1
U613, Institut National de la Santé et de la Recherche Médicale, Brest, France
2
Laboratory of Molecular Genetics and Histocompatibility, Centre Hospitalier Universitaire Brest, Brest, France
3
Institut Fédératif de Recherche 148, Brest University, Brest, France
4
Department of Cell and Developmental Biology, University College London, London, UK
Genetic investigations of X-linked mental retardation have demonstrated the implication of ARX in a wide spectrum of disorders extending from phenotypes with severe neuronal migration defects, such as lissencephaly, to mild or moderate forms of mental retardation without apparent brain abnormalities, but with associated features of dystonia and epilepsy. These investigations have in recent years directed attention to the role of this gene in brain development. Analysis of its spatio-temporal localization profile revealed expression in telencephalic structures at all stages of development, mainly restricted to populations of GABA-containing neurons. Furthermore, studies of the effects of ARX loss of function either in humans or in lines of mutant mice revealed varying defects, suggesting multiple roles of this gene during development. In particular, Arx has been shown to contribute to almost all fundamental processes of brain development: patterning, neuronal proliferation and migration, cell maturation and differentiation, as well as axonal outgrowth and connectivity. In this review, we will present and discuss recent findings concerning the role of ARX in brain development and how this information will be useful to better understand the pathophysiological mechanisms of mental retardation and epilepsy associated with ARX mutations.
In the last years, defects in transcription regulation have been linked to several monogenic neurodevelopmental disorders resulting, in some cases, in cerebral malformations, mental retardation and/or autism. Such defects may result from mutations located directly in genes encoding transcription factors such as ZIC2 (Zinc finger protein of the cerebellum 2), responsible for holoprosencephaly (Brown et al., 1998
). As these transcription factors often have precise spatio-temporal expression profiles as well as multiple trans-acting co-factors, mutations in these genes can have pleiotropic effects. This is, for example, the case for ARX (aristaless-related homeobox gene), which has been shown in humans to be responsible for a wide spectrum of brain disorders ranging from phenotypes with severe neuronal migration defects, such as lissencephaly, to milder forms of X-linked mental retardation (XLMR) often associated with epilepsy, but without apparent brain abnormalities (for review, see Gécz et al., 2006
).
Mental retardation is a heterogeneous group of disorders that result from a variety of acquired and genetic causes. The observation that mental retardation preferentially affects males, has led investigators to focus on genes located on the X-chromosome. XLMR may be: (i) syndromic, characterized by recognizable dysmorphic features, neurological complications and/or metabolic abnormalities, or (ii) non-syndromic, showing no specific features apart from an intelligence quotient (IQ) below 70 with a deficit in adaptive skills (for reviews, see Chelly and Mandel, 2001
; Gécz et al., 2009
). Lissencephaly, which represents the other end of the spectrum of brain disorders associated with ARX mutations, is also a heterogeneous group of cortical malformations resulting from mutations in at least five different genes: LIS1, DCX (doublecortin), RELN (reelin), ARX and TUBA1A (tubulin alpha1A) (Reiner et al., 1993
; des Portes et al., 1998
; Gleeson et al., 1998
; Hong et al., 2000
; Kitamura et al., 2002
; Keays et al., 2007
). Lissencephaly is caused by abnormal neuronal migration and is characterized by disrupted cytoarchitecture associated with an abnormally thick cortex and absence (agyria) or diminution (pachygyria) of gyri and sulci and, hence, a smooth brain surface (for reviews, see Francis et al., 2006
; Guerrini and Parrini, 2009
).
ARX encodes a transcription factor which belongs to the class of homeobox genes. Mutations in this class of genes were first described in Drosophila and result in the misexpression of body structures in different segments of the fly, demonstrating their important role in specifying the body segments. Since then, homeobox genes were shown to control many cellular processes including proliferation, differentiation, apoptosis, cell shape, cell adhesion, and migration (for review, see Pearson et al., 2005
). They are characterized by a 60-amino acid homeobox domain (or homeodomain), which is responsible for DNA-binding. In addition, they often contain other motifs that can contribute to DNA and/or co-factor binding to further define their target gene specificity. These additional motifs, as well as variations in the homeodomain, are used to divide the homeoprotein superfamily into families and subfamilies, such as Hox, Nk, Paired, Pax, Lim, or Six.
The ARX protein belongs to one of the three largest classes of homeoproteins, the paired (Prd) class. This class of homeobox genes are thought to be important regulators of essential events during vertebrate embryogenesis, including the development of the central and peripheral nervous systems (for reviews, see Meijlink et al., 1999
; Wigle and Eisenstat, 2008
). Three subclasses have been identified based on the nature of a key residue at position 50 within the homeodomain. A serine residue at position 50 (S50) of the homeodomain defines the Pax- or Paired-type subgroup, whereas a lysine residue at position 50 (K50) defines another subgroup including the Orthodenticle gene. The presence of a glutamine at position 50 (Q50) of the homeodomain defines a third subgroup, which contains the aristaless-related proteins (Galliot and Miller, 2000
). ARX has, in addition to the homeodomain [amino acid (aa) 334–377], a conserved domain located at the C-terminus called the aristaless domain (aa 527–542), an octapeptide domain (aa 27–34) located near the N-terminus, three nuclear localization sequences (aa 82–89, 325–333 and 378–386), a central acidic domain (aa 224–255) and four polyalanine tracts (aa 100–115, 144–155, 275–281 and 432–440) whose function is not well known (Figure 1
).
