 Lise Barbé1,2,3
Lise Barbé1,2,3 Steve Finkbeiner1,2,3*
Steve Finkbeiner1,2,3*- 1Center for Systems and Therapeutics, Gladstone Institutes, San Francisco, CA, United States
- 2Department of Neurology, University of California, San Francisco, San Francisco, CA, United States
- 3Department of Physiology, University of California, San Francisco, San Francisco, CA, United States
Repeat diseases, such as fragile X syndrome, myotonic dystrophy, Friedreich ataxia, Huntington disease, spinocerebellar ataxias, and some forms of amyotrophic lateral sclerosis, are caused by repetitive DNA sequences that are expanded in affected individuals. The age at which an individual begins to experience symptoms, and the severity of disease, are partially determined by the size of the repeat. However, the epigenetic state of the area in and around the repeat also plays an important role in determining the age of disease onset and the rate of disease progression. Many repeat diseases share a common epigenetic pattern of increased methylation at CpG islands near the repeat region. CpG islands are CG-rich sequences that are tightly regulated by methylation and are often found at gene enhancer or insulator elements in the genome. Methylation of CpG islands can inhibit binding of the transcriptional regulator CTCF, resulting in a closed chromatin state and gene down regulation. The downregulation of these genes leads to some disease-specific symptoms. Additionally, a genetic and epigenetic interplay is suggested by an effect of methylation on repeat instability, a hallmark of large repeat expansions that leads to increasing disease severity in successive generations. In this review, we will discuss the common epigenetic patterns shared across repeat diseases, how the genetics and epigenetics interact, and how this could be involved in disease manifestation. We also discuss the currently available stem cell and mouse models, which frequently do not recapitulate epigenetic patterns observed in human disease, and propose alternative strategies to study the role of epigenetics in repeat diseases.
Introduction
Nucleotide repeat expansions that extend the normal length of disease genes cause neuronal and neuromuscular diseases (Schmidt and Pearson, 2016). Most repeat diseases are caused by an expansion of trinucleotide repeats, typically a “CXG” expansion. Repeat diseases can originate from expansions in exons, introns, promoter regions, or the 5′ or 3′-UTR of genes (Ellerby, 2019). All classical repeat diseases involve neuronal dysfunction in patients, often accompanied by neuromuscular symptoms (Ellerby, 2019). Most repeat diseases are autosomal dominant, with an increase in disease severity and decrease in age of disease onset over successive generations (Ashizawa et al., 1992a,b). In addition, beyond classical repeat diseases, expanded tandem repeats are also thought to contribute to heritability in some cases of polygenic diseases, such as autism spectrum disorder (ASD) (Hannan, 2010).
Nucleotide repeats are found throughout the genome, but only certain repeat expansions cause disease (Sun et al., 2018). The mechanisms determining which repeat expansions lead to cellular dysfunction and disease remain unknown. In addition, the strength of the correlation of repeat expansion length to disease severity or age of onset varies and can be low. However, repeat length is not the only determining factor for the expression of symptoms in these diseases. Patients with similar repeat lengths can have very different ages of onset, progression and symptoms (Cobo et al., 1993; Barcelo et al., 1994; Clark et al., 1998; Langbehn et al., 2004; Wexler et al., 2004; De Antonio et al., 2016; Keum et al., 2016). Therefore, other mechanisms are thought to play a role in determining the effect repeat expansions have on cellular function and disease phenotype.
Methylation near a disease-implicated repeat region was first reported in Fragile X Syndrome, a trinucleotide repeat disease involving a CGG expansion in the 5′-UTR of the fragile X mental retardation I (FMR1) gene (Verkerk et al., 1991). Methylation at the promoter region of FMR1, upstream of the CGG repeat, silences the gene and causes neuronal dysfunction in patients (Oberle et al., 1991; Pieretti et al., 1991; Hansen et al., 1992; Sutcliffe et al., 1992; O’Donnell et al., 2002). Several other repeat diseases also show epigenetic changes in methylation, chromatin conformation and gene downregulation at the repeat regions.
In this review, we focus on six repeat diseases: myotonic dystrophy type I (DM1), fragile X syndrome (FXS), Friedreich ataxia (FRDA), amyotrophic lateral sclerosis (ALS), Huntington disease (HD), and spinocerebellar ataxias (SCA). Despite the different patterns of pathology, disease progression and onset, DNA methylation near repeat regions, CTCF binding and chromatin conformation changes are common in all these diseases (Table 1), and are linked to disease severity. Additionally, we discuss the interplay between genetics and epigenetics and their role in repeat instability and the evidence that expanded tandem repeats and increased methylation and chromatinization also contribute to ASD. Finally, we review the disease models that are currently available to study repeat diseases and the emerging importance of using transdifferentiated cells to study diseases that involve epigenetic mechanisms and aging.
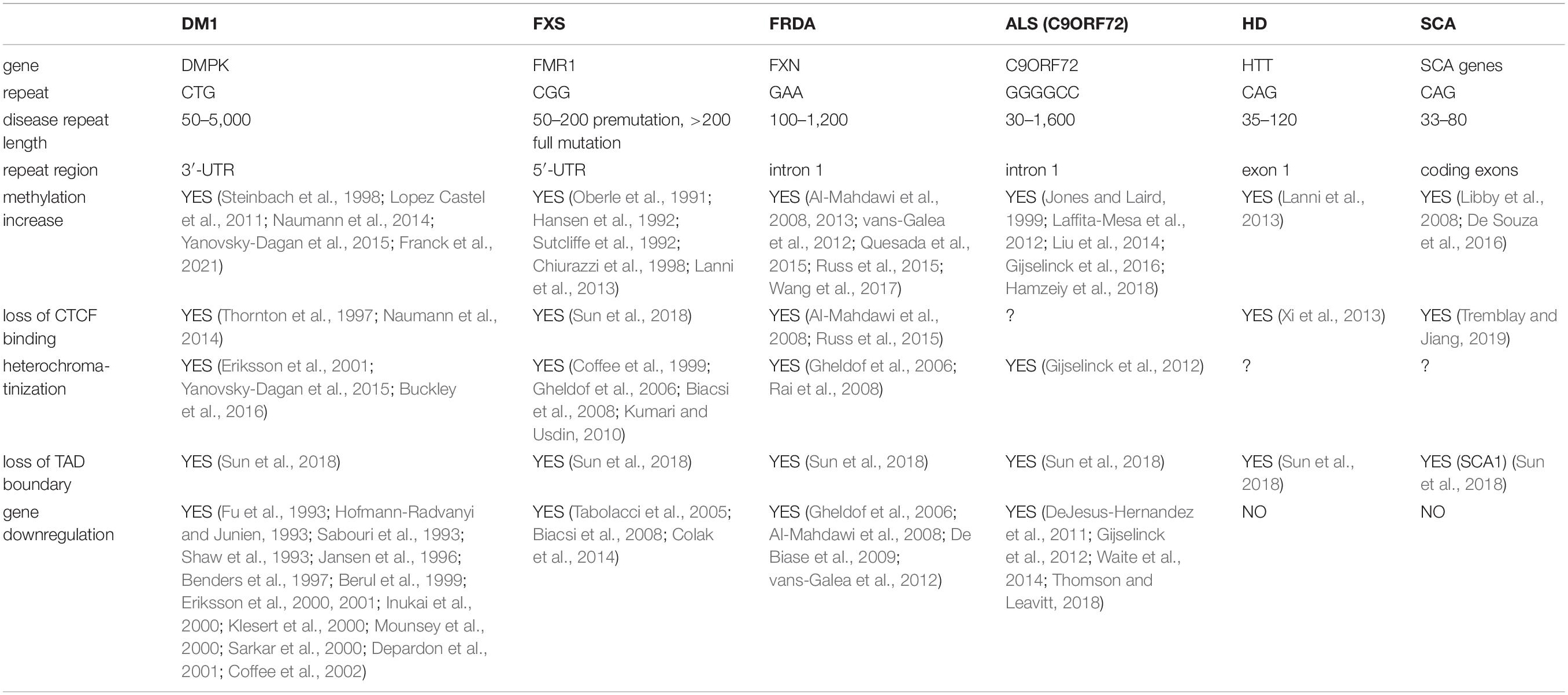
Table 1. A comparison of repeat diseases with different genetic characteristics shows a common epigenetic signature.
Genetics
Nucleotide repeat sequences called microsatellites are short stretches of bases that are repeated 5–50 times, and thousands of microsatellites are found across the genome (Richard et al., 2008). Microsatellites are useful for parental and cell line fingerprinting and cancer diagnosis (Gulcher, 2012). In repeat diseases, disease-specific microsatellites that occur in coding or non-coding regions are expanded past their typical length, with expansion exceeding a certain threshold resulting in disease. Repeat diseases show anticipation, a worsening of the disease and earlier disease onset in successive generations. Anticipation is explained by the inheritance of expanded repeats and an increase in repeat length in the offspring (Ridley et al., 1988; Ashizawa et al., 1992a,b; Shelbourne et al., 1992; Sutherland and Richards, 1992; Tsilfidis et al., 1992; Sano et al., 1994; Komure et al., 1995; La Spada, 1997; Nolin et al., 2003; Pearson, 2003; Hsiung et al., 2012). Furthermore, some individuals inherit pre-mutations, which are intermediate repeat lengths that do not induce disease symptoms in the individuals but do increase the chance of developing disease-causing expanded repeats in their offspring (Goldberg et al., 1993; Myers et al., 1993; Yamagata et al., 1994; Cossee et al., 1997; Stevanin et al., 1998; Tassone et al., 2000; Martorell et al., 2001; Aubry et al., 2008; Bourgeois et al., 2009; Nordin et al., 2015; Xi et al., 2015). Interruptions in the repetitive sequence have been found in a small subpopulation of patients for most repeat diseases. Interruptions tend to stabilize the repeat, and individuals with repeat interruptions show less dramatic disease phenotypes and later disease onset compared to patients with similar, but pure, repeat lengths (Matsuyama et al., 1999; Zuhlke et al., 2002; Stolle et al., 2008; Musova et al., 2009; Yrigollen et al., 2012; Hu et al., 2017; Ciosi et al., 2019; Genetic Modifiers of Huntington’s Disease (GeM-Hd) Consortium, 2019; Wright et al., 2019). Changes in repeat length within one individual are caused by slippage of DNA repair proteins on DNA loops, generated as a result of long repetitive sequences (Usdin et al., 2015; Schmidt and Pearson, 2016). This constant expansion and contraction of the repeat is called repeat instability, and results in different repeat lengths across affected tissues and even on a cell-to-cell basis within one tissue. The protective effect of repeat interruptions is likely due to increased repeat stability, since the interruptions prevent slippage of the DNA repair proteins (Gacy et al., 1995; Pearson et al., 1998a,b, 2005; Sobczak and Krzyzosiak, 2005; Kovtun and McMurray, 2008). Repeat size and instability is greater in disease-affected organs and tissues (Anvret et al., 1993; Ashizawa et al., 1993; Thornton et al., 1994; Chong et al., 1995; Dobkin et al., 1996; Takano et al., 1996; Watanabe et al., 2000; Kennedy et al., 2003; De Biase et al., 2007; Nordin et al., 2015; Mouro Pinto et al., 2020). Mismatch DNA repair proteins are thought to be the key regulators of repeat instability, and genetic variants in these genes are associated with a change in age of disease onset (Genetic Modifiers of Huntington’s Disease (GeM-Hd) Consortium, 2015, 2019; Bettencourt et al., 2016; Morales et al., 2016; Lee et al., 2017; Moss et al., 2017; Flower et al., 2019).
DM1 is caused by an expanded CTG repeat that exceeds 50 repeats in the 3′-UTR of the DM1 protein kinase (DMPK) gene (Aslanidis et al., 1992; Brook et al., 1992). Repeat size is somewhat correlated with disease severity (Yum et al., 2017). However, repeat lengths in individuals with the most severe form, congenital DM1, range from 500 to 3,000, which overlaps significantly with repeat length in individuals with the adult-onset form of the disease, which can range from 60 to 1,200 repeats (Cobo et al., 1993; Barcelo et al., 1994; Clark et al., 1998; De Antonio et al., 2016). This makes accurate prediction of disease phenotype and disease course based on repeat length impossible, and indicates that factors other than the repeat size play a role in disease phenotype and age of disease onset.
