 Bindu Diana Paul
Bindu Diana Paul- The Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, United States
The reverse transsulfuration pathway has emerged as a central hub that integrates the metabolism of sulfur-containing amino acids and redox homeostasis. Transsulfuration involves the transfer of sulfur from homocysteine to cysteine. Cysteine serves as the precursor for several sulfur-containing molecules, which play diverse roles in cellular processes. Recent evidence shows that disruption of the flux through the pathway has deleterious consequences. In this review article, I will discuss the actions and regulation of the reverse transsulfuration pathway and its links to other metabolic pathways, which are disrupted in Alzheimer’s disease (AD). The potential nodes of therapeutic intervention are also discussed, which may pave the way for the development of novel treatments.
Introduction
Alzheimer’s disease (AD) is a relentless, progressive neurodegenerative disorder predominantly affecting the hippocampus and the cortex leading to memory loss, cognitive deficits, and impaired executive function (Masters et al., 2015; Lane et al., 2018). AD is the most common form of dementia that afflicts 10–30% of the population 65 years or older, resulting in late-onset AD (LOAD) (Masters et al., 2015; Long and Holtzman, 2019; Alzheimer’s Association, 2020). AD may either be familial or sporadic, with either early onset (EOAD) or late-onset (LOAD) with familial causes constituting less than 5% of the total cases (Karch and Goate, 2015). The disease has a long prodromal and preclinical phase, and it may take years for symptoms to manifest, the most noticeable of which include memory impairments and language difficulties (Bateman et al., 2012; Villemagne et al., 2013; Vermunt et al., 2019). At the pathological level, AD is characterized by the accumulation of amyloid plaques, neurofibrillary tangles (NFTs), and paired helical filaments (PHFs) (Glenner and Wong, 1984; Masters et al., 1985, 2015; Grundke-Iqbal et al., 1986; Kosik et al., 1986; Wood et al., 1986).
Regardless of the cause of AD, elevated oxidative stress has been observed in AD, which contributes to disease progression (Torres et al., 2011; Martínez de Toda et al., 2019; Arslan et al., 2020). Oxidative stress refers to an imbalance in redox signaling, where physiological levels of reactive oxygen species (ROS) mediate signal transduction processes (oxidative eustress) and excess has deleterious effects (oxidative distress) (Sies et al., 2017). In AD, Aβ1–42, which is generated by the cleavage of amyloid precursor protein (APP), by enzymes such as β-secretase 1 (BACE1) and increases oxidative stress. Aβ1–42 elevates oxidative stress by increasing the formation of Fe2+ and Cu+, which generate hydroxyl radicals (•OH) via the Fenton reaction, which elicits oxidative damage (Imlay et al., 1988; Huang et al., 1999a,b). Additionally, Aβ1–42 activates the Jun N-terminal kinase (JNK) pathway, which is linked to upregulation of BACE1, which mediates increased cleavage of APP to produce more Aβ1–42, resulting in a vicious feed-forward cycle (Yao et al., 2005; Guglielmotto et al., 2011). A point to be noted is that Aβ peptides can act as antioxidants as well (Kontush, 2001). It has been proposed that the accumulation of Aβ peptides is protective (Smith et al., 2002). At lower concentrations, in the range of 0.1–1.0 nm, in body fluids, Aβ peptides prevent autooxidation of lipoproteins and plasma low-density lipoprotein (LDL) in the cerebrospinal fluid (CSF). Similar to Aβ1–40, the Aβ1–42 peptide is also capable of exerting antioxidant effects and the antioxidant activity has been attributed to its metal-chelating effect. At higher concentrations, however, its antioxidant action was abolished (Kontush et al., 2001).
Chronic oxidative stress also affects Tau to elicit neurotoxicity. Tau is hyperphosphorylated in response to oxidative stress, which leads to the formation of NFTs and neurotoxicity (Zhu et al., 2005; Su et al., 2010). Several studies report that oxidative stress may precede symptoms and pathology of AD (Nunomura et al., 2001; Zhu et al., 2004). Oxidative stress in AD is also linked to mitochondrial dysfunction, inflammation, and hypoxia, and these processes culminate in vicious cycles that contribute to neurodegeneration (Bonda et al., 2014; Oliver and Reddy, 2019; Merelli et al., 2020; Butterfield and Boyd-Kimball, 2020).
The reverse transsulfuration pathway plays a pivotal role in the maintenance of redox balance in cells. Transsulfuration involves the transfer of sulfur from homocysteine to form cysteine via cystathionine with connections to the transmethylation pathway and one-carbon metabolism (Figure 1). In addition to serving as a building block for protein synthesis, cysteine is channeled into multiple pathways to generate sulfur-containing molecules including glutathione, the cellular antioxidant, taurine, coenzyme A and lanthionine (Paul et al., 2018). The availability of cysteine is the rate-limiting step for the synthesis of glutathione (GSH) in cells, which maintains redox homeostasis. Diversion of cysteine from protein synthesis to synthesize glutathione is neuroprotective (Ratan et al., 1994). Also, cysteine is the substrate for the generation of the gaseous signaling molecule, hydrogen sulfide (H2S). Three enzymes, cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST) generate H2S in cells. Besides, CSE and CBS may also utilize homocysteine to generate H2S (Paul and Snyder, 2012; Paul et al., 2018). H2S, like nitric oxide (NO), modulates a myriad of cellular processes, including transcriptional regulation, response to stress, and mitochondrial function (Krishnan et al., 2011; Paul et al., 2018, 2020; Sbodio et al., 2018). Dysregulated transsulfuration has been observed in several pathological conditions and during aging. In this review, we focus on the status of the transsulfuration pathway in Alzheimer’s disease.