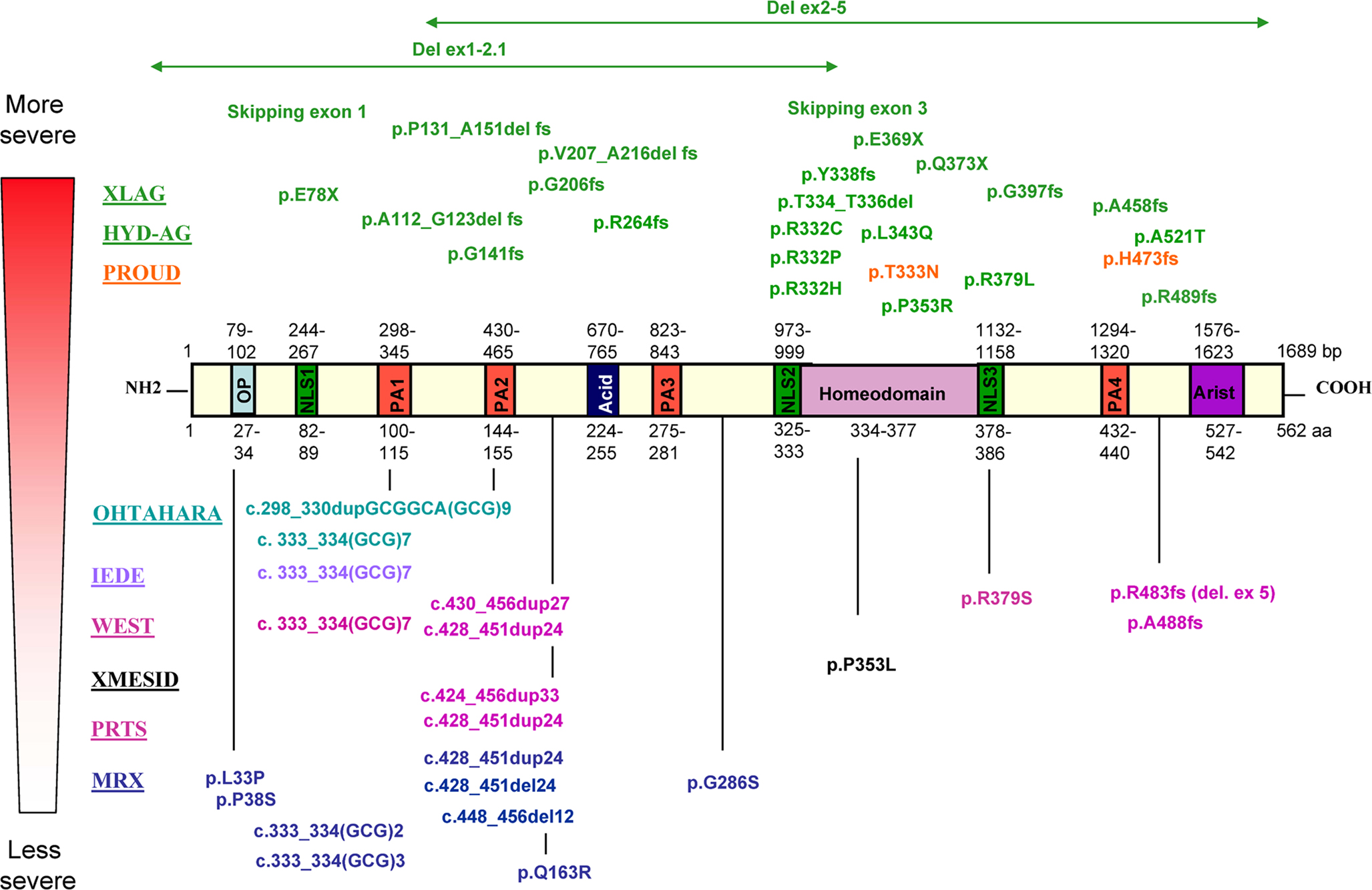
Figure 1. While non-malformation phenotypes [non-specific X-linked mental retardation (MRX), Partington (PRTS) and West syndromes, XMESID, IEDE and Ohtahara syndrome] tend to be caused by pathogenic variations outside the homeodomain or inside the first two polyalanines tracts of ARX, brain and genital malformation phenotypes [XLAG, hydranencephaly with abnormal genitalia (HYD-AG) and Proud syndrome] are associated with pathogenic variations that truncate the ARX protein or alter residues in the highly conserved homeodomain. Interestingly, a non-conservative missense mutation near the C-terminal aristaless domain (p.A521T) causes unusually severe XLAG with microcephaly and mild cerebellar hypoplasia.
One Single Gene Involved in Several Syndromes in Human
ARX, located on Xp22.13, was first identified in 2002 as being involved in non-syndromic XLMR (OMIM 300382 and 300419) (Bienvenu et al., 2002
) as well as in X-linked West syndrome [also called infantile spasms (ISSX)] (OMIM 308350) (Strømme et al., 2002
; Kato et al., 2003
). Since then, several other mutations have been described and broadened the spectrum of phenotypes resulting from ARX mutations (Figure 1
). These phenotypes can be divided into two groups: (1) a malformation group, which includes X-linked lissencephaly associated with abnormal genitalia (XLAG) (OMIM 300215) (Dobyns et al., 1999
; Ogata et al., 2000
; Kitamura et al., 2002
), hydranencephaly and abnormal genitalia (HYD-AG) (OMIM 300215) and Proud syndrome (OMIM 300004) (Kato et al., 2004
); and (2) a non-malformation group including non-syndromic XLMR (Bienvenu et al., 2002
), Partington syndrome (PRTS) (OMIM 309510) (Frints et al., 2002
; Strømme et al., 2002
), various forms of epilepsy including West syndrome (Strømme et al., 2002
; Kato et al., 2003
), X-linked myoclonic seizures, spasticity and intellectual disability (XMESID) (OMIM 308350) (Scheffer et al., 2002
; Strømme et al., 2002
), idiopathic infantile epileptic-dyskinetic encephalopathy (IEDE) (OMIM 308350) (Guerrini et al., 2007
) and early infantile epileptic encephalopathy with suppression-burst pattern (EIEE or Ohtahara’s syndrome) (OMIM 308350) (Kato et al., 2007
) (see Table 1
for a description of these syndromes).

Table 1. Short phenotypic description of the syndromes associated with ARX mutations.
Phenotype/genotype studies have suggested that there is a correlation between the genotype and the observed phenotype: premature termination mutations [large deletions, frameshift (fs), nonsense mutations or splice sites mutations] lead to the more severe phenotypes in the malformation group. In addition, non-conservative missense mutations within the homeobox or nuclear localization sequences (as for example p.R332C or p.P353R) also cause XLAG, while conservative substitutions in the homeodomain (p.T333N) cause Proud syndrome (Figure 1
). In contrast, missense mutations outside the homeobox or expansions/deletions of polyalanine tracts lead to the non-malformation group (Sherr, 2003
; Kato et al., 2004
). Although several studies have generally confirmed this correlation, recent case reports show evidence of strong phenotypic heterogeneity (Hartmann et al., 2004
; Van Esch et al., 2004
; Kato et al., 2007
; Wallerstein et al., 2008
; Absoud et al., 2009
; Fullston et al., 2010
), including in female carriers who have been reported to have in some cases partial or complete agenesis of the corpus callosum, cognitive levels spanning from normal to severely impaired and associated epilepsy (Bonneau et al., 2002
; Uyanik et al., 2003
; Kato et al., 2004
; Okazaki et al., 2008
; Wallerstein et al., 2008
; Marsh et al., 2009
). This phenotypic heterogeneity is probably due to differences in genetic and environmental backgrounds which are specific to each family.
ARX Mutations Expanding Polyalanine Tracts
The vast majority of ARX mutations identified so far affect the two first polyalanine tracts in the ARX protein, and most are expansions that result in highly variable phenotypes (Figure 1
). In particular, the most frequent and recurrent mutation (representing approximately 45% of all ARX mutations reported to date), the in-frame 24 bp duplication (c.428_451dup24), has been associated with PRTS, West syndrome and mental retardation with seizures or non-specific XLMR (Turner et al., 2002
; Szczaluba et al., 2006
; Guerrini et al., 2007
; Laperuta et al., 2007
). To date, various types of seizures have been reported in patients with this mutation, including infantile spasms (Strømme et al., 2002
; Turner et al., 2002
), tonic-clinic seizures (Turner et al., 2002
; Partington et al., 2004
; Szczaluba et al., 2006
), complex partial seizures (Partington et al., 2004
) and in one instance, absence of seizures (Szczaluba et al., 2006
). Two longer expansions have also been described, a 33- and 27-bp duplication (Demos et al., 2009
; Reish et al., 2009
). Interestingly, only the 27 bp duplication gives a more severe phenotype when compared to the spectrum of clinical presentations associated with the dup24 bp mutation (Reish et al., 2009
).