FXS is caused by a CGG repeat that exceeds 50 repeats in the 5′-UTR of the FRM1 gene (Verkerk et al., 1991). Disease phenotypes are strictly divided into two categories by a cut-off length of 200 repeats. Individuals with an expansion below 200 repeats develop fragile X-associated tremor/ataxia (FXTAS), with onset of muscular phenotypes followed by cognitive impairment in late adulthood. Expansion beyond 200 repeats is called a “full mutation,” and results in disease onset during childhood that includes excessive motor impairment and intellectual disability. Methylation at the FMR1 promoter region is specifically associated with full mutation alleles, and is the striking difference between FXTAS and FXS, highlighting the role epigenetics can play in the severity of disease manifestation (Jin and Warren, 2000).
FRDA is caused by a GAA repeat expansion beyond 100 repeats in the first intron of the frataxin (FXN) gene (Campuzano et al., 1996). In FRDA, repeat size is only minimally correlated with disease onset and disease phenotype. In fact, the shorter of the two alleles is more significantly correlated with age of disease onset than the expanded allele, and explains 25–50% of the variance in age of disease onset with a given expanded repeat (Filla et al., 1996; La Pean et al., 2008). Approximately 25% of patients have an unpredictable genotype-phenotype correlation. This is thought to be influenced by genetic background, interrupted GAA repeats, repeat instability and potentially other molecular signatures such as epigenetics (Montermini et al., 1997; Sharma et al., 2004; De Biase et al., 2007, 2009; Stolle et al., 2008).
One of the familial forms of ALS is caused by a hexanucleotide repeat GGGGCC (G4C2) repeat expansion in the first intron of the chromosome 9 open reading frame 72 (C9ORF72) gene (DeJesus-Hernandez et al., 2011). Interestingly, this same G4C2 expansion is also the single greatest genetic cause of frontotemporal dementia and the same families can get either disease or a mixture of symptoms of both (DeJesus-Hernandez et al., 2011). A phenotype-genotype correlation between repeat expansion size and ALS disease severity has not been established.
HD is caused by a CAG expansion of over 35 repeats in exon 1 of the HTT gene (MacDonald et al., 1993). Although correlation exists between repeat size and age of disease onset and death (Langbehn et al., 2004, 2010; Keum et al., 2016), only 60% of the age of onset can be predicted by CAG length itself (Wexler et al., 2004), and correlation between repeat length and disease progression is poor in common alleles (between 40 and 50 repeats) (Keum et al., 2016). Additionally, genetic variants in mismatch repair genes can change disease onset, potentially by influencing repeat instability (Genetic Modifiers of Huntington’s Disease (GeM-Hd) Consortium, 2015, 2019).
SCA is a collection of diseases, some of which are caused by expanded CAG repeats in the coding region of various genes. For ataxias SCA1, SCA2, SCA3, SCA6, SCA7, SCA17 and dentatorubral-pallidoluysian atrophy, expansion beyond 39, 33, 45, 20, 34, 41, and 35 repeats in the ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, TBP, and ATN1 genes, respectively, will cause disease. Pathologic repeat sizes are relatively small for most CAG repeat spinocerebellar ataxia disorders, and research on the correlation between molecular repeat characteristics and disease phenotype is limited. For SCA1, the number of CAG repeats is correlated with disease severity, and additional molecular characteristics, such as repeat interruption, are thought to play more of a role (Goldfarb et al., 1996; Matsuyama et al., 1999; Menon et al., 2013). Therefore, prediction of disease onset and severity is most accurate when considering the stretch of uninterrupted CAG repeats (Matsuyama et al., 1999). SCA17 patients typically have interrupted repeats, and the total length of the repeat is counted as a combination of the number of CAG and CAA repeats at the repeat locus. Atypical cases with uninterrupted repeats show increased disease severity, onset and anticipation of the disease in successive generations (Zuhlke et al., 2003).
ASD is predominantly a polygenic disorder, although some cases have been linked to mutations in the FMR1 gene, which is expanded in FXS (McCary and Roberts, 2013), and others to repeat expansions in DMPK, FXN, and HTT, which cause DM1, FRDA, and HD, respectively (Piras et al., 2020; Trost et al., 2020). In addition, an increase in expanded tandem repeats in genes related to neuronal development and function was recently described in children with ASD compared to controls (Trost et al., 2020; Mitra et al., 2021). Lastly, mutations in genes encoding proteins important in chromatin regulation and epigenetic machinery are also observed in higher prevalence in ASD patients (De Rubeis et al., 2014). Taken together, these studies indicate that diverse genetic alterations converge on pathological mechanisms similar to those implicated in classical repeat diseases to cause ASD.
In addition to differences in repeat size and genetic variants in repair genes, we and others have shown that epigenetics is a predictor of age of disease onset and disease severity. Epigenetic marks such as methylation and chromatinization are increased at the repeat loci of patients with severe disease forms and early age of disease onset (Castaldo et al., 2008; Godler et al., 2010; Lanni et al., 2013; Xi et al., 2013; Naumann et al., 2014; De Souza et al., 2016; Barbe et al., 2017; Wang et al., 2017). Next, we discuss epigenetic marks and regulation at the repeat region of repeat diseases, and their influence on gene expression and disease phenotype.
Methylation and CTCF Binding
DNA methylation is an epigenetic mark that changes the expression, regulation and organization of DNA. DNA methylation is essential for normal biological function and is associated with key processes important from early development to adulthood, such as X chromosome inactivation, gene imprinting, carcinogenesis and aging (Jones and Laird, 1999; Baylin et al., 2001; Robertson, 2001). Methylation occurs on cytosine residues, primarily those that are adjacent to guanine residues, and stretches of DNA with high incidence of CG sequences are most likely to be methylated. Such regions include promoters, enhancers and insulators, where methylation is typically associated with the inhibition of gene expression (Klose and Bird, 2006).
Stretches of hundreds of base pairs with a high percentage of CG sequences are called CpG islands. In many repeat disorders, the repeat expansions are located within or adjacent to CpG islands, thus DNA methylation can have an impact on disease phenotype. For many repeat diseases, CCCTC binding factor (CTCF) binding sites are located in the disease-associated CpG islands, and methylation results in an inhibition of CTCF binding. CTCF is a transcriptional repressor and often found at topologically associated domain (TAD) boundaries that regulate chromatin organization and gene expression (Nora et al., 2017). CTCF often co-localizes with cohesin, a protein important in the looping of the DNA during heterochromatin formation (Kagey et al., 2010).
Methylation up- and downstream of the CTG repeat has been noted in DM1 (Steinbach et al., 1998; Lopez Castel et al., 2011; Brouwer et al., 2013; Yanovsky-Dagan et al., 2015; Barbe et al., 2017), although almost exclusively in the most severe patients (Barbe et al., 2017). Two CTCF binding sites are located in the differentially methylated CpG islands up and downstream of the CTG repeat (Barbe et al., 2017; Franck et al., 2021), and are part of a regulatory element that controls DMPK, DMWD, and SIX5 expression (Fu et al., 1993; Sabouri et al., 1993; Hamshere et al., 1997; Thornton et al., 1997; Alwazzan et al., 1999; Inukai et al., 2000; Narang et al., 2000; Depardon et al., 2001; Filippova et al., 2001; Yanovsky-Dagan et al., 2015). Methylation upstream of the CTG repeat region has exclusively been detected in blood, chorionic villi samples and human embryonic stem cells from individuals with maternal inheritance of the disease (Barbe et al., 2017). Maternal transmission is also biased toward longer CTG repeat sizes and more severe disease forms compared to paternal transmission (Harper et al., 1972; Harley et al., 1993; Lavedan et al., 1993; Pearson, 2003; Lagrue et al., 2019).
A methylation boundary upstream of the CGG repeat is lost in FXS (Naumann et al., 2009, 2014). This methylation boundary is located 65 CpG sites upstream of the repeat, at a CTCF binding site, and inactivates the promoter region when methylated, silencing FMR1 (Oberle et al., 1991; Pieretti et al., 1991; Verkerk et al., 1991; Hansen et al., 1992; Sutcliffe et al., 1992; O’Donnell et al., 2002). Loss of FMR1 protein is the cause of FXS, and patients with atypically unmethylated promoter regions with full CGG repeat expansions are identified as “high functioning,” and lack FXS disease symptoms compared to patients with similar CGG expansions with methylation at the FMR1 promoter region, underscoring the importance of methylation in the disease manifestation (Godler et al., 2010; Lanni et al., 2013; Naumann et al., 2014). This understanding has led to the pursuit of methylation inhibitors, such as 5-aza-2-deoxycytidine, to reactivate FMR1 expression and potentially treat the disease (Chiurazzi et al., 1998; Pietrobono et al., 2002). However, toxicity has hampered the clinical roll-out of these approaches so far.
Methylation is increased in FRDA patients directly upstream of the GAA repeat and further upstream at a CpG island encompassing the promoter region, first exon and first intron of the FXN gene (Greene et al., 2007; Al-Mahdawi et al., 2008, 2013; Castaldo et al., 2008; vans-Galea et al., 2012; Quesada et al., 2015). CpG sites directly downstream of the repeat are hypomethylated in patients versus controls (Al-Mahdawi et al., 2008; vans-Galea et al., 2012). A subset of specific CpG sites in the FXN promoter and exon 1 are completely methylation-free in unaffected individuals but predominantly methylated in FRDA patients’ blood, brain, cerebellum and heart tissues (Greene et al., 2007; Al-Mahdawi et al., 2008). One of these CpG sites is located in a regulatory sequence that includes a CTCF binding sequence, and deletion of the regulatory sequence results in a drop in promoter activity (Greene et al., 2007), indicating that methylation could be responsible for gene expression changes. Increased levels of methylation are correlated with longer GAA repeats (Castaldo et al., 2008; vans-Galea et al., 2012) and earlier onset of disease (Castaldo et al., 2008).
Methylation is increased at the CpG island in the C9ORF72 promoter in ALS patients with expanded C9ORF72 hexanucleotide G4C2 repeats (Xi et al., 2013; Russ et al., 2015; Gijselinck et al., 2016; Hamzeiy et al., 2018), and higher methylation levels are correlated with shorter disease duration (Xi et al., 2013) and longer repeat lengths (Gijselinck et al., 2016). Healthy individuals and patients with short repeat sizes do not show any methylation (Xi et al., 2013; Russ et al., 2015) in the C9ORF72 promoter. The CpG island downstream of the G4C2 repeat remains unmethylated in patients, similar to control individuals (Xi et al., 2013; Liu et al., 2014).
In HD, there is a CpG methylation boundary > 700 bp upstream of the HTT promoter, around 1200 bp upstream of the CAG repeat. The methylation boundary mostly stays intact in HD patients (Naumann et al., 2014). However, tissue-specific methylation differences between HD patients and controls have been found in the cortex, where differential methylation is found at a CTCF binding site in the HTT proximal promoter and increased DNA methylation is associated with earlier age of disease onset (De Souza et al., 2016; Lu et al., 2020).
The promoter regions of ATXN2 and ATXN3 are methylated in SCA2 and SCA3 patients with expanded repeats (Laffita-Mesa et al., 2012; Wang et al., 2017), and increased levels of methylation are found in the CTCF binding domains of ATXN7 in SCA7 patients (Libby et al., 2008). In SCA3, methylation of the ATXN3 promoter region is correlated with younger age of disease onset, and families with intergenerational CAG repeat instability exhibit higher methylation levels (Wang et al., 2017).
Whole-exome and whole-genome sequencing has revealed a higher incidence of mutations in genes that regulate DNA methylation in ASD patients compared to controls (Tremblay and Jiang, 2019). This results in multiple differentially methylated regions in autism, primarily at promoter CpG islands (Ladd-Acosta et al., 2014; Nardone et al., 2014). In ASD, differential methylation is also found at intragenic sites predicted to alter splicing, which results in changes to transcript isoforms (Maunakea et al., 2013; Irimia et al., 2014; Zhu et al., 2014; Quesnel-Vallieres et al., 2016).
In summary, classical repeat diseases are characterized by remarkably similar methylation patterns, with increased methylation upstream of the repeat region, often at CpG islands in or near the promoter region of the affected gene. Also, all of these differentially methylated regions harbor a CTCF binding site that is inhibited by methylation, which could in turn impact chromatinization and gene expression, as discussed below.
Chromatin Organization and Gene Regulation
Chromatin organization regulates gene expression through DNA compaction. CTCF binding sites are disrupted upon increased methylation in repeat diseases, as discussed above, which results in chromatin changes in the repeat regions. In general, increased suppressing and decreased activating chromatin marks are found at the repeat regions for many repeat diseases, and disease-causing repeats are typically localized at TAD boundaries, which are disrupted by increased methylation and loss of CTCF binding. Chromatin compaction and loss of TAD boundaries results in gene downregulation in repeat diseases, which has been related to specific disease characteristics and symptoms.
