 Fei Yang
Fei Yang Liling Chen
Liling Chen Yanying Yu
Yanying Yu Tingwan Xu
Tingwan Xu Lu Chen
Lu Chen Wenqian Yang
Wenqian Yang Qian Wu
Qian Wu Yanbing Han
Yanbing Han- Department of Neurology, First Affiliated Hospital of Kunming Medical University, Kunming, China
Both Alzheimer’s disease (AD) and epilepsy are common chronic diseases in older people. Seizures and epileptiform discharges are very prevalent in AD and can occur since any stage of AD. Increasing evidence indicates that AD and epilepsy may be comorbid. Several factors may be related to the underlying mechanism of the comorbidity. Identifying seizures in patients with AD is a challenge because seizures are often clinically non-motor and may overlap with some AD symptoms. Not only seizures but also epileptiform discharges may exacerbate the cognitive decline in AD patients, highlighting the importance of early recognition and treatment. This review provides a comprehensive overview of seizures in AD from multiple aspects to provide more insight.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia. The AD patient population is rapidly expanding to 50 million people worldwide (Scearce-Levie et al., 2020). Most cases occur after age 65 and are referred to as late-onset AD (LOAD), while cases earlier than age 65 are much rarer, accounting for less than 5%, and are referred to as early-onset AD (EOAD) (Long and Holtzman, 2019). With the aging population, it is estimated that the incidence of AD will continue to increase, which will bring a heavy burden to society and families of AD patients (Teixeira et al., 2017; Jia et al., 2020; Scheltens et al., 2021).
Epilepsy is characterized by spontaneous seizures. Globally, epilepsy affects more than 70 million people, with the highest incidence in patients older than 65 years (Thijs et al., 2019). Neurocognitive disorders, including AD, are a common cause of new-onset epilepsy in older people. Recurrent seizures may be followed by cognitive decline, particularly in older patients (Sen et al., 2020; Wang et al., 2021).
Both AD and epilepsy have a serious impact on the patients’ health and reduce the quality of life of the patient and their family. Unfortunately, both can occur in one patient at the same time. Since Sjogren et al. (1952) reported that seizures were confirmed in patients with AD by pathological diagnosis, the relationship between AD and epilepsy has attracted an increasing amount of attention. Research on the relationship between seizures and AD has developed quickly.
High incidence of seizures and epileptiform discharges in Alzheimer’s disease
Table 1 lists the prevalence of seizures or epileptiform discharges in AD patients reported in various studies since 1952 (Sjogren et al., 1952; Hauser et al., 1986; Mendez et al., 1994; Mendez and Lim, 2003; Amatniek et al., 2006; Hommet et al., 2007; Bell et al., 2011; Vossel et al., 2013; Horváth et al., 2016; Nicastro et al., 2016; Vossel et al., 2016; Zarea et al., 2016; Beagle et al., 2017; Horváth et al., 2018; Rauramaa et al., 2018; Lam et al., 2020). These results are inconsistent and may be related to diverse study subjects, objectives, and strategies. The incidence of seizures in AD patients is much higher than that in the general population. Patients with AD were 5 to 10 times more likely to develop seizures than those without AD at the same age (Hauser et al., 1986; Larner, 2010; Corbett et al., 2017). During the course of AD, 10 to 22% of patients had at least one unprovoked seizure. Previously, it was generally believed that the first seizure occurred in the middle and late stages of AD, and the incidence increased with the progression of the disease (Hauser et al., 1986; Hesdorffer et al., 1996; Mendez and Lim, 2003; Scarmeas et al., 2009). The cumulative risk of seizures increased from 11 to 26% between 10 and 15 years after the diagnosis of AD (Mendez and Lim, 2003). In addition, the prevalence of seizures was higher in EOAD than in LOAD. People with EOAD were nearly twice as likely to have seizures (11%) as people with LOAD (6%) (Sulkava, 1982). In a large French cohort of autosomal dominant EOAD (ADEOAD), 47.7% of the patients suffered from at least one seizure during a mean follow-up of 8.4 years (Zarea et al., 2016). In fact, except for clinical seizures, electrophysiological (EEG) monitoring often shows subclinical and interictal epileptiform discharges in AD patients. Increasing evidence supports the emergence of epileptiform discharges in early AD (Vossel et al., 2013; Lam et al., 2020). Vossel et al. (2016) observed that up to 42.4% of AD patients had subclinical epileptiform activity, which was defined as paroxysmal sharp waveforms lasting 20 to 200 ms and disrupting background activity (Vossel et al., 2017), using overnight long-term monitoring with video-EEG (LTM-VEEG) or magnetoencephalography with simultaneous EEG (M/EEG). Thus, the incidence of epileptiform discharges in AD may be much higher than that of clinically noticeable seizures. How to identify seizures and epileptiform discharges early and understand the true incidence in AD deserve further attention. In order to evaluate AD patients effectively, clinicians and researchers should conduct sensitive and comprehensive neurophysiological assessments.
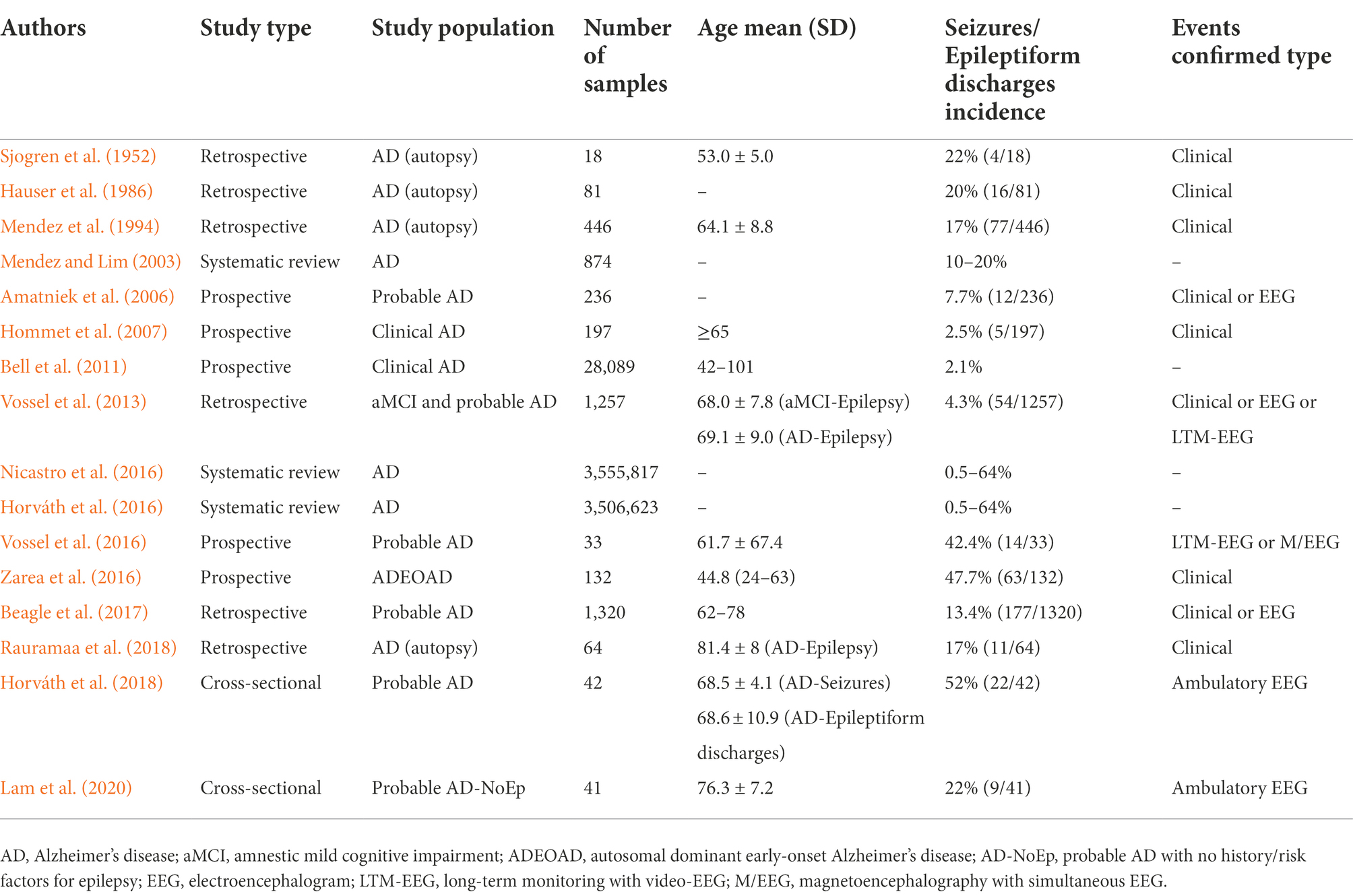
Table 1. Studies investigating seizures/ epileptiform discharges incidence in patients with Alzheimer’s disease.
