



- 1 Neurobiology Sector and IIT Unit, International School for Advanced Studies, Trieste, Italy
- 2 Spinal Person Injury Neurorehabilitation Applied Laboratory, Istituto di Medicina Fisica e Riabilitazione, Udine, Italy
Understanding the pathophysiological changes triggered by an acute spinal cord injury is a primary goal to prevent and treat chronic disability with a mechanism-based approach. After the primary phase of rapid cell death at the injury site, secondary damage occurs via autodestruction of unscathed tissue through complex cell-death mechanisms that comprise caspase-dependent and caspase-independent pathways. To devise novel neuroprotective strategies to restore locomotion, it is, therefore, necessary to focus on the death mechanisms of neurons and glia within spinal locomotor networks. To this end, the availability of in vitro preparations of the rodent spinal cord capable of expressing locomotor-like oscillatory patterns recorded electrophysiologically from motoneuron pools offers the novel opportunity to correlate locomotor network function with molecular and histological changes long after an acute experimental lesion. Distinct forms of damage to the in vitro spinal cord, namely excitotoxic stimulation or severe metabolic perturbation (with oxidative stress, hypoxia/aglycemia), can be applied with differential outcome in terms of cell types and functional loss. In either case, cell death is a delayed phenomenon developing over several hours. Neurons are more vulnerable to excitotoxicity and more resistant to metabolic perturbation, while the opposite holds true for glia. Neurons mainly die because of hyperactivation of poly(ADP-ribose) polymerase-1 (PARP-1) with subsequent DNA damage and mitochondrial energy collapse. Conversely, glial cells die predominantly by apoptosis. It is likely that early neuroprotection against acute spinal injury may require tailor-made drugs targeted to specific cell-death processes of certain cell types within the locomotor circuitry. Furthermore, comparison of network size and function before and after graded injury provides an estimate of the minimal network membership to express the locomotor program.
Introduction
The Scale of the Problem and Damage Quantification
Spinal cord injury (SCI) usually produces lifelong, devastating consequences and represents one of the most significant cause of mortality and disability worldwide (Rossignol et al., 2007; van den Berg et al., 2010). The mortality from acute SCI is between 48 and 79%, either at the time of the accident or on arrival at the hospital (Sekhon and Fehlings, 2001). The annual incidence of the survivors after traumatic SCI is of 15–40 cases per million population throughout the world, with an even higher incidence in developed countries (up to 53,4 per million; Sekhon and Fehlings, 2001; Rowland et al., 2008). Recent clinical data indicate that, in addition to typical traumatic causes of SCI (motor vehicle accidents, work accidents, community violence, recreational activities, war), there is growing etiopathogenetic importance for non-traumatic injuries (McKinley et al., 1999; van den Berg et al., 2010). The exact number of the non-traumatic SCI is difficult to determine because of their highly varied etiology, that implies different clinical settings for treatment. Non-traumatic lesions account for about 30–50% of spinal cord disorders and constitute a major risk factor for medical complications during rehabilitation (Nair et al., 2005). The most frequent causes of non-traumatic SCI are vertebral stenosis (54%; McKinley et al., 1999) and spine tumors (26%; Nair et al., 2005), while other causes are vascular, inflammatory, infective, degenerative, genetic, and metabolic diseases. Incidence and demographics of traumatic and non-traumatic SCI are very different: while traumatic SCI mostly occurs in young males (traumatic SCI is four times more common in men than in women), gender distribution is more equal in non-traumatic SCIs that are more often observed in the elderly (Sekhon and Fehlings, 2001). It is noteworthy that most animal models for experimental SCI are oriented to study SCI of traumatic origin.
No matter of its origin, the SCI can be complete, or more often incomplete when a degree of sensory–motor function below the level of spinal cord trauma remains (Dzidic and Moslavac, 1997). Non-traumatic SCIs are usually incomplete (McKinley et al., 1999; van den Berg et al., 2010). Understanding the pathological evolution and the potential recovery is complicated by the frequent assignment of patients with complete or incomplete deficits to the same treatment group, and that few animal studies examined the same pattern of injury encountered in man (Amar, 2007). The complex mechanisms regarding the pathology of incomplete lesions remain poorly understood because local ischemia with associated metabolic dysfunction is technically difficult to reproduce in animal models without compromising other tissues and organs and with the confounding influence of general anesthetics.
Lesion Amplification through Secondary Damage: An Avalanche Effect
If the mantra for stroke treatment is “time is brain” (Hill and Hachinski, 1998), it is most likely that “time is spinal cord” for acute SCI as well. Thus, great effort has been made to understand the pathophysiological changes underlying SCI in the hope of developing neuroprotective strategies and preventing disability. Despite its heterogeneous causes, SCI evolves into secondary damage affecting apparently spared areas, magnifying the disability and amplifying neurodegeneration (Rossignol et al., 2007). The challenge is to clarify why and when such a damage occurs.
The early stage of secondary injury is thought to start with excitotoxic damage due to massive release of glutamate together with a pathological cascade comprising nitric oxide, free oxygen radicals, and metabolic dysfunction due to ischemia/hypoxia, energy store collapse, acidosis, and edema triggered by loss of vascular tone autoregulation (Dumont et al., 2001; Norenberg et al., 2004; Amar, 2007). The secondary injury starts minutes after primary insult and can lasts up to weeks after injury. Extracellular glutamate levels are known to increase transiently within the first 3 h after SCI, with a likely second wave of glutamate release 2–3 days after injury (Park et al., 2004), probably due to delayed myelin destruction that compromises nearby axon integrity.
The events of the secondary injury phase can be divided into early and delayed stages (Rowland et al., 2008). The early phase comprises vasogenic and cytotoxic edema, necrosis, excitotoxicity, early demyelination, and systemic events like hypotension (2–48 h). Later, macrophage infiltration and initiation of glial scar occur. Within 2 weeks/6 months, glial scarring continues together with intraspinal cyst formation. Even later, profound pathological changes affect spinal networks through Wallerian degeneration, demyelination, aberrant plasticity with circuit rewiring leading to dysfunction like chronic pain and spasticity (Rowland et al., 2008).
The molecular cell-death pathways of SCI (and their effectors) remain largely elusive (Park et al., 2004). This condition makes it difficult to identify the best time window for satisfactory treatment of acute SCI with the aim of limiting (or even preventing) secondary damage. Nonetheless, the consensus is that the time to introduce effective neuroprotective strategies after SCI is short, probably restricted to the first hours after injury (Fehlings et al., 2001), in analogy with a similar situation for the brain (Hill and Hachinski, 1998). Studies of brain and spinal injuries support the theory that the central nervous system (CNS) responds to lesion in an archetypal fashion, regardless of the insult, and that similar pathological pathways and cell-death mechanisms may operate in the brain and spinal cord (Amar, 2007).
Protection of Locomotor Networks Needs Understanding Network Topography
Since paralysis (or paresis) is a hallmark of SCI, it is of particular interest to direct neuroprotective strategies to the circuits responsible for locomotion which is driven by intrinsic spinal networks, collectively called central pattern generator (CPG; Grillner et al., 1998; Heckmann et al., 2005; Kiehn, 2006; Boulenguez and Vinay, 2009). In vitro preparations of the spinal cord readily generate electrically oscillatory cycles (recorded from ventral roots) which possess all the hallmarks of locomotor patterns. Nonetheless, the absence of limbs makes necessary to refer to this pattern as fictive locomotion. Although the full membership of the locomotor CPG remains unclear, mouse genetics have provided substantial advances in the classification of propriospinal neurons involved in locomotion (Kiehn, 2006; Brownstone and Wilson, 2008; Grillner and Jessell, 2009; Ziskind-Conhaim et al., 2010).
Targeted neuroprotection might produce successful functional outcome as long as the minimal number of neurons (or other cells) essential for locomotor patterns would survive. In addition, any investigation to repair or rebuild locomotor networks needs reliable estimates of the minimal cell membership capable of producing the locomotor pattern. Otherwise, it would be like trying to rebuild a house damaged by a natural disaster without first calculating how many bricks and how much mortar should be ordered and how much of these materials must be actually employed to support the structure. It is essential to do a “quantitative survey” of undamaged and damaged spinal locomotor networks to supply precise information about the cells to which any neuroprotective strategy should be aimed.
The present review will focus on the molecular mechanisms involved in the death of cells comprising and controlling spinal locomotor networks after acute experimental injury. To this end, two in vitro animal SCI models have been used, namely the neonatal rat spinal cord preparation and rat organotypic slices (Taccola et al., 2008, 2010; Mazzone et al., 2010). These models allow testing novel experimental paradigms to mimic the consequences of strong or weak lesions (including those of non-traumatic origin or non-complete) taking as end point the functional activity of locomotor networks in relation to surviving cells, and investigating the processes that led to cell loss. By combining new data about cell-death mechanisms and neuronal networks involved in the control of locomotion during or shortly after acute SCI, it might be possible to provide proof-of-principle that neuroprotection is feasible and to design new therapeutic strategies to be tested on animal models in vivo with the ultimate goal to combat the consequences of SCI at the earliest possible stage. Furthermore, detailed analysis of surviving cells might help to delineate the minimal requirements (“network membership”) essential for the locomotor program. This information might be useful in future if a decision between rebuilding and replacing should ever be taken. Of course, the long-term functional outcome after SCI depends not only on the secondary lesion extent, but also on altered neuronal excitability due to upregulation of the persistent sodium current (Li and Bennett, 2003;ElBasiouny et al., 2010) that is important to control locomotor patterns (Tazerart et al., 2007; Zhong et al., 2007; Ziskind-Conhaim et al., 2008), and to the changes in synaptic inhibition caused by derailed chloride transport (Boulenguez et al., 2010). Both phenomena may contribute to the onset of spasticity. Furthermore, changes in extracellular Mg2+ are important to determine the functional outcome of the locomotor network (Margaryan et al., 2009), a subject often neglected by previous studies.
In vitro Spinal Cord Models to Study Cell-Death Mechanisms after SCI
In addition to in vivo animal models of SCI (Onifer et al., 2007), in vitro models (cell cultures, organotypic cultures, and isolated spinal cord preparations) can supply useful data because they simplify the complexity of in vivo SCI pathophysiology, and can point to the identification of specific injury processes without interference by general anesthesia or blood pressure changes.
