 Sepideh Goudarzi
Sepideh Goudarzi Asieh Hosseini2
Asieh Hosseini2- 1Department of Toxicology and Pharmacology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
- 2Razi Drug Research Center, Iran University of Medical Sciences, Tehran, Iran
- 3Department of Toxicology and Pharmacology, School of Pharmacy, and Toxicology and Diseases Group, Pharmaceutical Sciences Research Center, The Institute of Pharmaceutical Sciences, Tehran University of Medical Sciences, Tehran, Iran
- 4Pharmaceutical Sciences Research Center, Ardabil University of Medical Sciences, Ardabil, Iran
Background: Parkin-mediated mitophagy is the dominant mitophagy pathway of neural cells. Its restoration will result in prevention of cognitive decline, including Alzheimer's disease (AD). The role of this mitophagy pathway in neurodegenerative diseases has drawn attention in recent years. The two main pathological proteins in AD, amyloid β (Aβ) and human Tau (hTau), interfere with mitochondrial dynamics through several pathways. However, taking into consideration the specific interactions between Aβ/hTau and Parkin, special focus is required on this mitophagy pathway and AD. In this review, these interactions are fully discussed, and an overview of the neuroprotective drugs that enhance Parkin-mediated mitophagy is presented.
Methods: This systematic review was performed according to PRISMA guidelines, and a comprehensive literature search was done in the electronic databases up to September 2020, using search terms in the titles and abstracts to identify relevant studies. One hundred eighty-six articles were found, and 113 articles were screened by title and abstract. Finally, 25 articles were included in this systematic review according to our inclusion and exclusion criteria.
Results: Accumulation of Aβ and hTau affects mitophagy, including Parkin-mediated. Tau seems to prevent Parkin translocation directly. A Parkin level in the cell appears to be of importance in determining the damage caused by Aβ and hTau and in the future therapeutic approaches. Parkin controls the PINK1 level via the presenillins, suggesting that mutations in presenillins affect Parkin mitophagy.
Significance: Parkin mitophagy is a process affected by several AD pathological events multidimensionally.
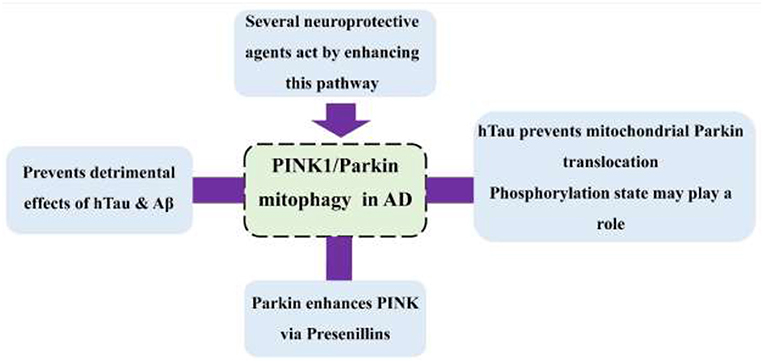
Graphical Abstract. Interactions of PINK1/Parkin pathway with AD-related proteins.
Introduction
Parkin (also known as “ubiquitin E3 ligase”) is a molecule of high interest in the recent decade due to its several fundamental roles in the cells, including mitochondrial homeostasis maintenance. It was first found as one of the proteins involved in the pathogenesis of Parkinson's disease, as its loss causes neuro-inflammation and degeneration. It is then not surprising that literature also indicates that this protein has a dominant role in reversing the cognition deficits and preventing Alzheimer's disease (AD) (Martín-Maestro et al., 2016; Fang et al., 2019). It appears that this protein is accumulated where most involved with AD pathology (hippocampus) and seems to be of great importance in the future AD treatments (Gao et al., 2020). During recent years, however, mitophagy on Parkinson's disease has been massively studied.
It is known that mitophagy effectively attenuates cognitive decline in AD (Fang et al., 2019). Although there is basal mitophagy activity as a normal function (McWilliams et al., 2018), this action becomes of great importance for stressed mitochondria. Upon mitochondrial stress outriding the anti-inflammatory defense system, mitochondria release further oxidative species, causing a vicious circle ending in a dysfunctional mitochondrion. Enough number of these waste bodies will result in cellular apoptosis, following their release of apoptotic factors, such as the cytochrome C. The key protection of the neuron in this condition is mitophagy, which corresponds to the removal of the dysfunctional mitochondria before the neuron is down to apoptosis. Although mitophagy pathways are not frequent, studies on enhancement of Parkin-mediated mitophagy have reported promising outcomes. This implies the Parkin-mediated pathway to be the dominant way of mitophagy under stress. Moreover, it is still not clear what the mechanisms of the others, including their induction pathways, would be (Villa et al., 2018).
Moreover, reviews of the role of this pathway in AD have not addressed the mechanisms of the specific proteins of Alzheimer's and the Parkin mitophagy. Additionally, there seems to be a lack of information in this specific area, while there is diffuse information on these pathways that urges us to bring them together to conclude how they influence each other. In this systematic review, characteristics of Parkin-mediated mitophagy in AD and the role of amyloid β (Aβ) and hTau and their related proteins in this pathway are discussed. Finally, potential enhancers of the pathway are reviewed.
Methodology
Study Protocol
The present systematic review study was performed based on a previous study (Moher et al., 2009). A comprehensive systematic search of the following electronic databases: PubMed, Embase, ProQuest, Scopus, and Web of Sciences according to the study search terms, which are based on the aims of the present study, including “Parkin RBR E3 ubiquitin protein ligase” or “PRKN” or “PDJ” or “AR-JP” or “LPRS2” or “PARK2” or “Park” or “Pdr-1” or “parkin” “PARK 2” and “mitochondrial degradation” or “mitophagy” and “Alzheimer's disease” or “Alzheimer” or “Alzheimer's disease” or “senile dementia” or “primary senile degenerative dementia” or “acute confusional senile dementia” or “senile dementia, acute confusional” or “dementia, presenile” or “presenile dementia,” was done up to September 27, 2020.