Risk factors for seizures in Alzheimer’s disease
Potential risk factors for seizures in AD patients include onset age of dementia, down syndrome (DS), epileptiform discharges on EEG, sharp decline in cognition, and medication (Spencer, 2014; Asadollahi et al., 2019). A study based on a representative database from the Nationwide Inpatient Sample among the elderly population older than 55 years revealed that the younger AD patients were, the more likely they were to have seizures (Sherzai et al., 2014). People with EOAD were nearly twice as likely to have seizures (11%) as people with LOAD (6%) (Sulkava, 1982). Seizures were also a common feature in patients with ADEOAD (Zarea et al., 2016). DS patients above 45 years were more likely to develop AD, and it was found that up to 84 percent of these patients suffered from seizures (Menéndez, 2005). Another study showed that patients with DS dementia were more likely to have seizures earlier (Gholipour et al., 2017). Researchers have suggested that epileptiform discharges might be a predictive factor of clinical seizures (Vossel et al., 2013; Asadollahi et al., 2019). Both seizures and epileptiform discharges might impair cognitive function and lead to a sharp decline in cognitive function in patients with AD (Volicer et al., 1995; Vossel et al., 2016). In turn, when AD patients experience sudden cognitive decline, they should be alert to the presence of seizures or epileptiform discharges. Moreover, some medications used in AD patients may decrease the seizure threshold. Antipsychotic drugs, such as clozapine and quetiapine, were found to increase the risk of seizures (Kumlien and Lundberg, 2010). Many studies have reported epileptic seizures in AD patients treated with these drugs (Wong and Delva, 2007; Shao et al., 2016). Therefore, patients with AD should be aware of seizures, particularly when they are using antipsychotic drugs. In addition, patients with AD may suffer from stroke, traumatic brain injury, encephalitis, metabolism and toxic damage, which are common risk factors that induce seizures in the general population (Beghi et al., 2010; Cretin et al., 2017).
Potential mechanisms for comorbidity of Alzheimer’s disease and epillepsy
In recent years, some researchers have found that there is a two-way relationship between epilepsy and dementia, especially AD (Giorgi et al., 2020; Stefanidou et al., 2020). Hippocampal atrophy is present in almost all patients with AD, while hippocampal sclerosis (HS) is also a frequent pathological change in epileptic patients, suggesting that AD and epilepsy may have a common anatomical basis. Furthermore, many elements involved in the pathogenesis of AD have been found to regulate neuronal excitability, and seizures result from excessive neuronal excitability (see Figure 1 and Table 2).
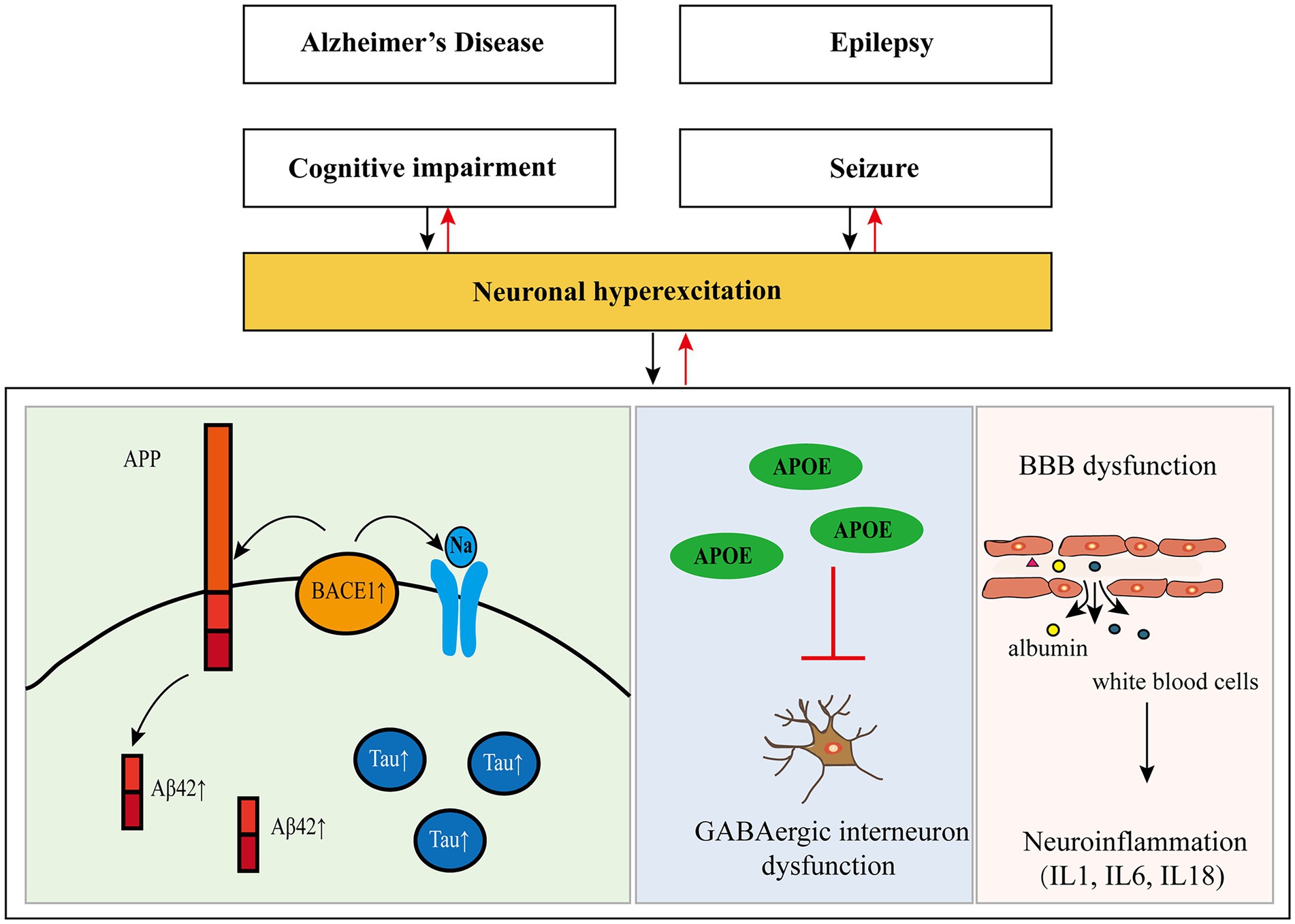
Figure 1. Pathogenesis of Alzheimer’s disease and epilepsy and their relationship. APP, amyloid precursor protein; Aβ, amyloid-β; Tau, protein tau; BACE1, β-site amyloid precursor protein cleaving enzyme 1; APOE, apolipoprotein E; GABA, gamma-aminobutyric acid; BBB, blood–brain barrier.
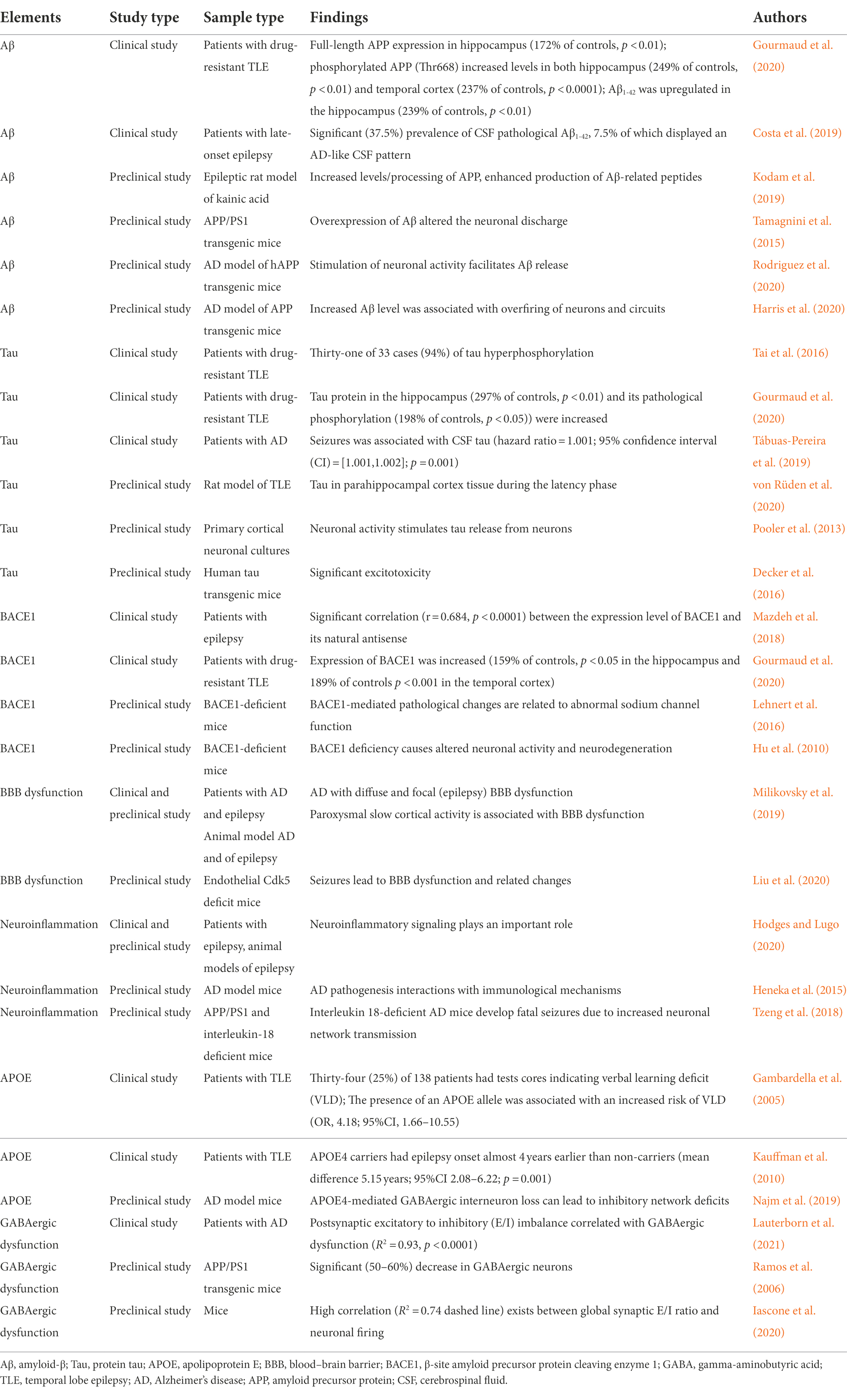
Table 2. Clinical and preclinical studies of elements involved in the pathogenesis of AD and epilepsy.