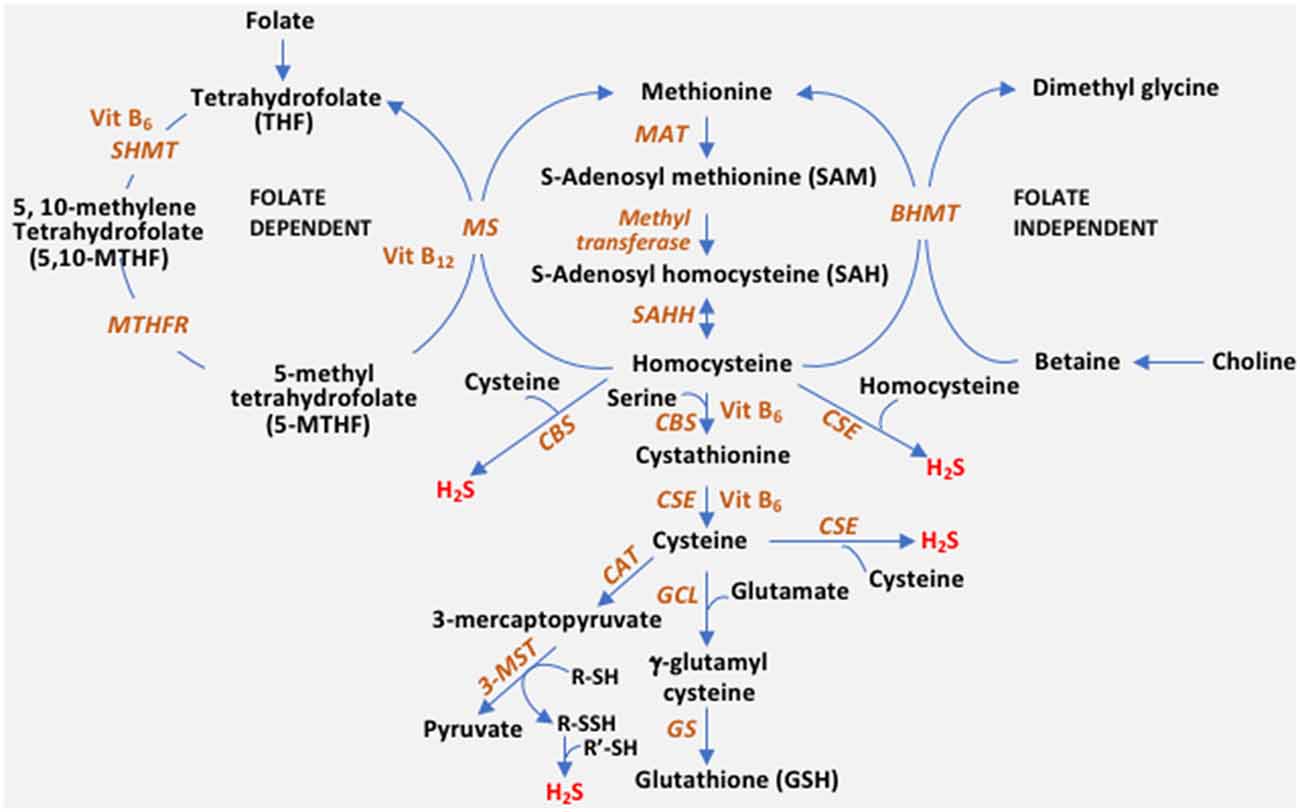
Figure 1. The reverse transsulfuration pathway and inputs from associated pathways. Transsulfuration refers to the transfer of sulfur from homocysteine to cysteine. Dietary methionine is converted to homocysteine, via S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH). Homocysteine is condensed with serine by cystathionine β-synthase (CBS) to form cystathionine, which is utilized by cystathionine γ-lyase (CSE) to generate cysteine. Cysteine can either enter the glutathione (GSH) biosynthetic pathway or be utilized as a substrate for hydrogen sulfide (H2S) biosynthesis. CBS generates H2S efficiently from a combination of cysteine and homocysteine, whereas CSE can utilize either cysteine or homocysteine by itself to generate the gaseous signaling molecule. A third enzyme, 3-mercaptopyruvate sulfurtransferase (3-MST) also generates H2S. 3-MST utilizes the 3-mercaptopyruvate generated by cysteine aminotransferase (CAT) by forming a persulfide on its active site (R-SH to R-SSH). The persulfide releases H2S in the presence of a reductant (R’-SH). Cysteine is converted to GSH by the sequential action of glutamyl cysteine ligase (GCL) and glutathione synthase (GS). Homocysteine is at the junction of the transsulfuration and remethylation pathway (where methionine is regenerated from homocysteine). Remethylation of homocysteine occurs in both a folate-independent and dependent pathway. In the folate-independent pathway, betaine/trimethylglycine (derived from choline) donates methyl groups in a reaction catalyzed by betaine–homocysteine methyltransferase (BHMT) to generate methionine. In the folate-dependent pathway, the vitamin B12-dependent enzyme, methionine synthase (MS), converts homocysteine to methionine and tetrahydrofolate (THF), where 5-Methyltetrahydrofolate (5-MTHF) acts as the methyl group donor. THF is converted to 5,10-methylenetetrahydrofolate (5,10-MTHF) by serine hydroxymethyltransferase (SHMT) which utilizes serine and vitamin B6. 5,10-methylenetetrahydrofolate reductase (MTHFR), then reduces 5,10-MTHF to 5-MTHF, which remethylates another molecule of homocysteine. MTHFR uses flavin adenine dinucleotide (FAD; the active form of vitamin B2) as a cofactor.
Elevated Homocysteine, Dementia, and Ad
Homocysteine is at the intersection of the transsulfuration and transmethylation pathway (Figure 1). Homocysteine may either be remethylated to methionine or utilized by the transsulfuration pathway to form cysteine. Hyperhomocysteinemia (elevated levels of homocysteine in the blood) is a risk factor for the development of cardiovascular disease and neurodegenerative diseases including dementia, AD, and Parkinson’s disease (PD) (Seshadri et al., 2002). Elevated homocysteine has also been observed in cases of epilepsy and neuropsychiatric disorders (Herrmann et al., 2007). The involvement of homocysteine in dementia and AD was noted more than 20 years ago using clinical and histochemical analyses (Clarke et al., 1998; Smith et al., 2018). The levels of homocysteine have been correlated with the severity of AD in several studies (Kitzlerová et al., 2014; Farina et al., 2017). Hyperhomocysteinemia has been linked to irreversible neurological deficits and could stem from deficits in levels of folate, vitamin B6, or vitamin B12 (Clarke et al., 1998; Smith and Refsum, 2016). Homocysteine mediates toxicity in multiple ways, all of which lead to conditions that pose a risk for developing AD. Homocysteine may elicit neurotoxicity by activation of the N-methyl D aspartate (NMDA) receptors to cause excitotoxicity, acting as an agonist at the glutamate binding site as well as a partial antagonist of the glycine co-agonist site (Lipton et al., 1997). Overactivation of the NMDA receptor has been reported to mediate brain damage in focal ischemia (Simon et al., 1984; Lipton and Rosenberg, 1994). Overstimulation of the NMDA receptor increases calcium influx leading to an imbalance in excitatory-inhibitory neurotransmission in the hippocampus of the brain and altered extracellular levels of neuroexcitatory (aspartate) and neuroinhibitory (GABA) neurotransmitters leading to excitotoxic neuronal death and seizures. Homocysteine also causes aberrant processing of the amyloid precursor protein (APP) by a mechanism involving hypomethylation (Lin et al., 2009). Additionally, homocysteine elicits the DNA damage response in neurons and promotes apoptosis and hypersensitivity to excitotoxicity (Kruman et al., 2000). Similarly, increased homocysteine levels were linked to DNA damage in patients deficient in CBS (Vanzin et al., 2014). Elevated homocysteine levels have been linked to traumatic brain injury (TBI) as well, which confers a greater risk for developing AD than stroke (Rahmani et al., 2016). Thus, homocysteine may not only mediate vascular injury, leading to stroke (a risk factor for AD), but also perpetuate the downstream neurotoxic responses. Conversion of homocysteine to homocysteic acid (HCA), a potent glutamate analog, and cysteine sulfinic acid (CSA), which are excitotoxins and generators of oxidative stress, also lead to neuronal damage (Bleich et al., 2000; Obeid and Herrmann, 2006).