Eligibility Criteria
After entering all the manuscripts searched from five databases into a reference, all the studies were initially screened based on the study aims in the title and abstract, including [1] all eligible articles that studied specific characteristics of Parkin-mediated mitophagy in AD (i.e., AD models) or studies on specific interactions between Aβ/hTau and Parkin [2], the studies with comprehensive and sufficient information, [3] the studies without limitation in in vitro and in vivo, and [4] the studies without restriction on the publication year. For exclusion criteria, we excluded [1] letters, [2] posters, [3] articles in languages else than English, [4] and studies on Alzheimer's disease and mitophagy, in which no conclusions were obtained exclusively on the Parkin pathway, e.g., mitochondrial dysfunction and AD.
Data Collection
After deleting duplicate articles by two reviewers (SG and HHA), the articles were screened independently based on the previously expressed keywords. The full texts of the articles were separately evaluated, and the related articles were entered based on the inclusion and exclusion of the relevant predefined criteria. Those inconsistent with the objectives and predefined criteria of the present study were removed upon agreement of the two reviewers.
Results
Literature Search and Screening
Based on our systematic search keywords, one hundred and eighty-six articles were found in electronic databases. After deleting the duplicate articles, 113 articles were screened based on prespecified keywords in the title and the abstract, and 62 articles were removed. Then, 51 full-text articles were evaluated for the second screening based on the predefined criteria. Eventually, 25 articles qualified to enter our systematic review. The process of searching in electronic databases and its two-step screening are shown in Figure 1.
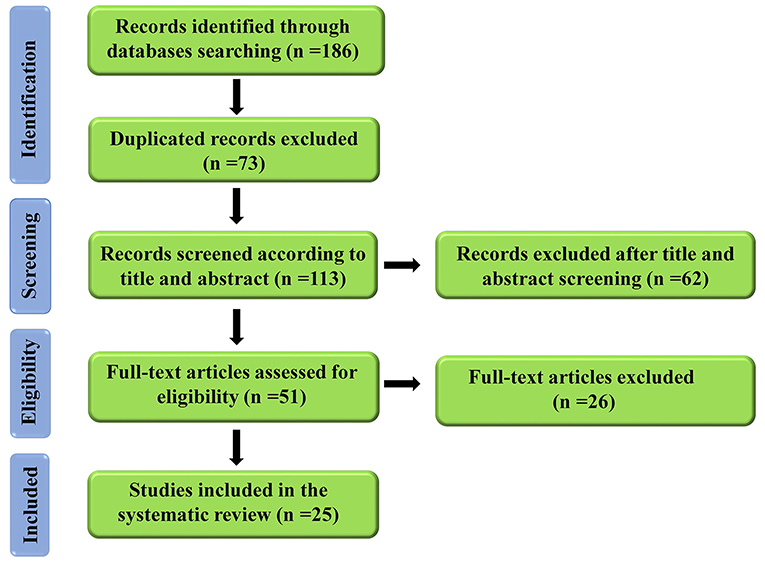
Figure 1. A flow diagram of the selection process for the present study.
Data Extraction
The data of each article were extracted by the author SG and compiled in Table 1, and rechecked by the author HHA under the following titles: (1) First author and year of publication, (2) models (in vitro or in vivo)/a route of administration, dosage, duration of treatment (3) in vivo assessments (4) the Parkin level in the AD model vs. control, (5) the Parkin level in the treated group vs. the AD model, (6) other related markers.
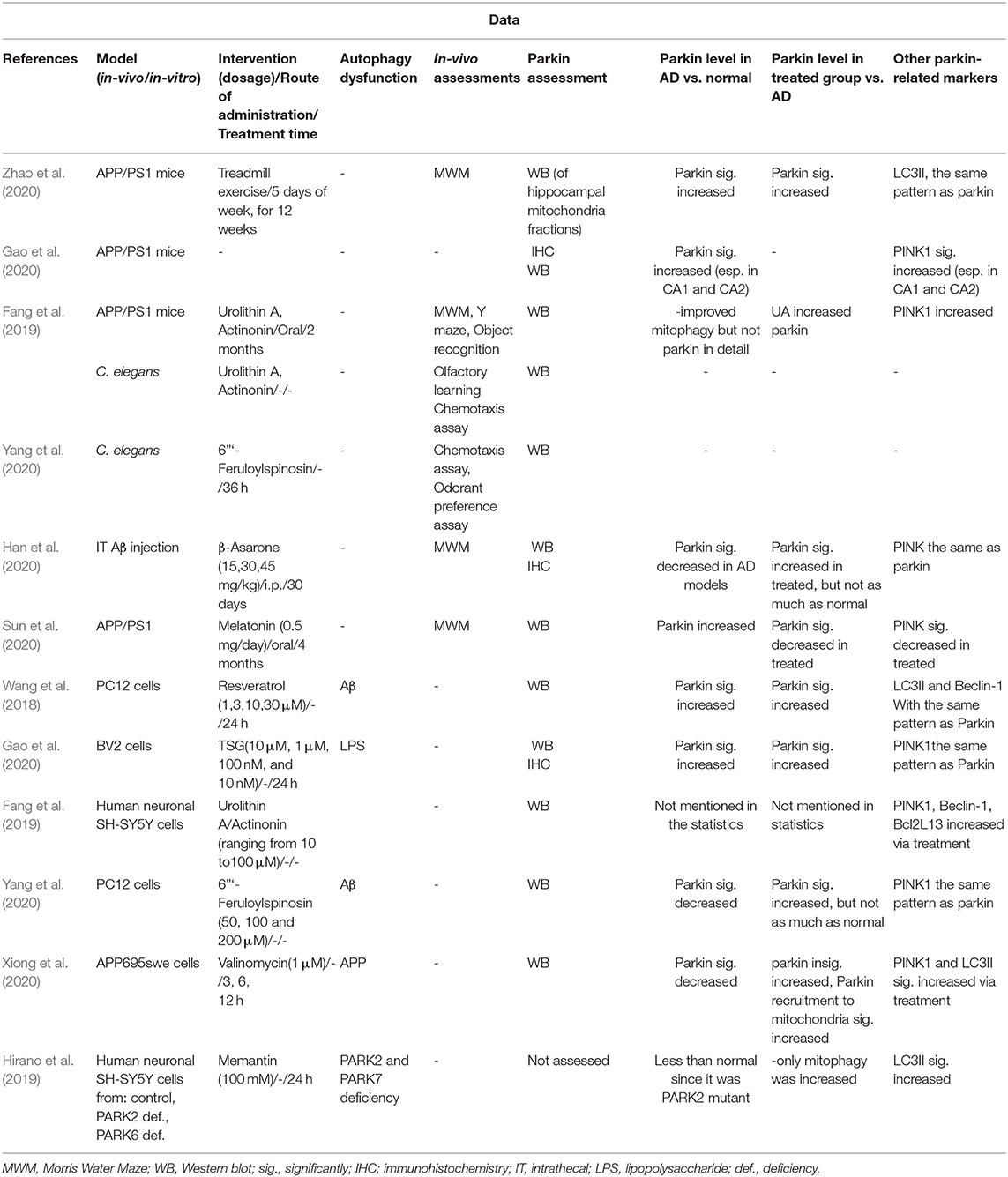
Table 1. Studies on Parkin level alterations.
