

- 1 Department of Molecular Neurobiology, Max-Planck Institute of Experimental Medicine, Göttingen, Germany
- 2 Emeritus Group Neurochemistry, Max-Planck Institute for Brain Research, Frankfurt/Main, Germany
Collybistin (Cb) is a brain-specific GDP/GTP-exchange factor, which interacts with the inhibitory receptor anchoring protein gephyrin. Data from mice carrying an inactivated Cb gene indicate that Cb is required for the formation and maintenance of gephyrin and gephyrin-dependent GABAA receptor (GABAAR) clusters at inhibitory postsynapses in selected regions of the mammalian forebrain. However, important aspects of how Cb’s GDP/GTP-exchange activity, structure, and regulation contribute to gephyrin and GABAAR clustering, as well as its role in synaptic plasticity, remain poorly understood. Here we review the current state of knowledge about Cb’s function and address open questions concerning its contribution to synapse formation, maintenance, plasticity, and adaptive changes in response to altered network activity.
Introduction
Fast synaptic transmission in the nervous system is mediated by ligand-gated ion channels, which are highly concentrated at postsynaptic membrane specializations (Figure 1). At inhibitory synapses, the scaffolding protein gephyrin has been shown to be essential for the synaptic localization of glycine receptors (GlyRs) and major GABAA receptor (GABAAR) subtypes (Kneussel and Betz, 2000; Moss and Smart, 2001). Ablation of gephyrin expression by either antisense depletion in cultured neurons or gene knockout (KO) in mice prevents the clustering of GlyRs (Kirsch and Betz, 1993; Feng et al., 1998) and α2- and γ2-subunit containing GABAARs (Essrich et al., 1998; Kneussel et al., 1999; Luscher and Keller, 2004) at developing postsynaptic sites. In gephyrin KO animals, the loss of gephyrin from inhibitory synapses leads to death on postnatal day 0 (P0; Feng et al., 1998). The generalized stiffness of the musculature seen in the newborn gephyrin-deficient mice resembles the rigid, hyperextended phenotype resulting from poisoning with the GlyR antagonist strychnine and is consistent with a loss of motoneuron inhibition (Feng et al., 1998). Besides its role in GlyR and GABAAR clustering, gephyrin has an essential function in molybdenum cofactor (MoCo) biosynthesis (Feng et al., 1998; Stallmeyer et al., 1999). However, MoCo-deficient mice (Lee et al., 2002) survive for up to 11 days after birth. Furthermore, transgenic expression of the plant homolog Cnx1 partially rescues MoCo synthesis in gephyrin KO mice but does not prolong their survival (Grosskreutz et al., 2003). The early postnatal lethality of the gephyrin KO mice thus is likely due to the lack of postsynaptic inhibitory receptors and not MoCo-deficiency.
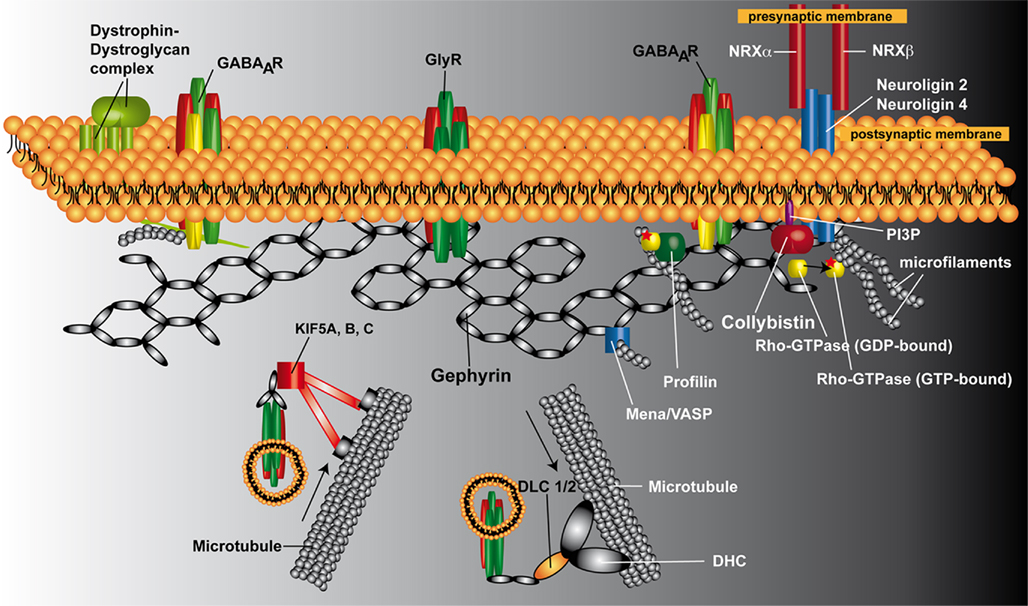
Figure 1. Schematic representation of the postsynaptic protein scaffold at inhibitory synapses. Gephyrin is proposed to form a hexagonal lattice beneath the plasma membrane, onto which inhibitory receptors (GlyRs and GABAARs) are anchored. Cb is required for the postsynaptic clustering of gephyrin at GABAergic synapses. Neuroligin 2 binds to Cb and functions as a specific activator of Cb, thus guiding membrane tethering of the gephyrin scaffold to inhibitory postsynaptic sites. However, GABAARs containing the α2 subunit in the cerebellar cortex require the dystrophin–dystroglycal complex and not gephyrin for their targeting to inhibitory postsynapses, suggesting the existence of dystrophin-dependent and gephyrin-independent mechanism for the clustering of selected GABAAR subtypes. At glycinergic synapses, gephyrin and GlyRs are targeted to the postsynapses independently of Cb. Newly synthesized GlyRs reach the plasma membrane through active transport along microtubules. Gephyrin interacts directly with KIF5 and GlyRs, and the gephyrin/KIF5/GlyR complex moves in anterograde directions toward the plus ends of microtubules. A GlyR/gephyrin/dynein transport complex mediates retrograde minus end-directed microtubule transport to intracellular compartments upon GlyR internalization. Of the known additional gephyrin-interacting factors, profilin and Mena/VASP are shown. Abbreviations: DHL, dynein heavy chain; DLC 1/2, dynein light chain 1/2; GABAAR, GABAA receptor; GlyR, glycine receptor; KIF5A, B, C, kinesin family protein 5A, B, and C; Mena, microfilament adaptors of the mammalian enabled; NRXα, neurexin α; NRXβ, neurexin β; PI3P, phosphatidylinositol-3-phosphate; VASP, vasodilator stimulated phosphoprotein.
Gephyrin is a 93-kDa protein that contains three major domains: an amino-terminal G-domain, a carboxyterminal E-domain and a central linker region. The crystal structures of the G- and E-domains have been determined. The G-domain is homologous to the Escherichia coli protein MogA and forms a trimer (Liu et al., 2000; Sola et al., 2001), whereas the E-domain is homologous to the E. coli protein MoeA and forms a dimer (Sola et al., 2004; Kim et al., 2006). Based on the oligomerization properties of the E- and G-domains, full-length gephyrin was proposed to form a hexagonal lattice beneath the plasma membrane onto which inhibitory receptors are anchored (Kneussel and Betz, 2000; Sola et al., 2004). Consistent with this model, disruption of G-domain trimerization and E-domain dimerization through the introduction of charged residues at the oligomerization interfaces impaired postsynaptic gephyrin clustering (Saiyed et al., 2007).
