 Stefania Zampatti1
Stefania Zampatti1 Cristina Peconi1
Cristina Peconi1 Rosa Campopiano2
Rosa Campopiano2 Stefano Gambardella2,3
Stefano Gambardella2,3 Carlo Caltagirone4
Carlo Caltagirone4 Emiliano Giardina1,5*
Emiliano Giardina1,5*- 1Genomic Medicine Laboratory UILDM, IRCCS Fondazione Santa Lucia, Rome, Italy
- 2IRCCS Neuromed, Pozzilli, Italy
- 3Department of Biomolecular Sciences, University of Urbino “Carlo Bo”, Urbino, Italy
- 4Department of Clinical and Behavioral Neurology, IRCCS Fondazione Santa Lucia, Rome, Italy
- 5Department of Biomedicine and Prevention, Tor Vergata University of Rome, Rome, Italy
Hexanucleotide expansion in C9orf72 has been related to several phenotypes to date, complicating the clinical recognition of these neurodegenerative disorders. An early diagnosis can improve the management of patients, promoting early administration of therapeutic supportive strategies. Here, we report known clinical presentations of C9orf72-related neurodegenerative disorders, pointing out suggestive phenotypes that can benefit the genetic characterization of patients. Considering the high variability of C9orf72-related disorder, frequent and rare manifestations are described, with detailed clinical, instrumental evaluation, and supportive therapeutical approaches. Furthermore, to improve the understanding of molecular pathways of the disease and potential therapeutical targets, a detailed description of the cellular mechanisms related to the pathological effect of C9orf72 is reported. New promising therapeutical strategies and ongoing studies are reported highlighting their molecular role in cellular pathological pathways of C9orf72. These therapeutic approaches are particularly promising because they seem to stop the disease before neuronal damage. The knowledge of clinical and molecular features of C9orf72-related neurodegenerative disorders improves the therapeutical application of known strategies and will lay the basis for the development of new potential therapies.
Introduction
Firstly discovered in 2011, the hexanucleotide G4C2 expansion in the noncoding region between exons 1a and 1b of C9orf72 gene was identified as the mutation related to the complex phenotype associated with the 9p21 locus (Vance et al., 2006). Characteristically, pedigrees in which the mutated allele of C9orf72 segregated from one generation to another, showed different affected phenotypes, ranging from amyotrophic lateral sclerosis (ALS) to frontotemporal dementia (FTD) sometimes with peculiar features (DeJesus-Hernandez et al., 2011; Renton et al., 2011). In detail, patients carrying the high-repeat expansion (HRE) in C9orf72 showed a pure ALS phenotype, a pure FTD phenotype (mostly behavior variant), or a combination of both. Interestingly, about 35% of patients with HRE C9orf72 receive a diagnosis at the onset of the disease that is different from ALS or FTD. This happens because about one in three patients shows an atypical presentation at onset, these different presentations often mime other neurodegenerative disorders (i.e., Alzheimer’s, Parkinson’s, and Huntington’s disease, dementia w/Lewy bodies, vascular dementia, atypical Parkinsonian syndromes, neuropsychiatric disorders, etc; Gossye et al., 2020; Moore et al., 2020). Clinically, there are rapid and slow progressive forms and the phenotype at the onset of the disease is sometimes misleading (van der Ende et al., 2021). The overall survival ranges between 2 months in rapidly progressive ALS to over 30 years in slowly progressive behavior variant of FTD (bvFTD).
The frequency of HRE C9orf72 is extremely variable worldwide, with a great prevalence in the European population, while it is almost absent in the Asian population. Frontotemporal dementia has a prevalence of 1–461:100,000. Among all FTD patients, a percentage of about 4%–29% carry an HRE C9orf72 (Hogan et al., 2016; Van Mossevelde et al., 2018). In detail, about 20%–25% of familial-FTDs and 6%–8% of sporadic-FTDs carries an HRE C9orf72 (van Es et al., 2017; van der Ende et al., 2021). Amyotrophic lateral sclerosis has a prevalence of 5–12:100,000. In detail, one in 10 cases of ALS has a positive family history, and about 30%–50% of ALS familial cases carry the HRE C9orf72 allele. Even in sporadic ALS, the HRE C9orf72 allele is greatly represented in (about 4%–10% of all sporadic ALS). Over 30% of patients manifesting both ALS and FTD phenotypes carried an HRE C9orf72 allele (van der Ende et al., 2021).
These frequencies support the phenotypic variability and incomplete penetrance of some HRE C9orf72 alleles (Oskarsson et al., 2018; Masrori and Van Damme, 2020). In particular, the HRE C9orf72 exhibits incomplete and age-dependent penetrance. A large study conducted by Murphy and coworkers reported that the median age at symptom onset is 58.0 years of age, with a younger age at onset for ALS than for FTD. Indeed, the youngest affected patient described was a 23-year-old patient with ALS. The penetrance increased with age and became nearly complete (99.5%) over 83 years of age (Murphy et al., 2017). Interestingly, the familial and sporadic cases showed the same penetrance of HRE C9orf72. In this scenario, it is hypothesized that almost all sporadic cases are unrecognized familial cases. This assumption is based on the increased life expectancy over the past decades. In fact, the average life expectancy in the United States was under 58 years until the 1930s. In the early decades of the 1900s, people with a HRE C9orf72 allele had the same probability of dying from a common disease rather than from a C9orf72 associated phenotype. This phenomenon is called Gompertzian inter-disease competition and explains the increased frequency of ALS in the past century, due to the effective increase in the susceptible population (Riggs, 1990). C9orf72 phenotypes showed great variability in age at the onset, even in the same family. This fact is perhaps due to the different possible phenotypes at onset. In fact, it is not surprising that motor disturbance (prevalent in ALS forms) is easily recognized if compared with language and/or behavior changes (prevalent in FTD forms).