The Included Studies Could Be Categorized Into Two Areas
Studies on how Amyloid β and hTau affect Parkin-mediated mitophagy and vice versa (Michiorri et al., 2010; Khandelwal et al., 2011; Corsetti et al., 2015; Wang X. et al., 2015; Ye et al., 2015; Hu et al., 2016; Martín-Maestro et al., 2016, 2017, 2019; Checler et al., 2017; Goiran et al., 2018; Castellazzi et al., 2019; Cummins et al., 2019; Fang et al., 2019; Reddy and Oliver, 2019; Wang et al., 2020). The main results were the following:
It seems that the PINK/Parkin pathway has several roles in the prevention of AD. Generally, accumulation of Aβ and hTau affects mitophagy, but Tau directly prevents Parkin translocation; however, the mechanism is not yet ascertained. The Parkin level seems to be a potential indicator of mitochondrial damage and the rate of cognitive decline. Not only does PINK1 recruit Parkin to the mitochondria, but Parkin enhances the PINK1 level via presenilins, and, thus, mutations in presenilins impair the PINK/Parkin mitophagy cycle.
Studies that evaluated potential neuroprotective agents in AD models that ameliorate neurodegeneration via enhancement of Parkin-mediated mitophagy (Wang et al., 2018; Fang et al., 2019; Hirano et al., 2019; Gao et al., 2020; Han et al., 2020; Sun et al., 2020; Xiong et al., 2020; Yang et al., 2020; Zhao et al., 2020, 2021). The studies used both in vivo and in vitro models of AD, and the outcomes mainly included increased the Parkin level or related proteins. The main results were the following: (Table 1 provides the details.).
PINK1 apparently starts to increase upon mitophagy enhancement prior to Parkin, taken from the fact that studies, which measured both the Parkin and PINK1 relative levels, reported a higher increase in PINK1 (Gao et al., 2020; Han et al., 2020; Xiong et al., 2020). In higher doses or exposures, effectiveness of these compounds seems to decrease, indicated by the PINK1 and Parkin levels (Gao et al., 2020; Xiong et al., 2020).
Discussion
This was the first study to bring together Parkin-mediated mitophagy characteristics focused on its interactions with proteins of AD. There seem to be complex interactions between these proteins, which indicate new roles contributing to pathology of AD, hence suggesting potential new targets for treatment.
Parkin-Mediated Mitophagy and AD
PINK1 (PTEN-induced kinase 1) is the well-known pivotal protein in the Parkin pathway, induced by PTEN (phosphatase and tensin homolog). Upon mitochondrial damage, depolarization of mitochondrial membrane causes translocation of cytosolic Parkin to the membrane, as well as PINK1. PINK1 not only induces translocation of Parkin but also phosphorylates it and activates its ligation. Ubiquitination of the outer membrane mitochondria (OMM) proteins will induce pathways, leading to autophagy of the organelle. Moreover, PINK1 seems to have interactions in autophagy pathways upstream to the Parkin, including induction of the protein Beclin-1 (Michiorri et al., 2010). Beclin-1 is one of the proteins involved in autophagy and is required to induce it. It appears to play a role in the PINK/Parkin mitophagy pathway. In an in vivo model of AD, it is reported that intracellular Aβ accumulation induces a beclin-independent autophagy pathway, which leads to accumulation of the dysfunctional mitochondria. However, by ubiquitinating the intracellular Aβ, Parkin induces the beclin-dependent pathway and leads to removal of the vesicles, containing mitochondria debris. Ubiquitination of the Aβ will also prevent the extracellular plaque formation (Khandelwal et al., 2011).
Aβ and Tau Impair Parkin-Mediated Mitophagy
Aβ and Tau cause disruptions in mitophagy (Fang et al., 2019) through abnormal interactions with mitochondrial fission proteins and the Parkin (Reddy and Oliver, 2019). In the study by Wang et al. cells treated with Aβ had a greater number of mitochondria than the normal, indicating the inability to remove dysfunctional mitochondria. Moreover, the cells exhibited more of the dysfunctional and misshaped ones and autophagosomes containing mitochondria (Wang et al., 2020).
A study by Hu et al. indicated that human N-terminal Tau (but not C-terminal, phosphorylated tau) is found in the outer mitochondrial membrane. This causes an increase in its membrane potential and hyperpolarizes the membrane, preventing Parkin translocation (Hu et al., 2016). Furthermore, phosphorylation state of hTau directly influences its membrane localization, where phosphorylation and mutations in the N terminal limit transport of tau within neuronal membrane (Pooler et al., 2012). Investigating further roles of this protein in this pathway by Cumminis et al. it was found that human Tau (hTau) protein prevented Parkin translocation by another mechanism. The team showed that cytosolic Parkin was trapped by hTau, although this was only observed in cytosol and not in the mitochondrial membrane. However, the authors declare that they did not observe OMM hyperpolarization by Tau (Cummins et al., 2019), compared with the study of Hu, neither have they discussed the phosphorylation state.