Causes of Hyperhomocysteinemia
Homocysteine can be consumed by either its conversion to methionine by the transmethylation pathway or by its conversion to cysteine via the reverse transsulfuration pathway (Figure 1). Since dietary folate and vitamin B12 influence the channeling of homocysteine into the reverse transsulfuration pathway, levels of these cofactors may modulate H2S production. Decreased expression or activities of the enzymes methylenetetrahydrofolate reductase (MTHFR), methionine synthase (MS), CBS, or CSE may lead to hyperhomocysteinemia. MTHFR is the rate-limiting step in the methyl cycle, catalyzing the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, a methyl donor for remethylation of homocysteine to methionine by MS, using NADPH as the reducing agent (Figure 1). MTHFR is the rate-limiting enzyme catalyzing folate production. Mutations in the MTHFR gene, which reduce its activity, confer a risk factor for developing AD and atherosclerosis. The mutations that elevate homocysteine include 677C > T, 1298A > C (Castro et al., 2003). The levels of homocysteine in the serum of AD patients and those with mild cognitive impairment (MCI) have been reported to be significantly higher than normal subjects (Joosten et al., 1997; Clarke et al., 1998; Kim et al., 2013; Ma et al., 2017). Similarly, mutations in CTH, the gene encoding CSE, are also linked to elevated homocysteine levels, for instance, the 1364G>T (Ser403Ile) and CTH 1364T/T homozygotes were associated with hyperhomocysteinemia (Wang and Hegele, 2003; Wang et al., 2004). The first report of CTH mutations in cystathioninuric individuals reported two frameshift mutations, c.940_941delCT (p.Leu262ThrfsX20), and c.1220delC (p.Thr355IlefsX18) and two missense mutations, c.200C>T (p.Thr67Ile) and c.718C>G (p.Gln240Glu) (Wang and Hegele, 2003). The single nucleotide polymorphism in CTH, originally described as c.1364G>T (p.S403Ile) was renamed as.c.1208G>T (p.Ser403Ile), according to the current convention (Kraus et al., 2009). Consistent with these observations, mice lacking CSE and CBS display increased homocysteine levels in both the blood and the CSF (Yang et al., 2008; Akahoshi et al., 2019). Increased homocysteine concentration compromises the integrity of the blood-brain barrier in mice as measured in the Cbs+/– mice (Kamath et al., 2006).
Various strategies have been utilized to counter the toxicity induced by elevated homocysteine levels, including supplementation with vitamin B6. Administering B6 (pyridoxine) may be effective where defective transsulfuration occurs, as the enzymes CSE and CBS utilize pyridoxal 5- phosphate (PLP) as a cofactor, which influences the flux through the reverse transsulfuration pathway (Lima et al., 2006; Gregory et al., 2016). Interestingly, in Huntington’s disease (HD), impaired PLP metabolism has been observed both in mouse models as well as in patients, which together with the impaired transsulfuration pathway, contributes to impaired cysteine and H2S metabolism (Sbodio et al., 2016; Sorolla et al., 2016). In several instances, supplementation with betaine has proven beneficial as betaine serves as the methyl donor in the remethylation of homocysteine to methionine and S-adenosylmethionine (SAM) (Chai et al., 2013; McBreairty et al., 2016). In cases, where mutations occur in the MTHFR gene, this strategy may not work. Vitamin B12 therapy has also proven beneficial in delaying symptoms of AD. Vitamin B12 (cobalamin) serves as a cofactor for methionine synthase, which is one of the enzymes involved in the conversion of homocysteine to methionine. Methylmalonyl-CoA mutase (MUT) is the only other enzyme identified, which utilizes vitamin B12 (Watkins and Rosenblatt, 1989; Froese et al., 2019). For cobalamin to be utilized, its efficient transit through the intracellular lysosomal compartment and subsequent delivery to the cytosol and mitochondria is required. Lysosomal function is derailed in Alzheimer’s disease (AD) and its utilization is compromised (Zhao et al., 2015).
In a randomized placebo-controlled trial in older men, supplementation with folate, vitamin B6 and vitamin B12 daily for 2 years decreased homocysteine levels and reduced the rate of increase in circulating levels of amyloid-β1–40 (Flicker et al., 2008). Similarly, folate supplementation for 3 years improved measures of cognitive function in men and women aged 50–70 years with raised plasma total homocysteine and normal serum vitamin B12 (Durga et al., 2007). Other studies reported increases in homocysteine levels in a cohort with mild to moderate AD as a function of disease progression, however, no significant decline in either dietary intake or blood levels of vitamin B12/folate was observed indicating that other reasons for hyperhomocysteinemia may operate (Farina et al., 2017).
Suboptimal Activity of the Reverse Transsulfuration Pathway
The reverse transsulfuration pathway leads to the production of cysteine, the availability of which is the rate-limiting step for glutathione (GSH) biosynthesis. Glutathione levels, as measured by in vivo proton magnetic resonance spectroscopy, were lower in hippocampi and frontal cortices of patients with AD compared to healthy controls where decreases in glutathione were correlated to decline in cognitive function. Cysteine is taken up by neurons by EAAC1/EAAT3, which exchanges cysteine for glutamate. Soluble Aβ can impair cysteine and glutathione metabolism in cells by inhibiting EAAT3 (Hodgson et al., 2013). Also, EAAT3 accumulates in the detergent-insoluble fraction of hippocampal neurons instead of its normal localization at the plasma membrane and its depletion causes age-dependent neurodegeneration (Aoyama et al., 2006; Duerson et al., 2009). Dysregulated cysteine metabolism has been observed in aging as well as neurodegenerative diseases such as Huntington’s disease (Dedeoglu et al., 2004; Paul et al., 2014; Zivanovic et al., 2019).
Glutathione Metabolism in Ad
In the APPTg2576 mouse model of AD, a decrease in GSH was observed at 19 months in the cortex of the brain (Dedeoglu et al., 2004). Similarly, in a mouse model of AD B6.Cg-Tg (APPSwe, PSEN1dE9), the ratio of reduced GSH to oxidized GSH (GSSG) decreased as a function of age (Zhang et al., 2012). In vivo proton, magnetic resonance spectroscopy studies revealed that GSH levels were decreased in the hippocampi and frontal cortices of patients with AD as compared to normal subjects and a decrease in glutathione was correlated to a decline in cognitive function (Mandal et al., 2015). A separate study reported sex-specific differences in GSH levels, where males with AD exhibited a decrease in blood cells as compared to their normal controls while samples from female subjects displayed no alterations in GSH content (Liu et al., 2005). A decrease in GSH and GSH/GSSG ratio was also observed in the plasma of patients with MCI and AD (Bermejo et al., 2008). Analysis of postmortem samples also revealed diminished GSH levels in mitochondrial, and synaptosomal fractions derived from the frontal cortices of MCI, mild and severe AD cases as compared to controls (Ansari and Scheff, 2010). Based on in vivo MRS studies, it was concluded that while hippocampal GSH levels could distinguish between MCI and elderly healthy controls with 87.5% sensitivity, 100% specificity, cortical GSH levels could differentiate MCI and AD with 91.7% sensitivity, 100% specificity (Mandal et al., 2012) and accordingly GSH has been proposed as a marker as well as therapy for AD (Mandal et al., 2019).
