 Hiroshi Ito1
Hiroshi Ito1 Sanae Hosomi1*
Sanae Hosomi1* Yoshihisa Koyama2,3
Yoshihisa Koyama2,3 Hisatake Matsumoto1Yukio Imamura1Hiroshi Ogura1
Hisatake Matsumoto1Yukio Imamura1Hiroshi Ogura1 Jun Oda1
Jun Oda1- 1Department of Traumatology and Acute Critical Medicine, Graduate School of Medicine, Osaka University, Osaka, Japan
- 2Department of Neuroscience and Cell Biology, Osaka University Graduate School of Medicine, Osaka, Japan
- 3Addiction Research Unit, Osaka Psychiatric Research Center, Osaka Psychiatric Medical Center, Osaka, Japan
Sepsis is defined as a life-threatening multi-organ dysfunction triggered by an uncontrolled host response to infectious disease. Systemic inflammation elicited by sepsis can cause acute cerebral dysfunction, characterized by delirium, coma, and cognitive dysfunction, known as septic encephalopathy. Recent evidence has reported the underlying mechanisms of sepsis. However, the reasons for the development of inflammation and degeneration in some brain regions and the persistence of neuroinflammation remain unclear. This mini-review describes the pathophysiology of region-specific inflammation after sepsis-associated encephalopathy (SAE), clinical features, and future prospects for SAE treatment. The hippocampus is highly susceptible to inflammation, and studies that perform treatments with antibodies to cytokine receptors, such as interleukin-1β, are in progress. Future development of clinically applicable therapies is expected.
Introduction
Sepsis is defined as a life-threatening multi-organ dysfunction triggered by an uncontrolled host response to infectious disease. Systemic inflammation elicited by sepsis can cause acute cerebral dysfunction, characterized by delirium, coma, and cognitive dysfunction, known as septic encephalopathy. Septic encephalopathy develops in 53% of patients with sepsis (Sonneville et al., 2017). In addition, septic encephalopathy is associated with increased mortality and with a 2.22-fold increased risk of developing dementia in the long term (Eidelman et al., 1996; Fritze et al., 2020). Septic encephalopathy is considered a diffuse brain dysfunction resulting from a systemic inflammatory response to infection, and not a symptom of direct central nervous system infection (Iacobone et al., 2009; Gao and Hernandes, 2021; Gu et al., 2021; Manabe and Heneka, 2021). Moreover, septic encephalopathy is an extremely critical condition that affects the functional prognosis and life expectancy of septic patients, and there is a strong need to elucidate and control its mechanisms.
Regarding the underlying mechanisms of sepsis, recent evidence suggests that endothelial cell degeneration, enhanced blood-brain barrier (BBB) permeability, and tight junction protein loss promote and trigger the influx of inflammatory mediators, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α, into the brain (Gao and Hernandes, 2021; Gu et al., 2021; Manabe and Heneka, 2021). However, the reasons why some brain regions develop inflammation and degeneration and the factors that determine the persistence of neuroinflammation remain unclear.
This mini-review describes the pathophysiology of region-specific inflammation after SAE, clinical features, and therapeutic measures attenuating SAE.
Characteristics of Septic Encephalopathy: A Region-Specific Perspective
Clinical Features in the Acute Stage of Sepsis
Acute and reversible deterioration of mental status is often associated with sepsis, and this disorder may lead to SAE. Diffuse cerebral dysfunction due to a systemic inflammatory response to infection is considered SAE, which does not include direct central nervous system infection (Iacobone et al., 2009). SAE is associated with abnormal biomarkers, such as elevated IL-1β levels, and by neuroradiologic imaging findings, such as hippocampus atrophy on brain magnetic resonance imaging (MRI). Moreover, it is assessed by various methods to identify which of the brain areas are affected (Table 1).