Gephyrin depends on both actin microfilaments and microtubules for synaptic targeting and submembranous scaffold formation (Kirsch and Betz, 1995; Allison et al., 2000; Bausen et al., 2006; Maas et al., 2006). These interactions are thought to be critical for the postsynaptic localization and intracellular transport of gephyrin (Kneussel, 2006; Fritschy et al., 2008; Figure 1). Furthermore, in dissociated hippocampal neurons small highly mobile, probably vesicle-associated gephyrin particles have been shown to be added to and removed from synaptic gephyrin clusters over a time scale of a few minutes (Maas et al., 2006). This, together with a direct interaction of gephyrin with the microtubule-associated motor proteins kinesin superfamily protein 5 (KIF5; Maas et al., 2009) and dynein light chains 1 and 2 (dlc1/2; Fuhrmann et al., 2002), provides evidence for an active intracellular transport of gephyrin. Gephyrin has also been shown to bind to mammalian enabled (Mena)/vasodilator stimulated phosphoprotein (VASP; Giesemann et al., 2003; Bausen et al., 2006). Furthermore, it has been shown that recombinant gephyrin colocalizes with “uncapped” actin and that this interaction requires ena/VASP as an adaptor (Bausen et al., 2006). In addition, cytochalasin D application induced a preferential loss of small gephyrin clusters from immature but not more differentiated hippocampal neurons, suggesting a transient and early role of the actin cytoskeleton in gephyrin scaffold formation (Bausen et al., 2006). Gephyrin binds also to polymerized tubulin (Kirsch et al., 1991). Whether this interaction involves adaptor proteins like the tubulin- and gephyrin-binding protein KIF5 (Maas et al., 2009) has not been investigated. However, exon 14 of the murine gephyrin gene encodes a 14 amino acid sequence that is 80 and 60%, respectively, identical to imperfect repeat motifs of MAP2 and tau (Ramming et al., 2000). In these microtubule-associated proteins, three such repeat motifs are known to be required for tubulin polymerization and microtubule binding. Thus, gephyrin trimers might directly bind to microtubules via these motifs.
Furthermore, gephyrin binds the cytoplasmic loop of the GlyR β-subunit (Meyer et al., 1995; Sola et al., 2004), the brain-specific guanine nucleotide exchange factor (GEF) collybistin (Cb; Kins et al., 2000), the postsynaptic adhesion molecule neuroligin 2 (NL2; Poulopoulos et al., 2009) and the actin-binding protein profilin (Mammoto et al., 1998). Thus, gephyrin clearly is not just a simple scaffolding protein.
In the present review we focus on the GDP/GTP-exchange factor Cb and summarize the current state of knowledge concerning its role in inhibitory synapse formation. In addition, we discuss unmet challenges and open questions which should be addressed to deepen our understanding of the molecular mechanisms involved in Cb-dependent clustering of gephyrin and GABAARs at inhibitory postsynapses.
Structure of Cb
A yeast two-hybrid screen with gephyrin originally identified two Cb splice variants (Cb I and Cb II) which both harbor a dbl-homology (DH) and a pleckstrin-homology (PH) domain connected by a short linker sequence (Figure 2A; Kins et al., 2000). Such DH/PH tandem domains are found in all known dbl-like GEFs and are considered a signature of this family of GEF. The DH-domain mediates the GDP/GTP-exchange activity of bdl-like oncoproteins, and the human homolog of Cb, hPEM-2, has been first shown to be a GEF specific for the small GTPase Cdc42 (Reid et al., 1999). The PH region is thought to regulate the attachment of GEFs to membranes by binding to phosphoinositides (Hyvonen et al., 1995), and the PH-domain of Cb has been shown to interact specifically with phosphatidylinositol-3-phosphate (PI3P; Kalscheuer et al., 2009).
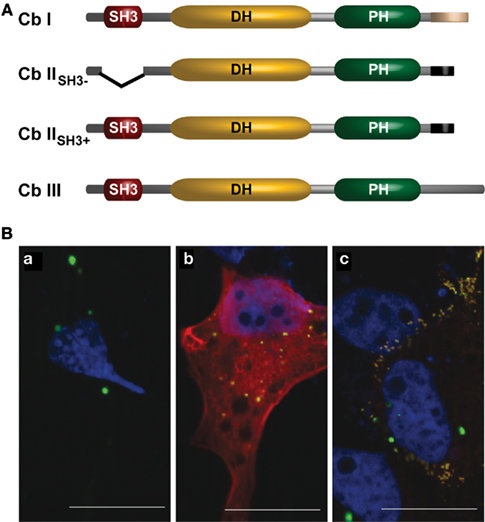
Figure 2. (A) Schematic representation of the domain structures of known Cb isoforms (according to Kins et al., 2000; Harvey et al., 2004). (B) Co-expression of Cb isoforms differentially affects gephyrin distribution in COS7 cells. COS7 cells expressing either (a) GFP–gephyrin alone (green), or together with (b) Myc-tagged CbIISH3+ (red), or (c) Myc-tagged CbIISH3− (red) are shown. Nuclei (blue) were visualized by DAPI-staining. Note that co-expression of Cb IISH3− and gephyrin induces the formation of numerous submembranous gephyrin clusters (c). Scale bars, 20 μm.
Murine Cb I and Cb II differ in their C-terminal regions (Kins et al., 2000). The longer variant Cb I contains a segment predicted to form a coiled coil, a structure known to mediate protein interactions. In 2004, Harvey et al. identified a third splice variant, Cb III (Figure 2A), which contains another C-terminus that shares 59 out of 60 residues with the C-terminus of human Cb (hPEM-2; Reid et al., 1999; Harvey et al., 2004).
A second region of variation is the N-terminus of Cb. The N-terminal region of Cb I contains an additional Src homology 3 (SH3) domain, whereas for Cb II two splice variants were detected, one containing (Cb IISH3+) and one lacking (Cb IISH3−) the SH3-domain (Figure 2A; Harvey et al., 2004). Since Cb IISH3− catalyzes nucleotide exchange on Cdc42 more efficiently than Cb I (Xiang et al., 2006), it is thought that similar to what is known for the homologous GEFs Asef 1 and 2 (Hamann et al., 2007; Mitin et al., 2007), the SH3-domain acts as an auto-inhibitory domain retaining Cb I, Cb IISH3+, and Cb III in an inactive conformation. To demonstrate an interaction of Cb with gephyrin in mammalian cells, Kins et al. (2000) used an assay previously employed to characterize the interaction of the GlyR β-subunit with gephyrin (Meyer et al., 1995). This assay was based on the observation that recombinant gephyrin forms large intracellular aggregates that “trap” hetero-oligomeric GlyRs (Kirsch and Betz, 1995) or green fluorescent protein (GFP) carrying a gephyrin-binding motif (Kneussel et al., 1999). These gephyrin aggregates have been shown to colocalize with the microtubule organizing center (MTOC) marker γ-tubulin (Maas et al., 2006), the actin uncapping protein ena/VASP as well as cotransfected γ-actin (Bausen et al., 2006), and thus appear to be linked to cytoskeletal structures. Figure 2Ba illustrates the large aggregates generated upon over-expression of GFP–gephyrin in mammalian cell lines. Notably, upon co-expression Cb IISH3+ colocalized with gephyrin in these large cytoplasmic aggregates (Figure 2Bb), whereas coexpressed Cb IISH3− not only colocalized but caused a redistribution of gephyrin into small (diameter, 0.2–0.5 μm) microclusters beneath the plasma membrane (Figure 2Bc). This observation led to the hypothesis that Cb IISH3− induces submembranous gephyrin microaggregates, which resemble the GlyR clusters found at the initial steps of postsynaptic membrane differentiation (Kins et al., 2000).
It is noteworthy that intracellular aggregates of neuronal endogenous gephyrin were also observed in vivo both at early stages of neuronal maturation (Colin et al., 1996, 1998; Sassoe-Pognetto and Wassle, 1997; Papadopoulos et al., 2008) and after partial denervation of the goldfish Mauthner cell, an identified neuron in the teleost brainstem (Seitanidou et al., 1992). This suggests that gephyrin accumulation in aggregates or “blobs” is not just an artifact caused by over-expression of recombinant gephyrin in heterologous cell lines. However, whether endogenously formed gephyrin aggregates colocalize with γ-tubulin (Maas et al., 2006), Mena (the neuronal ena/VASP isoform) and/or γ-actin (Bausen et al., 2006) or collybistin (Kins et al., 2000) remains to be elucidated.