Similarly, several pathogenic variations have been reported to expand the first polyalanine tract of ARX. In particular, an expansion of seven residues [c.333_334(GCG)7] has been reported to cause X-linked West syndrome (Strømme et al., 2002
), IEDE (Guerrini et al., 2007
) and Ohtahara syndrome (Absoud et al., 2009
). The largest reported expansion, which adds eleven alanines [c.298_330dupGCGGCA(GCG)9], also produces Ohtahara syndrome (Kato et al., 2007
). It is interesting to note that despite having only a short expansion of seven alanine residues, the patient described in Absoud’s report displays one of the most severe phenotype, Ohtahara syndrome with progressive and severe neurodegeneration, resulting in death during the first year of life. This clinical presentation and course represent a much more severe phenotype than previously described for this mutation, suggesting that there is no real correlation between the expansion length of the polyalanine tract and phenotypic severity.
It has been suggested that these mutations, although recessive, may cause protein aggregation, similar to other polyalanine disorders (for review, see Albrecht and Mundlos, 2005
). Indeed, some ex vivo data seem to confirm this hypothesis, at least for the c.333_334(GCG)7 mutation (Nasrallah et al., 2004
; Friocourt et al., 2006
; Shoubridge et al., 2007
). In vitro transfection of the c.333_334(GCG)7 construct causes protein aggregation, filamentous nuclear inclusions, and an increase in cell death. Similarly, cortical neurons, transfected with this mutant construct using whole-brain electroporation, form neuronal nuclear inclusions in vivo (Nasrallah et al., 2004
). More recently, Shoubridge et al. (2007)
showed that, in addition to increase the propensity of protein aggregation, the c.333_334(GCG)7 mutation results in a shift from nuclear to cytoplasmic localization of ARX protein. Interestingly, two recent reports of knock-in mice for the same mutation show the absence of neuronal inclusions in vivo (Kitamura et al., 2009
; Price et al., 2009
). However, Price et al. (2009)
reported that in the parietal cortex of the (GCG) + 7 mutant mice, 45% of cells show cytoplasmic localization of mutated Arx compared to only 28% in wild-type mice. These results suggest that, although protein aggregation is not detectable in vivo, polyalanine expansions may cause Arx protein mislocalization in the cytoplasm and thus, a partial loss of function, which may contribute to the pathogenesis.
Both in vitro and in vivo studies have demonstrated that Arx is a potent transcriptional repressor, but that it can also act as an activator (Collombat et al., 2003
; Seufert et al., 2005
; McKenzie et al., 2007
; Fullenkamp and El-Hodiri, 2008
). In particular, the highly conserved octapeptide domain and another C-terminal region (aa 432–483) including the fourth polyalanine tract, have transcriptional repressor activity while the aristaless-related domain (aa 527–542) has transcriptional activator activity (McKenzie et al., 2007
; Fullenkamp and El-Hodiri, 2008
). Some of Arx co-factors have even been identified: the Groucho/transducin-like enhancer (TLE) of split family of co-repressors interacts with Arx octapeptide, whereas repression by the second domain occurs through the interaction with C-terminal binding proteins (CtBPs) (Fullenkamp and El-Hodiri, 2008
). Moreover, it was shown that, although the domain encompassing polyalanine tracts 1 and 2 of ARX does not seem to significantly contribute to the repression activity, the expansions of either polyalanine tract 1 or 2 enhance transcriptional repression activity in a manner dependent on the length of the alanine expansion, suggesting that the expansion of ARX polyalanine tracts may be harmful to neurons in a size-dependent manner (McKenzie et al., 2007
). Thus, these results suggest that ARX dysfunction due to expanded polyalanine tracts may arise from increased repression activity of the mutant protein. Changes in the transcriptional activity of ARX may thus have subtle effects on neuronal organization and may contribute to the pathogenesis of ARX-related disorders.
Pyramidal neurons, the projection cells of the neocortex, derive from the primitive neuroepithelium and, between embryonic day 12 (E12) and the time of birth in mouse, migrate radially as sequential waves to take their positions in the developing cortex in an orderly fashion. The first wave of postmitotic neurons migrates to form a subpial preplate (or primitive plexiform zone). The second wave, which will form the cortical plate (CP), splits the preplate into the superficial molecular layer (or marginal zone) and the deep subplate. Then, the following waves of migrating neurons pass the subplate and generate cell layers in an “inside-out” sequence, such that neurons that are generated early reside in the deepest layers, whereas later-born cells migrate past the existing layers to form the more superficial layers (reviewed in Rakic, 1990
; Nadarajah and Parnavelas, 2002
; Kriegstein and Noctor, 2004
; Ayala et al., 2007
). The other neuronal cell type of the cortex, the GABA-containing interneurons, are generated for the most part from progenitors in the medial and caudal ganglionic eminences (MGE and CGE) of the ventral forebrain and reach the cortex by tangential migration (for reviews, see Marin and Rubenstein, 2003
; Métin et al., 2006
).
In mouse, Arx expression is first detectable at the 3-somite stage and, after the 10-somite stage, it appears confined to a specific area in the anterior neural plate (Colombo et al., 2004
). At later stages, it is widespread throughout telencephalic structures such as the GE, the cerebral cortex and the hippocampus (Miura et al., 1997
; Bienvenu et al., 2002
; Colombo et al., 2004
; Poirier et al., 2004
). No expression is detected in most of the mesencephalic and diencephalic structures, except the ventral thalamus (Bienvenu et al., 2002
; Poirier et al., 2004
). Outside the brain, Arx is detected in endocrine pancreas, developing testes as well as in heart, skeletal muscle, and liver (Miura et al., 1997
; Bienvenu et al., 2002
; Kitamura et al., 2002
; Collombat et al., 2003
; Biressi et al., 2008
). In the telencephalon, it is strongly expressed in the subventricular zones (SVZ) and mantle zones of the developing LGE and MGE, but not in the ventricular zones (VZ) of these structures. On the contrary, in the developing cortex, Arx is expressed in progenitors in the VZ/SVZ, as well as in tangentially migrating interneurons emanating from the GE, but not in radially migrating cells. In addition, double-labelling experiments, performed in both dissociated cultures and sections of embryonic or adult cortex, revealed extensive co-localization between Arx and GABA (Colombo et al., 2004
; Poirier et al., 2004
; Cobos et al., 2005
; Friocourt et al., 2006
, 2008
). The protein is still present in the adult, but is confined primarily in regions that are known to be rich in GABAergic neurons such as the amygdala and the olfactory bulb (Colombo et al., 2004
; Poirier et al., 2004
). Thus, it has been suggested that the observed seizures in the great majority of patients with ARX mutations, probably result from absence and/or dysfunction of GABAergic interneurons. Consistent with these results, Arx expression was shown to be regulated by several members of the Dlx (Distal-less) family of homeobox proteins, particularly Dlx2 (Cobos et al., 2005
; Colasante et al., 2008
). Indeed, Arx expression is ectopically induced following electroporation of plasmids expressing Dlx1, Dlx2 or Dlx5 in the chick neural tube and in mouse dorsal thalamus. These results are strengthened by the fact that, in vivo, Arx expression is severely reduced in Dlx1/2 double knockout mice (Cobos et al., 2005
).