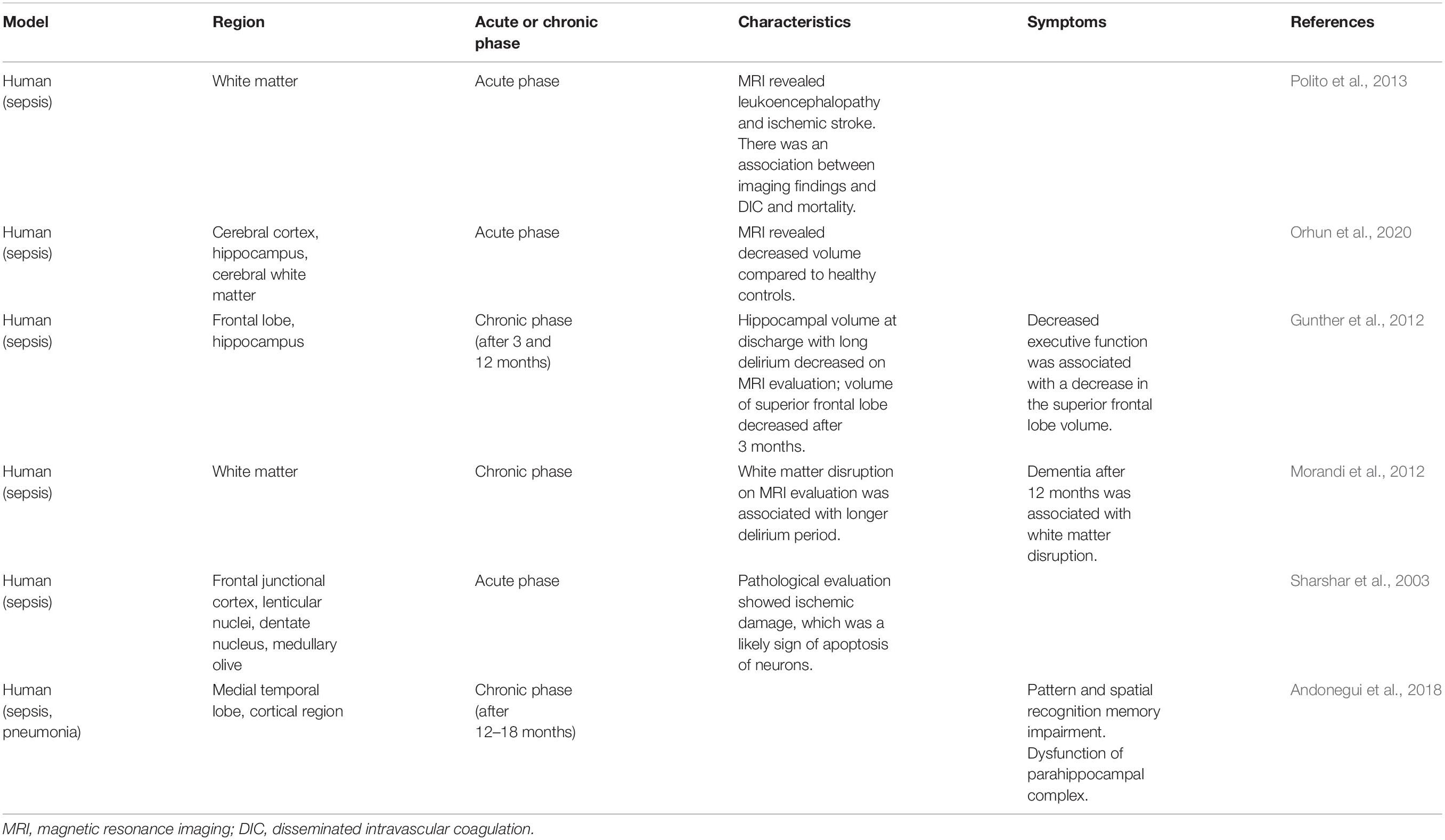
Table 1. Brain dysfunction in patients with sepsis.
In patients with septic shock who develop acute brain dysfunction, leukoencephalopathy (confluent or diffuse white matter lesions), and ischemic stroke can be detected on magnetic resonance imaging (MRI) (Polito et al., 2013). Ischemic stroke is associated with disseminated intravascular coagulation and increased mortality (Polito et al., 2013; Iba et al., 2019). The volumes of the cerebral cortex, hippocampus, amygdala, and cerebral white matter were smaller in patients with SAE than in healthy controls (Orhun et al., 2020). Among the non-survivors of SAE, the volume of the hippocampus was smaller than that of healthy controls and SAE survivors (Orhun et al., 2020).
Tissue samples from patients with delirium showed activated microglia and astrocytes and elevated levels of IL-6 in the hippocampus. This condition is associated with delirium in older patients, suggesting a relationship between delirium and inflammatory mechanisms (Munster et al., 2011).
Basic Studies on the Acute Phase of Sepsis
Various studies using mouse models of sepsis have been conducted on multiple brain regions (Table 2). In an in vivo study, microglia in the hippocampus showed morphological changes at 6 h after the systemic administration of lipopolysaccharide (LPS), which is a characteristic of activation. Staining for ionized calcium-binding adaptor molecule 1 (Iba1), a protein expressed in microglia and macrophages and a cytoskeletal component involved in migration, increased (Johansson et al., 2013). In previous works, the microglia and astrocytes were found to be activated in the cortex and hippocampus. Apoptosis of neurons in the cortex and hippocampus resulted in a decrease in the number of neurons (Lee et al., 2008; Semmler et al., 2008). In one such study, activation of microglia in the hippocampus in a mouse model of CLP was reported to be stronger than that in the cortex. This suggests that microglial activation by sepsis is different in a region-specific manner and that the hippocampus is more affected by sepsis (Moraes et al., 2015).
