 Sanghamitra Bandyopadhyay
Sanghamitra Bandyopadhyay- 1Developmental Toxicology Laboratory, Systems Toxicology & Health Risk Assessment Group, CSIR-Indian Institute of Toxicology Research (CSIR-IITR), Lucknow, India
- 2Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, India
Amyloidogenicity and vascular dysfunction are the key players in the pathogenesis of Alzheimer’s disease (AD), involving dysregulated cellular interactions. An intricate balance between neurons, astrocytes, microglia, oligodendrocytes and vascular cells sustains the normal neuronal circuits. Conversely, cerebrovascular diseases overlap neuropathologically with AD, and glial dyshomeostasis promotes AD-associated neurodegenerative cascade. While pathological hallmarks of AD primarily include amyloid-β (Aβ) plaques and neurofibrillary tangles, microvascular disorders, altered cerebral blood flow (CBF), and blood-brain barrier (BBB) permeability induce neuronal loss and synaptic atrophy. Accordingly, microglia-mediated inflammation and astrogliosis disrupt the homeostasis of the neuro-vascular unit and stimulate infiltration of circulating leukocytes into the brain. Large-scale genetic and epidemiological studies demonstrate a critical role of cellular crosstalk for altered immune response, metabolism, and vasculature in AD. The glia associated genetic risk factors include APOE, TREM2, CD33, PGRN, CR1, and NLRP3, which correlate with the deposition and altered phagocytosis of Aβ. Moreover, aging-dependent downregulation of astrocyte and microglial Aβ-degrading enzymes limits the neurotrophic and neurogenic role of glial cells and inhibits lysosomal degradation and clearance of Aβ. Microglial cells secrete IGF-1, and neurons show a reduced responsiveness to the neurotrophic IGF-1R/IRS-2/PI3K signaling pathway, generating amyloidogenic and vascular dyshomeostasis in AD. Glial signals connect to neural stem cells, and a shift in glial phenotype over the AD trajectory even affects adult neurogenesis and the neurovascular niche. Overall, the current review informs about the interaction of neuronal and glial cell types in AD pathogenesis and its critical association with cerebrovascular dysfunction.
Introduction
Alzheimer’s disease (AD) is a prevalent form of dementia, clinically manifested by loss in memory and cognitive decline. As reported, around 50 million people suffer from AD or age-associated dementia, which is projected to exceed 152 million globally by 2050 (Peprah and Mccormack, 2019). However, pure AD comprises less than half of the total cases, while most of the patients predominantly show features of cerebrovascular disease and mixed dementia (Barker et al., 2002). The amyloid β (Aβ) plaques and hyperphosphorylated-tau-induced neurofibrillary tangles (NFTs) in the hippocampus and cortical region have long been considered as the only key pathological features of AD (Jacobs et al., 1992; Jellinger and Bancher, 1998). Nonetheless, over the years significant association of AD with vascular dysfunction has been reported, involving brain metabolic derangements, deregulated Aβ clearance, and a subsequent loss in neuronal homeostasis (Cortes-Canteli and Iadecola, 2020; He et al., 2020a).
AD-related cerebrovascular pathology involves reduced episodic memory, without a loss in hippocampal volume. The vascular abnormalities primarily include cerebral micro- and macro-stroke, hemorrhages, lacunar infarcts, hypoxia, small vessel pathologies, embolism, microbleeds, periventricular and deep white matter hyperintensity (WMH), leukoencephalopathy, hypertensive intracerebral hemorrhages, enlarged perivascular spaces, ischemic lesions, and stroke (Pluta, 2004; Ashok et al., 2016; Kuhn and Sharman, 2020). It has been reported that the production and clearance rate of Aβ is 7.6% and 8.3% per hour respectively in humans, and a shift in this equilibrium towards Aβ production results in Aβ peptide and subsequent plaque deposition in the human brain (Bateman et al., 2006).
An immunohistochemical study in the cortical and hippocampal post-mortem samples of AD brain compared to non-demented elderly controls demonstrated intricate interaction between the neuronal and non-neuronal cells, such as astrocytes, microglia, and vascular endothelial cells in AD pathogenesis and an ultimate neurodegeneration (Kirabali et al., 2020). The astrocytes and microglia independently, as well as collectively, stimulate neuroinflammation and promote pro-inflammatory cytokine release and neuronal loss via cellular crosstalk in AD (Kaur et al., 2019). White matter damage, axonal loss, demyelination, and reduced differentiation of oligodendrocyte progenitor cells (OPCs) also participate in the AD-associated loss in synaptic plasticity (Butt et al., 2019; Vanzulli et al., 2020), and OPC damage comprises the initial signs of pathology, detected in the hippocampus of 3xTg-AD mice (Vanzulli et al., 2020). Additionally, observations in 3xTg-AD triple transgenic AD mice showed early white matter dystrophy and myelin damage in the hippocampus and entorhinal cortex, prior to Aβ and tau pathology or signs of cognitive decline (Desai et al., 2009). Although, the tau pathology correlates more strongly with neuronal cells, participation of glial activation in tau-induced neurodegeneration and tangle burden has been reported in the Tg4510 (rTg4510) mouse line (model of Tauopathy) overexpressing Chemokine C-C motif ligand (Joly-Amado et al., 2020). Decreased capillary vascular endothelial growth factor (VEGF) and endothelial nitric oxide (NO) synthase in the superior temporal and calcarine cortices also associated with Tau pathology in AD patients (Provias and Jeynes, 2008). Moreover, genes involved in immune modulation, inflammation, and the metabolism and functions of microglia, astrocytes, and oligodendrocytes have been implicated in neuronal loss and AD (Chen et al., 2020).
Vascular changes, in association with reactive glial cells, trigger oxidative stress, and reactive oxygen species (ROS) generation, which cause neurodegeneration (Bou Khalil et al., 2016; Govindpani et al., 2019). Build-up of amyloid on the cerebral vasculature in cerebral amyloid angiopathy (CAA) generates vascular oxidative stress, exacerbating undesired glial reactivity and cerebrovascular dysfunction (Carrano et al., 2011; Han et al., 2015). Additionally, observations in the aged Tg2576 mice (overexpressing human APP695) not only showed the participation of vascular oxidative stress in CAA but also indicated the significant role of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-mediated vascular oxidative stress in CAA-induced cerebrovascular changes (Han et al., 2015). CAA is a proven key pathological feature of AD, characterized by Aβ deposition in the wall of arteries, arterioles, capillaries, and blood vessels in the central nervous system (CNS; Revesz et al., 2002). Moreover, breakdown of the neurovascular unit (NVU) that comprises pericytes, neurons, and glia, together with blood-brain barrier (BBB) degeneration and reduced cerebral blood flow (CBF), exacerbates AD neuropathology (Zlokovic, 2011; Parkes et al., 2018).
Both amyloid and vascular concepts are well-studied in AD. Specifically, there have been exploratory studies discerning the cumulative involvements of ROS, inflammation, NVU dysfunction, and changes in the brain microvasculature on AD pathogenesis (He et al., 2020a; Ulamek-Koziol et al., 2020). However, a thorough understanding of the neuronal and vascular impairments, together with the participation of brain cells, in AD is yet awaited. The current review highlights the interaction among different cell types of the brain and enumerates the pathways stimulating AD pathogenesis, focusing on mixed dementia that includes AD-associated plaques, tangles, and vascular dementia.
Current Theories for AD
Several theories govern AD pathology, with the Aβ plaques and NFTs as the primary pathological hallmarks (Masters et al., 2006; Boscolo et al., 2007). Although the “amyloid hypothesis” appears most prominent in AD, NFT generation even seems common for diverse cerebral stress conditions (Khachaturian, 1985; Mirra et al., 1991; Felician and Sandson, 1999). Additionally, the vascular hypothesis emerged as a key theory, considering cardiovascular, cerebrovascular, and old age-related disorders (Kalaria, 1996; de la Torre, 2000b; Kalaria, 2002a). In reality, a synergism of factors augmenting Aβ plaque generation, NFT formation, and vascular abnormalities promotes neurodegeneration, culminating in AD pathology and severe cognitive impairment (de la Torre, 2000a, b; Iadecola, 2003a, b).
Amyloid and Tau Hypotheses
The amyloid cascade hypothesis is a dominant theory of AD pathology, proposed for the last 30 years. The hypothesis claims an amyloidogenic processing of Amyloid precursor protein (APP) by beta-secretase (BACE) to generate C-terminal fragment, CTFβ, and cleavage of the latter by gamma-secretase to release Aβ (more pathologic Aβ42 and soluble Aβ40; Hardy and Higgins, 1992). The amyloid cascade hypothesis focuses on Aβ deposit in brain parenchyma as a key component of senile plaques, with mutations in APP and presenilin (PSEN1 and PSEN2) genes that induce onset in familial AD (Nicolas et al., 2016). Moreover, AD being multifactorial, sporadic AD (whose cause is still elusive) involves the association of several etiological factors, mechanisms, and pathways in the brain (Armstrong, 2013; Gong et al., 2018). Further, a reduced non-amyloidogenic processing of APP by α-secretase [a disintegrin and metalloproteinase (ADAM)-10 and ADAM-17] has been shown as a key pathway promoting Aβ generation (Bandyopadhyay et al., 2006a, 2007).
Inflammatory pathways impact the onset and progression of AD involving metabolic and cellular alterations that trigger neurodegeneration, primarily in the hippocampus, entorhinal and temporoparietal cortex (Risacher and Saykin, 2013; DeTure and Dickson, 2019). The glial cells (that normally generate trophic factors for physiological requirements) undergo aberrant reactivity, resulting in the overproduction of pro-inflammatory mediators in the etiopathogenesis of AD. The altered inflammatory signaling pathways promote Aβ-peptide deposition and then oligomerization into senile plaques, tau hyperphosphorylation, and an ultimate synaptic and cognitive dysfunction (Zlokovic, 2011; Webers et al., 2020).
Both, oxidative stress and inflammation catalyzed Aβ plaque formation, where the 5′ untranslated region (UTR)-APPmRNA harboring the iron-responsive element (IRE) and interleukin-1 responsive element regulated APP translation (Rogers and Lahiri, 2004). Factors promoting ROS generation, lipid peroxidation, and pro-inflammatory cytokine levels stimulated amyloidogenic APP processing. They also induced an upregulation in the 5′UTR-mediated APP translation and neuronal Aβ through intricate participation of reactive astrocyte and microglia, observed in the hippocampus and frontal cortex of rats showing AD-like pathology (Ashok et al., 2015; Maurya et al., 2016). Hence, through studies on neuronal and astrocyte cell lines and in TgCRND8 mice, it has been claimed that targeting APP mRNA-5′UTR may be a potential strategy in limiting APP translation and attenuating AD pathogenesis (Bandyopadhyay et al., 2006b,c, 2010, 2013; Tucker et al., 2006; Bandyopadhyay and Rogers, 2014). AD is also characterized by the neuronal accumulation of toxic NFTs, formed from microtubule-associated hyperphosphorylated tau proteins as paired helical filaments (PHFs). The PHFs inhibit the normal microtubule assembly and stability, neuronal growth and morphology, and axonal and dendritic transport, inducing synaptic loss and neurodegeneration (Iqbal et al., 2010; Mietelska-Porowska et al., 2014; Alonso et al., 2018). The increased cerebral NFTs and cerebrospinal fluid (CSF) p-tau levels bear a prominent link with the severity of dementia and cognitive loss (Maccioni et al., 2001, 2006; Ghoshal et al., 2002). Hence, drugs targeted to the NFT attenuated the pathological symptoms of AD (Duff et al., 2010).
The toxic deposits of insoluble Aβ in the hippocampus and cortex promote the formation of neuritic senile plaques, and a neuron-Aβ interaction induces tau phosphorylation through tau kinase activation or by modulating phosphorylated state of tau, as seen in the primary septal and hippocampal neurons (Takashima et al., 1998; Zheng et al., 2002). Microtubule-associated tau proteins then undergo truncations and conformational shifts to form filamentous aggregates or NFTs (Binder et al., 2005). The neuritic plaques and aggregated tau induce proximal astroglial and microglial reactivity, neuritic dystrophy, and an increased Aβ plaque and NFT deposition in the hippocampal neurons. These conditions eventually induce neuronal death, degenerated nerve endings, cognitive decline, and dementia (Laurent et al., 2018; van Olst et al., 2020).
Aβ peptides also interact with neurons, inducing altered tau aggregations. A direct interaction of Aβ with the neuronal membranes and receptors [nAchR, N-methyl-D-aspartate (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor] trigger abnormal calcium influx and oxidative stress, which lead to the activation of glycogen synthase kinase-3β (GSK-3β), and hence increased tau phosphorylation (Wang et al., 2000; Stancu et al., 2014). In vitro as well as ex vivo studies in neuroblastoma cell lines and synaptosomes, respectively, demonstrated an enhanced Aβ peptide and α7 nAChR interaction, resulting in increased Tau pathology (Stancu et al., 2014). Additionally, cell culture (cultured hippocampal neurons) and animal-based (3xTg-AD) studies suggested the participation of reactive glial cells in acute and chronic inflammation, and further Aβ-mediated Tau pathology (Saez et al., 2006; Kitazawa et al., 2011; Sy et al., 2011).
Vascular Hypothesis
Aβ plaques and NFTs relate to neurodegeneration, supported by increased expression of these pathological hallmarks in the neuronal cell body and in abnormal neuronal processes adjoining hippocampal and cortical Aβ deposits (Duyckaerts et al., 1998; Lewis et al., 2001; Ribe et al., 2005; Cohen et al., 2013; Koss et al., 2016; Park et al., 2019). Alternatively, the vascular hypothesis claims that cerebrovascular diseases are prevalent comorbidities in AD, which induce cognitive impairment and dementia (Humpel and Marksteiner, 2005). AD involves a marked alteration in the structure and morphology of vascular cells and tissues, ensuing a dysregulated cortical blood flow, vascular fluid dynamics, and vessel integrity. Arterial spin-labeling magnetic resonance imaging (MRI) as well as ischemic vascular dementia demonstrated a loss in CBF in the cortex of AD patients (Schuff et al., 2009). A reduced blood supply in the occipital cortex of B6.PS2APP mouse model of AD also correlated with the findings in the cortex of AD patients (Weidensteiner et al., 2009).
Moreover, arterial spin labeling MRI [measured in AβPPSWE/PS1ΔE9 (APP/PS1) transgenic mice model of AD] revealed that alteration in CBF, mainly at the frontoparietal cortex and thalamus regions rather than the hippocampus and entorhinal cortex, had a significant link with early AD and vulnerability to AD pathology (Guo et al., 2019). Further, in vivo, time-lapse, multiphoton microscopy in the APP/PS1 mouse model of AD demonstrated that microvascular lesions regulate the accumulation and clearance of Aβ, indicating a key link between the dynamic morphology of Aβ plaques and vascular dysfunction (Zhang et al., 2020).
Aging factors, exacerbated by cardiovascular disorders and systemic vascular abnormalities, undergo a synergistic interaction, culminating in cerebrovascular damage and severe cognitive decline (Klohs, 2019). Studies in AD patients through single photon emission computed tomography (SPECT), arterial spin-labeling MRI, and [18F] fluorodeoxyglucose positron emission tomography (FDG-PET) indicated a key link between AD and altered metabolic parameters in the temporoparietal cortex (Bonte et al., 1986; Burns et al., 1989; Fazekas et al., 1989; Johnson et al., 2005; Alexopoulos et al., 2012). Hypoperfusion and hypometabolism also appeared as critical features in the angular gyrus (Fazekas et al., 1989; Alexopoulos et al., 2012) and posterior precuneus of parietal cortex in AD patients (Binnewijzend et al., 2016).
Vascular pathology starts with cerebral hypoperfusion, which stimulates glial reactivity and an ultimate neuronal cell death and atrophy, with the loss of CNS functions (Wang et al., 2016). Small vessel disease, structural white matter alterations, dysregulated cerebral vasculature, atherosclerosis, BBB dysfunction, and blood vessel rupture upregulate cortical and hippocampal Aβ levels and induce amyloid fibrillogenesis and neuropathological changes in patients at a risk for AD (Pantoni and Garcia, 1997; Kalaria, 2002b; Kovacic and Fuster, 2012). The vascular inflammatory processes also significantly engage in the pathogenesis and generation of AD. The microvascular endothelial cells that release pro-inflammatory cytokines and chemokines induce BBB disintegration, elevate neurotoxic thrombin release, and augment cerebrovascular oxidative stress, triggering neuronal death and cognitive loss in AD (He et al., 2020a; Iannucci et al., 2020).
An increased activity of the angiotensin-converting enzyme (ACE) that converts Angiotensin I (Ang I) to Angiotensin II (Ang II), together with enhanced vasoconstriction, has been reported in AD (Benigni et al., 2010). Ang II also activates NADPH-mediated oxidative stress that functions as a prime risk factor in AD (Ongali et al., 2014; Halliday et al., 2016; Tarafdar and Pula, 2018). Moreover, augmented perivascular ACE-1 levels promote pathological Aβ misfolding and correlate with the severity of AD, as discerned in post-mortem brain tissues from AD patients (from South West Dementia Brain Bank, University of Bristol) showing a frontal cortex Aβ load and signs of CAA (Miners et al., 2008). In terms of metabolic dysfunctions, individuals with obesity, together with type-II diabetes, insulin resistance, prolonged hyperinsulinemia, atherosclerosis, hypercholesterolemia, or hypertension are at a higher risk of developing AD (Polidori et al., 2012; Borshchev et al., 2019; Hayden, 2019; Lee et al., 2020). Ischemic infarcts, hemorrhagic stroke, capillary degeneration and microvascular damage [characterized by focal constriction and thickened vascular basement membrane in the cerebral leptomeningeal vessels and distal intraparenchymal arteries, arterioles, and capillaries of patients (Thal et al., 2002)] reduce CBF, impair functional hyperaemia, and participate in the amyloid pathogenesis (Lai et al., 2015; Tarantini et al., 2017; Hecht et al., 2018; Solis et al., 2020). Here, structural changes in the vascular basement membrane alter endothelial connectivity and adhesion, leading to BBB damage and a subsequent entry of secreted APP and neurotoxic plasma substances into the brain (Kelleher and Soiza, 2013; Govindpani et al., 2019). The ischemic conditions further promote BACE expression and activity, facilitating APP-amyloidogenic processing in the hippocampus and frontal cortex, as observed in rat and mice bilateral common carotid artery occlusion models of CCH (Ashok et al., 2016; Cai et al., 2017).