C9orf72 Phenotypes
Characteristically defined as ALS and FTD phenotypes, the clinical presentation of C9orf72 associated disorders may vary from pure ALS to pure FTD, with many intermediate phenotypes. In fact, the motor neuron disease–FTD continuum encompasses three ALS forms: ALS with executive dysfunction (ALS-eci), ALS with cognitive impairment (ALS-neci), and ALS with behavioral modifications (ALS-bi). Furthermore, FTD presentations may vary from the most frequent behavioral variant (bvFTD) to the primary progressive aphasias (PPAs), which are in turn subdivided in a non-fluent variant (nfvPPA), semantic variant (svPPA), and logopenic variant (van Es et al., 2017).
C9orf72-FTD Phenotype
Frontotemporal dementia represents a group of syndromes characterized by progressive impairment of behavior, language, and cognitive functions. The FTD is due to a frontotemporal lobar degeneration (FTLD) and is clinically subdivided into behavioral variant (bvFTD), semantic variant of primary progressive aphasia (svPPA), and non-fluent/agrammatical variant of primary progressive aphasias (nfvPPA). Among all FTD cases with an HRE C9orf72, there is a higher prevalence of bvFTD cases. The average age at onset is 57 years, regardless of the C9orf72 genotype. Progression is faster in C9-FTD cases if compared with non-C9-FTDs, with overall survival of 7 years in C9-FTD vs. 11 years in non-C9-FTDs, regardless of the occurrence of a concomitant ALS (van Es et al., 2017; van der Ende et al., 2021). Furthermore, up to 30% of C9-FTD patients develop motoneuron dysfunction, a substantially overlapping proportion to FTD cases regardless of C9 genotypes (Burrell et al., 2011).
Cognitive features associated with FTD involve early loss of executive functions, mnemonic deficits, and language impairment. Other clinical features may become evident with the disease progression like dyscalculia and apraxia, suggesting the involvement of parietal lobe (Van Mossevelde et al., 2018; Moore et al., 2020).
The clinical presentation of bvFTD includes early behavioral disinhibition, early apathy or inertia, early loss of sympathy or empathy, and early perseverative, stereotyped, or compulsive or ritualistic behavior. Patients often modify their food preferences, generally with a predilection for sweets. Other neuropsychiatric features are early delusions (mostly somatic) and hallucinations, psychosis, and anxiety. The PPA is characterized by early language deficits, with the impairment of the ability to understand oral and written language. Patients affected by svPPA forget word significance, with the inability to name the objects. On the other hand, patients affected by nfvPPA (agrammatical) lose the ability to apply grammar rules in oral and written language. They lose the ability to understand complex phrases and sometimes present pronunciation disturbance (apraxia) (Van Mossevelde et al., 2018; Moore et al., 2020).
At disease onset, about 90% of all C9-FTD patients are diagnosed as bvFTD, 5% as nfvPPA, and 3% as svPPA. The remaining 2% receive a diagnosis of other neurodegenerative disorders (corticobasal degeneration, progressive supranuclear palsy, etc.; Moore et al., 2020).
Although clinical presentation of C9-FTD and non-C9-FTD are very similar and no clinical criteria can support the differential diagnosis between the two groups, some differences have been described. In particular, mnemonic impairment is more frequent in C9-FTD patients and is often the onset symptom. Psychoses and bizarre behaviors are common in C9-FTD patients. Neuroimages show different degrees of atrophy ranging from the generalized symmetric cortical atrophy to the thalamus and cerebellum atrophy. Furthermore, some C9-FTD patients manifest quite a non-progressive benign variant of the disorder. The benign variant of bvFTD is slowly progressive, with an apparent steadiness of the clinical appearance, very few psychiatric symptoms, and cognitive decline. It is estimated that about 2% of patients with a benign variant of bvFTD carry an HRE C9orf72 allele (van der Ende et al., 2021).
The occurrence of mnemonic impairment at the onset of C9-FTD disease can confuse the diagnosis and lead to a misdiagnosis of Alzheimer’s disease (AD). It is important to consider that AD is a frequent disorder, and it is possible that C9 patients manifest FTD with concomitant AD. Furthermore, C9 patients may not manifest FTD in reason of an incomplete penetrance, independently from the occurrence of other dementing disorders. The small percentage (<1%) of HRE C9orf72 alleles described in AD patients is probably due to the phenotypic heterogeneity of C9-FTD (that involves mnemonic dysfunction) leading to misdiagnosis, and to the known incomplete penetrance (Cacace et al., 2013; Harms et al., 2013). In this scenario, it is important to consider C9orf72 expansion testing in dementing patients with a positive family history of ALS and/or FTD (Harms et al., 2013).
C9orf72-ALS Phenotype
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative progressive disorder that affects the brain and the spinal cord. Although ALS is traditionally considered a motoneuron disease, it can also involve frontal and temporal lobes. Furthermore, the clinical overlap has been demonstrated with other diseases (Paget’s disease of bone, inclusion body myopathy, etc.). Typically, the clinical picture involves a motor impairment with some symptoms of cognitive decline and/or behavior changes. ALS is a disorder with a high phenotypic heterogeneity regarding disease presentation, progression, and survival.
The prevalence of ALS is 5–12:100,000, with a slight reduction in European populations (2.6–3:100,000). Besides phenotype classifications (bulbar, spinal, pseudobulbar palsy, progressive spinal muscular atrophy), ALS can be divided into sporadic and familial forms according to the age at onset and the family history. The median age at onset of ALS is quite similar between sporadic and familial cases: about 65 years for sporadic ALS, and about 55 years for genetic-related familial ALS (Beck et al., 2013; Bieniek et al., 2014; Fahey et al., 2014; Block et al., 2016; Glasmacher et al., 2020). The hexanucleotide expansion in C9orf72 has been recognized as one of the most frequent causes of familial ALS, but it is frequent also in sporadic patients. The classical clinical onset of ALS is the occurrence of muscle weakness in a segment that successively progresses to the whole motor system. Clinically, the classic ALS (70% of cases) has a presentation at the onset that involves both upper motor neuron and lower motor neuron signs. The distribution of the disease is bulbar (33% of classic ALS) with dysarthria and dysphagia at onset, or spinal (66% of classic ALS) with lower and upper motor neuron involvement in the arms and legs. About 5%–15% of ALS patients manifest at onset cognitive or behavioral changes, and it is classified as ALS-FTD. Other rare phenotypes (10% of cases) present a generalized lower motor neuron involvement at onset (progressive spinal muscular atrophy) or an exclusive upper motor neuron involvement (primary lateral sclerosis) (van Es et al., 2017). As seen in the C9-FTD, the C9-ALS is clinically similar to non-C9-ALS, with a few differences in the frequency of some symptoms and signs between the two forms. In particular, the bulbar onset (dysarthria and dysphagia) is more frequent in C9-ALS patients than in non-C9-ALS, with about 30%–40% of all C9-ALS patients presenting bulbar signs at the onset. Neuroimages of C9-ALS patients show a major atrophy of non-motor frontal cortex areas if compared with non-C9-ALS. Cognitive decline is more common in C9-ALS patients than non-C9-ALS. Despite only 20% of ALS patients fulfill diagnostic criteria for FTD, another 20% of ALS patients show cognitive decline (generally in executive, social, or linguistic functions) and a further 10% present behavior changes. This clinical overlap confirmed the phenotype continuum between FTD and ALS.