Amyloid-β (Aβ) may contribute to seizures in AD by causing excitatory changes in neural networks. One of the pathological features of AD is the presence of extracellular Aβ, also known as Aβ plaques, which are clipped by human amyloid precursor protein (APP). A recent study showed that the expression and phosphorylation of APP and Aβ1-42 were upregulated in the brain tissues of patients with drug-resistant temporal lobe epilepsy (TLE). It was also found that the phosphorylation of APP in the hippocampus was inversely correlated with cognitive function (Gourmaud et al., 2020). Patients with late-onset epilepsy of unknown origin (LOEU) had a significant prevalence of cerebrospinal fluid (CSF) pathological Aβ1-42, 7.5% of which displayed an AD-like CSF pattern. Moreover, 17.5% of LOEU patients eventually progressed to AD. There was a hazard ratio of 3.4 for those patients with pathological Aβ1-42 converted to AD at further follow-up (Costa et al., 2019). In addition, some researchers suggested that Aβ might play a dual role in late-onset epilepsy and AD (Costa et al., 2018; Sen et al., 2018). These findings suggest that epilepsy and AD share the same pathological changes, providing a pathological basis for the comorbidity between the two.
In preclinical studies, APP/PS1 transgenic mice are commonly constructed as AD model mice, which can cause the overexpression of APP in the hippocampus and increase the expression of Aβ. After systemic injection of kainic acid, adult rats suffered from seizures, gliosis, and loss of hippocampal neurons, along with increased levels/processing of APP. The increased APP resulted in the enhanced production of Aβ-related peptides in the epileptic rats, which suggested that APP/Aβ peptides derived from astrocytes might have a role in epileptogenesis (Kodam et al., 2019). The upregulated Aβ level was closely related to abnormal epileptic activity and increased intrinsic excitability of CA1 hippocampal neurons. Moreover, overexpression of Aβ altered the neuronal discharge, action potential waveform, and capacitance of CA1 neurons in aged AD model mice (Tamagnini et al., 2015). Gureviciene et al. (2019) similarly identified several types of epileptic spikes in free-moving APP/PS1 transgenic mice using multiple cortical and subcortical electrodes. Elevated Aβ levels and non-motor seizures were also found in the cortical and hippocampal networks (Palop et al., 2007). Interestingly, stimulation of neuronal activity facilitated Aβ release from neurons in vivo (Rodriguez et al., 2020). Additionally, increased neuronal activity enhanced the production of Aβ from APP (Saito et al., 2012). The level of Aβ in hippocampal interstitial fluid (ISF) increased rapidly in APP transgenic mice, while tetrodotoxin (TTX) blocked neuronal activity and thus reduced the level of Aβ (Cirrito et al., 2005). A recent study also confirmed that the number of Aβ plaques was associated with the frequency of epileptiform discharges (Reyes-Marin and Nuñez, 2017). Moreover, the increased Aβ level was associated with hyperexcitability of neurons and circuits (Harris et al., 2020). Thus, we can see that Aβ may be involved in seizures in AD by regulating the excitability of neural networks.
Another key histopathological hallmark of AD is the neurofibrillary tangles of protein tau. Similar to Aβ, tau has also been found in pathological changes in patients with epilepsy. Tai et al. (2016) first performed pathological examination on tissue from 50-year-old to 65-year-old patients with drug-resistant TLE after surgery. They found that 94% of cases showed hyperphosphorylated tau pathology, which was one of the pathological characteristics of AD. Further analysis showed that modified tau scores were strongly negatively correlated with changes in cognitive test scores over 1 year (before and after temporal lobe resection). In a recent study, researchers reconfirmed that tau protein and its pathological phosphorylation were increased in patients with resistant TLE hippocampi, and both were inversely correlated with cognitive function (Gourmaud et al., 2020). Some scholars found that seizure risk in patients with AD was associated with high levels of protein tau in CSF and lower baseline Mini-Mental State Examination (MMSE) scores. They speculated that tau protein might also play a role in regulating neuronal excitability (Tábuas-Pereira et al., 2019). Another study also summarized the linking role of protein tau in AD and epilepsy. Researchers found that hyperphosphorylated tau was associated with the pathogenesis of epilepsy and AD, as well as with cognitive deficits. More interestingly, inhibition of tau not only minimized seizure and AD-like pathology but also improved cognitive decline in both epilepsy and AD (Paudel et al., 2019). These studies provide molecular and pathological evidence for the involvement of tau in AD and epilepsy.
In addition, seizures result from excessive neuronal excitability. Tau may also be involved in the formation of hyperexcitability in neurons. Proteomic analysis revealed the regulation of microtubule-associated tau in parahippocampal cortex tissue during the latency phase in a rat model of TLE (von Rüden et al., 2020). The stimulation of neuronal activity facilitated tau release both in vitro (Pooler et al., 2013) and in vivo (Yamada et al., 2014). Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-mediated tau release was significantly prevented when tetanus toxin blocked presynaptic vesicle release and when TTX inhibited neuronal activity (Pooler et al., 2013). Expressing human full-length Tau with the Tau mutation in transgenic mice showed significant excitotoxicity caused by dysfunction of increasing extracellular glutamate in hippocampal tissue slices. Significant axonal sprouting of mossy fibers and epileptiform discharges were also found in hippocampal slices (Decker et al., 2016). The sprouting of mossy fibers was shown to play a critical role in epileptogenesis, and tau was also related to the abnormal sprouting of mossy fibers in the pentylenetetrazol (PTZ) kindling model of epilepsy (Tian et al., 2010). Moreover, the severity and frequency of seizures were reduced in the PTZ kindling model of epilepsy after inhibition of protein tau production, which suggested that the reduction of protein tau prevented excitatory toxicity (Roberson et al., 2007). The increase in protein tau changes neuronal excitability and promotes seizure occurrence and the decline in cognitive function in patients with AD, while the decrease in protein tau can improve the cognitive function of epilepsy and AD. These findings may provide a new idea for the treatment of seizures in patients with AD.
β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) is very active in patients and animal models of AD. Aβ production is mediated by continuous cleavage of APP, and the extracellular domain of APP is mediated by BACE1. Therefore, BACE1 is necessary for the production of Aβ (Cole and Vassar, 2007; Ehlers, 2018). In a clinical study, researchers suggested that uncontrolled seizures increased tau, APP, and BACE1 protein levels, which accelerated Aβ42 production and tau pathology. They also found cognitive impairment and neuronal dysfunction in drug-resistant epilepsy (Gourmaud et al., 2020). There was a significant correlation between the expression level of BACE1 and its natural antisense (BACE1-AS) in the blood of patients with epilepsy (Mazdeh et al., 2018). In a preclinical study, evidence showed that BACE1-mediated pathological changes were related to abnormal sodium channel function (Kim et al., 2011; Lehnert et al., 2016). Abnormal sodium channels can cause abnormal firing of neurons and lead to seizures. The link between BACE1 and seizures was also demonstrated in animal studies (Hu et al., 2010). Although clinical trials of BACE1 inhibitors conducted to date have been discontinued due to inefficacy or safety reasons, BACE1 remains an effective therapeutic target for AD (Hampel et al., 2021), and researchers should examine whether BACE1 intervention can play a role in seizures in AD patients.
Blood–brain barrier (BBB) dysfunction may be another important link between AD and seizures. The results from patients and animal models of AD suggested that microvascular lesions, especially BBB dysfunction, played an important role (Sweeney et al., 2018). Recently, some scholars found that paroxysmal slow wave events (PSWEs) in patients with AD were recorded as bilateral, while PSWEs were usually focal and colocalized with BBB dysfunction in patients with epilepsy. They identified PSWEs as an EEG manifestation of non-motor seizures in patients with AD and suggested BBB dysfunction pathology. EEG recordings from animal models of AD with diffuse and focal (epilepsy) BBB dysfunction confirmed the occurrence of PSWEs (Milikovsky et al., 2019). Induced seizures, especially status epilepticus, can quickly lead to BBB dysfunction and related changes (Ndode-Ekane et al., 2010; Liu et al., 2020). BBB dysfunction also caused albumin extravasation and white blood cells from the blood into the brain parenchyma, which altered neuronal excitability and induced or promoted the occurrence of seizures (Marchi et al., 2016; Loscher and Friedman, 2020). BBB dysfunction may lead to neuronal hyperexcitability involved in seizures in AD, which could be an underlying mechanism and a promising therapeutic target.