Cerebrovascular damage and atherosclerotic lesions are also observed in AD patients and have often been considered as predisposing factors to developing the disease (Gupta and Iadecola, 2015; Alosco et al., 2018). White matter hyperintesity and AD-associated neuropathology were detected by MRI in participants from the National Alzheimer’s Coordinating Center’s Data Sets (Alosco et al., 2018). Studies in patients with subcortical stroke, cognitive impairment, and/or dementia or with radiologically defined lacunes revealed that subcortical lacunar infarcts from occluded and perforated arteries associate with reduced cerebral glucose metabolism, forming a contributory factor for the cognitive decline (Kwan et al., 1999; Reed et al., 2001). Hence, cerebrovascular insufficiency and vascular dementia co-exist with enhanced Aβ and senile plaques (Liu et al., 2015; Greenberg et al., 2020). Pathological amyloid accumulation also contributes to the development of subcortical intracerebral hemorrhage and hemorrhagic stroke in elderly AD patients (Vasilevko et al., 2010; Van Nostrand, 2016; Yanagawa et al., 2019). It distinctly connects with clinical conditions, such as intracranial bleeding, stroke, brain ischemia, ruptured aneurysm, head injury, and recurrent seizures. Additionally, diabetes, myocardial infarction, angina, stroke, arterial occlusion, hypertension, and microvascular diseases have strong vascular components and comprise key risk factors for AD (Petrie et al., 2018). Hence, an upregulated amyloidogenic pathway of Aβ generation, tau hyperphosphorylation, NFT formation, vascular damage, and reduced Aβ clearance from the brain are key pathogenic processes underlying AD pathology and related dementia (Figure 1).
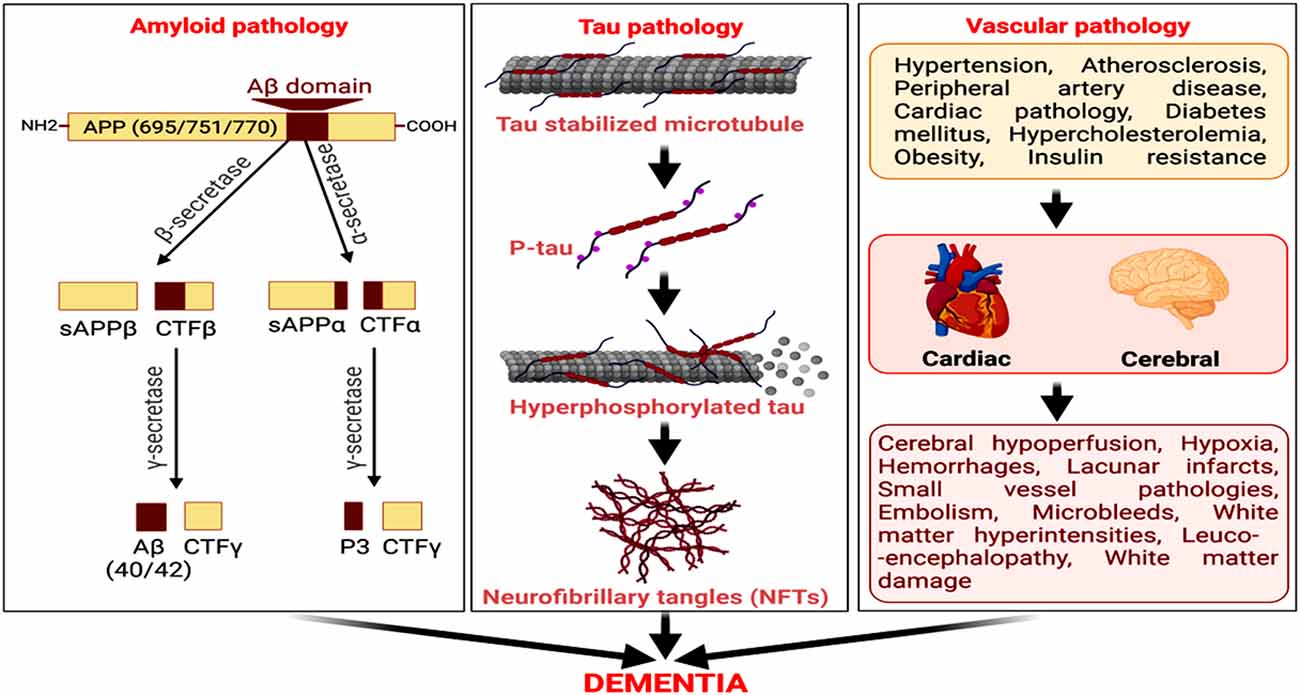
Figure 1. Amyloid, tau, and vascular pathologies as predominant factors for neurodegeneration and cognitive decline in Alzheimer’s disease (AD). The amyloidogenic processing of amyloid precursor protein (APP) by beta-secretase (BACE) and then γ-secretase generates amyloid-β (Aβ). The microtubule-associated protein, tau, undergoes hyperphosphorylation, and self-assemble to form neurofibrillary tangles (NFTs). Cerebrovascular diseases and cardiovascular disorders are also vital risk factors for AD, culminating in dementia.
Notably, BBB impairment appears as a key link between amyloid and vascular pathologies. Here, a dysregulated receptor for advanced glycation end product (RAGE; Aβ influx receptor) and low-density lipoprotein receptor-related protein-1 (LRP1; Aβ influx protein) expression and functioning play a central role in impaired vascular clearance of Aβ from the brain, together with the abnormal functioning of glial cells and neurons (Deane et al., 2004; Deane and Zlokovic, 2007). Activated matrix metaloproteinases (MMPs) induce BBB disruption, and the altered Aβ-RAGE and LRP1-interaction disrupt the tight junctions of BBB (Kook et al., 2012). It has also been shown that chronic cerebral hypoperfusion increased MMP9-mediated BBB damage and promoted the hippocampal amyloidogenic APP-processing towards Aβ (Ashok et al., 2016). Overall, the overlap between amyloid production and vascular aberrations accelerates neurodegeneration and AD pathogenesis, with the intricate participation of glial cells, inflammation, and altered cerebral microvasculature (Figure 2; Wanleenuwat et al., 2019).
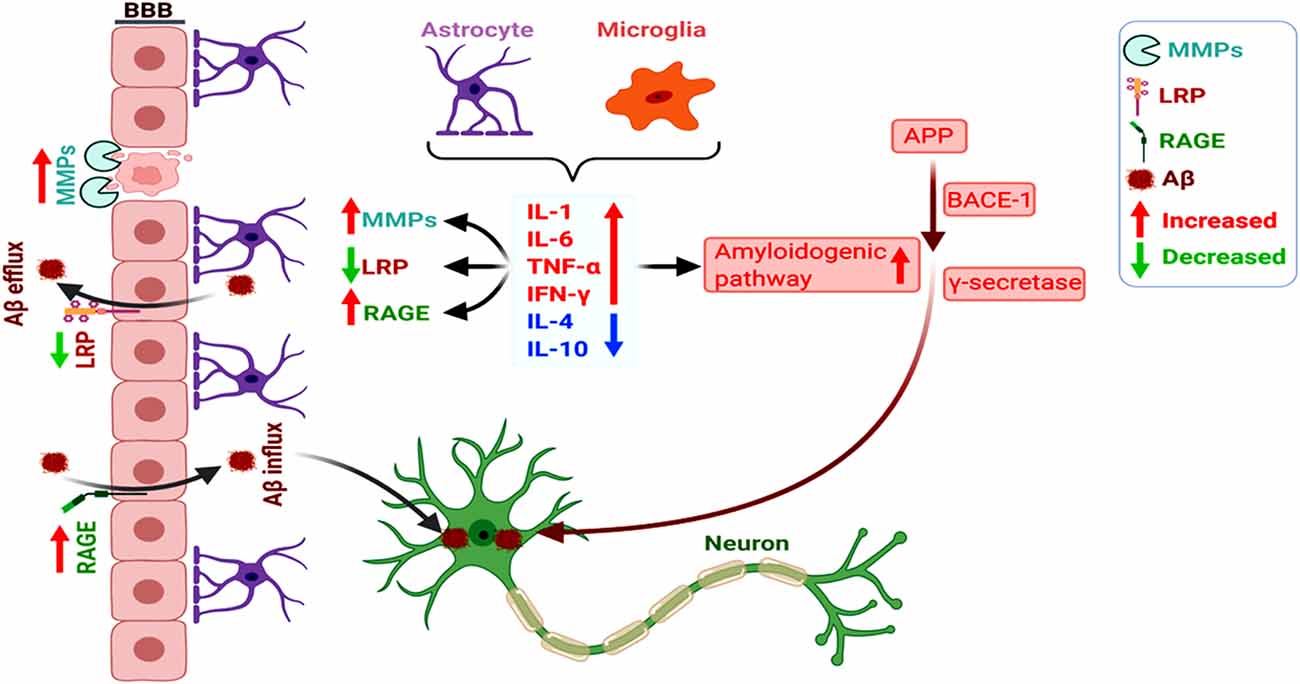
Figure 2. Increased amyloidogenicity, blood-brain barrier (BBB) damage, and dysregulated RAGE and LRP-1 expression induce neuronal Aβ deposition. The pro-inflammatory cytokines (as opposed to anti-inflammatory ones) secreted by the astrocytes and microglia promote the amyloidogenic pathway of APP processing towards Aβ. Glia-mediated inflammation also induces vascular dysfunction involving dysregulated activation of MMPs and altered RAGE and LRP-1-dependent Aβ clearance from the brain, overall increasing neuronal Aβ deposition.
Amyloid Hypothesis and Role of Neurons and Glia
Neuroinflammation
Neuroinflammation is one of the key factors promoting AD pathogenesis, and astrocytes and microglia have a significant contributory role in it. Aβ binds to microglial and astrocytic toll-like receptor 4 (TLR4), initiating MyD88 signaling cascade, which induces the tumor necrosis factor receptor-associated factor 6 (TRAF6) effector component (Okun et al., 2009; Shi et al., 2016; He et al., 2020c). It has also been reported that knockdown or silencing of LRP1 in WT, as well as APP/PS1 double-transgenic mice, resulted in the increased nuclear NF-κB p65 subunit expression, along with enhanced TLR4, MyD88, TRAF6, neuroinflammation, microgliosis and astrogliosis in the hippocampus and cerebral cortex (He et al., 2020b, c). Another microglial membrane receptor, CD33, undergoes an increased expression in the AD brain, where its interaction with the sialic acid residues of Aβ leads to a decreased Aβ clearance. Hence, CD33 knockout in the APP/PS1 mice attenuated Aβ plaque deposition and cognitive decline, respectively (Griciuc et al., 2013; Perea et al., 2020). Moreover, the type I transmembrane glycoprotein, TREM, expressed in the microglial cells, directly interacts with several forms of Aβ, with the strongest affinity for soluble Aβ42 oligomers (Lessard et al., 2018). The elevated TREM2 expression in the 5xFAD mouse model triggered a decrease in the amyloid burden and neurobehavioral deficits (Lee et al., 2018). On the other hand, microglia-mediated phagocytosis of Tau was found to have developed through its interaction with the microglial receptor, CX3CR1, where Tau competed with the neuronal ligand CX3CL1. Corroborating this, CX3CR1 deficient mice and microglial cells demonstrated an aberrant uptake and degradation of Tau (Bolos et al., 2017). Studies in Tg2576 transgenic mice models of AD, macrophage inflammatory protein-1α (MIP-1α−/−), and CC-chemokine receptor 5 (CCR5−/−)-deficient mice demonstrated that a subsequent NF-κB and mitogen-activated protein kinase (MAPK) pathways trigger a shift from the anti-inflammatory to pro-inflammatory phenotypes in astrocytes and microglia. This promoted the generation of cytokines, like interleukin (IL6, IL1α, and IL1β), tumor necrosis factor α (TNFα), granulocyte-macrophage colony-stimulating factor, and macrophage inflammatory proteins, which stimulated AD pathogenesis (Manczak et al., 2009; Passos et al., 2009; Kaur et al., 2019). These cytokines further induce Nitric oxide synthase activity in the glial cells, enhancing NO production, lipid peroxidation, and ultimate neuronal damage (Asiimwe et al., 2016; Figure 3). Astrocytes are one of the most well-studied cell types for neuroinflammation and AD, and the activation of its reactive phenotype involves an upregulated glial fibrillary acidic protein (GFAP) expression. It has also been seen that of the different GFAP isoforms, GFAPα and GFAPδ-immunoreactive astrocytes are usually in abundance near Aβ plaques (Kamphuis et al., 2014).
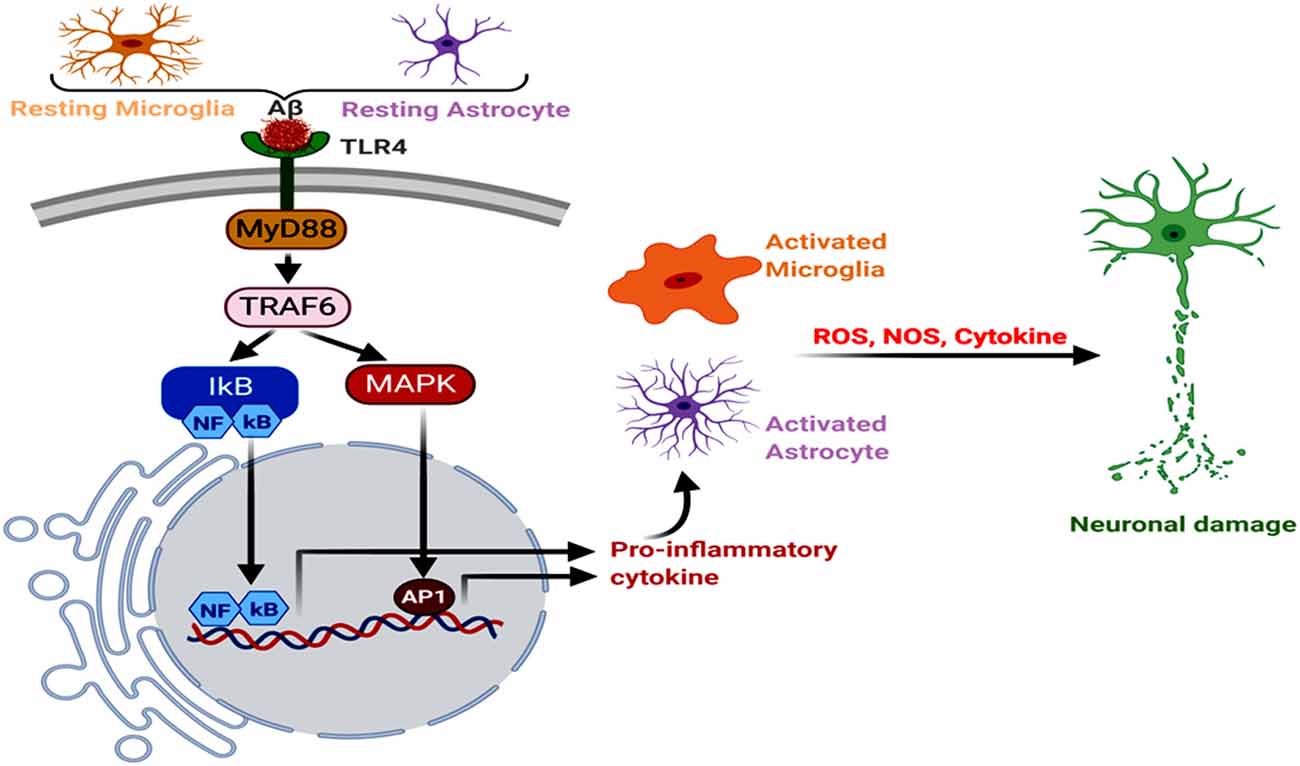
Figure 3. Neuroinflammation, glial Toll-like receptor 4 (TLR4), oxidative stress, and AD. The astroglial and microglial TLR4 interact with Aβ, triggering MyD88→TRAF6→MAPK to NFκB, AP1 and inflammatory signals. Further, increased NOS and reactive oxygen species (ROS) levels induce oxidative stress and neurodegeneration.
Microglia, that comprised the macrophage-like resident immune cells of the CNS, enter the brain during embryonic development and further participate in neuroinflammation, as well as AD pathogenesis in the elderly (Yin et al., 2017). Microglia induce an immune response to Aβ and migrate towards the dense-core senile plaques, with increased immunoreactivity for the reactive microglial markers, CD11b, CD68, complement receptor 3, and CD45 (Mandrekar-Colucci and Landreth, 2010). A distinct association has been reported between the different Aβ forms (monomers, oligomers, and fibril and microglia) and neuroinflammation through the microglial secretion of proinflammatory cytokines (Wang et al., 2015). The microglia-induced neuroinflammation and subsequent AD pathology also relate to the participation of glutamate-associated extrasynaptic NMDA receptor and GluR1 activation and an ultimate Aβ generation and neuronal cell death, studied in the hippocampal slices of mice (Wang et al., 2004; Heneka et al., 2013). Specifically, Nlrp3(−/−) or Casp1(−/−) mutations in WT and APP/PS1 mice demonstrated reduced hippocampal and cortical inflammation, apoptosis, and Aβ deposition, as well as decreased cognitive decline (Heneka et al., 2013). Moreover, an Aβ-associated microglial NLRP3 inflammasome induced the caspase-1-dependent cleavage of pro-IL-1β/pro-IL-18 into IL-1β/IL-18, as well as lysosomal damage and cathepsin B release, which further initiated pro-inflammatory and chemotactic mediators, inflammation cascade, and neurodegeneration (Hanslik and Ulland, 2020). Increased generation of Apolipoprotein E (APOE) associates with glia-derived cholesterol dyshomeostasis (Mauch et al., 2001), altering the communication between endoplasmic reticulum and mitochondria at the mitochondria-associated ER membranes. This impairs normal mitochondrial functioning and enhances a pro-inflammatory state in the brain, as also observed in an astrocyte-conditioned media model (Tambini et al., 2016; Tzioras et al., 2019). APOE affects inflammatory environments by controlling the expression of glial genes related to immune functions, which influence the central amyloid metabolism and the etiology and pathophysiology of AD (Liu et al., 2013). Moreover, Nuclear factor erythroid 2- (NF-E2-) related factor 2 (Nrf2), which acts through the Nrf2/antioxidant responsive element pathway, is a key neuroprotective factor in AD, having a distinct link with reactive astrogliosis and microglial reactivity in the hippocampus. Corroborating this, it was observed that inflammatory microglia infiltrated and surrounded the Aβ plaque following Nrf2 ablation in APP/PS1 transgenic mice, exacerbating the generation of pro-inflammatory cytokines and neurological impairments (Ren et al., 2020). Conversely, NRF2 activator, methysticin, attenuated the hippocampal microglial reactivity, astrogliosis, neuroinflammation, oxidative damage, and long–term memory deficit in the APP/PS1 mice (Fragoulis et al., 2017).