In mouse models of DM1, a decrease in the active histone mark histone 3 lysine 9/14 acetylation (H3K9/14ac) and an enrichment of histone 3 lysine 27 trimethylation (H3K27me3) results in decreased expression of DMPK and sine oculis homeobox homolog 5 gene (SIX5) (Brouwer et al., 2013). In addition, an increase in H3K9 and H3K4 methylation is found in human DM1 fibroblast cell lines (Cho et al., 2005). This altered chromatin pattern toward compaction was confirmed in disease-relevant tissues such as myoblasts, myotubes, skeletal muscle, heart, lung and osteoblasts (Buckley et al., 2016), and accordingly expression of DMPK and SIX5 has been reported to be decreased in these tissues in DM1 patients (Fu et al., 1993; Hofmann-Radvanyi and Junien, 1993; Sabouri et al., 1993; Thornton et al., 1997; Alwazzan et al., 1999; Eriksson et al., 2000, 2001; Buckley et al., 2016). DMPK is involved in muscle function, and decreased DMPK expression has been linked to muscle impairment and cardiac disease in patients and mouse models (Benders et al., 1997; Berul et al., 1999; Mounsey et al., 2000). A decrease of SIX5 expression is thought to cause cataracts in DM1 patients (Thornton et al., 1997; Alwazzan et al., 1999; Klesert et al., 2000; Sarkar et al., 2000). Male infertility has been associated with two genes further upstream in the repeat region, DMWD and RSPH6A, for which transcription is also reduced in DM1 patients (Shaw et al., 1993; Jansen et al., 1996; Alwazzan et al., 1999; Eriksson et al., 2001). Finally, a TAD boundary at the repeat region and CTCF binding site was recently shown to be disrupted in DM1, indicating that chromatin organization may be more broadly altered in the disease (Sun et al., 2018).
In FXS, expansion of the CGG repeat to > 200 repeats leads to a chromatin reorganization from euchromatin to heterochromatin. In this setting, heterochromatin marks H3K27me3, H4K20me3 and H3K9me2/3 are increased, and euchromatin marks H3 and H4 acetylation and H3K4me2 are decreased (Coffee et al., 1999, 2002; Gheldof et al., 2006; Kumari and Usdin, 2010). These chromatin changes result in the loss of a TAD boundary near the FMR1 gene, leading to dysregulated 3D chromatin structure with decreased genomic interactions directly at and downstream of the repeat region, and increased interactions of the FMR1 gene with upstream gene regions (Gheldof et al., 2006; Sun et al., 2018). The loss of the TAD boundary has directly been linked with the loss of CTCF binding at the FMR1 promoter region (Sun et al., 2018). As a result, FMR1 is silenced, and indeed reduced FMR1 protein is found in patient lymphoblast cells (Coffee et al., 1999). In a cell model, treatment with 5-aza-2′-deoxycytidine reinitiated acetylation on histones H3 and H4 and transcriptional reactivation of the FMR1 gene in lymphoblastoid cell lines (Coffee et al., 1999; Tabolacci et al., 2005; Biacsi et al., 2008). Interestingly, RNA-DNA duplexes of the expanded RNA binding to the repeat expansion in the DNA have been linked to silencing of the FMR1 gene, and blockage of this interaction reverts chromatin marks to a euchromatin state and reactivates FMR1 expression (Colak et al., 2014).
The GAA repeat region in FRDA is located near a TAD boundary, which is lost upon repeat expansion in FRDA patients (Sun et al., 2018). This is accompanied by loss of activating chromatin marks including acetylation of histones H3 and H4 (Herman et al., 2006). Heterochromatin marks H3K9me2/3 and H3K27me3 are increased in the repeat region, and the FXN gene is downregulated in patient cell lines, brain tissue and mouse models of FRDA (Greene et al., 2007; Al-Mahdawi et al., 2008; De Biase et al., 2009). Treatment of FRDA patient lymphoblast cells and FRDA mouse models with deacetylase inhibitors increases FXN expression (Herman et al., 2006; Rai et al., 2008; Sandi et al., 2011), suggesting a primary role of chromatin compaction in the regulation of FXN gene expression in disease. Also, removal of the full intron 1 sequence rather than the repeat region only was able to restore cellular and molecular deficits of the disease in dorsal root ganglia organoids, again suggesting that chromatin compaction of the larger region is involved in disease pathogenesis (Mazzara et al., 2020).
In C9ORF72 familial cases of ALS, there is also an increase in chromatin suppressive marks such as trimethylation at residues H3K9, H3K27, H3K79 and H4K20 (Belzil et al., 2013) and a TAD boundary is located at the C9ORF72 locus (Sun et al., 2018). C9ORF72 transcript and protein levels are downregulated in frontal cortices and cerebelli of C9ORF72 familial cases of ALS patients (DeJesus-Hernandez et al., 2011; Gijselinck et al., 2012; Belzil et al., 2013; Waite et al., 2014) and methylation inhibition of the chromatin lysine residues decreases methylation at the repeat region and restores C9ORF72 transcript levels in patient fibroblast cell lines (DeJesus-Hernandez et al., 2011; Gijselinck et al., 2012; Belzil et al., 2013).
In HD, a TAD boundary is located at the differentially methylated CTCF site upstream of the repeat which remodels chromatin compaction and can change gene-enhancer interactions (Sun et al., 2018). Although overall HTT levels are typically not reported to be downregulated in HD patients, supplementation of wild type HTT does decrease neuronal toxicity in mice (Leavitt et al., 2001; Thomson and Leavitt, 2018). Therefore, finding a balance between mutant and wild type HTT levels is key, and mutant HTT lowering strategies are well underway as an HD therapeutic.
A disrupted TAD boundary was recently identified at the repeat region of the ATXN1 gene in SCA1 patients (Sun et al., 2018) but an understanding of the chromatin regulation of the other spinocerebellar ataxia-causing genes remains lacking.
While ASD can have different genetic causes, the level of heterochromatin mark H3K4me3 (Shulha et al., 2012) and acetylation of H3K27 (Sun et al., 2016) are overall increased in post-mortem brain samples from patients with ASD.
Taken together, these studies indicate a common epigenetic signature across repeat diseases characterized by increased DNA methylation, loss of CTCF binding, loss of TAD boundaries and increased chromatinization at the repeat region (Figure 1).
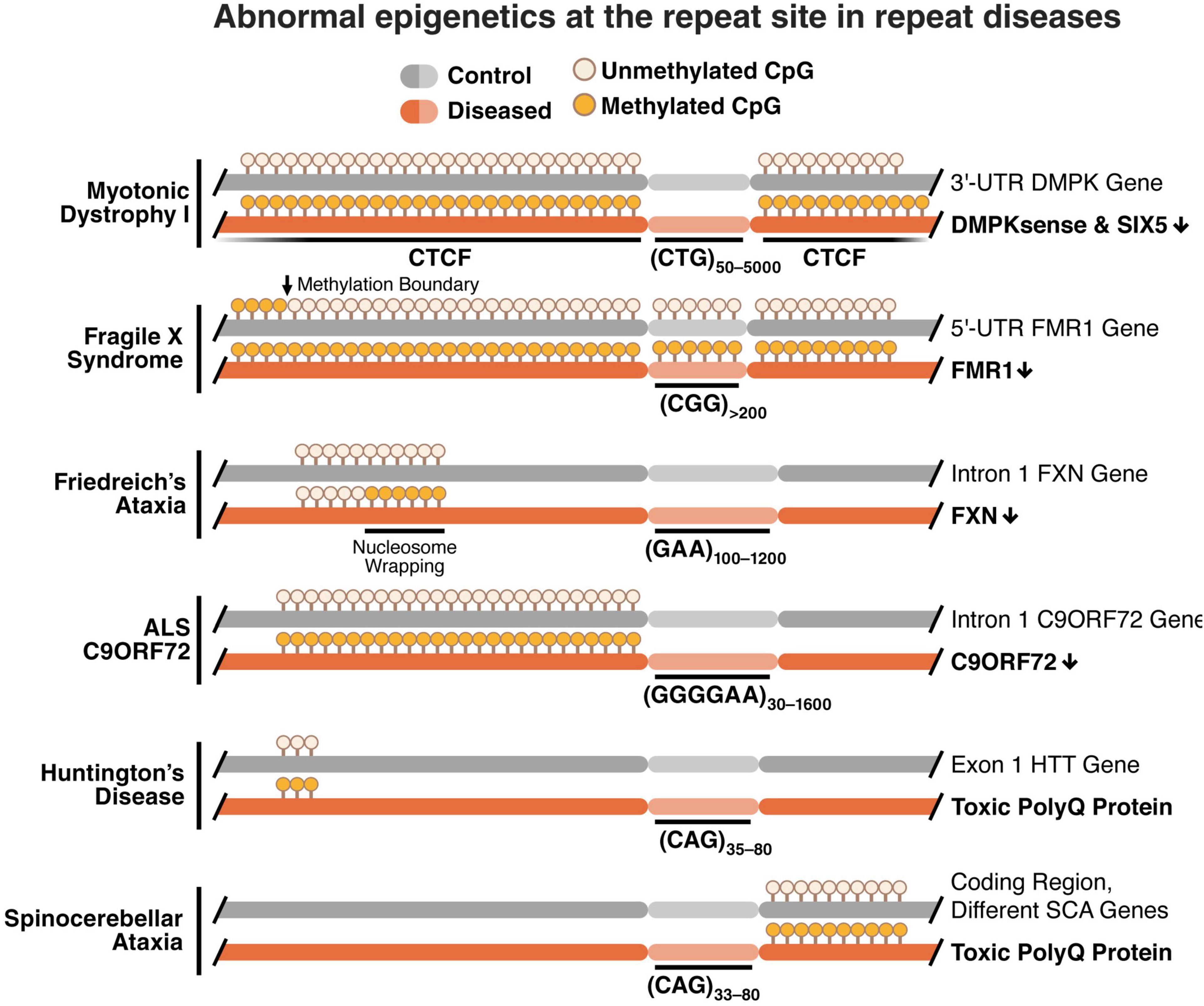
Figure 1. Methylation of the repeat locus is associated with epigenetic deregulation of the nucleotide repeat region with a loss in CTCF binding, loss of TAD boundaries, chromatin condensation, and gene down regulation. Increased methylation is typically found near the repeat region in CG-rich areas called CpG islands. These differentially methylated regions include CTCF binding sites, and methylation inhibits binding of the CTCF protein. With a loss of CTCF binding, TAD boundaries are also lost and chromatin folding is altered to a more condensed formation. This leads to down regulation of genes with promoters and enhancers at or near the repeat region in several repeat diseases.
Different epigenetic states may in part underlie the variability in disease manifestation across individuals with the same disease-causing mutation, and since environmental input can alter epigenetic state, it can also affect disease severity. There are several examples of such associations in the literature, such as the finding that cage enrichment delays disease onset in mouse models of HD (Van Dellen et al., 2000). Similar results were found in a mouse model of FXS, in which mice housed with cage enrichment showed behavioral and neuronal morphological recovery (Restivo et al., 2005). However, how these environmental exposures trigger epigenetic changes that interact with genetic mutations and affect disease phenotype are largely unknown.
The common signature of epigenetic changes that we found in classical repeat diseases are associated with disease severity and age of disease onset. Down regulation of genes in the repeat region could be one disease mechanism through which the epigenetic signature changes disease phenotype. Additionally, the epigenetic signature at the repeat region could directly or indirectly interact with the DNA and cause disease phenotypes through repeat instability, as discussed next.
Genetic and Epigenetic Interplay Through Repeat Instability
Repeat expansions are a critical element of repeat diseases. However, the repeat size alone does not explain the variation in disease severity and age of disease onset (Cobo et al., 1993; Barcelo et al., 1994; Clark et al., 1998; Langbehn et al., 2004; Wexler et al., 2004; De Antonio et al., 2016; Keum et al., 2016), and other factors like epigenetics may be better predictors of disease phenotype (Barbe et al., 2017). However, how genetics and epigenetics might interact to cause disease remains unknown.