Atypical Presentations
Psychiatric symptoms are very frequent in patients with a C9orf72-associated disorder if compared with non-C9-FTD and non-C9-ALS. In particular, 20%–60% of the patients carrying the HRE C9orf72 allele manifest psychotic signs, above all delusions (mostly somatic), and hallucinations (Benussi et al., 2021). Other psychiatric manifestations are behavior disturbance, obsessive-compulsive disorder, and catatonia. Sometimes psychiatric symptoms are the first manifestation of the C9orf72 disorder, preceding the clinical diagnosis even for many years. Attention must be paid to the atypical late onset of a psychiatric disorder (without prodromal signs at typical ages) because it can be misdiagnosed as late schizophrenia or bipolar disorder instead of C9orf72 disease. In fact, the identification of an HRE C9orf72 allele is very rare in primary psychiatric disorders (about 0.2%). On the other hand, the identification of psychotic symptoms in C9-FTD patients is very frequent, covering about 21%–56% of bvFTD and FTD-ALS patients (Silverman et al., 2019; van der Ende et al., 2021). In this scenario, the C9orf72 hexanucleotide expansion should be genotyped in all suspected late onset (over 40 years of age) psychiatric disorders in which the symptoms are accompanied by cognitive decline, cerebral or cerebellar atrophy, or familiar history of FTD, ALS, or psychiatric disorders.
Another atypical onset of C9orf72 disease is a Parkinsonian disorder. Although, parkinsonism is a common clinical sign of C9orf72-associated diseases, described in over 75% affected people, sometimes Parkinsonism is a unique sign at onset, or it is only accompanied by ataxia or apraxia. Some HRE C9orf72 carrying people show at onset bradykinesia, rigidity, postural instability, gaze paralysis, generally without tremors. Typical FTD or ALS signs became manifest several years after the onset of parkinsonism, leading to a misdiagnosis of Parkinson’s disease, progressive supranuclear palsy, corticobasal syndrome, or multisystemic atrophy (Wilke et al., 2016; van der Ende et al., 2021). Movement disorders are very frequent in patients with an HRE C9Orf72 allele: tremors and parkinsonism are described in over 60% of patients, myoclonus, dystonia, and chorea in 30%–40% of patients, and ataxia in less than 10% (Estevez-Fraga et al., 2021). Even in this scenario, a C9orf72hexanucleotide expansion genotyping should be considered in patients showing atypical parkinsonism and: (i) family history of ALS or FTD, or; (ii) motoneuron sign, and cognitive impairment with frontotemporal involvement.
Another interesting onset phenotype is the Huntington’s disease-like (HDL) syndrome. In particular, about 5% of patients with HDL syndrome (clinically indistinguishable from HD, but without the CAG expansion in HTT) carries an HRE C9orf72 allele. Although, a study has reported CAG expansions in FTD/ALS patients without HRE C9orf72 allele, further studies have to be performed in order to explain this peculiar overlap between C9orf72 and HTT-associated phenotypes (van der Ende et al., 2021).
Pathological Mechanisms
Three different etiopathological mechanisms have been proposed as responsible for the cellular impairment leading to C9orf72 disorder: haploinsufficiency (low expression of the HRE C9orf72 allele), RNA-binding protein (RBP) sequestration, and the toxic function of RNA-foci and DPRs (dipeptide repeats; Figure 1). These three mechanisms synergistically interplay. The hexanucleotide expansion of C9orf72 determines intron retention and haploinsufficiency of wild-type C9orf72. Furthermore, the HRE C9orf72 allele shows a hypermethylation of the promoter region with a consequent transcriptional silencing of the expanded allele and partial to complete absence of translation of the expanded allele (haploinsufficiency). The transcription of the HRE C9orf72 allele leads to the formation of RNA-DNA hybrids (R-loops). R-loops activate DNA damage response, which interferes with the expansion and is potentially responsible for the somatic variability of the C9orf72 expansion. Moreover, the long repeat RNA has a longer half-life than wild-type RNA, determining the sequestration of the RNA-binding proteins (RBPs; Malik et al., 2021). Sense and antisense long repeat RNAs and RBP interact to form RNA foci. RNA foci sequestrate RNA-binding proteins compromising the overall translation cellular activity. Moreover, intramolecular, and intermolecular interactions recognized in RNA foci (as G-quadruplexes) can further activate DNA damage response. Another mechanism of the disease involves the repeat associated non-AUG translation (RAN translation) of sense and antisense mRNAs from C9orf72 hexanucleotide (Figure 1). The RAN translation produces five different dipeptide repeat proteins (DPRs): polyGA (glycine–alanine), polyGR (glycine–arginine), polyGP (glycine–proline), polyPR (proline–arginine), and polyPA (proline–alanine). The DPRs exercise variable toxic function on tissues. In particular, polyRP shows the highest level of toxicity on several cellular substrates, on the other hand, polyPA shows the lowest level of toxicity (Semmelink et al., 2021).