Although the glial cells are key participants in neuroinflammation, the peripheral immune cells also infiltrate the brain and regulate AD pathology. In normal conditions, peripheral immune cells are present in the brain parenchyma. Conversely, a range of peripheral immune cells, such as macrophages, neutrophils, and lymphocytes infiltrate the brain in AD (Unger et al., 2020; Wyatt-Johnson and Brutkiewicz, 2020). Notably, the peripherally derived immune cells, such as monocytes, migrate (into the CNS) close to the Aβ deposits in AD, and further form macrophages to promote clearance of Aβ (Hohsfield and Humpel, 2015). A transgenic mouse model of AD showed that this monocyte trafficking was markedly controlled by the chemokine receptor, CCR2, which underwent a decrease in AD, enhancing cerebral Aβ deposition and cognitive decline (Naert and Rivest, 2011). Moreover, the infiltrating neutrophils served as a key source of inflammatory mediators, myeloperoxidase, ROS, and intravascular neutrophil extracellular traps (Zenaro et al., 2015; Rossi et al., 2020). The neutrophil migration into the CNS also induced damage to the vascular permeability and BBB integrity (Rossi et al., 2021). Additionally, the enhanced Aβ levels induced peripheral immune cell exhaustion, with the participation of regulatory T cells (Tregs) in amyloid clearance as well as amyloid pathology. While, the depletion of Tregs inhibited the microglial recruitment of Aβ towards amyloid deposits, selective amplification of Tregs increased the plaque-associated microglial count and spatial learning and memory loss in the transgenic APP/PS1 mice (Dansokho et al., 2016). Furthermore, the CD3+/CD4+ T-helper and CD3+/CD8+ cytotoxic T-cells were found in the brain of transgenic mice for AD (Ferretti et al., 2016; Merlini et al., 2018; Unger et al., 2018a, b). It has been reported that CD3+ T-cells are associated with Tau rather than Aβ amyloid plaque pathology (Merlini et al., 2018). A strong association between CD8+ T-cells in the brain parenchyma with microglia, neurons, and neuroinflammation has also been observed. The CD8+ T-cell population was regulated by microglia, with the increase in the CD8+ T-cells migration into the brain in microglia-depleted APP-PS1 transgenic mouse brain (Unger et al., 2018b). The removal of CD8+ T-cells triggered a significant change in neuronal and synaptic markers, indicating their potential participation in the AD-induced changes in neuronal functioning and synaptic plasticity (Unger et al., 2020). Moreover, Interferon gamma generated from the infiltrating Th1 cells increased plaque burden and AD pathology in the APP/PS1 mice model of AD (Browne et al., 2013).
Glutamate Signaling
Amyloid fibrils stimulate the activity of metabotropic glutamate receptor 1 (mGluRl) that plays a key role in the regulation of synaptic plasticity and cognition, as observed in the APPsw/PSEN1DE9 mice (Bie et al., 2019; Li and Selkoe, 2020). Astrocytes express glutamate transporters, participating in the active uptake of glutamate at the synaptic and extrasynaptic sites of the CNS (Rose et al., 2017). The astrocytic glutamate-aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1) reduce glutamate burden in the synaptic cleft and promote the synthesis of glutamine through increased glutamine synthase activity. Hence, a reduction in these astrocytic transporter levels accelerates the development of cognitive deficits in AD, as observed using a D-[3H]-aspartic acid uptake assay in cultured hippocampal astrocytes and neurons (Tong et al., 2017; Pajarillo et al., 2019). It has been reported that Aβ1–42 induces abnormal GLT-1 localization and internalization in astrocytes, inhibiting the clearance of synaptically released extracellular glutamate levels. This involves the participation of oxidative stress and inflammation pathways. Aβ1–42 also causes inflammation-related transcriptional alteration in GLT-1 and GLAST levels, predominantly in astrocytes, as observed in astrocytes from acute mouse hippocampal slices (Scimemi et al., 2013). A subsequent spread of glutamate, from one synaptic domain to another, enhances extrasynaptic NMDAR-mediated excitotoxicity, which restricts the normal neuronal survival, activity and networking and consequent synaptic transmission (Figure 4; Findley et al., 2019). An accompanying immediate neuronal Ca2+ influx, involving the activation of GluN2B-containing NMDARs, inhibits hippocampal long-term potentiation (Liu et al., 2019). Other than dyshomeostasis in extracellular glutamate level, a marked link between altered astrocytic GLT1 and complement proteins that participate in microglial synaptic remodeling has also been reported in AD. Aging involves an upregulated synaptic classical complement cascade in the CNS, initiated by C1q protein (observed in the aged C1q-deficient mice; Stephan et al., 2013) and microglia mediates the synaptic elimination through phagocytosis (Luchena et al., 2018; Lee and Chung, 2019). Additionally, GLT1 induces presynaptic metabotropic glutamate receptor activity and reduces the constitutive glutamatergic strength, amplitude and frequency of spontaneous miniature excitatory synaptic current in hippocampal CA1 neurons. The astrocyte-microglia interaction through complement activation here plays a vital role in amyloid pathology as reported in primary glial cells and hippocampal and cortical regions of APP transgenic mice (Lian et al., 2016). Moreover, the complement factors C3 and C1q that regulate innate immunity, microglia-mediated synaptic pruning, and neuronal survival undergo significant activation in the brain and promote AD pathogenesis (Hansen et al., 2018).
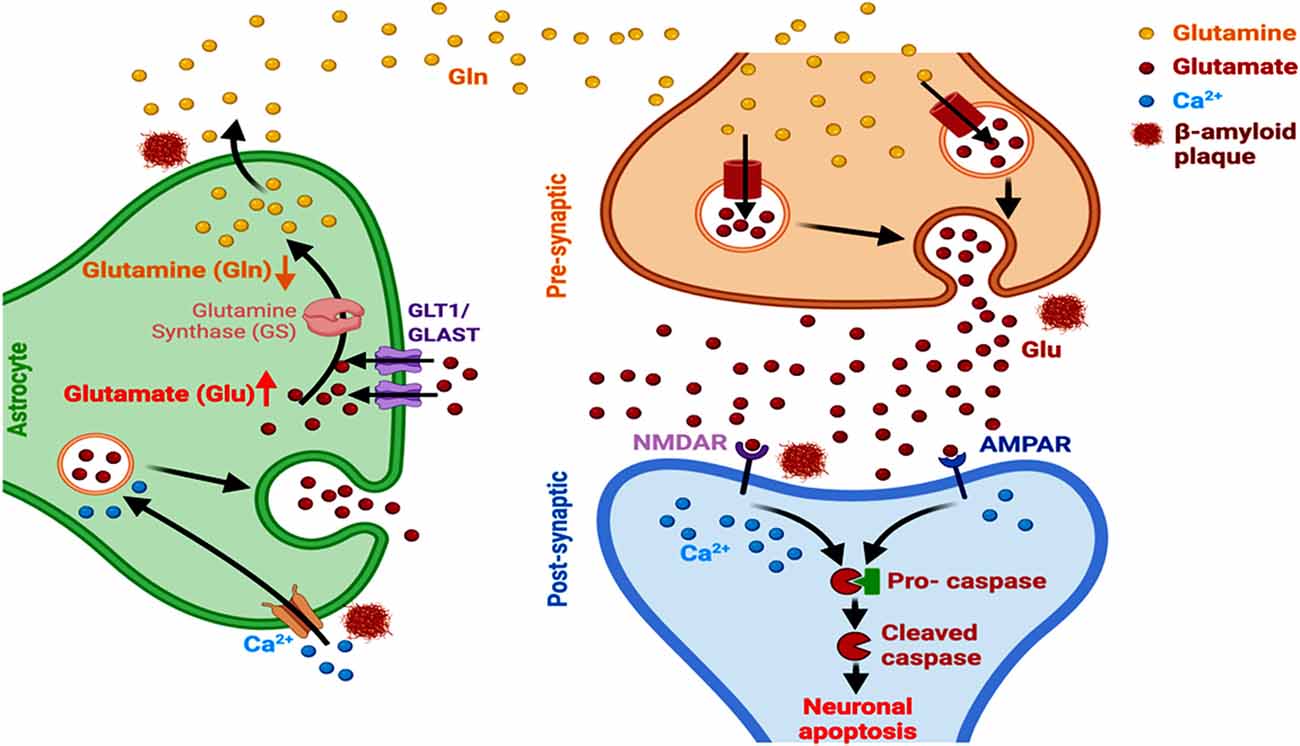
Figure 4. Astrocyte-dependent glutamate signaling and AD. Aβ alters the expression of GLT1 and GLAST transporters, alters glutamate-glutamine ratio, and induces astrocytic glutamate excitotoxicity and Ca2+ entry into the cells. This ultimately leads to a modulated activation and functioning of NMDARs and AMPAR, culminating in neuronal apoptosis in AD.
Lipoprotein Dysfunction
APP, PSEN1 or PSEN2 trigger familial AD, together with cholesterol and lipoprotein dysfunction, altered peripheral lipid metabolism and polymorphic gene expression (Dai et al., 2018; Huentelman et al., 2019). LRP1, that has a significant contribution in vascular dysfunctions (Ramanathan et al., 2015), modulates Aβ generation via altered regulation of proteases, growth factors and an eventual inflammation (Ashok et al., 2016; He et al., 2020b). LRP1 is abundant in brain cells, and a neuronal deletion of LRP1 stimulates astrogliosis and microglial reactivity and the NF-κB and MAPK signaling pathways. Corroborating this, a reduced hippocampal LRP1 expression has been observed in the transgenic AD mice (He et al., 2020b, c).
Unlike early-onset AD, that involves genetic mutations in APP, PSEN1 and PSEN2, late-onset AD (LOAD) that comprises 95% of AD cases, shows a distinct association with the isoforms of APOE gene (Corder et al., 1993). APOE-ε4 carrier in the brain is the key genetic risk factor for LOAD, and each APO-E-ε4 allele reduces the age of AD onset (Liu et al., 2013). The ApoE isoform shows a significant contribution in AD pathogenesis, characterized by increased gliosis, Aβ accumulation and tau hyperphosphorylation. As reported in the human subjects, this ε4 variant of APOE is predominantly secreted by the astroglial cells within the brain and associates with innate immune responses (Gale et al., 2014). Inducible expression of human ApoE4 in astrocytes of mouse models at the initial phase of amyloid development results in significant Aβ aggregation and fibrillar plaque formation in the cortex and hippocampus, together with neuritic dystrophy (Liu et al., 2017). The ApoE4 isoform reduces lipoprotein-mediated cholesterol transport from astrocytes to neurons, disrupting cholesterol homeostasis in neurons and glia. It leads to an aberrant Aβ accumulation, microglial inflammatory responses, altered synapse pruning activity in astrocytes, synaptic degeneration and neuronal cell death. APOE4-containing lipoproteins reduce the uptake of oligomeric and fibrillar Aβ in astrocytes and decrease microglial Aβ clearance, resulting in increased Aβ accumulation (Lin et al., 2018). A mouse model of tauopathy showed that the ApoE4 variant exacerbated tau pathology, independent of Aβ, involving microglial reactivity and upregulation of pro-inflammatory genes (Shi et al., 2017). The altered astrocytic APOE4 gene expression often appears as the reason for Aβ and tau-induced neural circuit hyperactivity in AD. An APOE4-mediated dysregulation of the membrane-lipid composition and endolysosomal homeostasis, in association with altered intracellular cholesterol distribution, Ca2+ signaling, and excitability, have been observed in the astrocytes of hippocampal slices from APOE3 and APOE4 gene-targeted replacement mice (Larramona-Arcas et al., 2020). The APOE allele also contributes to altered gliotransmission and neural-circuit stimulation in LOAD. This involves lysosome dysregulation-mediated Ca2+ excitability, changes in the membrane lipidome and distribution of intracellular cholesterol, and altered Ca2+ responses. An astrocytic APOE-cholesterol complex undergoes transport by ATP-binding cassette transporter ABCA1 and is then absorbed into neurons through LRP1 and metabolized to 24-hydroxycholesterol by Cytochrome P450 Family 46 Subfamily A Member 1 (CYP46A1; Czuba et al., 2017). This elevated 24-hydroxycholesterol level (essentially suppresses the expression of the transcription factors SREBP-2 and SREBP-1C, LDLR and HMG CoA-reductase, which restrict cholesterol levels in astrocytes), secreted from the brain into the plasma and CSF, marks the early-onset stages of AD (Loera-Valencia et al., 2019). The astrocyte and microglia-derived APOE4 stimulates the formation and deposition of Aβ in the neurons. Additionally, APOE4 obtained from the pericytes and other cells of the NVU induce BBB damage and dysfunction, as observed in the post-mortem paraffin-embedded human frontal cortex tissue samples acquired from the University of Southern California AD Research Center (Halliday et al., 2016). Of the different APOE isoforms, the AD pathogenicity follows the sequence of apoE4 > apoE3 > apoE2. Conversely, capacity for Aβ clearance and enzymatic degradation of Aβ appears reduced for apoE4 compared to the other isoforms (Figure 5).
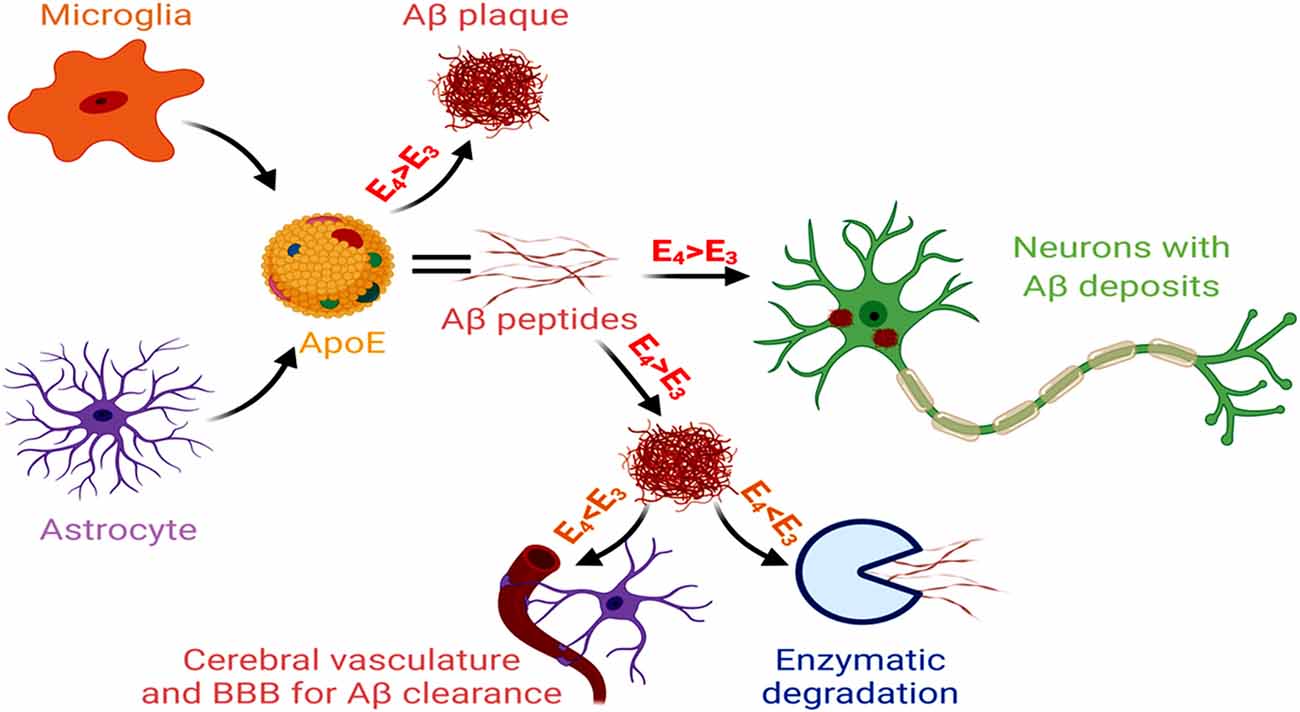
Figure 5. Glial cells, APOE and AD. Microglial and astrocytic APOE, with greater involvement of APOE4 > ApoE3, act as risk factors for Aβ generation and also participate in increased vascular damage. On the other hand, APOE3 > ApoE4 promotes Aβ clearance and degradation. Hence, the dysregulated APOE3 and APOE4 levels promote AD pathology.
A direct association between LRP1 and Tau has also been reported, where their potential interaction via the microtubule binding repeat region of tau regulated tau endocytosis in neurons. Moreover, LRP1 deficiency in neuroblastoma cell line and mice led to reduced LRP1 interaction, resulting in decreased neuronal propagation of tau (Rauch et al., 2020). Thus, LRP1 functions as a common factor which regulates the Aβ and tau pathology in AD.