Multiple Effects Resulting from ARX Loss of Expression in Mouse and Human
Arx knock-out mice have been shown to display a variety of defects (Kitamura et al., 2002
; Colombo et al., 2007
). For example, Arx-deficient brains were found to be dramatically altered, exhibiting poorly developed olfactory bulbs and reduced volumes of the cerebral cortex and hippocampus (Kitamura et al., 2002
). In particular, mutant brain sections revealed an accumulation of newly born interneurons near their proliferative zones in the MGE and CGE, resulting in a major loss of GABAergic interneurons in the cortex, hippocampus and striatum, similar to the human XLAG (Bonneau et al., 2002
; Uyanik et al., 2003
; Forman et al., 2005
; Okazaki et al., 2008
). Colombo et al. (2007)
showed that the early differentiation of the basal ganglia appeared normal, whereas subsequent differentiation was impaired, leading to the periventricular accumulation of immature neurons in both LGE and MGE. Neuronal migrations towards the cortex and basal ganglia were greatly reduced in mutants, causing a periventricular accumulation of NPY+ (neuropeptide Y) or calretinin+ neurons in the MGE. Altogether, these data suggest that Arx has major roles in promoting neuronal migration and regulating basal ganglia differentiation in mice. As a likely secondary effect of these malformations, Arx mutants also displayed altered connectivity between the cortex and thalamus. In particular, major axonal tracts failed to cross the LGE/MGE border, probably as a result of the loss of LGE corridor cells that guide thalamic axons through the MGE (López-Bendito et al., 2006
) and/or the severe reduction of the ventral thalamus and lack of eminentia thalami, an area connecting the diencephalon to the caudoventral telencephalon (Kitamura et al., 2002
; Colombo et al., 2007
).
In human, XLAG is typically characterized by severe congenital or postnatal microcephaly, lissencephaly with a posterior-to-anterior gradient, an agenesis of the corpus callosum and dysgenesis of the hippocampal dentate gyrus, midbrain malformations, hypothalamic dysfunction, neonatal-onset intractable epilepsy, severe hypotonia and ambiguous or underdeveloped genitalia in genotypic males (Dobyns et al., 1999
; Ogata et al., 2000
; Kitamura et al., 2002
). Histopathological studies revealed poorly delineated and atrophic basal ganglia with small fragmented caudate nuclei and hypodense cavitations within the striatum, the absence or hypoplasia of the olfactory bulbs, hypoplastic optic nerves, a selectively enlarged third ventricle and temporal lobe as well as hippocampal dysplasia. Histological analysis described a cortex organized into 3 distinct layers (a marginal layer, an intermediate layer and a less organized deep layer) instead of the usual six, and a strong decrease in the number, sometimes even a complete absence, of interneurons. Moreover, numerous heterotopic neurons can be observed in the white matter without any clear organization (Bonneau et al., 2002
; Uyanik et al., 2003
; Forman et al., 2005
; Okazaki et al., 2008
). These neuropathological features are very similar to those observed in Arx-/y mice (Kitamura et al., 2002
; Colombo et al., 2007
).
Role of ARX in Cell Proliferation and Neuronal Migration
The extensive co-localization between Arx and GABA, its absence of expression in radially migrating neurons, as well as the absence of interneurons documented in the cortex of XLAG patients (Bonneau et al., 2002
; Forman et al., 2005
; Okazaki et al., 2008
) and in Arx mutant mice (Kitamura et al., 2002
; Colombo et al., 2007
) have led to the idea that XLAG syndrome was the result of defective interneuron migration and function, and the proposal of a new term “interneuronopathy” to describe this pathology (Kato and Dobyns, 2005
). However, some data suggest that ARX expression in cortical progenitors may also be critical for radial migration (Friocourt et al., 2008
). Using in utero electroporation in mouse to knock-down or overexpress ARX in the developing cortex, we recently showed that targeted inhibition of the gene causes cortical progenitor cells to exit the cell cycle prematurely, whereas overexpression increases the length of the cell cycle, showing that ARX is important to maintain progenitor proliferation. In addition, RNA interference-mediated inactivation of ARX results in decreased neuronal motility, with an accumulation of cells in the SVZ/intermediate zone (IZ) (Friocourt et al., 2008
). These results are in agreement with the microcephaly and the misplacement of pyramidal neurons observed in the cortex of Arx knockout mice (Kitamura et al., 2002
), and in the cortex and white matter of XLAG syndrome (Bonneau et al., 2002
).
Interestingly, we observed that arrested cells due to ARX inactivation appeared oval or round in shape, with very few or no processes (Friocourt et al., 2008
), very different from the multipolar morphology normally exhibited when cells exit the VZ and enter the lower IZ (Tabata and Nakajima, 2003
). Since the first description of the multipolar stage during radial migration, several studies have suggested that the transition into and out of this stage is particularly vulnerable, and that it is disrupted in several disorders of neocortical development, including lissencephaly (LoTurco and Bai, 2006
). Cell morphology defects, very similar to those induced by RNAi-mediated inactivation of ARX, have been reported following inactivation of Filamin A (Nagano et al., 2004
), Rac1 (Chen et al., 2009
) or p27kip1 (Kawauchi et al., 2006
) and gain of function of MARK2 (Sapir et al., 2008
) or Rnd2 (Heng et al., 2008
). Interestingly, all these proteins interact with the actin cytoskeleton or are involved in neuronal polarity, suggesting that ARX may play a role in cell morphology through the regulation of the cytoskeleton. Consistently, we observed that ARX overexpression in radially migrating cells promotes tangentially orientated migration in the SVZ and lower IZ, although these cells do not express GABAergic markers (Figure 2
). Interestingly, these cells exhibit complex branching and very long processes, which confirm that ARX may have a role in cell morphology and especially in process formation (Friocourt et al., 2008
).