Signaling Mediated by Hydrogen Sulfide
In addition to its essential role as a building block in protein synthesis, cysteine is also the precursor of the gaseous signaling molecule, H2S, which participates in a multitude of physiological processes (Wang, 2012; Paul and Snyder, 2015). Three enzymes produce H2S endogenously. CSE utilizes cysteine to produce H2S, pyruvate, and ammonia (Stipanuk and Beck, 1982; Paul and Snyder, 2012, 2015). CBS condenses cysteine and homocysteine to produce H2S in addition to cystathionine (Chen et al., 2004; Paul and Snyder, 2012). 3-MST generates H2S in concert with cysteine aminotransferase (CAT). CAT metabolizes cysteine and α-ketoglutarate to form 3-mercaptopyruvate (3-MP). 3-MST acts on 3-MP formed to generate H2S and pyruvate. 3-MST also generates H2S using D-cysteine as a substrate in conjunction with D-amino acid oxidase (Shibuya et al., 2013). Over 150 mutations in the CBS protein have been reported, several of which cause its misfolding, and are linked to enzyme activity (Kozich et al., 2010). CBS and 3-MST occur mostly in the central nervous system, although these enzymes are also present in peripheral tissues (Shibuya et al., 2009). Similarly, CSE too is present in the brain. Accumulating studies have reported distinct spatial compartmentalization of the three enzymes in the central nervous system. While CBS is predominantly localized to astrocytes and glia, CSE is present in neurons (Enokido et al., 2005; Linden et al., 2008; Morikawa et al., 2012). 3-MST is also present in neurons and acts in conjunction with CAT to produce H2S. While CSE and CBS are predominantly cytosolic during basal conditions, they may translocate to the mitochondria or nucleus during stress (Paul et al., 2020). 3-MST is present in both the cytosolic and mitochondrial compartments.
H2S exerts its effects through a posttranslational modification termed persulfidation/sulfhydration, involving the conversion of -SH groups to persulfide or -SSH group (Mustafa et al., 2009; Paul and Snyder, 2012; Zivanovic et al., 2019). H2S and sulfhydration modulate several homeostatic processes in the central nervous system and its disruption occurs in several neurodegenerative diseases including Parkinson’s disease (PD) and HD (Vandiver et al., 2013; Paul et al., 2014; Paul and Snyder, 2018). H2S plays central role in processes regulating cognitive function and neuromodulation. The first study on the role of H2S in processes involved in learning and memory demonstrated that physiological concentrations of H2S selectively enhance NMDA receptor-mediated responses to modulate the induction of hippocampal long-term potentiation (LTP; Abe and Kimura, 1996). Clinical studies have reported a decrease in H2S levels in the plasma of AD patients, which correlated with the degree of cognitive decline (Liu et al., 2008). Additionally, S-adenosyl methionine (SAM), which allosterically activates CBS is depleted in the human AD brain as well as in the cerebrospinal fluid (Morrison et al., 1996; Linnebank et al., 2010). Dietary supplementation with SAM delayed amyloid plaque and Tau pathology in the 3xTg-AD mouse model of AD (Lee et al., 2012). Injection of Aβ1–42 into the hippocampi of rats induced cognitive impairment and reduction in H2S levels and expression of endogenous CBS and 3-MST and intraperitoneal injection of the H2S donor, NaSH, ameliorated cognitive deficits as well as neuroinflammation (Liu et al., 2015). Similarly, several studies have reported the protective effects of H2S in mouse models of AD, although the effects of sulfhydration on signaling pathways were not evaluated (Giuliani et al., 2013; Zhao et al., 2016; Vandini et al., 2019).
We have shown that H2S signaling is dysregulated in AD, and sulfhydration is diminished in the 3xTg-AD mouse model of AD as well as in human AD (Giovinazzo et al., 2021). The protein Tau, which is hyperphosphorylated and aggregated in Alzheimer’s disease, binds CSE and stimulates its catalytic activity. Interestingly, CSE does not bind mutant P301L Tau, which is present in the 3xTg-AD model of AD. Additionally, H2S produced by CSE sulfhydrates glycogen synthase kinase 3β (GSK3β) and inhibits its catalytic activity (Figure 2). Sulfhydration of GSK3β was decreased in the cortex of postmortem human AD samples as well. Finally, supplementing NaGYY, the sodium salt of GYY4137, a slow-releasing H2S donor, restored sulfhydration and ameliorated motor and cognitive deficits in the 3xTg-AD mice (Giovinazzo et al., 2021).
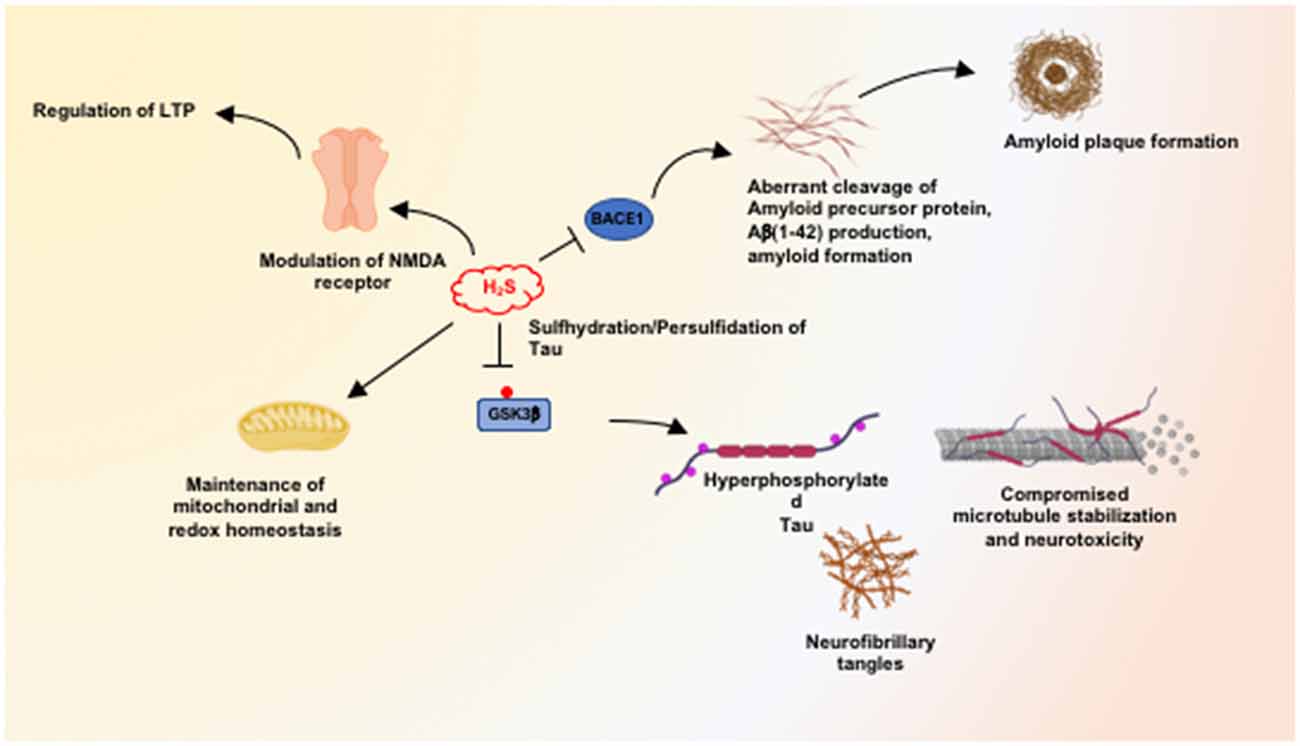
Figure 2. Neuroprotective processes influenced by hydrogen sulfide (H2S). H2S mediates neuroprotection by modulating multiple pathways, including maintenance of mitochondrial function and redox balance in the brain. At physiological concentrations, H2S facilitates long-term potentiation (LTP) in a manner dependent on the N-methyl D-aspartate (NMDA) receptors. H2S inhibits the expression and activity of beta-secretase 1 (BACE1) and thus inhibits amyloidogenic processing of the amyloid precursor protein (APP) to prevent the accumulation of β-amyloid plaques. H2S also mediates persulfidation/sulfhydration of glycogen synthase kinase 3β (GSK3β) and inhibits activity, preventing hyperphosphorylation of Tau which would prevent its aggregation into neurofibrillary tangles (NFTs).