Function of Cb: Insights from the Analysis of KO Mice
In vivo evidence for the requirement of Cb in gephyrin clustering was provided by inactivating the Cb gene in mice (Papadopoulos et al., 2007). This revealed that in the absence of this neuronal GEF, gephyrin and gephyrin-dependent GABAAR subtypes are lost from postsynaptic sites in selected regions of the mammalian forebrain (Figure 3 and not shown). As a consequence, reduced GABAergic transmission, altered hippocampal synaptic plasticity, increased anxiety scores and impaired spatial learning were observed in the Cb KO animals (Papadopoulos et al., 2007; Jedlicka et al., 2009). Furthermore, using the Cre-loxP strategy to specifically inactivate the Cb gene at different developmental stages, it could be shown that Cb is required for both the initial formation and the maintenance of GABAergic postsynapses in the mouse hippocampus (Papadopoulos et al., 2008). However, in the brainstem and spinal cord of Cb KO mice, glycinergic transmission and gephyrin clustering were indistinguishable from wildtype (WT) animals (Papadopoulos et al., 2007). These studies on Cb KO mice extended the diversity of regulatory mechanisms underlying the formation and maintenance of inhibitory postsynaptic membrane specializations by disclosing two types of gephyrin-dependent clustering reactions for inhibitory neurotransmitter receptors, Cb-dependent and Cb-independent ones. In addition, the unimpaired clustering of GABAARs containing the α2 and γ2 subunits in the cerebellum of the Cb KO mice, a region in which gephyrin cluster density was strongly affected, confirmed the existence of gephyrin-independent GABAAR clustering mechanisms as previously proposed (Knuesel et al., 1999; Fischer et al., 2000; Sassoe-Pognetto and Fritschy, 2000; Kneussel et al., 2001; Levi et al., 2004). The reasons for the region-specific roles of Cb are currently unknown but might reflect compensation or interaction specificity. Interestingly, Mdx mice lacking dystrophin exhibit a marked reduction in the clustering of GABAARs containing the α2 subunit but retain gephyrin clustering in the cerebellar cortex, suggesting a dystrophin-dependent and gephyrin-independent mechanism for the clustering of selected GABAAR subtypes (Knuesel et al., 1999). In line with this view, a rather recently published study demonstrates that synArf-GEF, a member of the brefeldin A-resistant Arf-GEF/IQSEC family, activates Arf6 and localizes preferentially at inhibitory postsynaptic specializations by forming a complex with the dystrophin-associated glycoprotein complex (DGC) and the synaptic scaffolding molecule, S-SCAM (Fukaya et al., 2011). S-SCAM was previously shown to interact directly with two inhibitory postsynaptic components, β-dystroglycan and NL2 (Sumita et al., 2007), suggesting that the interaction of synArf-GEF with dystrophin and S-SCAM enables synARf-GEF to activate Arf6 in the proximity of the DGC and NL2 at inhibitory synapses. Thus, in addition to Cb, synArf-GEF is the second known regulator of GTPases that shows a preferential localization at inhibitory postsynaptic sites.
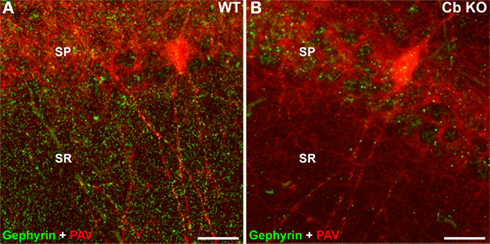
Figure 3. Reduced density of gephyrin clusters in the Cb KO hippocampus. Brain sections through the hippocampus [CA1, stratum pyramidale (SP), and stratum radiatum (SR)] of WT (A) and Cb KO (B) mice were stained with the gephyrin-specific mAb 7a (green) and an antibody specific for parvalbumin (PAV; red) and processed for confocal microscopy. Note that most of the remaining gephyrin clusters in sections derived from Cb KO mice are localized on dendrites of PAV-positive interneurons. Scale bars, 20 μm.
Similar to Cb-deficiency, deletion of NL2 in mice perturbs GABAergic transmission and leads to a loss of postsynaptic specializations (Poulopoulos et al., 2009). NLs have pivotal roles in organizing the assembly of different types of synapses (Varoqueaux et al., 2006) and ensuring proper synaptic connectivity (Shen and Scheiffele, 2010). NL2 binds to gephyrin through a conserved cytoplasmic motif and functions as a specific activator of Cb, thus guiding membrane tethering of the inhibitory postsynaptic scaffold (Poulopoulos et al., 2009). However, whereas Cb is required at both dendritic and perisomatic inhibitory synapses for proper clustering of gephyrin and gephyrin-dependent GABAARs (Papadopoulos et al., 2007), NL2 critically functions at perisomatic synapses to drive postsynaptic differentiation, suggesting that other proteins can compensate the loss of NL2 at dendritic inhibitory postsynaptic sites (Poulopoulos et al., 2009; Jedlicka et al., 2011). In agreement with this hypothesis, NL4 was recently identified as a second postsynaptic adhesion protein capable of interacting with gephyrin and Cb, whose co-expression in heterologous cells results in the formation of NL4, gephyrin and Cb submembranous microaggregates (Hoon et al., 2011). In the retina of NL2 KO mice, the number of NL4 immunoreactive puncta was significantly upregulated as compared to WT, suggesting that NL4 can replace NL2 at a subset of inhibitory synapses, and that NL4 and NL2 are functionally related (Hoon et al., 2011).
An interesting difference between the Cb KO and the NL2 KO mice emerged from analyzing the network activity in the dentate gyrus (DG) of Cb- and NL2-deficient mice after perforant-path (PP) stimulation in vivo (Jedlicka et al., 2009, 2011). Both in Cb KO and NL2 KO animals, an increase in granule cell (GC) excitability after PP stimulation in vivo was observed, as compared to WT. However, paired-pulse inhibition (PPI), which is a measure for somatic GABAergic network inhibition and depends on the activity of GABAergic interneurons in the dentate network (Sloviter, 1991; DiScenna and Teyler, 1994; Bronzino et al., 1997), was increased in Cb KO mice as compared to WT, consistent with enhanced GABAergic network inhibition in the DG of the Cb-deficient animals (Jedlicka et al., 2009). In contrast, in the DG of NL2-deficient mice PPI was severely impaired and associated with reduced perisomatic clustering of GABAARs containing the γ2 subunit, and the GABAAR-mediated miniature inhibitory postsynaptic currents recorded from NL2-deficient GCs were smaller (Jedlicka et al., 2011; Figure 4). As the reduction of gephyrin and GABAAR clusters containing the α2 and γ2 subunits seen in the DG was more pronounced in the Cb KO than the NL2 KO mice (Jedlicka et al., 2011), the enhanced PPI in the Cb KO animals was unexpected (Figure 4). A possible explanation could be non-linear network effects (Kapfer et al., 2007): reduced dendritic inhibition should make GCs more excitable, and hence GCs may recruit GABAergic feedback inhibition, mediated by soma-targeting interneurons, more effectively (Miles et al., 1996; Jedlicka et al., 2009; Figure 4B). Additional factors might also contribute to the compartment-specific alteration of GABAergic inhibition, e.g., a loss of Cb- and gephyrin-dependent GABAARs from soma-targeting interneurons (Simburger et al., 2001) leading to disinhibition. Furthermore, compartment-specific alterations of the GABAA reversal potential (EGABA) may significantly modulate the PPI of GCs, as altered expression of proteins involved in the regulation of chloride homeostasis has previously been reported to modulate PPI in the DG (Kang et al., 2006; Kwak et al., 2006).
