 Rosemarie Grantyn1,2,3*
Rosemarie Grantyn1,2,3* Christian Henneberger1,4
Christian Henneberger1,4 René Jüttner1,5
René Jüttner1,5 Jochen C. Meier1,6
Jochen C. Meier1,6 Sergei Kirischuk1,7
Sergei Kirischuk1,7
- 1 Institute of Neurophysiology, University Medicine Charité, Berlin, Germany
- 2 Cluster of Excellence “NeuroCure”, University Medicine Charité, Berlin, Germany
- 3 Department of Neurology, University Medicine Charité, Berlin, Germany
- 4 Department of Clinical and Experimental Epilepsy, Institute of Neurology, University College, London, UK
- 5 Developmental Neurobiology, Max Delbrück Center for Molecular Medicine, Berlin, Germany
- 6 RNA Editing and Hyperexcitability Disorders Group, Max Delbrück Center for Molecular Medicine, Berlin, Germany
- 7 Institute of Physiology and Pathophysiology, University Medical Center of the Johannes Gutenberg University Mainz, Germany
Functional impairment of the adult brain can result from deficits in the ontogeny of GABAergic synaptic transmission. Gene defects underlying autism spectrum disorders, Rett’s syndrome or some forms of epilepsy, but also a diverse set of syndromes accompanying perinatal trauma, hormonal imbalances, intake of sleep-inducing or mood-improving drugs or, quite common, alcohol intake during pregnancy can alter GABA signaling early in life. The search for therapeutically relevant endogenous molecules or exogenous compounds able to alleviate the consequences of dysfunction of GABAergic transmission in the embryonic or postnatal brain requires a clear understanding of its site- and state-dependent development. At the level of single synapses, it is necessary to discriminate between presynaptic and postsynaptic alterations, and to define parameters that can be regarded as both suitable and accessible for the quantification of developmental changes. Here we focus on the performance of GABAergic synapses in two brain structures, the hippocampus and the superior colliculus, describe some novel aspects of neurotrophin effects during the development of GABAergic synaptic transmission and examine the applicability of the following rules: (1) synaptic transmission starts with GABA, (2) nascent/immature GABAergic synapses operate in a ballistic mode (multivesicular release), (3) immature synaptic terminals release vesicles with higher probability than mature synapses, (4) immature GABAergic synapses are prone to paired pulse and tetanic depression, (5) synapse maturation is characterized by an increasing dominance of synchronous over asynchronous release, (6) in immature neurons GABA acts as a depolarizing transmitter, (7) synapse maturation implies inhibitory postsynaptic current shortening due to an increase in alpha1 subunit expression, (8) extrasynaptic (tonic) conductances can inhibit the development of synaptic (phasic) GABA actions.
Introduction
What does the construction of the very first synapses during embryonic development and the generation of new synapses after a stroke in the aged brain have in common? What enables or constrains the formation of new synapses after transplantation of exogenous neurons into an adult brain? How could we possibly stimulate inhibitory synapse formation in an epileptic cortex – or repair the sad consequences of alcohol consumption of a pregnant mother when too many inhibitory neurons are lost in her baby’s brain? – We are still far from an all-embracing answer to these questions, and more work is needed to understand the impact of synaptogenesis on the performance of each given functional system during a particular stage of development. Nonetheless we will try to delineate some basic principles of synapse development that can be regarded as common, at least in the sense that they are applicable to synaptic connections using the inhibitory γ-amino-butyric acid (GABA) as a neurotransmitter of both the hippocampus and the superior colliculus, i.e., two brain structures requiring intact GABAergic inhibition while fulfilling clearly different functions.
The hippocampus is involved in learning and memory formation, but apart from its functions, it has served as a classical model system for very many aspects of synaptic transmission and synapse development (see Ben Ari et al., 2007; McBain and Kauer, 2009). Likewise, the superior colliculus of mammals, or the optic tectum of avians, amphibians, and fish, is a structure that has become quite popular for its retina-, head-, and body-related sensory and motor maps and its role as novelty detectors (see Boehnke and Munoz, 2008; Stein et al., 2009 for recent review). As a nearly two-dimensional projection area of retinal and visual cortical afferents it served to identify a number of molecules relevant for the formation of orderly connections (Feldheim and O’Leary, 2010). It also appears to be well suited to study inhibitory synaptogenesis since the superior colliculus is reputed to contain the largest amounts of GABA and the highest density of GABAergic synaptic terminals in the brain (see Grantyn et al., 2004 for references).