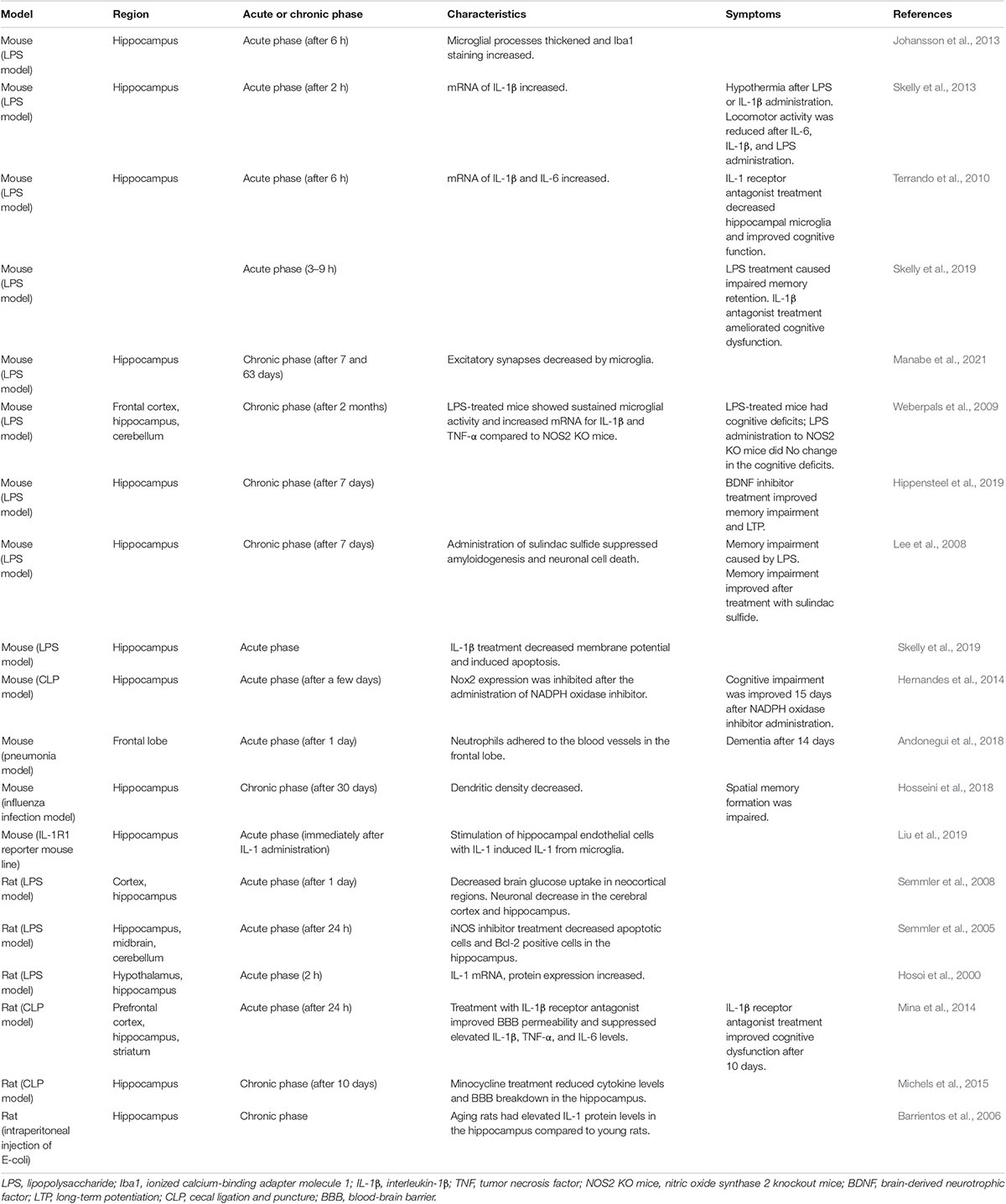
Table 2. Brain dysfunction in mouse and rat sepsis models.
In addition to these morphological changes, there were region-specific features related to cytokine expression. Under physiological conditions, brain IL-1 receptor 1 (IL-1R1) was expressed primarily in the choroid plexus and endothelial cells and rarely in astrocytes but was not expressed in the microglia.
In the microglia, where IL-1R1 expression was not originally found, IL-1ß mRNA was expressed when endothelial cells were stimulated with IL-1ß. In addition, IL-1R1 expression in the neurons was most strongly confirmed in the hippocampus, where multiple cell types expressing IL-1R1 mRNA have been identified in endothelial cells, astrocytes, ependymal cells, and choroid plexus cells (Liu et al., 2019). Peripheral administration of IL-1β and TNF-α significantly increased hippocampal IL-1β mRNA expression (Skelly et al., 2013), while a 6.5-fold increase in IL-1β and a 15-fold increase in IL-6 mRNA expression were noted in the hippocampus at 6 h after LPS administration (Terrando et al., 2010). IL-1β is secreted from microglia activated by LPS and can inhibit synaptogenesis (Moraes et al., 2015).
In another study, positron emission tomography revealed that in the LPS model mice, regional cerebral blood flow (rCBF) was reduced in the cerebral cortex and alpha activity on electroencephalography decreased. In addition, cerebral glucose uptake was maintained in the hippocampus and thalamus but decreased in all neocortical regions (Semmler et al., 2008). These findings suggested that there were differences in the regional vulnerability among brain areas.
In a report on functional impairment, locomotor activity clearly decreased at 2 h after administration of systemic IL-6, IL-1β, and LPS. The central nuclei of the amygdala are activated, and the frontal cortex is affected, which is thought to be associated with symptoms, such as increased anxiety, depression, and delirium (Skelly et al., 2013). Dysfunction of working memory is induced by LPS via circulating IL-1β. In addition, direct action of IL-1β on the hippocampus may result in neuronal dysfunction and promote neuronal cell death (Skelly et al., 2019). Indeed, the IL-1β level was found to be elevated on day 9 after CLP. Especially, IL-1β inhibited synaptogenesis, with excitatory synapses in the hippocampus being reduced on day 9. In behavioral experiments, the cognitive index also decreased on day 9, consistent with the symptom course (Moraes et al., 2015).