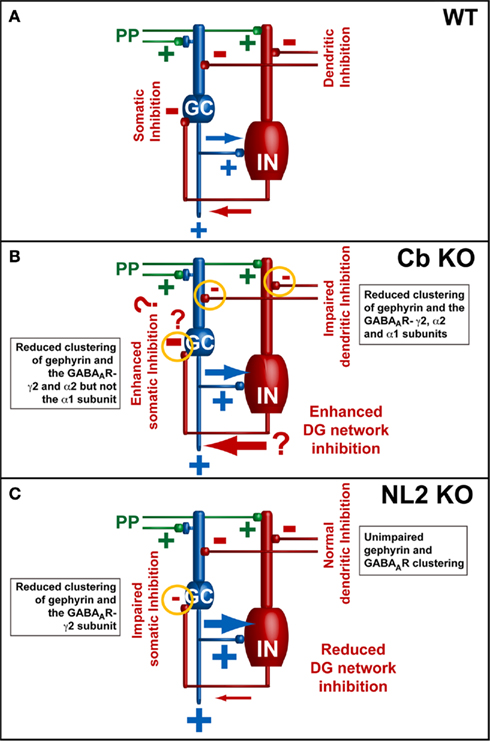
Figure 4. Models of the neuronal network in the dentate gyrus (modified from Jedlicka et al., 2009; Jedlicka et al., 2011). Basic dentate gyrus circuitry: PP, perforant-path; GC, granule cells; IN, GABAergic interneurons. (A) PP-stimulation initiates feed-forward excitation of GCs (PP → GC) along with feed-forward (PP → IN → GC) and feedback inhibition (PP → GC → IN → GC) responsible for the paired-pulse inhibition (PPI) of GC spikes in wildtype animals. (B,C) Electrophysiological data from Cb KO and NL2 KO mice indicate that the reduction of GABAergic inhibition leads to an enhanced ability of GCs to fire evoked action potentials [indicated by the larger blue arrows in (B,C)]. A previously published computational model of GABAAR-mediated PPI in the dentate gyrus indicated that PPI results mainly from a combination of perisomatic feed-forward and feedback inhibition of GCs by basket cells. Feed-forward inhibition mediated by basket cells appears to be the most significant source of PPI (Jedlicka et al., 2010). Thus, in the DG of NL2 KO mice (B), the observed reduction in PPI might be a consequence of changes in perisomatic inhibition, which lead to an overall increase of the excitation/inhibition ratio in the network. However, in the DG of Cb KO mice (A) PPI is increased (Jedlicka et al., 2009). This might be a consequence of changes in dendritic inhibition that lead to enhanced recruitment of somatic GABAergic feedback inhibition (indicated by the large red arrow). In addition, other factors leading to enhanced somatic inhibition (indicated by question marks) and contributing to homeostatic adaptation of the inhibitory network in the DG of Cb KO mice might also be involved in the regulation of the excitation/inhibition balance.
Electrophysiological recordings from the DG indicate that Cb KO mice display significantly higher field excitatory postsynaptic potentials (fEPSPs) than WT littermates, suggesting a higher efficacy of excitatory synapses in the Cb-deficient hippocampus (Jedlicka et al., 2009). Previous experiments in acute hippocampal slices in the CA1 region had yielded similar results, revealing a trend to higher fEPSP slope values (Papadopoulos et al., 2007). Notably, in a few Cb KO mice, generalized tonic-clonic convulsions were observed during routine handling (Papadopoulos, unpublished observations). In order to establish the origin of these seizures, the expression of c-fos was monitored; the upregulation of this transcription factor upon neuronal activation by epileptic seizures has been widely documented (Morgan et al., 1987; Figure 5). Double-labeling with c-fos and glutamate decarboxylase 65 (GAD65) antibodies disclosed that c-fos was mostly upregulated in GAD65-labeled interneurons (Figures 5D1–D0). This suggests a preferential activation of GABAergic interneurons in the DG 2 h after a seizure. However, epileptic seizures in Cb KO mice are very infrequent events, and therefore have not been characterized more thoroughly. Notably, Tuff et al. (1983) found enhanced PPI of GC in vivo following kindling, an animal model for temporal lobe epilepsy (Tuff et al., 1983). The GCs of the DG, which function as a gateway and rate-limiting control unit for the flow of information through the hippocampal trisynaptic circuit (Winson and Abzug, 1978), would be an appropriate site of endogenous modulation. Further work is needed to understand whether the compartment-specific alterations of GABAergic inhibition observed in the Cb KO hippocampus, might reflect a carefully orchestrated response of the brain to repeated seizures, designed to stabilize GC excitability and to reduce the likelihood of hippocampal activation and subsequent seizures.
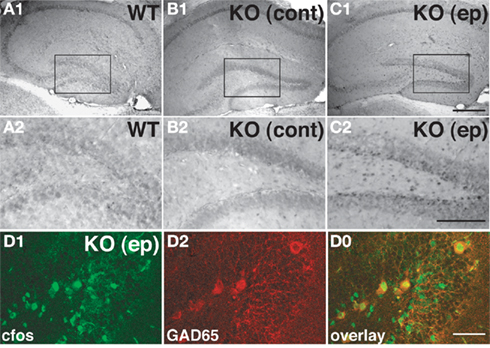
Figure 5. Induction of c-fos in the hippocampus of a Cb KO mouse after an epileptic seizure episode. (A–C) Analysis of sections derived from brains of a WT mouse, a Cb KO mouse that had not shown seizures at least 2 h before decapitation (control) and a Cb KO mouse that had an epileptic seizure (ep) 2 h before decapitation disclosed differences between the patterns of c-fos expression in the hippocampus. Specifically, in sections of the Cb KO (ep) brain (C1,C2) cells at the border between the GC layer and the hilus exhibited strong c-fos immunoreactivity as compared to control littermates (A1–B2). (A2–C2) Represent higher magnifications of the boxed areas indicated in (A1–C1), respectively. Scale bars, 400 μm (C1), 200 μm (C2). (D) Coronal section from the dentate gyrus of a Cb KO (ep) mouse double-stained for c-fos (D1) and GAD65 (D2). Note that GAD65-labeled cells show strong c-fos labeling in the overlay (D0). Scale bar, 50 μm.
Roles of Cb Domains in Gephyrin Clustering at Inhibitory Postsynapses
The SH3-Domain
As mentioned above, the SH3-domain of Cb is believed to act as an auto-inhibitory domain which retains Cb in an inactive conformation. Harvey et al. (2004) examined SH3 domain usage in the Cb II and Cb III splice variants and found that in postnatal rodent brain the “inactive” Cb IISH3+ and Cb IIISH3+ isoforms are predominant (Harvey et al., 2004). Moreover, in transfected cortical neurons both the SH3-containing and SH3-lacking splice variants of Cb were found to similarly redistribute gephyrin into synaptic clusters (Harvey et al., 2004). This suggested that in specific neuronal subpopulations SH3-containing Cb isoforms are locally “activated” by an SH3-interacting protein. In agreement with this hypothesis, NL2 and the α2 subunit of GABAARs were recently shown to interact with both gephyrin and the SH3-domain of Cb and to induce Cb-dependent gephyrin clustering by relieving SH3-mediated inhibition (Poulopoulos et al., 2009; Saiepour et al., 2010). This led to a model of postsynaptic membrane differentiation according to which the recruitment of NL2 under GABAergic presynaptic terminals is thought to create nucleation sites for gephyrin deposition and subsequent inhibitory receptor clustering by activating Cb through a mechanism involving NL2–Cb and NL2–gephyrin interactions (Poulopoulos et al., 2009).
Based on this model, the SH3 domain of Cb is now thought to function as trigger for a switch mechanism, which is specifically activated by NL2 (see Figure 6). Additionally, the inability of NL1 and NL3 to bind to and activate Cb (Poulopoulos et al., 2009) explains why these NL isoforms do not initiate inhibitory synaptic assembly. A more detailed structure-function analysis of the tripartite NL2–Cb–gephyrin complex is required for further validation and refinement of the signaling mechanism that underlies the assembly of the GABAergic inhibitory synapses. In addition, a more in-depth investigation of the NL2/NL4-dependent but Cb-independent clustering of GlyRs (Poulopoulos et al., 2009; Hoon et al., 2011) is needed in order to unravel how the formation of inhibitory synapses is triggered in brainstem and spinal cord, where glycinergic rather than GABAergic inhibition prevails.