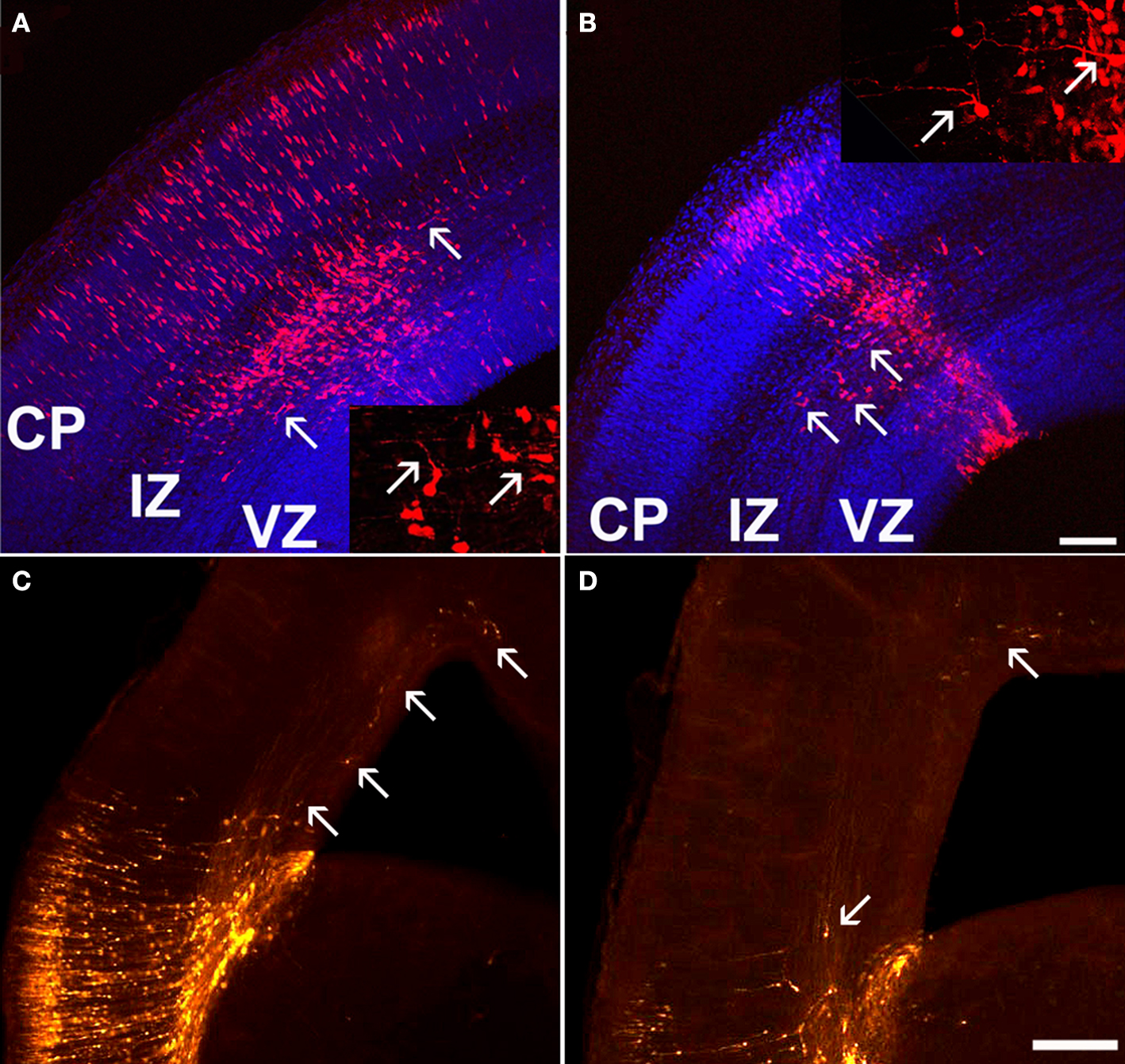
Figure 2. Tangential migration of a few ARX-overexpressing cells in the cortex. (A,B) Examination of coronal sections of E16.5 mouse brains electroporated at E13.5 with an ARX-overexpressing construct. Tangentially orientated cells migrating away from the site of electroporation are detectable in the IZ (see arrows). Some of these cells have long and complex processes, orientated tangentially. (C,D) Examination of E18.5 coronal sections of mouse brains electroporated at E13.5 with an ARX-overexpressing construct. Five days after electroporation, the number of tangentially orientated cells observed was reduced, but some had migrated long distances (see arrows). Scale bars: (A,B) 100 μm, (C,D) 200 μm.
In Arx-null embryos, migration from (i) the LGE to the striatum and other structures and (ii) the MGE to the cortical IZ and MZ are nearly absent, whereas migration through the cortical SVZ is only partially impaired. As a consequence, calbindin- and calretinin-expressing cells are severely reduced and NPY+ interneurons are nearly absent throughout the brain (Kitamura et al., 2002
; Colombo et al., 2007
). Defective tangential migration is similarly observed after electroporation into the MGE of rat brain slices (Friocourt et al., 2008
). Both inactivation and overexpression of ARX result in impairment of cortical interneuron migration from the MGE. Thus, these results suggest that this defect in tangential migration is cell autonomous and not the consequence of the absence of Arx earlier in development (Colombo et al., 2007
; Friocourt et al., 2008
). Indeed, studies on Arx knock-out mice described regional deficiencies and mis- and/or ectopic expression of several transcription factors, potentially important for migration and differentiation of certain population of neurons, as well as abnormal axonal tracts which may have been the reason for the impaired tangential migration (Kitamura et al., 2002
; Colombo et al., 2007
). Interestingly, we observed that many tangentially migrating neurons overexpressing ARX had one unusually long process (Friocourt et al., 2008
). Morphological defects were also observed in migrating interneurons derived from Arx mutant mice (Colombo et al., 2007
). These authors observed that Arx-mutant interneurons failed to migrate from small fragments of mutant LGE to the cortex both on mutant and wild-type slices. Similarly, in Matrigel experiments, Arx mutant explants exhibited more than 70% reduction in mean migration distance from the edge of the explant. In addition, the mutant leading processes exhibited an increase in length and branching, further suggesting that Arx may regulate the cytoskeleton dynamics during interneuron migration, similar to what was shown for Dcx and Lis1, two genes responsible for lissencephaly (McManus et al., 2004
; Kappeler et al., 2006
; Friocourt et al., 2007
). It is interesting to note that Arx has also been found to be necessary for neuronal migration in the rostral migratory stream, a structure that contains newly generated neurons migrating towards the olfactory bulb (Yoshihara et al., 2005
).
Recently, Colasante et al. (2009)
performed a gene expression profile analysis comparing E14.5 wild-type and Arx mutant ventral telencephalic tissues and identified Ebf3 among the 35 genes whose expression is consistently altered in Arx mutant GE. This gene is normally not (or only marginally) expressed in the developing telencephalon, but it was found strongly misexpressed in the MGE and LGE of Arx mutant mice (Colasante et al., 2009
). It is also known to be expressed in the developing hindbrain and spinal cord where it promotes neuronal differentiation and radial migration (Garcia-Dominguez et al., 2003
). More importantly, these authors observed that electroporation of a construct expressing Ebf3 into the MGE prevented neuronal tangential migration. Conversely, focal electroporation of a short hairpin RNA (shRNA) targeting Ebf3 into the MGE of brain slices taken from Arx mutants at E14.5 rescued the migration, although only partially. Electroporated cells were found to migrate away from the site of injection and spread following both a radial migration towards the mantle zone of the basal ganglia and a tangential route to the cortex. However, very few cells were able to move through the cortico-striatal boundary and reach the lateral cortex, suggesting that long distance migration capability was not rescued by this means. These results suggest the implication of Ebf3 in neuronal migration although the underlying mechanisms are still unknown.
Role of ARX in Neuronal Differentiation
Several studies have suggested that ARX may play important roles in neuronal maturation and/or differentiation. Indeed, Okazaki et al. (2008)
reported the presence of ectopic cells expressing nestin in the SVZ of a patient with XLAG, suggesting some neural maturation defects. Similarly, Colombo et al. (2007)
observed that many immature neurons produced after E11.5 in Arx mutants, that had failed to migrate out of the subpallial germinal layers, expressed only weakly MAP2, a marker of differentiated and mature neurons. These cells were also negative for various striatal markers such as Ebf1 or enkephalin mRNAs and calbindin, DARPP32 and dopamine 2 receptor, suggesting that they are unable to differentiate (Colombo et al., 2007
). Interestingly, these cells, when dissected from E18.5 LGE and MGE SVZ, expressed MAP2 as well as striatal markers (DARPP32, calbindin, and EBF), suggesting that Arx mutant SVZ cells of the LGE are capable of differentiation, and that the maturation defects observed in mutant mice may result from non-adapted local factors in the SVZ.