The Clinical Features in the Chronic Stage of Sepsis
The clinical profile of septic encephalopathy has been reported not only in the acute phase but also in the chronic phase. Sepsis survivors often exhibit prolonged cognitive impairment (Fritze et al., 2020).
Behavioral tests performed on patients recovering from sepsis due to pneumonia showed specific cognitive impairments in visuospatial memory function. This pattern of impairment suggests the possibility of dysfunction in the parahippocampal complex, which is highly dependent on environmental perturbations (Andonegui et al., 2018).
There are also reports on the associations among delirium duration, imaging findings, and clinical symptoms. Longer duration of delirium was related to smaller hippocampal volumes on computed tomography (indicative of hippocampal atrophy) at discharge. In addition, volume reduction in the superior frontal lobes was observed on follow-up scan at discharge and at 3 months later. Moreover, patients with reduced superior frontal lobe volume displayed reduced executive function and visual attention at 3 months after discharge (Gunther et al., 2012). MRI examinations also showed that the duration of delirium in the intensive care unit (ICU) caused white matter disruption on CT at discharge and at 3 months later; similarly, worse cognitive scores assessed by the Repeatable Battery for the Assessment of Neuropsychological Status at 12 months was associated with white matter disruption (Morandi et al., 2012).
Basic Studies on the Chronic Phase of Sepsis
Studies have been published on changes in neural tissue in the chronic phase using a sepsis model. In one such study, infection with influenza virus led to a reduction in dendritic density in the hippocampal subregions, both in the superior and inferior DG at 30 days after infection. With this reduction in density, activated microglia increased (DG-superior: 49.18%, DG-inferior: 55.96%) (one-way analysis of variance FDG–superior(3,76) = 17.57, p < 0.0001, and FDG–inferior(3,76) = 22.16, p < 0.0001), long-term potentiation (LTP) in the hippocampus decreased, and spatial memory formation was impaired. This finding indicates that inflammation caused by the influenza virus resulted in functional and structural alterations in the hippocampal network (Hosseini et al., 2018).
Subsequent studies have reported on mechanisms that result in functional and structural changes in the hippocampal network. In the LPS mouse model, no significant effects were observed in neurons and synapses at 7 and 63 days post-infection. However, in the CA3 region of the hippocampus, excitatory synapses and complement factor 3 punctuation showed a delayed, region-specific decrease at 63 days. This was associated with elevated CD11b (a marker of microglia or macrophages) levels in microglia for more than 1 week after LPS administration (E. coli LPS: 170.16 ± 3.99%, E. coli LPS vs. Veh: p < 0.0001), indicating that microglia were not affected until 63 days after infection by hippocampal CA 3 synapses involved in the loss of synapses (Manabe et al., 2021). Loss of synapses over a prolonged period after inflammation may have a functional impairment effect.
LPS administration to wild-type mice significantly increased the TNF-α and IL-1β mRNA levels in the cortex and hippocampus, as well as the TNF-α mRNA levels in the cerebellum (Weberpals et al., 2009). Treatment of aging mice with LPS significantly increased the hippocampal IL-1β levels compared to those of young mice, in addition to age-related effects on cognitive function (Barrientos et al., 2006).
The hippocampus is affected in a region-specific manner by blood factors, resulting in hippocampal dysfunction. In sepsis, the endothelial glycocalyx is degraded, and heparan sulfate fragments are released into the circulatory system. Heparan sulfate is sufficiently large and sulfated to bind to brain-derived neurotrophic factor (BDNF), and circulating heparan sulfate fragments enter the hippocampus. Heparan sulfate inhibits BDNF and is hypothesized to block BDNF-specific functions related to spatial memory formation. To examine these issues, Hippensteel et al. examined N- and 2-O-sulfated heparan sulfate and cognitive impairment in sepsis and found that the presence of circulating 2S-rich BDNF-avid heparan sulfate fragments at the onset of sepsis was associated with cognitive impairment by 2 weeks after ICU discharge. The entry of circulating heparan sulfate fragments into the hippocampus sequesters BDNF, impairs LTP, and causes septic cognitive impairment (Hippensteel et al., 2019).
Discussion
Why Is Region-Specific Inflammation Observed?
Some studies (Yokota et al., 2003; Fong et al., 2006) have reported a reduction in rCBF during acute delirium in the frontal, parietal, and occipital lobes. Reduced cerebral blood flow, if sufficiently persistent, can lead to cell death, neuronal loss, and brain atrophy (Sharshar et al., 2004; Semmler et al., 2005; Gunther et al., 2012).