In the following we shall highlight eight principles that appear to determine the performance of GABAergic synapses during embryonic and early postnatal development. Findings from the rodent superior colliculus will be discussed and, if available, compared to results from hippocampal preparations under the common assumption that fundamental principles of synapse development and function are shared among many brain regions despite regional tailoring of synapse parameters to the specific developmental and functional requirements. The focus of the discussion will be on functional parameters of synaptic transmission at the expense of molecular determinants of developmental changes. Building on this, we explore to what extent these principles are governed by neurotrophin signaling during development and how they predict characteristics of GABAergic synapses in lesioned brain tissue or during adult neurogenesis.
Functional Maturation of GABAergic Synapses
Synaptic Transmission Starts with GABA
The role of GABA as a “pioneer transmitter” (Ben Ari et al., 2007) implies a variety of paracrine actions of this neurotransmitter along with a lead in synaptogenesis. The rule of GABAergic lead during the formation of network activity is based on patch clamp recordings from hippocampal slices of newborn rats where AMPAR-mediated synaptic events were rare or entirely missing while GABAergic inhibitory postsynaptic currents (IPSCs) could either be induced by electrical stimulation or detected as spontaneously occurring events (Hollrigel and Soltesz, 1997; Tyzio et al., 1999).
In the superior colliculus, action potential-mediated spontaneous and unitary evoked synaptic currents did indeed occur in the absence of glutamatergic synaptic activity at E17 (Grantyn et al., 2004), i.e., at an age when inhibitory synaptic currents were first seen in the retina (Unsoeld et al., 2008). Furthermore, evaluation of double immunostaining using antibodies against synaptophysin and the vesicular GABA transporter vGAT showed that at E17 all presynaptic puncta were GABAergic (Figure 1A). Even at postnatal day (P) 0 the fraction of GABAergic terminals was as high as 87% (J. Walter and R. Grantyn, unpublished result).
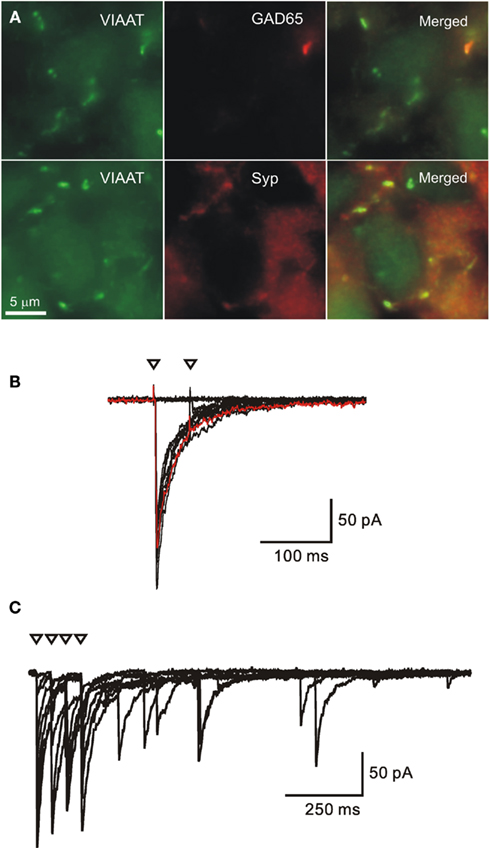
Figure 1. Early onset of GABAergic synaptic transmission and synapse formation in the embryonic superior colliculus (E17). (A) Specimens from sections of the immunostained superior colliculus. Most GABAergic synaptic terminals are double-immunoreactive for the vesicular inhibitory amino acid transporter (VIAAT) and synaptophysin (Syp) but few of them already stain for GAD65. (B,C) Electrophysiological recording of unitary synaptic responses in acute slices from the E17 mouse superior colliculus. Minimal stimulation with two (B) and four (C) pulses at an interval of 50 ms. Triangles denote time of stimulation. Note absence of second eIPSC in A and asynchronous delayed IPSCs in (B). (Modified from (Grantyn et al., 2004).