Glia Cells (Astrocytes and Microglia), PRR, TREM2, Complement Activation, and Aβ Degradation
Via cell-surface pattern recognition receptors (PRRs) that sense immune stimulation in the brain, microglial cells recognize the neurodegeneration-associated molecular patterns (NAMPs) in AD (Deczkowska et al., 2018). This triggers microglial transformation from an inactive “ramified” to an activated “hypertrophic or ameboid morphology.” A microglial deficit in clearing Aβ induces characteristics of LOAD, and a genome-wide association study in AD patients showed a distinct connection between reduced chances of AD and mutations in the microglial PRRs (Hickman et al., 2018). Microglial cells express the Triggering receptor expressed on myeloid cells 2 (TREM2) along with its adaptor protein and signal transduction partner, DNAX-activating protein of 12 kDa (DAP12), which control microglial reactivity and affect neuronal fate (Konishi and Kiyama, 2018). Under physiological conditions, a tyrosine kinase-mediated phosphorylation of DAP12 regulates cytoskeletal reorganization and cytokine release (Painter et al., 2015). Here, a normal TREM2/DAP12-interaction sustains the mammalian target of the rapamycin (mTOR) pathway. This is essential for the microglial biosynthetic metabolism, while an altered TREM2 expression deregulates the morphological and functional responses of microglial autophagy and its response to Aβ, as detected using the 5xFAD mouse model and AD patients, as well as in bone marrow-derived macrophages and microglia (Ulland et al., 2017). TREM2 binds to lipids, phospholipids, glycolipids (released from damaged neurons), as well as APOE and Aβ in the extracellular milieu of the AD brain (Mecca et al., 2018). It induces microglial reactivity, pro-inflammatory cytokine release, and the vicious AD pathogenicity, as studied in a series of AD patients, showing Aβ deposition in the leptomeningeal blood vessels, hippocampal neuritic plaques, and cortical NFTs. The findings resulted from a genome, exome, and Sanger sequencing followed by a meta-analysis-based study that analyzed the TREM2 genetic variability in AD (Guerreiro et al., 2013). The TREM2 and APOE interaction also induces a phenotypic switch from the homeostatic state to a “neurodegenerative” disease-associated one reported in the APP-PS1 mice (Krasemann et al., 2017). Moreover, reduced expression of the anti-amyloidogenic ADAMs inhibits the ADAM-mediated shedding of TREM2 ectodomain and generation of soluble TREM2 (sTREM2) into the CSF. The sTREM2 augments microglial viability through activated PI3K and NFκB pathways and helps in the physiological functions of microglia (Zhong et al., 2017). Insertion of sTREM2 in the brain of 5xFAD mice promotes Aβ uptake and degradation, and provides a neuroprotective role against amyloid pathology (Zhong et al., 2019). High-resolution confocal and in vivo two-photon imaging of brain slices (cortex, layer IV–VI) in the AD-like transgenic 5xFAD and CRND8 mice models showed that microglial cells generate a dynamic regulation of Aβ plaque morphology from less-compacted and fibrillar plaques to more compacted neurotoxic ones (Condello et al., 2015). This involves an altered TREM2 expression and deregulated microglia-mediated sustenance of the plaque morphology, reducing Aβ clearance and neuronal dystrophy (Condello et al., 2018; Cianciulli et al., 2020).
Receptors such as, sialic acid-binding immunoglobulin-like lectin (Siglec) or CD33 bind to glycoproteins and glycolipids within Aβ plaques and auto-trigger their signaling pathways, promoting AD-induced neuronal loss (Puigdellivol et al., 2020). The CD33 and CD22 activation also inhibit the functioning of immunoreceptor tyrosine-based activation motifs (ITAM) that participate in microglial phagocytosis, reducing Aβ clearance (Zhao, 2019). The enhanced microglial expression of C-Type lectin domain containing 7A (CLEC7A), that also functions through ITAMs, promotes AD pathology via reduced phagocytosis, as observed in TgCRND8 mice (Hansen et al., 2018; Rothman et al., 2018). Another PRR, RAGE, binds to Aβ, and an increased microglial expression of RAGE stimulates a MAPK (P38 and ERK1/2)-mediated microglial migration, viability, and growth, inducing activation of NF-kB pathway and upregulation of IL-1β and TNFα. Aβ accentuates RAGE expression through a positive feedback loop, which attenuates hippocampal expression of acetylcholinesterase-positive neurites and axonal activity, together with increased neuronal stress and cognitive deficit, as observed in the transgenic mice with the neuronal human mutant APP and microglial RAGE expression (Fang et al., 2010). An age-induced stimulation of the microglial receptors, CR3 and C1qR, that predominantly participate in synaptic pruning during brain development, often involves their increased binding with Aβ, causing hippocampal synaptic degeneration and learning memory loss (Hong et al., 2016). Conversely, CR3 deficiency enhances proteolytic degradation of Aβ, decreases neuronal loss and increases cognitive functioning, particularly in AD pathology (Czirr et al., 2017). Thus, unrestricted activation of the complement signaling induces synaptic degeneration and memory impairment, which may be inhibited upon blocking this pathway.
The degradation of monomeric, oligomeric, and fibrillar forms of Aβ is mediated by neprilysin (NEP), insulin-degrading enzyme (IDE), and ACE, which play a defensive role (Ries and Sastre, 2016). NEP deficiency involves an increased amyloidogenicity, with augmented neuronal and peripheral inflammation, and a normal or upregulated astrocyte and microglial NEP stimulate plaque degradation and clearance (Nalivaeva et al., 2012). Microglial cells induce an incomplete degradation of Aβ involving a decreased hydrolytic activity of one or more enzymes at the microtubule organizing center and lysosomes (Sole-Domenech et al., 2016). A significant link has also been seen between astroglial APOE and Aβ-degrading enzyme in a genotype-dependent manner, resulting in abnormal amyloid metabolism and altered immune function of glial cells, neuroinflammation and AD pathogenesis (Fernandez et al., 2019). The scavenger receptors, TLRs, induce changes in NEP expression, with TLR2, TLR3, and TLR9-dependent decrease in NEP and a TLR4-mediated and APOE genotype-dependent differential modulation of NEP expression. This has been studied in mice “humanized” for APOE, termed as targeted replacement (TR) mice and the primary glial cultures isolated from the TR APOE ε3 and TR APOE ε4 mice (Graykowski et al., 2020). In fact, the APOE genotype-independent reduction in NEP has been attributed to decreased Aβ clearance (Kanekiyo et al., 2014).
A receptor-mediated endocytosis and fluid-phase pinocytosis also participate in microglial Aβ degradation, as found in the immortalized BV-2 murine microglial cell line as well as Cx3cr1/Gfp++ mice (Mandrekar et al., 2009). Purinergic G protein-coupled receptors (P2Y2 and P2Y6) take part in microglial Aβ clearance, with greater efficiency in the internalization of Aβ protofibrils compared to monomers through phagocytosis and pinocytosis (Sole-Domenech et al., 2016). The microglial degradation of Aβ monomers also occurs via secreted IDE without involving lysosomal degradation (Qiu et al., 1997; Vepsalainen et al., 2008). This involves the participation of clathrin- and caveolae-dependent processes through ERK and p38 kinase/JNK pathways, ultimately stimulating endocytosis or endocytic biogenesis, as reported in the study using the HMO6 human microglial cell line (Jang et al., 2015). In addition, lysosomal biogenesis, pH, and hydrolase properties of lysosomes influence astrocyte-mediated fibrillar and oligomeric Aβ clearance, as reported in a study involving primary astrocytes, immortalized mouse neural progenitor cell line, C17.2 cells, and APP/PS1 transgenic mice (Xiao et al., 2014). Moreover, the sirtuin family members that regulate energy metabolism and senescence, mainly in relation to vascular pathology and atherogenesis, facilitate the degradation of oligomeric Aβ and Tau in the primary astrocytes via the lysosomal pathway (Li et al., 2018).
Genetic Factors
The rapid advancement of genome analysis technology has led to the recognition of risk factors for AD. CR1 is a complement receptor found on macrophages and microglia, and results in blood samples from AD and nondemented elderly subjects (from the National Institute on Aging Alzheimer’s Disease Centre, the Arizona AD Research Centre) suggested its strong link with AD, owing mainly to erythrocyte CR1-based mechanisms supporting pathogen clearance (Johansson et al., 2018). Granulin (GRN) is the precursor of the growth factor progranulin, expressed in neurons and microglia. GRN polymorphisms and deficiency induce aberration in lysosomal functioning, together with the enhanced and increased generation of complement factors. Increased C1q and C3 stimulate phagocytosis and alter microglial reactivity, which causes synaptic and cognitive loss in AD (Mendsaikhan et al., 2019). Polymorphism in the Interleukin (IL)-1 receptor accessory protein (IL1RAP) gene (rs12053868G), with functional receptor for IL-1α and IL-1β, stimulates microglial dysfunction. This results in reduced phagocytosis and clearance of Aβ, and increased plaque deposition in AD patients and the carriers of IL1RAP gene polymorphism (Hansen et al., 2018). Other genetic factors, such as the ATP-binding cassette transporter A7 (ABCA7; with a protective role in AD) has several variants, i.e., rs3764650, rs3752246, and rs115550680, and loss-of-function and nonsense mutations in the gene augment AD pathology. Pathogenic mechanisms that are induced during ABCA7 dysregulation also impact the properties and functioning of neurons and microglia (Aikawa et al., 2018). Other closely linked ABCA1 and ABCG1 cholesterol transporter genes (that encode the generation of high-density lipoprotein complexes) mediate the lipid-transporting activity for ApoE, indicating its participation in AD pathobiology (Koldamova et al., 2010). Additionally, ABCA1 and ABCA7 deficiencies modulate neuronal apoptosis and reduce the removal of cellular debris, promoting the accumulation of Aβ in the brain (Koldamova et al., 2010). The coding variant of phospholipase C-gamma-2 (PLCG2), expressed in microglia, regulates cytosolic Ca2+ influx and the inositol trisphosphate pathway, essential for suppressing AD pathomechanisms (Koldamova et al., 2010). The PI3K-regulated ABI family member 3 (ABI3) gene, which forms a key constituent of the Wiskott-Aldrich syndrome protein family verprolin-homologous protein regulatory complexes, controls cytoskeletal actin polymerization. Its coding variant (rs616338; p.S209F) shows a direct link with increased risk of LOAD (Kurisu and Takenawa, 2009; Sims et al., 2017). The ABI3 is expressed in the microglia of the AD brain, exhibiting a distinct association with amyloid plaques, as observed in the frontal cortex and the hippocampus of the autopsied brain from AD patients (Satoh et al., 2017). Microglial CELF1 gene variant rs1057233G, that demonstrates a link with an attenuated SP11, also reduced AD pathology through increased release of Aβ into the CSF from the brain (Hansen et al., 2018). This SPI1 encodes the transcription factor PU.1 that regulates the ABCA7, CD33, MS4A4A, MS4A6A, TREM2, TREML2 and TYROBP gene expression and controls the phagocytic activity of Aβ, seen through a genome-wide survival study in AD patients and controls from the International Genomics of Alzheimer’s Project Consortium (Huang et al., 2017). Microglial MEF2C gene, which shows a distinct connectivity with CX3C chemokine receptor 1 and tumor necrosis factor, is also a genetic risk factor for AD (Chen et al., 2016).
Oligodendrocytes and AD
Demyelination, characterized by reduced myelin basic protein (MBP), is prominent in AD, and has been considered as a predictor for AD onset and associated neurodegeneration (Nasrabady et al., 2018). Loss in myelin integrity within the hippocampus, detected in 3xTg-AD mice models, indicates that oligodendrocytes undergo a pathophysiological assault during the disease pathogenesis (Desai et al., 2010). Monocarboxylic acid transporter 1 (MCT1) protein helps oligodendrocytes in providing metabolic support to neurons, and AD models showed a significant reduction in the brain MCT1 levels, indicative of axon damage and neuron loss (Mot et al., 2018). MRI together with advanced positron emission tomography demonstrated increased white matter damage at frontal and temporal lobes of the brain during early, preclinical, and later stages of AD, prior to any apparent clinical signs (Khan, 2018). Additionally, impairment in the recruitment, migration, proliferation, differentiation, and regeneration into oligodendrocytes of the chondroitin sulfate proteoglycan Neural/glial antigen 2 (NG2)-expressing OPCs is a common occurrence in AD. The upregulated OPC proliferation plays a protective role, where the NG2 cells surrounding Aβ plaques undergo macropinocytosis-mediated engulfment and a subsequent autophagy-lysosome-dependent internalization and degradation of Aβ (Li et al., 2013). NG2 cells are also located spatially near the microglia and astrocytes, demonstrating a close link with AD-induced neuroinflammation (Nirzhor et al., 2018).
Lipopolysaccharides (LPS), that play a key regulatory role in myelination, also undergo co-localization with amyloid plaques and perivascular amyloid in the AD brain (Zhan et al., 2018). Impaired BBB in AD facilitates the entry of LPS into the brain, where it interacts with the TLR4/CD14 receptors on the peripheral monocytes/macrophages, neutrophils, and brain microglia. The ensuing NFκB activation triggers the generation of IL-1, IL-6, and TNFα cytokines, which degrade the axon and myelin sheath and enhance AD pathogenesis (Zhan et al., 2018). Together with decreased total protein, lipid, and cholesterol levels and reduced oligodendrocyte nucleus diameter, myelin sheath forms a key lesion site in AD. It has also been observed that an abnormal myelination precedes axon defects and axon transport prior to the appearance of Aβ and tau pathology (Cai and Xiao, 2016). Supporting this, MRI scanning on a unit of cognitively asymptomatic adults enriched for AD risk revealed a marked correlation between myelin water fraction (that depicts age-related myelin alterations in the cerebral white matter), brain white matter integrity, and the Aβ and p-tau levels, predominantly at the regions affected in AD (Dean et al., 2017). A primary reason for the observation appears to be a direct LPS binding to oligodendrocytes in the white matter as well as gray matter, augmenting cytokine and free radical generation. Moreover, very high levels of cytokines trigger the death of OPC and augment mature oligodendrocyte and myelin sheath injury, causing aberrant myelin aggregation in AD (Schmitz and Chew, 2008). LPS-induced neuroinflammation also enhances cerebral APP and Aβ, which bind to TLR4, further increasing the Aβ levels via a positive feedback loop (Zhan et al., 2018). Here, MBP binds and degrades APP and Aβ, reducing Aβ fibrillization, which often results from LPS-induction and myelin damage, where the latter fails to prevent Aβ aggregation owing to the inhibited autolytic activity of MBP (Liao et al., 2009). The injured oligodendrocytes and myelin sheath, degraded MBP, degenerated axons, and aggregated Aβ thereby promote Aβ plaque deposition (Papuc and Rejdak, 2020).
AD involves a disordered development of oligodendrocyte, characterized by an unevenly thick myelin sheath, reduced internodal gap, and enhanced NG2 and 2’,3’-Cyclic-nucleotide 3’-phosphodiesterase in the oligodendrocyte lineage cells. This has also been detected through histological and electrophysiological studies of the hippocampus and behavioral assays in the human platelet-derived growth factor beta polypeptide platelet-derived growth factor beta polypeptide (PDGFB)-APPSw.Ind transgenic mice (Ferreira et al., 2020). Oligodendrocyte development is critically linked to neuregulin expression from axons tightly regulated by caspase-6 activation. The AD-induced altered myelin morphology shows a critical association with caspase-6-dependent neuregulin type III cleavage (Hu et al., 2016). An increased neuregulin III expression induces hypermyelination, deregulates myelin morphology, alters electrical impulse along the axon and at the nerve terminals and induces an ultimate temporal and spatial progression of cognitive impairment (Hu et al., 2016). White matter alterations participate in AD pathogenesis, involving inflammatory responses. Presenilin and APP mutations enhance the susceptibility of oligodendrocytes towards stress and alter axon-mediated information to cells, affecting normal myelination (Nasrabady et al., 2018). It has also been hypothesized that myelin degeneration in the late-myelinating white matter tracts releases iron that enhances AD pathogenicity via oxidative stress (Raz and Daugherty, 2018).
Aβ, via an oxidative mechanism, triggers the activity of sphingomyelinase (NSMase)-ceramide in the cell membrane, releasing ceramide that has pro-apoptotic properties and a subsequent oligodendrocyte dysfunction (Jana et al., 2009). Moreover, inflammation and oxidative stress have been found to be the two key factors relating oligodendrocyte death to the NFTs (Cai and Xiao, 2016).
Clinical as well as histological studies demonstrated a distinct link between white matter degeneration and myelin atrophy in AD, where the white matter-induced neurodegeneration resulted from localized or remote cerebral injury (Nasrabady et al., 2018). A difference in the distribution pattern for white matter damage has been observed, and LOAD involved damage to the medial temporal region. Moreover, early-onset AD affected the white matter of the lateral temporal cortex, parietal cortex, cingulum, and corpus callosum (Migliaccio et al., 2012). Increased Aβ levels also demonstrated a distinct link with oligodendrocyte death, as a key feature of white matter demyelination, which further augmented neurodegeneration (Nasrabady et al., 2018).
AD features signs of leukoaraiosis in the white matter region, associated with demyelination, glial cell death, and intercellular edema in the epidermis (Rosenberg et al., 2016). The extent of leukoaraiosis appears correlated with the severity of cognitive fall and functional alteration in AD, as reported from a multicenter study comprising patients with AD and mild cognitive impairments (Sarabia-Cobo et al., 2014). WMH increases with age, hypertension and, the blood homocysteine, IL-1, and inflammation-induced C-reactive protein levels, having a distinct link with the pathobiology of AD. Moreover, APOE ε2 allele, IL-1β-511T allele homozygotes, and CRP-286T allele carriers that promote inflammation related to WMH, demonstrate a key link with AD pathology (Raz et al., 2012).