Primary cultures from spinal cord tissue (Seybold and Abrahams, 2004; Taylor et al., 2007) have been used in a wide range of experimental conditions, especially to reproduce in vitro excitotoxicity (Vandenberghe et al., 1998; Van Den et al., 2000) and ischemia (Kaushal and Schlichter, 2008). Nonetheless, these models suffer from the intrinsic disadvantage of unknown influences caused by the complex culturing media (Silani et al., 2000). Moreover, it is also difficult to precisely control the cell microenvironment, especially important for axon growth and regeneration (Abu-Rub et al., 2010). Likewise, acute slices of the spinal cord can be used for functional studies even though they have limited viability in vitro, and may pose barriers to drug diffusion (Lossi et al., 2009). In all these cases it is impossible to ascertain if there had been any specific damage to locomotor networks.
Neonatal Rat Spinal Cord Preparation
A novel model of in vitro SCI to investigate the rapid evolution of early secondary damage takes, as outcome, the operation of locomotor networks in relation to cell survival in the isolated spinal cord of the neonatal rat (Taccola et al., 2008). Despite the intrinsic limitation of this preparation (absence of immune system responses, lack of vascular supply, neonatal age), this model has the advantage of correlating the functional outcome of injury (with >24 h monitoring of locomotor-like activity, termed fictive locomotion) to the number, type, and topography of damaged or dead cells. Thus, it becomes feasible to unveil damage mechanisms because it does not introduce an “artificial neuroprotection” by general anesthesia at the time of injury, yet it retains cellular connections, networks, and activities, including fictive locomotion. The use of a neonatal preparation can also shed light on the issue of child spinal injury (Vitale et al., 2006; Achildi et al., 2007) characterized by high mortality and prevalence of cervical location (Cirak et al., 2004). The in vitro spinal cord model can be employed to produce (and compare) distinct forms of pharmacological spinal damage, namely strong excitotoxic stimulation presumed to arise from a trauma-like condition, or severe metabolic perturbation. These pathological events are believed to occur during the secondary phase of SCI (Park et al., 2004). Furthermore, the model can be applied to flexible protocols that comprise non-traumatic SCI, or incomplete SCI (Taccola et al., 2008, 2010; Kuzhandaivel et al., 2010a, b). One important conclusion arising from these studies is the diversity of cell-death pathways involved in neuronal and glial damage after SCI, as discussed in Sections “Apoptosis is Responsible for Glial Cell Death after Hypoxic/Ischemic Perturbation” and “Neuronal Cell Death after Excitotoxic Insult is Due to Parthanatos.”
Organotypic Cell Culture as Reliable in vitro Model to Study Cell Death
Organotypic cultures are an important tool to study developmental as well as chronic changes in network structure and function. This technique originally established by Gahwiler (1981) for brain culture slices, currently utilizes different tissue sources (embryonic, juvenile, or adult) (Spenger et al., 1991; Stoppini et al., 1991; Streit et al., 1991; Noraberg, 2004; Livera et al., 2006; Lossi et al., 2009). These studies have shown a clear correlation between in vivo and in vitro development (Livera et al., 2006). The common characteristic and the main advantage of the organotypic system is the maintenance of the basic cytoarchitecture of the in vivo tissue, retaining, for example, synaptic connectivity while supplying ready experimental access to structure and function. Spinal cord organotypic cultures are a good example of this approach (Spenger et al., 1991; Streit et al., 1991) as they maintain the dorsal–ventral orientation of spinal segments and fundamental properties of network dynamics related to distinct spinal regions in a bidemensional plane (see Figure 1A). Furthermore, they allow long-term studies in which plastic changes in network properties can be explored in relation to changes in the local environment (Sibilla and Ballerini, 2009), far in excess of the standard survival time of the rodent isolated spinal cord. Thus, organotypic cultures of the spinal cord are useful to investigate experimental neuronal lesions (Krassioukov et al., 2002; Guzman-Lenis et al., 2009; Mazzone et al., 2010), even though these cultures cannot generate locomotor-like patterns, and cannot readily relate molecular changes to complex network function.
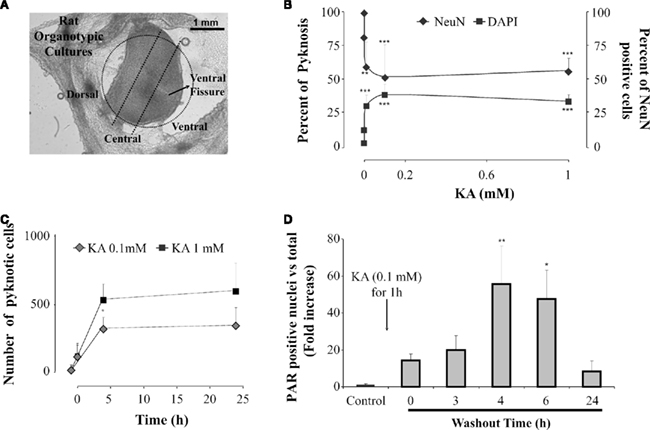
Figure 1. Neuronal damage induced by kainate on spinal cord organotypic cultures is a PARP-1 dependent process. (A) View of representative organotypic slice in which ventral,central, and dorsal regions, outlined with a dotted line, can be identified for cell counting. (B) Plots showing the percent of cells with pyknosis (left; filled squares) or percent of neurons (NeuN-positive cells; right; filled diamonds), in the central region of the slice, after 1 h application of various kainate (KA) concentrations. Data were collected after 24 h kainate washout from at least three different experiments, n = 4–12; **P < 0.01, ***P < 0.001 vs control (0 mM KA). (C) Plot of the number of cells, in the central region, showing pyknosis at various times points after kainate washout (0 h) at the concentration of 0.1 mM (gray diamonds) or 1 mM (filled squares). Data points before 0 time refer to pyknosis in control values (21 ± 23 cells). Note emergence of pyknosis at 4 h which is significantly (P = 0.0005) larger after 1 mM kainate. Average data are from three experiments, each one of them run in duplicate. (D) Histograms showing fold increase (with respect to control) in the number of PAR positive nuclei at various times after washing out kainate (0.1 mM). The data are from three experiments (n = 6); *P < 0.05, **P < 0.01 vs control.
Excitotoxicity and Metabolic Perturbation Damage Spinal Locomotor Networks through Different Cell-Death Pathways
Distinct Cell-Death Pathways Emerge after Experimental SCI
Understanding the complexity of cell-death mechanisms after SCI and their correct classification may be extremely important for therapeutic implications (Galluzzi et al., 2007). Cell death represents a highly heterogeneous process that can follow the activation of diverse, sometimes overlapping, and not fully understood biochemical cascades, manifesting with different morphological features (Kroemer et al., 2009). The traditional classification of cell death in apoptosis (programmed cell death dependent on caspase activation) and necrosis (non-programmed cell death) is now obsolete because necrosis can actually be programmed, apoptosis can occur even without caspase activation, and new types of cell-death processes have been reported (Galluzzi et al., 2007). In this review, the term apoptosis refers to a process with specific cell morphology characterized by round shape, cytoplasmic and nuclear condensation (pyknosis), DNA fragmentation, with minimal modifications of organelles, and preservation of plasma membrane until the final stage of destruction by phagocytes (Kerr et al., 1972; Kroemer et al., 2009). Activation of caspase, Apoptotic protease activating factor 1 (APAF-1), and of mitochondrial proteins termed the Bcl-2, are frequent, yet not essential markers for apoptosis (Yuan et al., 2003). For the purpose of this review, apoptosis will be identified only when shown to be caspase-dependent.
Extensive generation of poly-ADP-ribose (PAR) by hyperactivation of the PARP-1 enzyme has recently been proposed as a novel programmed cell death, termed “parthanatos” (Andrabi et al., 2006, 2008). It is thought that, in the attempt to repair strong DNA damage, PARP-1 catalyzes conversion of NAD+ to PAR polymers with loss of intracellular ATP and consequent energy depletion (Berger, 1985; Zhang et al., 1995). This mechanism may overload the energy handling process of mitochondria (Virag et al., 1998) and release the apoptosis inducing factor (AIF), a protein which enters into the nucleus to initiate lethal nuclear condensation (Yu et al., 2002). Additionally, PAR directly stimulates mitochondrial release of AIF to enhance the whole destructive process (Andrabi et al., 2006). It is noteworthy that the term AIF was first used when the cell-death process in which it was discovered was called caspase-independent apoptosis (Susin et al., 1999; Joza et al., 2009). Only later, it became clear that AIF was an intracellular effector of PARP-1 hyperactivity and a mediator of parthanatos (Yu et al., 2002; Joza et al., 2009). The traditional usage of the term AIF should not, therefore, imply that classical apoptosis is produced by this factor.
Necrosis is a process of cell death lacking the features of apoptosis or autophagy, the latter characterized by lack of chromatin condensation and by massive vacuolization of the cytoplasm. Characteristic morphological aspects of necrosis are cytoplasmic swelling, rupture of plasma membrane, swelling of cytoplasmic organelles, and moderate chromatin condensation (Galluzzi et al., 2007). Necrosis has been traditionally implicated as an early mechanism of cell death after injury (Golstein and Kroemer, 2007).
In in vivo and in vitro models of SCI, heterogenous cell-death mechanisms have been reported for the loss of spinal cells (Table A1 in Appendix). Because of the problems related to the exact terminology and classification of different cell-death pathways (Kroemer et al., 2009), it is difficult to compare results from different SCI studies and draw general conclusions. For example, not infrequently TUNEL staining (detecting DNA fragmentation) is taken as synonymous of apoptosis, even though DNA fragmentation can be a caspase-independent phenomenon (Belmokhtar et al., 2001; Zhang and Bhavnani, 2006) or cells can undergo apoptotic death without significant DNA degradation (Widlak and Garrard, 2009). Thus, results from different studies listed in Table A1 in Appendix, rely on the identification of the cell-death mechanism provided by the authors, as well as the assay to detect it.