These observations as well as the high degree of co-localization that exists between Arx and GABA in embryonic and adult brains, and the observation that Arx expression is regulated by Dlx genes which are strong candidates for regulating the differentiation of most, if not all, telencephalic GABAergic neurons (Stuhmer et al., 2002
), have led to the suggestion that Arx might be involved in the specification of the GABAergic phenotype. However, this hypothesis has not been confirmed. In a previous report, we observed that the co-localization of ARX and GABA in cortex or GE is not complete (Friocourt et al., 2008
). In particular, we observed that 61–65% of ARX-positive cells expressed GABA and 58–72% of GABAergic cells expressed ARX in dissociated cultures from E16 striatum. Moreover, ARX overexpression in cortical progenitors in vivo or in dissociated cultures from E16 rat cortex or GE does not induce GABA or calbindin expression, suggesting that even if ARX is involved in GABAergic specification, it does not appear to be sufficient by itself to induce the GABAergic phenotype and thus, it may act in combination with other genes (Friocourt et al., 2008
). In addition, the observation that the introduction of an ARX-overexpressing construct into the VZ of the dorsal telencephalon induces tangential migration within the IZ, although these cells do not express GABA (Figure 2
), suggests that ARX may be involved in migration rather than cell specification or differentiation. These results are further reinforced by recent findings which show that Arx re-expression in a Dlx1/2 mutant background is sufficient to rescue neuronal migration activity. In contrast, Dlx2 electroporation in Arx-deficient brain slices induces GAD65 expression, suggesting that Arx is necessary to promote Dlx-dependent GABAergic cell migration, but is dispensable for Dlx ability to induce GABAergic cell fate commitment (Colasante et al., 2008
).
However, it is still possible that ARX controls the specification of distinct subsets of GABAergic neurons in the subpallium. Indeed, different studies have reported a decrease in the number of cholinergic neurons in the basal forebrain of Arx mutants (Kitamura et al., 2002
; Colombo et al., 2007
), suggesting that ARX, uniquely or in combination with other transcription factors, may play a role in the specification of at least this subpopulation of GABAergic cells. In addition, the observation that ARX is still expressed in GABAergic cells in the mouse adult brain suggests that it may also have a role in more mature neurons. Interestingly, using a genome-wide transcriptomic screen to identify transcriptional changes in the subpallium between wild-type and Arx mutant mice, two recent studies isolated genes encoding calbindin and calretinin as up-regulated, suggesting that Arx may repress the expression of these genes (Fulp et al., 2008
; Colasante et al., 2009
). However, it is also possible that, in the case of calbindin, this up-regulation may result from the accumulation of interneurons which fail to reach the cortex, and that calbindin is not a direct target of Arx. On the contrary, the Lhx7/8 gene, which is required for cholinergic differentiation and maturation, was found down-regulated in Arx mutant striatum, suggesting that Arx normally promotes cholinergic neuron differentiation through the activation of this gene (Colasante et al., 2009
).
Recently, Kitamura et al. (2009)
published 3 knock-in mice for mutations found in human: P353L (a mutation responsible for XMESID), P353R (a mutation responsible for XLAG), as well as (GCG)+7 (expansion in the first polyalanine tract). They observed that the phenotype of the ArxP353R mutant mice is quite severe and very similar to Arx-null mice (Kitamura et al., 2002
; Colombo et al., 2007
). Similarly, the abnormal cortical layer formation, abnormal structure of the striatum, and deficiency of GABAergic neurons in the cortex and striatum caused by P353R mutation closely mimic XLAG (Bonneau et al., 2002
; Okazaki et al., 2008
). In contrast, the two other mutant lines survived and had milder phenotypes: mice with the (GCG)+7 mutation showed severe seizures and impaired learning performance, whereas mice with the P355L mutation exhibited mild seizures and only slightly impaired learning performance. All these mutants exhibited fewer GABAergic and cholinergic neurons in the striatum, medial septum and ventral forebrain nuclei when compared with wild-type mice (Table 2
).
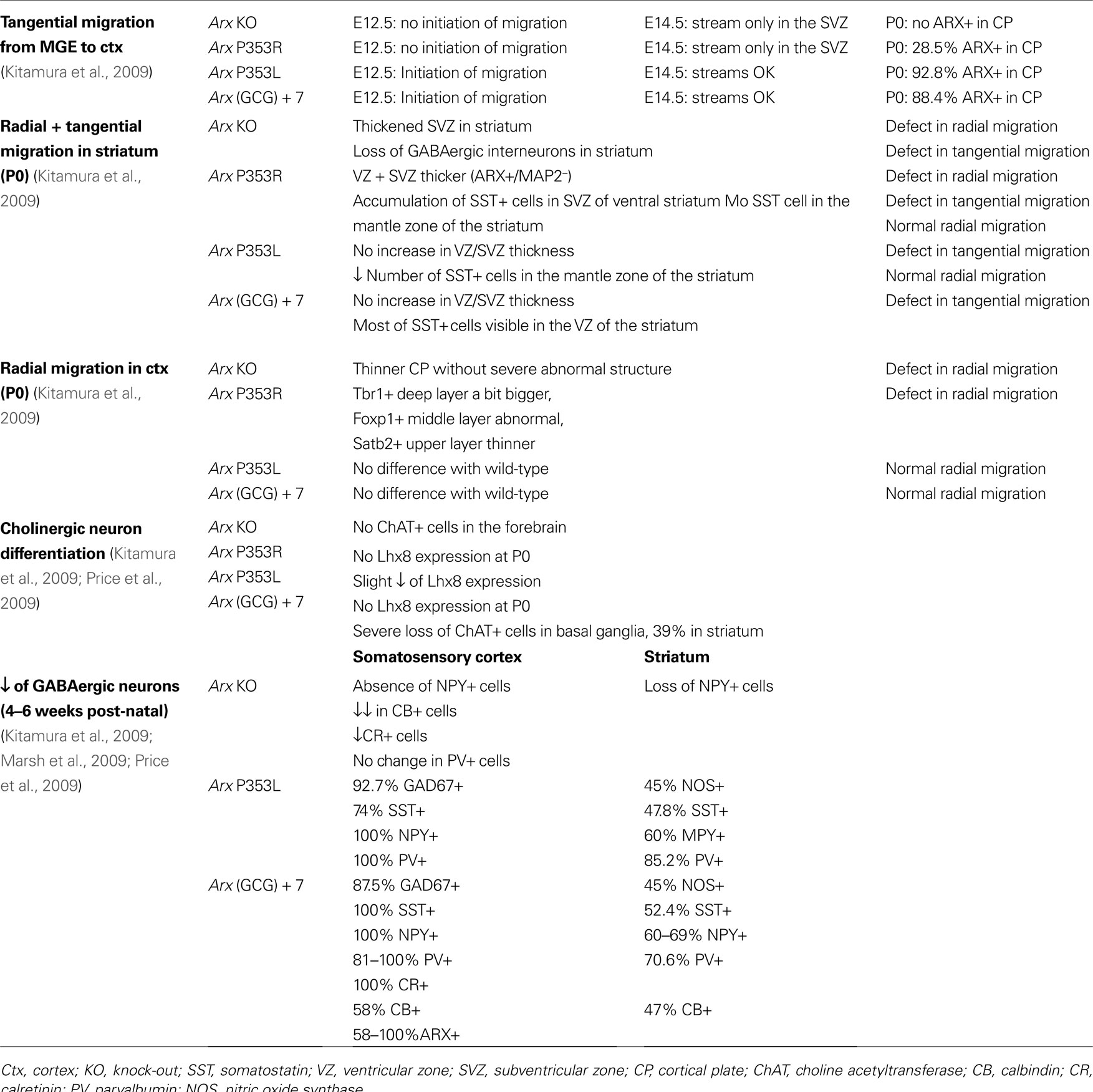
Table 2. Summary of the defects reported by studies on different Arx mutant lines. The percentages of cells are expressed by comparison to the number of cells in wild-type mice.