A recent in vitro study has pointed out the deteriorating synergistic effect of amyloid precursor protein (APP) and Tau on mitophagy. In their model, overexpression of Tau increased the basal Parkin level and prevented Parkin translocation, which was exacerbated via the presence of APP. The exciting finding was that overexpression of these two proteins did not interrupt proper stabilization of PINK1 in the mitochondrial membrane, while Parkin translocation was interrupted. This suggests the existence of unknown cross talks between PINK1 and Parkin, which is altered by the presence of APP and Tau (Martín-Maestro et al., 2016). This is in accordance with the findings of Hu et al. and Cumminis et al., in which Tau prevented Parkin translocation. Figure 2 depicts the probable disrupting mechanisms of Parkin translocation by Tau.
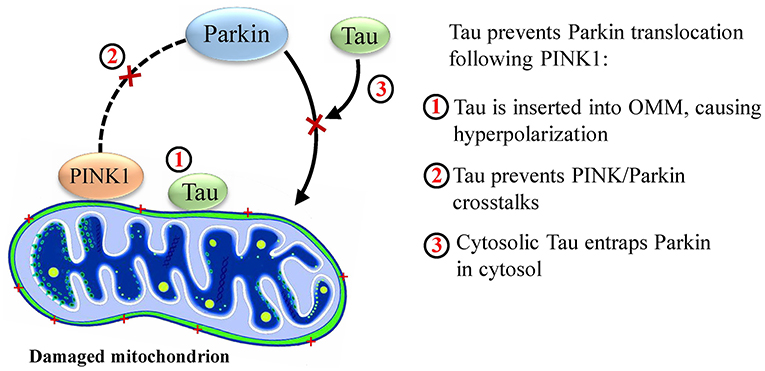
Figure 2. Probable mechanisms by which Tau disrupts mitophagy.
The effect of AD protein on mitophagy has also been approved clinically. In a recent study by Castellazzi et al., serum samples of patients at different stages of cognitive impairment due to amyloidopathies have had lower amounts of Parkin and Atg (autophagy-related proteins), being markers of autophagy and mitophagy. It was, therefore, suggested that these biomarkers could be of use in early diagnosis of the disease (Castellazzi et al., 2019).
Nevertheless, considering the alleviating mechanisms of mitophagy in neurodegeneration, it is not far from mind that mitophagy is a defense system of the cell against disruption of these proteins. An increase in Parkin, normally, is observed in the hippocampus of APP/PS1 transgenic mice, especially in the areas of CA1 and 2 (Wang X. et al., 2015). Wang et al. further explored alleviating the effects of Aβ on mitochondria through increasing the expression of Parkin. As expected, cells overexpressing Parkin treated with Aβ had better mitochondrial functions and less ROS production (Wang et al., 2020). On the other side, it is reported that an endogenous-truncated form of hTau (NH2-26-230 tau fragment) is found in synaptosomal mitochondria of AD patients much higher than in normal, and this causes excessive mitochondrial elimination due to over activation of the mitochondrial turnover, regulating systems by abundant Parkin recruitment (Corsetti et al., 2015). However, the authors do not discuss the phosphorylation state of this Tau form (which appears to be non-phosphorylated) in order for us to compare it with the previous statements on the effect of Tau on Parkin membrane localization, which were on the contrary to the present statement.
Up to this, it seems a circle has existed in which Aβ and Tau disrupt this mitophagy, and the mitophagy protects mitochondria from their actions. The fact is mitochondrial impairment and OMM depolarization that are caused by Aβ and hTau increase Parkin and Parkin translocation, whereas Aβ and hTau themselves prevent Parkin translocation and decrease Parkin. It appears that, in the first stages of pathology, the equation is, finally, in favor of increased Parkin, although not enough to defend mitochondrial homeostasis against AD pathological occurrences. However, in later stages, where abundance of Aβ and hTau exists; Parkin and the mitophagy mechanisms cannot overcome. In most of the molecular studies on Parkin in AD, it is mentioned that Parkin levels were significantly higher in AD mice compared with the healthy controls. In the study of Castellazzi et al., patients with MCI (mild cognitive impairment) have had higher maximum levels of Parkin even than the control of the same age. However, their median levels were as low as AD patients. This may be because of the gap between molecular and clinical observations, in which there might be substages to mild cognitive impairment as well (i.e., patients in later stages of MCI have had Parkin serum levels similar to patients with AD, whereas those in earlier stages had significantly higher).
Improved microglial activity and phagocytosis were also observed in an in vivo study on APP/PS1 transgenic mice, probably by enhancing energy production and transfer, following effective mitophagy (Fang et al., 2019).
The Role of Presenillins in Parkin-Mediated Mitophagy
Whether the APP is cleaved primarily by alpha or beta-secretase, it is then cleaved by gamma-secretase, whose active site is mainly constituted of Presenillins 1 and 2. These two proteins are shown to modulate PINK1 expression, independent of PTEN (Checler et al., 2017). Although PS1 may have several roles in the cell, it appears that its effect on PINK is dependent on its gamma-secretase activity since the effects were disrupted by inhibitors of this enzyme (Checler et al., 2017). Explorations revealed that the APP intracellular domain (AICD), a product of cleavage by PS1 in gamma-secretase, along with FOXO3, increases PINK1 promoter transactivation, a mRNA level, and protein expression (Checler et al., 2017; Goiran et al., 2018). This is while PS2 decreases the PINK1 level. What is surprising is that PARK2 modulates both PS1 and PS2 in a manner opposite of their effects on PINK1. This provides support for the hypothesis that PARK2 increases the PINK1 level via both the PS1 and PS2. It has been generally thought that PINK1 is an upstream protein of Parkin in its mitophagy pathway; while this finding proves that Parkin (and the gene PARK2) could modulate the levels of PINK1 upstream to the mitophagy, causing a cycle in which each enhances the other in the process of mitophagy (Figure 3).