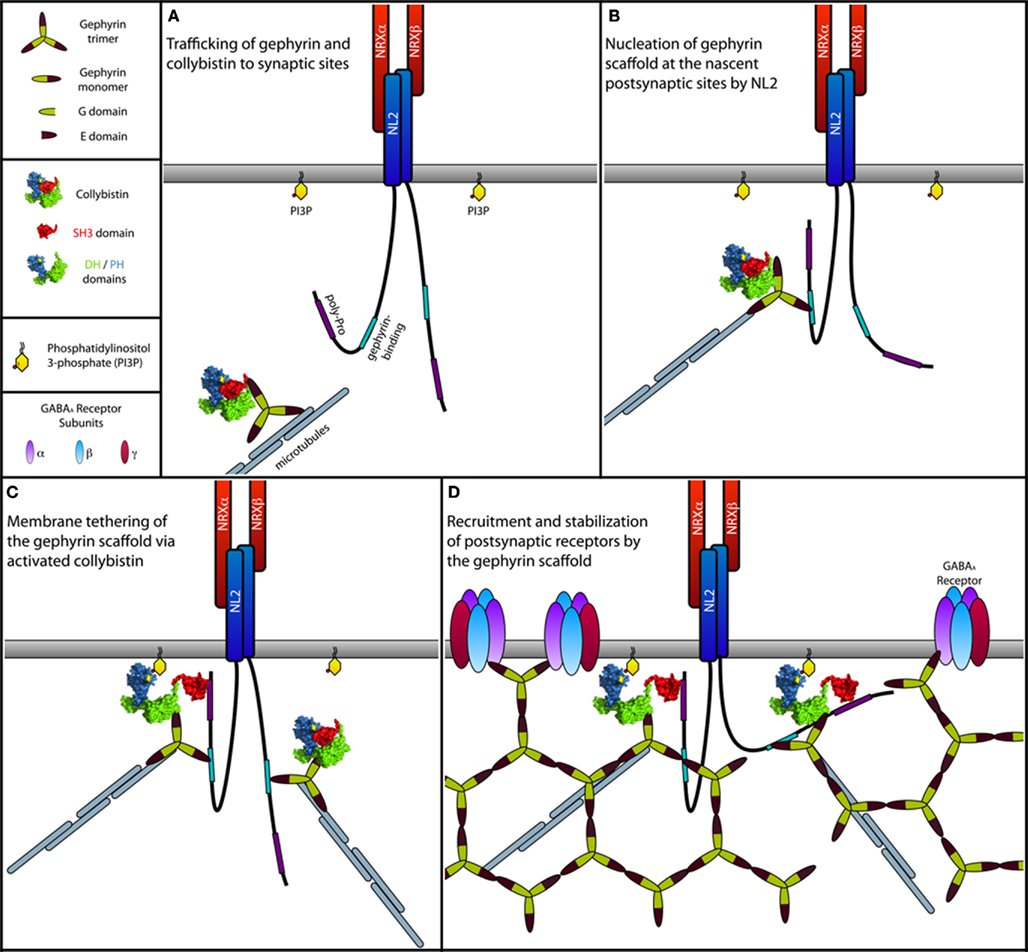
Figure 6. Model of the NL2-induced activation of Cb at inhibitory postsynapses. (A) Gephyrin trimers associated with Cb are transported to subsynaptic sites along the microtubules by forming complexes with motor proteins. For simplicity, motor proteins are not indicated. During transport, Cb adopts a closed conformation, in which the SH3 domain folds back onto the tandem DH/PH-domains, similar to what is known for the autoinhibited form of Asef, the closest homolog of Cb (Kawasaki et al., 2007; Mitin et al., 2007). In this closed inactive conformation, both the catalytic activity of the DH-domain and binding of the PH-domain to membrane phosphoinositides (see text below) are suppressed due to steric hindrance imposed by the SH3 domain. (B) At postsynaptic sites, NL2 molecules clustered by presynaptically expressed Neurexins (NRXs) interact transiently with the cytoplasmic gephyrin–Cb complex. This interaction brings the proline-rich cytoplasmic domain of NL2 into close vicinity of the SH3 domain of Cb. (C) A typical SH3 domain–ligand interaction mediated by a PXXP motif in the cytoplasmic domain of NL2 leads to a structural rearrangement in Cb, resulting in a more open conformation of this GEF. Similar to the APC mediated activation of Asef (Mitin et al., 2007), binding of NL2 is thought to interfere with the intramolecular interactions between the SH3 and the DH/PH-domains. This transition toward an open state now allows the PH-domain to bind PI3P-rich domains within the plasma membrane, which might be generated postsynaptically as a product of a yet unknown signaling mechanism (see text below) induced by either NL2, Cb, or other inhibitory synaptic molecules. Together with Cb, gephyrin is co-recruited to the PI3P-rich membrane domains where it assembles into the postsynaptic gephyrin scaffold. (D) GABAARs are then recruited to the growing postsynaptic scaffold.
The DH-Domain
Dbl-homology-domains are known to mediate the GDP/GTP-exchange activity of bdl-like oncoproteins (Hart et al., 1994). A hallmark of small GTPases is to undergo structural changes in response to alternate binding of GDP and GTP. The GDP-bound “off” state and the GTP-bound “on” state recognize different partner proteins, thereby allowing these G-proteins to function as molecular switches. The GTP-bound form interacts with effectors and activates pathways that affect cell morphology, trafficking, growth, differentiation and apoptosis. Inactivation of the GTP-bound form requires binding of the GTPase to another class of GTPase-interacting proteins, the GTPase activating proteins (GAPs; for review see Cherfils and Chardin, 1999). GEFs stimulate the dissociation of tightly bound GDP from small G-proteins in response to upstream signals. This reaction involves several stages: first, the GEF forms a low-affinity docking complex with the GDP-bound GTPase. GDP then dissociates from this initial complex, which results in a high-affinity binary GEF–G-protein complex. This intermediate does not accumulate in the cell because it is rapidly dissociated upon GTP binding (Cherfils and Chardin, 1999).
Although previous experiments have clearly shown that Cb’s gephyrin clustering function is “activated” by NL2, the signaling mechanisms through which Cb activation induces gephyrin recruitment remain enigmatic. As Cdc42 is the only GTPase known to be activated by Cb (Reid et al., 1999; Xiang et al., 2006), it was previously believed that the interaction of Cb with Cdc42 and activated Cdc42 are required for gephyrin clustering at inhibitory postsynaptic sites (Kneussel and Betz, 2000). However, the analysis of Cb mutants deficient in catalyzing GDP/GTP-exchange on Cdc42 and of Cdc42 conditional KO mice clearly demonstrated that Cdc42 is not essential for inhibitory synapse formation (Reddy-Alla et al., 2010). Importantly, inactivation of the Cdc42 gene in the mouse forebrain at early developmental stages had no effect on gephyrin and GABAAR cluster densities in the hippocampus (Reddy-Alla et al., 2010). These findings suggest that other, as yet unidentified Rho-like GTPases are activated by Cb at synaptic sites. Dbl-family proteins display varied selectivities, and the available analyses mainly focused on Cdc42, Rac1 and RhoA (Reid et al., 1999; Schmidt and Hall, 2002). Non-conserved residues that reside within the GTPase interaction sites of the DH-domains are likely candidates for determining specific coupling (Worthylake et al., 2000). A possible scenario thus could be that the DH-domain of Cb discriminates among different G-proteins, thereby regulating the spatio-temporal activation of more than one Rho–GTPase in response to distinct stimuli. Ultimately, different approaches will be needed to identify novel Cb cognate Rho–GTPases in the mouse brain and determine their importance for inhibitory synapse formation.
The PH-Domain
Deletion of the PH-domain of Cb has been previously shown to abolish PI3P binding without affecting the interaction of Cb with gephyrin (Kalscheuer et al., 2009). Furthermore, over-expression of a Cb IISH3− deletion mutant lacking the PH-domain interfered with gephyrin clustering at synaptic sites (Harvey et al., 2004). However, based on these results it was not clear whether the effects of PH-domain deletion are indeed due to impaired phosphoinositide binding or due to more global effects on protein structure. Reddy-Alla et al. (2010) analyzed the interaction of the Cb PH-domain with PI3P in more detail. In this study, a Cb IISH3− mutant was generated in which the arginine residues R303 and R304 were both replaced by asparagines. These residues reside within the β3–β4 loop of the Cb PH-domain, and their positively charged side chains are surface-exposed. The positively charged surfaces of the β1–β2 and β3–β4 loops of several other PH-domains are known to be involved in membrane lipid anchoring (Hyvonen et al., 1995; Yu et al., 2004; DiNitto and Lambright, 2006). Using immobilized PI3P and purified glutathione-S-transferase (GST)-tagged Cb IISH3− and GST–Cb IISH3−-RR303-304NN in a lipid overlay assay, the RR303-304 residues were confirmed to be essential for Cb IISH3− binding to this phosphoinositide. Co-expression of the Cb IISH3−-RR303-304NN mutant with GFP–gephyrin in NIH-3T3 cells led to the formation of large intracellular GFP–gephyrin aggregates, and its over-expression in cultured hippocampal neurons caused a strong reduction of synaptic GFP–gephyrin clusters colocalizing with epitope-tagged Cb (Reddy-Alla et al., 2010). Thus, Cb binding to PI3P appears to be essential for its efficient recruitment to postsynaptic sites together with gephyrin.