In general, local inflammation is the response of tissues to infection and damage. In the acute phase, dilation of vessels helps transport cells and molecules to the site of infection or damage to elicit a protective response. However, sepsis induces systemic hypotension, resulting in reduced cerebral perfusion. In addition, in the ICU, sedation worsens the situation (Egi et al., 2021). Especially in older adults, age-related decline in cardiovascular function may impair cerebral blood flow regulation, leading to the disruption of neuronal micro-environmental homeostasis (O’Rourke and Hashimoto, 2007).
Systemic inflammation, such as sepsis, produces nitric oxide (NO), which plays a key role in endotoxin-induced vasodilation and myocardial dysfunction. Endothelial injury results in microvascular thrombosis, vasodilation, and hypotension, which lead to tissue hypoxia and multi-organ dysfunction (Reinhart et al., 2002). Similar processes may occur in the brain, and if left untreated for an extended period, they can lead to neuronal cell death and brain atrophy (Sharshar et al., 2004). There is also a significant correlation between reduced cerebral blood flow, reduced glucose metabolism in the brain, and reduced neuronal cell death and neural activity (Semmler et al., 2008). Thus, septic encephalopathy involves cell death due to reduced cerebral blood flow and inflammation. Therefore, regions susceptible to ischemia, for example, lenticular nuclei, and medullary olives, are more likely to be damaged and may have impaired region-specific functions. However, the effects of septic inflammation are more significant than those of ischemia. A postmortem study suggested that during septic shock, autonomic neurons, including the amygdala, in the brain are subject to apoptosis by NO rather than ischemia (Sharshar et al., 2003).
In various experimental sepsis models, the complexity of inflammation and apoptosis-promoting factors and increased microglial activation and oxidative stress led to hippocampal destruction (Semmler et al., 2008; Zaghloul et al., 2017; Peng et al., 2018; Fu et al., 2019). Microglial activation has also been associated with reduced hippocampal volume and cognitive dysfunction in Alzheimer’s disease and other diseases (Orhun et al., 2020). Thus, the hippocampus is susceptible to inflammation due to its function. Stimulation of the vagus nerve promotes IL-1β transcription in the hippocampus, which improves memory formation and maintenance (Hosoi et al., 2000). The hippocampus is responsible for the majority of learning and memory processes, and IL-1 receptors are most highly expressed in the hippocampus. Accordingly, it has been reported that the hippocampus is highly susceptible to the unfavorable effects of neuroinflammation (Parnet et al., 1994; Gemma et al., 2005; Mina et al., 2014).
Area-specific symptoms are also observed in the chronic phase even after the acute inflammation subsides, possibly because activated microglia persist in the chronic phase, causing brain atrophy due to neuronal degeneration (Manabe et al., 2021).
Future Treatments
The pathophysiology of septic encephalopathy is becoming clearer, and studies have shown that knockout mice or antagonists are administered with several factors that affect the pathophysiology. In this issue, we focused on the hippocampus, which might be a key region, and introduced some of these reports.
Interleukin-1 Receptor Antagonist
Prophylactic treatment with an IL-1 receptor antagonist significantly reduces plasma cytokines and hippocampal microgliosis and improves working memory impairment and cognitive dysfunction. There was no effect on high-mobility group box-1 levels (Terrando et al., 2010; Mina et al., 2014; Skelly et al., 2019).
Sulindac Sulfide
Administration of the anti-inflammatory agent, sulindac sulfide, at 3 weeks before infection suppressed LPS-induced amyloidogenesis, memory impairment, and neuronal cell death (Lee et al., 2008).
Nitric Oxide Synthase 2 Knockout Mice and Apocynin
Reactive free radicals NO and reactive oxygen species (ROS) are produced in sepsis. Previous reports have stated that nitric oxide synthase 2 (NOS2) is related to NO synthesis and that nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) is involved in ROS synthesis. In a study using NOS2 knockout mice, LPS treatment resulted in sustained increased CD11b immunoreactivity in the hippocampus, frontal cortex, and cerebellum, with persistently increased glial activation in wild-type mice compared to that in NOS2 knockout mice. Expression analysis also showed that the brain mRNA levels of inflammatory factors were lower in LPS-treated NOS2 knockout mice than in wild-type mice and that no changes were observed in NOS2 knockout mice with respect to cognitive function (Weberpals et al., 2009).
In the cerebrum of cases with sepsis, microglia and astrocytes are activated and NO is produced; thus, resulting in apoptosis and necrosis in glial cells and neurons (Semmler et al., 2005).
NO induced by NOS2 may also cause neuroinflammatory responses and long-term changes in microglia and consequent cognitive dysfunction after LPS administration (Weberpals et al., 2009). Future studies are needed to clarify whether NO is detrimental to SAE and whether inhibition of NO may be neuroprotective (Semmler et al., 2005).
ROS produced by Nox2 cause oxidative damage to the hippocampus. This results in cognitive impairment after SAE and sepsis. Pharmacological inhibition of Nox2 (apocynin) or loss of Nox2 prevents glial cell activation, a central mechanism associated with SAE, suppressing oxidative stress in the hippocampus and preventing the development of long-term cognitive impairment (Hernandes et al., 2014).
Minocycline
Minocycline scavenges superoxide and peroxynitrite in non-neural in vitro assays (Whiteman and Halliwell, 1997; Zhang et al., 2003). Minocycline treatment reduces the cytokine levels and BBB decay in the hippocampus (Michels et al., 2015).
