 Julie Ouellette1,2
Julie Ouellette1,2 Baptiste Lacoste1,2,3*
Baptiste Lacoste1,2,3*- 1Ottawa Hospital Research Institute, Neuroscience Program, Ottawa, ON, Canada
- 2Department of Cellular and Molecular Medicine, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
- 3University of Ottawa Brain and Mind Research Institute, Ottawa, ON, Canada
Structural and functional integrity of the cerebral vasculature ensures proper brain development and function, as well as healthy aging. The inability of the brain to store energy makes it exceptionally dependent on an adequate supply of oxygen and nutrients from the blood stream for matching colossal demands of neural and glial cells. Key vascular features including a dense vasculature, a tightly controlled environment, and the regulation of cerebral blood flow (CBF) all take part in brain health throughout life. As such, healthy brain development and aging are both ensured by the anatomical and functional interaction between the vascular and nervous systems that are established during brain development and maintained throughout the lifespan. During critical periods of brain development, vascular networks remodel until they can actively respond to increases in neural activity through neurovascular coupling, which makes the brain particularly vulnerable to neurovascular alterations. The brain vasculature has been strongly associated with the onset and/or progression of conditions associated with aging, and more recently with neurodevelopmental disorders. Our understanding of cerebrovascular contributions to neurological disorders is rapidly evolving, and increasing evidence shows that deficits in angiogenesis, CBF and the blood-brain barrier (BBB) are causally linked to cognitive impairment. Moreover, it is of utmost curiosity that although neurodevelopmental and neurodegenerative disorders express different clinical features at different stages of life, they share similar vascular abnormalities. In this review, we present an overview of vascular dysfunctions associated with neurodevelopmental (autism spectrum disorders, schizophrenia, Down Syndrome) and neurodegenerative (multiple sclerosis, Huntington’s, Parkinson’s, and Alzheimer’s diseases) disorders, with a focus on impairments in angiogenesis, CBF and the BBB. Finally, we discuss the impact of early vascular impairments on the expression of neurodegenerative diseases.
Introduction
The human brain contains approximately 100 billion vessels (∼600 km), all of which are critical for the delivery of nutrients and oxygen to neural cells (Quaegebeur et al., 2011). Although the brain accounts for only 2% of the body’s mass, it consumes about a quarter of the body energy produced at rest (Attwell et al., 2010). This colossal energy consumption is elemental to maintain normal functioning of the brain. Such energy requirements make the brain heavily reliant on key vascular features: (i) a dense vasculature to sustain adequate perfusion, (ii) a functional blood-brain barrier (BBB) to maintain brain homeostasis, and (iii) the proper regulation of cerebral blood flow (CBF) to match metabolic demands (Figure 1). Thus, a healthy brain vasculature is essential to support neural cells and ensure normal brain maturation, function and aging (Attwell and Laughlin, 2001; Girouard and Iadecola, 2006; Andreone et al., 2015; Lacoste and Gu, 2015). This is accomplished in part via neurovascular coupling (NVC) mechanisms that regulate CBF to support energetic demands of brain cells (Hamel, 2006; Attwell et al., 2010; Kaplan et al., 2020). While most studies are describing neurovascular signaling at the level of the microvasculature, other vascular segments have received very little attention. There is evidence suggesting that different vascular segments play different roles during vascular responses which is involved in maintaining brain homeostasis. The concept of heterogeneous vascular modules has been extensively reviewed in Schaeffer and Iadecola (2021).
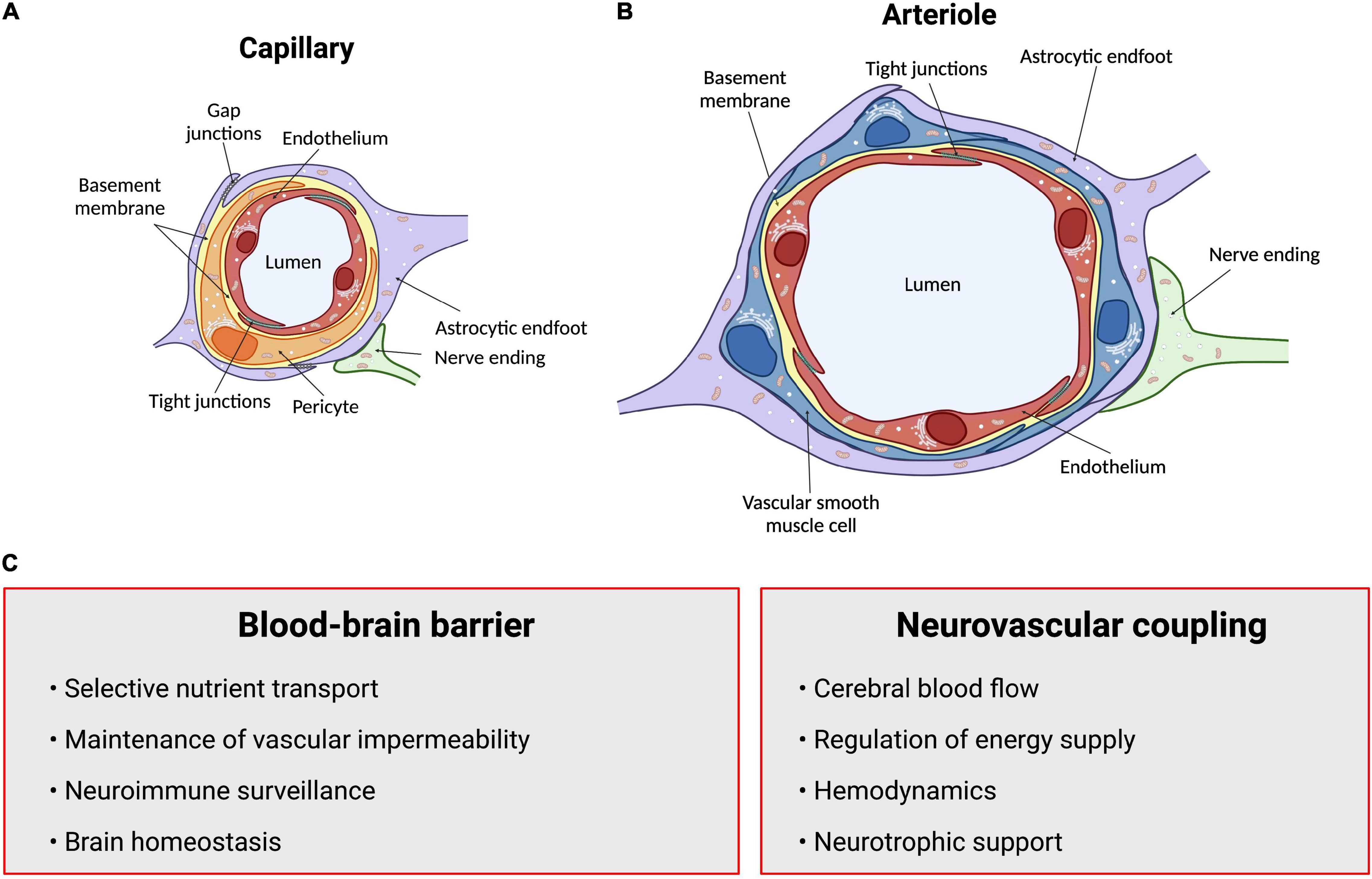
Figure 1. Summary of structures and functions of the neurovascular unit (NVU). The anatomical substrate for regulation of cerebral blood flow and the blood-brain barrier is a multicellular system consisting of neurons, pericytes, smooth muscle cells, astrocytes and endothelial cells known as the NVU. (A) Intracerebral capillaries lack vascular smooth muscle cells but are partly covered by contractile pericytes. (B) At the level of intracerebral arterioles, the endothelium is fully covered by a single layer of vascular smooth muscle cells, which provide contractile properties to the arteriole. Astrocytes send their processes called endfeet around both capillaries and arterioles, providing support as well as a functional connection to surrounding parenchyma. (C) Summary of functions of the blood-brain barrier and neurovascular coupling. Arrows point to neurovascular unit components. Figure made using BioRender.
The close anatomical apposition between the nervous and vascular systems supports a functionally integrated network (Attwell et al., 2010; Lecrux and Hamel, 2011; Hillman, 2014; Huneau et al., 2015; Kaplan et al., 2020). This involves modulating vascular tone by secretion of vasoconstrictor and vasodilator molecules. Initially, it was proposed that local metabolic factors released by neurons modulate local CBF (Sherrington, 1890; Friedland and Iadecola, 1991). Since then, several studies have introduced other cellular mediators of NVC which altogether form the neurovascular unit (NVU). This anatomical substrate of NVC indeed involves a multicellular system consisting of neurons, pericytes, smooth muscle cells, astrocytes, microglia and endothelial cells (ECs) that together orchestrate CBF, and thus brain function (Attwell et al., 2010; Andreone et al., 2015; Grubb et al., 2021; Figure 1). The cerebral cortex is innervated by projection neurons that release neurotransmitters including, but not limited to, acetylcholine, noradrenaline, serotonin and glutamate, involved in the regulation of vessel diameter (Sandoo et al., 2010). Pericytes, while having debated roles in NVC, possess contractile properties and regulate blood flow around capillaries (Attwell et al., 2010, 2016; Fernandez-Klett and Priller, 2015; Sweeney et al., 2018; Watson et al., 2020; Hartmann et al., 2021). Capillary pericytes are α-smooth muscle actin (SMA)-negative and only partially cover the vessel, while ensheathing pericytes are α-SMA-positive, occupy proximal branches of penetrating arteriole offshoots, and fully cover the vessels. However, they are classified as different from smooth muscle cells as they display an ovoid cell body (Grant et al., 2019). Vascular smooth muscle cells (SMCs), found on intracerebral arterioles and arteries, are absent from intracerebral capillaries. These cells are short, densely packed, ring-shaped, and essential for regulating vessel tone (Lacoste and Gu, 2015; Frosen and Joutel, 2018; Grant et al., 2019). Astrocytes occupy a critical position between blood vessels and neurons. They can modulate vessel tone via receptor-mediated increase in astrocytic Ca2+, resulting in the release of astrocyte-derived prostaglandins (PGE2), nitric oxide (NO), epoxyeicosatrienoic acids (EETs), glutamate, or adenosine, all of which can alter vascular diameter and tone (Attwell et al., 2010; Cauli and Hamel, 2010; Filosa and Iddings, 2013; Harada et al., 2015; Haidey et al., 2021), as reviewed in detailed elsewhere (Filosa and Iddings, 2013; Howarth, 2014; MacVicar and Newman, 2015; Mishra, 2017; McConnell et al., 2019; Stackhouse and Mishra, 2021). Whereas microglia are the main regulators of inflammatory processes in the brain, their role in NVC is not well defined. However, recently, they were suggested as essential in regulating CBF during neural activation (Császár et al., 2021). Brain ECs have unique morphological and functional features such as a lack of fenestration, the presence of tight junctions between cells, a low number of pinocytic vesicles that limit transcytosis, hence forming the first limiting layer of the BBB (Reese and Karnovsky, 1967; Stamatovic et al., 2008; Rizzo and Leaver, 2010; Salmina et al., 2014; Andreone et al., 2015). This highly selective barrier promotes a tightly regulated brain homeostasis to ensure proper neuronal function, protecting the brain from toxins, pathogens, inflammation, and injury (Weiss et al., 2009; Larsen et al., 2014; Daneman and Prat, 2015; Van Dyken and Lacoste, 2018). Furthermore, brain ECs regulate vascular tone by releasing vasodilators including endothelial-derived NO, endothelium-derived EETs, PGE2 and prostacyclin, as well as vasoconstrictors such as endothelin-1, thromboxane A2 and prostaglandin F2α (Mohan et al., 2012; Filosa and Iddings, 2013; Andreone et al., 2015; Kisler et al., 2016, 2017; Dabertrand et al., 2021). While the endothelium regulates vascular permeability and tone, it is also the main target of small vessel disease (SVD), which refers to a pathological process that damages arterioles, venules and brain capillaries. SVD has a major impact on CBF and cognition (Hakim, 2019). The NVU as a whole is also responsible for maintaining BBB integrity (Abbott et al., 2006; Zlokovic, 2008; Daneman, 2012; Kadry et al., 2020). Alterations in vascular patterning, CBF and BBB, either during development or later in life, contribute to the onset and/or progression of early- or late-onset neurological disorders (Figure 2).
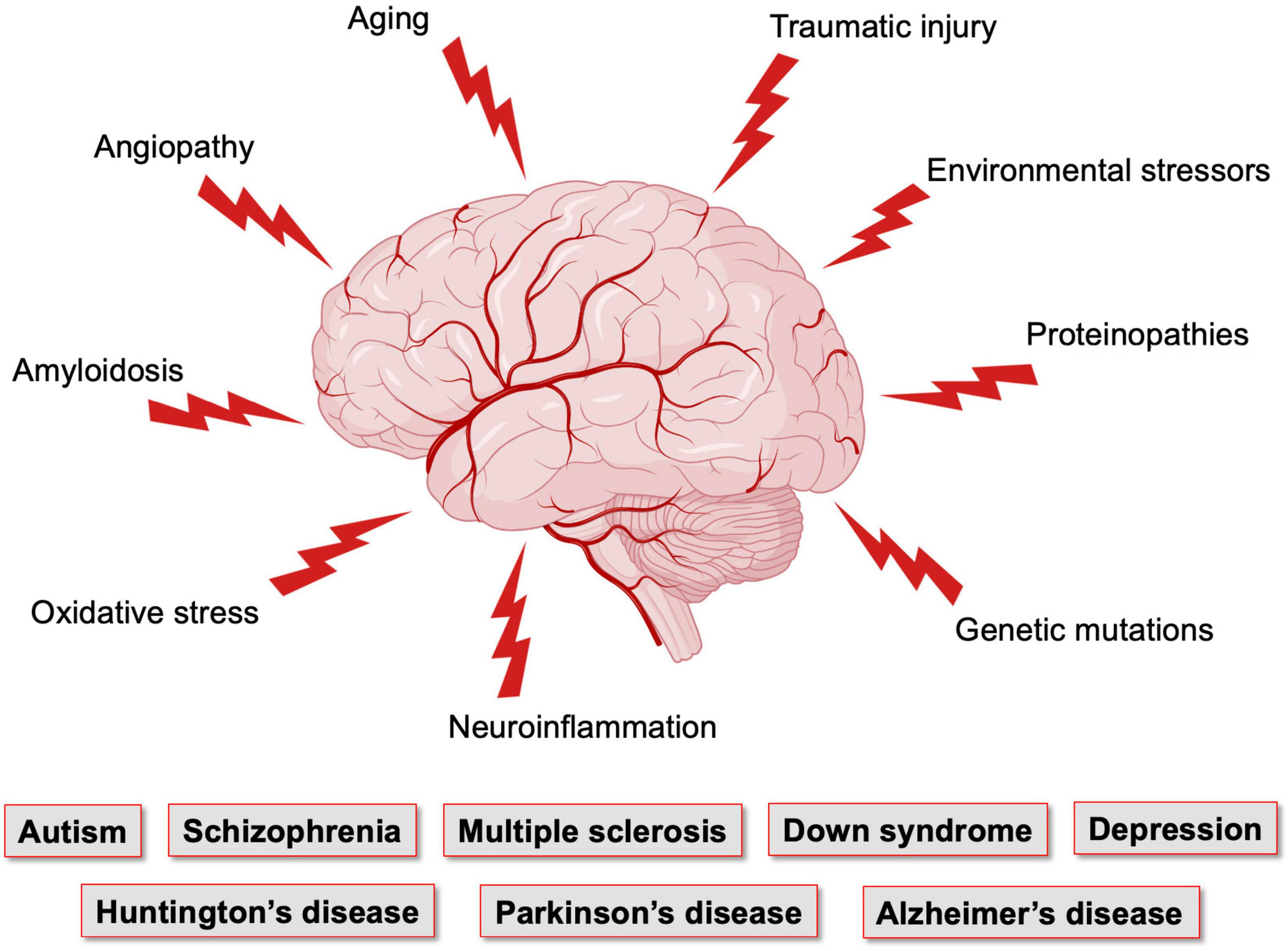
Figure 2. Factors affecting the brain vasculature and leading to neurological conditions. The brain, which has elevated metabolic needs but poor energy storage, is highly dependent on a continuous supply of nutrients and oxygen from the blood stream, and is thus dependent on the integrity of its vasculature. Vasculature of the brain is the most complex and dense in the human body. Yet, it is maintaining a very fragile equilibrium and is the target of numerous pathological conditions that affect neuronal maturation and function. Figure made using BioRender.
Well-balanced vascular and neuronal interactions are required to support brain function from early life. The shared spatial and temporal patterns of vascular and neuronal networks suggest an integrative role for vessels in neural development, and vice versa (Gu et al., 2005; Carmeliet and Jain, 2011; Andreone et al., 2015; Lacoste and Gu, 2015). Neurovascular crosstalk, which initially takes place during embryogenesis, supports the rising oxygen and nutrient demand of immature neurons as they require extensive energy to maintain normal course of development (De Filippis and Delia, 2011). The increased energy consumption by neurons creates a hypoxic environment acting as a signal for boosting blood vessel production to upsurge delivery of oxygen and metabolic substrates to the brain (Stone et al., 1995; Lacoste and Gu, 2015; Peguera et al., 2021). Hypoxia initiates vessel ingression into deep brain structures, followed by usage of vascular patterning cues (Lacoste and Gu, 2015; Tata et al., 2015; Tata and Ruhrberg, 2018; Okabe et al., 2020). Comparably, ECs instruct neural progenitors into dividing, differentiating or migrating through release of paracrine signals that regulate neuronal development in vascular niches (Hogan et al., 2004; Shen et al., 2004; Daneman et al., 2009; Goldman and Chen, 2011; Delgado et al., 2014; Lacoste and Gu, 2015; Licht and Keshet, 2015; Walchli et al., 2015; Tata and Ruhrberg, 2018; Peguera et al., 2021). Moreover, neuronal activity plays important roles in modulating postnatal brain angiogenesis (Lacoste et al., 2014; Whiteus et al., 2014; Biswas et al., 2020). As the brain matures, vascular networks remodel until the system consists of an extensive network that actively regulates blood flow to adequately sustain energy demands. The functional relationships between neurons and blood vessels ensures that NVC mechanisms progressively develop (Lacoste and Gu, 2015; Coelho-Santos and Shih, 2020). NVC becomes fully functional ∼3–4 weeks after birth in rodents, and 7–8 weeks in humans (Yamada et al., 2000; Muramuto et al., 2002; Kozberg et al., 2016).
These vascular features can become defective early in life, affecting brain maturation. Vascular susceptibilities can also emerge later in life, taking part in neurodegenerative processes. Indeed, NVU deficits play a role in both early- and late-onset neurological disorders (Figure 2). Mounting evidence shows that vascular impairments contribute to the pathophysiology of neurological conditions throughout life, including neurodevelopmental, metabolic, and neurodegenerative disorders (Nicolakakis and Hamel, 2011; Van Dyken and Lacoste, 2018; McConnell et al., 2019; Ouellette et al., 2020; Sharma and Brown, 2021). This suggests the existence of a vascular continuum between developmental conditions and illnesses of aging, which will be the focus of this review (Figure 3). A better understanding of mechanisms and key players involved in cerebrovascular impairments may lead to transformative therapeutic strategies at different stages of life.
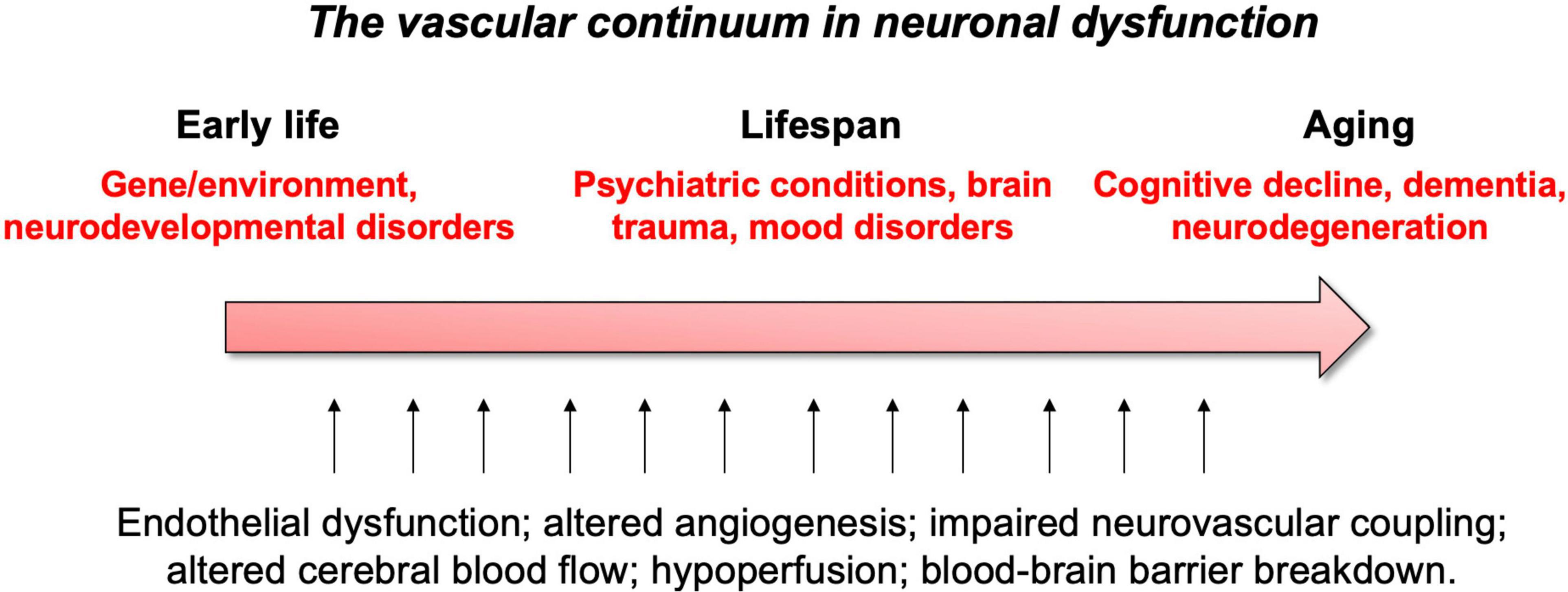
Figure 3. Summary of vascular links to neurological disease throughout life.
Cerebrovascular Deficits Associated With Neurodevelopmental Disorders
Neurodevelopmental disorders are considered a group of conditions with onset/diagnosis during infancy, childhood, or adolescence (Morris-Rosendahl and Crocq, 2020). They are defined by impairments in motor, social, cognitive, academic, and/or occupational functioning. Most studies focused on the neuronal contributions to these disorders; however, concomitant vascular impairments are starting to emerge (Ouellette et al., 2020). Here, we highlight vascular impairments identified in autism spectrum disorders (ASD) and schizophrenia.
Vascular Links to Autism Spectrum Disorders
ASD are pervasive neurodevelopmental disorders associated with social interaction deficits, speech and language impairments, as well as repetitive behaviors and restricted interests (Vijayakumar and Judy, 2016). These disorders have a prevalence of 1–2% in the general population and affect four times more boys than girls (Hogan et al., 2004; Daneman et al., 2009). Individuals with ASD show atypical behaviors associated with visual attention, imitation, social responses, and motor control by 12 months of age. By the age of 3, a child can be efficiently diagnosed with ASD (Park et al., 2016). While the underlying causes of ASD are enigmatic, both environmental and genetic origins have been found, leading to the identification of gene mutations within the ASD population (Hogan et al., 2004; James et al., 2009; Emerson et al., 2017). Although most studies have been neurocentric, ASD are now being associated with vascular vulnerabilities.
Altered Cerebral Blood Flow in Autism Spectrum Disorders
Neuroimaging techniques can map changes in CBF or blood oxygenation during various activities. Morphological and functional investigations using functional magnetic resonance imaging (fMRI), positron emission tomography (PET), single-photon emission computed tomography (SPECT), or Arterial Spin Labeling (ASL) are used to measure CBF changes in ASD children. CBF disruptions have been demonstrated in ASD patients when compared to healthy controls in different regions of the brain (Bjørklund et al., 2018). It has also been suggested that perfusion alterations are more pronounced in older children diagnosed with ASD. Cerebral hypoperfusion has been detected in nearly 75% of ASD children (Zilbovicius et al., 2000). As CBF impacts the delivery of oxygen and nutrients to neurons, hypoperfusion in ASD children has been associated with key ASD-related behaviors such as language deficits, impaired executive function and abnormal responses to sensory stimuli, as well as difficulty in facial perception (Siegel et al., 1992; Chiron et al., 1995; Ohnishi et al., 2000; Burroni et al., 2008; Reynell and Harris, 2013; Bjørklund et al., 2018; Yerys et al., 2018). These behaviors correlate with abnormal regional cerebral blood flow (rCBF) in the bilateral insula, superior temporal gyri and left prefrontal cortices, medial temporal lobe, supramarginal gyrus, right fusiform gyrus, and dorsal anterior cingulate cortex (Ohnishi et al., 2000; Zilbovicius et al., 2000; Burroni et al., 2008; Jann et al., 2015; Yerys et al., 2018). Studies are attempting to ameliorate these behavioral abnormalities using hyperbaric oxygen treatment (HBOT) to counteract cerebral hypoperfusion in children with ASD. There is some evidence that children who undertook 40 HBOT sessions of 60 min each showed improvements on selected psychosomatic parameters in the Autism Treatment Evaluation Checklist (ATEC) and Childhood Autism Rating Scale (CARS) (Kostiukow and Samborski, 2020). Currently, there is insufficient evidence to support the use of HBOT to treat children with ASD as there are many contradicting studies claiming no improvement in behaviors. Nevertheless, each study followed different protocols, consisted of patients with a large spectrum of behavioral impairments, and some lacked proper control groups, which could explain discrepancies. More research is required to determine if specific groups of children could benefit from HBOT treatment (Rossignol et al., 2012; Sakulchit et al., 2017).