PI3P was considered for a long time as a lipid constitutively present on endosomes, mainly produced by class III phosphatidylinositol 3-kinase (PI3K; Gillooly et al., 2000; Vicinanza et al., 2008), and without any dynamic roles in signaling. However, during the past few years it has become increasingly evident that pools of PI3P can be specifically generated upon cellular stimulation, and PI3P has emerged as a critical intracellular second messenger involved in different pathways (for review see Falasca and Maffucci, 2006). Notably, PI3P is known to promote plasma membrane translocation of the glucose transporter 4 (Maffucci et al., 2003). Whether Cb can bind to gephyrin associated with PI3P-containing intracellular vesicles, and whether this interaction might be crucial for the translocation of gephyrin and Cb to the plasma membrane remains to be explored. More recently, Falasca et al. (2007) showed that activation of a member of the class II PI3Ks, PI3K–C2α, but not activation of the class III PI3K, hVps34, generates PI3P in the plasma membrane (Falasca et al., 2007). In addition, previous studies on the differential sensitivities to PI3K inhibitors of the endosomal versus the plasma membrane pool of PI3P strongly suggest that multiple enzymes are involved in the generation of PI3P (Maffucci et al., 2003, 2005). The general consensus is that class III PI3Ks are responsible for the production of the constitutive endosomal pool of PI3P (Schu et al., 1993). Several lines of evidence suggest a potential role of class II PI3K isoforms in agonist-mediated regulations of cellular functions. This includes activation of PI3K–C2α by insulin, TNFα and leptin (Brown et al., 1999; Ktori et al., 2003), and of PI3K–C2β by insulin and LPA (Brown and Shepherd, 2001; Maffucci et al., 2005). It appears feasible that inhibitory synapse function involves a yet unknown PI3K enzyme that generates PI3P at specific sites of the neuronal plasma membrane, and thereby regulates Cb-mediated gephyrin deposition at synaptic sites. Clearly further work is needed to fully understand how Cb regulates gephyrin deposition at inhibitory postsynapses.
Perspectives
Unraveling the molecular mechanisms of the Cb-dependent transport, clustering, and maintenance of gephyrin and GABAARs at inhibitory synapses will be a key step toward a better understanding of GABAergic transmission in health and disease. This will require detailed knowledge of Cb’s specificity for Rho–GTPases expressed in the mammalian forebrain. Once novel candidate GTPases have been identified, defining how their Cb-dependent activation affects gephyrin postsynaptic clustering would be the next step toward understanding the role of this GEF in inhibitory synapse formation. Previous work indicates that different actin-associated proteins (reviewed in Kneussel and Betz, 2000; Moss and Smart, 2001), including Mena, the neuronal ena/VASP isoform (Giesemann et al., 2003), interact with gephyrin. However, whether Cb can regulate actin cytoskeleton dynamics by activating a Rho–GTPase other than Cdc42 and whether a Rho–GTPase activation is required for gephyrin postsynaptic clustering is currently not known.
Despite the recent progress, the mechanism of PI3P-mediated anchoring of Cb at the postsynaptic membrane remains poorly understood. Whether PI3P is interacting with Cb directly at the plasma membrane upon stimulated accumulation of this phosphoinositide, or whether Cb can bind to gephyrin associated with PI3P at intracellular vesicles remains to be further investigated.
Furthermore, structural analysis of the tripartite NL2–gephyrin–Cb complex would be required for complete understanding how the interaction of NL2 with Cb’s SH3-domain mediates Cb activation and subsequent gephyrin postsynaptic clustering.
In addition, a detailed spatio-temporal analysis on the expression of individual Cb isoforms in different regions of the mammalian brain, as well as an electron microscopic demonstration of the subcellular localization of the different isoforms during development will help to understand how the activation of Cb contributes to gephyrin postsynaptic clustering. Lastly, anatomical and electrophysiological studies with Cb-deficient mice will be required to identify the brain regions and neuron subclasses that contain Cb-dependent inhibitory postsynapses and to unravel the roles of Cb in inhibitory synaptic plasticity and adaptive responses to altered network activity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs Heinrich Betz, Nils Brose, Liam Tuffy, and Dilja Krueger for helpful discussions and comments on the manuscript. Work of the authors cited in this review was supported by grants from the Max-Planck Society, Deutsche Forschungsgemeinschaft, European Community, and Fonds der Chemischen Industrie.
References
Allison, D. W., Chervin, A. S., Gelfand, V. I., and Craig, A. M. (2000). Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J. Neurosci. 20, 4545–4554.
Bausen, M., Fuhrmann, J. C., Betz, H., and O’Sullivan, G. A. (2006). The state of the actin cytoskeleton determines its association with gephyrin: role of ena/VASP family members. Mol. Cell. Neurosci. 31, 376–386.
Bronzino, J. D., Blaise, J. H., and Morgane, P. J. (1997). The paired-pulse index: a measure of hippocampal dentate granule cell modulation. Ann. Biomed. Eng. 25, 870–873.
Brown, R. A., Domin, J., Arcaro, A., Waterfield, M. D., and Shepherd, P. R. (1999). Insulin activates the alpha isoform of class II phosphoinositide 3-kinase. J. Biol. Chem. 274, 14529–14532.
Brown, R. A., and Shepherd, P. R. (2001). Growth factor regulation of the novel class II phosphoinositide 3-kinases. Biochem. Soc. Trans. 29, 535–537.
Cherfils, J., and Chardin, P. (1999). GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem. Sci. 24, 306–311.
Colin, I., Rostaing, P., Augustin, A., and Triller, A. (1998). Localization of components of glycinergic synapses during rat spinal cord development. J. Comp. Neurol. 398, 359–372.
Colin, I., Rostaing, P., and Triller, A. (1996). Gephyrin accumulates at specific plasmalemma loci during neuronal maturation in vitro. J. Comp. Neurol. 374, 467–479.
DiNitto, J. P., and Lambright, D. G. (2006). Membrane and juxtamembrane targeting by PH and PTB domains. Biochim. Biophys. Acta 1761, 850–867.
DiScenna, P. G., and Teyler, T. J. (1994). Development of inhibitory and excitatory synaptic transmission in the rat dentate gyrus. Hippocampus 4, 569–576.
Essrich, C., Lorez, M., Benson, J. A., Fritschy, J. M., and Luscher, B. (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1, 563–571.
Falasca, M., Hughes, W. E., Dominguez, V., Sala, G., Fostira, F., Fang, M. Q., Cazzolli, R., Shepherd, P. R., James, D. E., and Maffucci, T. (2007). The role of phosphoinositide 3-kinase C2alpha in insulin signaling. J. Biol. Chem. 282, 28226–28236.
Falasca, M., and Maffucci, T. (2006). Emerging roles of phosphatidylinositol 3-monophosphate as a dynamic lipid second messenger. Arch. Physiol. Biochem. 112, 274–284.
Feng, G., Tintrup, H., Kirsch, J., Nichol, M. C., Kuhse, J., Betz, H., and Sanes, J. R. (1998). Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282, 1321–1324.
Fischer, F., Kneussel, M., Tintrup, H., Haverkamp, S., Rauen, T., Betz, H., and Wassle, H. (2000). Reduced synaptic clustering of GABA and glycine receptors in the retina of the gephyrin null mutant mouse. J. Comp. Neurol. 427, 634–648.
Fritschy, J. M., Harvey, R. J., and Schwarz, G. (2008). Gephyrin: where do we stand, where do we go? Trends Neurosci. 31, 257–264.
Fuhrmann, J. C., Kins, S., Rostaing, P., El Far, O., Kirsch, J., Sheng, M., Triller, A., Betz, H., and Kneussel, M. (2002). Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J. Neurosci. 22, 5393–5402.
Fukaya, M., Kamata, A., Hara, Y., Tamaki, H., Katsumata, O., Ito, N., Takeda, S., Hata, Y., Suzuki, T., Watanabe, M., Harvey, R. J., and Sakagami, H. (2011). SynArfGEF is a guanine nucleotide exchange factor for Arf6 and localizes preferentially at post-synaptic specializations of inhibitory synapses. J. Neurochem. 116, 1122–1137.
Giesemann, T., Schwarz, G., Nawrotzki, R., Berhorster, K., Rothkegel, M., Schluter, K., Schrader, N., Schindelin, H., Mendel, R. R., Kirsch, J., and Jockusch, B. M. (2003). Complex formation between the postsynaptic scaffolding protein gephyrin, profilin, and mena: a possible link of the microfilament system. J. Neurosci. 23, 8330–8339.