7,8-Dihydroxyflavone
Brain-derived neurotrophic factor, a nerve growth factor, has a significant role in hippocampal LTP and experimental BDNF sequestration induces cognitive dysfunction (Figurov et al., 1996). Moreover, 7,8-dihydroxyflavone, a selective agonist of BDNF receptor tyrosine kinase B, administered to endotoxin-infected mice prevents memory loss and normalizes contextual fear conditioning by day 7 (Hippensteel et al., 2019).
Study Limitations
Based on these findings, we should further examine whether targeting other cytokine receptors for treatment can prevent or ameliorate cognitive decline in the future (Terrando et al., 2010). If these studies can be applied to clinical practice, it is expected that in the real world, the treatment of patients with sepsis will not only treat acute delirium but also prevent dementia in the chronic phase over the long term.
In this study, we primarily reviewed whether treatment intervention improves symptoms of SAE. It is also necessary to examine whether the intervention improves SAE and systemic inflammation. The focus of treatment of SAE remains proper management of systemic infections, sepsis, and systemic inflammatory response syndrome (Egi et al., 2021). Treatment of systemic infections could be considered to avoid substantial morbidity and mortality associated with SAE. It has been reported that LPS affected inflammation in the brain even in the absence of systemic IL-1β activity, although systemic IL-1β action was suppressed and cytokines in the blood were reduced when mice were treated with an IL-1 receptor antagonist in the LPS treatment model (Terrando et al., 2010; Skelly et al., 2019). Thus, although antagonists have some effect in suppressing systemic inflammation, they act only on part of the complex inflammatory cascade, and their effect may be limited. Further research is needed to expand the clinical applicability of these findings.
Author Contributions
HI, SH, YK, HM, YI, and HO designed the study and wrote the manuscript. JO critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (18K08886 and 21K09016) to SH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andonegui, G., Zelinski, E. L., Schubert, C. L., Knight, D., Craig, L. A., Winston, B. W., et al. (2018). Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight. 3:e99364. doi: 10.1172/jci.insight.99364
Barrientos, R. M., Higgins, E. A., Biedenkapp, J. C., Sprunger, D. B., Wright-Hardesty, K. J., Watkins, L. R., et al. (2006). Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol. Aging. 27, 723–732. doi: 10.1016/j.neurobiolaging.2005.03.010
Egi, M., Ogura, H., Yatabe, T., Atagi, K., Inoue, S., Iba, T., et al. (2021). The Japanese clinical practice guidelines for management of sepsis and septic shock 2020 (J-SSCG 2020). Acute Med. Surg 8:e659.
Eidelman, L. A., Putterman, D., Putterman, C., and Sprung, C. L. (1996). The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 275, 470–473. doi: 10.1001/jama.1996.03530300054040
Figurov, A., Pozzo-Miller, L. D., Olafsson, P., Wang, T., and Lu, B. (1996). Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 381, 706–709. doi: 10.1038/381706a0
Fong, T. G., Bogardus, S. T., Daftary, A., Auerbach, E., Blumenfeld, H., Modur, S., et al. (2006). Cerebral perfusion changes in older delirious patients using 99mTc HMPAO SPECT. J. Gerontol. A Biol. Sci. Med. Sci. 61, 1294–1299. doi: 10.1093/gerona/61.12.1294
Fritze, T., Doblhammer, G., Widmann, C. N., and Heneka, M. T. (2020). Time course of dementia following sepsis in German health claims data. Neurol. Neuroimmunol. Neuroinflamm. 8:e911. doi: 10.1212/NXI.0000000000000911
Fu, Q., Wu, J., Zhou, X. Y., Ji, M. H., Mao, Q. H., Li, Q., et al. (2019). NLRP3/Caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation 42, 306–318. doi: 10.1007/s10753-018-0894-4
Gao, Q., and Hernandes, M. S. (2021). Sepsis-associated encephalopathy and blood–brain barrier dysfunction. Inflammation. 44, 2143–2150. doi: 10.1007/s10753-021-01501-3
Gemma, C., Fister, M., Hudson, C., and Bickford, P. C. (2005). Improvement of memory for context by inhibition of caspase-1 in aged rats. Eur. J. Neurosci. 22, 1751–1756. doi: 10.1111/j.1460-9568.2005.04334.x
Gu, M., Mei, X. L., and Zhao, Y. N. (2021). Sepsis and cerebral dysfunction: BBB damage, neuroinflammation, oxidative stress, apoptosis and autophagy as key mediators and the potential therapeutic approaches. Neurotox. Res. 39, 489–503. doi: 10.1007/s12640-020-00270-5