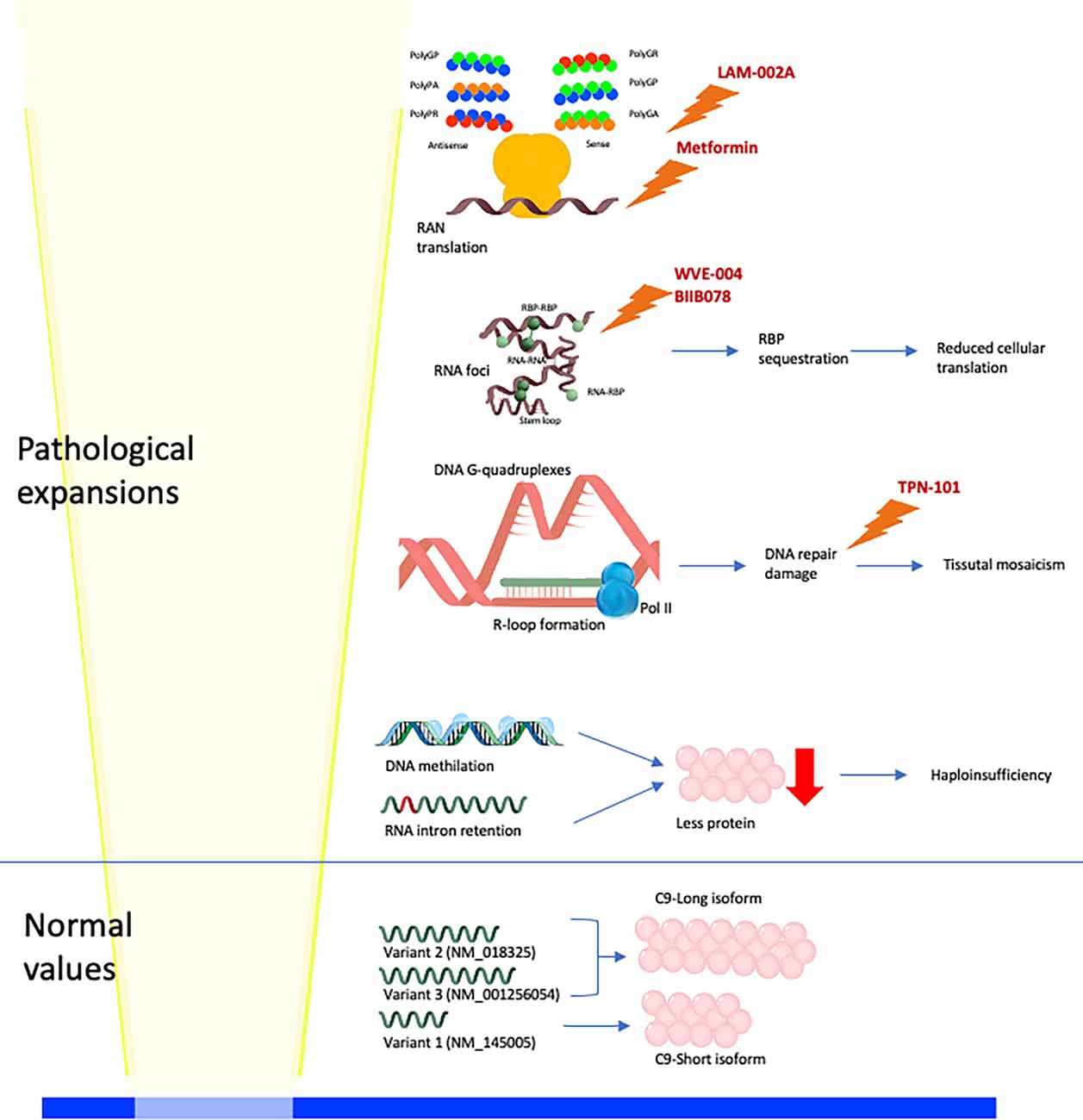
Figure 1. Physiological and pathological molecular mechanisms linked to C9orf72 expansion. Normal alleles lead to the production of three mRNA (V1, V2, and V3), translated into two C9 isoforms (long and short; Smeyers et al., 2021). High-repeat expansion (HRE) C9orf72 alleles lead to four different pathological mechanisms: (i) hypermethylation of DNA and intron retention in mRNA leading to haploinsufficiency; (ii) DNA G-quadruplexes and R-loop formation leading to activation of DNA repair damage response and somaticmosaicism; (iii) RNA foci formation, with intramolecular (stem-loop, and G-quadruplexes) and intermolecular (RNA-RNA, RNA-RNA-binding protein (RBP), and RBP-RBP) interactions (Malik et al., 2021), leading to RBP sequestration and reduced cellular translation; (iv) RAN translation of sense and antisense mRNAs produces five different DPRs (polyGA, polyGR, polyGP, polyPR, and polyPA). Drugs of which it is known or supposed molecular function are reported: metformin reduces RAN translation, LAM-002A interferes with DPRs formation, WVE-004 and BIIB078 promotes the degradation of C9orf72 expanded mRNAs, TPN-101 reduces the DNA repair damage response.
As described, the phenotypes associated with an HRE C9orf72 allele are variable. To date, one of the hypotheses at the basis of this great phenotypic variability is the somatic variability of hexanucleotide expansion. As seen, DNA damage response and maybe other molecular mechanisms determine the contraction or the expansion of the hexanucleotide alleles. In fact, a recent study evaluated the levels of expansion in C9orf72 on brain and blood tissues, finding no correlation between the blood and brain (frontal cortex and cerebellum) exists. In the same person, cells with small and large expansion size can coexist in different tissues (somatic mosaicism). A C9orf72-associated phenotype is typically observed in people with a large expansion in cerebral tissue. However, the sizing of the hexanucleotide repeat in the blood may not match the sizing in the brain. On the contrary, people with a large expansion in blood may have small expansion sizes in the cerebral tissues, resulting in milder C9orf72 phenotypes (Jackson et al., 2020).