Therapeutic Avenues
Analysis of clinical trials conducted so far, reveal a preference for non-amyloid targets, which include therapeutics targeting inflammation, synapse and neuronal protection, vascular factors, neurogenesis, and epigenetic modifications (Cummings et al., 2014). Additionally, there has been an increased interest in repurposed drugs. Several clinical trials involving the use of antioxidants have largely failed (Kim et al., 2015). Some of the antioxidants used only target specific reactive oxygen species (ROS) and thus do not affect other species. Others inhibit physiological processes such as autophagy, causing undesirable side effects (Underwood et al., 2010). Similarly, the timing and duration of intervention could also affect outcomes, as well as patient selection criteria. Thus, therapies which target multiple pathways without compromising normal cellular processes would be more effective. As the gasotransmitter H2S regulates a wide array of neuroprotective processes, therapies, and interventions involving either the use of H2S delivering drugs or agents which stimulate its production are gaining importance. In mice treated with homocysteine, supplementation of H2S donors ameliorated homocysteine-induced cerebrovascular pathology, cognitive deficits, and toxicity (Kamat et al., 2016). Supplementation of H2S donors was also reported to be beneficial in several mouse models of AD. Administering sodium hydrosulfide (NaSH), at 2.8 mg/kg, once a day for 3 months ameliorated memory deficits, reduced APP and BACE1, upregulated the master regulator of antioxidant response genes, nuclear factor erythroid-2-related factor 2 (Nrf2), heme oxygenase-1(HO-1) and glutathione S-transferase (GST) in the APP/PS1 model of AD (Liu et al., 2016). The mitochondria-targeted H2S donor, AP-39, maintained cellular bioenergetics and preserved mitochondrial function in the APP/PS1 mouse model of AD (Zhao et al., 2016). Other modes of action of H2S donors were also reported. For instance, NaSH afforded benefits in the APP/PS1 mice by acting via the GluN2B subunit of the NMDA receptor (Yang et al., 2016). Physiological concentrations of H2S also increased levels of the second messenger, cAMP, in primary cultures of brain cells, neuronal and glial cell lines, and Xenopus oocytes and activated the NMDA receptor in a protein kinase A (PKA)-dependent manner (Kimura, 2000). Thus, H2S modulates several neuroprotective pathways, and modulation of its production may be beneficial.
Future areas of research in this area would involve tunable delivery of H2S donors so that its concentration in cells and tissues can be modulated.
Author Contributions
BP conceptualized and wrote the review and prepared the figures.
Funding
This work was supported by the American Heart Association (AHA)/Paul Allen Frontiers Group; grant number 19PABH134580006 from the AHA/Allen Initiative in Brain Health and Cognitive Impairment team.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alzheimer’s Association. (2020). 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 16, 391–460. Available online at: https://www.alz.org/media/documents/alzheimers-facts-and-figures_1.pdf. Accessed November 1, 2020.
Abe, K., and Kimura, H. (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996
Akahoshi, N., Yokoyama, A., Nagata, T., Miura, A., Kamata, S., and Ishii, I. (2019). Abnormal amino acid profiles of blood and cerebrospinal fluid from cystathionine β-synthase-deficient mice, an animal model of homocystinuria. Biol. Pharm. Bull. 42, 1054–1057. doi: 10.1248/bpb.b19-00127
Ansari, M. A., and Scheff, S. W. (2010). Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 69, 155–167. doi: 10.1097/NEN.0b013e3181cb5af4
Aoyama, K., Suh, S. W., Hamby, A. M., Liu, J., Chan, W. Y., Chen, Y., et al. (2006). Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 9, 119–126. doi: 10.1038/nn1609
Arslan, J., Jamshed, H., and Qureshi, H. (2020). Early detection and prevention of Alzheimer’s disease: role of oxidative markers and natural antioxidants. Front. Aging Neurosci. 12:231. doi: 10.3389/fnagi.2020.00231
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Bermejo, P., Martin-Aragón, S., Benedí, J., Susín, C., Felici, E., Gil, P., et al. (2008). Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 42, 162–170. doi: 10.1080/10715760701861373
Bleich, S., Degner, D., Wiltfang, J., Maler, J. M., Niedmann, P., Cohrs, S., et al. (2000). Elevated homocysteine levels in alcohol withdrawal. Alcohol Alcohol. 35, 351–354. doi: 10.1093/alcalc/35.4.351
Bonda, D. J., Wang, X., Lee, H.-G., Smith, M. A., Perry, G., and Zhu, X. (2014). Neuronal failure in Alzheimer’s disease: a view through the oxidative stress looking-glass. Neurosci. Bull. 30, 243–252. doi: 10.1007/s12264-013-1424-x
Butterfield, D. A., and Boyd-Kimball, D. (2020). Mitochondrial oxidative and nitrosative stress and Alzheimer disease. Antioxidants 9:818. doi: 10.3390/antiox9090818
Castro, R., Rivera, I., Ravasco, P., Jakobs, C., Blom, H. J., Camilo, M. E., et al. (2003). 5,10-Methylenetetrahydrofolate reductase 677C–>T and 1298A–>C mutations are genetic determinants of elevated homocysteine. QJM 96, 297–303. doi: 10.1093/qjmed/hcg039
Chai, G. S., Jiang, X., Ni, Z. F., Ma, Z. W., Xie, A. J., Cheng, X. S., et al. (2013). Betaine attenuates Alzheimer-like pathological changes and memory deficits induced by homocysteine. J. Neurochem. 124, 388–396. doi: 10.1111/jnc.12094
Chen, X., Jhee, K.-H., and Kruger, W. D. (2004). Production of the neuromodulator H2S by cystathionine β-synthase via the condensation of cysteine and homocysteine. J. Biol. Chem. 279, 52082–52086. doi: 10.1074/jbc.C400481200
Clarke, R., Smith, A. D., Jobst, K. A., Refsum, H., Sutton, L., and Ueland, P. M. (1998). Folate, vitamin B12 and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 55, 1449–1455. doi: 10.1001/archneur.55.11.1449
Cummings, J. L., Morstorf, T., and Zhong, K. (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 6:37. doi: 10.1186/alzrt269
Dedeoglu, A., Choi, J.-K., Cormier, K., Kowall, N. W., and Jenkins, B. G. (2004). Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 1012, 60–65. doi: 10.1016/j.brainres.2004.02.079
Duerson, K., Woltjer, R. L., Mookherjee, P., Leverenz, J. B., Montine, T. J., Bird, T. D., et al. (2009). Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 19, 267–278. doi: 10.1111/j.1750-3639.2008.00186.x
Durga, J., van Boxtel, M. P., Schouten, E. G., Kok, F. J., Jolles, J., Katan, M. B., et al. (2007). Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet 369, 208–216. doi: 10.1016/S0140-6736(07)60109-3
