




- 1 Dipartimento di Neuroscienze, Sezione di Fisiologia, Università Politecnica delle Marche, Ancona, Italy
- 2 Fondazione di Medicina Molecolare, Università Politecnica delle Marche, Ancona, Italy
In monoamine-releasing terminals, neurotransmitter transporters – in addition to terminating synaptic transmission by clearing released transmitters from the extracellular space – are the primary mechanism for replenishing transmitter stores and thus regulate presynaptic homeostasis. Here, we analyze whether GAT-1, the main plasma membrane GABA transporter, plays a similar role in GABAergic terminals. Re-examination of existing literature and recent data gathered in our laboratory show that GABA homeostasis in GABAergic terminals is dominated by the activity of the GABA synthesizing enzyme and that GAT-1-mediated GABA transport contributes to cytosolic GABA levels. However, analysis of GAT-1 KO, besides demonstrating the effects of reduced clearance, reveals the existence of changes compatible with an impaired presynaptic function, as miniature IPSCs frequency is reduced by one-third and glutamic acid decarboxylases and phosphate-activated glutaminase levels are significantly up-regulated. Although the changes observed are less robust than those reported in mice with impaired dopamine, noradrenaline, and serotonin plasma membrane transporters, they suggest that in GABAergic terminals GAT-1 impacts on presynaptic GABA homeostasis, and may contribute to the activity-dependent regulation of inhibitory efficacy.
Introduction
In his Nobel Lecture, given on December 12, 1970, Julius Axelrod summarizes his seminal work on noradrenaline (NET) metabolism and uptake. In the paragraph entitled Effect of drugs on neuronal uptake he describes how blocking NET uptake using cocaine or other drugs resulted in a dramatic decrease in tissue [3H] NET and addresses the question of whether, in addition to blocking uptake, “these drugs could prevent storage or release of [3H] NET” (Axelrod, 1971). Ever since, the possible role of transport mechanisms on transmitter storage and release of neurotransmitters has been essentially neglected. Advent of molecular cloning and knock-out technologies made it possible to unveil the nature of proteins mediating neurotransmitter transport (transporters) and to investigate their functional role in vivo. Mice lacking dopamine (DAT), NET, or serotonin (SERT) transporters exhibit an increase of extracellular lifetime and levels of monoamines and a decrease of both their intracellular storage and release (Torres et al., 2003). These findings indicate that, besides terminating synaptic transmission by clearing released transmitters, monoamine transporters mediate replenishment of transmitter stores, thus regulating presynaptic homeostasis. Here, we analyze whether the same principle applies to GABAergic synapses1.
GABA Synthesis and Transport: A Complex Scenario
GABA Synthesis
Most GABA is synthesized from glutamate (Glu) by glutamic acid decarboxylase (GAD; Roberts and Frankel, 1950, 1951). In mammalian brain, GAD occurs in two molecular forms, GAD65 and GAD67; the former preferentially synthesizes GABA for vesicular release, the latter for cytoplasmic stores (Soghomonian and Martin, 1998). Indeed, ratio of GAD65 to GAD67 is higher in synaptic vesicle fractions than in cytosol (Solimena et al., 1993). GAD65 may be anchored to synaptic vesicles by forming a complex that includes the vesicular GABA transporter VGAT, an integral membrane protein of synaptic vesicles responsible for their filling (McIntire et al., 1997). This may provide a structural and functional coupling between synthesis and vesicular packaging of GABA (Hsu et al., 2000; Jin et al., 2003). Interestingly, [3H]GABA newly synthesized from [3H]Glu by synaptic vesicle-associated GAD is taken up preferentially into vesicles over cytosolic GABA (Jin et al., 2003). Minor sources of GABA, such as putrescine, spermine, spermidine, and ornithine, offer a negligible contribution to releasable GABA.
Glu used for GABA synthesis may originate from diverse sources.