Interestingly, the severity of the phenotype, as well as the decrease in specific GABAergic subpopulations in mutant brains, seem to correlate quite well with the decreased ability of transcriptional repression (Figure 3
), suggesting that even subtle transcriptional changes may be a potential basis for the neurological symptoms and cognitive impairment observed in the patients.
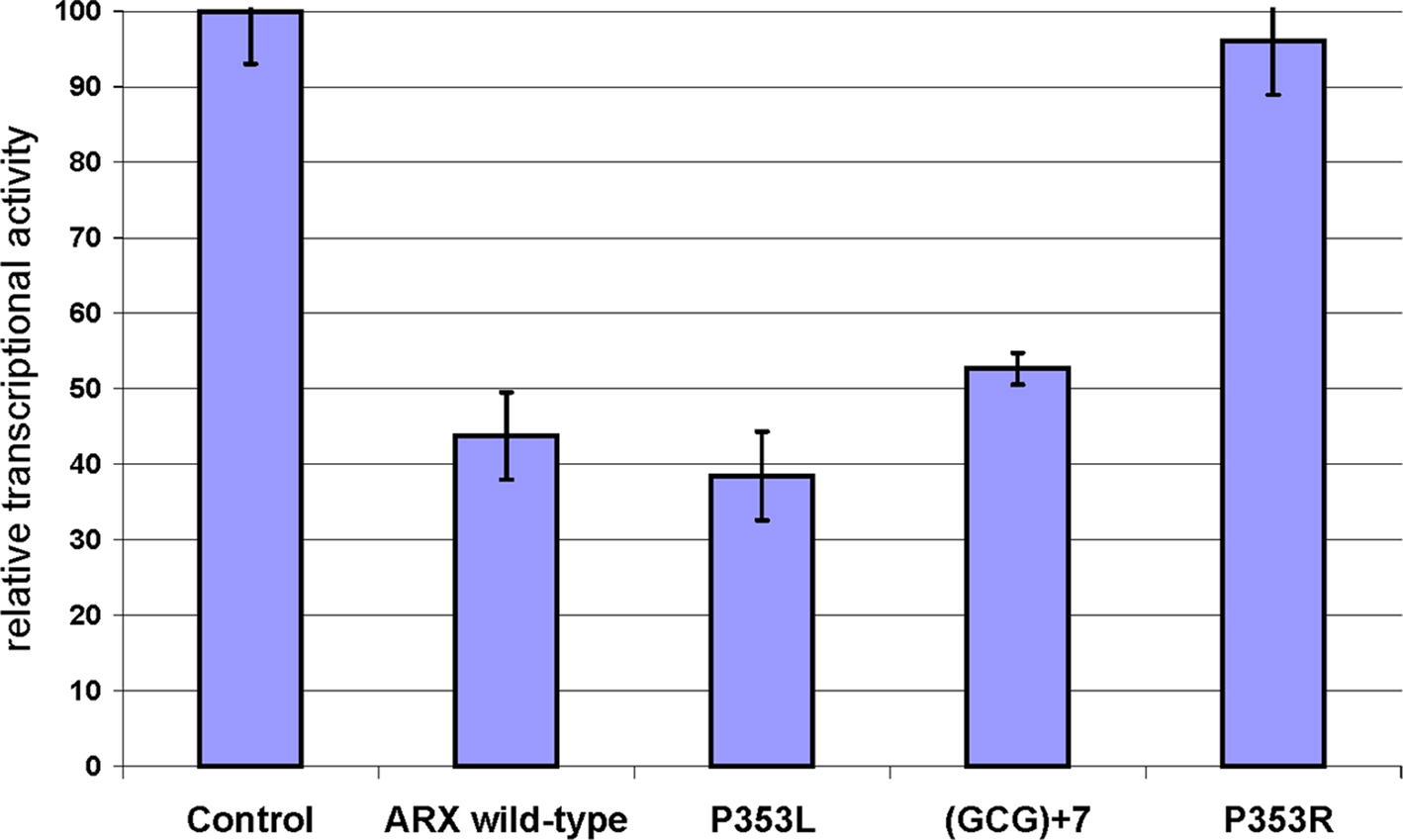
Figure 3. Measure of the transcriptional repression capacity of different ARX mutants corresponding to identified mutations in human. The capacity of transcriptional repression of mutant forms of ARX was tested on Lmo1 enhancer, which was cloned upstream TK-luciferase in a similar design as Fulp et al. (2008)
. ARX wild-type or mutant constructs were transfected in Neuro2a cells and the luciferase activity was measured. Luciferase data were normalized to Renilla expression and data are presented as the percentage of transcriptional activity compared to an empty vector control. Although P353L mutation does not have a detectable effect on ARX transcriptional repression, (GCG) + 7 and P353R both decrease ARX capacity to repress expression of the reporter gene.
To test the hypothesis that the epilepsy phenotype observed in children with ARX mutations results primarily from a deficit in forebrain cerebral cortical interneuron function, Marsh and colleagues recently generated a mouse line with a genetic ablation of Arx specifically in subpallial derived neurons (Marsh et al., 2009
). They found that both male and female mutant mice demonstrated early onset epilepsy resembling the one observed in patients with ARX mutations. Although the postnatal brains of these mutant mice appeared grossly normal, they identified interneuron subtype specific defects, again suggesting that Arx is necessary for the development of specific populations of interneurons in mice (Table 2
). The finding that these mutant mice, who have an Arx expression left intact in the developing neocortex, recapitulate many key characteristics of ARX-related disorders, suggests a critical role for interneurons in the pathogenesis of epilepsy in these patients and strongly support the concept of “interneuronopathy” (Kato and Dobyns, 2005
), as the cause of epilepsy, specifically infantile spasms. The proposed pathophysiological mechanism of the observed phenotype in these animals is thus a specific loss of interneurons resulting in an overall increase in excitation.