These tissue differences support the molecular diagnostic algorithm that defines only normal and pathological ranges, without a clear definition of intermediate alleles. It is hypothesized that the complexity to define a genotype-phenotype correlation among intermediate alleles is mainly due to the extreme variability of the size of the expansion in different cerebral and extra-cerebral tissues (van Blitterswijk et al., 2013 ; Van Mossevelde et al., 2017; Jackson et al., 2020).
Molecular Algorithms
To date, several molecular algorithms have been proposed to make a diagnosis in neurodegenerative disorders related to C9orf72. One of the most diffused involves the deep clinical evaluation of the patient and their family history, in order to administer a first-tier test that involves C9orf72 hexanucleotide evaluation. Patients without HRE C9orf72 alleles will be then evaluated for a multigene test (second tier) or whole-exome sequencing (third tier; Roggenbuck, 2020). This effective algorithm (Roggenbuck and Fong, 2020) must consider the different distribution of pathogenic mutations worldwide (Moore et al., 2020). In fact, although, in almost all Caucasian and Hispanic populations C9orf72 is an important cause of the disease, it is quite inexistent in Asian populations. In this scenario, the first evaluation of a patient with a clinical presentation that overlaps with typical or atypical C9orf72 phenotypes should consider not only the family history for related disorders but also the ethnical background, in order to prioritize the genetic test (C9orf72 hexanucleotide evaluation, multigene panel, or whole-exome sequencing).
Therapeutic Strategies
The management of C9orf72-related disorders requires a complete clinical and instrumental evaluation of the patient in order to identify appropriate treatment strategies. In detail, after the diagnosis, the patient should undergo: (i) neurologic examination (upper or lower motor neuron involvement, dysarthria, dysphagia), (ii) neuropsychological evaluation (cognitive assessment), (iii) ADL (activities of daily living) assessment (orthopedics, physiatrist), (iv) psychiatric evaluation (if history of psychiatric illness), (v) pulmonologist evaluation (if respiratory impairment). Therapeutic strategies involve the treatment of the disease manifestations: rehabilitation and riluzol/edaravone for UMN/LMN involvement and ADL, muscle stretching and baclofen/tizanidine/cannabinoids for spasticity, dietary supplements and drugs for muscular cramps, cognitive rehabilitation for cognitive impairment, neuropsychiatric therapies (pharmacological and psychological) for behavioral manifestations, rehabilitative treatment for dysphagia and dysarthria, pharmacological treatments for Parkinsonism, pseudobulbar signs, sialorrhea, bladder dysfunction (Gossye et al., 2020).
To date, 29 research studies on C9orf72-related disorders are recorded in ClinTrials.gov. Twelve of them investigate new and off-label drugs.
Off Label Drugs
Metformin
A Single-Center, Open Label Study at the University of Florida (NCT04220021) is studying the safety and potential efficacy of metformin for the treatment of C9-ALS/FTD. Metformin is a widely used drug for type 2 diabetes treatment. At a molecular level, metformin has demonstrated an activity to block the double-stranded RNA-dependent protein kinase (PKR) pathway (Zu et al., 2020). Furthermore, in a mouse model of C9-ALS/FTD metformin reduces neuronal RAN protein levels without modifications to total mRNA C9orf72 levels. A functional analysis of mouse models demonstrated that metformin treatment can improve the behavioral phenotype (Zu et al., 2020).
Deferiprone
A study on ALS patients (regardless of C9orf72 genotype) is evaluating the potential neuroprotective benefits of an iron chelator, deferiprone (NCT03293069). It is expected that deferiprone will reduce excess iron from brain regions, oxidative damage, and cell death.
Baricitinib
An open-label study (NCT05189106) is evaluating the potential effects of baricitinib therapy on patients with subjective cognitive disorder, mild cognitive impairment, Alzheimer’s disease (AD), Amyotrophic lateral sclerosis (ALS), or asymptomatic carriers of an ALS-related gene (as hexanucleotide expansion in the C9orf72), with evidence of abnormal inflammatory signaling in cerebrospinal fluid (CSF) at baseline. The objective of the study is to evaluate the ability of baricitiniband to reduce neuroinflammatory damage. Baricitinib is approved by the FDA in the United States for rheumatoid arthritis (2 mg per day) and has an emergency use authorization for COVID-19 (4 mg per day).
Triumeq
A Double-Blind Placebo-Controlled trial is evaluating the effectiveness and safety of triumeq (dolutegravir 50 mg, abacavir 600 mg, lamivudine 300 mg) in delaying the progression of ALS (NCT05193994). Triumeq therapy is expected to reduce neuroinflammation and human endogenous retroviruses (HERV) impairment, that may have a role in the pathophysiology of ALS. A preliminary study has reported a decline in ALS progression with triumeq therapy (Gold et al., 2019). Triumeq is approved in US and Europe for HIV treatment.
New Investigational Drugs
TPN-101
An ongoing phase 2a study is evaluating the safety and tolerability of TPN-101 on a small sample (40 participants) of C9orf72 ALS/FTD patients (NCT04993755). TPN-101 (OBP-601, BMS-986001, festinavir, censavudine; Smith et al., 2015) is a thymidine analog NRTI (nucleoside reverse transcriptase inhibitor) that shows major efficacy against HIV-2 and HIV-1 (Gupta et al., 2016). The active form of TPN-101 (BMS-986001-5’-triphosphate) is a substrate for incorporation by HIV-1 reverse transcriptase, inducing the termination of viral DNA synthesis (Smith et al., 2015).
LAM-002A
An ongoing phase 2a study on 12 adults with C9orf72-associated ALS (C9ALS) is evaluating the safety, tolerability, and biological effect of LAM-002A (NCT05163886). LAM-002A (apilimoddimesylate) reduce glutamate-induced neuronal loss inhibiting PIKFYVE [a lipid kinase that regulates endolysosomal trafficking, via conversion of phosphatidylinositol 3-phosphate PI3P in phosphtidylinositol (3,5)-bisphosphate PI(3,5) P2].