ASL-based measurements of cerebral perfusion showed that children with ASD presented a pattern of widespread hyperperfusion in frontotemporal regions including medial orbitofrontal cortex, bilateral inferior frontal operculum, left inferior/middle temporal gyrus and the right precentral gyrus (Jann et al., 2015). The medial orbitofrontal cortex is known to have extensive connections with the limbic system involved in socio-emotional cognition. Furthermore, hyperperfusion was detected throughout the frontal white matter and subcortical gray matter in ASD children, which correlated positively with severity of social deficits (Peterson et al., 2019). As shown by these studies, CBF abnormalities appear linked to clinical manifestations. Although opposing observations of CBF in ASD patients were reported, these further support the complexity of these disorders (Jann et al., 2015).
Neurovascular coupling alterations were also observed in ASD patients. Hemodynamic responses in children with ASD during a color-word task were significantly lower than the control group, especially in the dorsolateral prefrontal cortex (Uratani et al., 2019). Conversely, children displayed no difference in hemoglobin concentrations in the prefrontal cortex during a letter fluency task, while adults showed reduced responses (Kawakubo et al., 2009). Despite inter-study variability, there seems to be a consensus on the impact of altered CBF on the expression of behavioral impairments (Zilbovicius et al., 2000; Jann et al., 2015). But as ASD are heterogeneous, with various behavioral traits, genetic causes, medical co-morbidities and medications, these variables may have impacted neuroimaging results, which led to inconsistencies. Importantly, these studies take an important step toward the identification of key players in ASD pathophysiology, opening new opportunities for early diagnosis and treatment.
The relationship between CBF alterations and symptom profiles in ASD children provides insight into disease mechanisms that can be tested in animal models. As most pre-clinical studies have also focused on the neuronal aspects of ASD, very few have considered vascular contributions to these disorders in laboratory models. Recent studies using different ASD mouse models have reported alteration in CBF. A study by Abookasis et al. (2018) using inbred Black and Tan Brachyury (BTBR) T+tf/J reported decreased CBF in mutant mice using laser speckle imaging and laser Doppler flowmetry (LDF). Subsequently, work by Ouellette et al. (2020) using the 16p11.2 deletion mouse model of ASD (16p11.2df/+; Horev et al., 2011) demonstrated an increase in resting CBF as well as neurovascular uncoupling in adult (P50) 16p11.2df/+ mice compared to WT littermates using a combination of ultrasound imaging and LDF. No difference in CBF or NVC were observed between younger (P14) mutant and control mice (Ouellette et al., 2020). Results from this study in 16p11.2df/+ mice revealed the cause of these functional cerebrovascular impairments: an endothelial deficit. While normal vascular smooth muscle cell function was measured, defective endothelium-dependent vasodilation was found ex vivo following exposure to specific vasomodulators (Ouellette et al., 2020). This suggests that endothelial health plays an important role in the etiology of the 16p11.2 deletion ASD syndrome. Understanding the molecular and cellular factors that mediate CBF alterations in ASD could help design rescue approaches in animal models, as well as therapeutic approaches down the line.
Since MRI studies rely on Blood Oxygen Level Dependent (BOLD) signals as surrogates for neuronal activity (Hillman, 2014; Howarth et al., 2021; Moon et al., 2021), it is possible that changes in rCBF reflect changes in underlying neuronal activity. For instance, cerebral cortex hypoperfusion in ASD patients could reflect lower metabolic demands (Schifter et al., 1994). In the case 16p11.2df/+ mice, however, it is interesting to note that a neurovascular uncoupling was measured, with enhanced neuronal activation yet reduced vascular responses to whisker stimulations, which led to the discovery of endothelium-dependent deficits (Ouellette et al., 2020).
Altered Blood-Brain Barrier and Angiogenesis in Autism Spectrum Disorders
Cerebral vessels are central for the maintenance of brain homeostasis, sustaining proper neuronal function, and providing an effective protection against toxins and pathogens (Profaci et al., 2020). The BBB consists of specialized ECs lining the vessel wall to separate the peripheral blood from cerebral tissue. Brain (central) ECs are distinct from peripheral ECs, as they produce specific proteins to control the flux (entry and exit) of metabolites across vessels, to maintain low rates of trans-endothelial vesicular transport, and to form tight junctions to limit the para-cellular flow of material between adjacent ECs (Andreone et al., 2015; Chow and Gu, 2015; Kealy et al., 2020). Alterations in the BBB are at the core of the onset and/or progression of numerous neurological disorders (Daneman and Prat, 2015; Van Dyken and Lacoste, 2018; Profaci et al., 2020). Only few studies have investigated the components of the BBB in the context of ASD. Children diagnosed with ASD have been associated with reduced levels of adhesion molecules such as soluble Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1, or CD31) and P-selectin. Since these molecules are essential to modulate BBB permeability through signaling and leukocyte infiltration, it suggests that crucial BBB components may be at play in ASD pathophysiology (Onore et al., 2012). Furthermore, a post-mortem study, with a small sample size, demonstrated altered BBB integrity in ASD with increased gene expression of matrix metalloproteinase (MMP)-9 (Fiorentino et al., 2016). Studies have shown that MMP-9 regulates cell proliferation, adhesion, degradation of laminin and collagen, angiogenesis, oxidative injury, and is implicated in BBB breakdown (Lepeta and Kaczmarek, 2015; Turner and Sharp, 2016). Additionally, important components of BBB integrity displayed altered expression in ASD patients, including claudin-5 (CLDN5) and claudin-12 (CLDN12), as well as tricellulin (MARVD2), a component of tight junctions involved in decreased permeability to macromolecules in brain ECs (Fiorentino et al., 2016). In an older study, a small subset of ASD participants demonstrated higher levels of autoantibodies against brain ECs in the serum compared to typically developing individuals, suggesting an impact on the BBB (Connolly et al., 1999). Animal models have facilitated the study of BBB integrity in ASD. In a valproic acid rat model of autism, increased BBB permeability to Evans blue was found in the cerebellum, a phenotype attenuated by treatment with memantine, an NMDA receptor modulator. This BBB alteration was also attenuated using minocycline (antibiotic) and agomelatine (melatonin receptor) treatment (Kumar et al., 2015; Kumar and Sharma, 2016). Animal studies have investigated transendothelial transport mechanisms in ASD mouse models. Tarlungeanu et al. (2016) demonstrated that the large neutral amino acid transporter (LAT1, Slc7a5) localized at the BBB to maintain normal levels of brain branched chain amino acid (BCAA) was required for neurotypical development. Mice harboring an endothelial-specific deletion of Slc7a5 (Slc7a5ΔEC) displayed behaviors reminiscent of ASD, including motor dysfunctions consistent with a study in human patients harboring the constitutive mutation (Novarino et al., 2012; Tarlungeanu et al., 2016). Interestingly, administration of BCAA rescued ASD-like behaviors in Slc7a5ΔEC mice (Tarlungeanu et al., 2016).
Recently, a post-mortem analysis of brain tissue from individuals diagnosed with ASD revealed significantly higher levels of markers associated with pericytes, as well as increased vascular tortuosity, indirectly suggesting impairments in angiogenesis, a process through which new blood vessels are formed (Azmitia et al., 2016). A more recent study in 16p11.2df/+ mice revealed impaired cerebral angiogenesis in young (P14) 16p11.2df/+ male mice compared to sex-/age-matched littermates, a phenotype which was absent in adult mice. Defective angiogenic activity was also measured using primary brain ECs from P14 16p11.2df/+ males or ECs derived from human-induced pluripotent stem cells (hiPSCs) of 16p11.2 deletion carriers (Ouellette et al., 2020). Moreover, RNA-sequencing analysis of 16p11.2df/+ mouse brain EC transcriptome revealed changes in the expression of genes involved in angiogenesis (e.g., Grem1, Apln, Angpt2), while key genes involved in BBB regulation (e.g., Pecam1, Mfsd2a, Cldn5, Slc2a1) were not affected by the 16p11.2 deletion (Ouellette et al., 2020). Finally, this study generated a mouse model with endothelial-specific 16p11.2 haploinsufficiency which recapitulated ASD-related phenotypes, revealing a causal relationship between endothelial dysfunction and neuronal aspects of the 16p11.2 deletion syndrome (Ouellette et al., 2020).
Overall, these studies allude to the contribution (structural and functional) of a defective BBB and NVU in ASD, with an important role for endothelial impairments.
Vascular Links to Schizophrenia
Schizophrenia is a debilitating neurodevelopmental disorder affecting ∼1% of the population. It is associated with behavioral and cognitive symptoms that arise progressively. Memory and attention deficits appear in childhood, while positive symptoms (psychotic episodes) and negative symptoms (social and motivational deficits) emerge later in adolescence or early adulthood (Stachowiak et al., 2013). Although the incidence of schizophrenia is higher in men, women have a slightly later disease onset (Gogtay et al., 2011; Ochoa et al., 2012). While the behavioral aspects of schizophrenia have been described, the causes of this disorder are poorly known. As in ASD, both genetic and environmental origins are involved. Schizophrenia has been associated with genes essential for a wide range of functions including neuronal connectivity and patterning of brain structures, cell proliferation and differentiation, as well as cytoskeleton reorganization (Stachowiak et al., 2013; Clifton et al., 2019). As in most neurological disorders, the implication of neuronal alterations has been extensively studied, but research on vascular impairments in schizophrenia is starting to emerge.
Altered Cerebral Blood Flow in Schizophrenia
Cognitive impairments are often present before the first psychotic episode in patients with schizophrenia (Keefe and Harvey, 2012; Schuepbach et al., 2016) and deficits in executive functions are often parallel to changes in CBF. Several studies have linked altered CBF with schizophrenia-related symptoms (Sabri et al., 1997; Malaspina et al., 1999, 2004; Pinkham et al., 2011; Fujiki et al., 2013; Schuepbach et al., 2016; Stegmayer et al., 2017; Zhu et al., 2017). Interestingly, the manifestations of negative or positive symptoms correlate with different rCBF changes. In a study by Pinkham et al. (2011), CBF of 30 schizophrenia patients was measured using ASL perfusion MRI, which revealed a positive correlation between increased severity of positive symptoms and higher CBF in the cingulate and superior frontal gyri, but decreased CBF in precentral and middle frontal gyri. Patients who presented with severe negative symptoms also displayed reduced CBF in the superior temporal gyrus bilaterally, cingulate and left middle frontal gyri (Malaspina et al., 2004; Scheef et al., 2010; Pinkham et al., 2011; Liu et al., 2012). Most studies investigating CBF alterations in schizophrenia considered perfusion rates from medicated patients, and a small number of studies have measured CBF rates in neuroleptic-naïve patients. Using ASL in non-medicated patients, the schizophrenia group displayed resting-state hypoperfusion in the frontal lobes, anterior and medial cingulate gyri, as well as in the parietal lobes, while increased perfusion was measured in the cerebellum, brainstem and thalamus (Scheef et al., 2010). Sabri et al. (1997) measured rCBF using SPECT in non-medicated patients that have experienced positive symptoms, revealing that rCBF values varied depending on the severity of positive symptoms. Hyperperfusion was detected in the frontal, anterior cingulate as well as in both parietal and temporal cortices in patients who had scored high in severity for formal thought disorder (disturbance of the organization and expression of thought). In contrast, patients who scored high for delusions, hallucinations or distrust, with low scores for formal thought, displayed hypoperfusion in the same brain regions. No difference in rCBF was identified between control and schizophrenia groups after treatment (Andreasen et al., 1997; Sabri et al., 1997; Horn et al., 2009). Recent studies have detected hyperperfusion and hypoperfusion in brain regions from individuals with hallucinations. For instance, CBF increase was found in the right superior temporal gyrus and caudate nucleus, while CBF decrease was found bilaterally in the occipital and left parietal cortices (Zhuo et al., 2017). In another study, patients were classified based on the severity of three behavioral dimensions (language, affectivity, and motor) according to the Bern Psychopathology scale. Patients with altered affectivity were associated with increased CBF in the amygdala, while changes in language dimension were linked to increased CBF in Heschl’s gyrus (Stegmayer et al., 2017). While schizophrenia is classified as a neurodevelopmental disorder, its symptoms persist with age. Studies have identified significant bilateral temporal hypoperfusion related to aging and disease course. It has been suggested that this decrease in CBF with aging is paralleled with the degenerative changes observed in patients with schizophrenia (Schultz et al., 2002; Kawakami et al., 2014).
The polygenic risk of schizophrenia is an important dimension of this syndrome, and changes in CBF have been identified in patients diagnosed for either familial or sporadic schizophrenia. Sporadic schizophrenia patients were associated with hypofrontality (left frontal gyrus, orbitofrontal cortex, anterior cingulate, and paracingulate cortices), while familial schizophrenia patients had left temporoparietal hypoperfusion (posterior Sylvian fissure at the superior and inferior parietal lobules, angular, and supramarginal gyri). In both groups, positive symptoms are often associated with increased rCBF in the parahippocampal gyrus, cerebellum, and pons (Malaspina et al., 2004). Sporadic patients showed additional hyperperfusion in the fusiform gyrus, and familial patients the hippocampus, dentate, amygdala, thalamus, and putamen (Malaspina et al., 2004). In addition, the prefrontal cortex in schizophrenia has been associated with deficits of pericapillary oligodendrocytes, which could contribute to changes in CBF (Vostrikov et al., 2008; Uranova et al., 2010). Altogether, these studies support the idea that altered CBF is involved in schizophrenia pathophysiology.
In addition to studies investigating resting state CBF, there is evidence of altered NVC in schizophrenia whereby many reports demonstrate reduced hemodynamic response, reflecting reduced neuronal activity during processing of cognitive tasks, especially in the lateral prefrontal cortex and temporal regions (Ford et al., 1999, 2005; Mathalon et al., 2000; Carter et al., 2001; Hanlon et al., 2016; Pu et al., 2016). As with CBF reports, there are inconsistent hemodynamic responses associated with schizophrenia since increased hemodynamic responses in hippocampus, thalamus and prefrontal cortex have been identified (Tregellas et al., 2007). These conflicting results are translating to rodent models of schizophrenia whereby some models have revealed overall hypofrontality, hypoperfusion in the hippocampus or hyperperfusion in the somatosensory cortex (Finnerty et al., 2013; Song et al., 2013; Drazanova et al., 2019).
Altogether, these studies support the idea that altered CBF regulation is involved in schizophrenia pathophysiology. Moreover, it appears critical to consider the polygenic risk of disease, the category and severity of symptoms, as well as the age of patients when comparing CBF rates in schizophrenia. Although many studies have detected altered CBF using various methods, results thus far remain conflicting based on various stages of disease and pharmacological treatment (Drazanova et al., 2019).
Altered Blood-Brain Barrier and Angiogenesis in Schizophrenia
A dysfunctional BBB has been reported in schizophrenia, with increased permeability to damaging proteins (Müller and Ackenheil, 1995; Shcherbakova et al., 1999; Crockett et al., 2021). Studies are starting to decipher changes in cells associated with the BBB (for a detailed review, see Carrier et al., 2020). Briefly, evidence of schizophrenia-associated microvascular abnormalities in the neocortex include thickening and deformation of basal lamina, vacuolation of cytoplasm in ECs, basal lamina and astrocytic end-feet, swelling of astrocyte end-feet, activation of microglial cells in the prefrontal and visual cortex, as well as atypical vascular arborization (Uranova et al., 2010; Carrier et al., 2020).
Moreover, specific mutations are associated with schizophrenia, including alterations in the 22q11.2 deletion syndrome (22qDS) -strongest monogenic risk allele for this disorder, and polymorphisms in claudin-5, a densely expressed tight junction molecule (Gur et al., 2017; Greene et al., 2018; Carrier et al., 2020) altogether revealing barrier dysfunction in schizophrenia patients (Greene et al., 2018; Crockett et al., 2021). Post-mortem brain sections from 22qDS patients and animal models of 22qDS both demonstrate reduced claudin-5 expression in the BBB, which in turn compromised BBB function (Nishiura et al., 2017; Guo et al., 2020; Crockett et al., 2021; Usta et al., 2021). Additionally, altered levels of vascular endothelial (VE)-cadherin and occludin in ECs were identified in schizophrenia. These molecules regulate adherence of ECs and restrict movement of substances across the BBB (Cai et al., 2020). Furthermore, BBB hyperpermeability has been associated with another risk allele for schizophrenia. NDST3, expressed in the brain, encodes an enzyme involved in the metabolism of heparan sulfate, a component of basal lamina extracellular matrix that is required for BBB integrity (Khandaker et al., 2015).
Studies have documented primary vascular endothelial dysfunction in schizophrenia. Individuals carrying MTHER T and/or COMT Val risk allele have been associated with cerebrovascular endotheliopathy, as well as lower frontal executive functions (Grove et al., 2015). While endothelial dysfunction is possibly associated with schizophrenia, many studies are using peripheral endotheliopathy as a surrogate marker for endothelial dysfunction. For example, studies are using non-invasive peripheral arterial tonometry (RH-PAT) to assess peripheral arteriole endothelial-dependent vasodilation and revealed impaired peripheral arterial vasodilation in schizophrenia (Ellingrod et al., 2011; Burghardt et al., 2014). Notably, brain ECs have unique properties to maintain BBB integrity and brain homeostasis. Although altered endothelial function was found in the periphery, it does not represent a definite marker of brain (central) endothelial dysfunction. A critical regulator of angiogenesis, vascular endothelial growth factor (VEGF), and its receptor (VEGFR2) have been found upregulated in the prefrontal cortex of individuals diagnosed with schizophrenia (Hino et al., 2016). Findings of elevated VEGF could also be linked to vascular hyperpermeability, as VEGF not only regulates angiogenesis but increases BBB leakage (Mayhan, 1999; Zhang et al., 2000). Conversely, a different group revealed that a decreased production of VEGF predisposed individuals to develop this disorder and contributed to the severity of symptoms (Saoud et al., 2021). Another study investigated the impact of hiPSC-derived neural stem cells from schizophrenia patients on angiogenesis (Casas et al., 2018). This study found an imbalance in the expression and secretion of several angiogenic factors and non-canonical neuro-angiogenic guidance cues from neural stem cells from schizophrenic patients. Conditioned media from these cells induced impaired angiogenesis as evidenced by reduced number of sprouts and tubes formed in in vivo and in vitro models, as well as decreased neural stem cell migration compared to control conditioned media (Casas et al., 2018).
Cerebrovascular Deficits Associated With Neurodegenerative Disorders
CNS disorders are dichotomized as early onset neurodevelopmental disorders and late-onset neurodegenerative diseases (Taoufik et al., 2018). Neurodegenerative diseases consist of a group of heterogeneous disorders characterized by the progressive degeneration of structure and function in the CNS (Gitler et al., 2017). Although neurodegenerative and neurodevelopmental disorders are differentially classified, an accumulating body of work demonstrates significant similarities between these two groups of conditions. Here below, we cover cerebrovascular impairments reported in four neurodegenerative diseases that emerge throughout lifespan: multiple sclerosis (MS), Huntington’s disease (HD), Parkinson’s disease (PD), and Alzheimer’s disease (AD).
Vascular Links to Multiple Sclerosis
MS is a chronic autoimmune disease of the CNS, occurring when the immune system attacks its own nerve fibers and myelin sheaths (D’Haeseleer et al., 2013). The pathological hallmark of MS consists of perivenular inflammatory lesions, leading to demyelinating plaques and diffuse axonal degeneration throughout the CNS (Dobson and Giovannoni, 2019). It is characterized by the infiltration of T cells reactive against myelin in the CNS (Schwartz and Kipnis, 2005). This demyelinating disease has key features including inflammation, BBB disruption and neurodegeneration. MS has a prevalence of 0.5–1.5 per 100,000 individuals, whereby women are three times more affected than men (Harbo et al., 2013). The age of MS onset is situated between 20 and 40 years of age (Ortiz et al., 2014). General symptoms related to MS include, but are not limited to, tremors, lack of coordination as well as weakness in limbs. There are various types of MS including relapsing-remitting MS (RR-MS), secondary progressive MS (SP-MS) and primary progressive MS (PP-MS). RR-MS consists of unpredictable relapses or inflammatory flare-ups during which new symptoms appear or existing ones worsen (Adhya et al., 2006). Most people with RR-MS, transition to a disease phase known as SP-MS. In this phase, there is progressive worsening and fewer relapses. Active lesions with profound lymphocytic inflammation are mostly found in RR-MS (Dobson and Giovannoni, 2019). PP-MS is considered as a slow accumulation of disability without defined relapses. In this case, PP-MS is associated with an inactive lesion core surrounded by activated microglia and macrophages (Dobson and Giovannoni, 2019).
Altered Cerebral Blood Flow in Multiple Sclerosis
MS has been associated with functional cerebrovascular abnormalities including decreased cerebral perfusion and reduced CNS venous blood drainage, known as chronic cerebrospinal venous insufficiency (D’Haeseleer et al., 2011). SPECT, PET, and ASL imaging studies have reported decreased CBF in both gray and white matter of MS patients (Ge et al., 2005; D’Haeseleer et al., 2011). Widespread cerebral hypoperfusion has been revealed in SP-MS, RR-MS and PP-MS patients, while an ischemic threshold was not reached (Adhya et al., 2006; Ota et al., 2013; Monti et al., 2018). Gray matter hypoperfusion in MS suggests a reduction of metabolism due to the loss of cortical neurons (Peruzzo et al., 2013). Furthermore, studies have reported that CBF is globally impaired in normal appearing white matter (NAWM) of patients with early RR-MS (Law et al., 2004; Adhya et al., 2006). Of note, CBF was generally lower in PP-MS than in RR-MS in the periventricular and frontal white matter (Adhya et al., 2006). In the contrary, other studies have measured elevation of CBF and cerebral blood volume (CBV) in NAWM of patients with early RR-MS several weeks before signs of increased BBB permeability (Wuerfel et al., 2004). Although studies on different types of MS revealed changes in CBF, general active demyelinating lesion regions are associated with hyperperfusion while the more stable forms show hypoperfusion (Monti et al., 2018). Decreased CBF in cerebral NAWM, thalamus, and putamen was identified in patients whose symptoms emerged within the first 5 years of onset. This suggests that CBF alterations are present in the very early stages of the disease (Varga et al., 2009). Different mechanisms have been proposed to explain hypoperfusion in MS. A study suggested that decreased CBF is secondary to axonal degeneration, which leads to a decreased metabolic demand (Saindane et al., 2007). However, this hypothesis is yet to receive experimental support. A second mechanism that has been proposed is an impaired energy metabolism of astrocytes (De Keyser et al., 1999). In MS, astrocytes are deficient in β2-adrenergic receptors which regulate high energy-consuming activities, such as glycogenolysis and phosphocreatine metabolism (De Keyser et al., 1999). Reduced energy production in astrocytes could be contributing to altered CBF. A third mechanism suggested was increased release of vasoconstrictor endothelin-1 (ET-1) from reactive astrocytes, found in a post-mortem study on white matter samples of RR-MS patients (D’Haeseleer et al., 2013; Hostenbach et al., 2019). Hence, elevated levels of ET-1 could be involved in dysregulating CBF in MS. Interestingly, administration of ET-1 antagonist Bosentan restored CBF to control levels in MS patients (D’Haeseleer et al., 2013).
Impaired cerebral vascular reactivity was evidenced in MS patients exposed to hypercapnia, which has been suggested to contribute to neuronal death identified in this disorder (Marshall et al., 2014). This global deficit is thought to be associated with elevated levels of NO reported in MS (Trapp and Stys, 2009; Juurlink, 2013). These studies suggest that the overproduction of NO may desensitize endothelial and smooth muscle cell function, causing decreased vasodilatory capacity and limited blood supply for neurons that perform demanding tasks. Increased NO in MS may lead to neuronal activity-induced hypoxia leading to neurodegeneration (Marshall et al., 2014). Interestingly, high inflammatory MS lesion load has been associated with increased CBF. Therefore, perfusion changes may be sensitive to active inflammation (Bester et al., 2015). However, it remains unclear whether abnormal perfusion in MS is a precursor of lesions or occurs independently of lesion development (Marshall et al., 2014).
Notably, MS has been associated with cerebral SVD. It was demonstrated that younger MS cases are more severely impacted by cerebral SVD compared to older individuals (Geraldes et al., 2020). This suggests that the interaction between MS and cerebral SVD is affected by age, an assumption still under investigation (Geraldes et al., 2020).