The GABAergic lead during development has been attributed to earlier differentiation and functionality of GABAergic interneurons as opposed to glutamatergic principal cells (Gozlan and Ben Ari, 2003). As for the function of GABAergic pioneer synapses, one has to consider their depolarizing polarity (see below, part 6) and the associated capacity to generate local Ca2+ transients which make them a good substitute for the lack of glutamatergic inputs. Synaptic GABA release could also contribute to some initial steps of Ca2+-dependent neuron differentiation, such as dendrite and axon outgrowth (see Sernagor et al., 2010), and it might assist the formation of glutamatergic afferents by producing giant depolarizing potentials The latter concept of GABAergic excitation driving maturation of the glutamatergic synaptic phenotype, also termed “mènage à trois,” has attracted particular attention (Ben-Ari et al., 1997). Interestingly, GABAergic synaptic transmission could not only precede glutamatergic transmission within distinct sets of synapses, but one and the same synapse type was shown to shift from an initial GABAergic phenotype to a mixed glutamatergic/GABAergic and, later on, to a predominantly glutamatergic phenotype (Gutierrez, 2003; Sufiulina et al., 2010). While adult mossy fiber terminals are known to mediate a strong excitatory input to CA3 pyramidal neurons via postsynaptic AMPA-, NMDA-, and kainate receptors, at neonatal age these same terminals use GABA to induce depolarizing excitatory actions via GABA(A)Rs (Safiulina et al., 2006). Under pathophysiological conditions, such as the generation of seizures, the GABAergic phenotype can be re-expressed (Gutierrez and Heinemann, 2001).
It should be noted, however, that for many other brain areas the GABAergic lead in synaptogenesis is not yet firmly established, because recordings in situ were rarely performed early enough to catch the very onset of synaptogenesis and transmission at newborn synapses. In addition, different developmental scenarios are possible in the lower brainstem and spinal cord. For instance, in the rodent lateral superior olive glycinergic/GABAergic inhibitory synapses were shown to transiently use the neurotransmitter glutamate during the period of activity-dependent synapse refinement (Gillespie et al., 2005).
Nascent/Immature GABAergic Synapses Operate in a Ballistic Mode (Multivesicular Release)
A distinct feature of GABAergic synaptic transmission in the embryonic mouse brain is the large amplitude of action potential-mediated spontaneous or evoked IPSCs (sIPSCs, eIPSCs, respectively; Cohen et al., 2000; Kirischuk et al., 2005) and their pronounced fatigue under condition of rapid and repetitive activation. This is illustrated in Figure 1B. In an E17 superior colliculus slice, the well-defined eIPSCs induced by the first stimulus preceded a complete failure after the second. When applying a stimulus train larger eIPSCs were only generated after preceding failures, and there was a tendency for asynchronous release after the stimulus trains (Figure 1C), suggesting a protracted elevation of presynaptic Ca2+ concentration (Kirischuk and Grantyn, 2003).
Were these large responses induced by one or several synaptic terminals? – According to the “one site-one vesicle”-hypothesis of synaptic transmission (Korn and Faber, 1987) one might be tempted to suggest that the connections in the embryonic colliculus must be composed of multiple contacts and/or multiple active zones per ending. However, the electron microscopy (EM) images from P1 colliculi and evaluation of immunostained synaptic terminals in E17 tecta indicate that this is rather unlikely (Lund and Lund, 1972; Juettner et al., 2005). Taking into account that under the given experimental conditions miniature IPSC (mIPSC) amplitudes amounted to 25–30 pA we concluded that 10–15 vesicles may simultaneously be released from just one site.
The possibility of multivesicular release has also been considered for inhibitory synapses in hippocampal cultures (Fedulova and Veselovsky, 2002) and hippocampal slices (Biro et al., 2006) while data from small glutamatergic synapses appears contradictory (see Oertner et al., 2002; Huang et al., 2010 vs. Chen et al., 2004). Kirischuk et al. (1999) characterized the release of GABAergic from single synaptic terminals in dissociated cultures from the embryonic rat tectum. Selective stimulation of axon terminals after loading a Ca2+ indicator allowed us to record single-bouton-evoked stimulus-locked IPSCs (sbIPSCs) along with the respective asynchronous delayed IPSCs (dIPSCs; Figures 2A,B) and presynaptic Ca2+ transients. An estimate of quantal size (Q) can be extracted from amplitude distributions of sbIPSCs and dIPSCs (Figure 2C). Dividing maximal or mean sbIPSC amplitudes by the experimentally derived values of Q (Figures 2D,E) one directly obtains the maximal or mean quantal content (m) (Kirischuk and Grantyn, 2002). Even under physiological conditions ([Ca2+]/[Mg2+] = 2), the mean m value was larger than 1 in 30 out of 40 tested boutons (range: 0.4–6), and the maximal m reached 8–12. At any given synapse, the mean sbIPSC amplitudes changed with the third power of the presynaptic bulk Ca2+ concentration (Figure 2F). With few exceptions, these boutons had one active zone per terminal only, which lead us to suggest that these immature GABAergic terminals had the capacity to release several vesicles from just one docking site (Kirischuk and Grantyn, 2002).