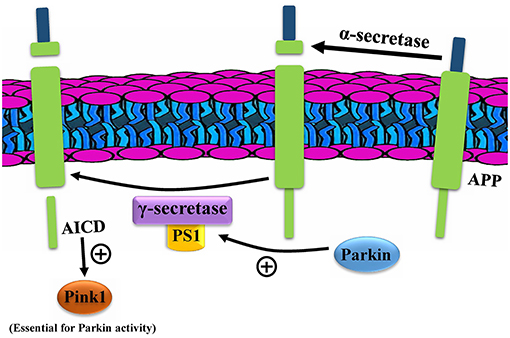
Figure 3. Parkin enhances PINK1 through presenillins (PS). AICD, APP intracellular domain; APP, amyloid precursor protein.
Mutations in these proteins, especially PS1, are the most frequent cause of familial AD due to unfancied cleavages, producing harmful Aβ peptides. Added to that are the disrupted PINK-Parkin cycle and mitophagy (Martín-Maestro et al., 2017).
Enhancing Parkin-Mediated Mitophagy Against AD
As mitophagy has been found to be a promising factor in alleviating cognitive decline, recent studies have assessed the effects of neuroprotective compounds and exercise on mitophagy, and, dominantly, Parkin-mediated mitophagy.
Exercise
Twelve weeks of treadmill exercise for APP/PS1 mice upregulated Parkin, enhanced mitophagy, and decreased Aβ levels (Zhao et al., 2020). However, PINK1 alteration patterns were not consistent with the rest (Zhao et al., 2020). NAD-dependent deacetylase sirtuin-1 (SIRT1) is an interesting protein with several protective roles in the body. It plays a role in alleviating inflammaging (chronic inflammation in the body that accelerates aging) via several mechanisms, including mitochondrial biogenesis and mitophagy through the PINK1/Parkin pathway (Zhao et al., 2021). It has recently been discovered that exercise, by increasing NAD+ levels, helps improve the function of SIRT1, thereby enhancing PINK1/Parkin mitophagy (Zhao et al., 2021).
Chemical Compounds
A 2017 in vitro study found that the neuroprotective role of resveratrol in AD results from its ability to induce mitophagy via upregulation of Parkin (Wang et al., 2018). In this study, cells treated with Aβ also showed an increase in Parkin compared with the control. In a recent study by Gao et al., tetrahydroxy stilbene glucoside (TSG) from the plant Polygonum multiflorum was shown to have neuroprotective effects via reducing the inflammatory cytokines and enhancing the Parkin-mediated mitophagy. Treating defective microglia with TSG upregulated both Parkin and PINK1 and, in higher doses, Beclin-1 as well. Out of both PINK1 and Parkin knockdown cells treated with TSG, those lacking PINK1 responded substantially less to TSG autophagy promotion. Moreover, this reduction was more than the case with Parkin knockdown cells, suggesting that TSG promoted PINK1 actions both directly and indirectly more than Parkin (Gao et al., 2020). However, higher levels of TSG decreased Parkin and PINK1 levels significantly (Gao et al., 2020), suggesting that excessive mitophagy may result in self-regulation. In another study, human neural cells treated with the mitophagy-inducing compound urolithin A showed higher amounts of parkin, PINK1, and Beclin-1. In vivo administration of this compound, along with another compound, actinonin, in APP/PS1 mice increased PINK-1(Fang et al., 2019). The interesting part was that urolithin A could inhibit Tau phosphorylation, and this action was interrupted in PINK1-knockdown cells, suggesting that this mitophagy could inhibit Tau phosphorylation (Fang et al., 2019). Moreover, both urolithin A and actinonin could prevent Tau phosphorylation via PINK1-dependent mitophagy (Fang et al., 2019). Finally, a study on an in vitro model of AD reported that valinomycin increased PINK1/Parkin mitophagy, enhanced Parkin translocation, and significantly reduced the number of mitochondria, as well as the Aβ level (Xiong et al., 2020). In this study, again, enhancement of PINK1 was higher than Parkin in the treated cells (relative significant increase vs. relative insignificant increase, respectively) (Xiong et al., 2020). The interesting finding in this study was that increasing valinomycin treatment time from 3 to 6 h increased the PINK1 and Parkin levels, while increasing the time from 6 to 12 h decreased them.
In a recent in vivo study, β-Asarone, a neuroprotective agent and the main component of Acorus tatarinowii, increased the expression of PINK1, Parkin, and Beclin-1 (Han et al., 2020). However, contrary to the above studies, lower levels of Parkin were found in the brains of AD models compared with control. Another study revealed that treatment of cells with Aβ downregulated the levels of Parkin and PINK1, while their studied compound, 6”‘-Feruloylspinosin, could prevent the detrimental effects of Aβ on Parkin and PINK1 overexpression (Yang et al., 2020). Relative expression of PINK1 was more enhanced with the compound (reached an insignificant difference with the control) than that of Parkin (difference with the control remained still significant), suggesting a higher effect on PINK than Parkin (Yang et al., 2020).
Melatonin was also shown to have neuroprotective effects via enhancement of mitophagy in APP/PS1 mice. However, contrary to the rest of the similar studies in this area, they observed a decrease in Parkin, as a sign of mitochondrial health; and the mice who received melatonin had better performances in memory tests and reduced levels of Parkin compared with AD controls (Sun et al., 2020).