Gunther, M. L., Morandi, A., Krauskopf, E., Pandharipande, P., Girard, T. D., Jackson, J. C., et al. (2012). The association between brain volumes, delirium duration, and cognitive outcomes in intensive care unit survivors: the VISIONS cohort magnetic resonance imaging study. Crit. Care Med. 40, 2022–2032. doi: 10.1097/CCM.0b013e318250acc0
Hernandes, M. S., D’Avila, J. C., Trevelin, S. C., Reis, P. A., Kinjo, E. R., Lopes, L. R., et al. (2014). The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J. Neuroinflammation. 11:36. doi: 10.1186/1742-2094-11-36
Hippensteel, J. A., Anderson, B. J., Orfila, J. E., McMurtry, S. A., Dietz, R. M., Su, G., et al. (2019). Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J. Clin. Invest. 129, 1779–1784. doi: 10.1172/JCI124485
Hosoi, T., Okuma, Y., and Nomura, Y. (2000). Electrical stimulation of afferent vagus nerve induces IL-1beta expression in the brain and activates HPA axis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R141–R147. doi: 10.1152/ajpregu.2000.279.1.R141
Hosseini, S., Wilk, E., Michaelsen-Preusse, K., Gerhauser, I., Baumgärtner, W., Geffers, R., et al. (2018). Long-term neuroinflammation induced by influenza a virus infection and the impact on hippocampal neuron morphology and function. J. Neurosci. 38, 3060–3080. doi: 10.1523/JNEUROSCI.1740-17.2018
Iacobone, E., Bailly-Salin, J., Polito, A., Friedman, D., Stevens, R. D., and Sharshar, T. (2009). Sepsis-associated encephalopathy and differential diagnoses Sepsis-associated encephalopathy and its differential diagnosis. Crit. Care Med. 37, S331–S336. doi: 10.1097/CCM.0b013e3181b6ed58
Iba, T., Umemura, Y., Watanabe, E., Wada, T., Hayashida, K., and Kushimoto, S. (2019). Diagnosis of sepsis-induced disseminated intravascular coagulation and coagulopathy. Acute Med. Surg. 6, 223–232. doi: 10.1002/ams2.411
Johansson, J. U., Pradhan, S., Lokteva, L. A., Woodling, N. S., Ko, N., Brown, H. D., et al. (2013). Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J. Neurosci. 33, 16016–16032. doi: 10.1523/JNEUROSCI.2203-13.2013
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of β-amyloid generation. J. Neuroinflammation. 5, 37. doi: 10.1186/1742-2094-5-37
Liu, X., Nemeth, D. P., McKim, D. B., Zhu, L., DiSabato, D. J., Berdysz, O., et al. (2019). Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity 50, 317.e–333.e. doi: 10.1016/j.immuni.2018.12.012
Manabe, T., and Heneka, M. T. (2021). Cerebral dysfunction caused by sepsis during aging. Nat. Rev. Immunol. 11, 1–15. doi: 10.1038/s41577-021-00643-7
Manabe, T., Rácz, I., Schwartz, S., Oberle, L., Santarelli, F., Emmrich, J. V., et al. (2021). Systemic inflammation induced the delayed reduction of excitatory synapses in the CA3 during ageing. J. Neurochem. 159, 525–542. doi: 10.1111/jnc.15491
Michels, M., Vieira, A. S., Vuolo, F., Zapelini, H. G., Mendonça, B., Mina, F., et al. (2015). The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav. Immun. 43, 54–59. doi: 10.1016/j.bbi.2014.07.002
Mina, F., Comim, C. M., Dominguini, D., Cassol, O. J. Jr., Dall Igna, D. M. D., Ferreira, G. K., et al. (2014). Il1-beta involvement in cognitive impairment after sepsis. Mol. Neurobiol 49, 1069–1076. doi: 10.1007/s12035-013-8581-9
Moraes, C. A., Santos, G., Spohr, T. C. L., D’Avila, J. C., Lima, F. R. S., Benjamin, C. F., et al. (2015). Activated microglia-induced deficits in excitatory synapses through il-1β: implications for cognitive impairment in sepsis. Mol. Neurobiol. 52, 653–663. doi: 10.1007/s12035-014-8868-5
Morandi, A., Rogers, B. P., Gunther, M. L., Merkle, K., Pandharipande, P., Girard, T. D., et al. (2012). The relationship between delirium duration, white matter integrity, and cognitive impairment in intensive care unit survivors as determined by diffusion tensor imaging: the VISIONS prospective cohort magnetic resonance imaging study. Crit. Care Med. 40, 2182–2189. doi: 10.1097/CCM.0b013e318250acdc
Munster, B. C., Aronica, E., Zwinderman, A. H., Eikelenboom, P., Cunningham, C., and Rooij, S. E. (2011). Neuroinflammation in delirium: a postmortem case-control study. Rejuvenation Res. 14, 615–622. doi: 10.1089/rej.2011.1185
O’Rourke, M. F., and Hashimoto, J. (2007). Mechanical factors in arterial aging: a clinical perspective. J. Am. Coll. Cardiol. 50, 1–13. doi: 10.1016/j.jacc.2006.12.050