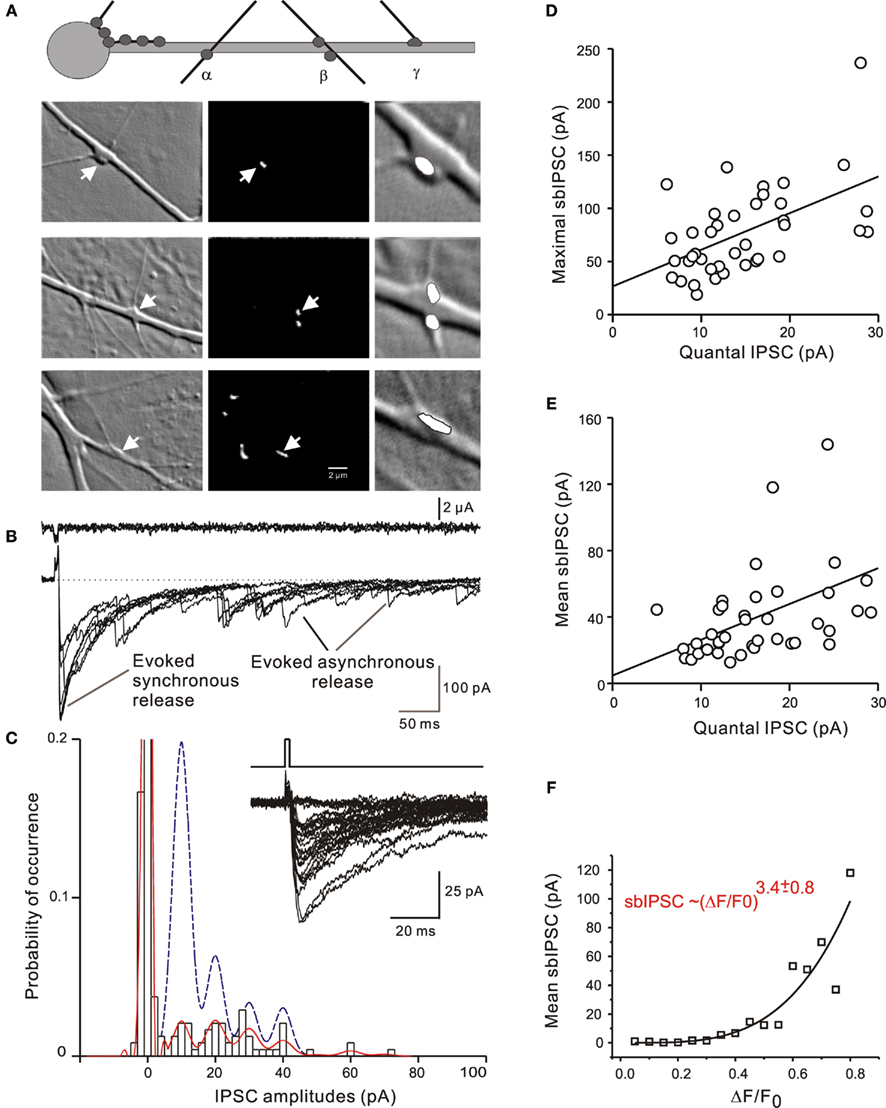
Figure 2. Multivesicular release from single GABAergic boutons in cultures from the E20 rat superior colliculus. (A) Types of synapses selected for direct application of depolarizing stimuli to single presynaptic terminals in the presence of action potential block with tetrodotoxin. Left panel: Phase contrast images; middle panels: fluorescent images showing same view fields after up-take of FM1-43; right panels: magnified synaptic sites with phase contrast optics. (B) Specimen records of single-bouton-activated IPSCs (sbIPSCS, lower trace) and respective stimulating current (upper trace). (C) Amplitude distribution and binomial fitting of sbIPSCs (bars and solide line) and dIPSCs (dashed line) for the solitary bouton illustrated in the upper row images of (A). dIPSCs were sampled during a period of 250–500 ms after the pulse. (D,E) Quantification of the results for maximal and mean sbIPSCs suggesting a quantal content >1. (F) Relationship between the mean sbIPSC amplitude and the maximal amplitude of the presynaptic bulk Ca2+ transient [Ca2+]pre recorded from a presynaptic area delineated on the basis of vesicular staining. (Modified from Kirischuk et al., 1999).