Repeat instability, whereby the repeat size expands and contracts within cells over time, has been identified in most repeat diseases. Overall, repeat instability and repeat expansions are higher in affected tissues and cell types (Anvret et al., 1993; Ashizawa et al., 1993; Thornton et al., 1994; Chong et al., 1995; Dobkin et al., 1996; Takano et al., 1996; Watanabe et al., 2000; Kennedy et al., 2003; De Biase et al., 2007; Nordin et al., 2015; Mouro Pinto et al., 2020). Repeat regions show an increase in secondary DNA structures, with increased DNA folding into DNA loops (Schmidt and Pearson, 2016). Repeat instability is thought to be caused by the incorrect repair of these secondary structures by mismatch repair proteins (Schmidt and Pearson, 2016). Whether the common epigenetic signature we describe in this review also plays a role in the formation of these secondary structures and repeat instability remains largely unknown (Figure 2).
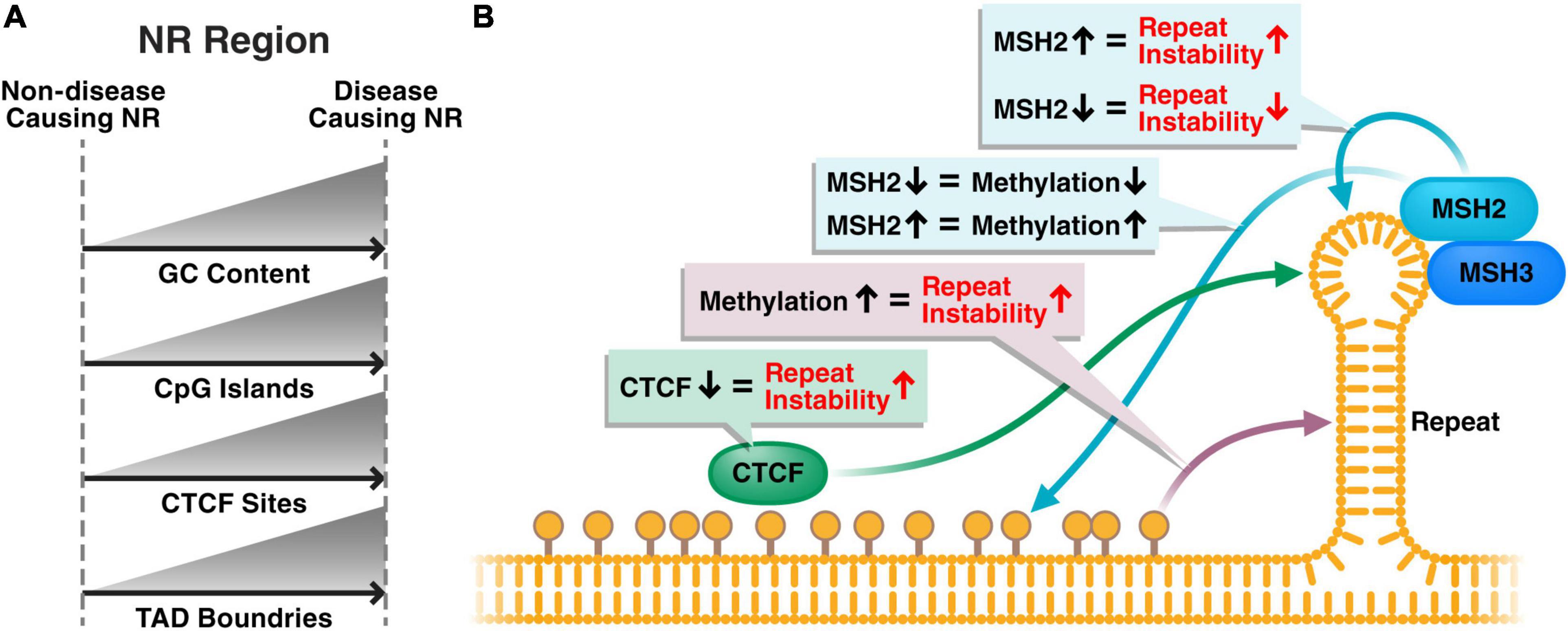
Figure 2. Hypothetical disease model of the role of the common epigenetic signature in repeat instability and disease phenotype. (A) Disease-causing nucleotide repeats typically have higher GC content, more CpG islands and more CTCF sites compared to non-disease-causing repeats in the genome. Typically, a TAD boundary will be present near disease-causing repeats. (B) Methylation near the nucleotide repeat results in increased repeat instability, potentially through a loss of CTCF binding, which is also associated with increased repeat instability. Additionally, the mismatch repair pathway, of which MSH2 and MSH3 are the main players at repeats, interacts with the hairpin structures formed at the repeat region and causes repeat instability. The close proximity of methylation and mismatch repair proteins to repeat regions could suggest that epigenetics and the mismatch repair machinery play a cooperative role regulating repeat instability.
Brock et al. (1999) identified repeat diseases to originate from loci rich in GC content. They found a strong correlation between “expandability” of the repeat and the GC content in the flanking regions, and showed that the most expandable repeats were located near CpG islands. Similarly, although repeat expansions happen throughout the genome, repeat diseases are primarily caused by expansions near a TAD domain, often located in GC-rich DNA sequences with CTCF binding sites (Sun et al., 2018).
In addition to altering expression of disease-causing genes, changes in DNA methylation may also enhance neurodegeneration and other neuronal disease phenotypes in repeat disease by increasing repeat instability and expansion. For example, knockdown of DNA methyltransferase 1 (DNMT1) in human cells and mouse models of SCA1 results in an increase in repeat instability (Dion et al., 2008). Additionally, high levels of DNA methylation upstream of the CGG repeat in the ovaries of SCA1 mice were correlated with low frequencies of stable repeat transmission to offspring (Dion et al., 2008). Franck et al. (2021) recently reported that downregulation of mismatch repair proteins, known to bind DNA loops formed at expanded CTG repeats, are associated with repeat stabilization and a loss of methylation at the repeat region in DM1 (Franck et al., 2021). They found that re-introduction of the mismatch repair proteins re-initiated repeat instability and partially restored methylation (Franck et al., 2021). These results provide direct evidence of an interplay between methylation and repeat instability at the expanded repeat region, however, a causal link between methylation and repeat instability has not been assessed.
CTCF binding is inhibited upon methylation of the CTCF binding site, which is thought to have an effect on chromatinization and thereby gene expression. Additionally, repeat instability has been linked to a loss of CTCF binding as a consequence of mutations in CTCF binding sites in SCA7 transgenic mouse models (Libby et al., 2008).
Many questions remain regarding the involvement of epigenetics in gene regulation, repeat instability and the consequences for cognitive symptoms and neurodegeneration. To address these questions, models that retain relevant epigenetic marks in disease-relevant cell types are necessary. Next, we discuss the disease models that are currently available to study the role of epigenetics in disease phenotype and how these compare to human pluripotent stem cell models.
Epigenetic Disease Modeling for Neurological Dysfunction in Repeat Diseases
Most current models for repeat diseases, including human induced pluripotent stem cell (hiPSC)-derived cells and animal models, primarily model the genetic aspects of the disease.
Animals are useful to model distinct sets of repeat disease pathologies and phenotypes. Because most repeat diseases do not naturally occur in rodents, animal models of the diseases are generally made by knocking-in abnormally large repeat lengths, and the resulting animals generally show fewer symptoms than typical patients (Jansen et al., 1996; Mangiarini et al., 1996; Matilla et al., 1998; Klesert et al., 2000; Sarkar et al., 2000; Puccio et al., 2001; Watase et al., 2002, 2008; Simon et al., 2004; Gomes-Pereira et al., 2007; Panaite et al., 2008; Menalled et al., 2009; Kazdoba et al., 2014; Chew et al., 2015; Jiang et al., 2016; Liu Y. et al., 2016). Alternatively, knock down or knock out models are used to eliminate expression of the disease-causing genes in DM1, FMR1 and SCA1 (Fu et al., 1993; The Dutch-Belgian Fragile X Consorthium et al., 1994; Matilla et al., 1998; Klesert et al., 2000; Huguet et al., 2012; Kazdoba et al., 2014). While these models have been useful to understand disease pathways and phenotypes, the epigenetic marks associated with the human diseases are often not expressed because of the knock-in and knock-down techniques used to induce repeat diseases in mice (Kovtun and McMurray, 2008). Therefore, these models are unfit to study the role epigenetic changes near the repeat region might have on disease.
Human post-mortem samples are widely used to study phenotypes related to repeat length (Greco et al., 2011; Lopez Castel et al., 2011; Seidel et al., 2012; Waldvogel et al., 2012; Esanov et al., 2016). However, these samples are typically only available from the most severe, fatal forms of the diseases, and give little information about early disease mechanisms that could be targeted with drugs. As an alternative, many research groups investigate repeat diseases using hiPSCs generated from fibroblasts or blood samples of patients. This extends the breadth of disease severities that can be studied, as well as the possibility to study family members who do not express disease phenotypes. Also, gene-corrected cell lines can be generated from the patient cell lines to specifically determine the effect of the repeat expansion in the same genetic background. However, modeling epigenetics using hiPSC models has been challenging because of the potential absence or loss of epigenetic and aging signatures during cell reprogramming to a pluripotent state (Tang et al., 2017; Victor et al., 2018).
Challenges to studying epigenetics in repeat diseases, and aging diseases overall, remain due to the inability of animal models to recapitulate disease phenotypes at human repeat lengths, the absence of temporal information and early disease phenotypes in post-mortem samples and the absence of epigenetic and aging signatures in hiPSC models (Patterson et al., 2012). Thus, models that retain epigenetic and aging marks have been proposed as alternative approaches to study the role of epigenetics and aging in repeat diseases and other neurodegenerative diseases.
Direct conversion of adult cells, also called transdifferentiation, is typically done from fibroblasts generated from skin biopsies (Vierbuchen et al., 2010) or from blood cells (Tanabe et al., 2018), and the method is furthest developed for neuronal conversion (Vierbuchen et al., 2010). Adult cells are converted to neurons with the use of small molecules, neuronal transcription factors, micro-RNAs and epigenetic modifiers and bypass the pluripotent stage by going through a unique intermediate state without transcriptional specification of either the donor or the target cell type (Treutlein et al., 2016; Chen et al., 2019). Converted neurons retain many of the epigenetic marks from the fibroblast state (Huh et al., 2016), and also appear to generate new epigenetic marks typical for the neuronal lineage (Luo et al., 2019; Traxler et al., 2019). The converted neurons have the same molecular age as the fibroblasts they originated from, as determined by DNA methylation marks of the epigenetic clock (Horvath, 2013; Huh et al., 2016). Converted neurons also retain other cellular properties of aging such as shorter telomere lengths, chromatin and nuclear organization, higher levels of DNA damage and increased oxidative stress (Huh et al., 2016; Tang et al., 2017), whereas hiPSC-derived neurons, generated from the same fibroblasts, adopt a younger, more embryonic-like state (Tang et al., 2017).
One approach to investigate epigenetic signatures in human cell models is to use direct conversion of patient cells to disease-relevant cell types. Direct conversion substantially retains the epigenetic signatures of the adult cell, an advantage for modeling diseases related to epigenetics and aging (Traxler et al., 2019). The direct conversion approach could also enable disease modeling at repeat sizes known to cause disease, with the flexibility to use samples from patients with different disease severities, and potentially without erasing epigenetic disease marks at the repeat region to enable studies of drug interventions and cause-effect relationships in cell culture. Epigenetic marks are generally established and maintained by enzymes and thus these enzymes are potentially druggable. For example, there are already FDA-approved cancer drugs that target epigenetic states including DNA methylation, chromatin remodeling and non-coding RNAs (Ghasemi, 2020). Additionally, these human cell models created by transdifferentiation could be used to study the impact of aging on disease phenotypes in which aging is a risk factor, such as HD and ALS.
A few studies have modeled repeat diseases with directly converted neurons and demonstrated disease phenotypes (Jovicic et al., 2015; Liu M. L. et al., 2016; Victor et al., 2018). For example, Jovicic et al. (2015) found that directly converted neurons from ALS patients showed mislocalization of the regulator of chromosome condensation 1 (RCC1) protein, a Ran guanine nucleotide exchange factor they used to determine nucleocytoplasmic exchange. Additionally, Liu et al. (Liu M. L. et al., 2016) found increased neurodegeneration of directly converted motor neurons from ALS patients, as evidenced by poor cell survival, a loss of neuromuscular junctions, reduced cell activity and soma shrinkage.
One study nicely compared HD phenotypes found in medium spiny neurons (MSNs) directly converted from fibroblasts to phenotypes in MSNs derived from iPSC-derived fibroblasts (Victor et al., 2018). This study showed that the directly converted MSNs expressed disease phenotypes that have been difficult to demonstrate in hiPSC-derived MSNs, including Huntingtin aggregates, a disease biomarker found in patient samples. In addition, the authors showed that cellular disease phenotypes were enhanced in the directly converted MSNs, such as increased DNA damage levels, mitochondrial dysfunction and neurodegeneration (Victor et al., 2018).