Vascular Pathology, Brain Cells, and AD
A marked overlap exists between cerebrovascular dysfunction, loss of neurovascular integrity, and AD, with reported cases of lacunar infarcts in more than 70% of AD patients (Govindpani et al., 2019). The hypoxia-induced insufficient CBF impairs the normal endothelial activity, inducing vasoconstriction. Thus, vascular complication involves the accumulation of toxic substances from the blood, resulting in decreased glucose and oxygen levels in the brain (Morikawa et al., 2012).
The BBB, comprised of microvessels and endothelial cells (in the brain capillaries) along with pericytes, astrocytic end-feet, microglia, and neuronal processes of NVU, restricts the undesired exchange of toxic materials between the CNS and systemic blood circulation. A dysfunctional NVU and BBB may precede AD pathology, involving extracellular Aβ-associated reactive astroglia and microglia, vascular wall thickening, reduction in smooth muscle cells, and neuritic loss (Soto-Rojas et al., 2021). The subsequent induction of astrocyte and microglia-induced inflammatory mediators, such as IL-1, TNFα, and the complement component subunit 1q (C1q; Liddelow et al., 2017), further enhance NVU and BBB damage through a feedback loop, with the accelerated generation of p-tau and NFT. The NVU-linked microglial cells associate with Aβ via CR1 (CD35) fAβ receptors, which appears as a critical reason for altered BBB and NVU integrity (Fonseca et al., 2016; Soto-Rojas et al., 2021). Microglial participation through increased contact size at the basement membrane is a key to the development of perivascular glia limitans surrounding the brain capillary vessels of the NVU, particularly during inflammation. This induces vascular permeability, endothelial phagocytosis, and altered leukocyte extravasation, as studied in a Middle cerebral artery occlusion model and wild-type C57Bl/6 mice and CX3CR1+/GFP mice (Jolivel et al., 2015; Joost et al., 2019). The reactive microglia promote BBB dysfunction, accompanied by dysregulated reorganization and signaling of tight junction proteins and controlled transendothelial electrical resistance in AD (Sumi et al., 2010; Zenaro et al., 2017). Additionally, the reactive microglia secrete excess IL-1β, which reduces BBB integrity, promotes vascular permeability and augments neutrophil trafficking through the BBB, as observed in AD patients (Zenaro et al., 2015). The VEGF that mediates a microglial chemotactic response undergoes an upregulation in AD, attracting microglia around blood vessels and parenchyma, which co-occurs with neovascularization and decreased blood flow (Ryu et al., 2009; Zhao et al., 2018). An increased VEGF expression is also correlated with angiogenesis and inflammation following Aβ peptide insertion in the rat brain hippocampus (Zand et al., 2005).
However, BBB integrity reduces with age and brain inflammation (Daneman and Prat, 2015). Since astrocytes are one of the fundamental cellular components of the BBB, reactive astrogliosis and a loss of end-feet processes in the aging brain act as contributing factors for BBB disruption and reduced Aβ efflux (Yamazaki and Kanekiyo, 2017). Astrocytes, via end-feet processes, associate with thousands of synapses and capillaries for neurovascular coupling, leading to a regulated CBF and signaling in response to neuronal activity (MacVicar and Newman, 2015). Moreover, astrocytes express LRP1 that causes Aβ efflux from the brain through the BBB (Sagare et al., 2012). Hence, a cerebrovascular injury-induced astrocyte damage and altered BBB permeability cause faulty clearance of Aβ, accentuating AD pathology (Erickson and Banks, 2013; Sweeney et al., 2018). Moreover, an altered expression, distribution, polarization, and mislocalization of aquaporin-4 (AQP4), at the astrocyte endfeet surrounding blood vessels and blood CSF barrier, lead to BBB impairment. AQP4 sustains the astroglial water transport, and its altered expression and functioning hinder the normal CSF flux into the parenchyma, hamper solute clearance through interstitial fluid (ISF) drainage and disturb cerebrovascular Aβ clearance. Increased thickening of the vascular basement membrane and altered cellular and extracellular constituents occur during aging, impeding the drainage of Aβ through the ISF.
Endothelial basement membrane anchors cerebral and vascular cells at the BBB. On the other hand, astrocytes help in the formation of the parenchymal basement membrane, rich in the extracellular matrix proteins, laminin α1, and α2 isoforms, that organize the spatial arrangement and physical integrity of the gliovascular interface (Thomsen et al., 2017). In addition to the astrocytes, pericytes associate with endothelial cells and regulate the soluble platelet-derived growth factor receptor β in the CSF. Thus, a disproportionate astrocyte-pericyte-endothelial interaction and pericyte deficit stimulate cerebral Aβ deposition and CAA formation (Yamazaki and Kanekiyo, 2017).
Vascular dysfunctions are normally observed at the initial stages of AD pathogenesis, with the local cerebral hypoxia playing a contributory role (Salminen et al., 2017). In vitro and in vivo studies showed that hypoxia enhances the expression and activity of BACE 1, reduces the functioning of α-secretase proteins (ADAM10 and ADAM17) and neuroprotective sAPPα levels. Hypoxia restricts NEP activity in neurons, astrocytes, and vascular cells, leading to enhanced Aβ generation and accumulation together with glutamate excitotoxicity in the hypoxic/ischemic brain (Auerbach and Vinters, 2006; Marshall et al., 2006; Sun et al., 2006, 2012; Fisk et al., 2007; Guglielmotto et al., 2009; Grammas et al., 2011; Pluta et al., 2016). Increased levels of Hypoxia inducible factor 1-alpha (HIF-1α) in the microcirculation of AD patients play a key contributory role in aggravating angiogenesis and vascular activation (Grammas et al., 2011). HIF-1α functions as a vital regulator of immune cell responses in inflamed tissues (Mcgettrick and O’neill, 2020). HIF-1α stimulates the activation of NFκB via cross-talk, resulting in neuroinflammation and generation of cytokines, ROS, and NO in the brain (Lukiw et al., 2003). This could resist the functioning of the insulin and insulin like growth factors (IGF)-1 and 2, alter glucose uptake and metabolism, trigger aberrant mitochondrial (mt) activity and reduce oxidative phosphorylation, which further triggers a mtDNA damage, reduces adenosine triphosphate formation, activates glycogen synthase kinase-3β (GSK-3β)-induced tau phosphorylation and increases Aβ formation (Ashok et al., 2017). Both hypoxia and AD associate with energy metabolic deficiencies and oxidative stress, and cerebral microvessels from AD patients release thrombin, VEGF, angiopoietin-2 (Ang-2), MMPs as well as inflammatory proteins, which correspond to the hypoxic responses (Grammas and Ovase, 2001; Grammas et al., 2006). The hypoxia-induced altered blood vessel function, capillary diameter, and endothelial metabolic properties synergize with AD pathology to induce dementia. The reactive astroglia, microglia, and inflammatory macrophages surround angiogenic factors in cerebral tissues, where the Prostaglandin E2 mediates the inflammatory responses and hypoxia-associated cellular angiogenesis in AD (Ko et al., 2015). The participation of inflammation in hypoxia-induced AD pathology is also evident from the study showing an IL-1β-mediated generation of VEGF gene expression in AD (Pogue and Lukiw, 2004; Grammas, 2011). Overall, reactive glial cells are abundant near parenchymal fibrillar amyloid plaques and inflammatory mediators (Mandrekar-Colucci and Landreth, 2010). Additionally, in CAA type-1, marked by the extension of vascular amyloid angiopathy and perivascular clearance, a strong expression of reactive astroglial cells and microglia has been observed around dyshoric Aβ-laden capillaries.
Conclusion
Damage to the vascular-neuronal axis leads to the pathogenesis of AD. The glial cells have a significant impact on AD pathogenesis, amyloid plaque formation, and cerebrovascular damage, involving diverse pathological pathways and mechanisms. The process results in dendritic loss, synaptic nerve damage, altered neurotransmission, and an overall hippocampal and cortical degeneration.
The neuronal plaques penetrate through the glial cells and macrophages, and further into the capillaries and cerebral parenchyma. This progression associates with glial scarring, which leads to increased fibrillogenesis and its infiltration into the cell membrane and cytosolic process, together with evidence of immune dysfunction and DNA damage as well. In association with this, a dysregulated functioning of neurovascular and gliovascular units, loss in BBB integrity, altered angiogenesis and neurogenesis, particularly in association with stroke, cardiovascular and cerebrovascular risk factors, cerebral and vascular injury, and neuroinflammation have been observed in AD. Hence, targeting the glial cells, immunomodulatory pathways, and NVU appears important in reducing neurodegeneration. Overall, novel therapies reducing glial dysfunction and compromised vascular functioning may have a significant role in improving neuronal integrity, and triggering increased cognitive function and reduced dementia in AD.
Author Contributions
I have written, compiled, and arranged the manuscript.
Funding
The author acknowledges grant support (GAP340 and GAP398) of Science and Engineering Research Board, Department of Science & Technology, Government of India.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SB also recognizes the help from Dr. Debabrata Ghosh, Senior Scientist, CSIR-Indian Institute of Toxicology Research in improving the Figures and Asmita Garg, UGC-JRF, CSIR-Indian Institute of Toxicology Research in the process of manuscript submission. The Institutional manuscript number is 3702.
References
Aikawa, T., Holm, M. L., and Kanekiyo, T. (2018). ABCA7 and pathogenic pathways of Alzheimer’s disease. Brain Sci. 8:27. doi: 10.3390/brainsci8020027
Alexopoulos, P., Sorg, C., Forschler, A., Grimmer, T., Skokou, M., Wohlschlager, A., et al. (2012). Perfusion abnormalities in mild cognitive impairment and mild dementia in Alzheimer’s disease measured by pulsed arterial spin labeling MRI. Eur. Arch. Psychiatry Clin. Neurosci. 262, 69–77. doi: 10.1007/s00406-011-0226-2
Alonso, A. D., Cohen, L. S., Corbo, C., Morozova, V., Elidrissi, A., Phillips, G., et al. (2018). Hyperphosphorylation of tau associates with changes in its function beyond microtubule stability. Front. Cell. Neurosci. 12:338. doi: 10.3389/fncel.2018.00338
Alosco, M. L., Sugarman, M. A., Besser, L. M., Tripodis, Y., Martin, B., Palmisano, J. N., et al. (2018). A clinicopathological investigation of white matter hyperintensities and Alzheimer’s disease neuropathology. J. Alzheimers Dis. 63, 1347–1360. doi: 10.3233/JAD-180017
Armstrong, R. A. (2013). What causes alzheimer’s disease? Folia Neuropathol. 51, 169–188. doi: 10.5114/fn.2013.37702
Ashok, A., Rai, N. K., Raza, W., Pandey, R., and Bandyopadhyay, S. (2016). Chronic cerebral hypoperfusion-induced impairment of Abeta clearance requires HB-EGF-dependent sequential activation of HIF1alpha and MMP9. Neurobiol. Dis. 95, 179–193. doi: 10.1016/j.nbd.2016.07.013
Ashok, A., Rai, N. K., Tripathi, S., and Bandyopadhyay, S. (2015). Exposure to As-, Cd- and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 143, 64–80. doi: 10.1093/toxsci/kfu208
Ashok, B. S., Ajith, T. A., and Sivanesan, S. (2017). Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 44, 327–334. doi: 10.1111/1440-1681.12717
Asiimwe, N., Yeo, S. G., Kim, M. S., Jung, J., and Jeong, N. Y. (2016). Nitric oxide: exploring the contextual link with Alzheimer’s disease. Oxid. Med. Cell. Longev. 2016:7205747. doi: 10.1155/2016/7205747
Auerbach, I. D., and Vinters, H. V. (2006). Effects of anoxia and hypoxia on amyloid precursor protein processing in cerebral microvascular smooth muscle cells. J. Neuropathol. Exp. Neurol. 65, 610–620. doi: 10.1097/00005072-200606000-00009
Bandyopadhyay, S., Cahill, C., Balleidier, A., Huang, C., Lahiri, D. K., Huang, X., et al. (2013). Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for down syndrome and Alzheimer’s disease. PLoS One 8:e65978. doi: 10.1371/journal.pone.0065978
Bandyopadhyay, S., Goldstein, L. E., Lahiri, D. K., and Rogers, J. T. (2007). Role of the APP non-amyloidogenic signaling pathway and targeting alpha-secretase as an alternative drug target for treatment of Alzheimer’s disease. Curr. Med. Chem. 14, 2848–2864. doi: 10.2174/092986707782360060
Bandyopadhyay, S., Hartley, D. M., Cahill, C. M., Lahiri, D. K., Chattopadhyay, N., and Rogers, J. T. (2006a). Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 84, 106–118. doi: 10.1002/jnr.20864
Bandyopadhyay, S., Huang, X., Cho, H., Greig, N. H., Youdim, M. B., and Rogers, J. T. (2006b). Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5′untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28 and a piperazine chelator. J. Neural Transm. Suppl. 71, 237–247. doi: 10.1007/978-3-211-33328-0_25
Bandyopadhyay, S., Ni, J., Ruggiero, A., Walshe, K., Rogers, M. S., Chattopadhyay, N., et al. (2006c). A high-throughput drug screen targeted to the 5′untranslated region of Alzheimer amyloid precursor protein mRNA. J. Biomol. Screen. 11, 469–480. doi: 10.1177/1087057106287271
Bandyopadhyay, S., Huang, X., Lahiri, D. K., and Rogers, J. T. (2010). Novel drug targets based on metallobiology of Alzheimer’s disease. Expert Opin. Ther. Targets 14, 1177–1197. doi: 10.1517/14728222.2010.525352
Bandyopadhyay, S., and Rogers, J. T. (2014). Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: maintenance of brain iron homeostasis. Biochem. Pharmacol. 88, 486–494. doi: 10.1016/j.bcp.2014.01.032
Barker, W. W., Luis, C. A., Kashuba, A., Luis, M., Harwood, D. G., Loewenstein, D., et al. (2002). Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 16, 203–212. doi: 10.1097/00002093-200210000-00001
Bateman, R. J., Munsell, L. Y., Morris, J. C., Swarm, R., Yarasheski, K. E., and Holtzman, D. M. (2006). Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861. doi: 10.1038/nm1438
Benigni, A., Cassis, P., and Remuzzi, G. (2010). Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol. Med. 2, 247–257. doi: 10.1002/emmm.201000080
Bie, B., Wu, J., Foss, J. F., and Naguib, M. (2019). Activation of mGluR1 mediates C1q-dependent microglial phagocytosis of glutamatergic synapses in Alzheimer’s rodent models. Mol. Neurobiol. 56, 5568–5585. doi: 10.1007/s12035-019-1467-8
Binder, L. I., Guillozet-Bongaarts, A. L., Garcia-Sierra, F., and Berry, R. W. (2005). Tau, tangles and Alzheimer’s disease. Biochim. Biophys. Acta 1739, 216–223. doi: 10.1016/j.bbadis.2004.08.014
Binnewijzend, M. A., Benedictus, M. R., Kuijer, J. P., Van Der Flier, W. M., Teunissen, C. E., Prins, N. D., et al. (2016). Cerebral perfusion in the predementia stages of Alzheimer’s disease. Eur. Radiol. 26, 506–514. doi: 10.1007/s00330-015-3834-9
Bolos, M., Llorens-Martin, M., Perea, J. R., Jurado-Arjona, J., Rabano, A., Hernandez, F., et al. (2017). Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 12:59. doi: 10.1186/s13024-017-0200-1
Bonte, F. J., Ross, E. D., Chehabi, H. H., and Devous, M. D., Sr (1986). SPECT study of regional cerebral blood flow in Alzheimer disease. J. Comput. Assist. Tomogr. 10, 579–583. doi: 10.1097/00004728-198607000-00005
Borshchev, Y. Y., Uspensky, Y. P., and Galagudza, M. M. (2019). Pathogenetic pathways of cognitive dysfunction and dementia in metabolic syndrome. Life Sci. 237:116932. doi: 10.1016/j.lfs.2019.116932
Boscolo, E., Folin, M., Nico, B., Grandi, C., Mangieri, D., Longo, V., et al. (2007). Beta amyloid angiogenic activity in vitro and in vivo. Int. J. Mol. Med. 19, 581–587. doi: 10.3892/ijmm.19.4.581
Bou Khalil, R., Khoury, E., and Koussa, S. (2016). Linking multiple pathogenic pathways in Alzheimer’s disease. World J. Psychiatry 6, 208–214. doi: 10.5498/wjp.v6.i2.208
Browne, T. C., Mcquillan, K., Mcmanus, R. M., O’reilly, J. A., Mills, K. H., and Lynch, M. A. (2013). IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 190, 2241–2251. doi: 10.4049/jimmunol.1200947
Burns, A., Philpot, M. P., Costa, D. C., Ell, P. J., and Levy, R. (1989). The investigation of Alzheimer’s disease with single photon emission tomography. J. Neurol. Neurosurg. Psychiatry 52, 248–253. doi: 10.1136/jnnp.52.2.248
Butt, A. M., De La Rocha, I. C., and Rivera, A. (2019). Oligodendroglial cells in Alzheimer’s disease. Adv. Exp. Med. Biol. 1175, 325–333. doi: 10.1007/978-981-13-9913-8_12
Cai, Z., Liu, Z., Xiao, M., Wang, C., and Tian, F. (2017). Chronic cerebral hypoperfusion promotes amyloid-beta pathogenesis via activating beta/gamma-secretases. Neurochem. Res. 42, 3446–3455. doi: 10.1007/s11064-017-2391-9
Cai, Z., and Xiao, M. (2016). Oligodendrocytes and Alzheimer’s disease. Int. J. Neurosci. 126, 97–104. doi: 10.3109/00207454.2015.1025778
Carrano, A., Hoozemans, J. J., Van Der Vies, S. M., Rozemuller, A. J., Van Horssen, J., and De Vries, H. E. (2011). Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 15, 1167–1178. doi: 10.1089/ars.2011.3895
Chen, W. T., Lu, A., Craessaerts, K., Pavie, B., Sala Frigerio, C., Corthout, N., et al. (2020). Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 182, 976.e919–991.e919. doi: 10.1016/j.cell.2020.06.038
Chen, P., Zhao, W., Guo, Y., Xu, J., and Yin, M. (2016). CX3CL1/CX3CR1 in Alzheimer’s disease: a target for neuroprotection. Biomed. Res. Int. 2016:8090918. doi: 10.1155/2016/8090918
Cianciulli, A., Porro, C., Calvello, R., Trotta, T., Lofrumento, D. D., and Panaro, M. A. (2020). Microglia mediated neuroinflammation: focus on PI3K modulation. Biomolecules 10:137. doi: 10.3390/biom10010137
Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., et al. (2013). A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta and frank neuronal loss. J. Neurosci. 33, 6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013
Condello, C., Yuan, P., and Grutzendler, J. (2018). Microglia-mediated neuroprotection, TREM2 and Alzheimer’s disease: evidence from optical imaging. Biol. Psychiatry 83, 377–387. doi: 10.1016/j.biopsych.2017.10.007
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 6:6176. doi: 10.1038/ncomms7176
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Cortes-Canteli, M., and Iadecola, C. (2020). Alzheimer’s disease and vascular aging: JACC focus seminar. J. Am. Coll. Cardiol. 75, 942–951. doi: 10.1016/j.jacc.2019.10.062
Czirr, E., Castello, N. A., Mosher, K. I., Castellano, J. M., Hinkson, I. V., Lucin, K. M., et al. (2017). Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J. Exp. Med. 214, 1081–1092. doi: 10.1084/jem.20162011
Czuba, E., Steliga, A., Lietzau, G., and Kowianski, P. (2017). Cholesterol as a modifying agent of the neurovascular unit structure and function under physiological and pathological conditions. Metab. Brain Dis. 32, 935–948. doi: 10.1007/s11011-017-0015-3
Dai, J., Johnson, E. C. B., Dammer, E. B., Duong, D. M., Gearing, M., Lah, J. J., et al. (2018). Effects of APOE genotype on brain proteomic network and cell type changes in Alzheimer’s disease. Front. Mol. Neurosci. 11:454. doi: 10.3389/fnmol.2018.00454
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
Dansokho, C., Ait Ahmed, D., Aid, S., Toly-Ndour, C., Chaigneau, T., Calle, V., et al. (2016). Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 139, 1237–1251. doi: 10.1093/brain/awv408
Dean, D. C., 3rd, Hurley, S. A., Kecskemeti, S. R., O’grady, J. P., Canda, C., Davenport-Sis, N. J., et al. (2017). Association of amyloid pathology with myelin alteration in preclinical Alzheimer disease. JAMA Neurol. 74, 41–49. doi: 10.1001/jamaneurol.2016.3232
Deane, R., Wu, Z., and Zlokovic, B. V. (2004). RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35, 2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1
Deane, R., and Zlokovic, B. V. (2007). Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 4, 191–197. doi: 10.2174/156720507780362245
de la Torre, J. C. (2000a). Critically attained threshold of cerebral hypoperfusion: can it cause Alzheimer’s disease? Ann. N Y Acad. Sci. 903, 424–436. doi: 10.1111/j.1749-6632.2000.tb06394.x
de la Torre, J. C. (2000b). Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol. Aging 21, 331–342. doi: 10.1016/s0197-4580(00)00111-1
Deczkowska, A., Keren-Shaul, H., Weiner, A., Colonna, M., Schwartz, M., and Amit, I. (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. doi: 10.1016/j.cell.2018.05.003
Desai, M. K., Mastrangelo, M. A., Ryan, D. A., Sudol, K. L., Narrow, W. C., and Bowers, W. J. (2010). Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 177, 1422–1435. doi: 10.2353/ajpath.2010.100087
Desai, M. K., Sudol, K. L., Janelsins, M. C., Mastrangelo, M. A., Frazer, M. E., and Bowers, W. J. (2009). Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 57, 54–65. doi: 10.1002/glia.20734
DeTure, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14:32. doi: 10.1186/s13024-019-0333-5
Duff, K., Kuret, J., and Congdon, E. E. (2010). Disaggregation of tau as a therapeutic approach to tauopathies. Curr. Alzheimer Res. 7, 235–240. doi: 10.2174/156720510791050885
Duyckaerts, C., Colle, M. A., Dessi, F., Piette, F., and Hauw, J. J. (1998). Progression of Alzheimer histopathological changes. Acta Neurol. Belg. 98, 180–185.