Altered Blood-Brain Barrier and Angiogenesis in Multiple Sclerosis
BBB dysfunction is considered a major hallmark of MS and is deemed a trigger of disease onset (McQuaid et al., 2009; Cramer et al., 2014). Intense focal disruption of the BBB associated with inflammation (identified by gadolinium-enhanced MRI at acute and chronic MS lesion sites; Saade et al., 2018) and diffuse extensive BBB disruption with a long-term pathological activity, are both found in MS patients (Bennett et al., 2010). Hyperpermeability of the BBB was evidenced by leukocyte passage across the BBB (Cramer et al., 2014). Increased BBB leakage was associated with decreased expression of tight junction proteins in brain capillary ECs in patients with active lesions, inactive lesions, as well as NAWM associated with fibrinogen leakage (Kirk et al., 2003; McQuaid et al., 2009; Bennett et al., 2010). More specifically, dysregulation of tight junction adaptor protein ZO-1, occludin and claudin-5 have been reported in both primary progressive and secondary progressive disease states (Kirk et al., 2003; Leech et al., 2007). Experimental autoimmune encephalomyelitis (EAE) in rodents is a disease model with clinical and pathological characteristics relevant to the study of MS. This model revealed reorganization of ZO-1 and actin in the presence of inflammatory factors in vitro, associated with increased permeability of an endothelial monolayer (Bennett et al., 2010). The EAE model also revealed increased expression of VEGF in ECs, astrocytes, monocytes and activated TH1 lymphocytes, all of which contribute to BBB permeability during the early phase of disease, while decreased expression VEGF was evident in the late phase (Girolamo et al., 2014). The increase in VEGF expression was also found in the brain of MS patients (Girolamo et al., 2014). Furthermore, junctional adhesion molecule-A, a component of tight junctions, was found abnormally distributed in active and inactive MS lesions, although adherent junction proteins were normally expressed and localized in MS tissue (Padden et al., 2007). In addition, levels of PECAM-1 were found increased in active gadolinium-enhancing MS lesions (Ortiz et al., 2014). While BBB leakage is evident in MS, the complex network of cellular and molecular players that lead to this dysfunction have yet to be fully understood. Targeting BBB defects in MS represent a therapeutic opportunity, for instance with MMP inhibitors, interferons, and corticosteroids (Minn et al., 2002; Ross et al., 2004; Pardridge, 2012; Ortiz et al., 2014). However, no current therapy addresses BBB deficits (Ortiz et al., 2014). For more details on BBB dysfunction in MS, the following reviews can be consulted (Girolamo et al., 2014; Kamphuis et al., 2015; Xiao et al., 2020).
ECs proliferation as well as an increase in vascular network density has been reported (Ludwin, 2006; Holley et al., 2010). Increased angiogenesis was suggested to contribute to disease progression as well as remission after relapses (Papadaki et al., 2014). In addition to increased VEGF levels, VEGFR2 is also expressed on ECs in active MS lesions (Seabrook et al., 2010). Other molecules, such as basic fibroblast growth factor, were increased in MS patients and involved in angiogenesis (Su et al., 2006). MS patients with activated lesions and NAWM show blood vessels with a glomeruloid morphology, hemorrhages and vessel wall hyalinization (Girolamo et al., 2014). Immunosuppressive therapies have been used in aggressive MS as they not only impact neuroinflammation but also have an anti-angiogenic effect. Further research is warranted to elucidate the vascular links to MS and identify new therapeutic targets, as disease modifying drugs have unfortunately little to no impact on MS progression (Girolamo et al., 2014).
Vascular Links to Huntington’s Disease
HD is an hereditary, autosomal dominant and neurodegenerative disorder (Davenport, 1915; Wasmuth et al., 1988; Bano et al., 2011) leading to altered muscle coordination and declined mental abilities (Paulsen, 2011; Ha and Fung, 2012). An expansion of trinucleotide CAG repeats on chromosome 4 within the Huntingtin gene (HTT) results in the production of an altered Huntingtin (Htt) protein which accumulates in specific brain regions. Aggregation of mutant Htt (mHtt) leads to increased neurotoxicity (Zheng and Diamond, 2012), particularly in subcortical brain structures such as the neostriatum (caudate and putamen) where GABAergic medium-spiny neurons are particularly vulnerable (Sieradzan and Mann, 2001; Walker, 2007; Ross and Tabrizi, 2011; Drouin-Ouellet et al., 2015; McColgan and Tabrizi, 2018). At the cellular level, mHtt results in neuronal dysfunction and death through disrupted mechanisms involved in proteostasis, transcription and mitochondrial function as well as toxicity from the mutant protein (McColgan and Tabrizi, 2018). Worldwide, 2.71 per 100,000 individuals suffer from HD (Rawlins et al., 2016; Kounidas et al., 2021). Both men and women are affected equally, and heterogeneous symptoms emerge at around 40 years of age. However, functional and structural brain alterations emerge a decade before symptoms manifest (Snowden, 2017). Carriers of CAG repeat expansions in HTT can be identified decades before clinical manifestation, allowing researchers to identify possible biomarkers in the premanifest stage of HD (preHD). With this comes the increasing interest to study cerebrovascular abnormalities in HD (Snowden, 2017).
Altered Cerebral Blood Flow in Huntington’s Disease
HD-related perfusion deficits have been mostly associated with cerebral hypoperfusion (Reid et al., 1988; Sotrel et al., 1991; Hasselbalch et al., 1992; Harris et al., 1999; Deckel and Duffy, 2000; Wild and Fox, 2009). There is evidence of reduced CBF in the basal ganglia in early HD, prior to gross structural changes and to motor symptoms. In these cases, severity of cortical hypoperfusion correlated with decreased functional capabilities (Sax et al., 1996; Harris et al., 1999). In preHD patients, classified as either near or far from motor symptom onset, displayed altered rCBF by MR-based perfusion imaging. Participants with preHDfar and preHDnear had lower rCBF in the medial prefrontal cortex and increased rCBF in the left precuneus. Of note, structure and function of the precuneus and hippocampus can be abnormal in very early HD (Feigin et al., 2006). PreHDnear participants had additional regions showing altered rCBF, including hypoperfusion in the medial and lateral prefrontal cortex and hyperperfusion in the right hippocampus (Wolf et al., 2011).
While resting CBF is affected, early manifest and premanifest HD patients also display altered neurovascular coupling during visual stimulation (Klinkmueller et al., 2021). After HD onset, a significant hypoperfusion in the HD group was identified in most of the cerebral cortex. During problem-solving activities, such as solving a maze or resting their eyes open while looking at a modified maze, patients with HD showed increased CBF in the caudate nucleus (Deckel and Duffy, 2000; Deckel et al., 2000). Following physical activity, HD patients were associated with CBF hyperperfusion compared to the control group (Steventon et al., 2020).
Animal models of HD (e.g., gene knock-in of a human exon 1 CAG140 expansion repeat) also revealed altered rCBF. In mice as in humans, different brain regions displayed either hypoperfusion (basal ganglia motor circuit, hippocampus and prefrontal area) or hyperperfusion (cerebellar-thalamic and somatosensory regions). This altered CBF was apparent at a presymptomatic stage (Wang et al., 2016).
While CBF is starting to emerge as a biomarker for HD, mounting evidence supports the utilization of CBV as an additional metric. Several studies have reported elevated CBV in preHD patients (Hua et al., 2014; Liu et al., 2020). In addition, there is evidence of increased CBV in cortical gray matter after HD onset (Drouin-Ouellet et al., 2015), suggesting that arteriolar CBV may be a sensitive biomarker for premanifest HD (Hua et al., 2014; Liu et al., 2020). From these studies it was suggest that imaging of CBF may be used to detect widespread functional abnormalities in HD, and possibly predict HD symptoms onset during premanifest stages.
Altered Blood-Brain Barrier and Angiogenesis in Huntington’s Disease
Increases in vessel density, BBB permeability and VEGF-A release were observed in HD patients and animal models of HD (Steventon et al., 2020). There is evidence that BBB leakage increases alongside disease progression (Drouin-Ouellet et al., 2015). Despite these observations, there seems to be discrepancies between mouse models of HD. For instance, the BACHD transgenic mice, a well-known model of HD expressing the full-length mutant human HTT, failed to develop BBB breakdown at 12 months of age despite robust motor deficits (Lin et al., 2013; Mantovani et al., 2016). BBB dysfunction in HD patients has been associated with decreased tight junction molecules such as occludin and claudin-5 (Drouin-Ouellet et al., 2015). Moreover, other markers associated with BBB permeability, including hepatocyte growth factor, interleukin-8 and tissue inhibitor of MMP-1, were found elevated in HD patients (Drouin-Ouellet et al., 2015). A transgenic mouse model of HD (R6/2 mice) confirmed elevated tight junction molecules similar to HD patients. The R6/2 mouse model of HD is the most commonly studied and harbors a mutant Htt with CAG repeat expansion in exon 1 (Li et al., 2005). R6/2 mice also displayed increased transcytosis and paracellular transport across the brain endothelium compared to control mice (Drouin-Ouellet et al., 2015). In R6/2 mice, tight junction imbalance and perturbed BBB homeostasis were perceptible at very early stage of the disease, in absence of symptoms (Di Pardo et al., 2017). At the structural level, mHtt aggregates were found in the basal membrane of cerebral blood vessels in HD patients (Drouin-Ouellet et al., 2015). Interestingly, mHtt aggregates were localized in ECs, smooth muscle cells and perivascular macrophages, consistent with observations in R6/2 mice.
Further research is needed to determine BBB impairments in preHD patients. Lim et al. (2017) reported that iPSCs-derived brain microvascular endothelial cells (BMECs) from HD patients exhibit increased angiogenesis and altered barrier properties associated with elevated transcytosis and paracellular permeability. An increased and unregulated angiogenic activity may lead blood vessels to become more permeable with a potential role in neurovascular dysfunction in HD. RNA-seq analysis revealed a significant number of affected gene that regulate both clathrin- and caveolin- mediated endocytosis, which could lead to changes in endo- and transcytosis across the brain endothelium. These genes include FABP4, DYNAMIN, and FILAMIN that play a role in vesicle formation and scission. In addition, higher levels of transcytosis-related genes such as CAV1 was detected in HD iPSCs-derived BMECs that also displayed impaired Wnt/β-catenin signaling (Lim et al., 2017). The Wnt/β-catenin pathway is essential for regulation of cell proliferation, cell determination and tissue homeostasis (Silva-Garcia et al., 2019). Furthermore, astrocytes from both HD patients and mouse models were associated with higher levels of VEGF-A, which may trigger proliferation of ECs and contributes to neurovascular changes in HD (Hsiao et al., 2015). Of note, sustained delivery of VEGF into the rat striatum via injectable hydrogels was neuroprotective in a lesioned model of HD; VEGF implants significantly protected against the quinolinic acid-induced loss of striatal neurons (Emerich et al., 2010). Moreover, neuroprotection induced by inhibition of hypoxia inducible factor (HIF) prolyl-4-hydroxylases in HD mice has been correlated with enhanced VEGF expression (Niatsetskaya et al., 2010). In post-mortem tissue, cerebral blood vessel density was greater in HD patients while no differences in diameter of small- or medium sized blood vessels have been observed (Drouin-Ouellet et al., 2015). Post-mortem tissue of HD patients revealed a higher proportion of small compared to medium-sized blood vessels in the putamen, an effect occurring in parallel with putamen degeneration. Notably, altered density of small blood vessels in HD patients was consistent with the R6/2 mouse model when brain vascular anomalies were restricted to smaller vessels (Drouin-Ouellet et al., 2015; St-Amour et al., 2015).
Vascular Links to Parkinson’s Disease
PD is the second most common neurodegenerative disorder after AD (Antony et al., 2013). It is characterized by the progressive degeneration of the nigrostriatal system, resulting in rigidity, bradykinesia, postural instability, and resting tremor (Antony et al., 2013; Pagano et al., 2016). The most affected cells are dopaminergic neurons from the substantia nigra pars compacta (SNc). The pathological hallmark of PD is the formation of Lewy bodies containing aggregated α-synuclein (Hijaz and Volpicelli-Daley, 2020). While increasing age is a risk factor for PD, the average age of onset is after 60 years old (Hindle, 2010; Parkinson Canada, 2010). The etiology of PD is multifactorial where genetics (familial PD) and environmental (sporadic PD) factors take part in disease onset (Klein and Westenberger, 2012). Familial PD accounts for 10–15% of all PD cases whereas the remainder is classified as sporadic PD (Verstraeten et al., 2015). Genetically linked PD is inherited in an autosomal dominant or recessive fashion (Ball et al., 2019). Research has identified seven causal genes for familial PD including phosphatase and tensing homolog-induced Kinase-1 (PINK1), Parkinson protein 7 (PARK7), parkin RBR E3 ubiquitin protein ligase (PARK2), vacuolar protein sorting-associated protein 35 (VPS35), alpha-synuclein (SNCA), glucocerebrosidase (GBA) and leucine-rich repeat Kinase 2 (LRRK2) (Verstraeten et al., 2015; Kalinderi et al., 2016; Ball et al., 2019). Conversely, sporadic PD may develop from gene-environment interactions (Benmoyal-Segal and Soreq, 2006). Environmental factors associated with PD includes but are not limited to pesticides, heavy metals, and illicit drugs (Kwakye et al., 2017). Notably, individuals may respond differently to environmental factors which results in diverse symptomology of PD, thus adding to the complexity of the disease (Ball et al., 2019).
Altered Cerebral Blood Flow in Parkinson’s Disease
Using non-invasive MRI in an heterogeneous PD patient population, studies revealed decreased CBF in the frontal, parietal and occipital areas, more specifically the posterior parieto-occipital cortex, cuneus, middle frontal gyri, putamen, anterior cingulate and post- and pre-central gyri (Kamagata et al., 2011; Melzer et al., 2011; Fernandez-Seara et al., 2012; Madhyastha et al., 2015). A study by Fernandez-Seara et al. (2012) reported a 20–40% decrease in CBF in PD patients compared to a control group. Studies are trying to determine if CBF changes are related to the presence of dementia in PD, or if it can be considered as a biomarker. Derejko et al. (2006) used SPECT in PD patients with dementia and demonstrated left temporo-parietal hypoperfusion compared to the group without dementia. This suggested that CBF differences between PD patients with or without dementia could represent a clinical biomarker for discriminating PD patients (Derejko et al., 2006). Another study revealed hypoperfusion in PD patients without dementia in posterior cortical regions (posterior cingulate/precuneus) compared to healthy individuals (Syrimi et al., 2017). Hypoperfusion was positively correlated with global cognitive performance and the level of motor impairment (Madhyastha et al., 2015; Syrimi et al., 2017). Melzer et al. (2011) and Fernandez-Seara et al. (2012) reported CBF reduction with parietal cortex thinning in mild PD patients without dementia and proposed that CBF alterations occur in the early stages of PD.
Although studies have identified hypoperfusion in PD patients, the mechanisms underlying these changes are unknown (Biju et al., 2020). One study used a mouse model of PD (α-synuclein transgenic mice), which overexpress human WT α-synuclein. α-synuclein pathology develops before clinical symptoms and is present in both sporadic and familial forms. Using ASL-MRI analysis in this PD mouse model, authors reported a 36.6% reduction in cortical CBF in mutant mice accompanied by motor coordination impairments and olfactory bulb atrophy/dysfunction (Biju et al., 2020).
Altered Blood-Brain Barrier and Angiogenesis in Parkinson’s Disease
The association of PD with altered vascular function has led studies to investigate possible players contributing to BBB (Al-Bachari et al., 2020). In animal studies, BBB disruption in the SNc has been reported (Barcia et al., 2005; Rite et al., 2007; Chao et al., 2009). While human studies investigating BBB in PD patients are sparse, there is evidence of BBB dysfunction with increased permeability in the post commissural putamen of PD patients (Kortekaas et al., 2005; Gray and Woulfe, 2015). Wardlaw et al. (2008) and Al-Bachari et al. (2020) revealed increased leakage of the BBB in PD using ASL and dynamic contrast enhanced -MRI (DCE-MRI). Authors compared PD patients with two other control groups: one with and one without known cerebrovascular disease. This comparison could determine if BBB changes are attributable to co-existing cerebrovascular disease in an aging population, or if a pattern of BBB alteration is specific to PD. Authors reported increased BBB leakage in the group with cerebrovascular disease compared to the group without cerebrovascular disease in regions previously associated with PD, including the substantia nigra, white matter, and posterior cortical regions (Al-Bachari et al., 2020).
Accumulation of α-synuclein in ECs may also contribute to BBB dysfunction and increased permeability (Elabi et al., 2021). Higher number of EC nuclei was found in the SNc of PD patients (Faucheux et al., 1999). Other EC dysfunctions were reported, such as down regulation of tight junction proteins (Kuan et al., 2016). In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD, down-regulation of tight junction protein ZO-1 and BBB leakage were measured in the substantia nigra (Patel et al., 2011). There is also evidence of string vessel formation in brain capillaries from human PD. String vessels are described as collapsed basement membrane without endothelium and no circulatory function. An altered basement membrane was also observed in PD mice (Yang et al., 2015). VEGF, a prominent growth factor promoting angiogenesis and BBB permeability, was upregulated in the substantia nigra, but not the striatum, of PD patients, while animal models of PD displayed parkinsonian traits following administration of exogenous VEGF into the substantia nigra (Barcia et al., 2005; Wada et al., 2006; Rite et al., 2007).
Guan et al. (2013) reported vascular degeneration in human PD, with formation of endothelial clusters, capillary network damage, and loss of capillary connections in the substantia nigra and brain stem nuclei. Authors found a larger vessel size in PD patients, while capillaries were shorter in average length, less in number and had fewer branches. These observations were also confirmed in an MPTP mouse model of PD (Guan et al., 2013; Sarkar et al., 2014). Furthermore, ultrastructural abnormalities were identified in cerebro-cortical microvessels of PD patients, including basement membrane thickening, vacuolization and pericyte degradation (Farkas et al., 2000). Structural alterations of the basement membrane can lead to pathophysiological consequences including compromised nutrient transport and cognitive disturbances (Farkas et al., 2000). Recently, a PD mouse model of α-synuclein overexpression was associated with altered vascular density at different stages of the disease (Elabi et al., 2021). The study reported that 8 month-old animals had increased vessel density compared to control mice, while 13 month-old PD mice displayed decreased vessel density, suggesting compensatory angiogenesis in the younger group (Elabi et al., 2021). Increased angiogenesis is considered an adaptative response to pathological conditions and is regulated by basement membrane proteins and their integrin receptors. These studies postulate that immature nascent vessels in PD could contribute to increased BBB permeability, as reviewed recently (Bogale et al., 2021).
Vascular Links to Alzheimer’s Disease
AD accounts for 60–80% of all diagnoses of dementia (Alzheimer’s Association, 2021). This progressive and debilitating neurodegenerative disease manifests with memory, attention, executive, visuospatial and perceptual impairments. AD is not only characterized by amyloid deposition, neuroinflammation, neurodegeneration and cognitive deficits, but also by cerebrovascular pathology. Indeed, an inadequate brain perfusion has been identified as an early event in the development and progression of AD (Nicolakakis and Hamel, 2011). The risk of developing AD is increased by age-associated vascular diseases such as hypercholesterolemia, hypertension, ischemic stroke, and diabetes (Kalaria, 1996; Roher et al., 2003; Casserly and Topol, 2004; Gorelick, 2004; Luchsinger et al., 2005). The AD brain is characterized by increased levels of soluble and insoluble amyloid-beta peptide (Aβ), derived from the amyloid protein precursor (APP), neurofibrillary tangles of hyperphosphorylated tau protein, neurodegeneration and neuroinflammation, and also linked with a cerebrovascular pathology (Selkoe, 2002; Iadecola, 2004; Querfurth and LaFerla, 2010). The latter is identified post-mortem by Aβ deposition in brain vessels (cerebral amyloid angiopathy, CAA), Aβ-induced oxidative stress, and alterations of the vessel wall that included fibrosis and degeneration of ECs (Buee et al., 1994; Vinters et al., 1994; Zarow et al., 1997; Farkas and Luiten, 2001; Humpel and Marksteiner, 2005). Various mouse models of AD have been developed, most mimicking the overproduction of Aβ through transgene expression of mutated human APP (hAPP) combined or not with the amyloidogenic presenilin (PS1) or the pathologic tau (Mucke et al., 2000; Oddo et al., 2003; Gotz and Ittner, 2008). These models recapitulate AD’s cerebrovascular pathology in addition to the cognitive deficits, senile plaques, Aβ-induced oxidative stress, neuroinflammation, cholinergic denervation, synaptic failure, and cerebral hypometabolism (Hsia et al., 1999; Palop et al., 2003; Aucoin et al., 2005; Tong et al., 2005; Nicolakakis et al., 2008; Iturria-Medina et al., 2016; Love and Miners, 2016; Liu et al., 2018; Czako et al., 2020). It is in fact estimated that up to 45% of all dementias worldwide are partly, or wholly, due to age-related SVD of the brain (Montagne et al., 2016; Clancy et al., 2021). This suggests that AD and vascular dementia share common grounds, which complicates their stratification. As such, it is of utmost importance to improve our understanding of vascular underpinnings of AD (Willis and Hakim, 2013). Clinical studies that attempted to reduce plaque load by blocking Aβ production, removing Aβ with antibodies, or preventing tau phosphorylation, have all failed to alleviate AD symptoms (Korte et al., 2020). However, mounting evidence demonstrates that the brain vasculature is the missing link (Sweeney et al., 2019). Early cerebrovascular dysfunction in AD leads to decreased Aβ clearance, vascular oxidative stress, inflammatory damage and impaired BBB function (Zlokovic, 2005). Here below we will succinctly describe vascular underpinnings of AD, from alterations in CBF to BBB dysfunction, topics that have been extensively reviewed elsewhere (Bell and Zlokovic, 2009; Zlokovic, 2011; Hamel, 2015; Hays et al., 2016; Nelson et al., 2016; Kisler et al., 2017; Korte et al., 2020; Solis et al., 2020; Soto-Rojas et al., 2021).
Cerebral Blood Flow Alterations in Alzheimer’s Disease
Numerous investigations on individuals diagnosed with AD observed reduced CBF (Prohovnik et al., 1988; Montaldi et al., 1990; Bressi et al., 1992; O’Brien et al., 1992; Smith et al., 1992; Minoshima et al., 1997; Mattsson et al., 2014; Mielke et al., 2014; Smith and Verkman, 2018). CBF decline can be detected prior to cognitive decline, but also before plaque deposition. The accumulation of soluble Aβ prior to plaque deposition has early pathogenic consequences in AD (Suo et al., 1998). Studies have demonstrated increased levels of soluble amyloid species including Aβ1–40 and Aβ1–42 in AD cases compared to age-matched controls (Suo et al., 1998; Smith and Greenberg, 2009). Both soluble Aβ1–40 and Aβ1–42 have been associated to abnormal vascular reactivity in the absence of plaque deposition or vessel wall dysfunction (Smith and Greenberg, 2009; Dietrich et al., 2010). In particular, studies have revealed that application of exogenous Aβ1–40 to mouse neocortex in vivo, or to healthy bovine blood vessels ex vivo, leads to endothelium-dependent vasoconstriction (Thomas et al., 1996; Niwa et al., 2000). In addition, increased levels of soluble amyloid species (Aβ1–40 and Aβ1–42) are associated with significantly reduced CBF, increased cerebral vascular resistance, decrease myogenic and vasodilator responses (Suo et al., 1998; Dietrich et al., 2010), where Aβ1–42 is equally potent to Aβ1–40 except at a higher concentration (Dietrich et al., 2010). Soluble Aβ impacts vascular function through increased production of reactive oxygen species (ROS). The reaction of ROS superoxide and excess NO produces peroxynitrite. Peroxynitrite is commonly known as a toxic oxidant which contributes to endothelial dysfunction, a mechanism relevant to AD but also to other neuroinflammatory and metabolic conditions (Beckman et al., 1990; Paris et al., 1998; Tan et al., 2004; Dietrich et al., 2010; Kelleher and Soiza, 2013; Salisbury and Bronas, 2015; Incalza et al., 2018). Both Aβ1–40 and Aβ1–42 have been shown to acutely increase ROS production in cultured rat cerebral microvascular endothelial and smooth muscle cells in a dose dependent manner (Dietrich et al., 2010). Interestingly, this response was inhibited by the ROS scavenger MnTBAP (Dietrich et al., 2010). Notably, Aβ1–40 is the predominant isoform found in cerebral vessel walls and is commonly associated with vascular deposits in CAA, which will be discussed later, while Aβ1–42 is the major isoform deposited in senile plaques (Suo et al., 1998). Although this concept is still controversial, it is thought that Aβ1–42 acts as a “seed” which initiates the formation of vascular Aβ deposit in CAA (McGowan et al., 2005; Gireud-Goss et al., 2020).