Neuroinflammation may play a role in seizures in patients with AD. Inflammation is a fundamental process in the development of epilepsy and continues to be susceptible to seizures. Neuroinflammatory signaling plays an important role in the development of epilepsy, both in patients with epilepsy and in animal models of acquired epilepsy and inherited epilepsy (Hodges and Lugo, 2020). Inflammatory markers were found in AD model mice (Choi et al., 2013). Increasing evidence suggests that AD pathogenesis is not only restricted to the neuronal compartment but also includes strong interactions with immunological mechanisms in the brain (Heneka et al., 2015). Recently, it was found that APP/PS1 mice deficient in the inflammatory body-derived cytokine interleukin-18 (IL18) showed a lower chemically induced epileptic threshold and a selective increase in gene expression associated with increased neuronal activity. It was suggested that IL18-deficient AD mice developed fatal seizures due to increased neuronal network transmission (Tzeng et al., 2018). These results suggest that neuroinflammation may indeed be involved in the mechanism of epileptic seizures in AD. In general, both AD and epilepsy cause abnormal neuroinflammation. Neuroinflammation, in turn, is a risk factor for seizures and AD progression.
The association between apolipoprotein E (APOE) and AD has been confirmed by many studies (Shi and Holtzman, 2018; Nguyen et al., 2020), while APOE is associated with neural network excitability. An alteration in cognitive performance as a function of the presence of the APOE4 allele was found in TLE patients (Gambardella et al., 2005). In addition, a study investigated the role of APOE4 as a moderator of age at TLE onset. It was confirmed that the onset of epilepsy occurred 4 years earlier in APOE4 carriers than in non-carriers (Kauffman et al., 2010). Gamma-aminobutyric acid (GABA)-ergic interneurons were selectively vulnerable to intracellularly produced APOE4 through a tau-dependent mechanism, which led to their dysfunction and eventual death. Furthermore, GABAergic interneuron loss causes dysregulation and hyperexcitability of neural networks in the hippocampus and cortex (Najm et al., 2019). APOE4 can induce abnormal excitability of neural networks in AD and epilepsy, suggesting that APOE4 may also be involved in mediating the mechanism of seizures in patients with AD.
GABAergic dysfunction may also contribute to seizures in AD. There is evidence of postsynaptic excitatory to inhibitory (E/I) imbalance in AD correlated with GABAergic dysfunction in humans (Lauterborn et al., 2021). A loss of GABAA receptors was reported in AD patients’ brains (Calvo-Flores Guzman et al., 2018). The CA1 region of the hippocampus of AD patients also had a significant reduction in GABAB receptor immunoreactivity (Iwakiri et al., 2005). In addition, GABAergic deficits and associated pathways seemed to be corrected by bumetanide. Bumetanide exposure was related to a significantly lower AD prevalence in individuals over age 65, and the treatment also rescued plasticity deficits and neuronal excitability in mice (Taubes et al., 2021). In a preclinical study, APP/PS1 transgenic mice exhibited a significant (50–60%) decrease in GABAergic neurons coexpressing somatostatin and neuropeptide cell loss (Ramos et al., 2006). Deficiencies in inhibitory GABAergic interneurons increase the excitability of neurons in many circuits by decreasing GABAergic inhibition (Wang et al., 2014). Moreover, a positive correlation existed between the global synaptic E/I ratio and neuronal firing (Iascone et al., 2020). Therefore, we cannot exclude the possibility that the synaptic E/I effect may be due to symptoms associated with AD, such as seizures (Amatniek et al., 2006).
Seizure types in Alzheimer’s disease
Generalized tonic–clonic seizures (GTCS) have been reported as a common seizure type, but they are probably focal seizures with secondary generalization in AD patients (Larner, 2010). Increasing evidence suggests that focal seizures might be underestimated in patients with AD (Mendez and Lim, 2003). Focal non-motor seizures should be the predominant seizure type in patients with AD. The seizure semiology can include déjà vu, jamais vu, confusion, unexplained emotions (e.g., fear or euphoria), amnestic spells, speech arrest or sensory phenomena (e.g., metallic taste or epigastric rising sensation), which may overlap with other symptoms of AD (Vossel et al., 2017).
Transient epileptic amnesia (TEA), an unusual seizure type, sometimes occurs in patients with AD. Rabinowicz et al. (2000) first reported TEA in AD patients. The two patients transiently got lost in familiar environments caused by paroxysmal amnestic wandering and disorientation. A subsequent study reported that four AD patients suffered from TEA. Their acute and transient memory dysfunction showed a clear-cut response to antiseizure medications (ASMs) (Cretin et al., 2014). Additionally, some patients with AD may present with myoclonic seizure, with a higher risk in the advanced stages of disease (Asadollahi et al., 2019). New onset myoclonic jerks were observed in two cohorts of elderly patients with AD who developed from DS. EEG detected generalized spike-waves and polyspike-waves. Valproate, levetiracetam, topiramate and lamotrigine all appeared efficacious for myoclonic seizures in these patients (De Simone et al., 2010; Aller-Alvarez et al., 2017).
It is challenging to accurately identify seizure types in patients with AD only by clinical symptoms. Both AD patients and caregivers often rarely provide a reliable history. However, the episodes can be distinguished as epileptic events by their paroxysmal, recurrent and stereotyped nature, and further confirmed by epileptiform discharges on ictal EEG. LTM-VEEG and high-density EEG may be helpful for differential diagnosis (Vossel et al., 2017).
Hazard of seizures and epileptiform discharges in Alzheimer’s disease
Seizures have adverse consequences on the natural course of AD and accelerate the decline of cognitive function in patients with AD. The results of a clinical retrospective study showed that 82% of patients with AD who suffered from seizures experienced sudden deterioration and required long-term care within 6 months of seizure onset. The study also revealed that approximately 50% of AD patients with seizures showed a significant decrease in language function compared to those without seizures (Volicer et al., 1995). In another study, patients with AD who had seizures began to experience a decline in cognitive function 5.5 years earlier than those without seizures (Vossel et al., 2013). In a recent study with similar results, the researchers also found that patients with AD who had seizures began to show a decline in cognitive function 3.6 years earlier than those without seizures (DiFrancesco et al., 2017). The presence of seizures in the memory clinic population predicted more severe impairments in activities of daily living (Baker et al., 2019). A preclinical study also showed that early seizure activity accelerated the depletion of hippocampal neural stem cells and impaired spatial discrimination in an AD model (Fu et al., 2019).
In addition, epileptiform discharges may also be associated with cognitive impairment. Hippocampal interictal epileptiform discharges might impair the maintenance and retrieval of memory (Kleen et al., 2013). Vossel et al. (2016) used the MMSE and executive function assessment and found that AD patients with subclinical epileptiform activity had a faster decline in overall cognitive ability than AD patients without. In a number of studies in AD model mice, epileptiform discharges have also been found to cause synaptic and cognitive impairment, which can be ameliorated by ASMs (Sanchez et al., 2012; Nygaard et al., 2015).
These findings suggest that both seizures and epileptiform discharges can cause cognitive impairment in patients with AD. Therefore, early and timely treatment may be necessary to prevent further deterioration of cognitive function in patients with AD.
Treatment of seizures in Alzheimer’s disease
There is controversy about whether ASMs are needed in AD patients with seizures. Previously, Larner (2010) proposed that antiseizure treatment might not always be necessary because seizures were often non-convulsive and infrequent in patients with AD. Moreover, side-effects related to ASMs are of particular concern. There was a nationwide cohort of 70,718 patients with clinically verified AD from 2005 to 2011 in Finland. Among those patients, 3,058–5,769 patients used ASMs for epilepsy, neuropathic pain, psychiatric or depression disorders (Sarycheva et al., 2018b). The researchers conducted a series of studies about adverse events associated with ASMs in AD patients. They found that ASM users had an increased risk of stroke (37%) (Sarycheva et al., 2018a), pneumonia (92%) (Taipale et al., 2019), hip fracture (17%) (Pisa et al., 2021), and hospitalization (12 days) (Lavikainen et al., 2019) compared with non-users during the time follow-up. However, further subgroup analysis revealed different results. Newer ASM users had a lower risk of hip fracture than older ASMs, even non-users [(incidence rate (IR): 1.4 (1.1–1.9) vs. 2.6 (2.1–3.3) vs. 1.8 (1.6–1.9); Pisa et al., 2021]. Users with gabapentin and pregabalin had a lower hospitalization than valproate users (Lavikainen et al., 2019). In fact, fractures, pneumonia, hospitalization and cognitive decline may also be due to seizures or falls in AD patients. Cognitive adverse effects (CAEs) may be caused by some ASMs (phenobarbital, carbamazepine, phenytoin, valproate, primidone, barbexaclone, ethosuximide, clonazepam, zonisamide, and topiramate). Occasional use of ASMs with CAEs was associated with an increased risk of incident dementia (20%) and AD (16%), and regular use was associated with a greater risk (28 and 15%, respectively). However, adverse effects were not observed in the users of other ASMs (including levetiracetam, oxcarbazepine, lamotrigine, gabapentin, vigabatrin, pregabalin, tiagabine, lacosamide), regardless of whether the usage was occasional or regular (Taipale et al., 2018). On the other hand, clinical and basic studies have indicated that proper ASMs might potentially improve the prognosis of AD patients with epilepsy (Volicer et al., 1995; DiFrancesco et al., 2017; Liu et al., 2018). Levetiracetam might improve cognitive performance, specifically oral fluency items and attention, in AD patients with epilepsy (Cumbo and Ligori, 2010). Topiramate significantly restored nest-building and social interaction and reduced Aβ deposition in AD mice (Owona et al., 2019). Therefore, AD patients with recurrent seizures should take ASMs to prevent adverse consequences related to unpredictable episodes.