Erickson, M. A., and Banks, W. A. (2013). Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 33, 1500–1513. doi: 10.1038/jcbfm.2013.135
Fang, F., Lue, L. F., Yan, S., Xu, H., Luddy, J. S., Chen, D., et al. (2010). RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 24, 1043–1055. doi: 10.1096/fj.09-139634
Fazekas, F., Alavi, A., Chawluk, J. B., Zimmerman, R. A., Hackney, D., Bilaniuk, L., et al. (1989). Comparison of CT, MR and PET in Alzheimer’s dementia and normal aging. J. Nucl. Med. 30, 1607–1615.
Felician, O., and Sandson, T. A. (1999). The neurobiology and pharmacotherapy of Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci. 11, 19–31. doi: 10.1176/jnp.11.1.19
Fernandez, C. G., Hamby, M. E., Mcreynolds, M. L., and Ray, W. J. (2019). The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front. Aging Neurosci. 11:14. doi: 10.3389/fnagi.2019.00014
Ferreira, S., Pitman, K. A., Wang, S., Summers, B. S., Bye, N., Young, K. M., et al. (2020). Amyloidosis is associated with thicker myelin and increased oligodendrogenesis in the adult mouse brain. J. Neurosci. Res. 98, 1905–1932. doi: 10.1002/jnr.24672
Ferretti, M. T., Merlini, M., Spani, C., Gericke, C., Schweizer, N., Enzmann, G., et al. (2016). T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 54, 211–225. doi: 10.1016/j.bbi.2016.02.009
Findley, C. A., Bartke, A., Hascup, K. N., and Hascup, E. R. (2019). Amyloid beta-related alterations to glutamate signaling dynamics during Alzheimer’s disease progression. ASN Neuro 11:1759091419855541. doi: 10.1177/1759091419855541
Fisk, L., Nalivaeva, N. N., Boyle, J. P., Peers, C. S., and Turner, A. J. (2007). Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurones and astrocytes. Neurochem. Res. 32, 1741–1748. doi: 10.1007/s11064-007-9349-2
Fonseca, M. I., Chu, S., Pierce, A. L., Brubaker, W. D., Hauhart, R. E., Mastroeni, D., et al. (2016). Analysis of the putative role of CR1 in Alzheimer’s disease: genetic association, expression and function. PLoS One 11:e0149792. doi: 10.1371/journal.pone.0149792
Fragoulis, A., Siegl, S., Fendt, M., Jansen, S., Soppa, U., Brandenburg, L. O., et al. (2017). Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol. 12, 843–853. doi: 10.1016/j.redox.2017.04.024
Gale, S. C., Gao, L., Mikacenic, C., Coyle, S. M., Rafaels, N., Murray Dudenkov, T., et al. (2014). APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 134, 127–134. doi: 10.1016/j.jaci.2014.01.032
Ghoshal, N., Garcia-Sierra, F., Wuu, J., Leurgans, S., Bennett, D. A., Berry, R. W., et al. (2002). Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp. Neurol. 177, 475–493. doi: 10.1006/exnr.2002.8014
Gong, C. X., Liu, F., and Iqbal, K. (2018). Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J. Alzheimers Dis. 64, S107–S117. doi: 10.3233/JAD-179921
Govindpani, K., Mcnamara, L. G., Smith, N. R., Vinnakota, C., Waldvogel, H. J., Faull, R. L., et al. (2019). Vascular dysfunction in Alzheimer’s disease: a prelude to the pathological process or a consequence of it? J. Clin. Med. 8:651. doi: 10.3390/jcm8050651
Grammas, P. (2011). Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 8:26. doi: 10.1186/1742-2094-8-26
Grammas, P., and Ovase, R. (2001). Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 22, 837–842. doi: 10.1016/s0197-4580(01)00276-7
Grammas, P., Samany, P. G., and Thirumangalakudi, L. (2006). Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J. Alzheimers Dis. 9, 51–58. doi: 10.3233/jad-2006-9105
Grammas, P., Tripathy, D., Sanchez, A., Yin, X., and Luo, J. (2011). Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 4, 616–627.
Graykowski, D., Kasparian, K., Caniglia, J., Gritsaeva, Y., and Cudaback, E. (2020). Neuroinflammation drives APOE genotype-dependent differential expression of neprilysin. J. Neuroimmunol. 346:577315. doi: 10.1016/j.jneuroim.2020.577315
Greenberg, S. M., Bacskai, B. J., Hernandez-Guillamon, M., Pruzin, J., Sperling, R., and Van Veluw, S. J. (2020). Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat. Rev. Neurol. 16, 30–42. doi: 10.1038/s41582-019-0281-2
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guglielmotto, M., Aragno, M., Autelli, R., Giliberto, L., Novo, E., Colombatto, S., et al. (2009). The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J. Neurochem. 108, 1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x
Guo, Y., Li, X., Zhang, M., Chen, N., Wu, S., Lei, J., et al. (2019). Age and brain regionassociated alterations of cerebral blood flow in early Alzheimer’s disease assessed in AbetaPPSWE/PS1DeltaE9 transgenic mice using arterial spin labeling. Mol. Med. Rep. 19, 3045–3052. doi: 10.3892/mmr.2019.9950
Gupta, A., and Iadecola, C. (2015). Impaired Abeta clearance: a potential link between atherosclerosis and Alzheimer’s disease. Front. Aging Neurosci. 7:115. doi: 10.3389/fnagi.2015.00115
Halliday, M. R., Rege, S. V., Ma, Q., Zhao, Z., Miller, C. A., Winkler, E. A., et al. (2016). Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 36, 216–227. doi: 10.1038/jcbfm.2015.44
Han, B. H., Zhou, M. L., Johnson, A. W., Singh, I., Liao, F., Vellimana, A. K., et al. (2015). Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. U S A 112, E881–890. doi: 10.1073/pnas.1414930112
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Hanslik, K. L., and Ulland, T. K. (2020). The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front. Neurol. 11:570711. doi: 10.3389/fneur.2020.570711
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hayden, M. R. (2019). Type 2 diabetes mellitus increases the risk of late-onset Alzheimer’s disease: ultrastructural remodeling of the neurovascular unit and diabetic gliopathy. Brain Sci. 9:262. doi: 10.3390/brainsci9100262
He, Y., Ruganzu, J. B., Jin, H., Peng, X., Ji, S., Ma, Y., et al. (2020b). LRP1 knockdown aggravates Abeta1–42-stimulated microglial and astrocytic neuroinflammatory responses by modulating TLR4/NF-kappaB/MAPKs signaling pathways. Exp. Cell Res. 394:112166. doi: 10.1016/j.yexcr.2020.112166
He, Y., Ruganzu, J. B., Zheng, Q., Wu, X., Jin, H., Peng, X., et al. (2020c). Silencing of LRP1 exacerbates inflammatory response via TLR4/NF-kappaB/MAPKs signaling pathways in APP/PS1 transgenic mice. Mol. Neurobiol. 57, 3727–3743. doi: 10.1007/s12035-020-01982-7
He, J. T., Zhao, X., Xu, L., and Mao, C. Y. (2020a). Vascular risk factors and Alzheimer’s disease: blood-brain barrier disruption, metabolic syndromes and molecular links. J. Alzheimers Dis. 73, 39–58. doi: 10.3233/JAD-190764
Hecht, M., Kramer, L. M., Von Arnim, C. A. F., Otto, M., and Thal, D. R. (2018). Capillary cerebral amyloid angiopathy in Alzheimer’s disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 135, 681–694. doi: 10.1007/s00401-018-1834-y
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hohsfield, L. A., and Humpel, C. (2015). Migration of blood cells to beta-amyloid plaques in Alzheimer’s disease. Exp. Gerontol. 65, 8–15. doi: 10.1016/j.exger.2015.03.002
Hong, S., Dissing-Olesen, L., and Stevens, B. (2016). New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134. doi: 10.1016/j.conb.2015.12.004
Hu, X., Fan, Q., Hou, H., and Yan, R. (2016). Neurological dysfunctions associated with altered BACE1-dependent Neuregulin-1 signaling. J. Neurochem. 136, 234–249. doi: 10.1111/jnc.13395
Huang, K. L., Marcora, E., Pimenova, A. A., Di Narzo, A. F., Kapoor, M., Jin, S. C., et al. (2017). A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 20, 1052–1061. doi: 10.1038/nn.4587
Huentelman, M., De Both, M., Jepsen, W., Piras, I. S., Talboom, J. S., Willeman, M., et al. (2019). Common BACE2 polymorphisms are associated with altered risk for Alzheimer’s disease and CSF amyloid biomarkers in APOE epsilon4 non-carriers. Sci. Rep. 9:9640. doi: 10.1038/s41598-019-45896-4
Humpel, C., and Marksteiner, J. (2005). Cerebrovascular damage as a cause for Alzheimer’s disease. Curr. Neurovasc. Res. 2, 341–347. doi: 10.2174/156720205774322610
Iadecola, C. (2003a). Atherosclerosis and neurodegeneration: unexpected conspirators in Alzheimer’s dementia. Arterioscler. Thromb. Vasc. Biol. 23, 1951–1953. doi: 10.1161/01.ATV.0000102660.99744.85
Iadecola, C. (2003b). Cerebrovascular effects of amyloid-beta peptides: mechanisms and implications for Alzheimer’s dementia. Cell. Mol. Neurobiol. 23, 681–689. doi: 10.1023/a:1025092617651
Iannucci, J., Renehan, W., and Grammas, P. (2020). Thrombin, a mediator of coagulation, inflammation and neurotoxicity at the neurovascular interface: implications for Alzheimer’s disease. Front. Neurosci. 14:762. doi: 10.3389/fnins.2020.00762
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi: 10.2174/156720510793611592
Jacobs, R. W., Duong, T., and Scheibel, A. B. (1992). Immunohistochemical analysis of the basal forebrain in Alzheimer’s disease. Mol. Chem. Neuropathol. 17, 1–20. doi: 10.1007/BF03159977
Jana, A., Hogan, E. L., and Pahan, K. (2009). Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J. Neurol. Sci. 278, 5–15. doi: 10.1016/j.jns.2008.12.010
Jang, S. K., Yu, J. M., Kim, S. T., Kim, G. H., Park, D. W., Lee, D. I., et al. (2015). An Abeta42 uptake and degradation via Rg3 requires an activation of caveolin, clathrin and Abeta-degrading enzymes in microglia. Eur. J. Pharmacol. 758, 1–10. doi: 10.1016/j.ejphar.2015.03.071
Jellinger, K. A., and Bancher, C. (1998). Neuropathology of Alzheimer’s disease: a critical update. J. Neural Transm. Suppl. 54, 77–95. doi: 10.1007/978-3-7091-7508-8_8
Johansson, J. U., Brubaker, W. D., Javitz, H., Bergen, A. W., Nishita, D., Trigunaite, A., et al. (2018). Peripheral complement interactions with amyloid beta peptide in Alzheimer’s disease: polymorphisms, structure and function of complement receptor 1. Alzheimers Dement. 14, 1438–1449. doi: 10.1016/j.jalz.2018.04.003
Johnson, N. A., Jahng, G. H., Weiner, M. W., Miller, B. L., Chui, H. C., Jagust, W. J., et al. (2005). Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234, 851–859. doi: 10.1148/radiol.2343040197
Jolivel, V., Bicker, F., Biname, F., Ploen, R., Keller, S., Gollan, R., et al. (2015). Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 129, 279–295. doi: 10.1007/s00401-014-1372-1
Joly-Amado, A., Hunter, J., Quadri, Z., Zamudio, F., Rocha-Rangel, P. V., Chan, D., et al. (2020). CCL2 overexpression in the brain promotes glial activation and accelerates tau pathology in a mouse model of tauopathy. Front. Immunol. 11:997. doi: 10.3389/fimmu.2020.00997
Joost, E., Jordao, M. J. C., Mages, B., Prinz, M., Bechmann, I., and Krueger, M. (2019). Microglia contribute to the glia limitans around arteries, capillaries and veins under physiological conditions, in a model of neuroinflammation and in human brain tissue. Brain Struct. Funct. 224, 1301–1314. doi: 10.1007/s00429-019-01834-8
Kalaria, R. N. (1996). Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol. Ther. 72, 193–214. doi: 10.1016/s0163-7258(96)00116-7
Kalaria, R. (2002a). Similarities between Alzheimer’s disease and vascular dementia. J. Neurol. Sci. 203–204, 29–34. doi: 10.1016/s0022-510x(02)00256-3
Kalaria, R. N. (2002b). Small vessel disease and Alzheimer’s dementia: pathological considerations. Cerebrovasc. Dis. 13, 48–52. doi: 10.1159/000049150
Kamphuis, W., Middeldorp, J., Kooijman, L., Sluijs, J. A., Kooi, E. J., Moeton, M., et al. (2014). Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol. Aging 35, 492–510. doi: 10.1016/j.neurobiolaging.2013.09.035
Kanekiyo, T., Xu, H., and Bu, G. (2014). ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron 81, 740–754. doi: 10.1016/j.neuron.2014.01.045
Kaur, D., Sharma, V., and Deshmukh, R. (2019). Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 27, 663–677. doi: 10.1007/s10787-019-00580-x
Kelleher, R. J., and Soiza, R. L. (2013). Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am. J. Cardiovasc. Dis. 3, 197–226.