Gillooly, D. J., Morrow, I. C., Lindsay, M., Gould, R., Bryant, N. J., Gaullier, J. M., Parton, R. G., and Stenmark, H. (2000). Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19, 4577–4588.
Grosskreutz, Y., Betz, H., and Kneussel, M. (2003). Rescue of molybdenum cofactor biosynthesis in gephyrin-deficient mice by a Cnx1 transgene. Biochem. Biophys. Res. Commun. 301, 450–455.
Hamann, M. J., Lubking, C. M., Luchini, D. N., and Billadeau, D. D. (2007). Asef2 functions as a Cdc42 exchange factor and is stimulated by the release of an autoinhibitory module from a concealed C-terminal activation element. Mol. Cell. Biol. 27, 1380–1393.
Hart, M. J., Eva, A., Zangrilli, D., Aaronson, S. A., Evans, T., Cerione, R. A., and Zheng, Y. (1994). Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J. Biol. Chem. 269, 62–65.
Harvey, K., Duguid, I. C., Alldred, M. J., Beatty, S. E., Ward, H., Keep, N. H., Lingenfelter, S. E., Pearce, B. R., Lundgren, J., Owen, M. J., Smart, T. G., Luscher, B., Rees, M. I., and Harvey, R. J. (2004). The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826.
Hoon, M., Soykan, T., Falkenburger, B., Hammer, M., Patrizi, A., Schmidt, K. F., Sassoe-Pognetto, M., Lowel, S., Moser, T., Taschenberger, H., Brose, N., and Varoqueaux, F. (2011). Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc. Natl. Acad. Sci. U.S.A. 108, 3053–3058.
Hyvonen, M., Macias, M. J., Nilges, M., Oschkinat, H., Saraste, M., and Wilmanns, M. (1995). Structure of the binding site for inositol phosphates in a PH domain. EMBO J. 14, 4676–4685.
Jedlicka, P., Deller, T., and Schwarzacher, S. W. (2010). Computational modeling of GABAA receptor-mediated paired-pulse inhibition in the dentate gyrus. J. Comput. Neurosci. 29, 509–519.
Jedlicka, P., Hoon, M., Papadopoulos, T., Vlachos, A., Winkels, R., Poulopoulos, A., Betz, H., Deller, T., Brose, N., Varoqueaux, F., and Schwarzacher, S. W. (2011). Increased dentate gyrus excitability in neuroligin-2-deficient mice in vivo. Cereb. Cortex 21, 357–367.
Jedlicka, P., Papadopoulos, T., Deller, T., Betz, H., and Schwarzacher, S. W. (2009). Increased network excitability and impaired induction of long-term potentiation in the dentate gyrus of collybistin-deficient mice in vivo. Mol. Cell. Neurosci. 41, 94–100.
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., Deas, E., Venkateswarlu, K., Menzel, C., Ullmann, R., Tommerup, N., Dalpra, L., Tzschach, A., Selicorni, A., Luscher, B., Ropers, H. H., Harvey, K., and Harvey, R. J. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68.
Kang, T. C., Kim, D. S., Kim, J. E., Kwak, S. E., Yoo, K. Y., Hwang, I. K., Jung, J. Y., Won, M. H., Kwon, O. S., and Choi, S. Y. (2006). Altered expression of K+ -Cl- cotransporters affects fast paired-pulse inhibition during GABA receptor activation in the gerbil hippocampus. Brain Res. 1072, 8–14.
Kapfer, C., Glickfeld, L. L., Atallah, B. V., and Scanziani, M. (2007). Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat. Neurosci. 10, 743–753.
Kawasaki, Y., Sagara, M., Shibata, Y., Shirouzu, M., Yokoyama, S., and Akiyama, T. (2007). Identification and characterization of Asef2, a guanine-nucleotide exchange factor specific for Rac1 and Cdc42. Oncogene 26, 7620–7267.
Kim, E. Y., Schrader, N., Smolinsky, B., Bedet, C., Vannier, C., Schwarz, G., and Schindelin, H. (2006). Deciphering the structural framework of glycine receptor anchoring by gephyrin. EMBO J. 25, 1385–1395.
Kins, S., Betz, H., and Kirsch, J. (2000). Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat. Neurosci. 3, 22–29.
Kirsch, J., and Betz, H. (1993). Widespread expression of gephyrin, a putative glycine receptor-tubulin linker protein, in rat brain. Brain Res. 621, 301–310.
Kirsch, J., and Betz, H. (1995). The postsynaptic localization of the glycine receptor-associated protein gephyrin is regulated by the cytoskeleton. J. Neurosci. 15, 4148–4156.
Kirsch, J., Langosch, D., Prior, P., Littauer, U. Z., Schmitt, B., and Betz, H. (1991). The 93-kDa glycine receptor-associated protein binds to tubulin. J. Biol. Chem. 266, 22242–22245.
Kneussel, M. (2006). Dynamic stabilization: structural plasticity at inhibitory postsynaptic sites. Traffic 7, 1604–1606.
Kneussel, M., and Betz, H. (2000). Clustering of inhibitory neurotransmitter receptors at developing postsynaptic sites: the membrane activation model. Trends Neurosci. 23, 429–435.
Kneussel, M., Brandstatter, J. H., Gasnier, B., Feng, G., Sanes, J. R., and Betz, H. (2001). Gephyrin-independent clustering of postsynaptic GABAA receptor subtypes. Mol. Cell. Neurosci. 17, 973–982.
Kneussel, M., Brandstatter, J. H., Laube, B., Stahl, S., Muller, U., and Betz, H. (1999). Loss of postsynaptic GABAA receptor clustering in gephyrin-deficient mice. J. Neurosci. 19, 9289–9297.
Knuesel, I., Mastrocola, M., Zuellig, R. A., Bornhauser, B., Schaub, M. C., and Fritschy, J. M. (1999). Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur. J. Neurosci. 11, 4457–4462.
Ktori, C., Shepherd, P. R., and O’Rourke, L. (2003). TNF-alpha and leptin activate the alpha-isoform of class II phosphoinositide 3-kinase. Biochem. Biophys. Res. Commun. 306, 139–143.
Kwak, S. E., Kim, J. E., Kim, D. S., Won, M. H., Lee, H. J., Choi, S. Y., Kwon, O. S., Kim, J. S., and Kang, T. C. (2006). Differential paired-pulse responses between the CA1 region and the dentate gyrus are related to altered CLC-2 immunoreactivity in the pilocarpine-induced rat epilepsy model. Brain Res. 1115, 162–168.
Lee, H.-J., Adham, I. M., Schwarz, G., Kneussel, M., Sass, J. O., Engel, W., and Reiss, J. (2002). Molybdenum cofactor-deficient mice resemble the phenotype of human patients. Hum. Mol. Genet. 11, 3309–3317.
Levi, S., Logan, S. M., Tovar, K. R., and Craig, A. M. (2004). Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J. Neurosci. 24, 207–217.
Liu, M. T., Wuebbens, M. M., Rajagopalan, K. V., and Schindelin, H. (2000). Crystal structure of the gephyrin-related molybdenum cofactor biosynthesis protein MogA from Escherichia coli. J. Biol. Chem. 275, 1814–1822.
Luscher, B., and Keller, C. A. (2004). Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol. Ther. 102, 195–221.
Maas, C., Belgardt, D., Lee, H. K., Heisler, F. F., Lappe-Siefke, C., Magiera, M. M., van Dijk, J., Hausrat, T. J., Janke, C., and Kneussel, M. (2009). Synaptic activation modifies microtubules underlying transport of postsynaptic cargo. Proc. Natl. Acad. Sci. U.S.A. 106, 8731–8736.
Maas, C., Tagnaouti, N., Loebrich, S., Behrend, B., Lappe-Siefke, C., and Kneussel, M. (2006). Neuronal cotransport of glycine receptor and the scaffold protein gephyrin. J. Cell Biol. 172, 441–451.
Maffucci, T., Brancaccio, A., Piccolo, E., Stein, R. C., and Falasca, M. (2003). Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 22, 4178–4189.
Maffucci, T., Cooke, F. T., Foster, F. M., Traer, C. J., Fry, M. J., and Falasca, M. (2005). Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J. Cell Biol. 169, 789–799.