Glu–GABA/glutamine cycle
Glutamate derived from glutamine (Gln) is an important GABA precursor (Bradford et al., 1983; Sonnewald et al., 1993). Released GABA is taken up by astrocytic transporters (i.e., GAT-3; Minelli et al., 1996), and catabolized to the tricarboxylic acid (TCA) cycle intermediate succinate by GABA transaminase and succinate semialdehyde dehydrogenase; the resulting α-ketoglutarate is then transformed to Glu which is converted to Gln by Gln synthetase (Martinez-Hernandez et al., 1977). Gln is then extruded from astrocytes by SNAT3, a system N transporter (Chaudry et al., 1999, 2002; Boulland et al., 2002), and taken up by axon terminals. Gln influx into neurons is thought to be mediated by SNAT1 and/or SNAT2, two system A transporters (Fricke et al., 2007); this view is compatible with expression of SNAT1 and SNAT2 in most GABAergic cells (Melone et al., 2004, 2006; Conti and Melone, 2006). In neurons, Gln is converted to Glu by phosphate-activated glutaminase (PAG; Kvamme et al., 2001). PAG immunoreactivity (ir) is detected in ∼18% of all puncta expressing VGAT, which is expressed in all GABAergic terminals (Chaudhry et al., 1998; Minelli et al., 2003), and electron microscope studies show that ∼20% of all PAG+ axon terminals making synaptic contacts form symmetric synapses (Figures 1A,B).
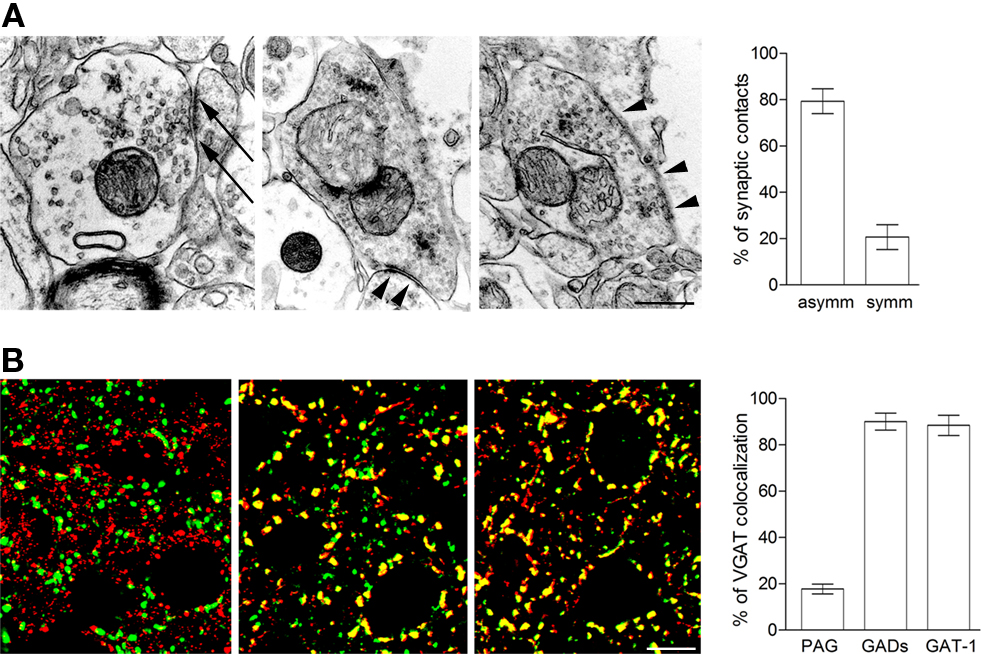
Figure 1. Anatomical insight into GABAergic presynaptic terminals. (A) Electron microscope studies of PAG immunoreactivity. Both axon terminals forming asymmetric (arrows) and symmetric synapses (arrowheads) contain PAG-ir. Layer II–III; parietal cortex. Rat brains perfused and post-fixed for 48 h with 1% PFA and antiproteolytic cocktail (Melone et al., 2006); vibratome sections processed with mouse anti-PAG primary antibodies (1:250; Kaneko et al., 1987) and avidin–biotin. Quantitative data from two animals. (B) Co-localization studies of VGAT/PAG (left), VGAT/GADs (middle), and VGAT/GAT-1 (right). In all series, green codes for VGAT. VGAT/PAG series: tissue preparation as in A; anti-VGAT antibodies (1:500; Synaptic System; No. 131003); anti-PAG antibodies (1:250; Kaneko et al., 1987). VGAT/GADs series: tissue perfused and post-fixed for 24 h with 4% PFA; anti-VGAT antibodies (1:50; Synaptic System; No. 131011); anti-GAD65/67 antibodies (1:800; Millipore; AB1511). VGAT/GAT-1 series: tissue perfused and post-fixed for 2 h with 4% PFA; anti-VGAT antibodies (1:50; Synaptic System; No. 131011); anti-GAT-1 antibodies (1:500; Minelli et al., 1995). For studies employing PAG antibodies, fixative conditions were selected on the basis of the results of preliminary studies performed to ascertain optimal conditions. Layer II–III; parietal cortex. Percentage of VGAT puncta co-localized with PAG, GADs, and GAT-1 defined by analysis of 60–80 microscopic cortical fields (from layers I–VI) from two animals for each antigen. Scale bars: 0.25 μm (upper row) and 5 μm (lower row).