Khachaturian, Z. S. (1985). Diagnosis of Alzheimer’s disease. Arch. Neurol. 42, 1097–1105. doi: 10.1001/archneur.1985.04060100083029
Khan, T. K. (2018). An algorithm for preclinical diagnosis of Alzheimer’s disease. Front. Neurosci. 12:275. doi: 10.3389/fnins.2018.00275
Kirabali, T., Rust, R., Rigotti, S., Siccoli, A., Nitsch, R. M., and Kulic, L. (2020). Distinct changes in all major components of the neurovascular unit across different neuropathological stages of Alzheimer’s disease. Brain Pathol. 30, 1056–1070. doi: 10.1111/bpa.12895
Kitazawa, M., Cheng, D., Tsukamoto, M. R., Koike, M. A., Wes, P. D., Vasilevko, V., et al. (2011). Blocking IL-1 signaling rescues cognition, attenuates tau pathology and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 187, 6539–6549. doi: 10.4049/jimmunol.1100620
Klohs, J. (2019). An integrated view on vascular dysfunction in Alzheimer’s disease. Neurodegener. Dis. 19, 109–127. doi: 10.1159/000505625
Ko, C. Y., Chu, Y. Y., Narumiya, S., Chi, J. Y., Furuyashiki, T., Aoki, T., et al. (2015). CCAAT/enhancer-binding protein delta/miR135a/thrombospondin 1 axis mediates PGE2-induced angiogenesis in Alzheimer’s disease. Neurobiol. Aging 36, 1356–1368. doi: 10.1016/j.neurobiolaging.2014.11.020
Koldamova, R., Fitz, N. F., and Lefterov, I. (2010). The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 1801, 824–830. doi: 10.1016/j.bbalip.2010.02.010
Konishi, H., and Kiyama, H. (2018). Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front. Cell. Neurosci. 12:206. doi: 10.3389/fncel.2018.00206
Kook, S. Y., Hong, H. S., Moon, M., Ha, C. M., Chang, S., and Mook-Jung, I. (2012). Abeta(1)(-)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J. Neurosci. 32, 8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012
Koss, D. J., Jones, G., Cranston, A., Gardner, H., Kanaan, N. M., and Platt, B. (2016). Soluble pre-fibrillar tau and beta-amyloid species emerge in early human Alzheimer’s disease and track disease progression and cognitive decline. Acta Neuropathol. 132, 875–895. doi: 10.1007/s00401-016-1632-3
Kovacic, J. C., and Fuster, V. (2012). Atherosclerotic risk factors, vascular cognitive impairment and Alzheimer disease. Mt. Sinai J. Med. 79, 664–673. doi: 10.1002/msj.21347
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566.e569–581.e569. doi: 10.1016/j.immuni.2017.08.008
Kurisu, S., and Takenawa, T. (2009). The WASP and WAVE family proteins. Genome Biol. 10:226. doi: 10.1186/gb-2009-10-6-226
Kwan, L. T., Reed, B. R., Eberling, J. L., Schuff, N., Tanabe, J., Norman, D., et al. (1999). Effects of subcortical cerebral infarction on cortical glucose metabolism and cognitive function. Arch. Neurol. 56, 809–814. doi: 10.1001/archneur.56.7.809
Lai, A. Y., Dorr, A., Thomason, L. A., Koletar, M. M., Sled, J. G., Stefanovic, B., et al. (2015). Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 138, 1046–1058. doi: 10.1093/brain/awv023
Larramona-Arcas, R., Gonzalez-Arias, C., Perea, G., Gutierrez, A., Vitorica, J., Garcia-Barrera, T., et al. (2020). Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol. Neurodegener. 15:35. doi: 10.1186/s13024-020-00382-8
Laurent, C., Buee, L., and Blum, D. (2018). Tau and neuroinflammation: what impact for Alzheimer’s disease and tauopathies? Biomed. J. 41, 21–33. doi: 10.1016/j.bj.2018.01.003
Lee, E., and Chung, W. S. (2019). Glial control of synapse number in healthy and diseased brain. Front. Cell. Neurosci. 13:42. doi: 10.3389/fncel.2019.00042
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L. L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032.e1035–1048.e1035. doi: 10.1016/j.neuron.2018.02.002
Lee, W. J., Liao, Y. C., Wang, Y. F., Lin, Y. S., Wang, S. J., and Fuh, J. L. (2020). Summative effects of vascular risk factors on the progression of Alzheimer disease. J. Am. Geriatr. Soc. 68, 129–136. doi: 10.1111/jgs.16181
Lessard, C. B., Malnik, S. L., Zhou, Y., Ladd, T. B., Cruz, P. E., Ran, Y., et al. (2018). High-affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol. Med. 10:e9027. doi: 10.15252/emmm.201809027
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. doi: 10.1126/science.1058189
Li, S., and Selkoe, D. J. (2020). A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J. Neurochem. 154, 583–597. doi: 10.1111/jnc.15007
Li, W., Tang, Y., Fan, Z., Meng, Y., Yang, G., Luo, J., et al. (2013). Autophagy is involved in oligodendroglial precursor-mediated clearance of amyloid peptide. Mol. Neurodegener. 8:27. doi: 10.1186/1750-1326-8-27
Li, M. Z., Zheng, L. J., Shen, J., Li, X. Y., Zhang, Q., Bai, X., et al. (2018). SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen. Res. 13, 2005–2013. doi: 10.4103/1673-5374.239449
Lian, H., Litvinchuk, A., Chiang, A. C., Aithmitti, N., Jankowsky, J. L., and Zheng, H. (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589. doi: 10.1523/JNEUROSCI.2117-15.2016
Liao, M. C., Ahmed, M., Smith, S. O., and Van Nostrand, W. E. (2009). Degradation of amyloid beta protein by purified myelin basic protein. J. Biol. Chem. 284, 28917–28925. doi: 10.1074/jbc.M109.050856
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141.e1147–1154.e1147. doi: 10.1016/j.neuron.2018.05.008
Liu, J., Chang, L., Song, Y., Li, H., and Wu, Y. (2019). The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 13:43. doi: 10.3389/fnins.2019.00043
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, W., Wong, A., Law, A. C., and Mok, V. C. (2015). Cerebrovascular disease, amyloid plaques and dementia. Stroke 46, 1402–1407. doi: 10.1161/STROKEAHA.114.006571
Liu, C. C., Zhao, N., Fu, Y., Wang, N., Linares, C., Tsai, C. W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron 96, 1024.e1023–1032.e1023. doi: 10.1016/j.neuron.2017.11.013
Loera-Valencia, R., Goikolea, J., Parrado-Fernandez, C., Merino-Serrais, P., and Maioli, S. (2019). Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 190, 104–114. doi: 10.1016/j.jsbmb.2019.03.003
Luchena, C., Zuazo-Ibarra, J., Alberdi, E., Matute, C., and Capetillo-Zarate, E. (2018). Contribution of neurons and glial cells to complement-mediated synapse removal during development, aging and in Alzheimer’s disease. Mediators Inflamm. 2018:2530414. doi: 10.1155/2018/2530414
Lukiw, W. J., Ottlecz, A., Lambrou, G., Grueninger, M., Finley, J., Thompson, H. W., et al. (2003). Coordinate activation of HIF-1 and NF-kappaB DNA binding and COX-2 and VEGF expression in retinal cells by hypoxia. Invest. Ophthalmol. Vis. Sci. 44, 4163–4170. doi: 10.1167/iovs.02-0655
Maccioni, R. B., Lavados, M., Guillon, M., Mujica, C., Bosch, R., Farias, G., et al. (2006). Anomalously phosphorylated tau and Abeta fragments in the CSF correlates with cognitive impairment in MCI subjects. Neurobiol. Aging 27, 237–244. doi: 10.1016/j.neurobiolaging.2005.01.011
Maccioni, R. B., Munoz, J. P., and Barbeito, L. (2001). The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 32, 367–381. doi: 10.1016/s0188-4409(01)00316-2
MacVicar, B. A., and Newman, E. A. (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7:a020388. doi: 10.1101/cshperspect.a020388
Manczak, M., Mao, P., Nakamura, K., Bebbington, C., Park, B., and Reddy, P. H. (2009). Neutralization of granulocyte macrophage colony-stimulating factor decreases amyloid beta 1–42 and suppresses microglial activity in a transgenic mouse model of Alzheimer’s disease. Hum. Mol. Genet. 18, 3876–3893. doi: 10.1093/hmg/ddp331
Mandrekar, S., Jiang, Q., Lee, C. Y., Koenigsknecht-Talboo, J., Holtzman, D. M., and Landreth, G. E. (2009). Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009
Mandrekar-Colucci, S., and Landreth, G. E. (2010). Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 9, 156–167. doi: 10.2174/187152710791012071
Marshall, A. J., Rattray, M., and Vaughan, P. F. (2006). Chronic hypoxia in the human neuroblastoma SH-SY5Y causes reduced expression of the putative alpha-secretases, ADAM10 and TACE, without altering their mRNA levels. Brain Res. 1099, 18–24. doi: 10.1016/j.brainres.2006.05.008
Masters, C. L., Cappai, R., Barnham, K. J., and Villemagne, V. L. (2006). Molecular mechanisms for Alzheimer’s disease: implications for neuroimaging and therapeutics. J. Neurochem. 97, 1700–1725. doi: 10.1111/j.1471-4159.2006.03989.x
Mauch, D. H., Nagler, K., Schumacher, S., Goritz, C., Muller, E. C., Otto, A., et al. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357. doi: 10.1126/science.294.5545.1354
Maurya, S. K., Mishra, J., Abbas, S., and Bandyopadhyay, S. (2016). Cypermethrin stimulates GSK3beta-dependent abeta and p-tau proteins and cognitive loss in young rats: reduced HB-EGF signaling and downstream neuroinflammation as critical regulators. Mol. Neurobiol. 53, 968–982. doi: 10.1007/s12035-014-9061-6
Mcgettrick, A. F., and O’neill, L. A. J. (2020). The role of HIF in immunity and inflammation. Cell Metab. 32, 524–536. doi: 10.1016/j.cmet.2020.08.002
Mecca, C., Giambanco, I., Donato, R., and Arcuri, C. (2018). Microglia and aging: the role of the TREM2-DAP12 and CX3CL1-CX3CR1 axes. Int. J. Mol. Sci. 19:318. doi: 10.3390/ijms19010318
Mendsaikhan, A., Tooyama, I., and Walker, D. G. (2019). Microglial progranulin: involvement in Alzheimer’s disease and neurodegenerative diseases. Cells 8:230. doi: 10.3390/cells8030230
Merlini, M., Kirabali, T., Kulic, L., Nitsch, R. M., and Ferretti, M. T. (2018). Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener. Dis. 18, 49–56. doi: 10.1159/000486200
Mietelska-Porowska, A., Wasik, U., Goras, M., Filipek, A., and Niewiadomska, G. (2014). Tau protein modifications and interactions: their role in function and dysfunction. Int. J. Mol. Sci. 15, 4671–4713. doi: 10.3390/ijms15034671
Migliaccio, R., Agosta, F., Possin, K. L., Rabinovici, G. D., Miller, B. L., and Gorno-Tempini, M. L. (2012). White matter atrophy in Alzheimer’s disease variants. Alzheimers Dement. 8, S78–S87. doi: 10.1016/j.jalz.2012.04.010
Miners, J. S., Ashby, E., Van Helmond, Z., Chalmers, K. A., Palmer, L. E., Love, S., et al. (2008). Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 34, 181–193. doi: 10.1111/j.1365-2990.2007.00885.x
Mirra, S. S., Heyman, A., Mckeel, D., Sumi, S. M., Crain, B. J., Brownlee, L. M., et al. (1991). The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. doi: 10.1212/wnl.41.4.479
Morikawa, T., Kajimura, M., Nakamura, T., Hishiki, T., Nakanishi, T., Yukutake, Y., et al. (2012). Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. U S A 109, 1293–1298. doi: 10.1073/pnas.1119658109
Mot, A. I., Depp, C., and Nave, K. A. (2018). An emerging role of dysfunctional axon-oligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin. Neurosci. 20, 283–292. doi: 10.31887/dcns.2018.20.4/amot
Naert, G., and Rivest, S. (2011). CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31, 6208–6220. doi: 10.1523/JNEUROSCI.0299-11.2011
Nalivaeva, N. N., Belyaev, N. D., Zhuravin, I. A., and Turner, A. J. (2012). The Alzheimer’s amyloid-degrading peptidase, neprilysin: can we control it? Int. J. Alzheimers Dis. 2012:383796. doi: 10.1155/2012/383796
Nasrabady, S. E., Rizvi, B., Goldman, J. E., and Brickman, A. M. (2018). White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6:22. doi: 10.1186/s40478-018-0515-3
Nicolas, G., Charbonnier, C., and Campion, D. (2016). From common to rare variants: the genetic component of Alzheimer disease. Hum. Hered. 81, 129–141. doi: 10.1159/000452256
Nirzhor, S. S. R., Khan, R. I., and Neelotpol, S. (2018). The biology of glial cells and their complex roles in Alzheimer’s disease: new opportunities in therapy. Biomolecules 8:93. doi: 10.3390/biom8030093
Okun, E., Griffioen, K. J., Lathia, J. D., Tang, S. C., Mattson, M. P., and Arumugam, T. V. (2009). Toll-like receptors in neurodegeneration. Brain Res. Rev. 59, 278–292. doi: 10.1016/j.brainresrev.2008.09.001
Ongali, B., Nicolakakis, N., Tong, X. K., Aboulkassim, T., Papadopoulos, P., Rosa-Neto, P., et al. (2014). Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol. Dis. 68, 126–136. doi: 10.1016/j.nbd.2014.04.018
Painter, M. M., Atagi, Y., Liu, C. C., Rademakers, R., Xu, H., Fryer, J. D., et al. (2015). TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 10:43. doi: 10.1186/s13024-015-0040-9
Pajarillo, E., Rizor, A., Lee, J., Aschner, M., and Lee, E. (2019). The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 161:107559. doi: 10.1016/j.neuropharm.2019.03.002
Pantoni, L., and Garcia, J. H. (1997). Cognitive impairment and cellular/vascular changes in the cerebral white matter. Ann. N Y Acad. Sci. 826, 92–102. doi: 10.1111/j.1749-6632.1997.tb48463.x
Papuc, E., and Rejdak, K. (2020). The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 16, 345–351. doi: 10.5114/aoms.2018.76863
Park, J. C., Han, S. H., Yi, D., Byun, M. S., Lee, J. H., Jang, S., et al. (2019). Plasma tau/amyloid-beta1–42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer’s disease. Brain 142, 771–786. doi: 10.1093/brain/awy347
Parkes, I., Chintawar, S., and Cader, M. Z. (2018). Neurovascular dysfunction in dementia - human cellular models and molecular mechanisms. Clin. Sci. (Lond) 132, 399–418. doi: 10.1042/CS20160720
Passos, G. F., Figueiredo, C. P., Prediger, R. D., Pandolfo, P., Duarte, F. S., Medeiros, R., et al. (2009). Role of the macrophage inflammatory protein-1alpha/CC chemokine receptor 5 signaling pathway in the neuroinflammatory response and cognitive deficits induced by beta-amyloid peptide. Am. J. Pathol. 175, 1586–1597. doi: 10.2353/ajpath.2009.081113
Peprah, K., and Mccormack, S. (2019). Medical Cannabis for the Treatment of Dementia: A Review of Clinical Effectiveness and Guidelines. Ottawa, ON: Canadian Agency for Drugs and Technologies in Health (CADTH).