These recent findings suggest that modeling repeat diseases with directly converted neurons may be preferred over hiPSC-derived neuronal cell models because of the increased number of disease phenotypes that can be detected, and because directly converted neurons express disease phenotypes that are similar to those observed in post-mortem patient samples. Moreover, a common epigenetic signature is present across repeat diseases, and disease models that retain epigenetic signatures are necessary to investigate the underlying mechanisms.
Conclusion
Repeat diseases are caused by genetic repeat expansions. However, the variation in age of disease onset and severity in repeat disease patients cannot be explained solely by differences in repeat size. Here, we describe a common epigenetic signature across repeat diseases, found to be correlated with disease severity and age of onset. This signature includes DNA methylation near the repeat region, including at CTCF binding sites, which results in the loss of CTCF binding, loss of a TAD boundary and increased chromatinization. This leads to altered expression of genes in the repeat region which can cause specific disease symptoms. Additionally, an interplay between genetics and epigenetics at the repeat region suggests that methylation contributes to repeat instability. Notably, these mechanisms likely also contribute to some polygenic disorders, such as ASD. Unfortunately, a broad understanding of how an altered epigenetic state can cause cellular dysfunction remains lacking, largely due to the inability to study cause-and-effect relationships in most current disease models. However, direct conversion of fibroblasts to disease-relevant cell types, with the retention of patient epigenetic and aging signatures, has set the stage to unravel the mechanisms by which epigenetic changes can cause disease phenotypes and study drug interventions in repeat diseases.
Author Contributions
LB and SF: conceptualization, writing–review, editing, and funding acquisition. LB: writing–original draft. Both authors contributed to the article and approved the submitted version.
Funding
This work was made possible with financial support from the NIH R37 NS101996-01 and RF1 AG058476, the Department of Defense (W81XWH-10-1-0095), the CHDI Foundation, and the Taube/Koret Center for Neurodegenerative Disease.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Mahdawi, S., Pinto, R. M., Ismail, O., Varshney, D., Lymperi, S., Sandi, C., et al. (2008). The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 17, 735–746. doi: 10.1093/hmg/ddm346
Al-Mahdawi, S., Sandi, C., Mouro Pinto, R., and Pook, M. A. (2013). Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS One 8:e74956. doi: 10.1371/journal.pone.0074956
Alwazzan, M., Newman, E., Hamshere, M. G., and Brook, J. D. (1999). Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum. Mol. Genet. 8, 1491–1497. doi: 10.1093/hmg/8.8.1491
Anvret, M., Ahlberg, G., Grandell, U., Hedberg, B., Johnson, K., and Edstrom, L. (1993). Larger expansions of the CTG repeat in muscle compared to lymphocytes from patients with myotonic dystrophy. Hum. Mol. Genet. 2, 1397–1400. doi: 10.1093/hmg/2.9.1397
Ashizawa, T., Dubel, J. R., Dunne, P. W., Dunne, C. J., Fu, Y. H., Pizzuti, A., et al. (1992a). Anticipation in myotonic dystrophy. II. Complex relationships between clinical findings and structure of the GCT repeat. Neurology 42, 1877–1883. doi: 10.1212/wnl.42.10.1877
Ashizawa, T., Dunne, C. J., Dubel, J. R., Perryman, M. B., Epstein, H. F., Boerwinkle, E., et al. (1992b). Anticipation in myotonic dystrophy. I. Statistical verification based on clinical and haplotype findings. Neurology 42, 1871–1877. doi: 10.1212/wnl.42.10.1871
Ashizawa, T., Dubel, J. R., and Harati, Y. (1993). Somatic instability of CTG repeat in myotonic dystrophy. Neurology 43, 2674–2678. doi: 10.1212/wnl.43.12.2674
Aslanidis, C., Jansen, G., Amemiya, C., Shutler, G., Mahadevan, M., Tsilfidis, C., et al. (1992). Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature 355, 548–551. doi: 10.1038/355548a0
Aubry, L., Bugi, A., Lefort, N., Rousseau, F., Peschanski, M., and Perrier, A. L. (2008). Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc. Natl. Acad. Sci. U.S.A. 105, 16707–16712. doi: 10.1073/pnas.0808488105
Barbe, L., Lanni, S., Lopez-Castel, A., Franck, S., Spits, C., Keymolen, K., et al. (2017). CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am. J. Hum. Genet. 100, 488–505. doi: 10.1016/j.ajhg.2017.01.033
Barcelo, J. M., Pluscauskas, M., MacKenzie, A. E., Tsilfidis, C., Narang, M., and Korneluk, R. G. (1994). Additive influence of maternal and offspring DM-kinase gene CTG repeat lengths in the genesis of congenital. Am. J. Hum. Genet. 54, 1124–1125.
Baylin, S. B., Esteller, M., Rountree, M. R., Bachman, K. E., Schuebel, K., and Herman, J. G. (2001). Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 10, 687–692. doi: 10.1093/hmg/10.7.687
Belzil, V. V., Bauer, P. O., Prudencio, M., Gendron, T. F., Stetler, C. T., Yan, I. K., et al. (2013). Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905. doi: 10.1007/s00401-013-1199-1
Benders, A. A., Groenen, P. J., Oerlemans, F. T., Veerkamp, J. H., and Wieringa, B. (1997). Myotonic dystrophy protein kinase is involved in the modulation of the Ca2+ homeostasis in skeletal muscle cells. J. Clin. Invest. 100, 1440–1447. doi: 10.1172/JCI119664
Berul, C. I., Maguire, C. T., Aronovitz, M. J., Greenwood, J., Miller, C., Gehrmann, J., et al. (1999). alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Invest. 103, R1–R7. doi: 10.1172/JCI5346
Bettencourt, C., Hensman-Moss, D., Flower, M., Wiethoff, S., Brice, A., Goizet, C., et al. (2016). DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 79, 983–990. doi: 10.1002/ana.24656
Biacsi, R., Kumari, D., and Usdin, K. (2008). SIRT1 inhibition alleviates gene silencing in fragile X mental retardation syndrome. PLoS Genet. 4:e1000017. doi: 10.1371/journal.pgen.1000017
Bourgeois, J. A., Coffey, S. M., Rivera, S. M., Hessl, D., Gane, L. W., Tassone, F., et al. (2009). A review of fragile X premutation disorders: expanding the psychiatric perspective. J. Clin. Psychiatry 70, 852–862. doi: 10.4088/JCP.08m04476
Brock, G. J., Anderson, N. H., and Monckton, D. G. (1999). Cis-acting modifiers of expanded CAG/CTG triplet repeat expandability: associations with flanking GC content and proximity to CpG islands. Hum. Mol. Genet. 8, 1061–1067. doi: 10.1093/hmg/8.6.1061
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 69:385.
Brouwer, J. R., Huguet, A., Nicole, A., Munnich, A., and Gourdon, G. (2013). Transcriptionally repressive chromatin remodelling and CpG methylation in the presence of expanded CTG-repeats at the DM1 locus. J. Nucleic Acids 2013:567435. doi: 10.1155/2013/567435
Buckley, L., Lacey, M., and Ehrlich, M. E. (2016). Epigenetics of the myotonic dystrophy-associated DMPK gene neighborhood. Epigenomics 8, 13–31. doi: 10.2217/epi.15.104
Campuzano, V., Montermini, L., Molto, M. D., Pianese, L., Cossee, M., Cavalcanti, F., et al. (1996). Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. doi: 10.1126/science.271.5254.1423
Castaldo, I., Pinelli, M., Monticelli, A., Acquaviva, F., Giacchetti, M., Filla, A., et al. (2008). DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 45, 808–812. doi: 10.1136/jmg.2008.058594
Chen, S., Zhang, J., Zhang, D., and Jiao, J. (2019). Acquisition of functional neurons by direct conversion: switching the developmental clock directly. J. Genet. Genomics 46, 459–465. doi: 10.1016/j.jgg.2019.10.003
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y. J., Castanedes-Casey, M., et al. (2015). Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi: 10.1126/science.aaa9344
Chiurazzi, P., Pomponi, M. G., Willemsen, R., Oostra, B. A., and Neri, G. (1998). In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum Mol Genet 7, 109–113. doi: 10.1093/hmg/7.1.109
Cho, D. H., Thienes, C. P., Mahoney, S. E., Analau, E., Filippova, G. N., and Tapscott, S. J. (2005). Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 20, 483–489. doi: 10.1016/j.molcel.2005.09.002
Chong, S. S., McCall, A. E., Cota, J., Subramony, S. H., Orr, H. T., Hughes, M. R., et al. (1995). Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 10, 344–350. doi: 10.1038/ng0795-344
Ciosi, M., Maxwell, A., Cumming, S. A., Hensman Moss, D. J., Alshammari, A. M., Flower, M. D., et al. (2019). A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 48, 568–580. doi: 10.1016/j.ebiom.2019.09.020
Clark, C., Petty, R. K., and Strong, A. M. (1998). Late presentation of myotonic dystrophy. Clin. Exp. Dermatol. 23, 47–48. doi: 10.1046/j.1365-2230.1998.00294.x
Cobo, A. M., Baiget, M., Lopez de Munain, A., Poza, J. J., Emparanza, J. I., and Johnson, K. (1993). Sex-related difference in intergenerational expansion of myotonic dystrophy gene. Lancet 341, 1159–1160. doi: 10.1016/0140-6736(93)93186-5
Coffee, B., Zhang, F., Ceman, S., Warren, S. T., and Reines, D. (2002). Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 71, 923–932. doi: 10.1086/342931
Coffee, B., Zhang, F., Warren, S. T., and Reines, D. (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 22, 98–101. doi: 10.1038/8807
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W. Y., Gerhardt, J., et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. doi: 10.1126/science.1245831
Cossee, M., Schmitt, M., Campuzano, V., Reutenauer, L., Moutou, C., Mandel, J. L., et al. (1997). Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: founder effect and premutations. Proc. Natl. Acad. Sci. U.S.A. 94, 7452–7457. doi: 10.1073/pnas.94.14.7452
De Antonio, M., Dogan, C., Hamroun, D., Mati, M., Zerrouki, S., Eymard, B., et al. (2016). Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev. Neurol. (Paris) 172, 572–580. doi: 10.1016/j.neurol.2016.08.003
De Biase, I., Chutake, Y. K., Rindler, P. M., and Bidichandani, S. I. (2009). Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One 4:e7914. doi: 10.1371/journal.pone.0007914
De Biase, I., Rasmussen, A., Endres, D., Al-Mahdawi, S., Monticelli, A., Cocozza, S., et al. (2007). Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann. Neurol. 61, 55–60. doi: 10.1002/ana.21052
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
De Souza, R. A., Islam, S. A., McEwen, L. M., Mathelier, A., Hill, A., Mah, S. M., et al. (2016). DNA methylation profiling in human Huntington’s disease brain. Hum. Mol. Genet. 25, 2013–2030. doi: 10.1093/hmg/ddw076
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Depardon, F., Cisneros, B., Alonso-Vilatela, E., and Montanez, C. (2001). Myotonic dystrophy protein kinase (DMPK) gene expression in lymphocytes of patients with myotonic dystrophy. Arch. Med. Res. 32, 123–128. doi: 10.1016/s0188-4409(01)00263-6
Dion, V., Lin, Y., Hubert, L. Jr., Waterland, R. A., and Wilson, J. H. (2008). Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum. Mol. Genet. 17, 1306–1317. doi: 10.1093/hmg/ddn019
Dobkin, C. S., Nolin, S. L., Cohen, I., Sudhalter, V., Bialer, M. G., Ding, X. H., et al. (1996). Tissue differences in fragile X mosaics: mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 64, 296–301. doi: 10.1002/(SICI)1096-8628(19960809)64:2<296::AID-AJMG13>3.0.CO;2-A
Ellerby, L. M. (2019). Repeat expansion disorders: mechanisms and therapeutics. Neurotherapeutics 16, 924–927. doi: 10.1007/s13311-019-00823-3
Eriksson, M., Ansved, T., Anvret, M., and Carey, N. (2001). A mammalian radial spokehead-like gene, RSHL1, at the myotonic dystrophy-1 locus. Biochem. Biophys. Res. Commun. 281, 835–841. doi: 10.1006/bbrc.2001.4465
Eriksson, M., Ansved, T., Edstrom, L., Wells, D. J., Watt, D. J., Anvret, M., et al. (2000). Independent regulation of the myotonic dystrophy 1 locus genes postnatally and during adult skeletal muscle regeneration. J. Biol. Chem. 275, 19964–19969. doi: 10.1074/jbc.M001592200
Esanov, R., Andrade, N. S., Bennison, S., Wahlestedt, C., and Zeier, Z. (2016). The FMR1 promoter is selectively hydroxymethylated in primary neurons of fragile X syndrome patients. Hum. Mol. Genet. 25, 4870–4880. doi: 10.1093/hmg/ddw311
Filippova, G. N., Thienes, C. P., Penn, B. H., Cho, D. H., Hu, Y. J., Moore, J. M., et al. (2001). CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 28, 335–343. doi: 10.1038/ng570
Filla, A., De Michele, G., Cavalcanti, F., Pianese, L., Monticelli, A., Campanella, G., et al. (1996). The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 59, 554–560.