It should be mentioned that simultaneous release of several transmitter quanta can even occur spontaneously, due to random elevations of presynaptic [Ca2+], as observed in basket cell terminals in slices of the postnatal rat cerebellum (Llano et al., 2000b). Lowering extracellular [Ca2+] would decompose “giant” mIPSCs and eventually limit spontaneous synaptic activity to monoquantal events.
Immature Synaptic Terminals Release Vesicles with Higher Probability than Mature Synapses
Presynaptically, synapse maturation is characterized by the formation of multiple release sites and the differentiation of the release machinery which encompasses a complex set of changes affecting docking, molecular and positional priming, fusion, site clearing, and several pathways for replenishment of the vesicle pool (Neher and Sakaba, 2008; Pang and Sudhof, 2010). Most commonly, changes in the release are characterized by invoking the statistical parameter “average probability of release” (Pr), often defined as the likelihood that any contact of a synaptic connection would liberate one quantum of the transmitter in response to a presynaptic action potential. In most cases Pr is not directly accessible for measurement, but determined by binomial fitting (for instance, Stricker et al., 1996), variance–mean analysis (Clements and Silver, 2000) or covariance analysis (Scheuss and Neher, 2001). In the frame of the binomial model of synaptic transmission, the unitary postsynaptic response (i.e., the response obtained by activation of just one presynaptic neuron) would reflect the product of N, Pr, and Q, N being the total number of synapses/active zones/docking sites formed by the presynaptic cell.
In synapses with multivesicular release, presynaptic differentiation can also be characterized on the basis of changes of m, the mean quantal content (Taschenberger et al., 2005). In view of the difficulties to accurately determine the number of active zones participating in release (or the structure-based “histological N”) it has become acceptable to disregard the site-dependent heterogeneity of individual release sites and to conceptually merge all the vesicles to one common pool, an approach first introduced for the calyx of Held (Sakaba et al., 2002) and later extended to general models of quantal synaptic transmission (Neher and Sakaba, 2008; Pan and Zucker, 2009), where N becomes the number of vesicles in the readily releasable pool (RRP). The average probability that a given vesicle is released from that pool is pves. (Heterogeneous vesicle pools and different modes of vesicle fusion are complexities to be considered in more elaborate reflections on presynaptic vesicle release).
As a first approximation, pves can be determined using experimental protocols that deplete the RRP, for example a high-frequency stimulation (Schneggenburger et al., 1999; Kirischuk and Grantyn, 2000; Hanse and Gustafsson, 2001). The GABAergic synapses of the postnatal mouse superior colliculus frequency needs to be adjusted for any given type of synapses. For neonatal and juvenile inhibitory synapses an appropriate stimulation frequency would be 50 Hz. After few pulses, the cumulative eIPSC amplitude exhibits a linear dependency on the pulse number, and back-extrapolation to stimulus 0 provides a value of RRP*Q (Figures 3A,B). Dividing the cumulative eIPSC value at y intercept by Q (as derived from the amplitude distribution of dIPSCs and mIPSCs) one obtains an estimate of RRP. This value corresponds to the maximal number of quanta that could be released at rest. Accordingly pves is eIPSC/RRP*Q. The applicability of this method can be verified by comparing the theoretical coefficient of variation of eIPSC amplitudes derived from binomial statistics (CV = [(1 − p)/RRP*p)]1/2) with the CV of the recorded eIPSCs (SD/mean amplitude; Figure 3C) and the relationship between the paired pulse ratio and pves(Figure 3D). This relatively simple approach was then applied to screen for developmental changes of GABAergic synapses in the superior colliculus of C57Bl6 mice between P0 and P22 (Kirischuk et al., 2005).