It is hypothesized that the inhibition of PIKFYVE activity may improve the autophagosome-lysosome fusion and increase the PI3P (phosphatidylinositol 3-phosphate) that leads to removal of glutamate receptors and DPRs. A study on human-induced motor neurons (iMNs) revealed that apilimod increases the iMNs survival of C9orf72 patients (Shi et al., 2018). Similarly, apilimod shows a significant activity in the reduction of the neuronal death in MAPT mutated cells (Bowles et al., 2021) and in ATP7A iMNs (Bakkar et al., 2021).
AL001
An ongoing clinical trial on patients with C9orf72-associated ALS (NCT05053035) is evaluating the safety, tolerability, pharmacokinetics, and pharmacodynamics of AL001. Another study (NCT03987295) is evaluating AL001 in symptomatic carriers of GRN mutation or hexanucleotide repeat expansion in C9orf72 causative of FTD. AL001 is an anti-sortilin antibody that binds sortilin 1 (SORT1), a transmembrane glycoprotein that improves progranulin (PGRN) clearance through cellular internalization. Anti-sortilin antibodies downregulates SORT1 inducing an up-regulation of PGRN (Miyakawa et al., 2020). Previous studies have shown the ability of AL001 to increase progranulin cerebral levels reducing the motor neuron damage TDP-43 associated. Similarly, GRN-related FTD AL001 reduces neurodegeneration (Haynes et al., 2020; Terryn et al., 2021).
WVE-004
WVE-004 is an antisense oligonucleotide (ASO) that binds the hexanucleotide expansion of C9orf72 promoting the degradation of C9orf72 expanded mRNAs. In this scenario, the reduction of mutated mRNA could improve many molecular mechanisms involved in the pathogenesis of C9orf72 disorders. In vitro and in vivo studies on patient-derived induced pluripotent stem cells (iPSC) and transgenic mice showed the reduction of expanded transcripts in iPSC motor neurons and in the spinal cord and cortex, respectively (Liu et al., 2022). An ongoing double bind, placebo-controlled, study (NCT04931862) is expected to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of WVE-004 in patients with C9orf72-associated ALS or FTD.
BIIB078
BIIB078 is an antisense oligonucleotide (ASO) targeting expanded C9orf72 mRNAs. BIIB078 accelerates the degradation of mutated mRNA, preventing the production of RNA foci and DPRs. Preclinical studies on cellular and murine models showed a reduction of sense RNA foci, and of poly(GP) and poly(GA) proteins. Furthermore, ASOs seems to attenuate behavioral impairment (Lagier-Tourenne et al., 2013; Jiang et al., 2016). An extension study on BIIB078 (NCT04288856) is ongoing on adults with C9orf72-associated ALS, previously treated in the double-bind study (NCT03626012).
PU-AD
PU-AD belongs to a class of drugs named epichaperome inhibitors. It is a molecular inhibitor of heat shock protein 90 (HSP90), a stabilizer of modified proteins in cancer and neurodegenerative disorders. In cellular and animal models of different neurodegenerative disorders, HSP90 inhibitors promote the degradation of pathogenic proteins (Carman et al., 2013). Epichaperomes are stable HSP90-centered chaperone complexes that incorporate proteins not physiologically associated with HSP90. Among these proteins, there are some involved in synaptic function, learning, and memory. PU-AD has demonstrated to be effective in disrupting the epichaperomes restoring physiological association protein-HSP90 (Inda et al., 2020).
A randomized double-blind placebo-controlled study (NCT04505358) is evaluating the effectiveness and safety of PU-AD therapy on ALS patients. It is a pilot study on 30 participants, randomized 3:2 (PU-AD vs. placebo, respectively).
Conclusions
From the first recognition of the hexanucleotide expansion in C9orf72 as the causative mutation of several neurodegenerative phenotypes, numerous studies have been conducted elucidating phenotype heterogeneity, penetrance, pathogenic molecular mechanisms, and treatment strategies. To date, a deep knowledge of the disorder is essential to improve the clinical practice, to provide an early diagnosis and new molecular therapies design. The knowledge of different onset phenotypes and the availability of a genetic test to confirm the diagnosis can improve the management of patients, providing an early administration of therapeutic strategies. Besides lifestyle interventions (Tsitkanou et al., 2019; Hautbergue et al., 2021; Julian et al., 2021), several therapeutic agents are being studied. Interestingly, many new interventional treatments are designed to act at a molecular level preventing neuronal damage even before the onset of symptoms. The potential effectiveness of these drugs in delaying disease onset in C9orf72 neurodegenerative disorders is designing an optimistic scenario for affected families. The advances in knowledge of clinical and molecular features of C9orf72-related neurodegenerative disorders will promote the designing of new molecular therapies, the early treatment administration, and the development of studies on possible off-label effectiveness of medications.