Following Aβ deposition, reduction of CBF was found in the frontal, parietal and temporal cortices from individuals carrying Apolipoprotein E4 (APOE4) gene, most prevalent genetic risk factor for AD (Thambisetty et al., 2010; Michels et al., 2016). In addition, ApoE4 allele carriers displayed early impairments in cerebrovascular reactivity to a memory task (Suri et al., 2015). BOLD-fMRI, which uses blood flow changes as a surrogate to neuronal activity, detected decreased activation in areas engaged during naming and fluency tasks in AD patients compared to individuals with no risk factors (Smith et al., 1999). Decreased BOLD-fMRI responses to different cognitive tasks in early stage of AD are region-specific (Kisler et al., 2017). Most studies investigating perfusion in AD reported either CBF or CBV alterations. However, CBF alterations appear before CBV deficits during AD progression (Lacalle-Aurioles et al., 2014).
Decreased CBF is associated with poor cognitive function, and evidence suggested that lower CBF is linked with faster cognitive decline in patients with AD (Benedictus et al., 2017). Zheng et al. (2019) investigated rCBF, functional activity and connectivity in AD by combining resting-state BOLD fMRI and ASL techniques. ASL revealed decreased rCBF in AD patients in the left posterior cingulate cortex, bilateral dorsolateral prefrontal cortex, left interior parietal lobule, right middle temporal gyrus, left middle occipital gyrus and left precuneus. In addition, they revealed decreased connectivity between regions in AD patients, which was associated with impaired cognitive performances (Alsop et al., 2000; Zheng et al., 2019). Brain regions affected by a reduction of CBF in AD patients (parietal, frontal, temporal and occipital cortices) are associated with cognitive impairment in all domains (language, global cognition, memory, attention, executive functioning and visuospatial functioning) (Leeuwis et al., 2017).
Blood flow reductions have also been identified in early preclinical AD, before Aβ plaque deposition (Nicolakakis and Hamel, 2011; Iturria-Medina et al., 2016; Szu and Obenaus, 2021). Early reduction of CBF has been reported in mouse models of AD, such as mice overexpressing mutant forms of APP (Niwa et al., 2002; Ongali et al., 2010; Lacoste et al., 2013) and in mice expressing the ApoE4 gene allele (Lin et al., 2017). In some brain areas, CBF reduction can reach over 50%. This CBF reduction has been associated with cognitive changes in mice, including a loss of ability to sustain attention (Marshall et al., 2001). Both ApoE4 transgenic and APP/PS1 mice revealed CBF reduction prior to neuronal and synaptic dysfunctions (Guo et al., 2019; Montagne et al., 2021).
While decreased CBF in AD is widely accepted, studies are only starting to identify underlying mechanisms, for example the involvement of pericytes. Pericytes have been linked to hypoperfusion and increased capillary constriction in AD (Bell et al., 2010; Korte et al., 2020). Pericyte-deficient transgenic mice with no Aβ pathology develop early CBF reduction in the gray matter, even with normal neuronal activity, endothelial-dependent vasodilation, astrocyte number and blood vessels coverage (Bell et al., 2010; Kisler et al., 2017). As these pericyte-deficient mice age, neuronal dysfunction and degeneration start to emerge. Another underlying mechanism was reported by Cruz Hernandez et al. (2019), demonstrating that capillaries become blocked by neutrophils, while another study revealed increased formation of occlusive thrombi in AD mice (Cortes-Canteli et al., 2019). Inhibiting neutrophils adhesion using an antibody against neutrophil-specific protein Ly6G in the APP/PSI mouse model led to rapid improvements in CBF (Cruz Hernandez et al., 2019). In a follow-up study, the same group assessed the impact of one treatment of anti-Ly6G on short-term memory function and reported increased CBF by 17% in 21–22 months old APP/PSI mice. Furthermore, they suggested that increased CBF improved cognition into late stages of AD mice (Bracko et al., 2020). Reduced neurovascular coupling and cerebrovascular reactivity have also been reported in AD mice (Girouard and Iadecola, 2006; Tong et al., 2019). Recently, impaired capillary endothelial inward rectifying Kir2.1 channel, playing a role in mediating blood delivery, has been associated with AD (Mughal et al., 2021). In a model of familial AD (5xFAD) where Kir2.1 channel function is impaired, systemic administration of the co-factor phosphatidylinositol 4,5-bisphosphate (PIP2), required for Kir2.1 activity, led to increased CBF and functional neurovascular coupling in 5xFAD mice (Mughal et al., 2021).
AD patients are often (80–90%) diagnosed with CAA, a vessel disorder (Gireud-Goss et al., 2020) and an important risk factor for intracerebral hemorrhage and cognitive impairment (Reijmer et al., 2016). CAA consist of vascular amyloid deposits similar to senile plaques in AD (Kumar-Singh et al., 2005). Neuropathological studies have revealed that CAA affects the outer leptomeningeal vessels on the surface of the brain as well as distal intraparenchymal arteries, arterioles, and capillaries (Gireud-Goss et al., 2020; Howe et al., 2020). APP23 mouse model and human AD brain revealed an association between CAA-related capillary occlusion with CBF disturbances, hypoperfusion, detected by magnetic resonance angiopathy (MRA), which could explain in part the changes in CBF measured in AD patients (Thal et al., 2009; Milner et al., 2014). As in AD, patients with CAA have been linked to altered hemodynamics during visual stimulation as evidenced by reduced amplitude of BOLD response (Smith et al., 2008; Dumas et al., 2012; Switzer et al., 2020).
Altered Blood-Brain Barrier and Angiogenesis in Alzheimer’s Disease
Early signs of BBB leakage in AD have been detected before dementia onset (Montagne et al., 2016). Neuroimaging techniques have evidenced BBB breakdown in AD in gray and white matter brain regions (Montagne et al., 2016; van de Haar et al., 2016). Aβ and tau pathologies contribute to increased BBB permeability in AD patients and mouse models (Park et al., 2011; Sagare et al., 2013; Alata et al., 2015). Several players involved in Aβ clearance, and closely related to the BBB, are reduced in AD patients, including phosphatidylinositol-binding clathrin assembly protein (PICALM, allows for Aβ exocytosis across the luminal part of the BBB), P-glycoprotein (expressed on both sided of the BBB) and glucose transporter (GLUT)1 (Mooradian et al., 1997; Chiu et al., 2015; Zhao et al., 2015). AD brain microvessel show diminished expression of LRP1, a major Aβ clearance receptor at the BBB (Deane et al., 2004; Donahue et al., 2006). LRP1 is an ApoE receptor and is expressed at the abluminal side of brain ECs and mediates the internalization of soluble Aβ (Deane et al., 2004). Endothelium-specific deletion of LRP1 leads to the acceleration of Aβ pathology in APP-overexpressing APPsw/0 mice (Storck et al., 2016). Moreover, studies have demonstrated low levels of GLUT1 in AD brain endothelium, which alters glucose transport (Kalaria and Harik, 1989; Simpson et al., 1994).
Several features lead to increased BBB permeability in AD, including reduced expression of tight junctions, perivascular accumulation of blood-derived products, degeneration of pericytes and ECs, as well as infiltration of circulating leukocytes (Sweeney et al., 2018; Huang et al., 2020). It was demonstrated that Aβ disrupts tight junctions and increases vascular permeability by suppressing expression of ZO-1, claudin-5 and occludin while increasing expression of MMP-2 and MMP-9 (Kook et al., 2012; Blair et al., 2015; Wan et al., 2015; Huang et al., 2020). Isolated rat cerebral cortical ECs treated with Aβ1–42 displayed decreased expression of occludin and redistribution of claudin-5 and ZO-2 in the cytoplasm while in untreated cells, both claudin-5 and ZO-2 were distributed along the plasma membrane at cell-cell contacts (Marco and Skaper, 2006). In addition, studies have reported leakage of blood-derived proteins (fibrinogen, thrombin, albumin, and IgG) around capillaries from post-mortem brain tissue in the prefrontal and entorhinal cortex as well as in hippocampus of AD patients (Ryu and McLarnon, 2009; Hultman et al., 2013; Sengillo et al., 2013). Furthermore, animal studies revealed that lacking pericyte-derived soluble factors, required for a healthy endothelium, can contribute to endothelial degeneration in AD (Bell et al., 2010). Finally, mouse models of AD have demonstrated that pericyte reduction is associated with BBB dysfunction as well as accelerated buildup of Aβ and tau pathology (Sagare et al., 2013). In human studies, there is also evidence of pericyte loss in the hippocampus and cortex of AD patients due in part to prolonged exposure to Aβ peptides (Sagare et al., 2013; Sengillo et al., 2013; Huang et al., 2020). Of note, pericytes play a role in Aβ clearance by internalizing different Aβ peptides using the LRP1 pathway (Sagare et al., 2013).
Evidence of reduced capillary length and basement membrane changes in AD patients have been reported (Salloway et al., 2002; Sengillo et al., 2013; Halliday et al., 2016). It was shown that AD patients display abnormal angiogenesis due to low expression of MEOX2, a regulator of vascular differentiation, as well as premature pruning of capillary networks resulting in reductions of CBF (Wu et al., 2005; Grammas, 2011). Endothelial degeneration including reduction of EC thickness, length and density of blood vessels were reported in brain tissue from AD patients (Sweeney et al., 2018). An increase of pro-angiogenic factors in the AD brain, without the increase in vasculature, was also reported (Grammas, 2011). Notably, the increased Aβ species and plaques in AD have anti-angiogenic effects (Parodi-Rullan et al., 2020), and impaired angiogenesis was identified in transgenic AD mice (Grammas, 2011). Emerging evidence suggest that dysfunction of the VEGF-A/VEGFR2 pathway may play an aggravating role in neurodegeneration and AD. For instance, sustained brain delivery of VEGF via injectable hydrogels was protective against quinolinic acid-induced neurodegeneration (Emerich et al., 2010), and low VEGF levels have been associated to another debilitating neurological disorder, spinocerebellar ataxia type 1 (Cvetanovic et al., 2011). Aβ acts as an antagonist of VEGF signaling via sequestration of VEGF-A in senile plaques, and also via inhibition of VEGFR2 tyrosine phosphorylation (Patel et al., 2010). Moreover, implantation of VEGF secreting microcapsules on the cerebral cortex of APP/PS1 mice attenuated both brain Aβ burden and cognitive impairments (Spuch et al., 2010). Whether impaired neural perfusion and increased neurotoxicity in AD correlate to a loss of VEGF function, and whether VEGF overexpression is neuroprotective in transgenic AD mice remains to be explored.
CAA is associated with increased BBB permeability and arterial stiffness (Magaki et al., 2018; Gireud-Goss et al., 2020). Aβ deposition in CAA has been found to occur on the cerebrovascular basement membrane of arteries, arterioles and on the basal lamina of capillaries as shown by electron microscopy (Gireud-Goss et al., 2020). Moreover, ultrastructural studies of CAA demonstrated a thinned endothelium, shrinkage and degeneration of ECs, as well as vessel occlusion, all of which can lead to CBF disturbances and microinfarcts (Attems and Jellinger, 2004; Thal et al., 2009; Magaki et al., 2018). Tight junction proteins in CAA-laden vessels are found decreased (Tai et al., 2010). After exposure to exogenous Aβ, human ECs showed decreased expression of occludin, while post-mortem brain tissue of CAA patients revealed decreased expression of claudin-5, ZO-1, CD31 and basement protein collagen IV (Tai et al., 2010; Carrano et al., 2011; Magaki et al., 2018). In addition, CAA patients displayed increased expression of MMP-2 and MMP-9, which may lead to basement membrane degradation and increased BBB permeability (Carrano et al., 2011). In the Tg2576 mouse model of CAA, BBB integrity was compromised due to decreased expression of claudin-5 and claudin-1 (Carrano et al., 2011). Moreover, TgSwDI mice, another model of CAA, revealed spontaneous hemorrhage and loss of BBB integrity (Davis et al., 2004). Soluble Aβ1–40, predominant amyloid isoform in vessel walls, also leads to tight junction redistribution at the BBB and decreased transendothelial electric resistance (Hartz et al., 2012; Gireud-Goss et al., 2020). Understanding the impact of Aβ in CAA and AD is essential for slowing cerebrovascular disease progression.
Additional Remarks: Vascular Deficits in Down Syndrome, Traumatic Brain Injury and Depression
In addition to neurodevelopmental disorders discussed earlier in this review, Down syndrome (DS), which results from trisomy of human chromosome 21, is a cause of early onset Alzheimer’s disease-dementia (AD-DS) (Ballard et al., 2016; Tosh et al., 2021). Two-thirds of individuals with DS will develop dementia by the age of 65 (Tosh et al., 2021). The onset of AD in DS patients parallels the development of the classic brain pathological lesions seen in AD patients without DS (Salehi et al., 2016). DS and AD disorders have genetic similarities, as individuals with DS possess a triplication of the gene encoding APP, while patients with familial AD have an extra copy of the APP gene (Salehi et al., 2016). In rodent studies of DS-AD, triplication of chromosome 21 genes other than APP demonstrated increased Aβ aggregation deposition and cognitive deficits (Wiseman et al., 2018). A recent study, focused on a model of DS comprising of a mutation in a Down syndrome critical region (Hsa21) on chromosome 21 encompassing 21q21–21q22.3 (Li et al., 2016; Tosh et al., 2021). This study crossed an Hsa21 mouse model of DS with partial trisomies other than APP with a transgenic APP mouse model and revealed that an additional copy of genes of the Hsa21 region modulates APP/Aβ biology, including Aβ aggregation and mortality (Tosh et al., 2021). Despite striking similarities between AD and DS in terms of genetics and symptoms onset, neurovascular impairments in DS have been largely overlooked. As such, studies aimed at elucidating vascular abnormalities in DS represent an unmet clinical need.
Early vascular insults following a traumatic brain injury (TBI) can also increase the risk of late-onset neurological diseases (Brett et al., 2021). TBI is a significant public health problem associated with long-term disabilities. Early chronic TBI may lead to secondary injury with pathophysiological changes similar to those observed in neurodegenerative diseases (Impellizzeri et al., 2016). For instance, neuroinflammation plays a fundamental role in TBI, including reactive microglia and astrocytes, as well as release of pro-inflammatory cytokines and chemokines that may hinder the brain’s ability to repair itself and lead to neurodegeneration following prolonged activation of these processes (Impellizzeri et al., 2016; Brett et al., 2021). Severe or repeated mild TBI can initiate long-term neurodegeneration with signs of AD (Mendez, 2017). For example, various contact-sport players developed TBI-associated dementia or parkinsonism years after retiring. TBI can induce acute BBB disruption through vascular shear stress, hemorrhages, edema, alterations in CBF and chronic inflammation, which is known to contribute to Aβ deposition and tau pathology (Iadecola, 2013; De Silva and Faraci, 2016). Autopsies of TBI patients show diffuse Aβ plaques similar to those identified in AD, as reviewed by Perry et al. (2016). The formation of Aβ in perivascular spaces following TBI may lead to an injury cascade consisting of cerebrovascular damage, oxidative stress and ECs dysfunction (Ramos-Cejudo et al., 2018). Interestingly, alterations in EC survival, BBB integrity and neuroinflammation are considered early events after TBI, all of which are characteristic of cerebrovascular damage involved in the progression of AD and impairment of Aβ clearance. Thus, these early vascular impairments promote the onset of neurodegenerative diseases (Ramos-Cejudo et al., 2018). Considering early vascular injuries in TBI, biomarker studies are integrating a variety of neuroimaging and molecular techniques to better understand the incidence of cerebrovascular dysfunction and the onset of neurodegenerative diseases, and therapeutic investigations have looked at ways to improve cerebrovascular function (Graham and Sharp, 2019; Martinez and Stabenfeldt, 2019).
One of the leading causes of mental illness worldwide, depression, has a tremendous impact on psychosocial behaviors and vascular health (Knight and Baune, 2017; Menard et al., 2017). Chronic stress is the primary environmental risk factor for depression. The nucleus accumbens (NAc) is one of the main players in regulating stress response (Russo and Nestler, 2013). Menard et al. (2017) have demonstrated that chronic social stress induces BBB leakiness in the NAc of mice, which leads to circulating proinflammatory mediators and depression-like behaviors such as helplessness, social avoidance and anhedonia. As seen in neurodevelopmental and neurodegenerative disorders, the increase in BBB permeability in the rodent model of chronic social stress was facilitated by the loss of tight junction protein claudin-5 (Menard et al., 2017). Furthermore, stress-induced BBB permeability has been linked to inflammation of the endothelium and up-regulation of an epigenetic repressor, hdac1, which is involved in reducing claudin-5 expression and loosening of tight junctions (Dudek et al., 2020). Consequently, these studies are highlighting mechanisms by which chronic stress impacts vascular health, which could have long-term consequences on brain maturation and aging.
The vascular system, as any other system, undergoes aging. It has been hypothesized that vascular aging leads to a progressive functional deterioration (Grunewald et al., 2021). During aging, the brain vasculature undergoes several changes including decreased capillary density, attenuation of neovascularization potential, increased BBB permeability and decreased CBF as reviewed in Watanabe et al. (2020) and Banks et al. (2021). A suggested mechanism of typical vascular aging consist of the inability of VEGF to replenish vessel loss. The mechanisms by which VEGF is involved in vascular aging are unknown. However, mice treated with VEGF have been shown to live longer, with extended multiorgan functionality (Grunewald et al., 2021). Furthermore, aging is associated with several vascular changes including aortic stiffness which has been linked to reduced blood flow in tissues leading to increased neuroinflammation and neurodegeneration later in life (Moore et al., 2021). Therefore, age-related changes in key vascular features may predispose to age-associated diseases (Banks et al., 2021). Improving early pathological conditions by protecting the brain vasculature is essential in preventing or modulating disease progression.
Conclusion
Vascular risk factors and co-morbidities take part in disease onset and/or exacerbate disease progression (Sweeney et al., 2018; Clancy et al., 2021). When it comes to alterations in CBF, BBB, and vascular patterning, neurodevelopmental and neurodegenerative disorders share interesting similarities (Table 1). While these disorders are siloed, mainly due to the age of onset, the commonalities in vascular alterations force to question the implication of early life vascular impairments on the expression of age-related neurodegenerative diseases. The vascular implications in middle-aged autistic adults have been largely overlooked, 10% of individuals diagnosed with ASD age between 40 and 60 years old will develop dementia, including AD within 15 years (Plana-Ripoll et al., 2019). In addition, there is a high frequency of parkinsonism among older ASD patients (Starkstein et al., 2015). The impact of altered brain perfusion and BBB integrity in ASD may contribute to the onset of neurodegenerative diseases due to the continuous vascular impairments associated with these diseases. Likewise, schizophrenia is associated with an elevated risk for developing Alzheimer’s and Parkinson’s diseases as they share core features including white matter abnormalities and cognitive deficits (Ribe et al., 2015; Kochunov et al., 2021; Kuusimaki et al., 2021).
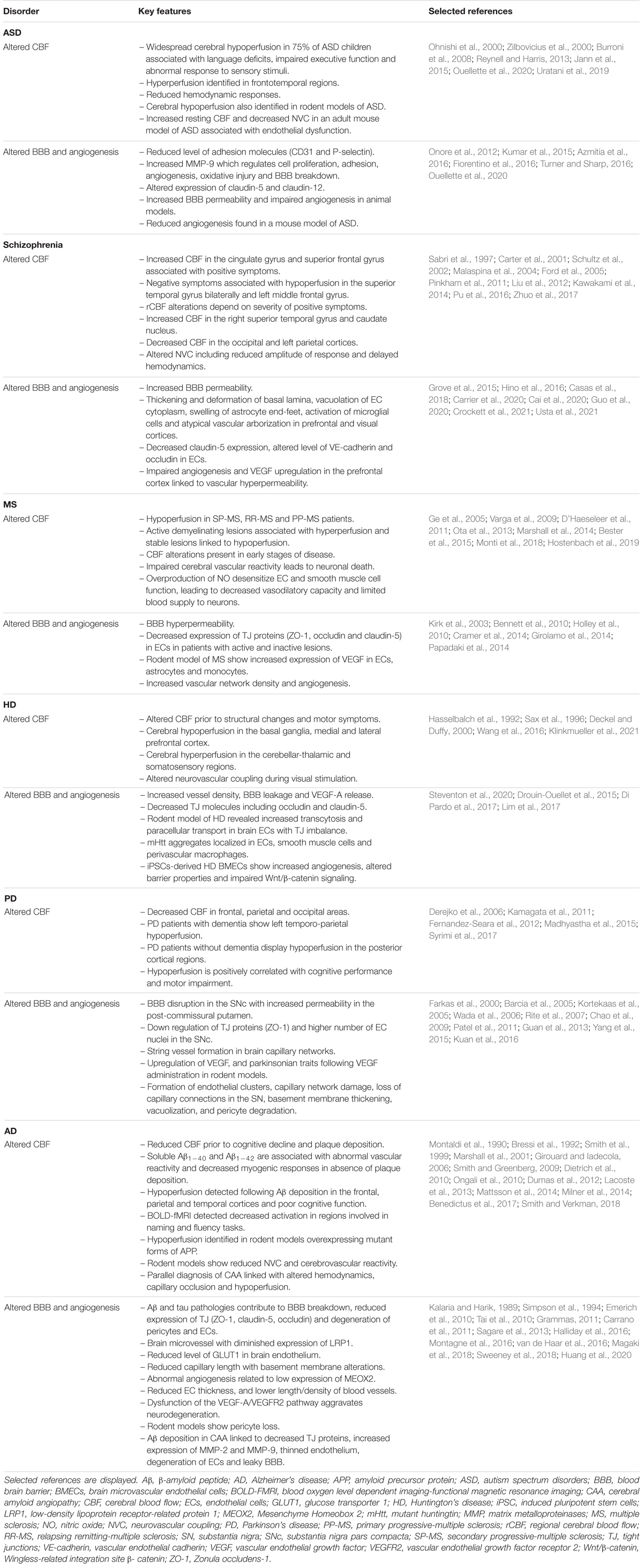
Table 1. Major altered features associated with CBF, BBB, and angiogenesis in neurodevelopmental and neurodegenerative disorders.
Since fast-growing evidence demonstrates the role of early vascular impairments in the onset and/or progression of numerous neurological conditions, more work is needed to identify therapeutic targets to promote healthy cerebrovascular maturation and aging, as well as hinder the progression of age-related dementia and neurodegeneration. This is primordial considering recent findings that ECs show limited turnover compared to other cells in the human body (Sender and Milo, 2021). For instance, it was estimated that the turnover rate of ECs is 0.1% per day, as opposed to much higher rates for erythrocytes (65%), neutrophils (18%) or gastrointestinal epithelial cells (12%). In addition, the turnover rates of cellular mass in the human body were estimated at 0.4% for ECs, 4% for skin cells and adipocytes, and 42% for gastrointestinal epithelial cells (Sender and Milo, 2021). Hence, as ECs are long-lived, they may carry on early structural and functional impairments into adulthood and throughout aging, altering organ function in the long term. This concept emphasizes the importance of infant screening for cerebrovascular abnormalities, and of continuous management of vascular risk factors during lifespan. As such, the vascular continuum between neurodevelopmental and neurodegenerative disease should represent a growing focus in modern neuroscience (Figure 3).
Author Contributions
JO wrote the draft following BL’s instructions. BL chose the theme and edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This publication was possible thanks to funding by the Canadian Institutes for Health Research (grant #388805) to BL and a Canadian Vascular Network scholarship to JO.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbott, N. J., Ronnback, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1038/nrn1824
Abookasis, D., Lerman, D., Roth, H., Tfilin, M., and Turgeman, G. (2018). Optically derived metabolic and hemodynamic parameters predict hippocampal neurogenesis in the BTBR mouse model of autism. J. Biophotonics 11:e201600322. doi: 10.1002/jbio.201600322
Adhya, S., Johnson, G., Herbet, J., Jaggi, H., Babb, J. S., Grossman, R. I., et al. (2006). Pattern of hemodynamic impairment in multiple sclerosis: dynamic susceptibility contrast perfusion MR imaging at 3.0 T. Neuroimage 33, 1029–1035. doi: 10.1016/j.neuroimage.2006.08.008
Al-Bachari, S., Naish, J. H., Parker, G. J. M., Emsley, H. C. A., and Parkes, L. M. (2020). Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 11:593026. doi: 10.3389/fphys.2020.593026
Alata, W., Ye, Y., St-Amour, I., Vandal, M., and Calon, F. (2015). Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab. 35, 86–94. doi: 10.1038/jcbfm.2014.172
Alsop, D. C., Detre, J. A., and Grossman, M. (2000). Assessment of cerebral blood flow in Alzheimer’s disease by spin-labeled magnetic resonance imaging. Ann. Neurol. 47, 93–100.
Alzheimer’s Association (2021). 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 17, 327–406. doi: 10.1002/alz.12328
Andreasen, N. C., O’Leary, D. S., Flaum, M., Nopoulos, P., Watkins, G. L., Boles Ponto, L. L., et al. (1997). Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naïve patients. Lancet 349, 1730–1734. doi: 10.1016/s0140-6736(96)08258-x
Andreone, B. J., Lacoste, B., and Gu, C. (2015). Neuronal and vascular interactions. Annu. Rev. Neurosci. 38, 25–46. doi: 10.1146/annurev-neuro-071714-033835
Antony, P. M., Diederich, N. J., Kruger, R., and Balling, R. (2013). The hallmarks of Parkinson’s disease. FEBS J. 280, 5981–5993. doi: 10.1111/febs.12335
Attems, J., and Jellinger, K. A. (2004). Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology–a pilot study. Acta Neuropathol. 107, 83–90. doi: 10.1007/s00401-003-0796-9
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Attwell, D., Mishra, A., Hall, C. N., O’Farrell, F. M., and Dalkara, T. (2016). What is a pericyte? J. Cereb. Blood Flow Metab. 36, 451–455. doi: 10.1177/0271678X15610340
Attwell, D. A., and Laughlin, S. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145.