Flower, M., Lomeikaite, V., Ciosi, M., Cumming, S., Morales, F., Lo, K., et al. (2019). MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain 142, 1876–1886. doi: 10.1093/brain/awz115
Franck, S., Barbe, L., Ardui, S., De Vlaeminck, Y., Allemeersch, J., Dziedzicka, D., et al. (2021). MSH2 knock-down shows CTG repeat stability and concomitant upstream demethylation at the DMPK locus in myotonic dystrophy type 1 human embryonic stem cells. Hum. Mol. Genet. 29, 3566–3577. doi: 10.1093/hmg/ddaa250
Fu, Y. H., Friedman, D. L., Richards, S., Pearlman, J. A., Gibbs, R. A., Pizzuti, A., et al. (1993). Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science 260, 235–238. doi: 10.1126/science.8469976
Gacy, A. M., Goellner, G., Juranic, N., Macura, S., and McMurray, C. T. (1995). Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81, 533–540. doi: 10.1016/0092-8674(95)90074-8
Genetic Modifiers of Huntington’s Disease (GeM-Hd) Consortium (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. doi: 10.1016/j.cell.2015.07.003
Genetic Modifiers of Huntington’s Disease (GeM-Hd) Consortium (2019). CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 178, 887–900.e14. doi: 10.1016/j.cell.2019.06.036
Ghasemi, S. (2020). Cancer’s epigenetic drugs: where are they in the cancer medicines? Pharmacogenomics J 20, 367–379. doi: 10.1038/s41397-019-0138-5
Gheldof, N., Tabuchi, T. M., and Dekker, J. (2006). The active FMR1 promoter is associated with a large domain of altered chromatin conformation with embedded local histone modifications. Proc. Natl. Acad. Sci. U.S.A. 103, 12463–12468. doi: 10.1073/pnas.0605343103
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/S1474-4422(11)70261-7
Gijselinck, I., Van Mossevelde, S., van der Zee, J., Sieben, A., Engelborghs, S., De Bleecker, J., et al. (2016). The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 21, 1112–1124. doi: 10.1038/mp.2015.159
Godler, D. E., Tassone, F., Loesch, D. Z., Taylor, A. K., Gehling, F., Hagerman, R. J., et al. (2010). Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 19, 1618–1632. doi: 10.1093/hmg/ddq037
Goldberg, Y. P., Kremer, B., Andrew, S. E., Theilmann, J., Graham, R. K., Squitieri, F., et al. (1993). Molecular analysis of new mutations for Huntington’s disease: intermediate alleles and sex of origin effects. Nat. Genet. 5, 174–179. doi: 10.1038/ng1093-174
Goldfarb, L. G., Vasconcelos, O., Platonov, F. A., Lunkes, A., Kipnis, V., Kononova, S., et al. (1996). Unstable triplet repeat and phenotypic variability of spinocerebellar ataxia type 1. Ann. Neurol. 39, 500–506. doi: 10.1002/ana.410390412
Gomes-Pereira, M., Foiry, L., Nicole, A., Huguet, A., Junien, C., Munnich, A., et al. (2007). CTG trinucleotide repeat “big jumps”: large expansions, small mice. PLoS Genet. 3:e52. doi: 10.1371/journal.pgen.0030052
Greco, C. M., Navarro, C. S., Hunsaker, M. R., Maezawa, I., Shuler, J. F., Tassone, F., et al. (2011). Neuropathologic features in the hippocampus and cerebellum of three older men with fragile X syndrome. Mol. Autism 2:2. doi: 10.1186/2040-2392-2-2
Greene, E., Mahishi, L., Entezam, A., Kumari, D., and Usdin, K. (2007). Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 35, 3383–3390. doi: 10.1093/nar/gkm271
Gulcher, J. (2012). Microsatellite markers for linkage and association studies. Cold Spring Harb. Protoc. 2012, 425–432. doi: 10.1101/pdb.top068510
Hamshere, M. G., Newman, E. E., Alwazzan, M., Athwal, B. S., and Brook, J. D. (1997). Transcriptional abnormality in myotonic dystrophy affects DMPK but not neighboring genes. Proc. Natl. Acad. Sci. U.S.A. 94, 7394–7399. doi: 10.1073/pnas.94.14.7394
Hamzeiy, H., Savas, D., Tunca, C., Sen, N. E., Gundogdu Eken, A., Sahbaz, I., et al. (2018). Elevated global DNA methylation is not exclusive to amyotrophic lateral sclerosis and is also observed in spinocerebellar ataxia types 1 and 2. Neurodegener. Dis. 18, 38–48. doi: 10.1159/000486201
Hannan, A. J. (2010). Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for ‘missing heritability’. Trends Genet. 26, 59–65. doi: 10.1016/j.tig.2009.11.008
Hansen, R. S., Gartler, S. M., Scott, C. R., Chen, S. H., and Laird, C. D. (1992). Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum. Mol. Genet. 1, 571–578. doi: 10.1093/hmg/1.8.571
Harley, H. G., Rundle, S. A., MacMillan, J. C., Myring, J., Brook, J. D., Crow, S., et al. (1993). Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum. Genet. 52, 1164–1174.
Harper, P., Penny, R., Foley, T. P. Jr., Migeon, C. J., and Blizzard, R. M. (1972). Gonadal function in males with myotonic dystrophy. J. Clin. Endocrinol. Metab. 35, 852–856. doi: 10.1210/jcem-35-6-852
Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S. L., and Gottesfeld, J. M. (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2, 551–558. doi: 10.1038/nchembio815
Hofmann-Radvanyi, H., and Junien, C. (1993). Myotonic dystrophy: over-expression or/and under-expression? A critical review on a controversial point. Neuromuscul. Disord. 3, 497–501. doi: 10.1016/0960-8966(93)90104-r
Hsiung, G. Y., DeJesus-Hernandez, M., Feldman, H. H., Sengdy, P., Bouchard-Kerr, P., Dwosh, E., et al. (2012). Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135, 709–722. doi: 10.1093/brain/awr354
Hu, Y., Hashimoto, Y., Ishii, T., Rayle, M., Soga, K., Sato, N., et al. (2017). Sequence configuration of spinocerebellar ataxia type 8 repeat expansions in a Japanese cohort of 797 ataxia subjects. J. Neurol. Sci. 382, 87–90. doi: 10.1016/j.jns.2017.08.3256
Huguet, A., Medja, F., Nicole, A., Vignaud, A., Guiraud-Dogan, C., Ferry, A., et al. (2012). Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8:e1003043. doi: 10.1371/journal.pgen.1003043
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. Elife 5:e18648. doi: 10.7554/eLife.18648
Inukai, A., Doyu, M., Kato, T., Liang, Y., Kuru, S., Yamamoto, M., et al. (2000). Reduced expression of DMAHP/SIX5 gene in myotonic dystrophy muscle. Muscle Nerve 23, 1421–1426. doi: 10.1002/1097-4598(200009)23:9<1421::aid-mus14>3.0.co;2-y
Irimia, M., Weatheritt, R. J., Ellis, J. D., Parikshak, N. N., Gonatopoulos-Pournatzis, T., Babor, M., et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523. doi: 10.1016/j.cell.2014.11.035
Jansen, G., Groenen, P. J., Bachner, D., Jap, P. H., Coerwinkel, M., Oerlemans, F., et al. (1996). Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 13, 316–324. doi: 10.1038/ng0796-316
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Jin, P., and Warren, S. T. (2000). Understanding the molecular basis of fragile X syndrome. Hum. Mol. Genet. 9, 901–908. doi: 10.1093/hmg/9.6.901
Jones, P. A., and Laird, P. W. (1999). Cancer epigenetics comes of age. Nature 21, 163–167. doi: 10.1038/5947
Jovicic, A., Mertens, J., Boeynaems, S., Bogaert, E., Chai, N., Yamada, S. B., et al. (2015). Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 18, 1226–1229. doi: 10.1038/nn.4085
Kagey, M. H., Newman, J. J., Bilodeau, S., Zhan, Y., Orlando, D. A., van Berkum, N. L., et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. doi: 10.1038/nature09380
Kazdoba, T. M., Leach, P. T., Silverman, J. L., and Crawley, J. N. (2014). Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res 3, 118–133. doi: 10.5582/irdr.2014.01024
Kennedy, L., Evans, E., Chen, C. M., Craven, L., Detloff, P. J., Ennis, M., et al. (2003). Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 12, 3359–3367. doi: 10.1093/hmg/ddg352
Keum, J. W., Shin, A., Gillis, T., Mysore, J. S., Abu Elneel, K., Lucente, D., et al. (2016). The HTT CAG-Expansion mutation determines age at death but not disease duration in Huntington disease. Am. J. Hum. Genet. 98, 287–298. doi: 10.1016/j.ajhg.2015.12.018
Klesert, T. R., Cho, D. H., Clark, J. J., Maylie, J., Adelman, J., Snider, L., et al. (2000). Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nature 25, 105–109. doi: 10.1038/75490
Klose, R. J., and Bird, A. P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97. doi: 10.1016/j.tibs.2005.12.008
Komure, O., Sano, A., Nishino, N., Yamauchi, N., Ueno, S., Kondoh, K., et al. (1995). DNA analysis in hereditary dentatorubral-pallidoluysian atrophy: correlation between CAG repeat length and phenotypic variation and the molecular basis of anticipation. Neurology 45, 143–149. doi: 10.1212/wnl.45.1.143
Kovtun, I. V., and McMurray, C. T. (2008). Features of trinucleotide repeat instability in vivo. Cell Res. 18, 198–213. doi: 10.1038/cr.2008.5
Kumari, D., and Usdin, K. (2010). The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 19, 4634–4642. doi: 10.1093/hmg/ddq394
La Pean, A., Jeffries, N., Grow, C., Ravina, B., and Di Prospero, N. A. (2008). Predictors of progression in patients with Friedreich ataxia. Mov. Disord. 23, 2026–2032. doi: 10.1002/mds.22248
La Spada, A. R. (1997). Trinucleotide repeat instability: genetic features and molecular mechanisms. Brain Pathol. 7, 943–963. doi: 10.1111/j.1750-3639.1997.tb00895.x
Ladd-Acosta, C., Hansen, K. D., Briem, E., Fallin, M. D., Kaufmann, W. E., and Feinberg, A. P. (2014). Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 19, 862–871. doi: 10.1038/mp.2013.114
Laffita-Mesa, J. M., Bauer, P. O., Kouri, V., Pena Serrano, L., Roskams, J., Almaguer Gotay, D., et al. (2012). Epigenetics DNA methylation in the core ataxin-2 gene promoter: novel physiological and pathological implications. Hum. Genet. 131, 625–638. doi: 10.1007/s00439-011-1101-y
Lagrue, E., Dogan, C., De Antonio, M., Audic, F., Bach, N., Barnerias, C., et al. (2019). A large multicenter study of pediatric myotonic dystrophy type 1 for evidence-based management. Neurology 92, e852–e865.