Perusal of the older literature indicates that, after SCI, early cell death is predominantly by necrosis, followed by a continuum of necrotic and apoptotic mechanisms (Liu et al., 1997; Baptiste and Fehlings, 2006). Later, different cell-death pathways have been proposed to mediate excitotoxicity, including caspase-dependent (Beattie et al., 2000; Yu et al., 2009) and caspase-independent pathways (Mandir et al., 2000; Yuan et al., 2003; Cho and Toledo-Pereyra, 2008), or sharing characteristics of both apoptosis and necrosis (Tan et al., 1998).
Parthanatos has recently been considered important for spinal cord neuronal cell-death in vitro (Scott et al., 2004) and in vivo (Genovese and Cuzzocrea, 2008; Wu et al., 2009) conditions. Furthermore, cell-death mechanisms involving calpain (Ray et al., 2003) or autophagy (Kanno et al., 2009) have been proposed to cause cell death after SCI (Table A1 in Appendix).
The time-scale through which cell death occurs is important. Even though axons and neurofilaments degrade as early as 15 min after experimental SCI (Park et al., 2004), axonal degeneration is a delayed process as loss of residual axonal tracts can continue for weeks (Wallerian degeneration), together with slow decline in impulse propagation of such fibers (Arvanian et al., 2009). Programmed cell death of white matter glial cells (observed with TUNEL staining) occurs in periodic waves with maximal intensity in the white matter tracts 1 week after injury (Shuman et al., 1997; Springer et al., 1999; Park et al., 2004). In contrast to oligodendrocyte cell loss, astrocytes survive and even proliferate after SCI by a process termed “reactive astrogliosis” (Park et al., 2004). In adult rats after traumatic SCI (Liu et al., 1997), TUNEL-positive glial cells appear between 4 h and 14 days after injury, with maximum presence within the lesion area at 24 h. Neuronal degeneration is faster as TUNEL-positive neurons are seen 4–24 h after injury, with a peak at 8 h. Motoneurons are particularly vulnerable to calcium-dependent glutamate excitotoxicity, because they lack certain calcium binding proteins, such as calbindinin-D(28k) and parvalbumin (Dekkers et al., 2004), and remain unprotected from the consequences of calcium overload. In the rabbit spinal cord most motoneurons survive for 2 days after ischemia, and then disappear via apoptosis (Hayashi et al., 1998) and autophagy (Baba et al., 2009) as result of co-activation of cell survival and cell-death pathways (Sakurai et al., 2003).
Different Experimental Paradigms to Mimic Excitotoxicity and Metabolic Perturbation
To clarify if excitotoxicity per se is sufficient to damage spinal networks responsible for locomotion and what is the contribution of metabolic perturbations to spinal cord damage, distinct protocols (based on the transient application of toxic solutions) have been developed. In fact, if one wishes to mimic clinical settings, it is necessary to consider that, after acute SCI, prompt hospital admission/treatment in intensive care involves correction of metabolic deficits, administration of neuroprotective agents, circulation support, and relief of any compressive lesion. In the best circumstances, this approach implies a delay of about 1 h after the primary injury (Rowland et al., 2008). Hence, recent experimental protocols were based on 1 h administration of a toxic solution with follow-up under optimal metabolic conditions in vitro for the subsequent 24 h. In this way, morphological and biochemical data can be correlated with the electrophysiological activity of spinal locomotor networks.
The protocol for excitotoxicity relies on kainate (Taccola et al., 2008), a potent glutamate receptor agonist which is not subjected to metabolic tissue destruction, and is not a substrate for glutamate transporters (Coyle, 1987). The cellular effects of kainate are mediated by a complex family of receptors, of which at least six forms are currently known to be expressed by the CNS at pre and postsynaptic level (Traynelis et al., 2010). In view of its strong depolarizing action on neurons, kainate produces excitotoxicity in a large range of animal models (Ben-Ari and Cossart, 2000) also indirectly since it releases glutamate (and other neurotransmitters) in addition to its direct excitatory effect. Thus, kainate strongly and persistently depolarizes rat spinal neurons in vitro (Taccola et al., 2008), and elicits a robust release of endogenous glutamate as measured with electrochemical detection (Mazzone and Nistri, 2011a), making it a suitable tool for evoking a sustained excitotoxic insult.
Another protocol is based on a type of dysmetabolic lesion evoked by a toxic solution (termed pathological medium; PM) that comprises many deleterious substances and conditions (NO, H2O2, low Mg2+, acidosis, aglycemia, hypoxia, edema) resembling the ones occurring shortly after acute SCI (Taccola et al., 2008).
By distinct or combined application of such protocols, different patterns of cell death and different changes in locomotor network activities emerge (Taccola et al., 2008, 2010; Kuzhandaivel et al., 2010a,2010b). Interestingly, there is a very narrow range of kainate concentrations (1–10 μM) through which locomotor network activity is still possible (albeit slower). Kainate (50 μM) is already sufficient to induce an irreversible loss of fictive locomotion even if spinal reflexes persist (Mazzone et al., 2010). These data confirm, with an in vitro model, that reflex amplitude is a poor predictor of locomotor function in line with clinical experience (Dietz et al., 1997; Hubli et al., 2010). The extent of neuronal damage by kainate is poorly related to this drug concentration as indicated by the occurrence of pyknosis and neuronal loss following a wide range (1–1000 μM) of kainate concentrations (Figure 1B). In fact, the main difference produced by changing doses is the speed of neuronal loss (Figure 1C; Mazzone et al., 2010): this observation may be important because it suggests that any attempt to arrest damage should start as early as possible and has a better outcome if the damage is treated when it is still limited. This notion is further supported by the demonstration that kainate toxicity does not imply nearly global neuronal loss since the number of surviving neurons is usually larger that the number of dead ones (Taccola et al., 2008; Mazzone et al., 2010). Furthermore, 24 h after kainate application, surviving networks are metabolically competent (Mazzone et al., 2010), and functionally active as they generate disinhibited bursting (although no fictive locomotion; Taccola et al., 2008).
Apoptosis is Responsible for Glial Cell Death after Hypoxic/Ischemic Perturbation
Apoptosis is a physiological process of cell elimination during normal development of the gray and white matter of the spinal cord (De Louw et al., 2002). After SCI, early neuronal cell death by apoptosis at the injury site is infrequent (Shuman et al., 1997; Emery et al., 1998; Li et al., 1999; Springer et al., 1999), because this process is mainly responsible for the delayed death of the oligodendrocytes locally (Li et al., 1999) and remotely (Li et al., 1999; Springer et al., 1999). It is interesting that also in other models of neurodegeneration, like in Alzheimer’s disease, status epilepticus, or brain ischemia, apoptotic neuronal death is rare, as apoptosis mainly involves glial cells (microglia, oligodendroglia, and astrocytes; Jellinger and Stadelmann, 2000; Shibata et al., 2000; Narkilahti et al., 2003).
In the neonatal rat spinal cord in vitro, pyknosis appears in neurons and, especially, in glia during first 24 h after hypoxic-dysmetabolic perturbation (Figure 2). This readily observable change in nuclear morphology is the result of chromatin condensation and can be either nucleolytic (with DNA fragmentation typical of apoptosis) or anucleolytic (without DNA fragmentation; see Burgoyne, 1999). As shown in Figure 2A, pyknosis is strongly found in the ventro-lateral white matter reaching a peak after 4–8 h from washout of PM application with low occurrence in other areas. Pyknosis is preceded by DNA fragmentation shown, already 2 h later, as positivity to phospho-histone H2A.X (Figure 2B; Widlak and Garrard, 2009; Kuzhandaivel et al., 2010a) and DNA laddering (Figure 2C; Loo and Rillema, 1998). Caspase-3 mediated apoptosis is largely detected during the following 24 h to reach a peak of approximately 60% pyknotic cells in the ventro-lateral white matter (Figure 2D). From a functional point of view, despite strong lesion of white matter elements, locomotor networks retain their activity even if the cycle period of locomotor patterns is clearly slower (Taccola et al., 2008). Activation of locomotor networks by dorsal afferent stimuli becomes, however, impossible (Taccola et al., 2008): thus, in this condition, despite the retained intrinsic ability to generate locomotor patterns, the continuous sensory feedback required to support locomotion (Barbeau et al., 1999) is lost. After PM treatment, morphological changes characteristic of necrotic death (gain in cell volume, organelle swelling, and disorganized dismantling of intracellular contents; Galluzzi et al., 2007) have not been routinely observed.
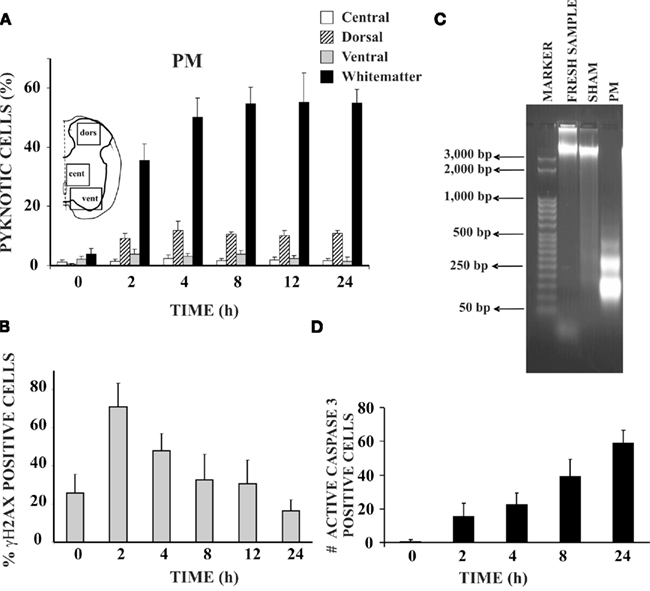
Figure 2. Hypoxic-dysmetabolic insult induces apoptosis in spinal cord in vitro. (A) Histograms plot percent of pyknosis (with respect to global number of DAPI positive cells) at various time points after washout (0 h) of PM. For each time point n = 3 spinal cords. Inset shows the schematic representation of the areas sampled for the cell count based on DAPI staining. (B) Histograms demonstrating percent occurrence (with respect to global DAPI stained cells) of γH2AX positive cells (n = 3) at different time points after 1 h of PM application. (C) Agarose gel electrophoresis of DNA samples from control spinal cords (freshly dissected, lane 2), from sham spinal cords (kept in vitro for 24 h in Krebs solution, lane 3), or PM treated spinal cords (1 h PM treatment followed by 24 h recovery in Krebs solution, lane 4). Note DNA laddering (due to internucleosomal DNA fragmentation) in the PM treated sample only (n = 3 for each sample). Lane 1 shows DNA ladder marker (50 bp steps). (D) Histograms indicate number of active caspase-3 positive cells in the white matter of the PM treated spinal cords (average of six slices from two spinal cords) at different time points after 1 h of PM application.