Aucoin, J. S., Jiang, P., Aznavour, N., Tong, X. K., Buttini, M., Descarries, L., et al. (2005). Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer’s disease. Neuroscience 132, 73–86. doi: 10.1016/j.neuroscience.2004.11.047
Azmitia, E. C., Saccomano, Z. T., Alzoobaee, M. F., Boldrini, M., and Whitaker-Azmitia, P. M. (2016). Persistent angiogenesis in the autism brain: an immunocytochemical study of postmortem cortex, brainstem and cerebellum. J. Autism Dev. Disord. 46, 1307–1318. doi: 10.1007/s10803-015-2672-6
Ball, N., Teo, W. P., Chandra, S., and Chapman, J. (2019). Parkinson’s disease and the environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Ballard, C., Mobley, W., Hardy, J., Williams, G., and Corbett, A. (2016). Dementia in Down’s syndrome. Lancet Neurol. 15, 622–636. doi: 10.1016/s1474-4422(16)00063-6
Banks, W. A., Reed, M. J., Logsdon, A. F., Rhea, E. M., and Erickson, M. A. (2021). Healthy aging and the blood-brain barrier. Nat. Aging 1, 243–254. doi: 10.1038/s43587-021-00043-5
Bano, D., Zanetti, F., Mende, Y., and Nicotera, P. (2011). Neurodegenerative processes in Huntington’s disease [Review]. Cell Death Dis. 2:e228. doi: 10.1038/cddis.2011.112
Barcia, C., Bautista, V., Sanchez-Bahillo, A., Fernandez-Villalba, E., Faucheux, B., Poza y Poza, M., et al. (2005). Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J Neural Transm. (Vienna) 112, 1237–1248. doi: 10.1007/s00702-004-0256-2
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624. doi: 10.1073/pnas.87.4.1620
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113. doi: 10.1007/s00401-009-0522-3
Benedictus, M. R., Leeuwis, A. E., Binnewijzend, M. A., Kuijer, J. P., Scheltens, P., Barkhof, F., et al. (2017). Lower cerebral blood flow is associated with faster cognitive decline in Alzheimer’s disease. Eur. Radiol. 27, 1169–1175. doi: 10.1007/s00330-016-4450-z
Benmoyal-Segal, L., and Soreq, H. (2006). Gene-environment interactions in sporadic Parkinson’s disease. J. Neurochem. 97, 1740–1755. doi: 10.1111/j.1471-4159.2006.03937.x
Bennett, J., Basivireddy, J., Kollar, A., Biron, K. E., Reickmann, P., Jefferies, W. A., et al. (2010). Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J. Neuroimmunol. 229, 180–191. doi: 10.1016/j.jneuroim.2010.08.011
Bester, M., Forkert, N. D., Stellmann, J. P., Sturner, K., Aly, L., Drabik, A., et al. (2015). Increased perfusion in normal appearing white matter in high inflammatory multiple sclerosis patients. PLoS One 10:e0119356. doi: 10.1371/journal.pone.0119356
Biju, K. C., Shen, Q., Hernandez, E. T., Mader, M. J., and Clark, R. A. (2020). Reduced cerebral blood flow in an alpha-synuclein transgenic mouse model of Parkinson’s disease. J. Cereb. Blood Flow Metab. 40, 2441–2453. doi: 10.1177/0271678X19895432
Biswas, S., Cottarelli, A., and Agalliu, D. (2020). Neuronal and glial regulation of CNS angiogenesis and barriergenesis. Development 147:dev182279. doi: 10.1242/dev.182279
Bjørklund, G., Kern, J. K., Urbina, M. A., Saad, K., El-Houfey, A. A., Geier, D. A., et al. (2018). Cerebral hypoperfusion in autism spectrum disorder. Acta Neurobiol. Exp. 78, 21–29. doi: 10.21307/ane-2018-005
Blair, L. J., Frauen, H. D., Zhang, B., Nordhues, B. A., Bijan, S., Lin, Y. C., et al. (2015). Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 3:8. doi: 10.1186/s40478-015-0186-2
Bogale, T. A., Faustini, G., Longhena, F., Mitola, S., Pizzi, M., and Bellucci, A. (2021). Alpha-Synuclein in the regulation of brain endothelial and perivascular cells: gaps and future perspectives. Front. Immunol. 12:611761. doi: 10.3389/fimmu.2021.611761
Bracko, O., Njiru, B. N., Swallow, M., Ali, M., Haft-Javaherian, M., and Schaffer, C. B. (2020). Increasing cerebral blood flow improves cognition into late stages in Alzheimer’s disease mice. J. Cereb. Blood Flow Metab. 40, 1441–1452. doi: 10.1177/0271678X19873658
Bressi, S., Volontè, M. A., Alberoni, M., Canal, N., and Franceschi, M. (1992). Transcranial doppler sonography in the early phase of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 3, 25–31. doi: 10.1159/000106990
Brett, B. L., Gardner, R. C., Godbout, J., Dams-O’Connor, K., and Keene, C. D. (2021). Traumatic brain injury and risk of neurodegenerative disorder. Biol. Psychiatry (in press). doi: 10.1016/j.biopsych.2021.05.025
Buee, L., Hof, P. R., Bouras, C., Delacourte, A., Perl, D. P., Morrison, J. H., et al. (1994). Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Acta Neuropathol. 87, 469–480.
Burghardt, K., Grove, T., and Ellingrod, V. (2014). Endothelial nitric oxide synthetase genetic variants, metabolic syndrome and endothelial function in schizophrenia. J. Psychopharmacol. 28, 349–356. doi: 10.1177/0269881113516200
Burroni, L., Orsi, A., Monti, L., Hayek, Y., Rocchi, R., and Vattimo, A. (2008). Regional cerebral blood flow in childhood autism: a SPET study with SPM evaluation. Nucl. Med. Commun. 29, 150–156. doi: 10.1097/MNM.0b013e3282f1bb8e
Cai, H. Q., Catts, V. S., Webster, M. J., Galletly, C., Liu, D., O’Donnell, M., et al. (2020). Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol. Psychiatry 25, 761–775. doi: 10.1038/s41380-018-0235-x
Carmeliet, P., and Jain, R. K. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nat. Review 473, 298–307. doi: 10.1038/nature10144REVIEWDLL4
Carrano, A., Hoozemans, J., van der Vies, S., Rozemuller, A., van Horssen, J., and de Vries, H. E. (2011). Amyloid beta induces oxidative stress-mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 15, 1167–1178. doi: 10.1089/ars.2011.3895
Carrier, M., Guilbert, J., Levesque, J. P., Tremblay, M. E., and Desjardins, M. (2020). Structural and functional features of developing brain capillaries, and their alteration in schizophrenia. Front. Cell. Neurosci. 14:595002. doi: 10.3389/fncel.2020.595002
Carter, C. S., MacDonald, A. W., Ross, L. L., and Stenger, V. A. (2001). Anterior cingulate cortex activity and impaired self-monitoring of performance in patients with schizophrenia: an event-related fMRI study. Am. J. Psychiatry 158, 1423–1428. doi: 10.1176/appi.ajp.158.9.1423
Casas, B. S., Vitoria, G., do Costa, M. N., Madeiro da Costa, R., Trindade, P., Maciel, R., et al. (2018). hiPSC-derived neural stem cells from patients with schizophrenia induce an impaired angiogenesis. Transl. Psychiatry 8:48. doi: 10.1038/s41398-018-0095-9
Casserly, I., and Topol, E. (2004). Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins [Review]. Lancet 363, 1139–1146. doi: 10.1016/S0140-6736(04)15900-X
Cauli, B., and Hamel, E. (2010). Revisiting the role of neurons in neurovascular coupling. Front. Neuroenergetics 2:9. doi: 10.3389/fnene.2010.00009
Chao, Y.-X., He, B.-P., and Wah Tay, S. S. (2009). Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson’s disease. J. Neuroimmunol. 216, 39–50. doi: 10.1016/j.jneuroim.2009.09.003
Chiron, C., Leboyer, M., Leon, F., Jambaque, L., Nuttin, C., and Syrota, A. (1995). SPECT of the Brain in childhood autism: evidence for a lack of normal hemispheric asymmetry. Dev. Med. Child Neurol. 37, 849–860. doi: 10.1111/j.1469-8749.1995.tb11938.x
Chiu, C., Miller, M. C., Monahan, R., Osgood, D. P., Stopa, E. G., and Silverberg, G. D. (2015). P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: preliminary observations. Neurobiol. Aging 36, 2475–2482. doi: 10.1016/j.neurobiolaging.2015.05.020
Chow, B. W., and Gu, C. (2015). The molecular constituents of the blood-brain barrier. Trends Neurosci. 38, 598–608. doi: 10.1016/j.tins.2015.08.003
Clancy, U., Gilmartin, D., Jochems, A. C. C., Knox, L., Doubal, F. N., and Wardlaw, J. M. (2021). Neuropsychiatric symptoms associated with cerebral small vessel disease: a systematic review and meta-analysis. Lancet Psychiatry 8, 225–236. doi: 10.1016/S2215-0366(20)30431-4
Clifton, N. E., Hannon, E., Harwood, J. C., Di Florio, A., Thomas, K. L., Holmans, P. A., et al. (2019). Dynamic expression of genes associated with schizophrenia and bipolar disorder across development. Transl. Psychiatry 9:74. doi: 10.1038/s41398-019-0405-x
Coelho-Santos, V., and Shih, A. Y. (2020). Postnatal development of cerebrovascular structure and the neurogliovascular unit. Wiley Interdiscip. Rev. Dev. Biol. 9:e363. doi: 10.1002/wdev.363
Connolly, A. M., Chez, M. G., Pestronk, A., Arnold, S. T., Mehta, S., and Deuel, R. K. (1999). Serum autoantibodies to brain in Landau-Kleffner variant, autism, and other neurologic disorders. J. Pediatr. 134, 607–613. doi: 10.1016/s0022-3476(99)70248-9
Cortes-Canteli, M., Kruyer, A., Fernandez-Nueda, I., Marcos-Diaz, A., Ceron, C., Richards, A. T., et al. (2019). Long-Term dabigatran treatment delays Alzheimer’s disease pathogenesis in the tgcrnd8 mouse model. J. Am. Coll. Cardiol. 74, 1910–1923. doi: 10.1016/j.jacc.2019.07.081
Cramer, S. P., Simonsen, H., Frederiksen, J. L., Rostrup, E., and Larsson, H. B. (2014). Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. Neuroimage Clin. 4, 182–189. doi: 10.1016/j.nicl.2013.12.001
Crockett, A. M., Ryan, S. K., Vasquez, A. H., Canning, C., Kanyuch, N., Kebir, H., et al. (2021). Disruption of the blood-brain barrier in 22q11.2 deletion syndrome. Brain 144, 1351–1360. doi: 10.1093/brain/awab055
Cruz Hernandez, J. C., Bracko, O., Kersbergen, C. J., Muse, V., Haft-Javaherian, M., Berg, M., et al. (2019). Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 22, 413–420. doi: 10.1038/s41593-018-0329-4
Császár, E., Lénárt, N., Cserép, C., Környei, Z., Fekete, R., Pósfai, B., et al. (2021). Microglia control cerebral blood flow and neurovascular coupling via P2Y12R-mediated actions. bioRXiv [Preprint] doi: 10.1101/2021.02.04.429741
Cvetanovic, M., Patel, J. M., Marti, H. H., Kini, A. R., and Opal, P. (2011). Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Nat. Med. 17, 1445–1447. doi: 10.1038/nm.2494
Czako, C., Kovacs, T., Ungvari, Z., Csiszar, A., Yabluchanskiy, A., Conley, S., et al. (2020). Retinal biomarkers for Alzheimer’s disease and vascular cognitive impairment and dementia (VCID): implication for early diagnosis and prognosis. Geroscience 42, 1499–1525. doi: 10.1007/s11357-020-00252-7
D’Haeseleer, M., Beelen, R., Fierens, Y., Cambron, M., Vanbinst, A. M., Verborgh, C., et al. (2013). Cerebral hypoperfusion in multiple sclerosis is reversible and mediated by endothelin-1. Proc. Natl. Acad. Sci. U.S.A. 110, 5654–5658. doi: 10.1073/pnas.1222560110
D’Haeseleer, M., Cambron, M., Vanopdenbosch, L., and De Keyser, J. (2011). Vascular aspects of multiple sclerosis. Lancet Neurol. 10, 657–666. doi: 10.1016/s1474-4422(11)70105-3
Dabertrand, F., Harraz, O. F., Masayo, K., Longden, T. A., Rosehart, A. C., Hill-Eubanks, D., et al. (2021). PIP 2 corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity. Proc. Natl. Acad. Sci. U.S.A. 118:e2025998118. doi: 10.1073/pnas.2025998118
Daneman, R. (2012). The blood–brain barrier in health and disease. Ann. Neurol. 72, 648–672. doi: 10.1002/ana.23648
Daneman, R., Agalliu, D., Zhou, L., Kuhnert, F., Kuo, C., and Barres, B. (2009). Wnt/ -catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 641–646.
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
Davenport, C. B. (1915). Huntington’s Chorea in relation to heredity and eugenics. Proc. Natl. Acad. Sci. U.S.A. 1, 283–285.
Davis, J., Xu, F., Deane, R., Romanov, G., Previti, M. L., Zeigler, K., et al. (2004). Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 279, 20296–20306. doi: 10.1074/jbc.M312946200
De Filippis, L., and Delia, D. (2011). Hypoxia in the regulation of neural stem cells. Cell. Mol. Life Sci. 68, 2831–2844. doi: 10.1007/s00018-011-0723-5
De Keyser, J., Wilczak, N., Leta, R., and Streetland, C. (1999). Astrocytes in multiple sclerosis lack beta-2 adrenergic receptors. Neurology 53, 1628–1633. doi: 10.1212/WNL.53.8.1628
De Silva, T. M., and Faraci, F. M. (2016). Microvascular dysfunction and cognitive impairment. Cell. Mol. Neurobiol. 36, 241–258. doi: 10.1007/s10571-015-0308-1
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
Deckel, A. W., and Duffy, J. D. (2000). Vasomotor hyporeactivity in the anterior cerebral artery during motor activation in Huntington’s disease patients. Brain Res. 872, 258–261. doi: 10.1016/S0006-8993(00)02506-3
Deckel, A. W., Weiner, R., Szigeti, D., Claark, V., and Vento, J. (2000). Altered patterns of regional cerebral blood flow in patients with Huntington’s disease: a SPECT study during rest and cognitive or motor activation. J. Nucl. Med. 41, 773–780.
Delgado, A. C., Ferron, S. R., Vicente, D., Porlan, E., Perez-Villalba, A., Trujillo, C. M., et al. (2014). Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron 83, 572–585. doi: 10.1016/j.neuron.2014.06.015
Derejko, M., Sławek, J., Wieczorek, D., Brockhuis, B., Dubaniewicz, M., and Lass, P. (2006). Regional cerebral blood flow in Parkinson’s disease as an indicator of cognitive impairment. Nucl. Med. Commun. 27, 945–951. doi: 10.1097/01.mnm.0000243370.18883.62
Di Pardo, A., Amico, E., Scalabri, F., Pepe, G., Castaldo, S., Elifani, F., et al. (2017). Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci. Rep. 7:41316. doi: 10.1038/srep41316
Dietrich, H. H., Xiang, C., Han, B. H., Zipfel, G. J., and Holtzman, D. M. (2010). Soluble amyloid-beta, effect on cerebral arteriolar regulation and vascular cells. Mol. Neurodegener. 5:15. doi: 10.1186/1750-1326-5-15
Dobson, R., and Giovannoni, G. (2019). Multiple sclerosis – a review. Eur. J. Neurol. 26, 27–40. doi: 10.1111/ene.13819
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A. III, Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 112, 405–415. doi: 10.1007/s00401-006-0115-3
Drazanova, E., Ruda-Kucerova, J., Kratka, L., Stark, T., Kuchar, M., Maryska, M., et al. (2019). Different effects of prenatal MAM vs. perinatal THC exposure on regional cerebral blood perfusion detected by Arterial Spin Labelling MRI in rats. Sci. Rep. 9:6062. doi: 10.1038/s41598-019-42532-z
Drouin-Ouellet, J., Sawiak, S. J., Cisbani, G., Lagace, M., Kuan, W. L., Saint-Pierre, M., et al. (2015). Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann. Neurol. 78, 160–177. doi: 10.1002/ana.24406
Dudek, K. A., Dion-Albert, L., Lebel, M., LeClair, K., Labrecque, S., Tuck, E., et al. (2020). Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. U.S.A. 117, 3326–3336. doi: 10.1073/pnas.1914655117
Dumas, A., Dierksen, G. A., Gurol, M. E., Halpin, A., Martinez-Ramirez, S., Schwab, K., et al. (2012). Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann. Neurol. 72, 76–81. doi: 10.1002/ana.23566
Elabi, O., Gaceb, A., Carlsson, R., Padel, T., Soylu-Kucharz, R., Cortijo, I., et al. (2021). Human alpha-synuclein overexpression in a mouse model of Parkinson’s disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Sci. Rep. 11:1120. doi: 10.1038/s41598-020-80889-8
Ellingrod, V. L., Taylor, S. F., Brook, R. D., Evans, S. J., Zollner, S. K., Grove, T. B., et al. (2011). Dietary, lifestyle and pharmacogenetic factors associated with arteriole endothelial-dependent vasodilatation in schizophrenia patients treated with atypical antipsychotics (AAPs). Schizophr. Res. 130, 20–26. doi: 10.1016/j.schres.2011.03.031
Emerich, D. F., Mooney, D. J., Storrie, H., Babu, R. S., and Kordower, J. H. (2010). Injectable hydrogels providing sustained delivery of vascular endothelial growth factor are neuroprotective in a rat model of Huntington’s disease. Neurotox. Res. 17, 66–74. doi: 10.1007/s12640-009-9079-0
Emerson, R. W., Adams, C., Nishino, T., Hazlett, H. C., Wolff, J. J., Zwaigenbaum, L., et al. (2017). Functional neuroimaging of high-risk 6-month-old infants predicts a diagnosis of autism at 24 months of age. Sci. Transl. Med. 9:eaag2882. doi: 10.1126/scitranslmed.aag2882
Farkas, E., De Jong, G. I., de Vos Ernst, R. A. I., Steur, J. N. H., and Luiten, P. G. M. (2000). Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 100, 395–402. doi: 10.1007/s004010000195
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease [Review]. Prog. Neurobiol. 64, 575–611.
Faucheux, B. A., Agid, Y., Hirsch, E. C., and Bonnet, A.-M. (1999). Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 353, 981–982. doi: 10.1016/s0140-6736(99)00641-8
Feigin, A., Ghilardi, M. F., Huang, C., Ma, Y., Carbon, M., Guttman, M., et al. (2006). Preclinical Huntington’s disease: compensatory brain responses during learning. Ann. Neurol. 59, 53–59. doi: 10.1002/ana.20684
Fernandez-Klett, F., and Priller, J. (2015). Diverse functions of pericytes in cerebral blood flow regulation and ischemia. J. Cereb. Blood Flow Metab. 35, 883–887. doi: 10.1038/jcbfm.2015.60
Fernandez-Seara, M. A., Mengual, E., Vidorreta, M., Aznarez-Sanado, M., Loayza, F. R., Villagra, F., et al. (2012). Cortical hypoperfusion in Parkinson’s disease assessed using arterial spin labeled perfusion MRI. Neuroimage 59, 2743–2750. doi: 10.1016/j.neuroimage.2011.10.033
Filosa, J. A., and Iddings, J. A. (2013). Astrocyte regulation of cerebral vascular tone. Am. J. Physiol. Heart Circ. Physiol. 305, H609–H619. doi: 10.1152/ajpheart.00359.2013
Finnerty, N. J., Bolger, F. B., Palsson, E., and Lowry, J. P. (2013). An investigation of hypofrontality in an animal model of schizophrenia using real-time microelectrochemical sensors for glucose, oxygen, and nitric oxide. ACS Chem. Neurosci. 4, 825–831. doi: 10.1021/cn4000567
Fiorentino, M., Sapone, A., Senger, S., Camhi, S. S., Kadzielski, S. M., Buie, T. M., et al. (2016). Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 7:49. doi: 10.1186/s13229-016-0110-z
Ford, J. M., Johnson, M. B., Whitfield, S. L., Faustman, W. O., and Mathalon, D. H. (2005). Delayed hemodynamic responses in schizophrenia. Neuroimage 26, 922–931. doi: 10.1016/j.neuroimage.2005.03.001
Ford, J. M., Roth, W. T., Menon, V., and Pfefferbaum, A. (1999). Failures of automatic and strategic processing in schizophrenia: comparisons of event-related brain potential and startle blink modification. Schizophr. Res. 37, 149–163. doi: 10.1016/s0920-9964(98)00148-0
Friedland, R. P., and Iadecola, C. (1991). Roy and Sherrington (1890): a centennial reexamination of “On the regulation of the blood-supply of the brain”. Neurology 41, 10–14. doi: 10.1212/wnl.41.1.10
Frosen, J., and Joutel, A. (2018). Smooth muscle cells of intracranial vessels: from development to disease. Cardiovasc. Res. 114, 501–512. doi: 10.1093/cvr/cvy002
Fujiki, R., Morita, K., Sato, M., Kamada, Y., Kato, Y., Inoue, M., et al. (2013). Reduced prefrontal cortex activation using the Trail Making Test in schizophrenia. Neuropsychiatr. Dis. Treat. 9, 675–685. doi: 10.2147/NDT.S43137
Ge, Y., Law, M., Johnson, G., Herbert, J., Babb, J. S., Mannon, L. J., et al. (2005). Dynamic susceptibility contrast perfusion MR imaging of multiple sclerosis lesions: characterizing hemodynamic impairment and inflammatory activity. AJNR Am. J. Neuroradiol. 26, 1539–1547.
Geraldes, R., Esiri, M. M., Perera, R., Yee, S. A., Jenkins, D., Palace, J., et al. (2020). Vascular disease and multiple sclerosis: a post-mortem study exploring their relationships. Brain 143, 2998–3012. doi: 10.1093/brain/awaa255
Gireud-Goss, M., Mack, A. F., McCullough, L. D., and Urayama, A. (2020). Cerebral amyloid angiopathy and blood-brain barrier dysfunction. Neuroscientist doi: 10.1177/1073858420954811 [Epub ahead of print].
Girolamo, F., Coppola, C., Ribatti, D., and Trojano, M. (2014). Angiogenesis in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2:84. doi: 10.1186/s40478-014-0084-z
Girouard, H., and Iadecola, C. (2006). Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. (1985) 100, 328–335. doi: 10.1152/japplphysiol.00966.2005
Gitler, A. D., Dhillon, P., and Shorter, J. (2017). Neurodegenerative disease: models, mechanisms, and a new hope. Dis. Model. Mech. 10, 499–502. doi: 10.1242/dmm.030205
Gogtay, N., Vyas, N. S., Testa, R., Wood, S. J., and Pantelis, C. (2011). Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr. Bull. 37, 504–513. doi: 10.1093/schbul/sbr030
Goldman, S. A., and Chen, Z. (2011). Perivascular instruction of cell genesis and fate in the adult brain. Nat. Neurosci. 14, 1382–1389. doi: 10.1038/nn.2963
Gorelick, P. B. (2004). Risk factors for vascular dementia and Alzheimer disease. Stroke 35(11 Suppl. 1), 2620–2622.
Gotz, J., and Ittner, L. M. (2008). Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 9, 532–544.