TCA cycle
Glutamate is synthesized in all cells from intermediates in the TCA cycle, and neurons are capable of de novo synthesis of Glu from TCA cycle intermediates, indicating that neuronal TCA cycle contribute significantly to Glu synthesis (Peng et al., 1993).
Glu transporters
Glutamate used for GABA synthesis could derive from the action of Glu transporters (GluT). EAAC1 is a neuronal GluT expressed also by GABAergic neurons (Rothstein et al., 1994; Conti et al., 1998a): in hippocampal slices from EAAC1 antisense-treated animals incubated in the presence of DON and gabaculine, newly synthesized [14C]GABA from [U–14C]Glu is lower in the presence of the Glu transport inhibitor DL-threo-β-hydroxy-aspartic acid; moreover, patch-clamp recordings of miniature IPSCs (mIPSCs) conducted in CA1 pyramidal neurons demonstrated a significant decrease in mIPSC amplitude, indicating decreased tonic inhibition (Sepkuty et al., 2002; see also Mathews and Diamond, 2003; Hartmann et al., 2008). The possible contribution of other GluTs (e.g., EAAT4 and GLT-1) to GABA synthesis remains to be verified.
GABA Transport
Four GABA transporters have been identified: GAT-1, GAT-2, GAT-3, and BGT-1 (Borden, 1996). GAT-1 is localized almost exclusively to axon terminals forming symmetric synaptic contacts and, in neocortex, to some astrocytic processes (Minelli et al., 1995; Conti et al., 1998b); GAT-2 is mainly expressed in the leptomeninges and in ependymal and choroid plexus cells, and to a lesser extent in neurons and astrocytes (Conti et al., 1999); GAT-3 is exclusively localized to distal astrocytic processes (Minelli et al., 1996); and BGT-1 is not localized to the CNS (Borden, 1996; Cherubini and Conti, 2001; Conti et al., 2004). Thus, the only GABA transporter that can contribute directly to GABA replenishment in terminals is GAT-1.
Are Mechanisms Subserving GABA Synthesis and Transport Expressed in all Gabaergic Terminals?
Crucial to the theme raised in this Perspective is whether the two mechanisms for GABA replenishment (synthesis and transport) are segregated in different terminals. Recent co-localization studies show that both GADs and GAT-1 are expressed in 90% of VGAT+ terminals (Figure 1B). Considering technical limitations, these values indicate that virtually all GABAergic terminals express both GADs and GAT-1 – i.e., that virtually all GABAergic neurons can synthesize GABA and take it up from extracellular milieu – and that GABAergic terminals cannot be differentiated on the basis of their mechanism of GABA replenishment.
Insights from KO Mice
GAT-1 deficient mice exhibit normal reproduction and life span, but have reduced body weight and higher body temperature fluctuations; they display motor disorders, including gait abnormality, 25–32 Hz tremor, reduced rotarod performance, and locomotor activity; they also display mild anxiety or nervousness (Jensen et al., 2003; Chiu et al., 2005).