Neuronal Cell Death after Excitotoxic Insult is Due to Parthanatos
In addition to the strong vulnerability of motoneurons (Mazzone et al., 2010), Figure 3A shows that the largest cell death after kainate is observed in the dorsal gray matter (which contains the highest density of kainate receptors; Tolle et al., 1993), leaving the white matter mostly intact. This phenomenon is accompanied by PARP-1 overexpression that peaks 8 h after washout of kainate (Figure 3B) and release of AIF (Figure 3B).
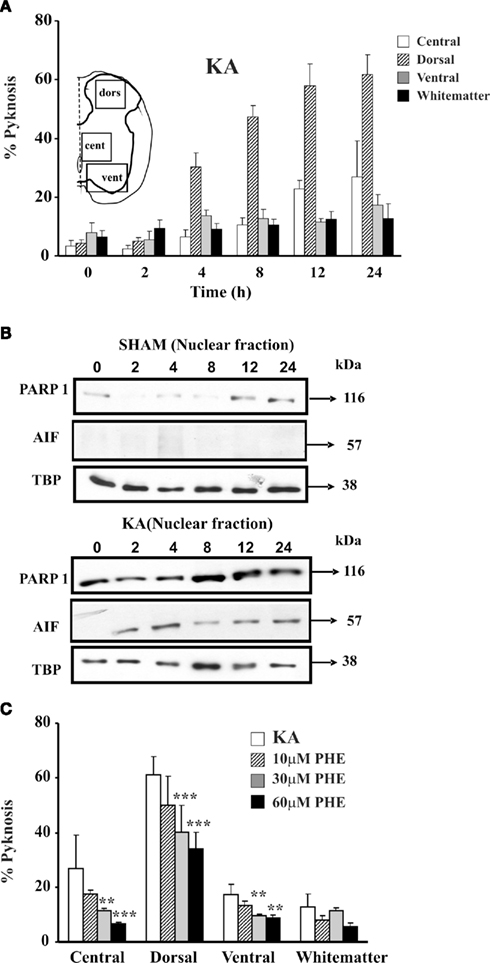
Figure 3. Kainate mediated excitotoxicity evokes PARP-1 dependent cell death (Parthanatos) in spinal cord in vitro. (A) Histograms plot percent occurrence of pyknosis detected at various time points after washout (0 h) of kainate (1 mM; 1 h application; KA). Inset shows the schematic representation of the areas sampled for the cell count based on DAPI staining. For each time point n = 3 spinal cords. (B) Example of Western immunoblotting of spinal cord samples obtained from sham (upper panels) or kainate treated (lower panels) preparations, and tested with PARP-1 or AIF specific antibody. In kainate treated spinal cords there is increased PARP-1 immunoreactivity, with early appearance immediately after kainate treatment (0 h) and rising up to 24 h after. AIF immunoreactivity appears in the nuclear fraction 2 h after kainate treatment. Nuclear loading assessed with TBP. (C) Histograms plot percent of pyknosis occurrence detected in different spinal cord regions 24 h after kainate treatment alone or followed by treatment with PARP-1 inhibitor PHE (three different concentrations). PHE significantly reduces (**P < 0.01; ***P < 0.001) the number of pyknotic cells in a dose-dependent manner in all four spinal cord regions. Pyknosis is normalized with respect to the total number of DAPI sensitive cells. For each time point n = 3 spinal cords.
The involvement of the PARP-1-dependent cell death after SCI was thought to be triggered by overproduction of nitric oxide and reactive oxygen species (Scott et al., 2004; Genovese et al., 2005; Wu et al., 2007; Genovese and Cuzzocrea, 2008). A similar phenomenon has been proposed to occur after ischemic and traumatic brain injury (Eliasson et al., 1997; Endres et al., 1997; Meli et al., 2003; David et al., 2009), including perinatal brain injury (Hagberg et al., 2004). Although the morphology of neuronal pyknosis after kainate is similar to the one detected in the white matter after PM, all tests for apoptosis have been negative (TUNEL, phospho-histone H2A.X staining, DNA laddering, and caspase-3; Kuzhandaivel et al., 2010b). Conversely, extensive PAR immunoreactivity has been found in gray matter neurons after kainate induced excitotoxicity to mediate translocation of mitochondrial AIF to the nucleus and cell death (Figures 1D,3B; Kuzhandaivel et al., 2010b). This effect becomes already apparent immediately after kainate washout with AIF nuclear translocation 2 h later, and correlates with the time- and dose-dependent onset of pyknosis (Mazzone et al., 2010).
Conversely, in PM treated preparations, modest PARP-1 activation (without extensive PAR generation) occurs as this enzyme is cleaved by active caspase-3 (Kuzhandaivel et al., 2010b). The apparent resistance of glial cells to kainate induced excitotoxicity has been already reported, for example, for the mature myelin basic protein-expressing oligodendrocytes (Rosenberg et al., 2003).
Electrophysiological studies indicate that, once the kainate concentration reaches 50 μM, fictive locomotor patterns are irreversibly lost within a few minutes from the start of the drug application (Taccola et al., 2008; Mazzone et al., 2010). Part of this functional loss is likely to be due to strong inactivation of voltage-gated conductances of network neurons due to the sustained depolarization. Nonetheless, network depolarization as well as endogenous glutamate release (Mazzone et al., 2010; Mazzone and Nistri, 2011a) subside at the end of kainate application, indicating that lack of locomotor patterns is not a merely functional deficit, but a structural damage of the spinal circuitry.
When PM and kainate are combined together, large-scale cell death in the gray and white matter appears with pyknosis as the primary morphological characteristic of damaged cells (Taccola et al., 2008). This combined application has been shown to be useful to produce focal lesions of the isolated spinal cord by restricting the administration of the toxic solution to a small number of segments with transverse barriers and testing its consequences on apparently unscathed segments (Taccola et al., 2010).
Figure 4 summarizes the principal cell-death pathways observed to mediate glial damage by PM (Figure 4A) or neuronal damage by kainate (Figure 4B). Apoptosis is the main process responsible for dysmetabolic lesion of glia via initial DNA damage, histone phosphorylation, and sequential activation of caspases, a phenomenon reinforced by further caspase-3 activation through damaged mitochondria and release of APAF-1 (Figure 4A). On the other hand, activation of glutamate receptors by kainate (Figure 4B) triggers through multiple processes including sustained elevation in intracellular free Ca2+, DNA damage which leads to hyperactivation of PARP-1, PAR production and mitochondrial damage with release of AIF and further nuclear damage.
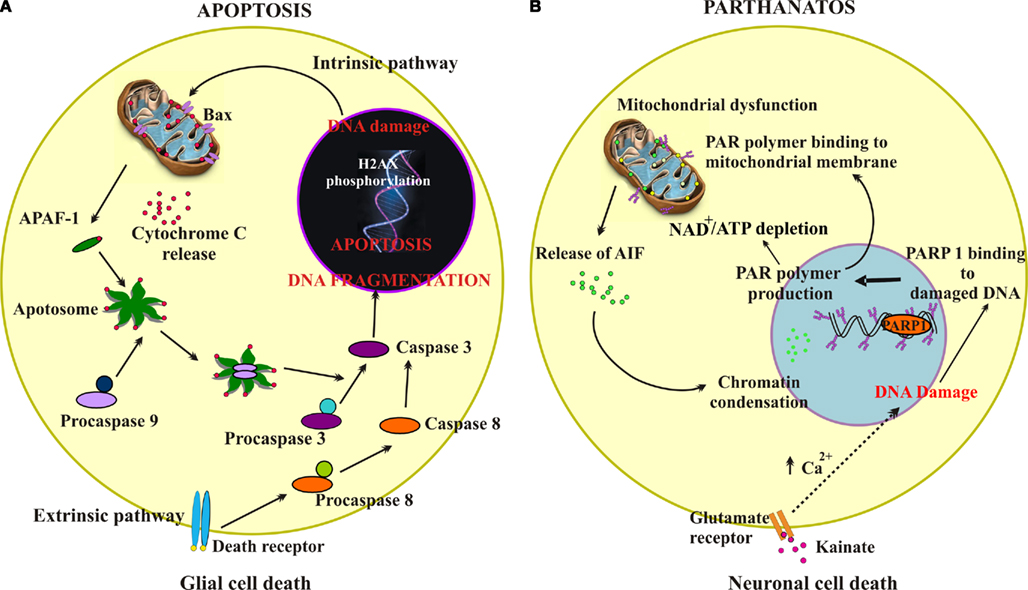
Figure 4. Schematic representation of apoptosis or parthanatos cell-death pathways. (A) Schematic representation demonstrating intrinsic and extrinsic pathways of apoptosis. The intrinsic pathway is initiated from within the cell usually in response to cellular signals resulting from DNA damage, hypoxia, or other types of severe cell stress. These signals stimulate proapoptotic proteins (e.g., Bax) in the mitochondrial membrane leading to mitochondrial membrane permeabilization, that allows translocation of cytochrome c from the mitochondrial intermembrane space to the cytosol. Cytochrome c binds the adaptor apoptotic protease activating factor-1 (APAF-1), forming a large multiprotein structure known as the apoptosome. The initiator caspase 9 is recruited into the apoptosome and activates the downstream effector caspases 3. The extrinsic pathway begins outside the cell through the activation of specific proapoptotic receptors on the cell surface by specific molecules known as proapoptotic ligands. Such a ligand binding induces receptor clustering and recruitment of the adaptor protein Fas-associated death domain and the initiator caspases 8 as procaspases, facilitating their autocatalytic processing and release into the cytoplasm where they activate the effector caspases 3, thereby converging on the intrinsic pathway. One of these pathways (or both) may operate in determining death of glial cells in the spinal cord subjected to PM application (dysmetabolic insult). (B) Schematic representation showing the parthanatos pathway mediated by PARP-1 hyperactivation. Following excitotoxicity (kainate and glutamate receptor overactivity), intracellular increase in free Ca2+ activates downstream cascades leading to peroxynitrite formation. This readily induces DNA damage, thereby activating PARP-1 that initiates the synthesis of PAR. PAR interacts with the mitochondrial membrane to change its membrane potential, thus allowing AIF translocation to the nucleus, a process leading cell death. An additional lethal mechanism may originate from excessive PARP-1 activation to deplete the NAD pool and cause severe ATP starvation and cell energy failure with the outcome of cell death.