Graham, N. S., and Sharp, D. J. (2019). Understanding neurodegeneration after traumatic brain injury: from mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 90, 1221–1233. doi: 10.1136/jnnp-2017-317557
Grammas, P. (2011). Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 8:26. doi: 10.1186/1742-2094-8-26
Grant, R. I., Hartmann, D. A., Underly, R. G., Berthiaume, A. A., Bhat, N. R., and Shih, A. Y. (2019). Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J. Cereb. Blood Flow Metab. 39, 411–425. doi: 10.1177/0271678X17732229
Gray, M. T., and Woulfe, J. M. (2015). Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 35, 747–750. doi: 10.1038/jcbfm.2015.32
Greene, C., Kealy, J., Humphries, M. M., Gong, Y., Hou, J., Hudson, N., et al. (2018). Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 23, 2156–2166. doi: 10.1038/mp.2017.156
Grove, T., Taylor, S., Dalack, G., and Ellingrod, V. (2015). Endothelial function, folate pharmacogenomics, and neurocognition in psychotic disorders. Schizophr. Res. 164, 115–121. doi: 10.1016/j.schres.2015.02.006
Grubb, S., Lauritzen, M., and Aalkjaer, C. (2021). Brain capillary pericytes and neurovascular coupling. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 254:110893. doi: 10.1016/j.cbpa.2020.110893
Grunewald, M., Kumar, S., Sharife, H., Volinsky, E., Gileles-Hillel, A., Licht, T., et al. (2021). Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 373:eabc8479. doi: 10.1126/science.abc8479
Gu, C., Yoshida, Y., Livet, J., Reimert, D., Mann, F., Merte, J., et al. (2005). Semaphorin 3E and Plexin-D1 control vascular pattern independently of neuropilins. Science 307, 265–268. doi: 10.1126/science.1105416
Guan, J., Pavlovic, D., Dalkie, N., Waldvogel, H., O’Carroll, S. J., Green, C. R., et al. (2013). Vascular degeneration in Parkinson’s disease. Brain Pathol. 23, 154–164. doi: 10.1111/j.1750-3639.2012.00628.x
Guo, Y., Li, X., Zhang, M., Chen, N., Wu, S., Lei, J., et al. (2019). Age and brain regionassociated alterations of cerebral blood flow in early Alzheimer’s disease assessed in AbetaPPSWE/PS1DeltaE9 transgenic mice using arterial spin labeling. Mol. Med. Rep. 19, 3045–3052. doi: 10.3892/mmr.2019.9950
Guo, Y., Singh, L. N., Zhu, Y., Gur, R. E., Resnick, A., Anderson, S. A., et al. (2020). Association of a functional Claudin-5 variant with schizophrenia in female patients with the 22q11.2 deletion syndrome. Schizophr. Res. 215, 451–452. doi: 10.1016/j.schres.2019.09.014
Gur, R. E., Bassett, A. S., McDonald-McGinn, D. M., Bearden, C. E., Chow, E., Emanuel, B. S., et al. (2017). A neurogenetic model for the study of schizophrenia spectrum disorders: the International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol. Psychiatry 22, 1664–1672. doi: 10.1038/mp.2017.161
Ha, A. D., and Fung, V. S. (2012). Huntington’s disease. Curr. Opin. Neurol. 25, 491–498. doi: 10.1097/WCO.0b013e3283550c97
Haidey, J. N., Peringod, G., Institoris, A., Gorzo, K. A., Nicola, W., Vandal, M., et al. (2021). Astrocytes regulate ultra-slow arteriole oscillations via stretch-mediated TRPV4-COX-1 feedback. Cell Rep. 36:109405. doi: 10.1016/j.celrep.2021.109405
Halliday, M. R., Rege, S. V., Ma, Q., Zhao, Z., Miller, C. A., Winkler, E. A., et al. (2016). Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 36, 216–227. doi: 10.1038/jcbfm.2015.44
Hamel, E. (2006). Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 100, 1059–1064. doi: 10.1152/japplphysiol.00954.2005
Hamel, E. (2015). Cerebral circulation function and dysfunction in Alzheimer’s disease. J. Cardiovasc. Pharmacol. 65, 317–324. doi: 10.1097/FJC.0000000000000177
Hanlon, F. M., Shaff, N. A., Dodd, A. B., Ling, J. M., Bustillo, J. R., Abbott, C. C., et al. (2016). Hemodynamic response function abnormalities in schizophrenia during a multisensory detection task. Hum. Brain Mapp. 37, 745–755. doi: 10.1002/hbm.23063
Harada, K., Kamiya, T., and Tsuboi, T. (2015). Gliotransmitter release from astrocytes: functional, developmental, and pathological implications in the brain. Front. Neurosci. 9:499. doi: 10.3389/fnins.2015.00499
Harbo, H. F., Gold, R., and Tintore, M. (2013). Sex and gender issues in multiple sclerosis. Ther. Adv. Neurol. Disord. 6, 237–248. doi: 10.1177/1756285613488434
Harris, G. J., Codoris, A. M., Lewis, R. F., Schmidt, E., Bedi, A., and Brandt, J. (1999). Reduced basal ganglia blood flow and volume in pre-sympotmatic, gene-tested persons at-risk for Huntington’s disease. Brain 122, 1667–1678. doi: 10.1093/brain/122.9.1667
Hartmann, D. A., Berthiaume, A. A., Grant, R. I., Harrill, S. A., Koski, T., Tieu, T., et al. (2021). Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 24, 633–645. doi: 10.1038/s41593-020-00793-2
Hartz, A. M., Bauer, B., Soldner, E. L., Wolf, A., Boy, S., Backhaus, R., et al. (2012). Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 43, 514–523. doi: 10.1161/STROKEAHA.111.627562
Hasselbalch, S. G., Øberg, G., Sørensen, S. A., Andersen, A. R., Waldemar, G., Schmidt, J. F., et al. (1992). Reduced regional cerebral blood flow in Huntington’s disease studied by SPECT. J. Neurol. Neurosurg. Psychiatry 55, 1018–1023. doi: 10.1136/jnnp.55.11.1018
Hays, C. C., Zlatar, Z. Z., and Wierenga, C. E. (2016). The utility of cerebral blood flow as a biomarker of preclinical Alzheimer’s disease. Cell. Mol. Neurobiol. 36, 167–179. doi: 10.1007/s10571-015-0261-z
Hijaz, B. A., and Volpicelli-Daley, L. A. (2020). Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 15:19. doi: 10.1186/s13024-020-00368-6
Hillman, E. M. (2014). Coupling mechanism and significance of the BOLD signal: a status report. Annu. Rev. Neurosci. 37, 161–181. doi: 10.1146/annurev-neuro-071013-014111
Hindle, J. V. (2010). Ageing, neurodegeneration and Parkinson’s disease. Age Ageing 39, 156–161. doi: 10.1093/ageing/afp223
Hino, M., Kunii, Y., Matsumoto, J., Wada, A., Nagaoka, A., Niwa, S., et al. (2016). Decreased VEGFR2 expression and increased phosphorylated Akt1 in the prefrontal cortex of individuals with schizophrenia. J. Psychiatry Res. 82, 100–108. doi: 10.1016/j.jpsychires.2016.07.018
Hogan, K. A., Ambler, C. A., Chapman, D. L., and Bautch, V. L. (2004). The neural tube patterns vessels developmentally using the VEGF signaling pathway. Development 131, 1503–1513. doi: 10.1242/dev.01039
Holley, J. E., Newcombe, J., Whatmore, J. L., and Gutowski, N. J. (2010). Increased blood vessel density and endothelial cell proliferation in multiple sclerosis cerebral white matter. Neurosci. Lett. 470, 65–70. doi: 10.1016/j.neulet.2009.12.059
Horev, G., Ellegood, J., Lerch, J. P., Son, Y. E., Muthuswamy, L., Vogel, H., et al. (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. U.S.A. 108, 17076–17081. doi: 10.1073/pnas.1114042108
Horn, H., Federspiel, A., Wirth, M., Muller, T. J., Wiest, R., Wang, J. J., et al. (2009). Structural and metabolic changes in language areas linked to formal thought disorder. Br. J. Psychiatry 194, 130–138. doi: 10.1192/bjp.bp.107.045633
Hostenbach, S., Pauwels, A., Michiels, V., Raeymaekers, H., Van Binst, A. M., Van Merhaeghen-Wieleman, A., et al. (2019). Role of cerebral hypoperfusion in multiple sclerosis (ROCHIMS): study protocol for a proof-of-concept randomized controlled trial with bosentan. Trials 20:164. doi: 10.1186/s13063-019-3252-4
Howarth, C. (2014). The contribution of astrocytes to the regulation of cerebral blood flow. Front. Neurosci. 8:103. doi: 10.3389/fnins.2014.00103
Howarth, C., Mishra, A., and Hall, C. N. (2021). More than just summed neuronal activity: how multiple cell types shape the BOLD response. Philos. Trans. R. Soc. Lond. B Biol. Sci. 376:20190630. doi: 10.1098/rstb.2019.0630
Howe, M. D., McCullough, L. D., and Urayama, A. (2020). The role of basement membranes in cerebral amyloid angiopathy. Front. Physiol. 11:601320. doi: 10.3389/fphys.2020.601320
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Proc. Natl. Acad. Sci. U.S.A. 96, 3228–3233.
Hsiao, H.-Y., Chen, Y.-C., Huang, C.-H., Chen, C.-C., Hsu, Y.-H., Chen, H.-M., et al. (2015). Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann. Neurol. 78, 178–192. doi: 10.1002/ana.24428
Hua, J., Unschuld, P. G., Margolis, R. L., van Zijl, P. C., and Ross, C. A. (2014). Elevated arteriolar cerebral blood volume in prodromal Huntington’s disease. Mov. Disord. 29, 396–401. doi: 10.1002/mds.25591
Huang, Z., Wong, L. W., Su, Y., Huang, X., Wang, N., Chen, H., et al. (2020). Blood-brain barrier integrity in the pathogenesis of Alzheimer’s disease. Front. Neuroendocrinol. 59:100857. doi: 10.1016/j.yfrne.2020.100857
Hultman, K., Strickland, S., and Norris, E. H. (2013). The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 33, 1251–1258. doi: 10.1038/jcbfm.2013.76
Humpel, C., and Marksteiner, J. (2005). Cerebrovascular damage as a cause for Alzheimer’s disease [Research Support, Non-U.S. Gov’t Review]. Curr. Neurovasc. Res. 2, 341–347.
Huneau, C., Benali, H., and Chabriat, H. (2015). Investigating human neurovascular coupling using functional neuroimaging: a critical review of dynamic models. Front. Neurosci. 9:467. doi: 10.3389/fnins.2015.00467
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Impellizzeri, D., Campolo, M., Bruschetta, G., Crupi, R., Cordaro, M., Paterniti, I., et al. (2016). Traumatic brain injury leads to development of Parkinson’s disease related pathology in mice. Front. Neurosci. 10:458. doi: 10.3389/fnins.2016.00458
Incalza, M. A., D’Oria, R., Natalicchio, A., Perrini, S., Laviola, L., and Giorgino, F. (2018). Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 100, 1–19. doi: 10.1016/j.vph.2017.05.005
Iturria-Medina, Y., Sotero, R. C., Toussaint, P. J., Mateos-Perez, J. M., Evans, A. C., and Alzheimer’s Disease Neuroimaging Initiative (2016). Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 7:11934. doi: 10.1038/ncomms11934
James, J. M., Gewolb, C., and Bautch, V. L. (2009). Neurovascular development uses VEGF-A signaling to regulate blood vessel ingression into the neural tube. Development 136, 833–841. doi: 10.1242/dev.028845
Jann, K., Hernandez, L. M., Beck-Pancer, D., McCarron, R., Smith, R. X., Dapretto, M., et al. (2015). Altered resting perfusion and functional connectivity of default mode network in youth with autism spectrum disorder. Brain Behav. 5:e00358. doi: 10.1002/brb3.358
Juurlink, B. H. (2013). The evidence for hypoperfusion as a factor in multiple sclerosis lesion development. Mult. Scler. Int. 2013:598093. doi: 10.1155/2013/598093
Kadry, H., Noorani, B., and Cucullo, L. (2020). A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 17:69. doi: 10.1186/s12987-020-00230-3
Kalaria, R. N. (1996). Cerebral vessels in ageing and Alzheimer’s disease [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S. Review]. Pharmacol. Ther. 72, 193–214.
Kalaria, R. N., and Harik, S. I. (1989). Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J. Neurochem. 53, 1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x
Kalinderi, K., Bostantjopoulou, S., and Fidani, L. (2016). The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurol. Scand. 134, 314–326. doi: 10.1111/ane.12563
Kamagata, K., Motoi, Y., Hori, M., Suzuki, M., Nakanishi, A., Shimoji, K., et al. (2011). Posterior hypoperfusion in Parkinson’s disease with and without dementia measured with arterial spin labeling MRI. J. Magn. Reson. Imaging 33, 803–807. doi: 10.1002/jmri.22515
Kamphuis, W. W., Derada Troletti, C., Reijerkerk, A., Romero, I. A., and de Vries, H. E. (2015). The blood-brain barrier in multiple sclerosis: microRNAs as key regulators. CNS Neurol. Disord. Drug Targets 14, 157–167. doi: 10.2174/1871527314666150116125246
Kaplan, L., Chow, B. W., and Gu, C. (2020). Neuronal regulation of the blood-brain barrier and neurovascular coupling. Nat. Rev. Neurosci. 21, 416–432. doi: 10.1038/s41583-020-0322-2
Kawakami, K., Wake, R., Miyaoka, T., Furuya, M., Liaury, K., and Horiguchi, J. (2014). The effects of aging on changes in regional cerebral blood flow in schizophrenia. Neuropsychobiology 69, 202–209. doi: 10.1159/000358840
Kawakubo, Y., Kuwabara, H., Watanabe, K., Minowa, M., Someya, T., Minowa, I., et al. (2009). Impaired prefrontal hemodynamic maturation in autism and unaffected siblings. PLoS One 4:e6881. doi: 10.1371/journal.pone.0006881
Kealy, J., Greene, C., and Campbell, M. (2020). Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 726:133664. doi: 10.1016/j.neulet.2018.06.033
Keefe, R. S. E., and Harvey, P. D. (2012). Cognitive Impairment in Schizophrenia, Vol. 213. Berlin: Springer, doi: 10.1007/978-3-642-25758-2_2
Kelleher, R. J., and Soiza, R. L. (2013). Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am. J. Cardiovasc. Dis. 3, 197–226.
Khandaker, G. M., Cousins, L., Deakin, J., Lennox, B. R., Yolken, R., and Jones, P. B. (2015). Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2, 258–270. doi: 10.1016/s2215-0366(14)00122-9
Kirk, J., Plumb, J., Mirakhur, M., and McQuaid, S. (2003). Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood–brain barrier leakage and active demyelination. J. Pathol. 201, 319–327. doi: 10.1002/path.1434
Kisler, K., Nelson, A. R., Montagne, A., and Zlokovic, B. V. (2017). Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 18, 419–434. doi: 10.1038/nrn.2017.48
Kisler, K., Nelson, A. R., Rege, S. V., Ramanathan, A., Wang, Y., Ahuja, A., et al. (2016). Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 20, 406–420. doi: 10.1038/nn.4489A
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Klinkmueller, P., Kronenbuerger, M., Miao, X., Bang, J., Ultz, K. E., Paez, A., et al. (2021). Impaired response of cerebral oxygen metabolism to visual stimulation in Huntington’s disease. J. Cereb. Blood Flow Metab. 41, 1119–1130. doi: 10.1177/0271678X20949286
Knight, M. J., and Baune, B. T. (2017). Psychosocial dysfunction in major depressive disorder-rationale, design, and characteristics of the cognitive and emotional recovery training program for depression (CERT-D). Front. Psychiatry 8:280. doi: 10.3389/fpsyt.2017.00280
Kochunov, P., Zavaliangos-Petropulu, A., Jahanshad, N., Thompson, P. M., Ryan, M. C., Chiappelli, J., et al. (2021). A white matter connection of schizophrenia and Alzheimer’s disease. Schizophr. Bull. 47, 197–206. doi: 10.1093/schbul/sbaa078
Kook, S. Y., Hong, H. S., Moon, M., Ha, C. M., Chang, S., and Mook-Jung, I. (2012). Abeta(1)(-)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J. Neurosci. 32, 8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012
Korte, N., Nortley, R., and Attwell, D. (2020). Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol. 140, 793–810. doi: 10.1007/s00401-020-02215-w
Kortekaas, R., Leenders, K. L., van Oostrom, J. C. H., Vaalburg, W., Bart, J., Willemsen, A. T. M., et al. (2005). Blood–brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 57, 176–179. doi: 10.1002/ana.20369
Kostiukow, A., and Samborski, W. (2020). The effectiveness of hyperbaric oxygen therapy (HBOT) in children with autism spectrum disorders. Pol. Merkur. Lekarski. 48, 15–18.
Kounidas, G., Cruickshank, H., Kastora, S., Sihlabela, S., and Miedzybrodzka, Z. (2021). The known burden of Huntington disease in the North of Scotland: prevalence of manifest and identified pre-symptomatic gene expansion carriers in the molecular era. J. Neurol. doi: 10.1007/s00415-021-10505-w
Kozberg, M. G., Ma, Y., Shaik, M. A., Kim, S. H., and Hillman, E. M. (2016). Rapid postnatal expansion of neural networks occurs in an environment of altered neurovascular and neurometabolic coupling. J. Neurosci. 36, 6704–6717. doi: 10.1523/JNEUROSCI.2363-15.2016
Kuan, W. L., Bennett, N., He, X., Skepper, J. N., Martynyuk, N., Wijeyekoon, R., et al. (2016). α-Synuclein pre-formed fibrils impair tight junction protein expression without affecting cerebral endothelial cell function. Exp. Neurol. 285(Pt A), 72–81. doi: 10.1016/j.expneurol.2016.09.003
Kumar, H., and Sharma, B. (2016). Memantine ameliorates autistic behavior, biochemistry & blood brain barrier impairments in rats. Brain Res. Bull. 124, 27–39. doi: 10.1016/j.brainresbull.2016.03.013
Kumar, H., Sharma, B. M., and Sharma, B. (2015). Benefits of agomelatine in behavioral, neurochemical and blood brain barrier alterations in prenatal valproic acid induced autism spectrum disorder. Neurochem. Int. 91, 34–45. doi: 10.1016/j.neuint.2015.10.007
Kumar-Singh, S., Pirici, D., McGowan, E., Serneels, S., Ceuterick, C., Hardy, J., et al. (2005). Dense-Core plaques in Tg2576 and PSAPP mouse models of Alzheimer’s disease are centered on vessel walls. Am. J. Pathol. 167, 527–543. doi: 10.1016/S0002-9440(10)62995-1
Kuusimaki, T., Al-Abdulrasul, H., Kurki, S., Hietala, J., Hartikainen, S., Koponen, M., et al. (2021). Increased risk of Parkinson’s disease in patients with schizophrenia spectrum disorders. Mov. Disord. 36, 1353–1361. doi: 10.1002/mds.28484
Kwakye, G. F., McMinimy, R. A., and Aschner, M. (2017). Disease-Toxicant interactions in Parkinson’s disease neuropathology. Neurochem. Res. 42, 1772–1786. doi: 10.1007/s11064-016-2052-4
Lacalle-Aurioles, M., Mateos-Perez, J. M., Guzman-De-Villoria, J. A., Olazaran, J., Cruz-Orduna, I., Aleman-Gomez, Y., et al. (2014). Cerebral blood flow is an earlier indicator of perfusion abnormalities than cerebral blood volume in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 34, 654–659. doi: 10.1038/jcbfm.2013.241
Lacoste, B., Comin, C. H., Ben-Zvi, A., Kaeser, P. S., Xu, X., Costa, L. F., et al. (2014). Sensory-Related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron 83, 1117–1130. doi: 10.1016/j.neuron.2014.07.034
Lacoste, B., and Gu, C. (2015). Control of cerebrovascular patterning by neural activity during postnatal development. Mech. Dev. 138(Pt 1), 43–49. doi: 10.1016/j.mod.2015.06.003
Lacoste, B., Tong, X. K., Lahjouji, K., Couture, R., and Hamel, E. (2013). Cognitive and cerebrovascular improvements following kinin B1 receptor blockade in Alzheimer’s disease mice. J. Neuroinflammation 10:57.