Enokido, Y., Suzuki, E., Iwasawa, K., Namekata, K., Okazawa, H., and Kimura, H. (2005). Cystathionine β-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 19, 1854–1856. doi: 10.1096/fj.05-3724fje
Farina, N., Jernerén, F., Turner, C., Hart, K., and Tabet, N. (2017). Homocysteine concentrations in the cognitive progression of Alzheimer’s disease. Exp. Gerontol. 99, 146–150. doi: 10.1016/j.exger.2017.10.008
Flicker, L., Martins, R. N., Thomas, J., Acres, J., Taddei, K., Vasikaran, S. D., et al. (2008). B-vitamins reduce plasma levels of β amyloid. Neurobiol. Aging 29, 303–305. doi: 10.1016/j.neurobiolaging.2006.10.007
Froese, D. S., Fowler, B., and Baumgartner, M. R. (2019). Vitamin B12, folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 42, 673–685. doi: 10.1002/jimd.12009
Giovinazzo, D., Bursac, B., Sbodio, J. I., Nalluru, S., Vignane, T., Snowman, A. M., et al. (2021). Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. U S A 118:e2017225118. doi: 10.1073/pnas.2017225118
Giuliani, D., Ottani, A., Zaffe, D., Galantucci, M., Strinati, F., Lodi, R., et al. (2013). Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 104, 82–91. doi: 10.1016/j.nlm.2013.05.006
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Gregory, J. F., DeRatt, B. N., Rios-Avila, L., Ralat, M., and Stacpoole, P. W. (2016). Vitamin B6 nutritional status and cellular availability of pyridoxal 5’-phosphate govern the function of the transsulfuration pathway’s canonical reactions and hydrogen sulfide production via side reactions. Biochimie 126, 21–26. doi: 10.1016/j.biochi.2015.12.020
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089. doi: 10.1016/S0021-9258(17)38495-8
Guglielmotto, M., Monteleone, D., Giliberto, L., Fornaro, M., Borghi, R., Tamagno, E., et al. (2011). Amyloid-β42 activates the expression of BACE1 through the JNK pathway. J. Alzheimers Dis. 27, 871–883. doi: 10.3233/JAD-2011-110884
Herrmann, W., Lorenzl, S., and Obeid, R. (2007). Review of the role of hyperhomocysteinemia and B-vitamin deficiency in neurological and psychiatric disorders–current evidence and preliminary recommendations. Fortschr. Neurol. Psychiatr. 75, 515–527. doi: 10.1055/s-2007-980112
Hodgson, N., Trivedi, M., Muratore, C., Li, S., and Deth, R. (2013). Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 36, 197–209. doi: 10.3233/JAD-130101
Huang, X., Atwood, C. S., Hartshorn, M. A., Multhaup, G., Goldstein, L. E., Scarpa, R. C., et al. (1999a). The A β peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38, 7609–7616. doi: 10.1021/bi990438f
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, J. D., Hanson, G. R., et al. (1999b). Cu(II) potentiation of Alzheimer aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116. doi: 10.1074/jbc.274.52.37111
Imlay, J. A., Chin, S. M., and Linn, S. (1988). Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240, 640–642. doi: 10.1126/science.2834821
Joosten, E., Lesaffre, E., Riezler, R., Ghekiere, V., Dereymaeker, L., Pelemans, W., et al. (1997). Is metabolic evidence for vitamin B-12 and folate deficiency more frequent in elderly patients with Alzheimer’s disease? J. Gerontol. A Biol. Sci. Med. Sci. 52, M76–M79. doi: 10.1093/gerona/52a.2.m76
Kamat, P. K., Kyles, P., Kalani, A., and Tyagi, N. (2016). Hydrogen sulfide ameliorates homocysteine-induced Alzheimer’s disease-like pathology, blood-brain barrier disruption, and synaptic disorder. Mol. Neurobiol. 53, 2451–2467. doi: 10.1007/s12035-015-9212-4
Kamath, A. F., Chauhan, A. K., Kisucka, J., Dole, V. S., Loscalzo, J., Handy, D. E., et al. (2006). Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 107, 591–593. doi: 10.1182/blood-2005-06-2506
Karch, C. M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Kim, G., Kim, H., Kim, K. N., Son, J. I., Kim, S. Y., Tamura, T., et al. (2013). Relationship of cognitive function with B vitamin status, homocysteine and tissue factor pathway inhibitor in cognitively impaired elderly: a cross-sectional survey. J. Alzheimers Dis. 33, 853–862. doi: 10.3233/JAD-2012-121345
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi: 10.5607/en.2015.24.4.325
Kimura, H. (2000). Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267, 129–133. doi: 10.1006/bbrc.1999.1915
Kitzlerová, E., Fisar, Z., Jirák, R., Zvěrová, M., Hroudová, J., Benaková, H., et al. (2014). Plasma homocysteine in Alzheimer’s disease with or without co-morbid depressive symptoms. Neuro Endocrinol. Lett. 35, 42–49.
Kontush, A. (2001). Amyloid-β: an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer’s disease. Free Radic. Biol. Med. 31, 1120–1131. doi: 10.1016/s0891-5849(01)00688-8
Kontush, A., Berndt, C., Weber, W., Akopyan, V., Arlt, S., Schippling, S., et al. (2001). Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic. Biol. Med. 30, 119–128. doi: 10.1016/s0891-5849(00)00458-5
Kosik, K. S., Joachim, C. L., and Selkoe, D. J. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U S A 83, 4044–4048. doi: 10.1073/pnas.83.11.4044
Kozich, V., Sokolová, J., Klatovská, V., Krijt, J., Janosík, M., Jelínek, K., et al. (2010). Cystathionine β-synthase mutations: effect of mutation topology on folding and activity. Hum. Mutat. 31, 809–819. doi: 10.1002/humu.21273
Kraus, J. P., Hasek, J., Kozich, V., Collard, R., Venezia, S., Janosíková, B., et al. (2009). Cystathionine γ-lyase: Clinical, metabolic, genetic, and structural studies. Mol. Genet. Metab. 97, 250–259. doi: 10.1016/j.ymgme.2009.04.001
Krishnan, N., Fu, C., Pappin, D. J., and Tonks, N. K. (2011). H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 4:ra86. doi: 10.1126/scisignal.2002329
Kruman, I. I., Culmsee, C., Chan, S. L., Kruman, Y., Guo, Z., Penix, L., et al. (2000). Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 20, 6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000
Lane, C. A., Hardy, J., and Schott, J. M. (2018). Alzheimer’s disease. Eur. J. Neurol. 25, 59–70. doi: 10.1111/ene.13439
Lee, S., Lemere, C. A., Frost, J. L., and Shea, T. B. (2012). Dietary supplementation with S-adenosyl methionine delayed amyloid-β and tau pathology in 3xTg-AD mice. J. Alzheimers Dis. 28, 423–431. doi: 10.3233/JAD-2011-111025
Lima, C. P., Davis, S. R., Mackey, A. D., Scheer, J. B., Williamson, J., Gregory, J. F., et al. (2006). Vitamin B-6 deficiency suppresses the hepatic transsulfuration pathway but increases glutathione concentration in rats fed AIN-76A or AIN-93G diets. J. Nutr. 136, 2141–2147. doi: 10.1093/jn/136.8.2141