Langbehn, D. R., Brinkman, R. R., Falush, D., Paulsen, J. S., Hayden, M. R., and International Huntington’s Disease Collaborative Group (2004). A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin. Genet. 65, 267–277. doi: 10.1111/j.1399-0004.2004.00241.x
Langbehn, D. R., Hayden, M. R., Paulsen, J. S., and The Predict-Hd Investigators of the Huntington Study Group (2010). CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 397–408. doi: 10.1002/ajmg.b.30992
Lanni, S., Goracci, M., Borrelli, L., Mancano, G., Chiurazzi, P., Moscato, U., et al. (2013). Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet 9:e1003601. doi: 10.1371/journal.pgen.1003601
Lavedan, C., Hofmann-Radvanyi, H., Rabes, J. P., Roume, J., and Junien, C. (1993). Different sex-dependent constraints in CTG length variation as explanation for congenital myotonic dystrophy. Lancet 341:237. doi: 10.1016/0140-6736(93)90097-z
Leavitt, B. R., Guttman, J. A., Hodgson, J. G., Kimel, G. H., Singaraja, R., Vogl, A. W., et al. (2001). Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am. J. Hum. Genet. 68, 313–324. doi: 10.1086/318207
Lee, J. M., Chao, M. J., Harold, D., Abu Elneel, K., Gillis, T., Holmans, P., et al. (2017). A modifier of Huntington’s disease onset at the MLH1 locus. Hum. Mol. Genet. 26, 3859–3867. doi: 10.1093/hmg/ddx286
Libby, R. T., Hagerman, K. A., Pineda, V. V., Lau, R., Cho, D. H., Baccam, S. L., et al. (2008). CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet. 4:e1000257. doi: 10.1371/journal.pgen.1000257
Liu, E. Y., Russ, J., Wu, K., Neal, D., Suh, E., McNally, A. G., et al. (2014). C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 128, 525–541. doi: 10.1007/s00401-014-1286-y
Liu, M. L., Zang, T., and Zhang, C. L. (2016). Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 14, 115–128. doi: 10.1016/j.celrep.2015.12.018
Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D. R., et al. (2016). C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90, 521–534. doi: 10.1016/j.neuron.2016.04.005
Lopez Castel, A., Nakamori, M., Tome, S., Chitayat, D., Gourdon, G., Thornton, C. A., et al. (2011). Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 20, 1–15. doi: 10.1093/hmg/ddq427
Lu, A. T., Narayan, P., Grant, M. J., Langfelder, P., Wang, N., Kwak, S., et al. (2020). DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat. Commun. 11:4529. doi: 10.1038/s41467-020-18255-5
Luo, C., Lee, Q. Y., Wapinski, O., Castanon, R., Nery, J. R., Mall, M., et al. (2019). Global DNA methylation remodeling during direct reprogramming of fibroblasts to neurons. Elife 8:e40197. doi: 10.7554/eLife.40197
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., and Srinidhi, L. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. the Huntington’s disease collaborative research group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-e
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. doi: 10.1016/s0092-8674(00)81369-0
Martorell, L., Monckton, D. G., Sanchez, A., Lopez De Munain, A., and Baiget, M. (2001). Frequency and stability of the myotonic dystrophy type 1 premutation. Neurology 56, 328–335. doi: 10.1212/wnl.56.3.328
Matilla, A., Roberson, E. D., Banfi, S., Morales, J., Armstrong, D. L., Burright, E. N., et al. (1998). Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J. Neurosci. 18, 5508–5516. doi: 10.1523/JNEUROSCI.18-14-05508.1998
Matsuyama, Z., Izumi, Y., Kameyama, M., Kawakami, H., and Nakamura, S. (1999). The effect of CAT trinucleotide interruptions on the age at onset of spinocerebellar ataxia type 1 (SCA1). J. Med. Genet. 36, 546–548.
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Mazzara, P. G., Muggeo, S., Luoni, M., Massimino, L., Zaghi, M., Valverde, P. T., et al. (2020). Frataxin gene editing rescues Friedreich’s ataxia pathology in dorsal root ganglia organoid-derived sensory neurons. Nat. Commun. 11:4178. doi: 10.1038/s41467-020-17954-3
McCary, L. M., and Roberts, J. E. (2013). Early identification of autism in fragile X syndrome: a review. J. Intellect. Disabil. Res. 57, 803–814. doi: 10.1111/j.1365-2788.2012.01609.x
Menalled, L., El-Khodor, B. F., Patry, M., Suarez-Farinas, M., Orenstein, S. J., Zahasky, B., et al. (2009). Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 35, 319–336. doi: 10.1016/j.nbd.2009.05.007
Menon, R. P., Nethisinghe, S., Faggiano, S., Vannocci, T., Rezaei, H., Pemble, S., et al. (2013). The role of interruptions in polyQ in the pathology of SCA1. PLoS Genet. 9:e1003648. doi: 10.1371/journal.pgen.1003648
Mitra, I., Huang, B., Mousavi, N., Ma, N., Lamkin, M., Yanicky, R., et al. (2021). Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250. doi: 10.1038/s41586-020-03078-7
Montermini, L., Richter, A., Morgan, K., Justice, C. M., Julien, D., Castellotti, B., et al. (1997). Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann. Neurol. 41, 675–682. doi: 10.1002/ana.410410518
Morales, F., Vasquez, M., Santamaria, C., Cuenca, P., Corrales, E., and Monckton, D. G. (2016). A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst.) 40, 57–66. doi: 10.1016/j.dnarep.2016.01.001
Moss, D. J. H., Pardiñas, A. F., Langbehn, D., Lo, K., Leavitt, B. R., Roos, R., et al. (2017). Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. 16, 701–711. doi: 10.1016/S1474-4422(17)30161-8
Mounsey, J. P., Mistry, D. J., Ai, C. W., Reddy, S., and Moorman, J. R. (2000). Skeletal muscle sodium channel gating in mice deficient in myotonic dystrophy protein kinase. Hum. Mol. Genet. 9, 2313–2320. doi: 10.1093/oxfordjournals.hmg.a018923
Mouro Pinto, R., Arning, L., Giordano, J. V., Razghandi, P., Andrew, M. A., Gillis, T., et al. (2020). Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet. 29, 2551–2567. doi: 10.1093/hmg/ddaa139
Musova, Z., Mazanec, R., Krepelova, A., Ehler, E., Vales, J., Jaklova, R., et al. (2009). Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. A 149A, 1365–1374. doi: 10.1002/ajmg.a.32987
Myers, R. H., MacDonald, M. E., Koroshetz, W. J., Duyao, M. P., Ambrose, C. M., Taylor, S. A., et al. (1993). De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat. Genet. 5, 168–173. doi: 10.1038/ng1093-168
Narang, M. A., Waring, J. D., Sabourin, L. A., and Korneluk, R. G. (2000). Myotonic dystrophy (DM) protein kinase levels in congenital and adult DM patients. Eur. J. Hum. Genet. 8, 507–512. doi: 10.1038/sj.ejhg.5200490
Nardone, S., Sams, D. S., Reuveni, E., Getselter, D., Oron, O., Karpuj, M., et al. (2014). DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 4:e433. doi: 10.1038/tp.2014.70
Naumann, A., Hochstein, N., Weber, S., Fanning, E., Doerfler, W., and distinct, A. (2009). DNA-methylation boundary in the 5’- upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am. J. Hum. Genet. 85, 606–616. doi: 10.1016/j.ajhg.2009.09.018
Naumann, A., Kraus, C., Hoogeveen, A., Ramirez, C. M., and Doerfler, W. (2014). Stable DNA methylation boundaries and expanded trinucleotide repeats: role of DNA insertions. J. Mol. Biol. 426, 2554–2566. doi: 10.1016/j.jmb.2014.04.025
Nolin, S. L., Brown, W. T., Glicksman, A., Houck, G. E. Jr., Gargano, A. D., Sullivan, A., et al. (2003). Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 72, 454–464. doi: 10.1086/367713
Nora, E. P., Goloborodko, A., Valton, A. L., Gibcus, J. H., Uebersohn, A., Abdennur, N., et al. (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22. doi: 10.1016/j.cell.2017.05.004
Nordin, A., Akimoto, C., Wuolikainen, A., Alstermark, H., Jonsson, P., Birve, A., et al. (2015). Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 24, 3133–3142. doi: 10.1093/hmg/ddv064
Oberle, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi: 10.1126/science.252.5009.1097
O’Donnell, W. T., Warren, S. T., and decade, A. (2002). of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 25, 315–338.
Panaite, P. A., Gantelet, E., Kraftsik, R., Gourdon, G., Kuntzer, T., and Barakat-Walter, I. (2008). Myotonic dystrophy transgenic mice exhibit pathologic abnormalities in diaphragm neuromuscular junctions and phrenic nerves. J. Neuropathol. Exp. Neurol. 67, 763–772. doi: 10.1097/NEN.0b013e318180ec64
Patterson, M., Chan, D. N., Ha, I., Case, D., Cui, Y., Van Handel, B., et al. (2012). Defining the nature of human pluripotent stem cell progeny. Cell Res. 22, 178–193. doi: 10.1038/cr.2011.133
Pearson, C. E. (2003). Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol. Med. 9, 490–495. doi: 10.1016/j.molmed.2003.09.006
Pearson, C. E., Eichler, E. E., Lorenzetti, D., Kramer, S. F., Zoghbi, H. Y., Nelson, D. L., et al. (1998a). Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation. Biochemistry 37, 2701–2708. doi: 10.1021/bi972546c
Pearson, C. E., Wang, Y. H., Griffith, J. D., and Sinden, R. R. (1998b). Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n. (CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res. 26, 816–823. doi: 10.1093/nar/26.3.816
Pearson, C. E., Nichol Edamura, K., and Cleary, J. D. (2005). Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 6, 729–742. doi: 10.1038/nrg1689
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi: 10.1016/0092-8674(91)90125-i
Pietrobono, R., Pomponi, M. G., Tabolacci, E., Oostra, B., Chiurazzi, P., and Neri, G. (2002). Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 30, 3278–3285. doi: 10.1093/nar/gkf434
Piras, I. S., Picinelli, C., Iennaco, R., Baccarin, M., Castronovo, P., Tomaiuolo, P., et al. (2020). Huntingtin gene CAG repeat size affects autism risk: family-based and case-control association study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 183, 341–351. doi: 10.1002/ajmg.b.32806
Puccio, H., Simon, D., Cossee, M., Criqui-Filipe, P., Tiziano, F., Melki, J., et al. (2001). Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27, 181–186. doi: 10.1038/84818
Quesada, M. P., Jones, J., Rodriguez-Lozano, F. J., Moraleda, J. M., and Martinez, S. (2015). Novel aberrant genetic and epigenetic events in Friedreich’s ataxia. Exp. Cell Res. 335, 51–61. doi: 10.1016/j.yexcr.2015.04.013
Quesnel-Vallieres, M., Dargaei, Z., Irimia, M., Gonatopoulos-Pournatzis, T., Ip, J. Y., Wu, M., et al. (2016). Misregulation of an activity-dependent splicing network as a common mechanism underlying autism spectrum disorders. Mol. Cell 64, 1023–1034. doi: 10.1016/j.molcel.2016.11.033
Rai, M., Soragni, E., Jenssen, K., Burnett, R., Herman, D., Coppola, G., et al. (2008). HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One 3:e1958. doi: 10.1371/journal.pone.0001958
Restivo, L., Ferrari, F., Passino, E., Sgobio, C., Bock, J., Oostra, B. A., et al. (2005). Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 11557–11562. doi: 10.1073/pnas.0504984102
Richard, G. F., Kerrest, A., and Dujon, B. (2008). Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 72, 686–727. doi: 10.1128/MMBR.00011-08
Ridley, R. M., Frith, C. D., Crow, T. J., and Conneally, P. M. (1988). Anticipation in Huntington’s disease is inherited through the male line but may originate in the female. J. Med. Genet. 25, 589–595. doi: 10.1136/jmg.25.9.589
Robertson, K. D. (2001). DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155. doi: 10.1038/sj.onc.1204341
Russ, J., Liu, E. Y., Wu, K., Neal, D., Suh, E., Irwin, D. J., et al. (2015). Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 129, 39–52. doi: 10.1007/s00401-014-1365-0
Sabouri, L. A., Mahadevan, M. S., Narang, M., Lee, D. S., Surh, L. C., and Korneluk, R. G. (1993). Effect of the myotonic dystrophy (DM) mutation on mRNA levels of the DM gene. Nat. Genet. 4, 233–238. doi: 10.1038/ng0793-233
Sandi, C., Pinto, R. M., Al-Mahdawi, S., Ezzatizadeh, V., Barnes, G., Jones, S., et al. (2011). Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol. Dis. 42, 496–505. doi: 10.1016/j.nbd.2011.02.016
Sano, A., Yamauchi, N., Kakimoto, Y., Komure, O., Kawai, J., Hazama, F., et al. (1994). Anticipation in hereditary dentatorubral-pallidoluysian atrophy. Hum. Genet. 93, 699–702.