Indeed GABA, the principal inhibitory neurotransmitter, has been shown to synchronize activity in cortical neuronal circuits, reduce cell hyperexcitability, and prevent epileptiform activity in the cerebral cortex and hippocampus. In the developing and newborn rodent brains, it has been shown that this neurotransmitter was excitatory (for review, see Represa and Ben-Ari, 2005
). This excitatory action is supposed to play important roles during brain maturation and may transiently regulate neuronal growth, cell proliferation in the germinative zones, neuronal migration and cell differentiation (for reviews, see Represa and Ben-Ari, 2005
; Heng et al., 2007
). Abnormalities of GABAergic function have already been associated with epilepsy in humans and subcortical structures rich in GABAergic neurons, such as the basal ganglia, have also been implicated in the generation of epileptic spasms (for review, see Dulac, 2001
). So, in conclusion, although the precise mechanisms of action of ARX and the signalling pathways involved are still unknown, both the descriptions of the patient clinical conditions and the GABAergic defects characterized in different Arx mutant lines strongly suggest a functional impairment of GABAergic interneurons in the basal ganglia as the major cause of ARX-related phenotypes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank the Inserm, Fondation Bettencourt-Schueller, Fondation Jerome Lejeune, le Fonds Européen de Développement Régional and the Wellcome Trust (grant number 074549) for support of the work on ARX. We also acknowledge the valuable collaboration of Dr S. Kanatani and his colleagues in many of our experiments.
Bienvenu, T., Poirier, K., Friocourt, G., Bahi, N., Beaumont, D., Fauchereau, F., Ben Jeema, L., Zemni, R., Vinet, M. C., Francis, F., Couvert, P., Gomot, M., Moraine, C., van Bokhoven, H., Kalscheuer, V., Frints, S., Gecz, J., Ohzaki, K., Chaabouni, H., Fryns, J. P., des Portes, V., Beldjord, C., and Chelly, J. (2002). ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum. Mol. Genet. 11, 981–991.
Biressi, S., Messina, G., Collombat, P., Tagliafico, E., Monteverde, S., Benedetti, L., Cusella De Angelis, M. G., Mansouri, A., Ferrari, S., Tajbakhsh, S., Broccoli, V., and Cossu, G. (2008). The homeobox gene Arx is a novel positive regulator of embryonic myogenesis. Cell Death Differ. 15, 94–104.
Bonneau, D., Toutain, A., Laquerrière, A., Marret, S., Saugier-Veber, P., Barthez, M. A., Radi, S., Biran-Mucignat, V., Rodriguez, D., and Gélot, A. (2002). X-linked lissencephaly with absent corpus callosum and ambigous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann. Neurol. 51, 340–349.
Colasante, G., Collombat, P., Raimondi, V., Bonanomi, D., Ferrai, C., Maira, M., Yoshikawa, K., Mansouri, A., Valtorta, F., Rubenstein, J. L., and Broccoli, V. (2008). Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J. Neurosci. 28, 10674–10686.
Colombo, E., Collombat, P., Colasante, G., Bianchi, M., Long, J., Mansouri, A., Rubenstein, J. L., and Broccoli, V. (2007). Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J. Neurosci. 27, 4786–4798.
des Portes, V., Pinard, J. M., Billuart, P., Vinet, M. C., Koulakoff, A., Carrie, A., Gelot, A., Dupuis, E., Motte, J., Berwald-Netter, Y., Catala, M., Kahn, A., Beldjord, C., and Chelly, J. (1998). A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92, 51–61.
Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., Cooper, E. C., Dobyns, W. B., Minnerath, S. R., Ross, M. E., and Walsh, C. A. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72.
Hartmann, H., Uyanik, G., Gross, C., Hehr, U., Lücke, T., Arslan-Kirchner, M., Antosch, B., Das, A. M., and Winkler, J. (2004). Agenesis of the corpus callosum, abnormal genitalia and intractable epilepsy due to a novel familial mutation in the Aristaless-related homeobox gene. Neuropediatrics 35, 157–160.
Kato, M., Das, S., Petras, K., Kitamura, K., Morohashi, K., Abuelo, D. N., Barr, M., Bonneau, D., Brady, A. F., Carpenter, N. J., Cipero, K. L., Frisone, F., Fukuda, T., Guerrini, R., Iida, E., Itoh, M., Feldman Lewanda, A., Nanba, Y., Oka, A., Proud, V. K., Saugier-Veber, P., Schelley, S. L., Selicorni, A., Shaner, R., Silengo, M., Stewart, F., Sugiyama, N., Toyama, J., Toutain, A., Lia Vargas, A., Yanazawa, M., Zackai, E. H., and Dobyns, W. B. (2004). Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 23, 147–159.
Kato, M., Saitoh, S., Kamei, A., Shiraishi, H., Ueda, Y., Akasaka, M., Tohyama, J., Akasaka, N., and Hayasaka, K. (2007). A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 81, 361–366.
Keays, D. A., Tian, G., Poirier, K., Huang, G. J., Siebold, C., Cleak, J., Oliver, P. L., Fray, M., Harvey, R. J., Molnár, Z., Piñon, M. C., Dear, N., Valdar, W., Brown, S. D., Davies, K. E., Rawlins, J. N., Cowan, N. J., Nolan, P., Chelly, J., and Flint, J. (2007). Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45–57.
Kitamura, K., Itou, Y., Yanazawa, M., Ohsawa, M., Suzuki-Migishima, R., Umeki, Y., Hohjoh, H., Yanagawa, Y., Shinba, T., Itoh, M., Nakamura, K., and Goto, Y. (2009). Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Hum. Mol. Genet. 18, 3708–3724.
Kitamura, K., Yanazawa, M., Sugiyama, N., Miura, H., Iizuka-Kogo, A., Kusaka, M., Omichi, K., Suzuki, R., Kato-Fukui, Y., Kamiirisa, K., Matsuo, M., Kamijo, S. I., Kasahara, M., Yoshioka, H., Ogata, T., Fukuda, T., Kondo, I., Kato, M., Dobyns, W. B., Yokoyama, M., and Morohashi, K. I. (2002). Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 32, 359–369.
Marsh, E., Fulp, C., Gomez, E., Nasrallah, I., Minarcik, J., Sudi, J., Christian, S. L., Mancini, G., Labosky, P., Dobyns, W., Brooks-Kayal, A., and Golden, J. A. (2009). Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 132, 1563–1576.
McKenzie, O., Ponte, I., Mangelsdorf, M., Finnis, M., Colasante, G., Shoubridge, C., Stifani, S., Gécz, J., and Broccoli V. (2007). Aristaless-related homeobox gene, the gene responsible for West syndrome and related disorders, is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. Neuroscience 146, 236–247.
Okazaki, S., Ohsawa, M., Kuki, I., Kawawaki, H., Koriyama, T., Ri, S., Ichiba, H., Hai, E., Inoue, T., Nakamura, H., Goto, Y., Tomiwa, K., Yamano, T., Kitamura, K., and Itoh, M. (2008). Aristaless-related homeobox gene disruption leads to abnormal distribution of GABAergic interneurons in human neocortex: evidence based on a case of X-linked lissencephaly with abnormal genitalia (XLAG). Acta Neuropathol. 116, 453–462.
Price, M. G., Yoo, J. W., Burgess, D. L., Deng, F., Hrachovy, R. A., Frost, J. D. Jr., and Noebels, J. L. (2009). A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10 + 7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J. Neurosci. 29, 8752–8763.