Lin, H.-C., Hsieh, H.-M., Chen, Y.-H., and Hu, M.-L. (2009). S-Adenosylhomocysteine increases β-amyloid formation in BV-2 microglial cells by increased expressions of β-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology 30, 622–627. doi: 10.1016/j.neuro.2009.03.011
Linden, D. R., Sha, L., Mazzone, A., Stoltz, G. J., Bernard, C. E., Furne, J. K., et al. (2008). Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J. Neurochem. 106, 1577–1585. doi: 10.1111/j.1471-4159.2008.05502.x
Linnebank, M., Popp, J., Smulders, Y., Smith, D., Semmler, A., Farkas, M., et al. (2010). S-adenosylmethionine is decreased in the cerebrospinal fluid of patients with Alzheimer’s disease. Neurodegener. Dis. 7, 373–378. doi: 10.1159/000309657
Lipton, S. A., Kim, W. K., Choi, Y. B., Kumar, S., D’Emilia, D. M., Rayudu, P. V., et al. (1997). Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U S A 94, 5923–5928. doi: 10.1073/pnas.94.11.5923
Lipton, S. A., and Rosenberg, P. A. (1994). Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 330, 613–622. doi: 10.1056/NEJM199403033300907
Liu, H., Deng, Y., Gao, J., Liu, Y., Li, W., Shi, J., et al. (2015). Sodium hydrosulfide attenuates β-amyloid-induced cognitive deficits and neuroinflammation via modulation of MAPK/NF-kappaB pathway in Rats. Curr. Alzheimer Res. 12, 673–683. doi: 10.2174/1567205012666150713102326
Liu, Y., Deng, Y., Liu, H., Yin, C., Li, X., and Gong, Q. (2016). Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: a novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 150–151, 207–216. doi: 10.1016/j.pbb.2016.11.002
Liu, H., Harrell, L. E., Shenvi, S., Hagen, T., and Liu, R. M. (2005). Gender differences in glutathione metabolism in Alzheimer’s disease. J. Neurosci. Res. 79, 861–867. doi: 10.1002/jnr.20424
Liu, X.-Q., Liu, X.-Q., Jiang, P., Huang, H., and Yan, Y. (2008). Plasma levels of endogenous hydrogen sulfide and homocysteine in patients with Alzheimer’s disease and vascular dementia and the significance thereof. Zhonghua Yi Xue Za Zhi 88, 2246–2249. doi: 10.3321/j.issn.0376-2491.2008.32.004
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Ma, F., Wu, T., Zhao, J., Ji, L., Song, A., Zhang, M., et al. (2017). Plasma homocysteine and serum folate and vitamin B12 levels in mild cognitive impairment and Alzheimer’s disease: a case-control study. Nutrients 9:725. doi: 10.3390/nu9070725
Mandal, P. K., Saharan, S., Tripathi, M., and Murari, G. (2015). Brain glutathione levels—a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 78, 702–710. doi: 10.1016/j.biopsych.2015.04.005
Mandal, P. K., Shukla, D., Tripathi, M., and Ersland, L. (2019). Cognitive improvement with glutathione supplement in Alzheimer’s disease: a way forward. J. Alzheimers Dis. 68, 531–535. doi: 10.3233/JAD-181054
Mandal, P. K., Tripathi, M., and Sugunan, S. (2012). Brain oxidative stress: detection and mapping of anti-oxidant marker ’Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 417, 43–48. doi: 10.1016/j.bbrc.2011.11.047
Martínez de Toda, I., Miguélez, L., Vida, C., Carro, E., and De la Fuente, M. (2019). Altered redox state in whole blood cells from patients with mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 71, 153–163. doi: 10.3233/JAD-190198
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers 1:15056. doi: 10.1038/nrdp.2015.56
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U S A 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
McBreairty, L. E., Robinson, J. L., Harding, S. V., Randell, E. W., Brunton, J. A., and Bertolo, R. F. (2016). Betaine is as effective as folate at re-synthesizing methionine for protein synthesis during moderate methionine deficiency in piglets. Eur. J. Nutr. 55, 2423–2430. doi: 10.1007/s00394-015-1049-0
Merelli, A., Repetto, M., Lazarowski, A., and Auzmendi, J. (2020). Hypoxia, oxidative stress, and inflammation: three faces of neurodegenerative diseases. J. Alzheimers Dis. [Epub ahead of print]. doi: 10.3233/JAD-201074
Morikawa, T., Kajimura, M., Nakamura, T., Hishiki, T., Nakanishi, T., Yukutake, Y., et al. (2012). Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. U S A 109, 1293–1298. doi: 10.1073/pnas.1119658109
Morrison, L. D., Smith, D. D., and Kish, S. J. (1996). Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 67, 1328–1331. doi: 10.1046/j.1471-4159.1996.67031328.x
Mustafa, A. K., Gadalla, M. M., Sen, N., Kim, S., Mu, W., Gazi, S. K., et al. (2009). H2S signals through protein S-sulfhydration. Sci. Signal. 2:ra72. doi: 10.1126/scisignal.2000464
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Obeid, R., and Herrmann, W. (2006). Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 580, 2994–3005. doi: 10.1016/j.febslet.2006.04.088
Oliver, D. M. A., and Reddy, P. H. (2019). Molecular basis of Alzheimer’s disease: focus on mitochondria. J. Alzheimers Dis. 72, S95–S116. doi: 10.3233/JAD-190048
Paul, B. D., Sbodio, J. I., and Snyder, S. H. (2018). Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol. Sci. 39, 513–524. doi: 10.1016/j.tips.2018.02.007
Paul, B. D., Sbodio, J. I., Xu, R., Vandiver, M. S., Cha, J. Y., Snowman, A. M., et al. (2014). Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 509, 96–100. doi: 10.1038/nature13136
Paul, B. D., and Snyder, S. H. (2012). H2S signaling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507. doi: 10.1038/nrm3391
Paul, B. D., and Snyder, S. H. (2015). Modes of physiologic H2S signaling in the brain and peripheral tissues. Antioxid. Redox Signal. 22, 411–423. doi: 10.1089/ars.2014.5917
Paul, B. D., and Snyder, S. H. (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 149, 101–109. doi: 10.1016/j.bcp.2017.11.019
Paul, B. D., Snyder, S. H., and Kashfi, K. (2020). Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 38:101772. doi: 10.1016/j.redox.2020.101772
Rahmani, A., Hatefi, M., Dastjerdi, M. M., Zare, M., Imani, A., and Shirazi, D. (2016). Correlation between serum homocysteine levels and outcome of patients with severe traumatic brain injury. World Neurosurg. 87, 507–515. doi: 10.1016/j.wneu.2015.09.016
Ratan, R. R., Murphy, T. H., and Baraban, J. M. (1994). Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J. Neurosci. 14, 4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2016). Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. U S A 113, 8843–8848. doi: 10.1073/pnas.1608264113
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2018). Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc. Natl. Acad. Sci. U S A 115, 780–785. doi: 10.1073/pnas.1717877115
