edaravone attenuates brain edema and neurologic deficits in a rat model of acute intracerebral hemorrhage. Stroke 39, 463–469. doi: 10.1161/STROKEAHA.107.486654
Nakamura, T., and Lipton, S. A. (2011). Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 18, 1478–1486. doi: 10.1038/cdd.2011.65
Nighoghossian, N., Berthezene, Y., Mechtouff, L., Derex, L., Cho, T. H., Ritzenthaler, T., et al. (2015). Cyclosporine in acute ischemic stroke. Neurology 84, 2216–2223. doi: 10.1212/WNL.0000000000001639
Pekkurnaz, G., Trinidad, J. C., Wang, X., Kong, D., and Schwarz, T. L. (2014). Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158, 54–68. doi: 10.1016/j.cell.2014.06.007
Picca, A., Guerra, F., Calvani, R., Bucci, C., Lo Monaco, M. R., Bentivoglio, A. R., et al. (2019). Mitochondrial dysfunction and aging: insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 20:805. doi: 10.3390/ijms20040805
Porter, G. A., and Beutner, G. (2018). Cyclophilin D, somehow a master regulator of mitochondrial function. Biomolecules 8:176. doi: 10.3390/biom8040176
Qu, J., Chen, W., Hu, R., and Feng, H. (2016). The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxid. Med. Cell Longev. 2016:2592935. doi: 10.1155/2016/2592935
Rong, R., Xia, X., Peng, H., Li, H., You, M., Liang, Z., et al. (2020). Cdk5-mediated Drp1 phosphorylation drives mitochondrial defects and neuronal apoptosis in radiation-induced optic neuropathy. Cell Death Dis. 11:720. doi: 10.1038/s41419-020-02922-y
Sabirzhanov, B., Stoica, B. A., Zhao, Z., Loane, D. J., Wu, J., Dorsey, S. G., et al. (2016). miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 23, 654–668. doi: 10.1038/cdd.2015.132
Salihu, A. T., Muthuraju, S., Idris, Z., Izaini Ghani, A. R., and Abdullah, J. M. (2016). Functional outcome after intracerebral haemorrhage–a review of the potential role of antiapoptotic agents. Rev. Neurosci. 27, 317–327. doi: 10.1515/revneuro-2015-0046
Shi, R. Y., Zhu, S. H., Li, V., Gibson, S. B., Xu, X. S., and Kong, J. M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Shinohara, Y., Saito, I., Kobayashi, S., and Uchiyama, S. (2009). Edaravone (radical scavenger) versus sodium ozagrel (antiplatelet agent) in acute noncardioembolic ischemic stroke (EDO trial). Cerebrovasc. Dis. 27, 485–492. doi: 10.1159/000210190
Sidlauskaite, E., Gibson, J. W., Megson, I. L., Whitfield, P. D., Tovmasyan, A., Batinic-Haberle, I., et al. (2018). Mitochondrial ROS cause motor deficits induced by synaptic inactivity: implications for synapse pruning. Redox Biol. 16, 344–351. doi: 10.1016/j.redox.2018.03.012
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi: 10.1146/annurev-biochem-061516-045037
Song, L., Xu, L. F., Pu, Z. X., and Wang, H. H. (2019). IL-10 inhibits apoptosis in brain tissue around the hematoma after ICH by inhibiting proNGF. Eur. Rev. Med. Pharmacol. Sci. 23, 3005–3011. doi: 10.26355/eurrev_201904_17582
Tang, X., Yan, K., Wang, Y., Wang, Y., Chen, H., Xu, J., et al. (2020). Activation of PPAR-beta/delta attenuates brain injury by suppressing inflammation and apoptosis in a collagenase-induced intracerebral hemorrhage mouse model. Neurochem. Res. 45, 837–850. doi: 10.1007/s11064-020-02956-w
Tomasello, F., Messina, A., Lartigue, L., Schembri, L., Medina, C., Reina, S., et al. (2009). Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 19, 1363–1376. doi: 10.1038/cr.2009.98
Tschoe, C., Bushnell, C. D., Duncan, P. W., Alexander-Miller, M. A., and Wolfe, S. Q. (2020). Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J. Stroke 22, 29–46. doi: 10.5853/jos.2019.02236
Wang, X., Mori, T., Sumii, T., and Lo, E. H. (2002). Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke 33, 1882–1888. doi: 10.1161/01.STR.0000020121.41527.5D
Wang, Z., Zhou, F., Dou, Y., Tian, X., Liu, C., Li, H., et al. (2018). Melatonin alleviates intracerebral hemorrhage-induced secondary brain injury in rats via suppressing apoptosis, inflammation oxidative stress, DNA damage, and mitochondria injury. Transl. Stroke Res. 9, 74–91. doi: 10.1007/s12975-017-0559-x
Weir, H. J., Yao, P., Huynh, F. K., Escoubas, C. C., Goncalves, R. L., Burkewitz, K., et al. (2017). Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26, 884–896 e885. doi: 10.1016/j.cmet.2017.09.024
Wu, X., Cui, W., Guo, W., Liu, H., Luo, J., Zhao, L., et al. (2020a). Acrolein aggravates secondary brain injury after intracerebral hemorrhage through Drp1-mediated mitochondrial oxidative damage in mice. Neurosci. Bull. 36, 1158–1170. doi: 10.1007/s12264-020-00505-7