In CA1 pyramidal cells, cerebellar granule and Purkinje cells, and neocortical layer II/III pyramids of KO mice, GABAA receptors (R)-mediated tonic conductance is significantly increased. Frequency, amplitude, and kinetics of spontaneous IPSCs are unchanged, whereas the decay time of evoked IPSCs is prolonged in KO mice (Jensen et al., 2003; Chiu et al., 2005; Bragina et al., 2008). In neocortex, high-frequency stimulation of GABAergic terminals induces large GABAAR-mediated inward currents associated with a reduction in amplitude and decay time of IPSCs; the recovery process is slower in KO than in WT mice (Bragina et al., 2008). Thus, GAT-1 has a prominent role in both tonic and phasic GABAAR-mediated inhibition, in particular during sustained neuronal activity (Bragina et al., 2008). The effects reported in KO mice are ascribable to impaired GABA uptake and subsequent reduction of GABA clearance from the synaptic cleft, a conclusion that would rule out any significant role of GAT-1 on presynaptic homeostasis.
However, analysis of mIPSCs in hippocampal pyramidal cells recorded in the presence of tetrodotoxin show that whereas mIPSCs have similar rise times, decay time constants, and amplitudes in WT and GAT-1 KO, their frequency in GAT-1 KO animals is reduced to about one-third of that recorded in control animals (Jensen et al., 2003; Figure 2A). Since reduced frequency of miniature events reflects presynaptic changes of quantal transmission (del Castillo and Katz, 1954; Clements, 1993), these findings indicate that in GAT-1 KO mice a presynaptic deficit does exist. Interestingly, simultaneous application of the GAT-1 inhibitor NNC-711 and of TBOA reduces mIPSC amplitudes, whereas high-frequency stimulation of CA1 afferents determines an activity-dependent increase in mIPSCs quantal size, that is produced by GAT-1-mediated GABA uptake and EAAC1-mediated Glu uptake in terminals of inhibitory interneurons (Hartmann et al., 2008).
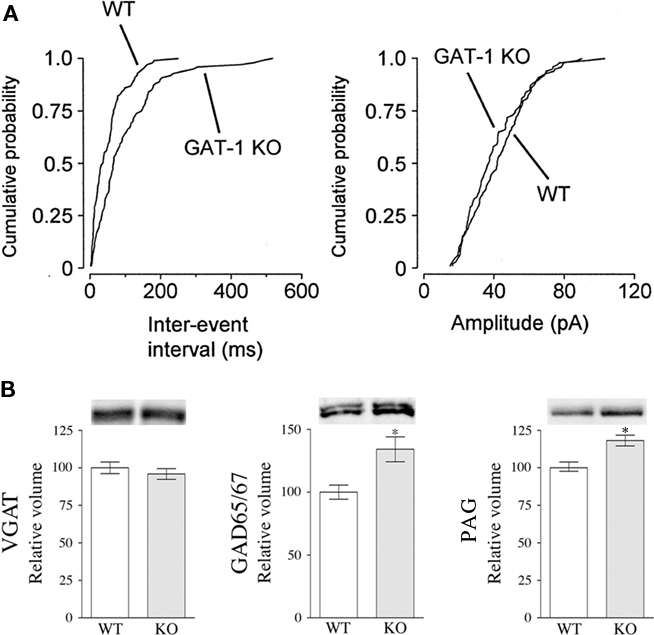
Figure 2. GAT-1 may contribute to presynaptic GABA storage and release. (A) mIPSC inter-event intervals (left) and amplitudes (right) in WT and GAT-1 KO hippocampal neurons. Note the longer inter-event intervals for GAT-1 KO mIPSCs compared to WT (p < 0.01; From Jensen et al., 2003). (B) Western blotting studies on cortical cellular extracts show increased expression of GAD65/67 and PAG, but not VGAT, in GAT-1 KO mice. Age of all animals was comparable. VGAT and GAD65/67 data from Bragina et al. (2008). For PAG analysis, homogenates (10 μg/μl) were exposed to anti-PAG antibodies (1:1000; Akiyama et al., 1990).