Age and drug interactions must also be considered when deciding to treat seizures. Most patients with AD are older and might be more prone to potential liver, kidney and heart toxicity. More importantly, due to their property to induce or inhibit cytochrome P450 enzymes (e.g., phenytoin, phenobarbital, carbamazepine and valproate), older ASMs may interact with concomitant medications. For example, some common medications of AD (donepezil, galantamine, rivastigmine, and ginkgo biloba) might inhibit hepatic enzymes and slow down the metabolism of ASMs, which may increase the risk of patient intoxication (Larner, 2010). In addition, sudden withdrawal of cholinesterase inhibitors might increase epileptic susceptibility and induce seizures in AD patients (Piecoro et al., 1998). Hence, older ASMs should problely be avoided in AD patients with epilepsy.
Currently, there is much evidence that levetiracetam and lamotrigine are used to control seizures in AD patients. A small-sample prospective observational study showed that levetiracetam (1,000 mg/d–2,000 mg/d) had an approximately 80% seizure-free rate for at least 1 year and a 14% withdrawal rate for adverse effects in late-stage AD patients with new onset epilepsy (Belcastro et al., 2007). For aMCI or AD patients with seizures, levetiracetam (250 mg/d–3,000 mg/d) and lamotrigine (50 mg/d–600 mg/d) exhibited better efficacy and tolerability than phenytoin (100 mg/d–600 mg/d) (Vossel et al., 2013). Similarly, compared with lamotrigine (25 mg/d–100 mg/d) and phenobarbital (50 mg/d–100 mg/d), levetiracetam (500 mg/d–2,000 mg/d) had fewer adverse events (28% for lamotrigine vs. 43.4% for phenobarbital and 17% for levetiracetam) in a prospective, randomized, case–control study of AD patients with epilepsy. At the 12-month visit, levetiracetam increased the MMSE score (−0.64 for lamotrigine vs. −1.57 for phenobarbital and + 0.23 for levetiracetam), and lamotrigine improved mood in the score for Cornell scale for depression (−0.72 for lamotrigine vs. +1.74 for phenobarbital and + 0.20 for levetiracetam) (Cumbo and Ligori, 2010). Furthermore, levetiracetam and lamotrigine have broad-spectrum efficacy in treating focal seizures, myoclonic seizures and generalized or unclassifiable seizures (Kanner and Bicchi, 2022). Lacosamide and eslicarbazepine are also prescribed ASMs in the setting of cognitive impairment, while limited data are available for their use in patients with AD. Considering the above reasons, levetiracetam and lamotrigine are recommended as current optimal therapeutic options for comorbid epilepsy in patients with AD (Cretin, 2018).
Given that Aβ and tau may be involved in epileptogenesis in AD, targeted therapy for Aβ or tau seems to be a rational choice for antiepilepsy therapy in AD. An animal experiment suggested that berberine restored Aβ-induced neurotoxicity and cognitive impairments (Haghani et al., 2015). Intracerebroventricular infusion of Aβ-specific antibody could prevent early-onset seizures in young ArcticAβ mice (Gschwind et al., 2018). Excitability of dentate gyrus granule cells in hippocampi from tau−/−, htau mice was significantly reduced with deletion of functional tau in an in vitro electrophysiological study (Cloyd et al., 2021). Another in vivo study showed that targeted reduction of tauopathy alleviated seizures and improved spatial learning and memory (Gao et al., 2020). Moreover, several clinical studies targeting Aβ- and tau-dependent pathways with immunotherapy are ongoing, but it is unclear whether therapeutic strategies can exhibit good antiepileptic efficacy (Liu and Wang, 2021).
Recently, the approaches deserving attention for treating epilepsy in patients with AD also include non-pharmacotherapeutic strategies, such as ketogenic diet therapies (KDTs), deep brain stimulation (DBS), and transcranial magnetic stimulation (TMS). KDTs were originally developed as a treatment for patients with intractable epilepsy, but studies have expanded their potential to treat other neurological disorders, such as AD, in recent years (Augustin et al., 2018; Koppel and Swerdlow, 2018). KDTs could improve cerebral blood flow and cognitive function in patients with AD (McDonald and Cervenka, 2019), which provides a potential for dietary management in AD patients with epilepsy. Additionally, chronic DBS might reduce neuronal loss and synaptic dysfunction in AD and epilepsy (McKinnon et al., 2019), and TMS could be used to improve cognitive function in dementia and to control seizures in epilepsy (Chen et al., 2008).
Conclusion and prospects
Accumulating studies have revealed that seizures and epileptiform discharges are prevalent and persist throughout the course of AD, with a high probability of onset in the early stages. AD and epilepsy have a bidirectional connection. Epilepsy as a comorbidity of AD has been increasingly recognized, and a variety of potential mechanisms have been discovered. Various seizure types may occur in patients with AD, of which non-motor seizures are the most common. In addition, both seizures and epileptiform discharges are detrimental to the natural course of AD and may lead to a further decline in cognitive function in AD patients. Therefore, clinicians in the management of AD patients need to identify and intervene in seizures and epileptiform discharges early. At present, levetiracetam and lamotrigine are recommended as optimal ASMs for AD patients because of their preferable antiseizure efficacy and tolerability. Novel therapeutic strategies are worthy of attention. If the exact comorbidity mechanism of epilepsy in AD can be determined, relevant targeted treatment will substantially improve clinical outcomes.
Author contributions
FY wrote and edited the manuscript. YH designed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81260199, 81660228, and 82160261), Yunnan Province Talent Training Program (Nos. L-2019019 and H-2018056), Yunnan High-Level Talent Training Support Program Famous Doctor Special Project (No. RLMY20200005), and Project of Nanchong Science and Technology Bureau (No. 19SXHZ0051).
Acknowledgments
We would like to thank Ting Wang and Sunbing Du for their support of this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aller-Alvarez, J. S., Menéndez-González, M., Ribacoba-Montero, R., Salvado, M., Vega, V., Suárez-Moro, R., et al. (2017). Myoclonic epilepsy in down syndrome and Alzheimer’s disease. Neurologia 32, 69–73. doi: 10.1016/j.nrl.2014.12.008
Amatniek, J. C., Hauser, W. A., DelCastillo-Castaneda, C., Jacobs, D. M., Marder, K., Bell, K., et al. (2006). Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia 47, 867–872. doi: 10.1111/j.1528-1167.2006.00554.x
Asadollahi, M., Atazadeh, M., and Noroozian, M. (2019). Seizure in Alzheimer's disease: An underestimated phenomenon. Am. J. Alzheimers Dis. Other Dement. 34, 81–88. doi: 10.1177/1533317518813551
Augustin, K., Khabbush, A., Williams, S., Eaton, S., Orford, M., Cross, J. H., et al. (2018). Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 17, 84–93. doi: 10.1016/s1474-4422(17)30408-8
Baker, J., Libretto, T., Henley, W., and Zeman, A. (2019). The prevalence and clinical features of epileptic seizures in a memory clinic population. Seizure 71, 83–92. doi: 10.1016/j.seizure.2019.06.016
Beagle, A. J., Darwish, S. M., Ranasinghe, K. G., La, A. L., Karageorgiou, E., and Vossel, K. A. (2017). Relative incidence of seizures and myoclonus in Alzheimer's disease, dementia with Lewy bodies, and frontotemporal dementia. J. Alzheimers Dis. 60, 211–223. doi: 10.3233/jad-170031
Beghi, E., Carpio, A., Forsgren, L., Hesdorffer, D. C., Malmgren, K., Sander, J. W., et al. (2010). Recommendation for a definition of acute symptomatic seizure. Epilepsia 51, 671–675. doi: 10.1111/j.1528-1167.2009.02285.x