The difference in targeted cell type and cell-death pathways between metabolic dysfunction and excitotoxicity suggests the need of specific tools to combat the consequences of lesion arising from distinct causes.
Pharmacological Neuroprotection of Network Damage
In vitro models of SCI can be suitable for preclinical exploration of neuroprotective drug activity. In fact, in terms of locomotor patterns and cell numbers, neuroprotection with glutamate antagonists such as 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX) and D-aminophosphonovalerate (APV) is possible against PM even when these antagonists are applied after PM washout (Margaryan et al., 2010). On the other hand, such antagonist administration is ineffective against excitotoxicity when started after washout of kainate, and poorly efficient when co-applied, as only a minority of preparations show locomotor patterns 1 day later (Margaryan et al., 2010). These data raise a number of interesting issues: overactivation of glutamate receptors by PM is relatively limited (in accordance with biochemical and morphological data for glial damage) and can be contrasted by glutamate antagonists even applied late. Thus, any excitotoxicity arising from the effects of PM is likely subthreshold for substantial neuronal loss and is manifested as a moderate dysfunction of spinal networks consequent to metabolic distress, from which recovery is indeed possible.
Full-blown excitotoxicity due to direct stimulation of glutamate receptors can be poorly arrested by glutamate antagonists because of their slow pharmacokinetics in comparison with the speed of kainate effects (Margaryan et al., 2010). Furthermore, delayed application of antagonists cannot reverse damage which is apparently induced by downstream mechanisms like parthanatos and proceeds independently from glutamate receptor activation (Kuzhandaivel et al., 2010b; Mazzone et al., 2010). This realization suggests studies of neuroprotection targeted to cell-death processes downstream of glutamate receptors.
As a delayed pharmacological approach becomes desirable to combat excitotoxicity, PARP-1 inhibitors like 6,5-(H)phenanthridinone (PHE) and 2-(dimethylamino)-N-(5,6-dihydro-6-oxophenanthridin-2yl) acetamide (PJ-34; Abdelkarim et al., 2001) have been tested (Nasrabady et al., 2011a, b; Mazzone and Nistri, 2011b). As shown in Figure 3C, PHE exerts histological neuroprotection, but it cannot preserve locomotor network function (Kuzhandaivel et al., 2010b; Nasrabady et al., 2011a). PJ-34 (60 μM) applied 30 min after the start of kainate administration and maintained for 24 h can preserve spinal network histology with return of locomotor patterns only when the excitotoxic stimulus is moderate (Nasrabady et al., 2011b). Delayed application of PJ-34 in coincidence with kainate washout consistently fails to generate neuroprotection (Mazzone and Nistri, 2011b).
The poor outcome observed with PARP-1 inhibitors suggests that, in addition (or alongside) to PARP-1 hyperactivation, other cell-death mechanisms (for instance PARP-2; Moroni, 2008; Moroni et al., 2009) have been triggered to evoke excitotoxicity. Full elucidation of these mechanisms will be important to devise more effective treatments. It is, however, possible that, after the initial excitotoxic stimulus, spinal networks are structurally protected, yet functionally inhibited because of an unknown form of downregulation of their motor output which might wane only after days, namely a temporal target currently untestable with these preparations.
In the attempt to circumvent this difficulty, it becomes important to establish the number of surviving neurons and glia necessary to express locomotor activity, because this information can be a predictor of locomotor function and a target to be achieved for neuroprotection. Whilst this objective is difficult to reach in vivo because of the yet-undefined description of locomotor networks, it seems feasible to calculate the minimal membership of the network to support locomotor pattern expression in vitro.
Minimal Network Membership for Locomotor Function
The divergence between histological and functional outcome of the experimentally induced SCI, implies a narrow borderline between neuronal numbers compatible or not with fictive locomotion (Margaryan et al., 2009, 2010; Nistri et al., 2010). Hence, comparison of the immunohistochemical data from sham preparations with those experimentally damaged and/or otherwise protected (in functional terms) by CNQX plus APV, can be exploited to formulate a preliminary estimate of the minimal network membership required for expressing locomotor patterns in the rat lumbar spinal cord.
The locomotor CPG related to the rodent hindlimbs is primarily localized to the rostral lumbar segments L1–L3 (Cazalets et al., 1992, 1995; Kjaerulff and Kiehn, 1996; Cowley and Schmidt, 1997; Kiehn et al., 2008), and comprises cells (on each side) in the ventro-medial part of laminae VII, VIII, and IX, while dorsal horn regions appear to supply just modulatory inputs to the CPG operation (Kiehn, 2006; Nistri et al., 2006; Taccola and Nistri, 2006). As exemplified in Figure 5A, in the neonatal rat spinal cord the axial length of these segments is approximately 3 mm. Histological analysis requires, as a routine, circa 100 sections (30 μm each) to be processed with NeuN (neuronal marker) or SMI-32 (motoneuronal marker) immunoreactivity (see right panels in Figure 5A). Figure 5B shows an example of the ventro-medial spinal gray matter area containing a fixed region of interest (Figure 5C; 350 × 350 μm) for immunochemical determination of neuronal numbers. Thus, on one side of a 30 μm section, there are, on average, approximately 315 NeuN-positive neurons. Multiplying this value for the number of sections in three segments will provide, for one side of the spinal cord, the value of ∼31,500 NeuN-positive cells, that becomes a total of 63,000 NeuN-positive cells for both spinal sides. The number of motoneurons in the same segments can be calculated as 3,600 bilaterally, starting from 18 SMI32 positive cells in one hemisection of 30 μm. These results are consistent with previous measurements (3,500 motoneurons) for the neonatal rat spinal cord obtained by Oppenheim (1986) who did not find any postnatal fall in the number of such cells. It is noteworthy that Tomlinson and Irving (1977) have reported an average of 60,000 anterior horn cells in the whole human lumbosacral spinal cord with no change from youth up to the age of 60.
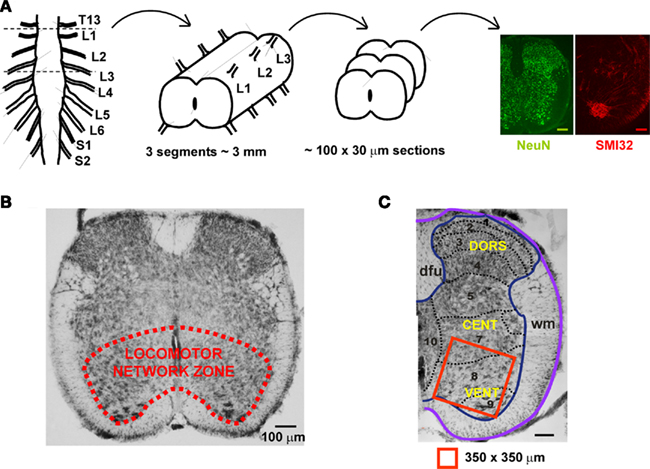
Figure 5. Schematic representation of the locomotor networks in rat neonatal spinal cord. (A) The main locomotor network for the rat hindlimb is thought to be localized to spinal cord lumbar segments L1–L3. In the neonatal animal, this region is about 3 mm long, from which approximately 100 slices (each 30 μm thick) are obtained for experimental purpose. Representative immuno-stainings with the neuronal marker NeuN (green) and the motoneuronal marker SMI32 (red) are shown. Scale bars = 100 μm. (B) The locomotor network zone is outlined in the ventro-medial area of a 30 μm section from the upper lumbar region. (C) Example of hemisected section comprising the location of locomotor networks (laminae 7, 8, and 9 shown in red box of 350 × 350 μm) used for counting of the number of neurons and motoneurons. DORS, dorsal; CENT, central; VENT, ventral; dfu, dorsal funiculus; wm, white matter.
Applying the same analysis to the experiments with neuroprotection by CNQX and APV (Margaryan et al., 2010), it appears that fictive locomotion could still be observed when 64% of NeuN-positive cells are present and it is lost when the number falls to 45% in the ventro-medial area. Thus, the minimal membership of ventral horn neurons necessary for fictive locomotion can be estimated between 28,000 and 40,000 cells in the three segments. As far as motoneurons are concerned, fictive locomotion is still present when 88% motoneurons remain, and is absent with 68%. Hence, the minimal number of motoneurons essential to express fictive locomotion of the hindlimbs may be estimated between 2,370 and 3,050 in the three lumbar segments. Similar suggestions on the membership size have been supplied by experiments with changes in extracellular Mg2+ and its consequences on fictive locomotion and ventral horn histology (Margaryan et al., 2009). Conversely, experimental paradigms that induce extensive white matter damage show that fictive locomotion can still be expressed despite depressed reflex activity (Taccola et al., 2008; Margaryan et al., 2009). Notwithstanding the approximate nature of the current neuronal estimates, these numbers have a heuristic value because they can supply a minimal target for future studies aimed at rebuilding damaged networks or at constraining damage. Once the network membership falls below such a threshold, the surviving circuits (which are still metabolically viable; Mazzone et al., 2010) can produce disinhibited bursting (that is known to be localized to the ventral horn quadrant; Bracci et al., 1996), indicating that the basic connectivity necessary to express network rhythmicity is still present.