Moreover, western blotting studies from neocortex of WT and GAT-1 KO mice show that whereas VGAT levels are unchanged, GAD65/67 and PAG levels are increased by 35% and 18%, respectively, in GAT-1 KOs compared to WTs (Bragina et al., 2008; Figure 2B). These studies show that in conditions in which GAT-1-mediated GABA uptake is lacking, the biosynthetic machinery is up-regulated. Interestingly, although expressed in a limited number of GABAergic neurons, PAG expression increases significantly, indicating that the Glu–GABA/Gln cycle may play a not negligible role in GABA synthesis. Most importantly, by showing that in GAT-1 KO mice the levels of GADs, the fundamental enzyme for GABA biosynthesis, and PAG, an enzyme responsible for the generation of some of the Glu used for GABA synthesis, are up-regulated, these results suggest that the lack of GAT-1 reduces intraterminal GABA, which in turn triggers the increased expression of PAG and GAD. These observations strengthen the hypothesis that in physiological conditions GAT-1 exerts an influence on GABA homeostasis in GABAergic terminals.
Conclusion
In monoamine-releasing terminals, neurotransmitter transporters are crucial for presynaptic homeostasis, and this notion prompted us to verify whether GAT-1 plays a similar role in GABAergic terminals. Re-examination of existing literature from this perspective and some recent data gathered in our laboratory permits some initial conclusions: (1) in physiological conditions, GABA homeostasis in GABAergic terminals is dominated by the activity of GADs; conceivably, GABA transported by GAT-1 contributes to the cytosolic stores of GABA; (2) analysis of GAT-1 KO, besides demonstrating the effects of reduced clearance, reveals the existence of changes (altered mIPSCs frequency; GADs and PAG up-regulation) compatible with an impaired presynaptic function; (3) notwithstanding the paucity of data, it appears safe to conclude that in GABAergic terminals GAT-1 impacts on presynaptic homeostasis, though less so than DAT, NET, and SERT in monoamine-releasing terminals; (4) these conclusions open new and interesting problems; among these, the following appear of some interest: Where and how is GAT-1-derived GABA compartmentalized in GABAergic terminals? What is the relationship between GAT-1 transported GABA and vesicle filling? How dynamic relationships between GABA taken up by GAT-1 and GABA derived from other sources are regulated in diverse physiological conditions, including activity-dependent plasticity? What is, at both the mechanistic and functional level, the link between GAT-1 and mechanisms regulating GABA release? How does the presynaptic role of GAT-1 contribute to the pathophysiology of neuropsychiatric diseases such as epilepsy and schizophrenia?
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by MIUR and Università Politecnica delle Marche. We are indebted to the colleagues that collaborated in the original studies, NC Brecha (Los Angeles, CA, USA) and T Kaneko (Kyoto, Japan) for providing us with GAT-1 and PAG antibodies, and Henry Lester (Pasadena, CA, USA) for GAT-1 KO mice.
Footnote
- ^Space constraints prevent a detailed analysis of all brain regions; accordingly, most of the studies reported and the unpublished material presented here concern the cerebral cortex.
References
Akiyama, H., Kaneko, T., Mizuno, N., and McGeer, P. L. (1990). Distribution of phosphate-activated glutaminase in the human cerebral cortex. J. Comp. Neurol. 297, 239–252.
Borden, L. A. (1996). GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem. Int. 29, 335–356.
Boulland, J. L., Osen, K. K., Levy, L. M., Danbolt, N. C., Edwards, R. H., Storm-Mathisen, J., and Chaudhry, F. A. (2002). Cell-specific expression of the glutamine transporter SN1 suggests differences in dependence on the glutamine cycle. Eur. J. Neurosci. 15, 1615–1631.
Bradford, H. F., Ward, H. K., and Thanki, C. M. (1983). “Glutamine as neurotransmitter precursor: complementary studies in vivo and in vitro of the synthesis and release of transmitter glutamate and GABA,” in Glutamine, Glutamate and GABA in the Central Nervous Sytem, eds L. Hertz, E. Kvamme, E. G. McGeer, and A. Schousboe (New York: Alan R. Liss), 249–260.
Bragina, L., Marchionni, I., Omrani, A., Cozzi, A., Pellegrini-Giampietro, D. E., Cherubini, E., and Conti, F. (2008). GAT-1 regulates both tonic and phasic GABA(A) receptor-mediated inhibition in the cerebral cortex. J. Neurochem. 105, 1781–1793.
Chaudhry, F. A., Reimer, R. J., Bellocchio, E. E., Danbolt, N. C., Osen, K. K., Edwards, R. H., and Storm-Mathisen, J. (1998). The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J. Neurosci. 18, 9733–9750.