Wu, X., Luo, J., Liu, H., Cui, W., Guo, K., Zhao, L., et al. (2020b). Recombinant adiponectin peptide ameliorates brain injury following intracerebral hemorrhage by suppressing astrocyte-derived inflammation via the inhibition of Drp1-mediated mitochondrial fission. Transl. Stroke Res. 11, 924–939. doi: 10.1007/s12975-019-00768-x
Wu, X., Luo, J., Liu, H., Cui, W., Guo, W., Zhao, L., et al. (2020c). Recombinant adiponectin peptide promotes neuronal survival after intracerebral haemorrhage by suppressing mitochondrial and ATF4-CHOP apoptosis pathways in diabetic mice via Smad3 signalling inhibition. Cell Prolif. 53:e12759. doi: 10.1111/cpr.12759
Xu, S., Wang, P., Zhang, H., Gong, G., Gutierrez Cortes, N., Zhu, W., et al. (2016). CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun. 7:13189. doi: 10.1038/ncomms13189
Yang, J., Cui, X., Li, J., Zhang, C., Zhang, J., and Liu, M. (2015). Edaravone for acute stroke: meta-analyses of data from randomized controlled trials. Dev. Neurorehabil. 18, 330–335. doi: 10.3109/17518423.2013.830153
Yang, J., Liu, M., Zhou, J., Zhang, S., Lin, S., and Zhao, H. (2011). Edaravone for acute intracerebral haemorrhage. Cochrane Database Syst. Rev. CD007755. doi: 10.1002/14651858.CD007755.pub2
You, Y., Hou, Y., Zhai, X., Li, Z., Li, L., Zhao, Y., et al. (2016). Protective effects of PGC-1alpha via the mitochondrial pathway in rat brains after intracerebral hemorrhage. Brain Res. 1646, 34–43. doi: 10.1016/j.brainres.2016.04.076
Yu, J., Zheng, J., Lu, J., Sun, Z., Wang, Z., and Zhang, J. (2019). AdipoRon protects against secondary brain injury after intracerebral hemorrhage via alleviating mitochondrial dysfunction: possible involvement of AdipoR1-AMPK-PGC1alpha pathway. Neurochem. Res. 44, 1678–1689. doi: 10.1007/s11064-019-02794-5
Zandalinas, S. I., and Mittler, R. (2018). ROS-induced ROS release in plant and animal cells. Free Radic. Biol. Med. 122, 21–27. doi: 10.1016/j.freeradbiomed.2017.11.028
Zhang, T., Wu, P., Budbazar, E., Zhu, Q., Sun, C., Mo, J., et al. (2019). Mitophagy reduces oxidative stress via Keap1 (kelch-like epichlorohydrin-associated protein 1)/Nrf2 (nuclear factor-E2-related factor 2)/PHB2 (prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke 50, 978–988. doi: 10.1161/STROKEAHA.118.021590
Zheng, J., Shi, L., Liang, F., Xu, W., Li, T., Gao, L., et al. (2018). Sirt3 ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Front. Neurosci. 12:414. doi: 10.3389/fnins.2018.00414
Zhou, H., Zhu, P., Wang, J., Toan, S., and Ren, J. (2019). DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct. Target Ther. 4:56. doi: 10.1038/s41392-019-0094-1
Zhou, Z. D., and Tan, E. K. (2020). Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson's disease. Ageing Res. Rev. 62:101107. doi: 10.1016/j.arr.2020.101107
Zhuo, F., Qiu, G., Xu, J., Yang, M., Wang, K., Liu, H., et al. (2016). Both endoplasmic reticulum and mitochondrial pathways are involved in oligodendrocyte apoptosis induced by capsular hemorrhage. Mol. Cell Neurosci. 72, 64–71. doi: 10.1016/j.mcn.2016.01.009
Zia, M. T., Csiszar, A., Labinskyy, N., Hu, F., Vinukonda, G., Lagamma, E. F., et al. (2009). Oxidative-nitrosative stress in a rabbit pup model of germinal matrix hemorrhage: role of NAD(P)H oxidase. Stroke 40, 2191–2198. doi: 10.1161/STROKEAHA.108.544759
Zille, M., Karuppagounder, S. S., Chen, Y., Gough, P. J., Bertin, J., Finger, J., et al. (2017). Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48, 1033–1043. doi: 10.1161/STROKEAHA.116.015609
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950. doi: 10.1152/physrev.00026.2013
Keywords: intracerebral hemorrhage, mitochondrial protection, secondary brain injury, mitochondrial membrane potential, stroke
Citation: Chen W, Guo C, Feng H and Chen Y (2021) Mitochondria: Novel Mechanisms and Therapeutic Targets for Secondary Brain Injury After Intracerebral Hemorrhage. Front. Aging Neurosci. 12:615451. doi: 10.3389/fnagi.2020.615451
Received: 09 October 2020; Accepted: 28 December 2020;
Published: 27 January 2021.
Edited by:
Feng Yan, Zhejiang University, ChinaReviewed by:
Zhen-Ni Guo, First Affiliated Hospital of Jilin University, ChinaAnatol Manaenko, University Hospital Erlangen, Germany
Copyright © 2021 Chen, Guo, Feng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujie Chen, eXVqaWVjaGVuNjg4NkBmb3htYWlsLmNvbQ==; orcid.org/0000-0002-9905-9138
†These authors have contributed equally to this work