Author Contributions
SZ and EG designed the theme of the manuscript. SZ, CP, and RC contributed by writing all the sections and creating the figure. EG, SG, and CC conducted critical revisions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is partially supported by the Italian IRCCS Network of Neuroscience and Neurorehabilitation, the National Virtual Institute for dementing disorders, and the Ricerca Corrente (Italian Ministry of Health).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bakkar, N., Starr, A., Rabichow, B. E., Lorenzini, I., McEachin, Z. T., Kraft, R., et al. (2021). The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol. Dis. 149:105228. doi: 10.1016/j.nbd.2020.105228
Beck, J., Poulter, M., Hensman, D., Rohrer, J. D., Mahoney, C. J., Adamson, G., et al. (2013). Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92, 345–353. doi: 10.1016/j.ajhg.2013.01.011
Benussi, A., Premi, E., Gazzina, S., Brattini, C., Bonomi, E., Alberici, A., et al. (2021). Progression of behavioral disturbances and neuropsychiatric symptoms in patients with genetic frontotemporal dementia. JAMA Netw. Open 4:e2030194. doi: 10.1001/jamanetworkopen.2020.30194
Bieniek, K. F., van Blitterswijk, M., Baker, M. C., Petrucelli, L., Rademakers, R., and Dickson, D. W. (2014). Expanded C9ORF72 hexanucleotide repeat in depressive pseudodementia. JAMA Neurol. 71, 775–781. doi: 10.1001/jamaneurol.2013.6368
Block, N. R., Sha, S. J., Karydas, A. M., Fong, J. C., De May, M. G., Miller, B. L., et al. (2016). Frontotemporal dementia and psychiatric illness: emerging clinical and biological links in gene carriers. Am. J. Geriatr. Psychiatry 24, 107–116. doi: 10.1016/j.jagp.2015.04.007
Bowles, K. R., Silva, M. C., Whitney, K., Bertucci, T., Berlind, J. E., Lai, J. D., et al. (2021). ELAVL4, splicing and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 184, 4547–4563.e17. doi: 10.1016/j.cell.2021.07.003
Burrell, J. R., Kiernan, M. C., Vucic, S., and Hodges, J. R. (2011). Motor neuron dysfunction in frontotemporal dementia. Brain 134, 2582–2594. doi: 10.1093/brain/awr195
Cacace, R., Van Cauwenberghe, C., Bettens, K., Gijselinck, I., van der Zee, J., Engelborghs, S., et al. (2013). C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 34, 1712.e1–1717. doi: 10.1016/j.neurobiolaging.2012.12.019
Carman, A., Kishinevsky, S., Koren, J., 3rd, Lou, W., and Chiosis, G. (2013). Chaperone-dependent neurodegeneration: a molecular perspective on therapeutic intervention. J. Alzheimers Dis. Parkinsonism 2013:007. doi: 10.4172/2161-0460.S10-007
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Estevez-Fraga, C., Magrinelli, F., Hensman Moss, D., Mulroy, E., Di Lazzaro, G., Latorre, A., et al. (2021). Expanding the spectrum of movement disorders associated with C9orf72 hexanucleotide expansions. Neurol. Genet. 7:e575. doi: 10.1212/NXG.0000000000000575
Fahey, C., Byrne, S., McLaughlin, R., Kenna, K., Shatunov, A., Donohoe, G., et al. (2014). Analysis of the hexanucleotide repeat expansion and founder haplotype at C9ORF72 in an Irish psychosis case-control sample. Neurobiol. Aging 35, 1510.e1–1510.e5. doi: 10.1016/j.neurobiolaging.2013.12.003
Glasmacher, S. A., Wong, C., Pearson, I. E., and Pal, S. (2020). Survival and prognostic factors in C9orf72 repeat expansion carriers: a systematic review and meta-analysis. JAMA Neurol. 77, 367–376. doi: 10.1001/jamaneurol.2019.3924
Gold, J., Rowe, D. B., Kiernan, M. C., Vucic, S., Mathers, S., van Eijk, R. P. A., et al. (2019). Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: the Lighthouse trial. Amyotroph. Lateral Scler. Frontotemporal Degeneration 20, 595–604. doi: 10.1080/21678421.2019.1632899
Gossye, H., Engelborghs, S., Van Broeckhoven, C., and van der Zee, J. (2020). “C9orf72 frontotemporal dementia and/or amyotrophic lateral sclerosis,” in GeneReviews, eds M. P. Adam H. H. Ardinger R. A. Pagon S. E. Wallace L. J. H. Bean K. W. Gripp et al. (Seattle, WA: University of Washington, Seattle), 1993–2022.
Gupta, S. K., McComsey, G. A., Lombaard, J., Echevarría, J., Orrell, C., Avihingsanon, A., et al. (2016). Efficacy, safety, bone and metabolic effects of HIV nucleoside reverse transcriptase inhibitor BMS-986001 (AI467003): a phase 2b randomized, controlled, partly blinded trial. Lancet 3, e13–e22. doi: 10.1016/S2352-3018(15)00231-3
Harms, M., Benitez, B. A., Cairns, N., Cooper, B., Cooper, P., Mayo, K., et al. (2013). C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 70, 736–741. doi: 10.1001/2013.jamaneurol.537
Hautbergue, G. M., Cleary, J. D., and Guo, S. (2021). Therapeutic strategies for C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 34, 748–755. doi: 10.1097/WCO.0000000000000984
Haynes, B. A., Rhinn, H., Yeh, F., Long, H., Ward, M., Mackenzie, H., et al. (2020). AL001 restores CSF PGRN levels and normalizes disease-associated biomarkers in individuals with frontotemporal dementia due to heterozygous mutations in the progranulin gene. Alzheimers Dement. 16:e046114. doi: 10.1002/alz.046114
Hogan, D. B., Jetté, N., Fiest, K. M., Roberts, J. I., Pearson, D., Smith, E. E., et al. (2016). The prevalence and incidence of frontotemporal dementia: a systematic review. Can. J. Neurol. Sci. 43, S96–S109. doi: 10.1017/cjn.2016.25
Inda, M. C., Joshi, S., Wang, T., Bolaender, A., Gandu, S., Koren Iii, J., et al. (2020). The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nat. Commun. 11:319. doi: 10.1038/s41467-019-14082-5
Jackson, J. L., Finch, N. A., Baker, M. C., Kachergus, J. M., DeJesus-Hernandez, M., Pereira, K., et al. (2020). Elevated methylation levels, reduced expression levels and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol. Neurodegener. 15:7. doi: 10.1186/s13024-020-0359-8
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Julian, T. H., Glascow, N., and Barry, A. D. F. (2021). Physical exercise is a risk factor for amyotrophic lateral sclerosis: convergent evidence from Mendelian randomization, transcriptomics and risk genotypes. EBioMedicine 68:103397. doi: 10.1016/j.ebiom.2021.103397