Belcastro, V., Costa, C., Galletti, F., Pisani, F., Calabresi, P., and Parnetti, L. (2007). Levetiracetam monotherapy in Alzheimer patients with late-onset seizures: a prospective observational study. Eur. J. Neurol. 14, 1176–1178. doi: 10.1111/j.1468-1331.2007.01907.x
Bell, J. S., Lönnroos, E., Koivisto, A. M., Lavikainen, P., Laitinen, M. L., Soininen, H., et al. (2011). Use of antiepileptic drugs among community-dwelling persons with Alzheimer's disease in Finland. J. Alzheimers Dis. 26, 231–237. doi: 10.3233/jad-2011-110200
Calvo-Flores Guzman, B., Vinnakota, C., Govindpani, K., Waldvogel, H. J., Faull, R. L. M., and Kwakowsky, A. (2018). The GABAergic system as a therapeutic target for Alzheimer's disease. J. Neurochem. 146, 649–669. doi: 10.1111/jnc.14345
Chen, R., Cros, D., Curra, A., Di Lazzaro, V., Lefaucheur, J. P., Magistris, M. R., et al. (2008). The clinical diagnostic utility of transcranial magnetic stimulation: report of an IFCN committee. Clin. Neurophysiol. 119, 504–532. doi: 10.1016/j.clinph.2007.10.014
Choi, S. H., Aid, S., Caracciolo, L., Minami, S. S., Niikura, T., Matsuoka, Y., et al. (2013). Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer's disease. J. Neurochem. 124, 59–68. doi: 10.1111/jnc.12059
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Cloyd, R. A., Koren, J. 3rd, Abisambra, J. F., and Smith, B. N. (2021). Effects of altered tau expression on dentate granule cell excitability in mice. Exp. Neurol. 343:113766. doi: 10.1016/j.expneurol.2021.113766
Cole, S. L., and Vassar, R. (2007). The Alzheimer's disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2:22. doi: 10.1186/1750-1326-2-22
Corbett, B. F., You, J. C., Zhang, X., Pyfer, M. S., Tosi, U., Iascone, D. M., et al. (2017). ΔFosB regulates gene expression and cognitive dysfunction in a mouse model of Alzheimer's disease. Cell Rep. 20, 344–355. doi: 10.1016/j.celrep.2017.06.040
Costa, C., Romoli, M., and Calabresi, P. (2018). Late onset epilepsy and Alzheimer's disease: exploring the dual pathogenic role of amyloid-β. Brain 141:e60. doi: 10.1093/brain/awy162
Costa, C., Romoli, M., Liguori, C., Farotti, L., Eusebi, P., Bedetti, C., et al. (2019). Alzheimer's disease and late-onset epilepsy of unknown origin: two faces of beta amyloid pathology. Neurobiol. Aging 73, 61–67. doi: 10.1016/j.neurobiolaging.2018.09.006
Cretin, B. (2018). Pharmacotherapeutic strategies for treating epilepsy in patients with Alzheimer's disease. Expert. Opin. Pharmacother. 19, 1201–1209. doi: 10.1080/14656566.2018.1496237
Cretin, B., Philippi, N., Bousiges, O., Dibitonto, L., Sellal, F., Martin-Hunyadi, C., et al. (2017). Do we know how to diagnose epilepsy early in Alzheimer's disease? Rev. Neurol. 173, 374–380. doi: 10.1016/j.neurol.2017.03.028
Cretin, B., Philippi, N., Sellal, F., Dibitonto, L., Martin-Hunyadi, C., and Blanc, F. (2014). Can the syndrome of transient epileptic amnesia be the first feature of Alzheimer's disease? Seizure 23, 918–920. doi: 10.1016/j.seizure.2014.07.008
Cumbo, E., and Ligori, L. D. (2010). Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer's disease. Epilepsy Behav. 17, 461–466. doi: 10.1016/j.yebeh.2010.01.015
De Simone, R., Puig, X. S., Gélisse, P., Crespel, A., and Genton, P. (2010). Senile myoclonic epilepsy: delineation of a common condition associated with Alzheimer's disease in down syndrome. Seizure 19, 383–389. doi: 10.1016/j.seizure.2010.04.008
Decker, J. M., Kruger, L., Sydow, A., Dennissen, F. J., Siskova, Z., Mandelkow, E., et al. (2016). The tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 17, 552–569. doi: 10.15252/embr.201541439
DiFrancesco, J. C., Tremolizzo, L., Polonia, V., Giussani, G., Bianchi, E., Franchi, C., et al. (2017). Adult-onset epilepsy in presymptomatic Alzheimer's disease: a retrospective study. J. Alzheimers Dis. 60, 1267–1274. doi: 10.3233/jad-170392
Ehlers, M. D. (2018). Peripheral activity and central substrates of BACE1: therapeutic implications for Alzheimer's disease. Biol. Psychiatry 83, 393–394. doi: 10.1016/j.biopsych.2017.12.005
Fu, C. H., Iascone, D. M., Petrof, I., Hazra, A., Zhang, X., Pyfer, M. S., et al. (2019). Early seizure activity accelerates depletion of hippocampal neural stem cells and impairs spatial discrimination in an Alzheimer's disease model. Cell Rep. 27, 3741–3751.e4. doi: 10.1016/j.celrep.2019.05.101
Gambardella, A., Aguglia, U., Chifari, R., Labate, A., Manna, I., Serra, P., et al. (2005). ApoE epsilon4 allele and disease duration affect verbal learning in mild temporal lobe epilepsy. Epilepsia 46, 110–117. doi: 10.1111/j.0013-9580.2005.15804.x
Gao, Y., Zheng, J., Jiang, T., Pi, G., Sun, F., Xiong, R., et al. (2020). Targeted reducing of tauopathy alleviates epileptic seizures and spatial memory impairment in an optogenetically inducible mouse model of epilepsy. Front. Cell Dev. Biol. 8:633725. doi: 10.3389/fcell.2020.633725
Gholipour, T., Mitchell, S., Sarkis, R. A., and Chemali, Z. (2017). The clinical and neurobehavioral course of down syndrome and dementia with or without new-onset epilepsy. Epilepsy Behav. 68, 11–16. doi: 10.1016/j.yebeh.2016.12.014
Giorgi, F. S., Saccaro, L. F., Busceti, C. L., Biagioni, F., and Fornai, F. (2020). Epilepsy and Alzheimer's disease: potential mechanisms for an association. Brain Res. Bull. 160, 107–120. doi: 10.1016/j.brainresbull.2020.04.009
Gourmaud, S., Shou, H., Irwin, D. J., Sansalone, K., Jacobs, L. M., Lucas, T. H., et al. (2020). Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 143, 191–209. doi: 10.1093/brain/awz381
Gschwind, T., Lafourcade, C., Gfeller, T., Zaichuk, M., Rambousek, L., Knuesel, I., et al. (2018). Contribution of early Alzheimer's disease-related pathophysiology to the development of acquired epilepsy. Eur. J. Neurosci. 47, 1534–1562. doi: 10.1111/ejn.13983
Gureviciene, I., Ishchenko, I., Ziyatdinova, S., Jin, N., Lipponen, A., Gurevicius, K., et al. (2019). Characterization of epileptic spiking associated with brain amyloidosis in APP/PS1 mice. Front. Neurol. 10:1151. doi: 10.3389/fneur.2019.01151
Haghani, M., Shabani, M., and Tondar, M. (2015). The therapeutic potential of berberine against the altered intrinsic properties of the CA1 neurons induced by abeta neurotoxicity. Eur. J. Pharmacol. 758, 82–88. doi: 10.1016/j.ejphar.2015.03.016
Hampel, H., Vassar, R., De Strooper, B., Hardy, J., Willem, M., Singh, N., et al. (2021). The beta-secretase BACE1 in Alzheimer's disease. Biol. Psychiatry 89, 745–756. doi: 10.1016/j.biopsych.2020.02.001
Harris, S. S., Wolf, F., De Strooper, B., and Busche, M. A. (2020). Tipping the scales: peptide-dependent dysregulation of neural circuit dynamics in Alzheimer's disease. Neuron 107, 417–435. doi: 10.1016/j.neuron.2020.06.005
Hauser, W. A., Morris, M. L., Heston, L. L., and Anderson, V. E. (1986). Seizures and myoclonus in patients with Alzheimer's disease. Neurology 36, 1226–1230. doi: 10.1212/wnl.36.9.1226
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lan. Neurol. 14, 388–405. doi: 10.1016/s1474-4422(15)70016-5
Hesdorffer, D. C., Hauser, W. A., Annegers, J. F., Kokmen, E., and Rocca, W. A. (1996). Dementia and adult-onset unprovoked seizures. Neurology 46, 727–730. doi: 10.1212/wnl.46.3.727
Hodges, S. L., and Lugo, J. N. (2020). Therapeutic role of targeting mTOR signaling and neuroinflammation in epilepsy. Epilepsy Res. 161:106282. doi: 10.1016/j.eplepsyres.2020.106282
Hommet, C., Hureaux, R., Barré, J., Constans, T., and Berrut, G. (2007). Epileptic seizures in clinically diagnosed Alzheimer's disease: report from a geriatric medicine population. Aging Clin. Exp. Res. 19, 430–431. doi: 10.1007/bf03324726
Horváth, A., Szűcs, A., Barcs, G., Noebels, J. L., and Kamondi, A. (2016). Epileptic seizures in Alzheimer disease: a review. Alzheimer Dis. Assoc. Disord. 30, 186–192. doi: 10.1097/WAD.0000000000000134
Horváth, A., Szűcs, A., Hidasi, Z., Csukly, G., Barcs, G., and Kamondi, A. (2018). Prevalence, semiology, and risk factors of epilepsy in Alzheimer's disease: An ambulatory EEG study. J. Alzheimers Dis. 63, 1045–1054. doi: 10.3233/jad-170925