Orhun, G., Tüzün, E., Bilgiç, B., Özcan, P., Sencer, S., Barburoǧlu, M., et al. (2020). Brain volume changes in patients with acute brain dysfunction due to sepsis. Neurocrit. Care. 32, 459–468. doi: 10.1007/s12028-019-00759-8
Parnet, P., Amindari, S., Wu, C., Brunke-Reese, D., Goujon, E., Weyhenmeyer, J. A., et al. (1994). Expression of type I and type II interleukin-1 receptors in mouse brain. Brain Res. Mol. Brain Res. 27, 63–70. doi: 10.1016/0169-328x(94)90185-6
Peng, Q. Y., Wang, Y. M., Chen, C. X., Zou, Y., Zhang, L. N., Deng, S. Y., et al. (2018). Inhibiting the CD38/cADPR pathway protected rats against sepsis associated brain injury. Brain Res. 1678, 56–63. doi: 10.1016/j.brainres.2017.09.029
Polito, A., Eischwald, F., Maho, A. L., Polito, A., Azabou, E., Annane, D., et al. (2013). Pattern of brain injury in the acute setting of human septic shock. Crit. Care. 17:R204. doi: 10.1186/cc12899
Reinhart, K., Bayer, O., Brunkhorst, F., and Meisner, M. (2002). Markers of endothelial damage in organ dysfunction and sepsis. Crit. Care Med 30, S302–S312. doi: 10.1097/00003246-200205001-00021
Semmler, A., Hermann, S., Mormann, F., Weberpals, M., Paxian, S. A., Okulla, T., et al. (2008). Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J. Neuroinflammation. 5:38. doi: 10.1186/1742-2094-5-38
Semmler, A., Okulla, T., Sastre, M., Dumitrescu-Ozimek, L., and Heneka, M. T. (2005). Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J. Chem. Neuroanat. 30, 144–157. doi: 10.1016/j.jchemneu.2005.07.003
Sharshar, T., Annane, D., de la Grandmaison, G. L., Brouland, J. P., Hopkinson, N. S., and Françoise, G. (2004). The neuropathology of septic shock. Brain Pathol 14, 21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x
Sharshar, T., Gray, F., Lorin de la Grandmaison, G. L., Hopkinson, N. S., Ross, E., Dorandeu, A., et al. (2003). Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 362, 1799–1805. doi: 10.1016/S0140-6736(03)14899-4
Skelly, D. T., Griffin, ÉW., Murray, C. L., Harney, S., O’Boyle, C., Hennessy, E., et al. (2019). Acute transient cognitive dysfunction and acute brain injury induced by systemic inflammation occur by dissociable IL-1-dependent mechanisms. Mol. Psychiatry 24, 1533–1548. doi: 10.1038/s41380-018-0075-8
Skelly, D. T., Hennessy, E., Dansereau, M. A., and Cunningham, C. (2013). A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1β, [corrected] TNF-α and IL-6 challenges in C57BL/6 mice. PLoS One. 8:e69123. doi: 10.1371/journal.pone.0069123
Sonneville, R., de Montmollin, E., Poujade, J., Garrouste-Orgeas, M., Souweine, B., Darmon, M., et al. (2017). Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 43, 1075–1084. doi: 10.1007/s00134-017-4807-z
Terrando, N., Rei Fidalgo, A., Vizcaychipi, M., Cibelli, M., Ma, D., Monaco, C., et al. (2010). The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit. Care. 14:R88. doi: 10.1186/cc9019
Weberpals, M., Hermes, M., Hermann, S., Kummer, M. P., Terwel, D., Semmler, A., et al. (2009). NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J. Neurosci. 29, 14177–14184. doi: 10.1523/JNEUROSCI.3238-09.2009
Whiteman, M., and Halliwell, B. (1997). Prevention of peroxynitrite-dependent tyrosine nitration and inactivation of alpha1-antiproteinase by antibiotics. Free Radic. Res. 26, 49–56. doi: 10.3109/10715769709097783
Yokota, H., Ogawa, S., Kurokawa, A., and Yamamoto, Y. (2003). Regional cerebral blood flow in delirium patients. Psychiatry Clin. Neurosci. 57, 337–339. doi: 10.1046/j.1440-1819.2003.01126.x
Zaghloul, N., Addorisio, M. E., Silverman, H. A., Patel, H. L., Valdés-Ferrer, S. I., Ayasolla, K. R., et al. (2017). Forebrain cholinergic dysfunction and systemic and brain inflammation in murine sepsis survivors. Front. Immunol. 8:1673. doi: 10.3389/fimmu.2017.01673
Keywords: sepsis-associated encephalopathy, hippocampus, interleukin-1β, delirium, dementia
Citation: Ito H, Hosomi S, Koyama Y, Matsumoto H, Imamura Y, Ogura H and Oda J (2022) Sepsis-Associated Encephalopathy: A Mini-Review of Inflammation in the Brain and Body. Front. Aging Neurosci. 14:912866. doi: 10.3389/fnagi.2022.912866
Received: 05 April 2022; Accepted: 06 May 2022;
Published: 27 May 2022.
Edited by:
Guanghui Wang, Soochow University, ChinaReviewed by:
Joana C. D’Avila, Universidade Iguaçu, BrazilNeera Chaudhry, Vardhman Mahavir Medical College and Safdarjung Hospital, India
Copyright © 2022 Ito, Hosomi, Koyama, Matsumoto, Imamura, Ogura and Oda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanae Hosomi, cy1ob3NvbWlAaHAtZW1lcmcubWVkLm9zYWthLXUuYWMuanA=