Seshadri, S., Beiser, A., Selhub, J., Jacques, P. F., Rosenberg, I. H., D’Agostino, R. B., et al. (2002). Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 346, 476–483. doi: 10.1056/NEJMoa011613
Shibuya, N., Koike, S., Tanaka, M., Ishigami-Yuasa, M., Kimura, Y., Ogasawara, Y., et al. (2013). A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 4:1366. doi: 10.1038/ncomms2371
Shibuya, N., Tanaka, M., Yoshida, M., Ogasawara, Y., Togawa, T., Ishii, K., et al. (2009). 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714. doi: 10.1089/ars.2008.2253
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi: 10.1146/annurev-biochem-061516-045037
Simon, R. P., Swan, J. H., Griffiths, T., and Meldrum, B. S. (1984). Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 226, 850–852. doi: 10.1126/science.6093256
Smith, M. A., Casadesus, G., Joseph, J. A., and Perry, G. (2002). Amyloid-β and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic. Biol. Med. 33, 1194–1199. doi: 10.1016/s0891-5849(02)01021-3
Smith, A. D., and Refsum, H. (2016). Homocysteine, B vitamins, and cognitive impairment. Annu. Rev. Nutr. 36, 211–239. doi: 10.1146/annurev-nutr-071715-050947
Smith, A. D., Refsum, H., Bottiglieri, T., Fenech, M., Hooshmand, B., McCaddon, A., et al. (2018). Homocysteine and dementia: an international consensus statement. J. Alzheimers Dis. 62, 561–570. doi: 10.3233/JAD-171042
Sorolla, M. A., Rodríguez-Colman, M. J., Vall-Llaura, N., Vived, C., Fernández-Nogales, M., Lucas, J. J., et al. (2016). Impaired PLP-dependent metabolism in brain samples from Huntington disease patients and transgenic R6/1 mice. Metab. Brain Dis. 31, 579–586. doi: 10.1007/s11011-015-9777-7
Stipanuk, M. H., and Beck, P. W. (1982). Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 206, 267–277. doi: 10.1042/bj2060267
Su, B., Wang, X., Lee, H. G., Tabaton, M., Perry, G., Smith, M. A., et al. (2010). Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 468, 267–271. doi: 10.1016/j.neulet.2009.11.010
Torres, L. L., Quaglio, N. B., de Souza, G. T., Garcia, R. T., Dati, L. M., Moreira, W. L., et al. (2011). Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 26, 59–68. doi: 10.3233/JAD-2011-110284
Underwood, B. R., Imarisio, S., Fleming, A., Rose, C., Krishna, G., Heard, P., et al. (2010). Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 19, 3413–3429. doi: 10.1093/hmg/ddq253
Vandini, E., Ottani, A., Zaffe, D., Calevro, A., Canalini, F., Cavallini, G. M., et al. (2019). Mechanisms of hydrogen sulfide against the progression of severe Alzheimer’s disease in transgenic mice at different ages. Pharmacology 103, 50–60. doi: 10.1159/000494113
Vandiver, M. S., Paul, B. D., Xu, R., Karuppagounder, S., Rao, F., Snowman, A. M., et al. (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 4:1626. doi: 10.1038/ncomms2623
Vanzin, C. S., Manfredini, V., Marinho, A. E., Biancini, G. B., Ribas, G. S., Deon, M., et al. (2014). Homocysteine contribution to DNA damage in cystathionine β-synthase-deficient patients. Gene 539, 270–274. doi: 10.1016/j.gene.2014.02.015
Vermunt, L., Sikkes, S. A. M., van den Hout, A., Handels, R., Bos, I., van der Flier, W. M., et al. (2019). Duration of preclinical, prodromal. and dementia stages of Alzheimer’s disease in relation to age, sex and APOE genotype. Alzheimers Dement. 15, 888–898. doi: 10.1016/j.jalz.2019.04.001
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., et al. (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367. doi: 10.1016/S1474-4422(13)70044-9
Wang, R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896. doi: 10.1152/physrev.00017.2011
Wang, J., and Hegele, R. A. (2003). Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine γ-lyase (CTH). Hum. Genet. 112, 404–408. doi: 10.1007/s00439-003-0906-8
Wang, J., Huff, A. M., Spence, J. D., and Hegele, R. A. (2004). Single nucleotide polymorphism in CTH associated with variation in plasma homocysteine concentration. Clin. Genet. 65, 483–486. doi: 10.1111/j.1399-0004.2004.00250.x
Watkins, D., and Rosenblatt, D. S. (1989). Functional methionine synthase deficiency (cblE and cblG): clinical and biochemical heterogeneity. Am. J. Med. Genet. 34, 427–434. doi: 10.1002/ajmg.1320340320
Wood, J. G., Mirra, S. S., Pollock, N. J., and Binder, L. I. (1986). Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. U S A 83, 4040–4043. doi: 10.1073/pnas.83.11.4040
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Yang, Y.-J., Zhao, Y., Yu, B., Xu, G.-G., Wang, W., Zhan, J.-Q., et al. (2016). GluN2B-containing NMDA receptors contribute to the beneficial effects of hydrogen sulfide on cognitive and synaptic plasticity deficits in APP/PS1 transgenic mice. Neuroscience 335, 170–183. doi: 10.1016/j.neuroscience.2016.08.033
Yao, M., Nguyen, T. V., and Pike, C. J. (2005). β-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 25, 1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005
Zhang, C., Rodriguez, C., Spaulding, J., Aw, T. Y., and Feng, J. (2012). Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 28, 655–666. doi: 10.3233/JAD-2011-111244
Zhao, F. L., Fang, F., Qiao, P. F., Yan, N., Gao, D., and Yan, Y. (2016). AP39, a mitochondria-targeted hydrogen sulfide donor, supports cellular bioenergetics and protects against Alzheimer’s disease by preserving mitochondrial function in APP/PS1 mice and neurons. Oxid. Med. Cell. Longev. 2016:8360738. doi: 10.1155/2016/8360738
Zhao, H., Li, H., Ruberu, K., and Garner, B. (2015). Impaired lysosomal cobalamin transport in Alzheimer’s disease. J. Alzheimers Dis. 43, 1017–1030. doi: 10.3233/JAD-140681
Zhu, X., Lee, H. G., Casadesus, G., Avila, J., Drew, K., Perry, G., et al. (2005). Oxidative imbalance in Alzheimer’s disease. Mol. Neurobiol. 31, 205–217. doi: 10.1385/MN:31:1-3:205
Zhu, X., Raina, A. K., Perry, G., and Smith, M. A. (2004). Alzheimer’s disease: the two-hit hypothesis. Lancet Neurol. 3, 219–226. doi: 10.1016/S1474-4422(04)00707-0
Keywords: Alzheimer’s disease, hydrogen sulfide, cysteine, redox, sulfhydration/persulfidation, transsulfuration
Citation: Paul BD (2021) Neuroprotective Roles of the Reverse Transsulfuration Pathway in Alzheimer’s Disease. Front. Aging Neurosci. 13:659402. doi: 10.3389/fnagi.2021.659402
Received: 27 January 2021; Accepted: 22 February 2021;
Published: 16 March 2021.
Edited by:
P. Hemachandra Reddy, Texas Tech University Health Sciences Center, United StatesReviewed by:
Bobby Thomas, Medical University of South Carolina, United StatesMichael Lardelli, University of Adelaide, Australia
Copyright © 2021 Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bindu Diana Paul, YnBhdWw4QGpobWkuZWR1