Larsen, J., Martin, D. R., and Byrne, M. (2014). Recent advances in delivery through the blood-brain barrier. Curr. Top. Med. Chem. 14, 1148–1160. doi: 10.2174/1568026614666140329230311
Law, M., Saindane, A. M., Ge, Y., Babb, J. S., Johnson, G., Mannon, L. J., et al. (2004). Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter. Radiology 231, 645–652. doi: 10.1148/radiol.2313030996
Lecrux, C., and Hamel, E. (2011). The neurovascular unit in brain function and disease. Acta Physiol. (Oxf.) 203, 47–59. doi: 10.1111/j.1748-1716.2011.02256.x
Leech, S., Kirk, J., Plumb, J., and McQuaid, S. (2007). Persistent endothelial abnormalities and blood-brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol. Appl. Neurobiol. 33, 86–98. doi: 10.1111/j.1365-2990.2006.00781.x
Leeuwis, A. E., Benedictus, M. R., Kuijer, J. P. A., Binnewijzend, M. A. A., Hooghiemstra, A. M., Verfaillie, S. C. J., et al. (2017). Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimers Dement. 13, 531–540. doi: 10.1016/j.jalz.2016.08.013
Lepeta, K., and Kaczmarek, L. (2015). Matrix Metalloproteinase-9 as a Novel Player in Synaptic Plasticity and Schizophrenia. Schizophr. Bull. 41, 1003–1009. doi: 10.1093/schbul/sbv036
Li, J. Y., Popovic, N., and Brundin, P. (2005). The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroTherapeutics 2, 447–464. doi: 10.1602/neurorx.2.3.447
Li, S. S., Qu, Z., Haas, M., Ngo, L., Heo, Y. J., Kang, H. J., et al. (2016). The HSA21 gene EURL/C21ORF91 controls neurogenesis within the cerebral cortex and is implicated in the pathogenesis of Down Syndrome. Sci. Rep. 6:29514. doi: 10.1038/srep29514
Licht, T., and Keshet, E. (2015). The vascular niche in adult neurogenesis. Mech. Dev. 138(Pt 1), 56–62. doi: 10.1016/j.mod.2015.06.001
Lim, R. G., Quan, C., Reyes-Ortiz, A. M., Lutz, S. E., Kedaigle, A. J., Gipson, T. A., et al. (2017). Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. 19, 1365–1377. doi: 10.1016/j.celrep.2017.04.021
Lin, A. L., Jahrling, J. B., Zhang, W., DeRosa, N., Bakshi, V., Romero, P., et al. (2017). Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 37, 217–226. doi: 10.1177/0271678X15621575
Lin, C.-Y., Hsu, Y.-H., Lin, M.-H., Yang, T.-H., Chen, H.-M., Chen, Y.-C., et al. (2013). Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp. Neurol. 250, 20–30. doi: 10.1016/j.expneurol.2013.08.019
Liu, H., Zhang, C., Xu, J., Jin, J., Cheng, L., Wu, Q., et al. (2020). HTT silencing delays onset and slows progression of Huntington’s disease like phenotype: monitoring with a novel neurovascular biomarker. bioRXiv [Preprint] doi: 10.1101/2020.11.17.386631
Liu, Y., Braidy, N., Poljak, A., Chan, D. K. Y., and Sachdev, P. (2018). Cerebral small vessel disease and the risk of Alzheimer’s disease: a systematic review. Ageing Res. Rev. 47, 41–48. doi: 10.1016/j.arr.2018.06.002
Liu, Y., Cao, Y., Zhang, W., Bergmeier, S., Qian, Y., Akbar, H., et al. (2012). A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 11, 1672–1682. doi: 10.1158/1535-7163.MCT-12-0131
Love, S., and Miners, J. S. (2016). Cerebral Hypoperfusion and the Energy Deficit in Alzheimer’s Disease. Brain Pathol. 26, 607–617. doi: 10.1111/bpa.12401
Luchsinger, J. A., Reitz, C., Honig, L. S., Tang, M. X., Shea, S., and Mayeux, R. (2005). Aggregation of vascular risk factors and risk of incident Alzheimer disease [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Neurology 65, 545–551. doi: 10.1212/01.wnl.0000172914.08967.dc
Ludwin, S. K. (2006). The pathogenesis of multiple sclerosis: relating human pathology to experimental studies. J. Neuropathol. Exp. Neurol. 65, 305–318. doi: 10.1097/01.jnen.0000225024.12074.80
MacVicar, B. A., and Newman, E. A. (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7:a020388. doi: 10.1101/cshperspect.a020388
Madhyastha, T. M., Askren, M. K., Boord, P., Zhang, J., Leverenz, J. B., and Grabowski, T. J. (2015). Cerebral perfusion and cortical thickness indicate cortical involvement in mild Parkinson’s disease. Mov. Disord. 30, 1893–1900. doi: 10.1002/mds.26128
Magaki, S., Tang, Z., Tung, S., Williams, C. K., Lo, D., Yong, W. H., et al. (2018). The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging 70, 70–77. doi: 10.1016/j.neurobiolaging.2018.06.004
Malaspina, D., Harkavy-Friedman, J., Corcoran, C., Mujica-Parodi, L., Printz, D., Gorman, J. M., et al. (2004). Resting neural activity distinguishes subgroups of schizophrenia patients. Biol. Psychiatry 56, 931–937. doi: 10.1016/j.biopsych.2004.09.013
Malaspina, D., Storer, S., Furman, V., Esser, P., Printz, D., Berman, A., et al. (1999). SPECT study of visual fixation in schizophrenia and comparison subjects. Soc. Biol. Psychiatry 46, 89–93. doi: 10.1016/s0006-3223(98)00306-0
Mantovani, S., Gordon, R., Li, R., Christie, D. C., Kumar, V., and Woodruff, T. M. (2016). Motor deficits associated with Huntington’s disease occur in the absence of striatal degeneration in BACHD transgenic mice. Hum. Mol. Genet. 25, 1780–1791. doi: 10.1093/hmg/ddw050
Marco, S., and Skaper, S. D. (2006). Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 401, 219–224. doi: 10.1016/j.neulet.2006.03.047
Marshall, O., Lu, H., Brisset, J. C., Xu, F., Liu, P., Herbert, J., et al. (2014). Impaired cerebrovascular reactivity in multiple sclerosis. JAMA Neurol. 71, 1275–1281. doi: 10.1001/jamaneurol.2014.1668
Marshall, R. S., Lazar, R. M., Pile-Spellman, J., Young, W. L., Hoang Duong, D., Joshi, S., et al. (2001). Recovery of brain function during induced cerebral hypoperfusion. Brain 124, 1208–1217. doi: 10.1093/brain/124.6.1208
Martinez, B. I., and Stabenfeldt, S. E. (2019). Current trends in biomarker discovery and analysis tools for traumatic brain injury. J. Biol. Eng. 13:16. doi: 10.1186/s13036-019-0145-8
Mathalon, D. H., Ford, J. M., and Pfefferbaum, A. (2000). Trait and state aspects of P300 amplitude reduction in schizophrenia: a retrospective longitudinal study. Biol. Psychiatry 47, 434–449. doi: 10.1016/s0006-3223(99)00277-2
Mattsson, N., Tosun, D., Insel, P. S., Simonson, A., Jack, C. R. Jr., Beckett, L. A., et al. (2014). Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 137(Pt 5), 1550–1561. doi: 10.1093/brain/awu043
Mayhan, W. G. (1999). VEGF increases permeability of the blood-brain barrier via a nitric oxide synthase/cGMP-dependent pathway. Am. J. Physiol. 276, C1148–C1153. doi: 10.1152/ajpcell.1999.276.5.C1148
McColgan, P., and Tabrizi, S. J. (2018). Huntington’s disease: a clinical review. Eur. J. Neurol. 25, 24–34. doi: 10.1111/ene.13413
McConnell, H. L., Li, Z., Woltjer, R. L., and Mishra, A. (2019). Astrocyte dysfunction and neurovascular impairment in neurological disorders: correlation or causation? Neurochem. Int. 128, 70–84. doi: 10.1016/j.neuint.2019.04.005
McGowan, E., Pickford, F., Kim, J., Onstead, L., Eriksen, J., Yu, C., et al. (2005). Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47, 191–199. doi: 10.1016/j.neuron.2005.06.030
McQuaid, S., Cunnea, P., McMahon, J., and Fitzgerald, U. (2009). The effects of blood–brain barrier disruption on glial cell function in multiple sclerosis. Biochem. Soc. Trans. 37(Pt 1), 329–331. doi: 10.1042/BST0370329
Melzer, T. R., Watts, R., MacAskill, M. R., Pearson, J. F., Rueger, S., Pitcher, T. L., et al. (2011). Arterial spin labelling reveals an abnormal cerebral perfusion pattern in Parkinson’s disease. Brain 134(Pt 3), 845–855. doi: 10.1093/brain/awq377
Menard, C., Pfau, M. L., Hodes, G. E., Kana, V., Wang, V. X., Bouchard, S., et al. (2017). Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760. doi: 10.1038/s41593-017-0010-3
Mendez, M. F. (2017). What is the relationship of traumatic brain injury to dementia? J. Alzheimers Dis. 57, 667–681. doi: 10.3233/JAD-161002
Michels, L., Warnock, G., Buck, A., Macauda, G., Leh, S. E., Kaelin, A. M., et al. (2016). Arterial spin labeling imaging reveals widespread and Abeta-independent reductions in cerebral blood flow in elderly apolipoprotein epsilon-4 carriers. J. Cereb. Blood Flow Metab. 36, 581–595. doi: 10.1177/0271678X15605847
Mielke, M. M., Vemuri, P., and Rocca, W. A. (2014). Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin. Epidemiol. 6, 37–48. doi: 10.2147/CLEP.S37929
Milner, E., Zhou, M. L., Johnson, A. W., Vellimana, A. K., Greenberg, J. K., Holtzman, D. M., et al. (2014). Cerebral amyloid angiopathy increases susceptibility to infarction after focal cerebral ischemia in Tg2576 mice. Stroke 45, 3064–3069. doi: 10.1161/STROKEAHA.114.006078
Minn, A., Leclerc, S., Heydel, J.-M., Minn, A.-L., Denizcot, C., Cattarelli, M., et al. (2002). Drug transport into the mammalian brain: the nasal pathway and its specific metabolic barrier. J. Drug Target 10, 285–296. doi: 10.1080/713714452
Minoshima, S., Giordani, B., Berent, S., Frey, K. A., Foster, N. L., and Kuhl, D. E. (1997). Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 42, 85–94. doi: 10.1002/ana.410420114
Mishra, A. (2017). Binaural blood flow control by astrocytes: listening to synapses and the vasculature. J. Physiol. 595, 1885–1902. doi: 10.1113/JP270979
Mohan, S., Ahmad, A. S., Glushakov, A. V., Chambers, C., and Dore, S. (2012). Putative role of prostaglandin receptor in intracerebral hemorrhage. Front. Neurol. 3:145. doi: 10.3389/fneur.2012.00145
Montagne, A., Nation, D. A., Pa, J., Sweeney, M. D., Toga, A. W., and Zlokovic, B. V. (2016). Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 131, 687–707. doi: 10.1007/s00401-016-1570-0
Montagne, A., Nikolakopoulou, A. M., Huuskonen, M. T., Sagare, A. P., Lawson, E. J., Lazic, D., et al. (2021). APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 1, 506–520. doi: 10.1038/s43587-021-00073-z
Montaldi, D., Brooks, D. N., McColl, J. H., Wyper, D., Patterson, J., Barron, E., et al. (1990). Measurement of regional cerebral blood flow and cognitive performance in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 53, 33–38. doi: 10.1136/jnnp.53.1.33
Monti, L., Morbidelli, L., and Rossi, A. (2018). Impaired cerebral perfusion in multiple sclerosis: relevance of endothelial factors. Biomark. Insights 13, 1–10. doi: 10.1177/1177271918774800
Moon, H. S., Jiang, H., Vo, T. T., Jung, W. B., Vazquez, A. L., and Kim, S. G. (2021). Contribution of excitatory and inhibitory neuronal activity to BOLD fMRI. Cereb. Cortex 31, 4053–4067. doi: 10.1093/cercor/bhab068
Mooradian, A. D., Chung, H. C., and Shah, G. N. (1997). GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 18, 469–474. doi: 10.1016/s0197-4580(97)00111-5
Moore, E. E., Liu, D., Li, J., Schimmel, S. J., Cambronero, F. E., Terry, J. G., et al. (2021). Association of aortic stiffness with biomarkers of neuroinflammation, synaptic dysfunction, and neurodegeneration. Neurology 97, e329–e340. doi: 10.1212/WNL.0000000000012257
Morris-Rosendahl, D. J., and Crocq, M. A. (2020). Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin. Neurosci. 22, 65–72. doi: 10.31887/DCNS.2020.22.1/macrocq
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058.
Mughal, A., Harraz, O. F., Gonzales, A. L., Hill-Eubanks, D., and Nelson, M. T. (2021). PIP2 improves cerebral blood flow in a mouse model of Alzheimer’s disease. Function (Oxf) 2:zqab010. doi: 10.1093/function/zqab010
Müller, N., and Ackenheil, M. (1995). Immunoglobulin and albumin content of cerebrospinal fluid in schizophrenic patients: relationship to negative symptomatology. Schizophr. Res. 14, 223–228. doi: 10.1016/0920-9964(94)00045-a
Muramuto, S., Yamada, H., Sadato, N., Kimura, H., Konishi, Y., Kimura, K., et al. (2002). Age-dependent change in metabolic re- sponse to photic stimulation of the primary visual cortex in infants: functional mag- netic resonance imaging study. J. Comput. Assist. Tomogr. 26, 894–901.
Nelson, A. R., Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2016). Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 1862, 887–900. doi: 10.1016/j.bbadis.2015.12.016
Niatsetskaya, Z., Basso, M., Speer, R. E., McConoughey, S. J., Coppola, G., Ma, T. C., et al. (2010). HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington’s disease and Alzheimer’s disease [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Antioxid. Redox Signal. 12, 435–443. doi: 10.1089/ars.2009.2800
Nicolakakis, N., Aboulkassim, T., Ongali, B., Lecrux, C., Fernandes, P., Rosa-Neto, P., et al. (2008). Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J. Neurosci. 28, 9287–9296.
Nicolakakis, N., and Hamel, E. (2011). Neurovascular function in Alzheimer’s disease patients and experimental models. J. Cereb. Blood Flow Metab. 31, 1354–1370. doi: 10.1038/jcbfm.2011.43
Nishiura, K., Ichikawa-Tomikawa, N., Sugimoto, K., Kunii, Y., Kashiwagi, K., Tanaka, M., et al. (2017). PKA activation and endothelial claudin-5 breakdown in the schizophrenic prefrontal cortex. Oncotarget 7, 93382–93391. doi: 10.18632/oncotarget.21850
Niwa, K., Carlson, G. A., and Iadecola, C. (2000). Exogenous Ab1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J. Cereb. Blood Flow Metab. 20, 1659–1668. doi: 10.1097/00004647-200012000-00005
Niwa, K., Kazama, K., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002). Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol. Dis. 9, 61–68. doi: 10.1006/nbdi.2001.0460
Novarino, G., El-Fishawy, P., Kayserili, H., Meguid, N. A., Scott, E. M., Schroth, J., et al. (2012). Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338, 394–397. doi: 10.1126/science.1224631
O’Brien, J. T., Eagger, S., Syed, G. M., Sahakian, B. J., and Levy, R. (1992). A study of regional cerebral blood flow and cognitive performance in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 55, 1182–1187. doi: 10.1136/jnnp.55.12.1182
Ochoa, S., Usall, J., Cobo, J., Labad, X., and Kulkarni, J. (2012). Gender differences in schizophrenia and first-episode psychosis: a comprehensive literature review. Schizophr. Res. Treat. 2012:916198. doi: 10.1155/2012/916198
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction [Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Neuron 39, 409–421.
Ohnishi, T., Matsuda, H., Hashimoto, T., Kunihiro, T., Nishikawa, M., Uema, T., et al. (2000). Abnormal regional cerebral blood flow in childhood autism. Brain 123(Pt 9), 1838–1844.
Okabe, K., Fukada, H., Tai-Nagara, I., Ando, T., Honda, T., Nakajima, K., et al. (2020). Neuron-derived VEGF contributes to cortical and hippocampal development independently of VEGFR1/2-mediated neurotrophism. Dev. Biol. 459, 65–71. doi: 10.1016/j.ydbio.2019.11.016
Ongali, B., Nicolakakis, N., Lecrux, C., Aboulkassim, T., Rosa-Neto, P., Papadopoulos, P., et al. (2010). Transgenic mice overexpressing APP and transforming growth factor-beta1 feature cognitive and vascular hallmarks of Alzheimer’s disease. Am. J. Pathol. 177, 3071–3080. doi: 10.2353/ajpath.2010.100339
Onore, C., Careaga, M., and Ashwood, P. (2012). The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 26, 383–392. doi: 10.1016/j.bbi.2011.08.007
Ortiz, G. G., Pacheco-Moises, F. P., Macias-Islas, M. A., Flores-Alvarado, L. J., Mireles-Ramirez, M. A., Gonzalez-Renovato, E. D., et al. (2014). Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 45, 687–697. doi: 10.1016/j.arcmed.2014.11.013
Ota, T., Sato, N., Nakata, Y., Ito, K., Kamiya, K., Maikusa, N., et al. (2013). Abnormalities of cerebral blood flow in multiple sclerosis: a pseudocontinuous arterial spin labeling MRI study. Magn. Reson. Imaging 31, 990–995. doi: 10.1016/j.mri.2013.03.016
Ouellette, J., Toussay, X., Comin, C. H., Costa, L. D. F., Ho, M., Lacalle-Aurioles, M., et al. (2020). Vascular contributions to 16p11.2 deletion autism syndrome modeled in mice. Nat. Neurosci. 23, 1090–1101. doi: 10.1038/s41593-020-0663-1
Padden, M., Leech, S., Craig, B., Kirk, J., Brankin, B., and McQuaid, S. (2007). Differences in expression of junctional adhesion molecule-A and beta-catenin in multiple sclerosis brain tissue: increasing evidence for the role of tight junction pathology. Acta Neuropathol. 113, 177–186. doi: 10.1007/s00401-006-0145-x
Pagano, G., Ferrara, N., Brooks, D. J., and Pavese, N. (2016). Age at onset and Parkinson disease phenotype. Neurology 86, 1400–1407. doi: 10.1212/WNL.0000000000002461
Palop, J. J., Jones, B., Kekonius, L., Chin, J., Yu, G. Q., Raber, J., et al. (2003). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 100, 9572–9577.
Papadaki, E. Z., Simos, P. G., Mastorodemos, V. C., Panou, T., Maris, T. G., Karantanas, A. H., et al. (2014). Regional MRI perfusion measures predict motor/executive function in patients with clinically isolated syndrome. Behav. Neurol. 2014:252419. doi: 10.1155/2014/252419
Pardridge, W. M. (2012). Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 1959–1972. doi: 10.1038/jcbfm.2012.126
Paris, D., Parker, T. A., Suo, Z., Fang, C., Humphrey, J., Crawford, F., et al. (1998). Role of peroxynitrite in the vasoactive and cytotoxic effects of Alzheimer’s beta-amyloid1-40 peptide. Exp. Neurol. 152, 116–122. doi: 10.1006/exnr.1998.6828
Park, H. R., Lee, J. M., Moon, H. E., Lee, D. S., Kim, B. N., Kim, J., et al. (2016). A short review on the current understanding of autism spectrum disorders. Exp. Neurobiol. 25, 1–13. doi: 10.5607/en.2016.25.1.1
Park, L., Wang, G., Zhou, P., Zhou, J., Pitstick, R., Previti, M. L., et al. (2011). Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc. Natl. Acad. Sci. U.S.A. 108, 5063–5068. doi: 10.1073/pnas.1015413108
Parkinson Canada (2010). A Manual for People Living with Parkinson’s Disease. Available online at: https://www.parkinson.ca/gated/parkinsons-disease-an-introductory-guide/ (accessed July 5, 2021).
Parodi-Rullan, R., Ghiso, J., Cabrera, E., Rostagno, A., and Fossati, S. (2020). Alzheimer’s amyloid beta heterogeneous species differentially affect brain endothelial cell viability, blood-brain barrier integrity, and angiogenesis. Aging Cell 19:e13258. doi: 10.1111/acel.13258
Patel, A., Toia, G. V., Colletta, K., Bradaric, B. D., Carvey, P. M., and Hendey, B. (2011). An angiogenic inhibitor, cyclic RGDfV, attenuates MPTP-induced dopamine neuron toxicity. Exp. Neurol. 231, 160–170. doi: 10.1016/j.expneurol.2011.06.004
Patel, N. S., Mathura, V. S., Bachmeier, C., Beaulieu-Abdelahad, D., Laporte, V., Weeks, O., et al. (2010). Alzheimer’s beta-amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. J. Neurochem. 112, 66–76. doi: 10.1111/j.1471-4159.2009.06426.x
Paulsen, J. S. (2011). Cognitive impairment in Huntington disease: diagnosis and treatment [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Curr. Neurol. Neurosci. Rep. 11, 474–483. doi: 10.1007/s11910-011-0215-x
Peguera, B., Segarra, M., and Acker-Palmer, A. (2021). Neurovascular crosstalk coordinates the central nervous system development. Curr. Opin. Neurobiol. 69, 202–213. doi: 10.1016/j.conb.2021.04.005
Perry, D. C., Sturm, V. E., Peterson, M. J., Pieper, C. F., Bullock, T., Boeve, B. F., et al. (2016). Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J. Neurosurg. 124, 511–526. doi: 10.3171/2015.2.JNS14503
Peruzzo, D., Castellaro, M., Calabrese, M., Veronese, E., Rinaldi, F., Bernardi, V., et al. (2013). Heterogeneity of cortical lesions in multiple sclerosis: an MRI perfusion study. J. Cereb. Blood Flow Metab. 33, 457–463. doi: 10.1038/jcbfm.2012.192
Peterson, B. S., Zargarian, A., Peterson, J. B., Goh, S., Sawardekar, S., Williams, S. C. R., et al. (2019). Hyperperfusion of frontal white and subcortical gray matter in autism spectrum disorder. Biol. Psychiatry 85, 584–595. doi: 10.1016/j.biopsych.2018.11.026
Pinkham, A., Loughead, J., Ruparel, K., Wu, W. C., Overton, E., Gur, R., et al. (2011). Resting quantitative cerebral blood flow in schizophrenia measured by pulsed arterial spin labeling perfusion MRI. Psychiatry Res. 194, 64–72. doi: 10.1016/j.pscychresns.2011.06.013
Plana-Ripoll, O., Pedersen, C. B., Holtz, Y., Benros, M. E., Dalsgaard, S., de Jonge, P., et al. (2019). Exploring comorbidity within mental disorders among a danish national population. JAMA Psychiatry 76, 259–270. doi: 10.1001/jamapsychiatry.2018.3658
Profaci, C. P., Munji, R. N., Pulido, R. S., and Daneman, R. (2020). The blood-brain barrier in health and disease: important unanswered questions. J. Exp. Med. 217:e20190062. doi: 10.1084/jem.20190062
Prohovnik, I., Mayeux, R., Sackeim, H. A., Smith, G., Stern, Y., and Alderson, P. O. (1988). Cerebral perfusion as a diagnostic marker of early Alzheimer’s disease. Neurology 38, 931–937. doi: 10.1212/WNL.38.6.931
Pu, S., Nakagome, K., Yamada, T., Itakura, M., Yamanashi, T., Yamada, S., et al. (2016). Social cognition and prefrontal hemodynamic responses during a working memory task in schizophrenia. Sci. Rep. 6:22500. doi: 10.1038/srep22500
Quaegebeur, A., Lange, C., and Carmeliet, P. (2011). The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron 71, 406–424. doi: 10.1016/j.neuron.2011.07.013
Ramos-Cejudo, J., Wisniewski, T., Marmar, C., Zetterberg, H., Blennow, K., de Leon, M. J., et al. (2018). Traumatic brain injury and Alzheimer’s disease: the cerebrovascular link. EBioMedicine 28, 21–30. doi: 10.1016/j.ebiom.2018.01.021
Rawlins, M. D., Wexler, N. S., Wexler, A. R., Tabrizi, S. J., Douglas, I., Evans, S. J., et al. (2016). The prevalence of Huntington’s disease. Neuroepidemiology 46, 144–153. doi: 10.1159/000443738
Reese, T. S., and Karnovsky, M. (1967). Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 34, 207–217. doi: 10.1083/jcb.34.1.207
Reid, I. C., Besson, J. A. O., Best, P. V., Sharp, P. F., Gemmell, H. G., and Smith, F. W. (1988). Imaging of cerebral blood flow markers in Huntington’s disease using single photon emission computed tomography. J. Neurol. Neurosurg. Psychiatry 51, 1264–1268. doi: 10.1136/jnnp.51.10.1264
Reijmer, Y. D., van Veluw, S. J., and Greenberg, S. M. (2016). Ischemic brain injury in cerebral amyloid angiopathy. J. Cereb. Blood Flow Metab. 36, 40–54. doi: 10.1038/jcbfm.2015.88
Reynell, C., and Harris, J. J. (2013). The BOLD signal and neurovascular coupling in autism. Dev. Cogn. Neurosci. 6, 72–79. doi: 10.1016/j.dcn.2013.07.003
Ribe, A. R., Laursen, T. M., Charles, M., Katon, W., Fenger-Gron, M., Davydow, D., et al. (2015). Long-term Risk of dementia in persons with Schizophrenia: a Danish population-based cohort study. JAMA Psychiatry 72, 1095–1101. doi: 10.1001/jamapsychiatry.2015.1546
Rite, I., Machado, A., Cano, J., and Venero, J. L. (2007). Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J. Neurochem. 101, 1567–1582. doi: 10.1111/j.1471-4159.2007.04567.x
Rizzo, M. T., and Leaver, H. A. (2010). Brain endothelial cell death: modes, signaling pathways, and relevance to neural development, homeostasis, and disease. Mol. Neurobiol. 42, 52–63. doi: 10.1007/s12035-010-8132-6
Roher, A. E., Esh, C., Kokjohn, T. A., Kalback, W., Luehrs, D. C., Seward, J. D., et al. (2003). Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler. Thromb. Vasc. Biol. 23, 2055–2062.
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/S1474-4422(10)70245-3
Ross, T. M., Martinez, P. M., Renner, J. C., Thorne, R. G., Hanson, L. R., and Frey, W. H. II (2004). Intranasal administration of interferon beta bypasses the blood–brain barrier to target the central nervous system and cervical lymph nodes: a non-invasive treatment strategy for multiple sclerosis. J. Neuroinflammation 151, 66–77. doi: 10.1016/j.jneuroim.2004.02.011
Rossignol, D. A., Bradstreet, J. J., Van Dyke, K., Schneider, C., Freedenfeld, S. H., O’Hara, N., et al. (2012). Hyperbaric oxygen treatment in autism spectrum disorders. Med. Gas Res. 2:16.
Russo, S. J., and Nestler, E. J. (2013). The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625. doi: 10.1038/nrn3381
Ryu, J. K., and McLarnon, J. G. (2009). A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 13, 2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x
Saade, C., Bou-Fakhredin, R., Yousem, D. M., Asmar, K., Naffaa, L., and El-Merhi, F. (2018). Gadolinium and multiple sclerosis: vessels, barriers of the brain, and glymphatics. AJNR Am. J. Neuroradiol. 39, 2168–2176. doi: 10.3174/ajnr.A5773
Sabri, O., Erkwoh, R., Schreckenberger, M., Owega, A., Sass, H., and Buell, U. (1997). Correlation of positive symptoms exclusively to hyperperfusion or hypoperfusion of cerebral cortex in never-treated schizophrenics. Lancet 349, 1735–1739. doi: 10.1016/s0140-6736(96)08380-8
Sagare, A. P., Bell, R. D., Zhao, Z., Ma, Q., Winkler, E. A., Ramanathan, A., et al. (2013). Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 4:2932. doi: 10.1038/ncomms3932
Saindane, A. M., Law, M., Ge, Y., Johnson, G., Babb, J. S., and Grossman, R. I. (2007). Correlation of diffusion tensor and dynamic perfusion MR imaging metrics in normal- appearing corpus callosum: support for primary hypoperfusion in multiple sclerosis. Am. J. Neuroradiol. 28, 767–772.
Sakulchit, T., Ladish, C., and Goldman, R. D. (2017). Hyperbaric oxygen therapy for children with autism spectrum disorder. Can. Fam. Physician 63, 446–448.
Salehi, A., Ashford, J. W., and Mufson, E. J. (2016). The link between Alzheimer’s disease and down syndrome. a historical perspective. Curr. Alzheimer Res. 13, 2–6. doi: 10.2174/1567205012999151021102914
Salisbury, D., and Bronas, U. (2015). Reactive oxygen and nitrogen species: impact on endothelial dysfunction. Nurs. Res. 64, 53–66. doi: 10.1097/NNR.0000000000000068
Salloway, S., Gur, T., Berzin, T., Zipser, B., Correia, S., Hovanesian, V., et al. (2002). Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J. Neurol. Sci. 203-204, 183–187. doi: 10.1016/s0022-510x(02)00288-5
Salmina, A. B., Morgun, A. V., Kuvacheva, N. V., Pozhilenkova, E. A., Solonchuk, Y. R., Lopatina, O. L., et al. (2014). Endothelial progenitor cells in cerebral endothelium development and repair. Curr. Technol. Med. 6, 213–221. doi: 10.1186/s13041-016-0193-7
Sandoo, A., Veldhuijzen van Zanten, J., Metsios, G., Carroll, D., and Kitas, G. (2010). The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 4, 302–312.