Hu, X., Zhou, X., He, W., Yang, J., Xiong, W., Wong, P., et al. (2010). BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 30, 8819–8829. doi: 10.1523/JNEUROSCI.1334-10.2010
Iascone, D. M., Li, Y., Sumbul, U., Doron, M., Chen, H., Andreu, V., et al. (2020). Whole-neuron synaptic mapping reveals spatially precise excitatory/inhibitory balance limiting dendritic and somatic spiking. Neuron 106, 566–578 e568. doi: 10.1016/j.neuron.2020.02.015
Iwakiri, M., Mizukami, K., Ikonomovic, M. D., Ishikawa, M., Hidaka, S., Abrahamson, E. E., et al. (2005). Changes in hippocampal GABABR1 subunit expression in Alzheimer's patients: association with Braak staging. Acta Neuropathol. 109, 467–474. doi: 10.1007/s00401-005-0985-9
Jia, L., Quan, M., Fu, Y., Zhao, T., Li, Y., Wei, C., et al. (2020). Dementia in China: epidemiology, clinical management, and research advances. Lan. Neurol. 19, 81–92. doi: 10.1016/s1474-4422(19)30290-x
Kanner, A. M., and Bicchi, M. M. (2022). Antiseizure medications for adults with epilepsy: a review. JAMA 327, 1269–1281. doi: 10.1001/jama.2022.3880
Kauffman, M. A., Consalvo, D., Moron, D. G., Lereis, V. P., and Kochen, S. (2010). ApoE epsilon4 genotype and the age at onset of temporal lobe epilepsy: a case-control study and meta-analysis. Epilepsy Res. 90, 234–239. doi: 10.1016/j.eplepsyres.2010.05.007
Kim, D. Y., Gersbacher, M. T., Inquimbert, P., and Kovacs, D. M. (2011). Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J. Biol. Chem. 286, 8106–8116. doi: 10.1074/jbc.M110.134692
Kleen, J. K., Scott, R. C., Holmes, G. L., Roberts, D. W., Rundle, M. M., Testorf, M., et al. (2013). Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology 81, 18–24. doi: 10.1212/WNL.0b013e318297ee50
Kodam, A., Ourdev, D., Maulik, M., Hariharakrishnan, J., Banerjee, M., Wang, Y., et al. (2019). A role for astrocyte-derived amyloid β peptides in the degeneration of neurons in an animal model of temporal lobe epilepsy. Brain Pathol. 29, 28–44. doi: 10.1111/bpa.12617
Koppel, S. J., and Swerdlow, R. H. (2018). Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem. Int. 117, 114–125. doi: 10.1016/j.neuint.2017.05.019
Kumlien, E., and Lundberg, P. O. (2010). Seizure risk associated with neuroactive drugs: data from the WHO adverse drug reactions database. Seizure 19, 69–73. doi: 10.1016/j.seizure.2009.11.005
Lam, A. D., Sarkis, R. A., Pellerin, K. R., Jing, J., Dworetzky, B. A., Hoch, D. B., et al. (2020). Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology 95, e2259–e2270. doi: 10.1212/wnl.0000000000010612
Larner, A. J. (2010). Epileptic seizures in AD patients. NeuroMol. Med. 12, 71–77. doi: 10.1007/s12017-009-8076-z
Lauterborn, J. C., Scaduto, P., Cox, C. D., Schulmann, A., Lynch, G., Gall, C. M., et al. (2021). Increased excitatory to inhibitory synaptic ratio in parietal cortex samples from individuals with Alzheimer's disease. Nat. Commun. 12:2603. doi: 10.1038/s41467-021-22742-8
Lavikainen, P., Taipale, H., Tanskanen, A., Koponen, M., Tiihonen, J., Hartikainen, S., et al. (2019). Antiepileptic drugs and accumulation of hospital days among persons with Alzheimer's disease. J. Am. Med. Dir. Assoc. 20, 751–758. doi: 10.1016/j.jamda.2018.11.012
Lehnert, S., Hartmann, S., Hessler, S., Adelsberger, H., Huth, T., and Alzheimer, C. (2016). Ion channel regulation by beta-secretase BACE1–enzymatic and non-enzymatic effects beyond Alzheimer's disease. Channels 10, 365–378. doi: 10.1080/19336950.2016.1196307
Liu, J., and Wang, L. N. (2021). Treatment of epilepsy for people with Alzheimer's disease. Cochrane Database Syst. Rev. 2021:CD011922. doi: 10.1002/14651858.CD011922.pub4
Liu, J., Wang, L. N., Wu, L. Y., and Wang, Y. P. (2018). Treatment of epilepsy for people with Alzheimer's disease. Cochrane Database Syst. Rev. 12:Cd011922. doi: 10.1002/14651858.CD011922.pub3
Liu, X. X., Yang, L., Shao, L. X., He, Y., Wu, G., Bao, Y. H., et al. (2020). Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 217:e20180992. doi: 10.1084/jem.20180992
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cells 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Loscher, W., and Friedman, A. (2020). Structural, molecular, and functional alterations of the blood-brain barrier during epileptogenesis and epilepsy: a cause, consequence, or both? Int. J. Mol. Sci. 21:591. doi: 10.3390/ijms21020591
Marchi, N., Banjara, M., and Janigro, D. (2016). Blood-brain barrier, bulk flow, and interstitial clearance in epilepsy. J. Neurosci. Methods 260, 118–124. doi: 10.1016/j.jneumeth.2015.06.011
Mazdeh, M., Komaki, A., Omrani, M. D., Gharzi, V., Sayad, A., Taheri, M., et al. (2018). Expression analysis of beta-secretase 1 (BACE1) and its naturally occurring antisense (BACE1-AS) in blood of epileptic patients. Neurol. Sci. 39, 1565–1569. doi: 10.1007/s10072-018-3458-3
McDonald, T. J. W., and Cervenka, M. C. (2019). Lessons learned from recent clinical trials of ketogenic diet therapies in adults. Curr. Opin. Clin. Nutr. Metab. Care 22, 418–424. doi: 10.1097/mco.0000000000000596
McKinnon, C., Gros, P., Lee, D. J., Hamani, C., Lozano, A. M., Kalia, L. V., et al. (2019). Deep brain stimulation: potential for neuroprotection. Ann. Clin. Transl. Neurol. 6, 174–185. doi: 10.1002/acn3.682
Mendez, M. F., Catanzaro, P., Doss, R. C. R. A. R., and Frey, W. H. 2nd. (1994). Seizures in Alzheimer's disease: clinicopathologic study. J. Geriatr. Psychiatry Neurol. 7, 230–233. doi: 10.1177/089198879400700407
Mendez, M., and Lim, G. (2003). Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging 20, 791–803. doi: 10.2165/00002512-200320110-00001
Menéndez, M. (2005). Down syndrome, Alzheimer's disease and seizures. Brain Dev. 27, 246–252. doi: 10.1016/j.braindev.2004.07.008
Milikovsky, D. Z., Ofer, J., Senatorov, V. V. Jr., Friedman, A. R., Prager, O., Sheintuch, L., et al. (2019). Paroxysmal slow cortical activity in Alzheimer's disease and epilepsy is associated with blood-brain barrier dysfunction. Sci. Transl. Med. 11:521. doi: 10.1126/scitranslmed.aaw8954
Najm, R., Jones, E. A., and Huang, Y. (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer's disease. Mol. Neurodegener. 14:24. doi: 10.1186/s13024-019-0324-6
Ndode-Ekane, X. E., Hayward, N., Grohn, O., and Pitkanen, A. (2010). Vascular changes in epilepsy: functional consequences and association with network plasticity in pilocarpine-induced experimental epilepsy. Neuroscience 166, 312–332. doi: 10.1016/j.neuroscience.2009.12.002
Nguyen, A. T., Wang, K., Hu, G., Wang, X., Miao, Z., Azevedo, J. A., et al. (2020). APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer's disease. Acta Neuropathol. 140, 477–493. doi: 10.1007/s00401-020-02200-3
Nicastro, N., Assal, F., and Seeck, M. (2016). From here to epilepsy: the risk of seizure in patients with Alzheimer's disease. Epileptic Disord. 18, 1–12. doi: 10.1684/epd.2016.0808
Nygaard, H. B., Kaufman, A. C., Sekine-Konno, T., Huh, L. L., Going, H., Feldman, S. J., et al. (2015). Brivaracetam, but not ethosuximide, reverses memory impairments in an Alzheimer's disease mouse model. Alzheimers Res. Ther. 7:25. doi: 10.1186/s13195-015-0110-9
Owona, B. A., Zug, C., Schluesener, H. J., and Zhang, Z. Y. (2019). Amelioration of behavioral impairments and neuropathology by antiepileptic drug Topiramate in a transgenic Alzheimer's disease model mice, APP/PS1. Int. J. Mol. Sci. 20:3003. doi: 10.3390/ijms20123003
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Paudel, Y. N., Angelopoulou, E., Jones, N. C., O'Brien, T. J., Kwan, P., Piperi, C., et al. (2019). Tau related pathways as a connecting link between epilepsy and Alzheimer's disease. ACS Chem. Neurosci. 10, 4199–4212. doi: 10.1021/acschemneuro.9b00460
Piecoro, L. T., Wermeling, D. P., Schmitt, F. A., and Ashford, J. W. (1998). Seizures in patients receiving concomitant antimuscarinics and acetylcholinesterase inhibitor. Pharmacotherapy 18, 1129–1132.