It should be pointed out that these calculations cannot obviously reflect the rat physiological ability to walk, since the animal functional activity as well as the electromyography of skeletal limb muscles have not been measured. The present scheme suggests the size of the locomotor network, but it does not address the precise distribution of the locomotor CPG and the location of its intrinsic components like the neurons operating as the rhythm clock or as pattern formation (McCrea and Rybak, 2008). Nevertheless, these data indicate a cell number essential to express the locomotor program: falling below the minimal membership cannot allow generation of the locomotor patterns. Future studies are required to clarify the role of various premotoneuron types identified on the basis of their genetic markers (Kiehn, 2006; Grossmann et al., 2010) in the network locomotor activity after lesion.
Studies of the locomotor network size in man are few and primarily obtained from post-mortem examination (Kaelan et al., 1988), since even functional magnetic resonance imaging has provided scant evidence for a clear link between spinal lesions and disability (Stankiewicz et al., 2009). From human observations it emerges that transynaptic degeneration of ventral horn neurons does not occur following complete corticospinal tract lesions (Kaelan et al., 1988), and that locomotor activity was present shortly before death of patients who at necroscopy examination had fewer than 30,000 ventral horn neurons against an average control of approximately 60,000 for the whole lumbosacral enlargement (Tomlinson and Irving, 1977). Less certain is the number of interneurons making up the CPG network: nonetheless, this value is likely to be even larger because in most vertebrate species the ratio of interneurons to motoneurons is 5:1 (Walløe et al., 2011). These estimates do not provide a conclusive size of the human locomotor network, but they do indicate the daunting size that any reconstructive and repairing attempt to re-establish locomotion after SCI must meet.
Acknowledgments
This study was supported by grants from the government of the Friuli Venezia Giulia Region, the Italian Institute of Technology and the Italian Ministry for Education and Research (MIUR) under their PRIN program. Miranda Mladinic is a research biologist of the Local Health Authority (ASS4 MedioFriuli).
References
Abdelkarim, G. E., Gertz, K., Harms, C., Katchanov, J., Dirnagl, U., Szabo, C., and Endres, M. (2001). Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int. J. Mol. Med. 7, 255–260.
Abu-Rub, M., McMahon, S., Zeugolis, D. I., Windebank, A., and Pandit, A. (2010). Spinal cord injury in vitro: modelling axon growth inhibition. Drug Discov. Today 15, 436–443.
Achildi, O., Betz, R. R., and Grewal, H. (2007). Lapbelt injuries and the seatbelt syndrome in pediatric spinal cord injury. J. Spinal Cord Med. 30(Suppl. 1), S21–S24.
Amar, A. P. (ed.) (2007). “Pathogenesis of acute spinal cord injury and theoretical bases of neurological recovery,” in Surgical management of spinal cord injury: controversies and consensus (Oxford: Blackwell Publishing, Ltd), 1–17.
Andrabi, S. A., Dawson, T. M., and Dawson, V. L. (2008). Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y. Acad. Sci. 1147, 233–241.
Andrabi, S. A., Kim, N. S., Yu, S. W., Wang, H., Koh, D. W., Sasaki, M., Klaus, J. A., Otsuka, T., Zhang, Z., Koehler, R. C., Hurn, P. D., Poirier, G. G., Dawson, V. L., and Dawson, T. M. (2006). Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U.S.A. 103, 18308–18313.
Arvanian, V. L., Schnell, L., Lou, L., Golshani, R., Hunanyan, A., Ghosh, A., Pearse, D. D., Robinson, J. K., Schwab, M. E., Fawcett, J. W., and Mendell, L. M. (2009). Chronic spinal hemisection in rats induces a progressive decline in transmission in uninjured fibers to motoneurons. Exp. Neurol. 216, 471–480.
Baba, H., Sakurai, M., Abe, K., and Tominaga, R. (2009). Autophagy-mediated stress response in motor neuron after transient ischemia in rabbits. J. Vasc. Surg. 50, 381–387.
Baptiste, D. C., and Fehlings, M. G. (2006). Pharmacological approaches to repair the injured spinal cord. J. Neurotrauma 23, 318–334.
Barbeau, H., McCrea, D. A., O’Donovan, M. J., Rossignol, S., Grill, W. M., and Lemay, M. A. (1999). Tapping into spinal circuits to restore motor function. Brain Res. Brain Res. Rev. 30, 27–51.
Beattie, M. S., Farooqui, A. A., and Bresnahan, J. C. (2000). Review of current evidence for apoptosis after spinal cord injury. J. Neurotrauma 17, 915–925.
Belmokhtar, C. A., Hillion, J., and Ségal-Bendirdjian, E. (2001). Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20, 3354–3362.
Ben-Ari, Y., and Cossart, R. (2000). Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 23,580–587.
Berger, N. A. (1985). Poly(ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 101, 4–15.
Boulenguez, P., Liabeuf, S., Bos, R., Bras, H., Jean-Xavier, C., Brocard, C., Stil, A., Darbon, P., Cattaert, D., Delpire, E., Marsala, M., and Vinay, L. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307.
Boulenguez, P., and Vinay, L. (2009). Strategies to restore motor functions after spinal cord injury. Curr. Opin. Neurobiol. 19, 587–600.
Bracci, E., Ballerini, L., and Nistri, A. (1996). Localization of rhythmogenic networks responsible for spontaneous bursts induced by strychnine and bicuculline in the rat isolated spinal cord. J. Neurosci. 16, 7063–7076.
Brownstone, R. M., and Wilson, J. M. (2008). Strategies for delineating spinal locomotor rhythm-generating networks and the possible role of Hb9 interneurones in rhythmogenesis. Brain Res. Rev. 57, 64–76.
Burgoyne, L. A. (1999). The mechanisms of pyknosis: hypercondensation and death. Exp. Cell Res. 248, 214–222.
Cazalets, J. R., Borde, M., and Clarac, F. (1995). Localization and organization of the central pattern generator for hindlimb locomotion in newborn rat. J. Neurosci. 15(7 Pt 1), 4943–4951.
Cazalets, J. R., Sqalli-Houssaini, Y., and Clarac, F. (1992). Activation of the central pattern generators for locomotion by serotonin and excitatory amino acids in neonatal rat. J. Physiol. 455, 187–204.
Cho, B. B., and Toledo-Pereyra, L. H. (2008). Caspase-independent programmed cell death following ischemic stroke. J. Invest. Surg. 21, 141–147.
Cirak, B., Ziegfeld, S., Knight, V. M., Chang, D., Avellino, A. M., and Paidas, C. N. (2004). Spinal injuries in children. J. Pediatr. Surg. 39, 607–612.
Cowley, K. C., and Schmidt, B. J. (1997). Regional distribution of the locomotor pattern-generating network in the neonatal rat spinal cord. J. Neurophysiol. 77, 247–259.
Coyle, J. T. (1987). Kainic acid: insights into excitatory mechanisms causing selective neuronal degeneration. Ciba Found. Symp. 126, 186–203.
David, K. K., Andrabi, S. A., Dawson, T. M., and Dawson, V. L. (2009). Parthanatos, a messenger of death. Front. Biosci. 14, 1116–1128.
De Louw, A. J., Van De Berg, W. D., De Vente, J., Blanco, C. E., Gavilanes, A. W., Steinbusch, H. P., Steinbusch, H. W., Troost, J., and Vles, J. S. (2002). Developmental apoptosis in the spinal cord white matter in neonatal rats. Glia 37, 89–91.
Dekkers, J., Bayley, P., Dick, J. R., Schwaller, B., Berchtold, M. W., and Greensmith, L. (2004). Over-expression of parvalbumin in transgenic mice rescues motoneurons from injury-induced cell death. Neuroscience 123, 459–466.
Dietz, V., Wirz, M., and Jensen, L. (1997). Locomotion in patients with spinal cord injuries. Phys. Ther. 77, 508–516.
Dumont, R. J., Okonkwo, D. O., Verma, S., Hurlbert, R. J., Boulos, P. T., Ellegala, D. B., and Dumont, A. S. (2001). Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin. Neuropharmacol. 24, 254–264.
Dzidic, I., and Moslavac, S. (1997). Functional skills after the rehabilitation of spinal cord injury patients; observation period of 3 years. Spinal Cord 35, 620–623.
ElBasiouny, S. M., Schuster, J. E., and Heckman, C. J. (2010). Persistent inward currents in spinal motoneurons: important for normal function but potentially harmful after spinal cord injury and in amyotrophic lateral sclerosis. Clin. Neurophysiol. 121, 1669–1679.
Eliasson, M. J., Sampei, K., Mandir, A. S., Hurn, P. D., Traystman, R. J., Bao, J., Pieper, A., Wang, Z. Q., Dawson, T. M., Snyder, S. H., and Dawson, V. L. (1997). Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 3, 1089–1095.
Emery, E., Aldana, P., Bunge, M. B., Puckett, W., Srinivasan, A., Keane, R. W., Bethea, J., and Levi, A. D. (1998). Apoptosis after traumatic human spinal cord injury. J. Neurosurg. 89, 911–920.
Endres, M., Wang, Z. Q., Namura, S., Waeber, C., and Moskowitz, M. A. (1997). Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 17, 1143–1151.
Fehlings, M. G., Sekhon, L. H., and Tator, C. (2001). The role and timing of decompression in acute spinal cord injury: what do we know? What should we do? Spine 26(24 Suppl), S101-110.
Gahwiler, B. H. (1981). Organotypic monolayer cultures of nervous tissue. J. Neurosci. Methods 4, 329–342.
Galluzzi, L., Maiuri, M. C., Vitale, I., Zischka, H., Castedo, M., Zitvogel, L., and Kroemer, G. (2007). Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 14, 1237–1243.
Genovese, T., and Cuzzocrea, S. (2008). Role of free radicals and poly(ADP-ribose)polymerase-1 in the development of spinal cord injury: new potential therapeutic targets. Curr. Med. Chem. 15, 477–487.
Genovese, T., Mazzon, E., Muia, C., Patel, N. S., Threadgill, M. D., Bramanti, P., De Sarro, A., Thiemermann, C., and Cuzzocrea, S. (2005). Inhibitors of poly(ADP-ribose) polymerase modulate signal transduction pathways and secondary damage in experimental spinal cord trauma. J. Pharmacol. Exp. Ther. 312, 449–457.
Golstein, P., and Kroemer, G. (2007). Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37–43.