Perea, J. R., Bolos, M., and Avila, J. (2020). Microglia in Alzheimer’s disease in the context of tau pathology. Biomolecules 10:1439. doi: 10.3390/biom10101439
Petrie, J. R., Guzik, T. J., and Touyz, R. M. (2018). Diabetes, hypertension and cardiovascular disease: clinical insights and vascular mechanisms. Can. J. Cardiol. 34, 575–584. doi: 10.1016/j.cjca.2017.12.005
Pluta, R. (2004). From brain ischemia-reperfusion injury to possible sporadic Alzheimer’s disease. Curr. Neurovasc. Res. 1, 441–453. doi: 10.2174/1567202043361839
Pluta, R., Kocki, J., Ulamek-Koziol, M., Petniak, A., Gil-Kulik, P., Januszewski, S., et al. (2016). Discrepancy in expression of beta-secretase and amyloid-beta protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J. Alzheimers Dis. 51, 1023–1031. doi: 10.3233/JAD-151102
Pogue, A. I., and Lukiw, W. J. (2004). Angiogenic signaling in Alzheimer’s disease. Neuroreport 15, 1507–1510. doi: 10.1097/01.wnr.0000130539.39937.1d
Polidori, M. C., Pientka, L., and Mecocci, P. (2012). A review of the major vascular risk factors related to Alzheimer’s disease. J. Alzheimers Dis. 32, 521–530. doi: 10.3233/JAD-2012-120871
Provias, J., and Jeynes, B. (2008). Neurofibrillary tangles and senile plaques in Alzheimer’s brains are associated with reduced capillary expression of vascular endothelial growth factor and endothelial nitric oxide synthase. Curr. Neurovasc. Res. 5, 199–205. doi: 10.2174/156720208785425729
Puigdellivol, M., Allendorf, D. H., and Brown, G. C. (2020). Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 14:162. doi: 10.3389/fncel.2020.00162
Qiu, W. Q., Ye, Z., Kholodenko, D., Seubert, P., and Selkoe, D. J. (1997). Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 272, 6641–6646. doi: 10.1074/jbc.272.10.6641
Ramanathan, A., Nelson, A. R., Sagare, A. P., and Zlokovic, B. V. (2015). Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Rauch, J. N., Luna, G., Guzman, E., Audouard, M., Challis, C., Sibih, Y. E., et al. (2020). LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385. doi: 10.1038/s41586-020-2156-5
Raz, N., and Daugherty, A. M. (2018). Pathways to brain aging and their modifiers: free-radical-induced energetic and neural decline in senescence (FRIENDS) model - a mini-review. Gerontology 64, 49–57. doi: 10.1159/000479508
Raz, N., Yang, Y., Dahle, C. L., and Land, S. (2012). Volume of white matter hyperintensities in healthy adults: contribution of age, vascular risk factors and inflammation-related genetic variants. Biochim. Biophys. Acta 1822, 361–369. doi: 10.1016/j.bbadis.2011.08.007
Reed, B. R., Eberling, J. L., Mungas, D., Weiner, M., and Jagust, W. J. (2001). Frontal lobe hypometabolism predicts cognitive decline in patients with lacunar infarcts. Arch. Neurol. 58, 493–497. doi: 10.1001/archneur.58.3.493
Ren, P., Chen, J., Li, B., Zhang, M., Yang, B., Guo, X., et al. (2020). Nrf2 ablation promotes Alzheimer’s disease-like pathology in APP/PS1 transgenic mice: the role of neuroinflammation and oxidative stress. Oxid. Med. Cell. Longev. 2020:3050971. doi: 10.1155/2020/3050971
Revesz, T., Holton, J. L., Lashley, T., Plant, G., Rostagno, A., Ghiso, J., et al. (2002). Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 12, 343–357. doi: 10.1111/j.1750-3639.2002.tb00449.x
Ribe, E. M., Perez, M., Puig, B., Gich, I., Lim, F., Cuadrado, M., et al. (2005). Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis. 20, 814–822. doi: 10.1016/j.nbd.2005.05.027
Ries, M., and Sastre, M. (2016). Mechanisms of Abeta clearance and degradation by glial cells. Front. Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
Risacher, S. L., and Saykin, A. J. (2013). Neuroimaging biomarkers of neurodegenerative diseases and dementia. Semin. Neurol. 33, 386–416. doi: 10.1055/s-0033-1359312
Rogers, J. T., and Lahiri, D. K. (2004). Metal and inflammatory targets for Alzheimer’s disease. Curr. Drug Targets 5, 535–551. doi: 10.2174/1389450043345272
Rose, C. R., Felix, L., Zeug, A., Dietrich, D., Reiner, A., and Henneberger, C. (2017). Astroglial glutamate signaling and uptake in the hippocampus. Front. Mol. Neurosci. 10:451. doi: 10.3389/fnmol.2017.00451
Rosenberg, G. A., Wallin, A., Wardlaw, J. M., Markus, H. S., Montaner, J., Wolfson, L., et al. (2016). Consensus statement for diagnosis of subcortical small vessel disease. J. Cereb. Blood Flow Metab. 36, 6–25. doi: 10.1038/jcbfm.2015.172
Rossi, B., Constantin, G., and Zenaro, E. (2020). The emerging role of neutrophils in neurodegeneration. Immunobiology 225:151865. doi: 10.1016/j.imbio.2019.10.014
Rossi, B., Santos-Lima, B., Terrabuio, E., Zenaro, E., and Constantin, G. (2021). Common peripheral immunity mechanisms in multiple sclerosis and Alzheimer’s disease. Front. Immunol. 12:639369. doi: 10.3389/fimmu.2021.639369
Rothman, S. M., Tanis, K. Q., Gandhi, P., Malkov, V., Marcus, J., Pearson, M., et al. (2018). Human Alzheimer’s disease gene expression signatures and immune profile in APP mouse models: a discrete transcriptomic view of Abeta plaque pathology. J. Neuroinflammation 15:256. doi: 10.1186/s12974-018-1265-7
Ryu, J. K., Cho, T., Choi, H. B., Wang, Y. T., and Mclarnon, J. G. (2009). Microglial VEGF receptor response is an integral chemotactic component in Alzheimer’s disease pathology. J. Neurosci. 29, 3–13. doi: 10.1523/JNEUROSCI.2888-08.2009
Saez, E. T., Pehar, M., Vargas, M. R., Barbeito, L., and Maccioni, R. B. (2006). Production of nerve growth factor by beta-amyloid-stimulated astrocytes induces p75NTR-dependent tau hyperphosphorylation in cultured hippocampal neurons. J. Neurosci. Res. 84, 1098–1106. doi: 10.1002/jnr.20996
Sagare, A. P., Deane, R., and Zlokovic, B. V. (2012). Low-density lipoprotein receptor-related protein 1: a physiological Abeta homeostatic mechanism with multiple therapeutic opportunities. Pharmacol. Ther. 136, 94–105. doi: 10.1016/j.pharmthera.2012.07.008
Salminen, A., Kauppinen, A., and Kaarniranta, K. (2017). Hypoxia/ischemia activate processing of amyloid precursor protein: impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 140, 536–549. doi: 10.1111/jnc.13932
Sarabia-Cobo, C. M., Perez, V., Hermosilla, C., Nunez, M. J., and De Lorena, P. (2014). Apathy and leukoaraiosis in mild cognitive impairment and Alzheimer’s disease: multicenter diagnostic criteria according to the latest studies. Dement. Geriatr. Cogn. Dis. Extra 4, 228–235. doi: 10.1159/000363227
Satoh, J. I., Kino, Y., Yanaizu, M., Tosaki, Y., Sakai, K., Ishida, T., et al. (2017). Microglia express ABI3 in the brains of Alzheimer’s disease and Nasu-Hakola disease. Intractable Rare Dis. Res. 6, 262–268. doi: 10.5582/irdr.2017.01073
Schmitz, T., and Chew, L. J. (2008). Cytokines and myelination in the central nervous system. ScientificWorldJournal 8, 1119–1147. doi: 10.1100/tsw.2008.140
Schuff, N., Matsumoto, S., Kmiecik, J., Studholme, C., Du, A., Ezekiel, F., et al. (2009). Cerebral blood flow in ischemic vascular dementia and Alzheimer’s disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimers Dement. 5, 454–462. doi: 10.1016/j.jalz.2009.04.1233
Scimemi, A., Meabon, J. S., Woltjer, R. L., Sullivan, J. M., Diamond, J. S., and Cook, D. G. (2013). Amyloid-beta1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 33, 5312–5318. doi: 10.1523/JNEUROSCI.5274-12.2013
Shi, S., Liang, D., Chen, Y., Xie, Y., Wang, Y., Wang, L., et al. (2016). Gx-50 reduces beta-amyloid-induced TNF-alpha, IL-1beta, NO and PGE2 expression and inhibits NF-kappaB signaling in a mouse model of Alzheimer’s disease. Eur. J. Immunol. 46, 665–676. doi: 10.1002/eji.201545855
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Sims, R., Van Der Lee, S. J., Naj, A. C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., et al. (2017). Rare coding variants in PLCG2, ABI3 and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384. doi: 10.1038/ng.3916
Sole-Domenech, S., Cruz, D. L., Capetillo-Zarate, E., and Maxfield, F. R. (2016). The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 32, 89–103. doi: 10.1016/j.arr.2016.07.002
Solis, E., Hascup, K. N., and Hascup, E. R. (2020). Alzheimer’s disease: the link between amyloid-beta and neurovascular dysfunction. J. Alzheimers Dis. 76, 1179–1198. doi: 10.3233/JAD-200473
Soto-Rojas, L. O., Pacheco-Herrero, M., Martinez-Gomez, P. A., Campa-Cordoba, B. B., Apatiga-Perez, R., Villegas-Rojas, M. M., et al. (2021). The neurovascular unit dysfunction in Alzheimer’s disease. Int. J. Mol. Sci. 22:2022. doi: 10.3390/ijms22042022
Stancu, I. C., Vasconcelos, B., Terwel, D., and Dewachter, I. (2014). Models of beta-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol. Neurodegener. 9:51. doi: 10.1186/1750-1326-9-51
Stephan, A. H., Madison, D. V., Mateos, J. M., Fraser, D. A., Lovelett, E. A., Coutellier, L., et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013
Sumi, N., Nishioku, T., Takata, F., Matsumoto, J., Watanabe, T., Shuto, H., et al. (2010). Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell. Mol. Neurobiol. 30, 247–253. doi: 10.1007/s10571-009-9446-7
Sun, X., Bromley-Brits, K., and Song, W. (2012). Regulation of beta-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s disease. J. Neurochem. 120, 62–70. doi: 10.1111/j.1471-4159.2011.07515.x
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U S A 103, 18727–18732. doi: 10.1073/pnas.0606298103
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Sy, M., Kitazawa, M., Medeiros, R., Whitman, L., Cheng, D., Lane, T. E., et al. (2011). Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. 178, 2811–2822. doi: 10.1016/j.ajpath.2011.02.012
Takashima, A., Honda, T., Yasutake, K., Michel, G., Murayama, O., Murayama, M., et al. (1998). Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 31, 317–323. doi: 10.1016/s0168-0102(98)00061-3
Tambini, M. D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., et al. (2016). ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 17, 27–36. doi: 10.15252/embr.201540614
Tarafdar, A., and Pula, G. (2018). The role of NADPH oxidases and oxidative stress in neurodegenerative disorders. Int. J. Mol. Sci. 19:3824. doi: 10.3390/ijms19123824
Tarantini, S., Fulop, G. A., Kiss, T., Farkas, E., Zolei-Szenasi, D., Galvan, V., et al. (2017). Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. Geroscience 39, 465–473. doi: 10.1007/s11357-017-9980-z
Thal, D. R., Ghebremedhin, E., Rub, U., Yamaguchi, H., Del Tredici, K., and Braak, H. (2002). Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293. doi: 10.1093/jnen/61.3.282
Thomsen, M. S., Routhe, L. J., and Moos, T. (2017). The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 37, 3300–3317. doi: 10.1177/0271678X17722436
Tong, H., Zhang, X., Meng, X., Xu, P., Zou, X., and Qu, S. (2017). Amyloid-beta peptide decreases expression and function of glutamate transporters in nervous system cells. Int. J. Biochem. Cell Biol. 85, 75–84. doi: 10.1016/j.biocel.2017.01.017
Tucker, S., Ahl, M., Cho, H. H., Bandyopadhyay, S., Cuny, G. D., Bush, A. I., et al. (2006). RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin and N-acetyl cysteine. Curr. Alzheimer Res. 3, 221–227. doi: 10.2174/156720506777632835
Tzioras, M., Davies, C., Newman, A., Jackson, R., and Spires-Jones, T. (2019). Invited review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 45, 327–346. doi: 10.1111/nan.12529
Ulamek-Koziol, M., Czuczwar, S. J., Januszewski, S., and Pluta, R. (2020). Proteomic and genomic changes in tau protein, which are associated with Alzheimer’s disease after ischemia-reperfusion brain injury. Int. J. Mol. Sci. 21:892. doi: 10.3390/ijms21030892
Ulland, T. K., Song, W. M., Huang, S. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649.e613–663.e613. doi: 10.1016/j.cell.2017.07.023
Unger, M. S., Li, E., Scharnagl, L., Poupardin, R., Altendorfer, B., Mrowetz, H., et al. (2020). CD8(+) T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav. Immun. 89, 67–86. doi: 10.1016/j.bbi.2020.05.070
Unger, M. S., Marschallinger, J., Kaindl, J., Klein, B., Johnson, M., Khundakar, A. A., et al. (2018a). Doublecortin expression in CD8+ T-cells and microglia at sites of amyloid-beta plaques: a potential role in shaping plaque pathology? Alzheimers Dement. 14, 1022–1037. doi: 10.1016/j.jalz.2018.02.017
Unger, M. S., Schernthaner, P., Marschallinger, J., Mrowetz, H., and Aigner, L. (2018b). Microglia prevent peripheral immune cell invasion and promote an anti-inflammatory environment in the brain of APP-PS1 transgenic mice. J. Neuroinflammation 15:274. doi: 10.1186/s12974-018-1304-4
Van Nostrand, W. E. (2016). The influence of the amyloid ss-protein and its precursor in modulating cerebral hemostasis. Biochim. Biophys. Acta 1862, 1018–1026. doi: 10.1016/j.bbadis.2015.10.020
van Olst, L., Verhaege, D., Franssen, M., Kamermans, A., Roucourt, B., Carmans, S., et al. (2020). Microglial activation arises after aggregation of phosphorylated-tau in a neuron-specific P301S tauopathy mouse model. Neurobiol. Aging 89, 89–98. doi: 10.1016/j.neurobiolaging.2020.01.003
Vanzulli, I., Papanikolaou, M., De-La-Rocha, I. C., Pieropan, F., Rivera, A. D., Gomez-Nicola, D., et al. (2020). Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 94, 130–139. doi: 10.1016/j.neurobiolaging.2020.05.016
Vasilevko, V., Passos, G. F., Quiring, D., Head, E., Kim, R. C., Fisher, M., et al. (2010). Aging and cerebrovascular dysfunction: contribution of hypertension, cerebral amyloid angiopathy and immunotherapy. Ann. N Y Acad. Sci. 1207, 58–70. doi: 10.1111/j.1749-6632.2010.05786.x
Vepsalainen, S., Hiltunen, M., Helisalmi, S., Wang, J., Van Groen, T., Tanila, H., et al. (2008). Increased expression of Abeta degrading enzyme IDE in the cortex of transgenic mice with Alzheimer’s disease-like neuropathology. Neurosci. Lett. 438, 216–220. doi: 10.1016/j.neulet.2008.04.025
Wang, L., Du, Y., Wang, K., Xu, G., Luo, S., and He, G. (2016). Chronic cerebral hypoperfusion induces memory deficits and facilitates Abeta generation in C57BL/6J mice. Exp. Neurol. 283, 353–364. doi: 10.1016/j.expneurol.2016.07.006
Wang, H. Y., Lee, D. H., D’andrea, M. R., Peterson, P. A., Shank, R. P., and Reitz, A. B. (2000). Beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 275, 5626–5632. doi: 10.1074/jbc.275.8.5626
Wang, Q., Rowan, M. J., and Anwyl, R. (2004). Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J. Neurosci. 24, 6049–6056. doi: 10.1523/JNEUROSCI.0233-04.2004
Wang, W. Y., Tan, M. S., Yu, J. T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
Wanleenuwat, P., Iwanowski, P., and Kozubski, W. (2019). Alzheimer’s dementia: pathogenesis and impact of cardiovascular risk factors on cognitive decline. Postgrad. Med. 131, 415–422. doi: 10.1080/00325481.2019.1657776
Webers, A., Heneka, M. T., and Gleeson, P. A. (2020). The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 98, 28–41. doi: 10.1111/imcb.12301
Weidensteiner, C., Metzger, F., Bruns, A., Bohrmann, B., Kuennecke, B., and Von Kienlin, M. (2009). Cortical hypoperfusion in the B6.PS2APP mouse model for Alzheimer’s disease: comprehensive phenotyping of vascular and tissular parameters by MRI. Magn. Reson. Med. 62, 35–45. doi: 10.1002/mrm.21985
Wyatt-Johnson, S. K., and Brutkiewicz, R. R. (2020). The complexity of microglial interactions with innate and adaptive immune cells in Alzheimer’s disease. Front. Aging Neurosci. 12:592359. doi: 10.3389/fnagi.2020.592359
Xiao, Q., Yan, P., Ma, X., Liu, H., Perez, R., Zhu, A., et al. (2014). Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 34, 9607–9620. doi: 10.1523/JNEUROSCI.3788-13.2014
Yamazaki, Y., and Kanekiyo, T. (2017). Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 18:1965. doi: 10.3390/ijms18091965
Yanagawa, T., Takao, M., Yasuda, M., Kamide, T., Sato, H., Suzuki, K., et al. (2019). Clinical and neuropathologic analysis of intracerebral hemorrhage in patients with cerebral amyloid angiopathy. Clin. Neurol. Neurosurg. 176, 110–115. doi: 10.1016/j.clineuro.2018.11.020
Yin, J., Valin, K. L., Dixon, M. L., and Leavenworth, J. W. (2017). The role of microglia and macrophages in cns homeostasis, autoimmunity and cancer. J. Immunol. Res. 2017:5150678. doi: 10.1155/2017/5150678
Zand, L., Ryu, J. K., and Mclarnon, J. G. (2005). Induction of angiogenesis in the beta-amyloid peptide-injected rat hippocampus. Neuroreport 16, 129–132. doi: 10.1097/00001756-200502080-00011
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56. doi: 10.1016/j.nbd.2016.07.007
Zenaro, E., Pietronigro, E., Della Bianca, V., Piacentino, G., Marongiu, L., Budui, S., et al. (2015). Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. doi: 10.1038/nm.3913
Zhan, X., Stamova, B., and Sharp, F. R. (2018). Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: a review. Front. Aging Neurosci. 10:42. doi: 10.3389/fnagi.2018.00042
Zhang, Y., Bander, E. D., Lee, Y., Muoser, C., Schaffer, C. B., and Nishimura, N. (2020). Microvessel occlusions alter amyloid-beta plaque morphology in a mouse model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 40, 2115–2131. doi: 10.1177/0271678X19889092
Zhao, L. (2019). CD33 in Alzheimer’s disease - biology, pathogenesis and therapeutics: a mini-review. Gerontology 65, 323–331. doi: 10.1159/000492596
Zhao, X., Eyo, U. B., Murugan, M., and Wu, L. J. (2018). Microglial interactions with the neurovascular system in physiology and pathology. Dev. Neurobiol. 78, 604–617. doi: 10.1002/dneu.22576
Zheng, W. H., Bastianetto, S., Mennicken, F., Ma, W., and Kar, S. (2002). Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201–211. doi: 10.1016/s0306-4522(02)00404-9
Zhong, L., Chen, X. F., Wang, T., Wang, Z., Liao, C., Wang, Z., et al. (2017). Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 214, 597–607. doi: 10.1084/jem.20160844
Zhong, L., Xu, Y., Zhuo, R., Wang, T., Wang, K., Huang, R., et al. (2019). Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 10:1365. doi: 10.1038/s41467-019-09118-9
Keywords: astrocyte, microglia, amyloid beta, neurofibrillary tangles, neurovascular unit, neurodegeneration, cognition
Citation: Bandyopadhyay S (2021) Role of Neuron and Glia in Alzheimer’s Disease and Associated Vascular Dysfunction. Front. Aging Neurosci. 13:653334. doi: 10.3389/fnagi.2021.653334
Received: 14 January 2021; Accepted: 05 May 2021;
Published: 15 June 2021.
Edited by:
Ignacio Torres-Aleman, Achucarro Basque Center for Neuroscience, SpainReviewed by:
Marie-Ève Tremblay, University of Victoria, CanadaJavier Vitorica, Sevilla University, Spain
Copyright © 2021 Bandyopadhyay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanghamitra Bandyopadhyay,c2FuZ2htaXRyYUBpaXRyLnJlcy5pbg==