Saoud, H., Aflouk, Y., Ben Afia, A., Gaha, L., and Bel Hadj Jrad, B. (2021). Association of VEGF and KDR polymorphisms with the development of schizophrenia. medRxiv [preprint] doi: 10.1101/2021.08.06.21261566
Sarkar, S., Raymick, J., Mann, D., Bowyer, J. F., Hanig, J. P., Schmued, L. C., et al. (2014). Neurovascular changes in acute, sub-acute and chronic mouse models of Parkinson’s disease. Curr. Neurovasc. Res. 11, 48–61. doi: 10.2174/1567202610666131124234506
Sax, D. S., Powsner, R., Kim, A., Tilak, S., Bhatia, R., Cupples, L. A., et al. (1996). Evidence of cortical metabolic dysfunction in early Huntington’s disease by single-photon-emission computed tomography. Mov. Disord. 11, 671–677. doi: 10.1002/mds.870110612
Schaeffer, S., and Iadecola, C. (2021). Revisiting the neurovascular unit. Nat. Neurosci. 24, 1198–1209. doi: 10.1038/s41593-021-00904-7
Scheef, L., Manka, C., Daamen, M., Kühn, K., Maier, W., Schild, H., et al. (2010). Resting-State perfusion in nonmedicated schizophrenic patients: a continuous arterial spin-labeling 3.0-T MR study. Radiology 256, 253–260. doi: 10.1148/radiol.10091224
Schifter, T., Hoffman, J. M., Hatten, P. Jr., Hanson, M. W., Coleman, E., and DeLong, R. (1994). Neuroimaging in infantile autism. J. Child Neurol. 9, 155–161. doi: 10.1177/088307389400900210
Schuepbach, D., Egger, S. T., Boeker, H., Duschek, S., Vetter, S., Seifritz, E., et al. (2016). Determinants of cerebral hemodynamics during the Trail Making Test in schizophrenia. Brain Cogn. 109, 96–104. doi: 10.1016/j.bandc.2016.09.002
Schultz, S. K., O’Leary, D. S., Boles Ponto, L. L., Arndt, S., Magnotta, V., Leonard Watkins, G., et al. (2002). Age and regional cerebral blood flow in schizophrenia: age effects in anterior cingulate, frontal, and parietal cortex. J. Neuropsychiatry Clin. Neurosci. 14, 19–24. doi: 10.1176/jnp.14.1.19
Schwartz, M., and Kipnis, J. (2005). Protective autoimmunity and neuroprotection in inflammatory and noninflammatory neurodegenerative diseases. J. Neurol. Sci. 233, 163–166. doi: 10.1016/j.jns.2005.03.014
Seabrook, T. J., Littlewood-Evans, A., Brinkmann, V., Pöllinger, B., Schnell, C., and Hiestand, P. C. (2010). Angiogenesis is present in experimental autoimmune encephalomyelitis and pro- angiogenic factors are increased in multiple sclerosis lesions. J. Neuroinflammation 7:95. doi: 10.1186/1742-2094-7-95
Sender, R., and Milo, R. (2021). The distribution of cellular turnover in the human body. Nat. Med. 27, 45–48. doi: 10.1038/s41591-020-01182-9
Sengillo, J. D., Winkler, E. A., Walker, C. T., Sullivan, J. S., Johnson, M., and Zlokovic, B. V. (2013). Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 23, 303–310. doi: 10.1111/bpa.12004
Sharma, S., and Brown, C. E. (2021). Microvascular basis of cognitive impairment in type 1 diabetes. Pharmacol. Therap. 107929. doi: 10.1016/j.pharmthera.2021.107929
Shcherbakova, I., Neshkova, E., Dotsenko, V., Platonova, T., Shcherbakova, E., and Yarovaya, G. (1999). The possible role of plasma kallikrein-kinin system and leukocyte elastase in pathogenesis of schizophrenia. Immunopharmacology 43, 273–279. doi: 10.1016/s0162-3109(99)00099-5
Shen, Q., Goderie, S. K., Jin, L., Karanth, N., Sun, Y., Abramova, N., et al. (2004). Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 304, 1338–1340. doi: 10.1126/science.1095505
Siegel, B. V., Tanguay, P., Call, J. D., Abel, L., Ho, A., Lott, I., et al. (1992). Regional cerebral glucose metabolism and attention in adults with a history of childhood autism. J. Neuropsychiatry Clin. Neurosci. 4, 406–414. doi: 10.1176/jnp.4.4.406
Sieradzan, K. A., and Mann, D. M. (2001). The selective vulnerability of nerve cells in Huntington’s disease [Review]. Neuropathol. Appl. Neurobiol. 27, 1–21.
Silva-Garcia, O., Valdez-Alarcon, J. J., and Baizabal-Aguirre, V. M. (2019). Wnt/beta-Catenin signaling as a molecular target by pathogenic bacteria. Front. Immunol. 10:2135. doi: 10.3389/fimmu.2019.02135
Simpson, I. A., Chundu, K., Davies-Hill, T., Honer, W., and Davies, P. (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s diseas. Ann. Neurol. 35, 546–551. doi: 10.1002/ana.410350507
Smith, A. J., and Verkman, A. S. (2018). The “glymphatic” mechanism for solute clearance in Alzheimer’s disease: game changer or unproven speculation? FASEB J. 32, 543–551. doi: 10.1096/fj.201700999
Smith, C. D., Andersen, A. H., Kryscio, R. J., Schmitt, F. A., Kindy, M. S., Blonder, L. X., et al. (1999). Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology 53, 581–595.
Smith, E. E., and Greenberg, S. M. (2009). Beta-amyloid, blood vessels, and brain function. Stroke 40, 2601–2606. doi: 10.1161/STROKEAHA.108.536839
Smith, E. E., Vijayappa, M., Lima, F., Delgado, P., Wendell, L., Rosand, J., et al. (2008). Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology 71, 1424–1430. doi: 10.1212/01.wnl.0000327887.64299.a4
Smith, G. S., de Leon, M. J., George, A. E., Kluger, A., Volkow, N. D., McRae, T., et al. (1992). Topography of cross-sectional and longitudinal glucose metabolic deficits in Alzheimer’s disease. Arch. Neurol. 49, 1142–1150. doi: 10.1001/archneur.1992.00530350056020
Snowden, J. S. (2017). The neuropsychology of Huntington’s disease. Arch. Clin. Neuropsychol. 32, 876–887. doi: 10.1093/arclin/acx086
Solis, E. Jr., Hascup, K. N., and Hascup, E. R. (2020). Alzheimer’s disease: the link between amyloid-beta and neurovascular dysfunction. J. Alzheimers Dis. 76, 1179–1198. doi: 10.3233/JAD-200473
Song, C., Schwarzkopf, D. S., Lutti, A., Li, B., Kanai, R., and Rees, G. (2013). Effective connectivity within human primary visual cortex predicts interindividual diversity in illusory perception. J. Neurosci. 33, 18781–18791. doi: 10.1523/JNEUROSCI.4201-12.2013
Soto-Rojas, L. O., Pacheco-Herrero, M., Martinez-Gomez, P. A., Campa-Cordoba, B. B., Apatiga-Perez, R., Villegas-Rojas, M. M., et al. (2021). The neurovascular unit dysfunction in Alzheimer’s disease. Int. J. Mol. Sci. 22:2022. doi: 10.3390/ijms22042022
Sotrel, A., Paskevich, P. A., Kiely, D. K., Bird, E. D., Williams, R. S., and Myers, R. H. (1991). Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology 41, 1117–1123. doi: 10.1212/wnl.41.7.1117
Spuch, C., Antequera, D., Portero, A., Orive, G., Hernandez, R. M., Molina, J. A., et al. (2010). The effect of encapsulated VEGF-secreting cells on brain amyloid load and behavioral impairment in a mouse model of Alzheimer’s disease [Research Support, Non-U.S. Gov’t]. Biomaterials 31, 5608–5618. doi: 10.1016/j.biomaterials.2010.03.042
St-Amour, I., Aubé, B., Rieux, M., and Cicchetti, F. (2015). Targeting cerebrovascular impairments in Huntington’s disease: a novel treatment perspective. Future Med. 5, 389–393. doi: 10.2217/nmt.15.41
Stachowiak, M. K., Kucinski, A., Curl, R., Syposs, C., Yang, Y., Narla, S., et al. (2013). Schizophrenia: a neurodevelopmental disorder — Integrative genomic hypothesis and therapeutic implications from a transgenic mouse model. Schizophr. Res. 143, 367–376. doi: 10.1016/j.schres.2012.11.004
Stackhouse, T. L., and Mishra, A. (2021). Neurovascular coupling in development and disease: focus on astrocytes. Front. Cell Dev. Biol. 9:702832. doi: 10.3389/fcell.2021.702832
Stamatovic, S., Keep, R., and Andjelkovic, A. (2008). Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr. Neuropharmacol. 6, 179–192. doi: 10.2174/157015908785777210
Starkstein, S., Gellar, S., Parlier, M., Payne, L., and Piven, J. (2015). High rates of parkinsonism in adults with autism. J. Neurodev. Disord. 7:29. doi: 10.1186/s11689-015-9125-6
Stegmayer, K., Strik, W., Federspiel, A., Wiest, R., Bohlhalter, S., and Walther, S. (2017). Specific cerebral perfusion patterns in three schizophrenia symptom dimensions. Schizophr. Res. 190, 96–101. doi: 10.1016/j.schres.2017.03.018
Steventon, J. J., Furby, H., Ralph, J., O’Callaghan, P., Rosser, A. E., Wise, R. G., et al. (2020). Altered cerebrovascular response to acute exercise in patients with Huntington’s disease. Brain Commun. 2:fcaa044. doi: 10.1093/braincomms/fcaa044
Stone, J., Itin, A., Alon, T., Pe’er, J., Gnessin, H., Chan-Ling, T., et al. (1995). Development of retinal vasculature is mediated by hypoxia- induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 15, 4738–4747.
Storck, S. E., Meister, S., Nahrath, J., Meissner, J. N., Schubert, N., Di Spiezio, A., et al. (2016). Endothelial LRP1 transports amyloid-beta(1-42) across the blood-brain barrier. J. Clin. Invest. 126, 123–136. doi: 10.1172/JCI81108
Su, J. J., Osoegawa, M., Matsuoka, T., Minohara, M., Tanaka, M., Ishizu, T., et al. (2006). Upregulation of vascular growth factors in multiple sclerosis: correlation with MRI findings. J. Neurol. Sci. 243, 21–30. doi: 10.1016/j.jns.2005.11.006
Suo, Z., Humphrey, J., Kundtz, A., Sethi, F., Placzek, A., Crawford, F., et al. (1998). Soluble Alzheimers beta-amyloid constricts the cerebral vasculature in vivo. Neurosci. Lett. 257, 77–80. doi: 10.1016/s0304-3940(98)00814-3
Suri, S., Mackay, C. E., Kelly, M. E., Germuska, M., Tunbridge, E. M., Frisoni, G. B., et al. (2015). Reduced cerebrovascular reactivity in young adults carrying the APOE epsilon4 allele. Alzheimers Dement. 11, 648–657 e641. doi: 10.1016/j.jalz.2014.05.1755
Sweeney, M. D., Kisler, K., Montagne, A., Toga, A. W., and Zlokovic, B. V. (2018). The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 21, 1318–1331. doi: 10.1038/s41593-018-0234-x
Sweeney, M. D., Montagne, A., Sagare, A. P., Nation, D. A., Schneider, L. S., Chui, H. C., et al. (2019). Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 15, 158–167. doi: 10.1016/j.jalz.2018.07.222
Switzer, A. R., Cheema, I., McCreary, C. R., Zwiers, A., Charlton, A., Alvarez-Veronesi, A., et al. (2020). Cerebrovascular reactivity in cerebral amyloid angiopathy, Alzheimer disease, and mild cognitive impairment. Neurology 95, e1333–e1340. doi: 10.1212/WNL.0000000000010201
Syrimi, Z. J., Vojtisek, L., Eliasova, I., Viskova, J., Svatkova, A., Vanicek, J., et al. (2017). Arterial spin labelling detects posterior cortical hypoperfusion in non-demented patients with Parkinson’s disease. J. Neural Transm. (Vienna) 124, 551–557. doi: 10.1007/s00702-017-1703-1
Szu, J. I., and Obenaus, A. (2021). Cerebrovascular phenotypes in mouse models of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 41, 1821–1841. doi: 10.1177/0271678X21992462
Tai, L. M., Holloway, K. A., Male, D. K., Loughlin, A. J., and Romero, I. A. (2010). Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J. Cell. Mol. Med. 14, 1101–1112. doi: 10.1111/j.1582-4934.2009.00717.x
Tan, K. H., Harrington, S., Purcell, W. M., and Hurst, R. D. (2004). Peroxynitrite mediates nitric oxide–induced blood–brain barrier damage. Neurochem. Res. 29, 579–587. doi: 10.1023/b:nere.0000014828.32200.bd
Taoufik, E., Kouroupi, G., Zygogianni, O., and Matsas, R. (2018). Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: an overview of induced pluripotent stem-cell-based disease models. Open Biol. 8:180138. doi: 10.1098/rsob.180138
Tarlungeanu, D. C., Deliu, E., Dotter, C. P., Kara, M., Janiesch, P. C., Scalise, M., et al. (2016). Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494 e1418. doi: 10.1016/j.cell.2016.11.013
Tata, M., and Ruhrberg, C. (2018). Cross-talk between blood vessels and neural progenitors in the developing brain. Neuronal Signal. 2:NS20170139. doi: 10.1042/NS20170139
Tata, M., Ruhrberg, C., and Fantin, A. (2015). Vascularisation of the central nervous system. Mech. Dev. 138(Pt 1), 26–36. doi: 10.1016/j.mod.2015.07.001
Thal, D. R., Capetillo-Zarate, E., Larionov, S., Staufenbiel, M., Zurbruegg, S., and Beckmann, N. (2009). Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 30, 1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017
Thambisetty, M., Beason-Held, L., An, Y., Kraut, M. A., and Resnick, S. M. (2010). APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol. 67, 93–98. doi: 10.1001/archneurol.2009.913
Thomas, T., Thomas, G., McLendon, C., Sutton, T., and Mullan, M. (1996). β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380, 168–171. doi: 10.1038/380168a0
Tong, X. K., Nicolakakis, N., Kocharyan, A., and Hamel, E. (2005). Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J. Neurosci 25, 11165–11174.
Tong, Y., Hocke, L. M., and Frederick, B. B. (2019). Low frequency systemic hemodynamic “noise” in resting state BOLD fMRI: characteristics, causes, implications, mitigation strategies, and applications. Front. Neurosci. 13:787. doi: 10.3389/fnins.2019.00787
Tosh, J. L., Rhymes, E. R., Mumford, P., Whittaker, H. T., Pulford, L. J., Noy, S. J., et al. (2021). Genetic dissection of down syndrome-associated alterations in APP/amyloid-beta biology using mouse models. Sci. Rep. 11:5736. doi: 10.1038/s41598-021-85062-3
Trapp, B. D., and Stys, P. K. (2009). Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 8, 280–291. doi: 10.1016/S1474-4422(09)70043-2
Tregellas, J. R., Davalos, D. B., Rojas, D. C., Waldo, M. C., Gibson, L., Wylie, K., et al. (2007). Increased hemodynamic response in the hippocampus, thalamus and prefrontal cortex during abnormal sensory gating in schizophrenia. Schizophr. Res. 92, 262–272. doi: 10.1016/j.schres.2006.12.033
Turner, R. J., and Sharp, F. R. (2016). Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front. Cell. Neurosci. 10:56. doi: 10.3389/fncel.2016.00056
Uranova, N. A., Zimina, I. S., Vikhreva, O. V., Krukov, N. O., Rachmanova, V. I., and Orlovskaya, D. D. (2010). Ultrastructural damage of capillaries in the neocortex in schizophrenia. World J. Biol. Psychiatry 11, 567–578. doi: 10.3109/15622970903414188
Uratani, M., Ota, T., Iida, J., Okazaki, K., Yamamuro, K., Nakanishi, Y., et al. (2019). Reduced prefrontal hemodynamic response in pediatric autism spectrum disorder measured with near-infrared spectroscopy. Child Adolesc. Psychiatry Ment. Health 13:29. doi: 10.1186/s13034-019-0289-9
Usta, A., Kilic, F., Demirdas, A., Isik, U., Doguc, D. K., and Bozkurt, M. (2021). Serum zonulin and claudin-5 levels in patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 271, 767–773. doi: 10.1007/s00406-020-01152-9
van de Haar, H., Burgmans, S., Jansen, J., van Osch, M., van Buchem, M., Muller, M., et al. (2016). Blood-Brain barrier leakage in patients with early Alzheimer disease. Radiology 281, 527–535. doi: 10.1148/radiol.2016152244
Van Dyken, P., and Lacoste, B. (2018). Impact of metabolic syndrome on neuroinflammation and the blood–brain barrier. Front. Neurosci. 12:930. doi: 10.3389/fnins.2018.00930
Varga, A. W., Johnson, G., Babb, J. S., Herbert, J., Grossman, R. I., and Inglese, M. (2009). White matter hemodynamic abnormalities precede sub-cortical gray matter changes in multiple sclerosis. J. Neurol. Sci. 282, 28–33. doi: 10.1016/j.jns.2008.12.036
Verstraeten, A., Theuns, J., and Van Broeckhoven, C. (2015). Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 31, 140–149. doi: 10.1016/j.tig.2015.01.004
Vijayakumar, N. T., and Judy, M. V. (2016). Autism spectrum disorders: integration of the genome, transcriptome and the environment. J. Neurol. Sci. 364, 167–176. doi: 10.1016/j.jns.2016.03.026
Vinters, H. V., Secor, D. L., Read, S. L., Frazee, J. G., Tomiyasu, U., Stanley, T. M., et al. (1994). Microvasculature in brain biopsy specimens from patients with Alzheimer’s disease: an immunohistochemical and ultrastructural study [Research Support, U.S. Gov’t, P.H.S.]. Ultrastruct. Pathol. 18, 333–348.
Vostrikov, V., Orlovskaya, D. D., and Uranova, N. A. (2008). Deficit of pericapillary oligodendrocytes in the prefrontal cortex in schizophrenia. World J. Biol. Psychiatry 9, 34–42. doi: 10.1080/15622970701210247
Wada, K., Arai, H., Takahashi, M., Fukae, J., Oizumi, H., Yasuda, T., et al. (2006). Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport 17, 705–709. doi: 10.1097/01.wnr.0000215769.71657.65
Walchli, T., Wacker, A., Frei, K., Regli, L., Schwab, M. E., Hoerstrup, S. P., et al. (2015). Wiring the vascular network with neural cues: a CNS perspective. Neuron 87, 271–296. doi: 10.1016/j.neuron.2015.06.038
Walker, F. O. (2007). Huntington’s disease [Review]. Lancet 369, 218–228. doi: 10.1016/S0140-6736(07)60111-1
Wan, W., Cao, L., Liu, L., Zhang, C., Kalionis, B., Tai, X., et al. (2015). Abeta(1-42) oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 134, 382–393. doi: 10.1111/jnc.13122
Wang, T., Zhan, W., Chen, Q., Chen, N., Zhang, J., Liu, Q., et al. (2016). Altered resting-state ascending/descending pathways associated with the posterior thalamus in migraine without aura. Neuroreport 27, 257–263. doi: 10.1097/WNR.0000000000000529
Wardlaw, J. M., Farrall, A., Armitage, P. A., Carpenter, T., Chappell, F., Doubal, F., et al. (2008). Changes in background blood-brain barrier integrity between lacunar and cortical ischemic stroke subtypes. Stroke 39, 1327–1332. doi: 10.1161/STROKEAHA.107.500124
Wasmuth, J. J., Hewitt, J., Smith, B., Allard, D., Haines, J. L., Skarecky, D., et al. (1988). A highly polymorphic locus very tightly linked to the Huntington’s disease gene [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Nature 332, 734–736. doi: 10.1038/332734a0
Watanabe, C., Imaizumi, T., Kawai, H., Suda, K., Honma, Y., Ichihashi, M., et al. (2020). Aging of the vascular system and neural diseases. Front. Aging Neurosci. 12:557384. doi: 10.3389/fnagi.2020.557384
Watson, A. N., Berthiaume, A.-A., Faino, A. V., McDowell, K. P., Bhat, N. R., Hartmann, D. A., et al. (2020). Mild pericyte deficiency is associated with aberrant brain microvascular flow in aged PDGFRβ+/- mice. J. Cereb. Blood Flow Metab. 40, 2387–2400. doi: 10.1177/0271678X19900543
Weiss, N., Miller, F., Cazaubon, S., and Couraud, P. O. (2009). The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 1788, 842–857. doi: 10.1016/j.bbamem.2008.10.022
Whiteus, C., Freitas, C., and Grutzendler, J. (2014). Perturbed neural activity disrupts cerebral angiogenesis during a postnatal critical period. Nature 505, 407–411. doi: 10.1038/nature12821
Wild, E. J., and Fox, N. C. (2009). Serial volumetric MRI in Parkinsonian disorders. Mov. Disord. 24(Suppl. 2), S691–S698. doi: 10.1002/mds.22500
Willis, K. J., and Hakim, A. M. (2013). Stroke prevention and cognitive reserve: emerging approaches to modifying risk and delaying onset of dementia. Front. Neurol. 4:13. doi: 10.3389/fneur.2013.00013
Wiseman, F. K., Pulford, L. J., Barkus, C., Liao, F., Portelius, E., Webb, R., et al. (2018). Trisomy of human chromosome 21 enhances amyloid-beta deposition independently of an extra copy of APP. Brain 141, 2457–2474. doi: 10.1093/brain/awy159
Wolf, R. C., Gron, G., Sambataro, F., Vasic, N., Wolf, N. D., Thomann, P. A., et al. (2011). Magnetic resonance perfusion imaging of resting-state cerebral blood flow in preclinical Huntington’s disease. J. Cereb. Blood Flow Metab. 31, 1908–1918. doi: 10.1038/jcbfm.2011.60
Wu, Z., Guo, H., Chow, N., Sallstrom, J., Bell, R. D., Deane, R., et al. (2005). Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 11, 959–965. doi: 10.1038/nm1287
Wuerfel, J., Bellmann-Strobl, J., Brunecker, P., Aktas, O., McFarland, H., Villringer, A., et al. (2004). Changes in cerebral perfusion precede plaque formation in multiple sclerosis: a longitudinal perfusion MRI study. Brain 127(Pt 1), 111–119. doi: 10.1093/brain/awh007
Xiao, M., Xiao, Z. J., Yang, B., Lan, Z., and Fang, F. (2020). Blood-Brain barrier: more contributor to disruption of central nervous system homeostasis than victim in neurological disorders. Front. Neurosci. 14:764. doi: 10.3389/fnins.2020.00764
Yamada, H., Sadato, N., Konishi, Y., Muramuto, S., Kimura, K., Tanaka, M., et al. (2000). A milestone for normal development of the infantile brain detected by functional MRI. Neurology 55, 218–223.
Yang, P., Pavlovic, D., Waldvogel, H., Dragunow, M., Synek, B., Turner, C., et al. (2015). String vessel formation is increased in the brain of Parkinson disease. J. Parkinsons Dis. 5, 821–836. doi: 10.3233/JPD-140454
Yerys, B. E., Herrington, J. D., Bartley, G. K., Liu, H. S., Detre, J. A., and Schultz, R. T. (2018). Arterial spin labeling provides a reliable neurobiological marker of autism spectrum disorder. J. Neurodev. Disord. 10:32. doi: 10.1186/s11689-018-9250-0
Zarow, C., Barron, E., Chui, H. C., and Perlmutter, L. S. (1997). Vascular basement membrane pathology and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 147–160.
Zhang, Z. G., Zhang, L., Jiang, Q., Zhang, R., Davies, K., Powers, C., et al. (2000). VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Invest. 106, 829–838. doi: 10.1172/JCI9369
Zhao, Z., Sagare, A. P., Ma, Q., Halliday, M. R., Kong, P., Kisler, K., et al. (2015). Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci. 18, 978–987. doi: 10.1038/nn.4025
Zheng, W., Cui, B., Han, Y., Song, H., Li, K., He, Y., et al. (2019). Disrupted regional cerebral blood flow, functional activity and connectivity in Alzheimer’s disease: a combined ASL perfusion and resting state fMRI Study. Front. Neurosci. 13:738. doi: 10.3389/fnins.2019.00738
Zheng, Z., and Diamond, M. I. (2012). Huntington disease and the huntingtin protein [Review]. Prog. Mol. Biol. Transl. Sci. 107, 189–214. doi: 10.1016/B978-0-12-385883-2.00010-2
Zhu, J., Zhuo, C., Xu, L., Liu, F., Qin, W., and Yu, C. (2017). Altered coupling between resting-state cerebral blood flow and functional connectivity in schizophrenia. Schizophr. Bull. 43, 1363–1374. doi: 10.1093/schbul/sbx051
Zhuo, C., Zhu, J., Qin, W., Qu, H., Ma, X., and Yu, C. (2017). Cerebral blood flow alterations specific to auditory verbal hallucinations in schizophrenia. Br. J. Psychiatry 210, 209–215. doi: 10.1192/bjp.bp.115.174961
Zilbovicius, M., Boddaert, N., Belin, P., Poline, J.-B., Remy, P., Mangin, J.-F., et al. (2000). Temporal lobe dysfunction in childhood autism: a PET study. Am. J. Psychiatry 157, 1988–1993.
Zlokovic, B. V. (2005). Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 28, 202–208. doi: 10.1016/j.tins.2005.02.001
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Keywords: cerebrovascular abnormalities, neurodevelopment and intellectual disabilities, aging, neurodegeneration, cerebral blood flow, angiogenesis, blood-brain barrier
Citation: Ouellette J and Lacoste B (2021) From Neurodevelopmental to Neurodegenerative Disorders: The Vascular Continuum. Front. Aging Neurosci. 13:749026. doi: 10.3389/fnagi.2021.749026
Received: 28 July 2021; Accepted: 13 September 2021;
Published: 20 October 2021.
Edited by:
Anusha Mishra, Oregon Health and Science University, United StatesReviewed by:
Barbara Lykke Lind, University of Copenhagen, DenmarkRavi L. Rungta, Université de Montréal, Canada
Vanessa Coelho-Santos, Seattle Children’s Research Institute, United States
Copyright © 2021 Ouellette and Lacoste. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baptiste Lacoste, YmxhY29zdGVAdW90dGF3YS5jYQ==