Normally, it is believed that an agent or act, which increases the expression of Parkin, triggers mitophagy and helps restore mitochondrial homeostasis. What underlies this conflict cannot be fully understood without experiments; however, it might be the case of time, as once discussed in the previous section: elevation of Parkin at early stages of the disease in order to defend and protect, but failing the detrimental multidimensional effects of Aβ and hTau. Here, again, at early stages of compound administration (or exercise, respectively), the Parkin expression level increases, restoring mitophagy. Then, as the compound effects progress, dysfunctional mitochondria decrease in a load and quantity, allowing for Parkin levels back to normal (Gao et al., 2020; Xiong et al., 2020). The determining factor here is the time when assessments were done, especially that this study designed a long-term treatment, and treatment days were maximum among other similar studies. However, as melatonin is not a complete cure to AD, this time effect cannot be fully accepted, as the damage will never be totally removed (the decrease cannot reach the normal states).
These inconsistencies between the results of the studies cause controversies over analyzing Parkin level changes. Another factor might be that whether the Parkin level was assessed in only mitochondria or the whole cell. Except for one study, all other studies used whole cell or tissue for protein level assessment, and the controversies were within these studies (Table 1), ruling out the effects of Parkin assessment variations.
Finally, memantine is a well-known drug used in AD. In addition to its other roles against this disease, it appears that this drug helps restore mitophagy in both neurons lacking the Parkin gene and healthy controls, indicating a Parkin-independent pathway (Hirano et al., 2019).
Having a glance at the chemical structure of these compounds, it is implied that most of the structures follow a pattern of 3-Phenylethylene or its bioisosteres (Figure 4). Nevertheless, it is clear that data on their chemical-structure properties still extremely lack. The provided compounds are all either selected based on traditional medicine suggestions or have shown enhancing effects on autophagy in general, e.g., resveratrol was shown to activate SIRT1 in human umbilical vein endothelial cells (Guo et al., 2013). Since PINK1 is required to initiate the cascade on one hand, and these studies report increased PINK1 and Parkin by these agents on the other, it would be suggestable that increase in PINK1 and Parkin would compensate the lacks of the proteins in a brain undergoing rapid inflammation and oxidative stress. What happens in the cases of patients with neurodegeneration is innate insufficiency of these proteins, which cannot compete rapid aging of the brain, aside from deteriorating effects of amyloid beta and Tau on Parkin. It is, therefore, hypothesized that the effective agents increase PINK1 and Parkin to assist initiation of mitophagy. The exception is valinomycin, which has a structure close to aminoglycosides and supposedly exerts effects by depolarizing mitochondrial membrane, a supplementary effect to increase the PINK1 and Parkin levels (Rakovic et al., 2019). However, we are not aware of the exact site of action of these compounds that influences the Parkin pathway, nor have we yet found a compound that specifically interacts with this pathway and not the rest, since neuroactive compounds usually interact with many active sites in the brain. Moreover, the similarity of 3-phenylethylene with other neuroactive compounds, such as dopamine, indicates that development of a drug based on this SAR (structure activity relationship) may not be feasible, since many adverse reactions should be avoided, considering the fact that the origin of these compounds, which were, therefore, assumed to have effects on the Parkin pathway, was experimental, either through traditional medicine or recent discoveries about neuroprotective effects.
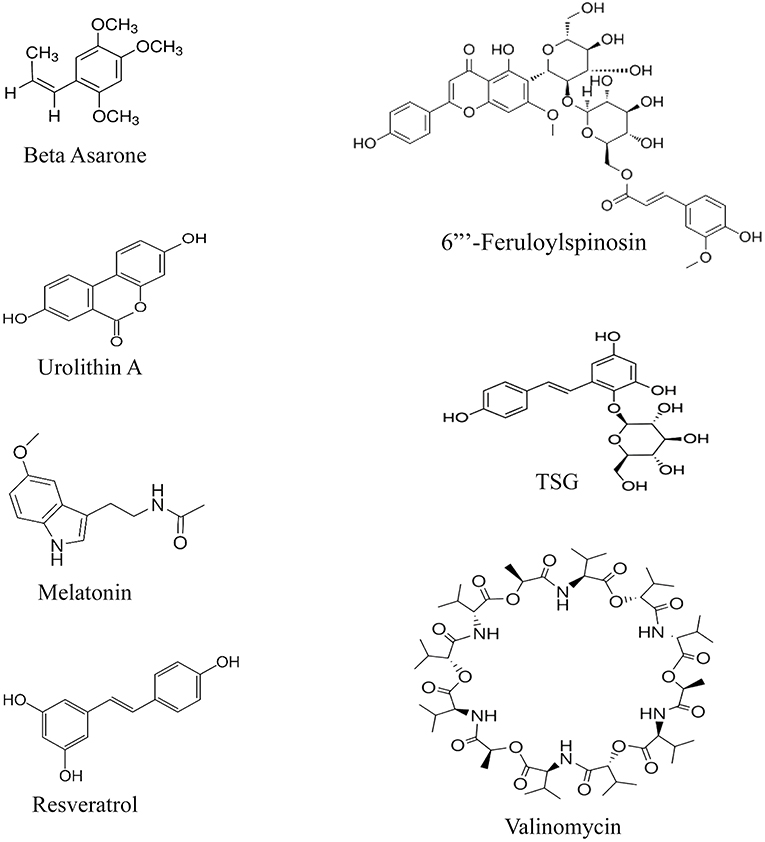
Figure 4. Chemical structures of the enhancers of the PINK/Parkin pathway.
Deubiquitination, a Checkpoint to Parkin-Mediated Mitophagy
Since Parkin is a ubiquitin, and since mitophagy is a programmed pathway in the cell, deubiquitinating enzymes [ubiquitin-specific peptidases (USPs)] play as the checkpoints to mitophagy. This is done to prevent excess mitophagy under stress conditions. An in vitro study by Wang et al. has investigated the role of USP30 and 35 in regulating Parkin-mediated mitophagy (Wang Y. et al., 2015). The mitofusin protein 2 (MFN2), from a class of proteins responsible for mitochondrial content exchange, is thought to send signals for Parkin translocation. This translocation is the most known sign for the initiation of Parkin-mediated mitophagy. The translocase of outer mitochondrial membrane 20 (TOMM20) is the other molecule responsible for Parkin translocation, which acts in association with PINK1. The USP30 is hypothesized to regulate the levels of these proteins (Wang Y. et al., 2015). It is further reported that this protein has a role in the protection of other OMM from ubiquitination. Finally, upon chemical mitophagy induction, this protein delayed translocation of Parkin to the OMM, which is a consequence of MFN2 prevention, they hypothesize (Wang Y. et al., 2015).