Grillner, S., Ekeberg, O., El Manira, A., Lansner, A., Parker, D., Tegnér, J., and Wallén, P. (1998). Intrinsic function of a neuronal network – a vertebrate central pattern generator. Brain Res. Brain Res. Rev. 26, 184–197.
Grillner, S., and Jessell, T. M. (2009). Measured motion: searching for simplicity in spinal locomotor networks. Curr. Opin. Neurobiol. 19, 572–586.
Grossmann, K. S., Giraudin, A., Britz, O., Zhang, J., and Goulding, M. (2010). Genetic dissection of rhythmic motor networks in mice. Prog. Brain Res. 187, 19–37.
Guzman-Lenis, M. S., Navarro, X., and Casas, C. (2009). Drug screening of neuroprotective agents on an organotypic-based model of spinal cord excitotoxic damage. Restor. Neurol. Neurosci. 27, 335–349.
Hagberg, H., Wilson, M. A., Matsushita, H., Zhu, C., Lange, M., Gustavsson, M., Poitras, M. F., Dawson, T. M., Dawson, V. L., Northington, F., and Johnston, M. V. (2004). PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J. Neurochem. 90, 1068–1075.
Hayashi, T., Sakurai, M., Abe, K., Sadahiro, M., Tabayashi, K., and Itoyama, Y. (1998). Apoptosis of motor neurons with induction of caspases in the spinal cord after ischemia. Stroke 29, 1007–1012.
Heckmann, C. J., Gorassini, M. A., and Bennett, D. J. (2005). Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31, 135–156.
Hubli, M., Bolliger, M., and Dietz, V. (2010). Neuronal dysfunction in chronic spinal cord injury. Spinal Cord 49, 582–587.
Jellinger, K. A., and Stadelmann, C. H. (2000). The enigma of cell death in neurodegenerative disorders. J. Neural Transm. Suppl. 60, 21–36.
Joza, N., Pospisilik, J. A., Hangen, E., Hanada, T., Modjtahedi, N., Penninger, J. M., and Kroemer, G. (2009). AIF: not just an apoptosis-inducing factor. Ann. N. Y. Acad. Sci. 1171, 2–11.
Kaelan, C., Jacobsen, P. F., and Kakulas, B. A. (1988). An investigation of possible transynaptic neuronal degeneration in human spinal cord injury. J. Neurol. Sci. 86, 231–237.
Kanno, H., Ozawa, H., Sekiguchi, A., and Itoi, E. (2009). The role of autophagy in spinal cord injury. Autophagy 5, 390–392.
Kaushal, V., and Schlichter, L. C. (2008). Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 28, 2221–2230.
Kerr, J. F., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257.
Kiehn, O. (2006). Locomotor circuits in the mammalian spinal cord. Annu. Rev. Neurosci. 29, 279–306.
Kiehn, O., Quinlan, K. A., Restrepo, C. E., Lundfald, L., Borgius, L., Talpalar, A. E., and Endo, T. (2008). Excitatory components of the mammalian locomotor CPG. Brain Res. Rev. 57, 56–63.
Kjaerulff, O., and Kiehn, O. (1996). Distribution of networks generating and coordinating locomotor activity in the neonatal rat spinal cord in vitro: a lesion study. J. Neurosci. 16, 5777–5794.
Krassioukov, A. V., Ackery, A., Schwartz, G., Adamchik, Y., Liu, Y., and Fehlings, M. G. (2002). An in vitro model of neurotrauma in organotypic spinal cord cultures from adult mice. Brain Res. Brain Res. Protoc. 10, 60–68.
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., El-Deiry, W. S., Golstein, P., Green, D. R., Hengartner, M., Knight, R. A., Kumar, S., Lipton, S. A., Malorni, W., Nuñez, G., Peter, M. E., Tschopp, J., Yuan, J., Piacentini, M., Zhivotovsky, B., and Melino, G. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 16, 3–11.
Kuzhandaivel, A., Margaryan, G., Nistri, A., and Mladinic, M. (2010a). Extensive occurrence of glial apoptosis develops early after hypoxic-dysmetabolic insult to the neonatal rat spinal cord in vitro. Neuroscience 169, 325–338.
Kuzhandaivel, A., Nistri, A., and Mladinic, M. (2010b). Kainate-mediated excitotoxicity induces neuronal death in the rat spinal cord in vitro via a PARP-1 dependent cell death pathway (parthanatos). Cell. Mol. Neurobiol. 30, 1001–1012.
Li, G. L., Farooque, M., Holtz, A., and Olsson, Y. (1999). Apoptosis of oligodendrocytes occurs for long distances away from the primary injury after compression trauma to rat spinal cord. Acta Neuropathol. 98, 473–480.
Li, Y., and Bennett, D. J. (2003). Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J. Neurophysiol. 90, 857–869.
Liu, X. Z., Xu, X. M., Hu, R., Du, C., Zhang, S. X., McDonald, J. W., Dong, H. X., Wu, Y. J,. Fan, G. S., Jacquin, M. F., Hsu, C. Y., and Choi, D. W. (1997). Neuronal and glial apoptosis after traumatic SCI. J. Neurosci. 17, 5395–5406.
Livera, G., Delbes, G., Pairault, C., Rouiller-Fabre, V., and Habert, R. (2006). Organotypic culture, a powerful model for studying rat and mouse fetal testis development. Cell Tissue Res. 324, 507–521.
Lossi, L., Alasia, S., Salio, C., and Merighi, A. (2009). Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog. Neurobiol. 88, 221–245.
Mandir, A. S., Poitras, M. F., Berliner, A. R., Herring, W. J., Guastella, D. B., Feldman, A., Poirier, G., Wang, Z. Q., Dawson, T. M., and Dawson, V. L. (2000). NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J. Neurosci. 20, 8005–8011.
Margaryan, G., Mattioli, C., Mladinic, M., and Nistri, A. (2010). Neuroprotection of locomotor networks after experimental injury to the neonatal rat spinal cord in vitro. Neuroscience 165, 996–1010.
Margaryan, G., Mladinic, M., Mattioli, C., and Nistri, A. (2009). Extracellular Mg2+ enhances the damage to locomotor networks produced by metabolic perturbation mimicking spinal injury in the neonatal rat spinal cord in vitro. Neuroscience 163, 669–682.
Mazzone, G. L., Margaryan, G., Kuzhandaivel, A., Nasrabady, S. E., Mladinic, M., and Nistri, A. (2010). Kainate-induced delayed onset of excitotoxicity with functional loss unrelated to the extent of neuronal damage in the in vitro spinal cord. Neuroscience 168, 451–462.
Mazzone, G. L., and Nistri, A. (2011a). Electrochemical detection of endogenous glutamate release from rat spinal cord organotypic slices as a real-time method to monitor excitotoxicity. J. Neurosci. Methods 197, 128–132.
Mazzone, G. L., and Nistri, A. (2011b). Effect of the PARP-1 inhibitor PJ 34 on excitotoxic damage evoked by kainate on rat spinal cord organotypic slices. Cell. Mol. Neurobiol. 31, 469–478.
McCrea, D. A., and Rybak, I. A. (2008). Organization of mammalian locomotor rhythm and pattern generation. Brain Res. Rev. 57, 134–146.
McKinley, W. O., Seel, R. T., and Hardman, J. T. (1999). Nontraumatic spinal cord injury: incidence, epidemiology, and functional outcome. Arch. Phys. Med. Rehabil. 80, 619–623.
Meli, E., Pangallo, M., Baronti, R., Chiarugi, A., Cozzi, A., Pellegrini-Giampietro, D. E., and Moroni, F. (2003). Poly(ADP-ribose) polymerase as a key player in excitotoxicity and post-ischemic brain damage. Toxicol. Lett. 139, 153–162.
Moroni, F. (2008). Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr. Opin. Pharmacol. 8, 96–103.
Moroni, F., Formentini, L., Gerace, E., Camaioni, E., Pellegrini-Giampietro, D. E., Chiarugi, A., and Pellicciari, R. (2009). Selective PARP-2 inhibitors increase apoptosis in hippocampal slices but protect cortical cells in models of post-ischaemic brain damage. Br. J. Pharmacol. 157, 854–862.
Nair, K. P., Taly, A. B., Maheshwarappa, B. M., Kumar, J., Murali, T., and Rao, S. (2005). Nontraumatic spinal cord lesions: a prospective study of medical complications during in-patient rehabilitation. Spinal Cord 43, 558–564.
Narkilahti, S., Pirttilä, T. J., Lukasiuk, K., Tuunanen, J., and Pitkänen, A. (2003). Expression and activation of caspase 3 following status epilepticus in the rat. Eur. J. Neurosci. 18, 1486–1496.
Nasrabady, S. E., Kuzhandaivel, A., Mladinic, M., and Nistri, A. (2011a). A. Effects of 6(5H)-phenanthridinone, an inhibitor of poly(ADP-ribose)polymerase-1 activity (PARP-1), on locomotor networks of the rat isolated spinal cord. Cell. Mol. Neurobiol. 31, 503–508.
Nasrabady, S. E., Kuzhandaivel, A., and Nistri, A. (2011b). Studies of locomotor network neuroprotection by the selective PARP-1 inhibitor PJ-34 against excitotoxic injury to the rat spinal cord in vitro. Eur. J. Neurosci. doi: 10.1111/j.1460-9568.2011.07714.x [Epub ahead of print].
Nistri, A., Ostroumov, K., Sharifullina, E., and Taccola, G. (2006). Tuning and playing a motor rhythm: how metabotropic glutamate receptors orchestrate generation of motor patterns in the mammalian central nervous system. J. Physiol. 572(Pt 2), 323–334.
Nistri, A., Taccola, G., Mladinic, M., Margaryan, G., and Kuzhandaivel, A. (2010). Deconstructing locomotor networks with experimental injury to define their membership. Ann. N. Y. Acad. Sci. 1198, 242–251.
Noraberg, J. (2004). Organotypic brain slice cultures: an efficient and reliable method for neurotoxicological screening and mechanistic studies. Altern. Lab. Anim. 32, 329–337.
Norenberg, M. D., Smith, J., and Marcillo, A. (2004). The pathology of human spinal cord injury: defining the problems. J. Neurotrauma 21, 429–440.