Sarkar, P. S., Appukuttan, B., Han, J., Ito, Y., Ai, C., Tsai, W., et al. (2000). Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat. Genet. 25, 110–114. doi: 10.1038/75500
Schmidt, M. H. M., and Pearson, C. E. (2016). Disease-associated repeat instability and mismatch repair. DNA Repair (Amst.) 38, 117–126. doi: 10.1016/j.dnarep.2015.11.008
Seidel, K., Siswanto, S., Brunt, E. R., den Dunnen, W., Korf, H. W., and Rub, U. (2012). Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 124, 1–21. doi: 10.1007/s00401-012-1000-x
Sharma, R., De Biase, I., Gomez, M., Delatycki, M. B., Ashizawa, T., and Bidichandani, S. I. (2004). Friedreich ataxia in carriers of unstable borderline GAA triplet-repeat alleles. Ann. Neurol. 56, 898–901. doi: 10.1002/ana.20333
Shaw, D. J., McCurrach, M., Rundle, S. A., Harley, H. G., Crow, S. R., Sohn, R., et al. (1993). Genomic organization and transcriptional units at the myotonic dystrophy locus. Genomics 18, 673–679. doi: 10.1016/s0888-7543(05)80372-6
Shelbourne, P., Winqvist, R., Kunert, E., Davies, J., Leisti, J., Thiele, H., et al. (1992). Unstable DNA may be responsible for the incomplete penetrance of the myotonic dystrophy phenotype. Hum. Mol. Genet. 1, 467–473. doi: 10.1093/hmg/1.7.467
Shulha, H. P., Cheung, I., Whittle, C., Wang, J., Virgil, D., Lin, C. L., et al. (2012). Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch. Gen. Psychiatry 69, 314–324. doi: 10.1001/archgenpsychiatry.2011.151
Simon, D., Seznec, H., Gansmuller, A., Carelle, N., Weber, P., Metzger, D., et al. (2004). Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 24, 1987–1995. doi: 10.1523/JNEUROSCI.4549-03.2004
Sobczak, K., and Krzyzosiak, W. J. (2005). CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J. Biol. Chem. 280, 3898–3910. doi: 10.1074/jbc.M409984200
Steinbach, P., Glaser, D., Vogel, W., Wolf, M., and Schwemmle, S. (1998). The DMPK gene of severely affected myotonic dystrophy patients is hypermethylated proximal to the largely expanded CTG repeat. Am. J. Hum. Genet. 62, 278–285. doi: 10.1086/301711
Stevanin, G., Giunti, P., Belal, G. D., Durr, A., Ruberg, M., Wood, N., et al. (1998). De novo expansion of intermediate alleles in spinocerebellar ataxia 7. Hum. Mol. Genet. 7, 1809–1813. doi: 10.1093/hmg/7.11.1809
Stolle, C. A., Frackelton, E. C., McCallum, J., Farmer, J. M., Tsou, A., Wilson, R. B., et al. (2008). Novel, complex interruptions of the GAA repeat in small, expanded alleles of two affected siblings with late-onset Friedreich ataxia. Mov. Disord. 23, 1303–1306. doi: 10.1002/mds.22012
Sun, J. H., Zhou, L., Emerson, D. J., Phyo, S. A., Titus, K. R., Gong, W., et al. (2018). Disease-associated short tandem repeats co-localize with chromatin domain boundaries. Cell 175, 224–238.e15. doi: 10.1016/j.cell.2018.08.005
Sun, W., Poschmann, J., Cruz-Herrera Del Rosario, R., Parikshak, N. N., Hajan, H. S., Kumar, V., et al. (2016). Histone acetylome-wide association study of autism spectrum disorder. Cell 167, 1385–1397.e11. doi: 10.1016/j.cell.2016.10.031
Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400. doi: 10.1093/hmg/1.6.397
Sutherland, G. R., and Richards, R. I. (1992). Anticipation legitimized: unstable DNA to the rescue. Am. J. Hum. Genet. 51, 7–9.
Tabolacci, E., Pietrobono, R., Moscato, U., Oostra, B. A., Chiurazzi, P., and Neri, G. (2005). Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 13, 641–648. doi: 10.1038/sj.ejhg.5201393
Takano, H., Onodera, O., Takahashi, H., Igarashi, S., Yamada, M., Oyake, M., et al. (1996). Somatic mosaicism of expanded CAG repeats in brains of patients with dentatorubral-pallidoluysian atrophy: cellular population-dependent dynamics of mitotic instability. Am. J. Hum. Genet. 58, 1212–1222.
Tanabe, K., Ang, C. E., Chanda, S., Olmos, V. H., Haag, D., Levinson, D. F., et al. (2018). Transdifferentiation of human adult peripheral blood T cells into neurons. Proc. Natl. Acad. Sci. U.S.A. 115, 6470–6475. doi: 10.1073/pnas.1720273115
Tang, Y., Liu, M. L., Zang, T., and Zhang, C. L. (2017). Direct Reprogramming rather than iPSC-based reprogramming maintains aging hallmarks in human motor neurons. Front. Mol. Neurosci. 10:359. doi: 10.3389/fnmol.2017.00359
Tassone, F., Hagerman, R. J., Taylor, A. K., Gane, L. W., Godfrey, T. E., and Hagerman, P. J. (2000). Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 66, 6–15. doi: 10.1086/302720
The Dutch-Belgian Fragile X Consorthium, Bakker, C. E., Verheij, C., Willemsen, R., van der Helm, R., Oerlemans, F., et al. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33.
Thomson, S. B., and Leavitt, B. R. (2018). Transcriptional regulation of the huntingtin gene. J. Huntingtons. Dis. 7, 289–296. doi: 10.3233/jhd-180331
Thornton, C. A., Johnson, K., and Moxley, R. T. III (1994). Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol. 35, 104–107. doi: 10.1002/ana.410350116
Thornton, C. A., Wymer, J. P., Simmons, Z., McClain, C., and Moxley, R. T. III (1997). Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 16, 407–409. doi: 10.1038/ng0897-407
Traxler, L., Edenhofer, F., and Mertens, J. (2019). Next-generation disease modeling with direct conversion: a new path to old neurons. FEBS Lett. 593, 3316–3337. doi: 10.1002/1873-3468.13678
Tremblay, M. W., and Jiang, Y. H. (2019). DNA methylation and susceptibility to autism spectrum disorder. Annu. Rev. Med. 70, 151–166. doi: 10.1146/annurev-med-120417-091431
Treutlein, B., Lee, Q. Y., Camp, J. G., Mall, M., Koh, W., Shariati, S. A., et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. doi: 10.1038/nature18323
Trost, B., Engchuan, W., Nguyen, C. M., Thiruvahindrapuram, B., Dolzhenko, E., Backstrom, I., et al. (2020). Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86. doi: 10.1038/s41586-020-2579-z
Tsilfidis, C., MacKenzie, A. E., Mettler, G., Barcelo, J., and Korneluk, R. G. (1992). Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat. Genet. 1, 192–195. doi: 10.1038/ng0692-192
Usdin, K., House, N. C., and Freudenreich, C. H. (2015). Repeat instability during DNA repair: insights from model systems. Crit. Rev. Biochem. Mol. Biol. 50, 142–167. doi: 10.3109/10409238.2014.999192
Van Dellen, A., Blakemore, C., Deacon, R., York, D., and Hannan, A. J. (2000). Delaying the onset of Huntington’s in mice. Nature 404, 721–722. doi: 10.1038/35008142
vans-Galea, M. V. E., Carrodus, N., Rowley, S. M., Corben, L. A., Tai, G., Saffery, R., et al. (2012). FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 71, 487–497. doi: 10.1002/ana.22671
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-h
Victor, M. B., Richner, M., Olsen, H. E., Lee, S. W., Monteys, A. M., Ma, C., et al. (2018). Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 21, 341–352. doi: 10.1038/s41593-018-0075-7
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Sudhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Waite, A. J., Baumer, D., East, S., Neal, J., Morris, H. R., Ansorge, O., et al. (2014). Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35, 1779.e5–1779.e13. doi: 10.1016/j.neurobiolaging.2014.01.016
Waldvogel, H. J., Kim, E. H., Thu, D. C., Tippett, L. J., and Faull, R. L. (2012). New perspectives on the neuropathology in Huntington’s disease in the human brain and its relation to symptom variation. J. Huntingtons Dis. 1, 143–153. doi: 10.3233/JHD-2012-120018
Wang, C., Peng, H., Li, J., Ding, D., Chen, Z., Long, Z., et al. (2017). Alteration of methylation status in the ATXN3 gene promoter region is linked to the SCA3/MJD. Neurobiol Aging 53, 192.e5–192.e10. doi: 10.1016/j.neurobiolaging.2016.12.014
Watanabe, H., Tanaka, F., Doyu, M., Riku, S., Yoshida, M., Hashizume, Y., et al. (2000). Differential somatic CAG repeat instability in variable brain cell lineage in dentatorubral pallidoluysian atrophy (DRPLA): a laser-captured microdissection (LCM)-based analysis. Hum. Genet. 107, 452–457. doi: 10.1007/s004390000400
Watase, K., Barrett, C. F., Miyazaki, T., Ishiguro, T., Ishikawa, K., Hu, Y., et al. (2008). Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl. Acad. Sci. U.S.A. 105, 11987–11992. doi: 10.1073/pnas.0804350105
Watase, K., Weeber, E. J., Xu, B., Antalffy, B., Yuva-Paylor, L., Hashimoto, K., et al. (2002). A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34, 905–919. doi: 10.1016/s0896-6273(02)00733-x
Wexler, N. S., Lorimer, J., Porter, J., Gomez, F., Moskowitz, C., Shackell, E., et al. (2004). Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. U.S.A. 101, 3498–3503. doi: 10.1073/pnas.0308679101
Wright, G. E. B., Collins, J. A., Kay, C., McDonald, C., Dolzhenko, E., Xia, Q., et al. (2019). Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am. J. Hum. Genet. 104, 1116–1126. doi: 10.1016/j.ajhg.2019.04.007
Xi, Z., van Blitterswijk, M., Zhang, M., McGoldrick, P., McLean, J. R., Yunusova, Y., et al. (2015). Jump from pre-mutation to pathologic expansion in C9orf72. Am. J. Hum. Genet. 96, 962–970. doi: 10.1016/j.ajhg.2015.04.016
Xi, Z., Zinman, L., Moreno, D., Schymick, J., Liang, Y., Sato, C., et al. (2013). Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 92, 981–989. doi: 10.1016/j.ajhg.2013.04.017
Yamagata, H., Miki, T., Sakoda, S., Yamanaka, N., Davies, J., Shelbourne, P., et al. (1994). Detection of a premutation in Japanese myotonic dystrophy. Hum. Mol. Genet. 3, 819–820. doi: 10.1093/hmg/3.5.819
Yanovsky-Dagan, S., Avitzour, M., Altarescu, G., Renbaum, P., Eldar-Geva, T., Schonberger, O., et al. (2015). Uncovering the role of hypermethylation by CTG expansion in myotonic dystrophy type 1 using mutant human embryonic stem cells. Stem Cell Rep. 5, 221–231. doi: 10.1016/j.stemcr.2015.06.003
Yrigollen, C. M., Durbin-Johnson, B., Gane, L., Nelson, D. L., Hagerman, R., Hagerman, P. J., et al. (2012). AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 14, 729–736. doi: 10.1038/gim.2012.34
Yum, K., Wang, E. T., and Kalsotra, A. (2017). Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr. Opin. Genet. Dev. 44, 30–37. doi: 10.1016/j.gde.2017.01.007
Zhu, L., Wang, X., Li, X. L., Towers, A., Cao, X., Wang, P., et al. (2014). Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 23, 1563–1578. doi: 10.1093/hmg/ddt547
Zuhlke, C., Dalski, A., Hellenbroich, Y., Bubel, S., Schwinger, E., and Burk, K. (2002). Spinocerebellar ataxia type 1 (SCA1): phenotype-genotype correlation studies in intermediate alleles. Eur. J. Hum. Genet. 10, 204–209. doi: 10.1038/sj.ejhg.5200788
Keywords: epigenetics, genetics, nucleotide repeat disease, trinucleotide repeat disease, methylation, repeat instability, chromatinization, CTCF
Citation: Barbé L and Finkbeiner S (2022) Genetic and Epigenetic Interplay Define Disease Onset and Severity in Repeat Diseases. Front. Aging Neurosci. 14:750629. doi: 10.3389/fnagi.2022.750629
Received: 30 July 2021; Accepted: 01 March 2022;
Published: 03 May 2022.
Edited by:
Andrew Yoo, Washington University in St. Louis, United StatesReviewed by:
Anthony John Hannan, University of Melbourne, AustraliaRicardo Mouro Pinto, Harvard Medical School, United States
Copyright © 2022 Barbé and Finkbeiner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steve Finkbeiner, c2ZpbmtiZWluZXJAZ2xhZHN0b25lLnVjc2YuZWR1