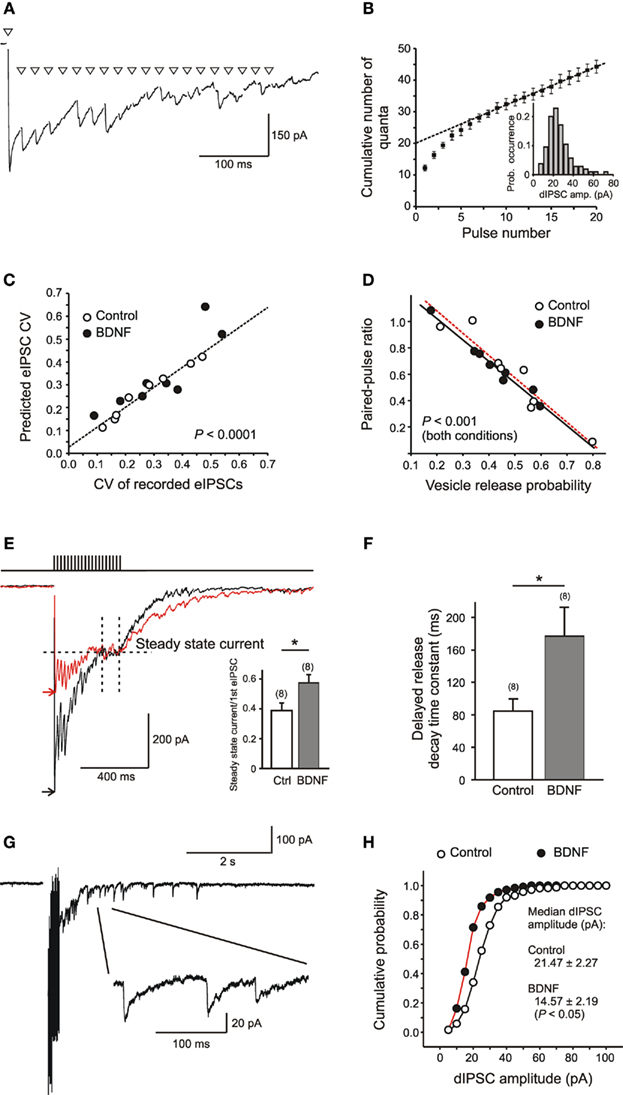
Figure 3. Measurement of RRP, pves, and Q in collicular neurons, and the effects of BDNF. (A) Specimen trace of eIPSCs induced by high-frequency (HF) stimulation (20 pulses at 50 Hz) of a single GABAergic axon. For clarity stimulus artifacts are replaced by triangles. (B) Cumulative plot of eIPSC amplitudes vs. stimulus number. The eIPSC amplitudes were normalized to the median dIPSC amplitude of the same unitary connection (inset). Back-extrapolation to the y intercept indicates RRP. (C,D) Tests for applicability of the estimates of pves. (E) Sample records to illustrate BDNF effects on IPSCs induced by HF stimulation. To obtain the amplitude of the steady state current, the current integral was normalized to the total time of integration for the last five stimulus intervals. Arrow heads denote the peak level of the eIPSC after the first pulse in the train. Note that BDNF does not affect the steady state current but significantly reduces the ratio between steady state current and first eIPSC (inset). (F) BDNF increases the time constant of decay of the “synaptic tail current,” i.e., the current produced by dIPSCs after the stimulus train, as estimated by single exponential fit. (G) Sample record of the postsynaptic response to HF stimulation of a single GABAergic axon. Note the presence of dIPSCs (inset: enlarged) after the end of stimulation. (H) Reduction of dIPSC amplitudes as evidence for a depressant postsynaptic effect of BDNF, in contrast to the absence of significant changes in the range of coefficient of variation (C), paired pulse ratio, and pves(D). (Modified from Henneberger et al., 2005b).
It turned out that pves was high at P1, but dropped to a significantly lower value shortly after. The fact that pves undergoes an initial developmental decrease was at odds with expectations that nascent synapses release their transmitter ineffectively (Dumas and Foster, 1995; Aizenman and Cline, 2007), but in line with studies of glutamatergic connections in the hippocampus (Bolshakov and Siegelbaum, 1995; Wasling et al., 2004), where a developmental decrease of P, albeit at older age, had been described.