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H. R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U S A 110, E4530–E4539. doi: 10.1073/pnas.1318835110
Liu, Y., Andreucci, A., Iwamoto, N., Yin, Y., Yang, H., Liu, F., et al. (2022). Preclinical evaluation of WVE-004, an investigational stereopure oligonucleotide for the treatment of C9orf72-associated ALS or FTD. Mol. Ther. Nucleic Acids 28, 558–570. doi: 10.1016/j.omtn.2022.04.007
Malik, I., Kelley, C. P., Wang, E. T., and Todd, P. K. (2021). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607. doi: 10.1038/s41580-021-00382-6
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27, 1918–1929. doi: 10.1111/ene.14393
Miyakawa, S., Sakuma, H., Warude, D., Asanuma, S., Arimura, N., Yoshihara, T., et al. (2020). Anti-sortilin1 antibody Up-regulates progranulin via sortilin1 down-regulation. Front. Neurosci. 14:586107. doi: 10.3389/fnins.2020.586107
Moore, K. M., Nicholas, J., Grossman, M., McMillan, C. T., Irwin, D. J., Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 19, 145–156. doi: 10.1016/S1474-4422(19)30394-1
Murphy, N. A., Arthur, K. C., Tienari, P. J., Houlden, H., Chiò, A., and Traynor, B. J. (2017). Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 7:2116. doi: 10.1038/s41598-017-02364-1
Oskarsson, B., Gendron, T. F., and Staff, N. P. (2018). Amyotrophic lateral sclerosis: an update for 2018. Mayo Clin. Proc. 93, 1617–1628. doi: 10.1016/j.mayocp.2018.04.007
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riggs, J. E. (1990). Longitudinal Gompertzian analysis of amyotrophic lateral sclerosis mortality in the U. S. 1977–1986: evidence for an inherently susceptible population subset. Mech Ageing Dev. 55, 207–220. doi: 10.1016/0047-6374(90)90149-a
Roggenbuck, J. (2020). C9orf72 and the care of the patient with ALS or FTD: progress and recommendations after 10 years. Neurol. Genet. 7:e542. doi: 10.1212/NXG.0000000000000542
Roggenbuck, J., and Fong, J. C. (2020). Genetic testing for amyotrophic lateral sclerosis and frontotemporal dementia: impact on clinical management. Clin. Lab. Med. 40, 271–287. doi: 10.1016/j.cll.2020.05.002
Semmelink, M. F. W., Steen, A., and Veenhoff, L. M. (2021). Measuring and interpreting nuclear transport in neurodegenerative disease-the example of C9orf72 ALS. Int. J. Mol. Sci. 22:9217. doi: 10.3390/ijms22179217
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Silverman, H. E., Goldman, J. S., and Huey, E. D. (2019). Links between the C9orf72 repeat expansion and psychiatric symptoms. Curr. Neurol. Neurosci. Rep. 19:93. doi: 10.1007/s11910-019-1017-9
Smeyers, J., Banchi, E. G., and Latouche, M. (2021). C9ORF72: what it is, what it does and why it matters. Front. Cell Neurosci. 15:661447. doi: 10.3389/fncel.2021.661447
Smith, R. A., Raugi, D. N., Wu, V. H., Leong, S. S., Parker, K. M., Oakes, M. K., et al. (2015). The nucleoside analog BMS-986001 shows greater in vitro activity against HIV-2 than against HIV-1. Antimicrob. Agents Chemother. 59, 7437–7446. doi: 10.1128/AAC.01326-15
Terryn, J., Verfaillie, C. M., and Van Damme, P. (2021). Tweaking progranulin expression: therapeutic avenues and opportunities. Front. Mol. Neurosci. 14:713031. doi: 10.3389/fnmol.2021.713031
Tsitkanou, S., Della Gatta, P., Foletta, V., and Russell, A. (2019). The role of exercise as a nonpharmacological therapeutic approach for amyotrophic lateral sclerosis: beneficial or detrimental. Front. Neurol. 10:783. doi: 10.3389/fneur.2019.00783
van Blitterswijk, M., DeJesus-Hernandez, M., Niemantsverdriet, E., Murray, M. E., Heckman, M. G., Diehl, N. N., et al. (2013). Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. doi: 10.1016/S1474-4422(13)70210-2
van der Ende, E. L., Jackson, J. L., White, A., Seelaar, H., van Blitterswijk, M., and Van Swieten, J. C. (2021). Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J. Neurol. Neurosurg. Psychiatry 92, 502–509. doi: 10.1136/jnnp-2020-325377
van Es, M. A., Hardiman, O., Chio, A., Al-Chalabi, A., Pasterkamp, R. J., Veldink, J. H., et al. (2017). Amyotrophic lateral sclerosis. Lancet 390, 2084–2098. doi: 10.1016/S0140-6736(17)31287-4
Van Mossevelde, S., Engelborghs, S., van der Zee, J., and Van Broeckhoven, C. (2018). Genotype-phenotype links in frontotemporal lobar degeneration. Nat. Rev. Neurol. 14, 363–378. doi: 10.1038/s41582-018-0009-8
Van Mossevelde, S., van der Zee, J., Cruts, M., and Van Broeckhoven, C. (2017). Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 44, 117–124. doi: 10.1016/j.gde.2017.02.008
Vance, C., Al-Chalabi, A., Ruddy, D., Smith, B. N., Hu, X., Sreedharan, J., et al. (2006). Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 129, 868–876. doi: 10.1093/brain/awl030
Wilke, C., Pomper, J. K., Biskup, S., Puskás, C., Berg, D., and Synofzik, M. (2016). Atypical parkinsonism in C9orf72 expansions: a case report and systematic review of 45 cases from the literature. J. Neurol. 263, 558–574. doi: 10.1007/s00415-016-8021-7
Keywords: neurodegeneration, C9orf72, ALS, FTD, therapeutic strategies, molecular mechanisms
Citation: Zampatti S, Peconi C, Campopiano R, Gambardella S, Caltagirone C and Giardina E (2022) C9orf72-Related Neurodegenerative Diseases: From Clinical Diagnosis to Therapeutic Strategies. Front. Aging Neurosci. 14:907122. doi: 10.3389/fnagi.2022.907122
Received: 29 March 2022; Accepted: 16 May 2022;
Published: 10 June 2022.
Edited by:
Roberta Ghidoni, San Giovanni di Dio Fatebenefratelli Center (IRCCS), ItalyReviewed by:
Maria Rosario Almeida, University of Coimbra, PortugalJulie van der Zee, University of Antwerp, Belgium
Copyright © 2022 Zampatti, Peconi, Campopiano, Gambardella, Caltagirone and Giardina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emiliano Giardina, ZW1pbGlhbm8uZ2lhcmRpbmFAdW5pcm9tYTIuaXQ=