Chaudry, F. A., Reimer, R. J., and Edwards, R. H. (2002). The glutamine commute: take the N line and transfer to the A. J. Cell Biol. 157, 349–355.
Chaudry, F. A., Reimer, R. J., Krizaj, I., Barber, D., Storm-Mathisen, J., Copenhagen, D. R., and Edwards, R. H. (1999). Molecular analysis of system N suggests novel physiological roles in nitrogen metabolism and synaptic transmission. Cell 99, 769–780.
Cherubini, E., and Conti, F. (2001). Generating diversity at GABAergic synapses. Trends Neurosci. 24, 155–162.
Chiu, C. S., Brickley, S., Jensen, K., Southwell, A., Mckinney, S., Cull-Candy, S., Mody, I., and Lester, H. A. (2005). GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J. Neurosci. 25, 3234–3245.
Clements, J. D. (1993). “Presynaptic receptors and quantal models of synaptic transmission,” in Presynaptic Receptors in the Mammalian Brain, eds T. V. Dunwiddie and D. M. Lovinger (Boston: Birkhauser), 180–196.
Conti, F., DeBiasi, S., Minelli, A., Rothstein, J. D., and Melone, M. (1998a). EAAC1, a high-affinity glutamate transporter, is localized to astrocytes and GABAergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb. Cortex 8, 108–116.
Conti, F., Melone, M., De Biasi, S., Ducati, A., Minelli, A., and Brecha, N. C. (1998b). Neuronal and glial localization of GAT-1, a high-affinity GABA plasma membrane transporter, in the human cerebral cortex. J. Comp. Neurol. 396, 51–63.
Conti, F., and Melone, M. (2006). The glutamine commute: lost in the tube? Neurochem. Int. 48, 459–464.
Conti, F., Minelli, A., and Melone, M. (2004). GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res. Brain Res. Rev. 45, 196–212.
Conti, F., Vitellaro-Zuccarello, L., Barbaresi, P., Minelli, A., Brecha, N. C., and Melone, M. (1999). Neuronal, glial, and epithelial localization of g-aminobutyric acid transporter-2, a high-affinity g-aminobutyric acid plasma membrane transporter, in the cerebral cortex and neighboring structures. J. Comp. Neurol. 409, 482–494.
del Castillo, J., and Katz, B. (1954). Quantal components of the end-plate potential. J. Physiol. 124, 560–573.
Fricke, M. N., Jones-Davis, D. M., and Mathews, G. C. (2007). Glutamine uptake by System A transporters maintains neurotransmitter GABA synthesis and inhibitory synaptic transmission. J. Neurochem. 102, 1895–1904.
Hartmann, K., Bruehl, C., Golovko, T., and Draguhn, A. (2008). Fast homeostatic plasticity of inhibition via activity-dependent vesicular filling. PLoS ONE 3, e2979. doi: 10.1371/journal.pone.0002979
Hsu, C. C., Davis, K. M., Jin, H., Foos, T., Floor, E., Chen, W. Q., Tyburski, J. B., Yang, C. Y., Schloss, J. V., and Wu, J. Y. (2000). Association of L-glutamic acid decarboxylase to the 70-kDa heat shock protein as a potential anchoring mechanism to synaptic vesicles. J. Biol. Chem. 275, 20822–20828.
Jensen, K., Chiu, C. S., Sokolova, I., Lester, H. A., and Mody, I. (2003). GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J. Neurophysiol. 90, 2690–2701.
Jin, H., Wu, H., Osterhaus, G., Wei, J., Davis, K., Sha, D., Floor, E., Hsu, C. C., Kopke, R. D., and Wu, J. Y. (2003). Demonstration of functional coupling between gamma-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 100, 4293–4298.
Kaneko, T., Urade, Y., Watanabe, Y., and Mizuno, N. (1987). Production, characterization, and immunohistochemical application of monoclonal antibodies to glutaminase purified from rat brain. J. Neurosci. 7, 302–309.
Kvamme, E., Torgner, I. A., and Roberg, B. (2001). Kinetics and localization of brain phosphate activated glutaminase. J. Neurosci. Res. 66, 951–958.