Pisa, F., Reinold, J., Lavikainen, P., Koponen, M., Taipale, H., Tanskanen, A., et al. (2021). Hip fracture risk in antiepileptic drug initiators and non-initiators with Alzheimer's disease. Clin. Epidemiol. 13, 295–307. doi: 10.2147/CLEP.S278306
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Rabinowicz, A. L., Starkstein, S. E., Leiguarda, R. C., and Coleman, A. E. (2000). Transient epileptic amnesia in dementia: a treatable unrecognized cause of episodic amnestic wandering. Alzheimer Dis. Assoc. Disord. 14, 231–233. doi: 10.1097/00002093-200010000-00008
Ramos, B., Baglietto-Vargas, D., del Rio, J. C., Moreno-Gonzalez, I., Santa-Maria, C., Jimenez, S., et al. (2006). Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer's disease. Neurobiol. Aging 27, 1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022
Rauramaa, T., Saxlin, A., Lohvansuu, K., Alafuzoff, I., Pitkänen, A., and Soininen, H. (2018). Epilepsy in neuropathologically verified Alzheimer's disease. Seizure 58, 9–12. doi: 10.1016/j.seizure.2018.03.014
Reyes-Marin, K. E., and Nuñez, A. (2017). Seizure susceptibility in the APP/PS1 mouse model of Alzheimer's disease and relationship with amyloid β plaques. Brain Res. 1677, 93–100. doi: 10.1016/j.brainres.2017.09.026
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rodriguez, G. A., Barrett, G. M., Duff, K. E., and Hussaini, S. A. (2020). Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces abeta and tau pathology in the hippocampus. PLoS Biol. 18:e3000851. doi: 10.1371/journal.pbio.3000851
Saito, Y., Inoue, T., Zhu, G., Kimura, N., Okada, M., Nishimura, M., et al. (2012). Hyperpolarization-activated cyclic nucleotide gated channels: a potential molecular link between epileptic seizures and Aβ generation in Alzheimer's disease. Mol. Neurodegener. 7:50. doi: 10.1186/1750-1326-7-50
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc. Natl. Acad. Sci. U. S. A. 109, E2895–E2903. doi: 10.1073/pnas.1121081109
Sarycheva, T., Lavikainen, P., Taipale, H., Tiihonen, J., Tanskanen, A., Hartikainen, S., et al. (2018a). Antiepileptic drug use and the risk of stroke among community-dwelling people with Alzheimer disease: a matched cohort study. J. Am. Heart Assoc. 7:e009742. doi: 10.1161/JAHA.118.009742
Sarycheva, T., Taipale, H., Lavikainen, P., Tiihonen, J., Tanskanen, A., Hartikainen, S., et al. (2018b). Incidence and prevalence of antiepileptic medication use in community-dwelling persons with and without Alzheimer's disease. J. Alzheimers Dis. 66, 387–395. doi: 10.3233/jad-180594
Scarmeas, N., Honig, L. S., Choi, H., Cantero, J., Brandt, J., Blacker, D., et al. (2009). Seizures in Alzheimer disease: who, when, and how common? Arch. Neurol. 66, 992–997. doi: 10.1001/archneurol.2009.130
Scearce-Levie, K., Sanchez, P. E., and Lewcock, J. W. (2020). Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov. 19, 447–462. doi: 10.1038/s41573-020-0065-9
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chetelat, G., Teunissen, C. E., et al. (2021). Alzheimer's disease. Lancet 397, 1577–1590. doi: 10.1016/S0140-6736(20)32205-4
Sen, A., Capelli, V., and Husain, M. (2018). Cognition and dementia in older patients with epilepsy. Brain 141, 1592–1608. doi: 10.1093/brain/awy022
Sen, A., Jette, N., Husain, M., and Sander, J. W. (2020). Epilepsy in older people. Lancet 395, 735–748. doi: 10.1016/s0140-6736(19)33064-8
Shao, S. C., Wu, W. H., Yang, Y. K., and Lai, E. C. (2016). Quetiapine-induced absence seizures in a dementia patient. Geriatr. Gerontol. Int. 16, 1168–1171. doi: 10.1111/ggi.12683
Sherzai, D., Losey, T., Vega, S., and Sherzai, A. (2014). Seizures and dementia in the elderly: Nationwide inpatient sample 1999-2008. Epilepsy Behav. 36, 53–56. doi: 10.1016/j.yebeh.2014.04.015
Shi, Y., and Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772. doi: 10.1038/s41577-018-0051-1
Sjogren, T., Sjogren, H., and Lindgren, A. G. (1952). Morbus Alzheimer and morbus pick; a genetic, clinical and patho-anatomical study. Acta Psychiatr. Neurol. Scand. Suppl. 82, 1–152.
Spencer, D. (2014). Seizures and epileptiform activity in early Alzheimer disease: how hard should we be looking? Epilepsy Curr. 14, 73–75. doi: 10.5698/1535-7597-14.2.73
Stefanidou, M., Beiser, A. S., Himali, J. J., Peng, T. J., Devinsky, O., Seshadri, S., et al. (2020). Bi-directional association between epilepsy and dementia: the Framingham heart study. Neurology 95, e3241–e3247. doi: 10.1212/WNL.0000000000011077
Sulkava, R. (1982). Alzheimer's disease and senile dementia of Alzheimer type: A comparative study. Acta Neurol. Scand. 65, 636–650. doi: 10.1111/j.1600-0404.1982.tb03117.x
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Tábuas-Pereira, M., Durães, J., Lopes, J., Sales, F., Bento, C., Duro, D., et al. (2019). Increased CSF tau is associated with a higher risk of seizures in patients with Alzheimer's disease. Epilepsy Behav. 98, 207–209. doi: 10.1016/j.yebeh.2019.06.033
Tai, X. Y., Koepp, M., Duncan, J. S., Fox, N., Thompson, P., Baxendale, S., et al. (2016). Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 139, 2441–2455. doi: 10.1093/brain/aww187
Taipale, H., Gomm, W., Broich, K., Maier, W., Tolppanen, A. M., Tanskanen, A., et al. (2018). Use of antiepileptic drugs and dementia risk-an analysis of Finnish health register and German health insurance data. J. Am. Geriatr. Soc. 66, 1123–1129. doi: 10.1111/jgs.15358
Taipale, H., Lampela, P., Koponen, M., Tanskanen, A., Tiihonen, J., Hartikainen, S., et al. (2019). Antiepileptic drug use is associated with an increased risk of pneumonia among community-dwelling persons with Alzheimer's disease-matched cohort study. J. Alzheimers Dis. 68, 127–136. doi: 10.3233/JAD-180912
Tamagnini, F., Novelia, J., Kerrigan, T. L., Brown, J. T., Tsaneva-Atanasova, K., and Randall, A. D. (2015). Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front. Cell. Neurosci. 9:372. doi: 10.3389/fncel.2015.00372
Taubes, A., Nova, P., Zalocusky, K. A., Kosti, I., Bicak, M., Zilberter, M. Y., et al. (2021). Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer's disease. Nat. Aging 1, 932–947. doi: 10.1038/s43587-021-00122-7
Teixeira, F. B., Saito, M. T., Matheus, F. C., Prediger, R. D., Yamada, E. S., Maia, C. S. F., et al. (2017). Periodontitis and Alzheimer's disease: a possible comorbidity between Oral chronic inflammatory condition and Neuroinflammation. Front. Aging Neurosci. 9:327. doi: 10.3389/fnagi.2017.00327
Thijs, R. D., Surges, R., O'Brien, T. J., and Sander, J. W. (2019). Epilepsy in adults. Lancet 393, 689–701. doi: 10.1016/S0140-6736(18)32596-0
Tian, F. F., Zeng, C., Ma, Y. F., Guo, T. H., Chen, J. M., Chen, Y., et al. (2010). Potential roles of Cdk5/p35 and tau protein in hippocampal mossy fiber sprouting in the PTZ kindling model. Clin. Lab. 56, 127–136.
Tzeng, T. C., Hasegawa, Y., Iguchi, R., Cheung, A., Caffrey, D. R., Thatcher, E. J., et al. (2018). Inflammasome-derived cytokine IL18 suppresses amyloid-induced seizures in Alzheimer-prone mice. Proc. Natl. Acad. Sci. U. S. A. 115, 9002–9007. doi: 10.1073/pnas.1801802115
Volicer, L., Smith, S., and Volicer, B. J. (1995). Effect of seizures on progression of dementia of the Alzheimer type. Dementia 6, 258–263. doi: 10.1159/000106956
von Rüden, E. L., Zellinger, C., Gedon, J., Walker, A., Bierling, V., Deeg, C. A., et al. (2020). Regulation of Alzheimer's disease-associated proteins during epileptogenesis. Neuroscience 424, 102–120. doi: 10.1016/j.neuroscience.2019.08.037
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Vossel, K. A., Ranasinghe, K. G., Beagle, A. J., Mizuiri, D., Honma, S. M., Dowling, A. F., et al. (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Ann. Neurol. 80, 858–870. doi: 10.1002/ana.24794
Vossel, K. A., Tartaglia, M. C., Nygaard, H. B., Zeman, A. Z., and Miller, B. L. (2017). Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol. 16, 311–322. doi: 10.1016/s1474-4422(17)30044-3
Wang, R. R., Jin, J. H., Womack, A. W., Lyu, D., Kokane, S. S., Tang, N., et al. (2014). Neonatal ketamine exposure causes impairment of long-term synaptic plasticity in the anterior cingulate cortex of rats. Neuroscience 268, 309–317. doi: 10.1016/j.neuroscience.2014.03.029
Wang, X., Loi, S. M., Foster, E., Chen, Z., Velakoulis, D., and Kwan, P. (2021). Predictors of new-onset epilepsy in people with younger-onset neurocognitive disorders. Front. Aging Neurosci. 13:637260. doi: 10.3389/fnagi.2021.637260
Wong, J., and Delva, N. (2007). Clozapine-induced seizures: recognition and treatment. Can. J. Psychiatr. 52, 457–463. doi: 10.1177/070674370705200708
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem.20131685
Keywords: Alzheimer’s disease, epilepsy, comorbidity, seizure, cognition
Citation: Yang F, Chen L, Yu Y, Xu T, Chen L, Yang W, Wu Q and Han Y (2022) Alzheimer’s disease and epilepsy: An increasingly recognized comorbidity. Front. Aging Neurosci. 14:940515. doi: 10.3389/fnagi.2022.940515
Edited by:
Long Meng, Shenzhen Institutes of Advanced Technology (CAS), ChinaReviewed by:
Agenor Limon, University of Texas Medical Branch at Galveston, United StatesAndres Kanner, University of Miami, United States
Copyright © 2022 Yang, Chen, Yu, Xu, Chen, Yang, Wu and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanbing Han, eW5oeWJAMTYzLmNvbQ==