The USP 35, however, exerts its regulatory effects through unknown mechanisms. However, as the protein dissociates from the mitochondria at the time of mitochondrial depolarization (suggestive of mitophagy time), the authors suggest that this molecule routinely regulates MFN2 activity, preventing Parkin translocation (Wang Y. et al., 2015).
Other USP molecules are found to have a role in deubiquitination; however, data on their detailed mechanisms in mitophagy and Parkinson's and Alzheimer's disease seem to be extremely insufficient. Although the USPs perform several roles in the body, targeting one of such pathways could potentially help enhance the Parkin mitophagy via removing the regulation.
Future Prospects
The effects of Aβ and hTau on Parkin regulation seem yet to have controversies and remain to be further explored. Moreover, data on the role of mitophagy in microglia activation and enhancement in AD models seem to be extremely limited. Since both Aβ/hTau and Parkin have contradictory roles against each other, their roles should be studied in detail.
Considering the probable role of SIRT 3, 4, and 5 proteins in mitophagy, it is recommended that future studies on neurodegeneration focus on the correlation between mitochondrial sirtuins and the Parkin pathway.
Almost all the non-clinical studies did not include gender differences in memory retention, although this is an interfering factor.
It appears that neuroprotective treatments targeting Parkin-mediated mitophagy are found with an increase in Parkin, which, again, requires exploration. The certain result could add a dimension in the evaluation of future neuroprotective agents.
Conclusion
Studies on Parkin-mediated mitophagy seem to provide a promising future in ameliorating neurodegeneration and cognitive decline in AD (Moloudizargari et al., 2017). However, there is a considerable lack of experimental data on this area. It seems that, in molecular studies, an increase in Parkin is the result of damage to mitochondria and is observed in AD compared with healthy controls. However, by the time Aβ and hTau overcome compensatory systems of body, including Parkin's, the level significantly drops. This should be the case of what is observed at the clinical level. Certainty in this area needs further investigations. Moreover, it seems that a proper range/optimum dose exists for Parkin-enhancer compounds since higher exposures have yielded opposite results in some studies.
In summary, it appears that Parkin is a multifunctional protein preventing pathogenesis of AD, whose interactions with AD proteins are not yet fully understood. Discovering the gaps is strongly encouraged since it would help with a more profound and earlier diagnosis of Alzheimer's disease.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
SG: idea suggestion, final screen, data extraction, writing of the manuscript, and extracting figures and tables. HH-A: systematic search and screen, sketching the graphs, and revision of the manuscript. MA: supervision. AH: systematic search and screen. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to sincerely thank the Institute of Pharmaceutical Sciences for their support.
References
Castellazzi, M., Patergnani, S., Donadio, M., Giorgi, C., Bonora, M., Bosi, C., et al. (2019). Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer's disease and mild cognitive impairment. Sci. Rep. 9:20009. doi: 10.1038/s41598-019-56614-5
Checler, F., Goiran, T., and Alves da Costa, C. (2017). Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: implication for neurodegenerative diseases. Autophagy 13, 2004–2005. doi: 10.1080/15548627.2017.1363950
Corsetti, V., Florenzano, F., Atlante, A., Bobba, A., Ciotti, M. T., Natale, F., et al. (2015). NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer's disease. Hum. Mol. Genet. 24, 3058–3081. doi: 10.1093/hmg/ddv059
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., and Götz, J. (2019). Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 38:e99360. doi: 10.15252/embj.201899360
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Gao, Y., Li, J., Li, J., Hu, C., Zhang, L., Yan, J., et al. (2020). Tetrahydroxy stilbene glycoside alleviated inflammatory damage by mitophagy via AMPK related PINK1/Parkin signaling pathway. Biochem. Pharmacol. 177:113997. doi: 10.1016/j.bcp.2020.113997
Goiran, T., Duplan, E., Chami, M., Bourgeois, A., El Manaa, W., Rouland, L., et al. (2018). β-amyloid precursor protein intracellular domain controls mitochondrial function by modulating phosphatase and tensin homolog-induced kinase 1 transcription in cells and in Alzheimer mice models. Biol. Psychiatry. 83, 416–427. doi: 10.1016/j.biopsych.2017.04.011
Guo, H., Chen, Y., Liao, L., and Wu, W. (2013). Resveratrol protects HUVECs from oxidized-LDL induced oxidative damage by autophagy upregulation via the AMPK/SIRT1 pathway. Cardiovasc. Drugs Ther. 27, 189–198. doi: 10.1007/s10557-013-6442-4
Han, Y., Wang, N., Kang, J., and Fang, Y. (2020). β-Asarone improves learning and memory in Aβ(1-42)-induced Alzheimer's disease rats by regulating PINK1-Parkin-mediated mitophagy. Metab. Brain Dis. 35, 1109–1117. doi: 10.1007/s11011-020-00587-2
Hirano, K., Fujimaki, M., Sasazawa, Y., Yamaguchi, A., Ishikawa, K. I., Miyamoto, K., et al. (2019). Neuroprotective effects of memantine via enhancement of autophagy. Biochem. Biophys. Res. Commun. 518, 161–170. doi: 10.1016/j.bbrc.2019.08.025