Onifer, S. M., Rabchevsky, A. G., and Scheff, S. W. (2007). Rat models of traumatic spinal cord injury to assess motor recovery. ILAR J. 48, 385–395.
Oppenheim, R. W. (1986). The absence of significant postnatal motoneuron death in the brachial and lumbar spinal cord of the rat. J. Comp. Neurol. 246, 281–286.
Park, E., Velumian, A. A., and Fehlings, M. G. (2004). The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J. Neurotrauma 21, 754–774.
Ray, S. K., Hogan, E. L., and Banik, N. L. (2003). Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res. Brain Res. Rev. 42, 169–185.
Rosenberg, P. A., Dai, W., Gan, X. D., Ali, S., Fu, J., Back, S. A., Sanchez, R. M., Segal, M. M., Follett, P. L., Jensen, F. E., and Volpe, J. J. (2003). Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J. Neurosci. Res. 71, 237–245.
Rossignol, S., Schwab, M., Schwartz, M., and Fehlings, M. G. (2007). Spinal cord injury: time to move? J. Neurosci. 27, 11782–11792.
Rowland, J. W., Hawryluk, G. W., Kwon, B., and Fehlings, M. G. (2008). Current status of acute spinal cord injury pathophysiology and emerging therapies: promise on the horizon. Neurosurg. Focus 25, E2.
Sakurai, M., Nagata, T., Abe, K., Horinouchi, T., Itoyama, Y., and Tabayashi, K. (2003). Survival and death-promoting events after transient spinal cord ischemia in rabbits: induction of Akt and caspase3 in motor neurons. J. Thorac. Cardiovasc. Surg. 125, 370–377.
Scott, G. S., Szabó, C., and Hooper, D. C. (2004). Poly(ADP-ribose) polymerase activity contributes to peroxynitrite-induced spinal cord neuronal cell death in vitro. J. Neurotrauma 21, 1255–1263.
Sekhon, L. H., and Fehlings, M. G. (2001). Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine 26, S2–S12.
Seybold, V. S., and Abrahams, L. G. (2004). Primary cultures of neonatal rat spinal cord. Methods Mol. Med. 99, 203–213.
Shibata, M., Hisahara, S., Hara, H., Yamawaki, T., Fukuuchi, Y., Yuan, J., Okano, H., and Miura, M. (2000). Caspases determine the vulnerability of oligodendrocytes in the ischemic brain. J. Clin. Invest. 106, 643–653.
Shuman, S. L., Bresnahan, J. C., and Beattie, M. S. (1997). Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J. Neurosci. Res. 50, 798–808.
Sibilla, S., and Ballerini, L. (2009). GABAergic and glycinergic interneuron expression during spinal cord development: dynamic interplay between inhibition and excitation in the control of ventral network outputs. Prog. Neurobiol. 89, 46–60.
Silani, V., Braga, M., Ciammola, A., Cardin, V., and Scarlato, G. (2000). Motor neurones in culture as a model to study ALS. J. Neurol. 247(Suppl. 1), I28–I36.
Spenger, C., Braschler, U. F., Streit, J., and Luscher, H. R. (1991). An organotypic spinal cord – dorsal root ganglion – skeletal muscle coculture of embryonic rat. I. The morphological correlates of the spinal reflex arc. Eur. J. Neurosci. 3, 1037–1053.
Springer, J. E., Azbill, R. D., and Knapp, P. E. (1999). Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat. Med. 5, 943–946.
Stankiewicz, J. M., Neema, M., Alsop, D. C., Healy, B. C., Arora, A., Buckle, G. J., Chitnis, T., Guttmann, C. R., Hackney, D., and Bakshi, R. (2009). Spinal cord lesions and clinical status in multiple sclerosis: a 1.5 T and 3 T MRI study. J. Neurol. Sci. 279, 99–105.
Stoppini, L., Buchs, P. A., and Muller, D. (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182.
Streit, J., Spenger, C., and Luscher, H. R. (1991). An organotypic spinal cord – dorsal root ganglion – skeletal muscle coculture of embryonic rat. II. Functional evidence for the formation of spinal reflex arcs in vitro. Eur. J. Neurosci. 3, 1054–1068.
Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R., Siderovski, D. P., Penninger, J. M., and Kroemer, G. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446.
Taccola, G., Margaryan, G., Mladinic, M., and Nistri, A. (2008). Kainate and metabolic perturbation mimicking spinal injury differentially contribute to early damage of locomotor networks in the in vitro neonatal rat spinal cord. Neuroscience 155, 538–555.
Taccola, G., Mladinic, M., and Nistri, A. (2010). Dynamics of early locomotor network dysfunction following a focal lesion in an in vitro model of spinal injury. Eur. J. Neurosci. 31, 60–78.
Taccola, G., and Nistri, A. (2006). Oscillatory circuits underlying locomotor networks in the rat spinal cord. Crit. Rev. Neurobiol. 18, 25–36.
Tan, S., Wood, M., and Maher, P. (1998). Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. 71, 95–105.
Taylor, A. R., Robinson, M. B., and Milligan, C. E. (2007). In vitro methods to prepare astrocyte and motoneuron cultures for the investigation of potential in vivo interactions. Nat. Protoc. 2, 1499–1507.
Tazerart, S., Viemari, J. C., Darbon, P., Vinay, L., and Brocard, F. (2007). Contribution of persistent sodium current to locomotor pattern generation in neonatal rats. J. Neurophysiol. 98, 613–628.
Tolle, T. R., Berthele, A., Zieglgänsberger, W., Seeburg, P. H., and Wisden, W. (1993). The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J. Neurosci. 13, 5009–5028.
Tomlinson, B. E., and Irving, D. (1977). The numbers of limb motor neurons in the human lumbosacral cord throughout life. J. Neurol. Sci. 34, 213–219.
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., Hansen, K. B., Yuan, H., Myers, S. J., and Dingledine, R. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Van Den, B. L., Vandenberghe, W., Klaassen, H., Van Houtte, E., and Robberecht, W. (2000). Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 180, 29–34.
van den Berg, M. E., Castellote, J. M., Mahillo-Fernandez, I., and de Pedro-Cuesta, J. (2010). Incidence of spinal cord injury worldwide: a systematic review. Neuroepidemiology 34, 184–192.
Vandenberghe, W., Van Den, B. L., and Robberecht, W. (1998). Glial cells potentiate kainate-induced neuronal death in a motoneuron-enriched spinal coculture system. Brain Res. 807, 1–10.
Virag, L., Salzman, A. L., and Szabo, C. (1998). Poly(ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J. Immunol. 161, 3753–3759.
Vitale, M. G., Goss, J. M., Matsumoto, H., and Roye, D. P. Jr. (2006). Epidemiology of pediatric spinal cord injury in the United States: years 1997 and 2000. J. Pediatr. Orthop. 26, 745–749.
Walløe, S., Nissen, U. V., Berg, R. W., Hounsgaard, J., and Pakkenberg, B. (2011). Stereological estimate of the total number of neurons in spinal segment D9 of the red-eared turtle. J. Neurosci. 31, 2431–2435.
Widlak, P., and Garrard, W. T. (2009). Roles of the major apoptotic nuclease-DNA fragmentation factor-in biology and disease. Cell. Mol. Life Sci. 66, 263–274.
Wu, K. L., Hsu, C., and Chan, J. Y. (2007). Impairment of the mitochondrial respiratory enzyme activity triggers sequential activation of apoptosis-inducing factor-dependent and caspase-dependent signaling pathways to induce apoptosis after spinal cord injury. J. Neurochem. 101, 1552–1566.
Wu, K. L., Hsu, C., and Chan, J. Y. (2009). Nitric oxide and superoxide anion differentially activate poly(ADP-ribose) polymerase-1 and Bax to induce nuclear translocation of apoptosis-inducing factor and mitochondrial release of cytochrome c after spinal cord injury. J. Neurotrauma 26, 965–977.
Yu, S. W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., Poirier, G. G., Dawson, T. M., and Dawson, V. L. (2002). Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263.
Yu, W. R., Liu, T., Fehlings, T. K., and Fehlings, M. G. (2009). Involvement of mitochondrial signaling pathways in the mechanism of Fas-mediated apoptosis after spinal cord injury. Eur. J. Neurosci. 29, 114–131.
Yuan, J., Lipinski, M., and Degterev, A. (2003). Diversity in the mechanisms of neuronal cell death. Neuron 40, 401–413.
Zhang, J., Pieper, A., and Snyder, S. H. (1995). Poly(ADP-ribose) synthetase activation: an early indicator of neurotoxic DNA damage. J. Neurochem. 65, 1411–1414.
Zhang, Y., and Bhavnani, B. R. (2006). Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 7, 49. doi: 10.1186/1471-2202-7-49
Zhong, G., Masino, M. A., and Harris-Warrick, R. M. (2007). Persistent sodium currents participate in fictive locomotion generation in neonatal mouse spinal cord. J. Neurosci. 27, 4507–4518.
Ziskind-Conhaim, L., Mentis, G. Z., Wiesner, E. P., and Titus, D. J. (2010). Synaptic integration of rhythmogenic neurons in the locomotor circuitry: the case of Hb9 interneurons. Ann. N. Y. Acad. Sci. 1198, 72–84.
Keywords: motoneuron, isolated spinal cord, fictive locomotion, synaptic transmission, spinal cord injury, organotypic cultures, apoptosis, parthanatos
Citation: Kuzhandaivel A, Nistri A, Mazzone GL and Mladinic M (2011) Molecular mechanisms underlying cell death in spinal networks in relation to locomotor activity after acute injury in vitro. Front. Cell. Neurosci. 5:9. doi: 10.3389/fncel.2011.00009
Received: 29 April 2011; Paper pending published: 16 May 2011;
Accepted: 08 June 2011; Published online: 17 June 2011.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Laurent Vinay, CNRS and Aix-Marseille Université, FranceDaniela Tropea, Trinity College Dublin, Ireland
Copyright: © 2011 Kuzhandaivel, Nistri, Mazzone and Mladinic. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Andrea Nistri, Neurobiology Sector, International School for Advanced Studies, Via Bonomea 265, 34136 Trieste, Italy. e-mail:bmlzdHJpQHNpc3NhLml0