Martinez-Hernandez, A., Bell, K. P., and Norenberg, M. D. (1977). Glutamine synthetase: glial localization in brain. Science 195, 1356–1358.
Mathews, G. C., and Diamond, J. S. (2003). Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J. Neurosci. 23, 2040–2048.
McIntire, S. L., Reimer, R. J., Schuske, K., Edwards, R. H., and Jorgensen, E. M. (1997). Identification and characterization of the vesicular GABA transporter. Nature 389, 870–876.
Melone, M., Quagliano, F., Barbaresi, P., Varoqui, H., Erickson, J. D., and Conti, F. (2004). Localization of the glutamine transporter SNAT1 in rat cerebral cortex and neighboring structures, with a note on its localization in human cortex. Cereb. Cortex 14, 562–574.
Melone, M., Varoqui, H., Erickson, J. D., and Conti, F. (2006). Localization of the amino acid transporter SNAT2 in the cerebral cortex. Neuroscience 140, 281–292.
Minelli, A., Alonso-Nanclares, L., Edwards, R. H., DeFelipe, J., and Conti, F. (2003). Postnatal development of the vesicular GABA transporter in rat cerebral cortex. Neuroscience 117, 337–346.
Minelli, A., Brecha, N. C., Karschin, C., DeBiasi, S., and Conti, F. (1995). GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci. 15, 7734–7746.
Minelli, A., DeBiasi, S., Brecha, N. C., and Conti, F. (1996). GAT-3, a high affinity GABA plasma membrane transporter, is localized exclusively to astrocytic processes in the cerebral cortex. J. Neurosci. 16, 6255–6264.
Peng, L., Hertz, L., Huang, R., Sonnewald, U., Petersen, S. B., Westergaard, N., Larsson, O., and Schousboe, A. (1993). Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev. Neurosci. 15, 367–377.
Roberts, E., and Frankel, S. (1950). γ-Aminobutyric acid in brain: its formation from glutamic acid. J. Biol. Chem. 187, 55–63.
Roberts, E., and Frankel, S. (1951). Glutamic acid decarboxylase in brain. J. Biol. Chem. 188, 789–795.
Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., Nash, N., and Kuncl, R. W. (1994). Localization of neuronal and glial glutamate transporters. Neuron 13, 713–725.
Sepkuty, J. P., Cohen, A. S., Eccles, C., Rafiq, A., Behar, K., Ganel, R., Coulter, D. A., and Rothstein, J. D. (2002). A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J. Neurosci. 22, 6372–6379.
Soghomonian, J. J., and Martin, D. L. (1998). Two isoforms of glutamate decarboxylase: why? Trends Pharmacol. Sci. 19, 500–505.
Solimena, M., Aggujaro, D., Muntzel, C., Dirkx, R. Jr., Butler, M., De Camilli, P., and Hayday, A. (1993). Association of GAD-65, but not of GAD-67, with the Golgi complex of transfected Chinese hamster ovary cells mediated by the N-terminal region. Proc. Natl. Acad. Sci. U.S.A. 90, 3073–3077.
Sonnewald, U., Westergaard, N., Schousboe, A., Svendsen, J. S., Unsgard, G., and Petersen, S. B. (1993). Direct demonstration by [13C]NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem. Int. 22, 19–29.
Keywords: GABA, GABA transporters, GAT-1, mIPSCs, knock-out mice
Citation: Conti F, Melone M, Fattorini G, Bragina L and Ciappelloni S (2011) A role for GAT-1 in presynaptic GABA homeostasis? Front. Cell. Neurosci. 5:2. doi: 10.3389/fncel.2011.00002
Received: 16 March 2011;
Paper pending published: 24 March 2011;
Accepted: 25 March 2011;
Published online: 05 April 2011.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Michela Matteoli, University of Milano, ItalyNicholas Brecha, University of California Los Angeles School of Medicine, USA
Copyright: © 2011 Conti, Melone, Fattorini, Bragina and Ciappelloni. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Fiorenzo Conti, Dipartimento di Neuroscienze, Sezione di Fisiologia, Università Politecnica delle Marche, Via Tronto 10/A, Torrette di Ancona, I-60020 Ancona, Italy. e-mail:Zi5jb250aUB1bml2cG0uaXQ=