Hu, Y., Li, X. C., Wang, Z. H., Luo, Y., Zhang, X., Liu, X. P., et al. (2016). Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 7, 17356–17368. doi: 10.18632/oncotarget.7861
Khandelwal, P. J., Herman, A. M., Hoe, H. S., Rebeck, G. W., and Moussa, C. E. (2011). Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated abeta in AD models. Hum. Mol. Genet. 20, 2091–2102. doi: 10.1093/hmg/ddr091
Martín-Maestro, P., Gargini, R., A. A. S., García, E., Antón, L. C., Noggle, S., et al. (2017). Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer's disease-associated presenilin 1 mutation. Front. Mol. Neurosci. 10:291. doi: 10.3389/fnmol.2017.00291
Martín-Maestro, P., Gargini, R., García, E., Simón, D., Avila, J., and García-Escudero, V. (2019). Mitophagy failure in APP and Tau overexpression model of Alzheimer's disease. J. Alzheimer's Dis. 70, 525–540. doi: 10.3233/JAD-190086
Martín-Maestro, P., Gargini, R., Perry, G., Avila, J., and García-Escudero, V. (2016). PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer's disease. Hum. Mol. Genet. 25, 792–806. doi: 10.1093/hmg/ddv616
McWilliams, T. G., Prescott, A. R., Montava-Garriga, L., Ball, G., Singh, F., Barini, E., et al. (2018). Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–49.e5. doi: 10.1016/j.cmet.2017.12.008
Michiorri, S., Gelmetti, V., Giarda, E., Lombardi, F., Romano, F., Marongiu, R., et al. (2010). The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 17, 962–974. doi: 10.1038/cdd.2009.200
Moher, D., Liberati, A., Tetzlaff, J., and Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6:e1000097. doi: 10.1371/journal.pmed.1000097
Moloudizargari, M., Asghari, M. H., Ghobadi, E., Fallah, M., Rasouli, S., and Abdollahi, M. (2017). Autophagy, its mechanisms and regulation: implications in neurodegenerative diseases. Ageing Res. Rev. 40, 64–74. doi: 10.1016/j.arr.2017.09.005
Pooler, A. M., Usardi, A., Evans, C. J., Philpott, K. L., Noble, W., and Hanger, D. P. (2012). Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging. 33, 431.e27–38. doi: 10.1016/j.neurobiolaging.2011.01.005
Rakovic, A., Ziegler, J., Mårtensson, C. U., Prasuhn, J., Shurkewitsch, K., König, P., et al. (2019). PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 26, 1428–1441. doi: 10.1038/s41418-018-0219-z
Reddy, P. H., and Oliver, D. M. (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer's Disease. Cells. 8:488. doi: 10.3390/cells8050488
Sun, C., Qiu, X., Wang, Y., Liu, J., Li, Q., Jiang, H., et al. (2020). Long-term oral melatonin alleviates memory deficits, reduces amyloid-β deposition associated with downregulation of BACE1 and mitophagy in APP/PS1 transgenic mice. Neurosci. Lett. 735:135192. doi: 10.1016/j.neulet.2020.135192
Villa, E., Marchetti, S., and Ricci, J. E. (2018). No parkin zone: mitophagy without parkin. Trends Cell Biol. 28, 882–895. doi: 10.1016/j.tcb.2018.07.004
Wang, H., Jiang, T., Li, W., Gao, N., and Zhang, T. (2018). Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer's disease. Toxicol. Lett. 282, 100–108. doi: 10.1016/j.toxlet.2017.10.021
Wang, H., Zhang, T., Ge, X., Chen, J., Zhao, Y., and Fu, J. (2020). Parkin overexpression attenuates Aβ-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 244:117322. doi: 10.1016/j.lfs.2020.117322
Wang, X., Zhao, X. L., Xu, L. L., Wang, C. F., Wei, L. F., Liu, Z., et al. (2015). Mitophagy in APPsw/PS1dE9 transgenic mice and APPsw stably expressing in HEK293 cells. Eur. Rev. Med. Pharmacol. Sci. 19, 4595–4602.
Wang, Y., Serricchio, M., Jauregui, M., Shanbhag, R., Stoltz, T., Di Paolo, C. T., et al. (2015). Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11, 595–606. doi: 10.1080/15548627.2015.1034408
Xiong, X., Li, S., Han, T. L., Zhou, F., Zhang, X., Tian, M., et al. (2020). Study of mitophagy and ATP-related metabolomics based on β-amyloid levels in Alzheimer's disease. Exp. Cell Res. 396:112266. doi: 10.1016/j.yexcr.2020.112266
Yang, T., Zhao, X., Zhang, Y., Xie, J., and Zhou, A. (2020). 6”'-Feruloylspinosin alleviated beta-amyloid induced toxicity by promoting mitophagy in Caenorhabditis elegans (GMC101) and PC12 cells. Sci. Total Environ. 715:136953. doi: 10.1016/j.scitotenv.2020.136953
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum. Mol. Genet. 24, 2938–2951. doi: 10.1093/hmg/ddv056
Zhao, N., Xia, J., and Xu, B. (2021). Physical exercise may exert its therapeutic influence on Alzheimer's disease through the reversal of mitochondrial dysfunction via SIRT1-FOXO1/3-PINK1-Parkin-mediated mitophagy. J. Sport Health Sci. 10, 1–3. doi: 10.1016/j.jshs.2020.08.009
Keywords: Alzheimer's disease, amyloid–beta, mitophagy, PINK 1, Parkin (PARK2)
Citation: Goudarzi S, Hosseini A, Abdollahi M and Haghi-Aminjan H (2021) Insights Into Parkin-Mediated Mitophagy in Alzheimer's Disease: A Systematic Review. Front. Aging Neurosci. 13:674071. doi: 10.3389/fnagi.2021.674071
Received: 28 February 2021; Accepted: 28 June 2021;
Published: 29 July 2021.
Edited by:
Allison B. Reiss, New York University, United StatesReviewed by:
Karen Schmitt, Central Institute of Mental Health (ZI), GermanyGargi Mahapatra, Wake Forest School of Medicine, United States
Copyright © 2021 Goudarzi, Hosseini, Abdollahi and Haghi-Aminjan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hamed Haghi-Aminjan, aGFtZWRoYWdoaS5hQGdtYWlsLmNvbQ==