Mammoto, A., Sasaki, T., Asakura, T., Hotta, I., Imamura, H., Takahashi, K., Matsuura, Y., Shirao, T., and Takai, Y. (1998). Interactions of drebrin and gephyrin with profilin. Biochem. Biophys. Res. Commun. 243, 86–89.
Meyer, G., Kirsch, J., Betz, H., and Langosch, D. (1995). Identification of a gephyrin binding motif on the glycine receptor beta subunit. Neuron 15, 563–572.
Miles, R., Toth, K., Gulyas, A. I., Hajos, N., and Freund, T. F. (1996). Differences between somatic and dendritic inhibition in the hippocampus. Neuron 16, 815–823.
Mitin, N., Betts, L., Yohe, M. E., Der, C. J., Sondek, J., and Rossman, K. L. (2007). Release of autoinhibition of Asef by APC leads to Cdc42 activation and tumor suppression. Nat. Struct. Mol. Biol. 14, 814–823.
Morgan, J. I., Cohen, D. R., Hempstead, J. L., and Curran, T. (1987). Mapping patterns of c-fos expression in the central nervous system after seizure. Science 237, 192–197.
Moss, S. J., and Smart, T. G. (2001). Constructing inhibitory synapses. Nat. Rev. Neurosci. 2, 240–250.
Papadopoulos, T., Eulenburg, V., Reddy-Alla, S., Mansuy, I. M., Li, Y., and Betz, H. (2008). Collybistin is required for both the formation and maintenance of GABAergic postsynapses in the hippocampus. Mol. Cell. Neurosci. 39, 161–169.
Papadopoulos, T., Korte, M., Eulenburg, V., Kubota, H., Retiounskaia, M., Harvey, R. J., Harvey, K., O’Sullivan, G. A., Laube, B., Hulsmann, S., Geiger, J. R., and Betz, H. (2007). Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 26, 3888–3899.
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Papadopoulos, T., Zhang, M., Paarmann, I., Fuchs, C., Harvey, K., Jedlicka, P., Schwarzacher, S. W., Betz, H., Harvey, R. J., Brose, N., Zhang, W., and Varoqueaux, F. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642.
Ramming, M., Kins, S., Werner, N., Hermann, A., Betz, H., and Kirsch, J. (2000). Diversity and phylogeny of gephyrin: tissue-specific splice variants, gene structure, and sequence similarities to molybdenum cofactor synthesizing and cytoskeleton-associated proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 10266–10271.
Reddy-Alla, S., Schmitt, B., Birkenfeld, J., Eulenburg, V., Dutertre, S., Bohringer, C., Gotz, M., Betz, H., and Papadopoulos, T. (2010). PH-domain-driven targeting of collybistin but not Cdc42 activation is required for synaptic gephyrin clustering. Eur. J. Neurosci. 31, 1173–1184.
Reid, T., Bathoorn, A., Ahmadian, M. R., and Collard, J. G. (1999). Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J. Biol. Chem. 274, 33587–33593.
Saiepour, L., Fuchs, C., Patrizi, A., Sassoe-Pognetto, M., Harvey, R. J., and Harvey, K. (2010). Complex role of collybistin and gephyrin in GABAA receptor clustering. J. Biol. Chem. 285, 29623–29631.
Saiyed, T., Paarmann, I., Schmitt, B., Haeger, S., Sola, M., Schmalzing, G., Weissenhorn, W., and Betz, H. (2007). Molecular basis of gephyrin clustering at inhibitory synapses: role of G- and E-domain interactions. J. Biol. Chem. 282, 5625–5632.
Sassoe-Pognetto, M., and Fritschy, J. M. (2000). Mini-review: gephyrin, a major postsynaptic protein of GABAergic synapses. Eur. J. Neurosci. 12, 2205–2210.
Sassoe-Pognetto, M., and Wassle, H. (1997). Synaptogenesis in the rat retina: subcellular localization of glycine receptors, GABAA receptors, and the anchoring protein gephyrin. J. Comp. Neurol. 381, 158–174.
Schmidt, A., and Hall, A. (2002). Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609.
Schu, P. V., Takegawa, K., Fry, M. J., Stack, J. H., Waterfield, M. D., and Emr, S. D. (1993). Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260, 88–91.
Seitanidou, T., Nicola, M. A., Triller, A., and Korn, H. (1992). Partial glycinergic denervation induced transient changes in the distribution of a glycine receptor-associated protein in a central neuron. J. Neurosci. 12, 116–131.
Shen, K., and Scheiffele, P. (2010). Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 33, 473–507.
Simburger, E., Plaschke, M., Fritschy, J. M., and Nitsch, R. (2001). Localization of two major GABAA receptor subunits in the dentate gyrus of the rat and cell type-specific up-regulation following entorhinal cortex lesion. Neuroscience 102, 789–803.
Sloviter, R. S. (1991). Feedforward and feedback inhibition of hippocampal principal cell activity evoked by perforant path stimulation: GABA-mediated mechanisms that regulate excitability in vivo. Hippocampus 1, 31–40.
Sola, M., Bavro, V. N., Timmins, J., Franz, T., Ricard-Blum, S., Schoehn, G., Ruigrok, R. W. H., Paarmann, I., Saiyed, T., O’Sullivan, G. A., Schmitt, B., Betz, H., and Weissenhorn, W. (2004). Structural basis of dynamic glycine receptor clustering by gephyrin. EMBO J. 23, 2510–2519.
Sola, M., Kneussel, M., Heck, I. S., Betz, H., and Weissenhorn, W. (2001). X-ray crystal structure of the trimeric N-terminal domain of gephyrin. J. Biol. Chem. 276, 25294–25301.
Stallmeyer, B., Schwarz, G., Schulze, J., Nerlich, A., Reiss, J., Kirsch, J., and Mendel, R. R. (1999). The neurotransmitter receptor-anchoring protein gephyrin reconstitutes molybdenum cofactor biosynthesis in bacteria, plants, and mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 96, 1333–1338.
Sumita, K., Sato, Y., Iida, J., Kawata, A., Hamano, M., Hirabayashi, S., Ohno, K., Peles, E., and Hata, Y. (2007). Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J. Neurochem. 100, 154–166.
Tuff, L. P., Racine, R. J., and Mishra, R. K. (1983). The effects of kindling on GABA-mediated inhibition in the dentate gyrus of the rat. II. Receptor binding. Brain Res. 277, 91–98.
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., Zhang, W., Südhof, T. C., and Brose, N. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754.
Vicinanza, M., D’Angelo, G., Di Campli, A., and De Matteis, M. A. (2008). Function and dysfunction of the PI system in membrane trafficking. EMBO J. 27, 2457–2470.
Winson, J., and Abzug, C. (1978). Neuronal transmission through hippocampal pathways dependent on behavior. J. Neurophysiol. 41, 716–732.
Worthylake, D. K., Rossman, K. L., and Sondek, J. (2000). Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature 408, 682–688.
Xiang, S., Kim, E. Y., Connelly, J. J., Nassar, N., Kirsch, J., Winking, J., Schwarz, G., and Schindelin, H. (2006). The crystal structure of Cdc42 in complex with collybistin II, a gephyrin-interacting guanine nucleotide exchange factor. J. Mol. Biol. 359, 35–46.
Keywords: inhibitory synapse, GEF, GABA(A) receptor, GlyR, synaptogenesis, neuroligin, SH3-domain, PI3P
Citation: Papadopoulos T and Soykan T (2011) The role of collybistin in gephyrin clustering at inhibitory synapses: facts and open questions. Front. Cell. Neurosci. 5:11. doi: 10.3389/fncel.2011.00011
Received: 29 March 2011; Paper pending published: 24 April 2011;
Accepted: 13 June 2011; Published online: 24 June 2011.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Matthias Kneussel, Zentrum für Molekulare Neurobiologie Hamburg, GermanyRichard Olsen, University of California Los Angeles, USA
Copyright: © 2011 Papadopoulos and Soykan. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Theofilos Papadopoulos, Max-Planck Institute of Experimental Medicine, Department of Molecular Neurobiology, Hermann-Rein Str. 3, 37075 Göttingen, Germany. e-mail:cGFwYWRvcG91bG9zQGVtLm1wZy5kZQ==