The RRP, too, experiences a decrease around P3, but increases again with age. Similar results on the developmental changes of RRP were reported for hippocampal synapses (Mozhayeva et al., 2002). Thus, a relatively large RRP containing high release probability vesicles is temporary replaced by a relatively small RRP of low release probability vesicles, resulting in a decrease of eIPSC amplitude. At P11–15 PPR size and eIPSC amplitudes increase, but the release of low probability vesicles is more tightly locked to the presynaptic action potential. As this coincides with IPSC shortening (see below), IPSCs are now fit for better temporal resolution which may play a role in the acquisition of pattern vision after eye opening.
This leaves us with the following question: is the multivesicular release/high probability of vesicle exocytosis at nascent GABAergic synapses merely a functional deficit to be overcome in the course of further development? Or does it serve a useful role in the development of neuronal networks? – The answer remains open, but one may speculate that a ballistic mode of operation of immature synaptic terminals could be the most effective way to recruit postsynaptic receptors and to stabilize them via a postsynaptic local Ca2+ signal (see below). The fact that in the developing superior colliculus the decrease of pves and the reduction of RRP were paralleled by the disappearance of depolarizing GABA action at P2–3 is at least in line with this idea (see below).
Immature GABAergic Synapses are Prone to Paired Pulse and Tetanic Depression
As already mentioned, at immature stages stimulus-locked responses tend to undergo depression if two or more presynaptic depolarizations occur at short intervals. The mechanisms underlying the use-dependent depression in immature GABAergic synapses have been studied at some detail in our lab using single-bouton activation in the presence of TTX or Ca2+ channel blockers to suppress co-activation of other boutons in contact with the postsynaptic cell (Kirischuk et al., 2002). This approach has the advantage that one and the same synaptic terminal is reliably stimulated with each trial, presynaptic depolarizations can be graded and presynaptic Ca2+ transients could serve as indicators of presynaptic activation (Kirischuk et al., 1999). The experiments showed that at short intervals (<100 ms) different depressant mechanisms occur simultaneously but differ in their recovery kinetics.
The rapidly recovering paired pulse depression (PPDfast), as seen at interstimulus intervals of 25–50 ms, is release-dependent (the amplitude of the second eIPSC being inversely proportional to the amplitude of the first one) and strongly affected by the extracellular Ca2+ concentration (Jensen et al., 1999; Chen et al., 2004). Developmental changes of PPDfast can be expected due to the maturation of presynaptic Ca2+ buffering (for instance, Llano et al., 2000a) or changes in the patterns of presynaptic G-protein coupled receptor expression. Both mechanisms account for neuron-specific differences of paired pulse plasticity in the adult brain (see, for instance, Senn et al., 1998; Poncer et al., 2000) while at the onset of GABAergic synaptogenesis modulatory diversity is low. Even a GABA(B)R-mediated contribution to paired pulse plasticity is missing in immature GABAergic synapses (Wilcox and Dichter, 1994; Jensen et al., 1999; Kirischuk et al., 2002), and the predominant type of paired pulse behavior is depression.
With further synapse maturation the amount of PPD decreases (Juettner et al., 2001), and this change can be ascribed to a decrease in pves (Kirischuk et al., 2005). Raising the presynaptic Ca2+ influx by increasing extracellular Ca2+ levels (Senn et al., 1998), prolonging presynaptic depolarization (Kirischuk et al., 2002) or blocking G-protein mediated depression by N-ethylmaleimide (NEM)(Kirmse and Kirischuk, 2006) can enhance pves and turn an already established paired pulse facilitation (PPF) into PPD.
A much more slowly recovering form of PPD can be isolated at intervals of 1 s (PPDslow). In contrast to PPDfast, PPDslow was found to be calcium- and release-independent. This type of depression has first been described for neuromuscular junctions (Betz, 1970), the squid giant synapse (Hsu et al., 1996), the calyx of Held (Bellingham and Walmsley, 1999; Borst and Sakmann, 1999), and a synapse formed by the Mauthner cell axon in the goldfish (Waldeck et al., 2000). PPDslow has been incorporated in the contemporary models of transmitter release under the term “transient refractoriness” (Zucker and Regehr, 2002; Pan and Zucker, 2009) or “site clearing” (Neher and Sakaba, 2008). Despite its prominence, at least in immature synapses (see Kirischuk et al., 2002), it still awaits detailed characterization at a molecular level along with a more systematic testing for developmental changes.
It should be noted that the probability of obtaining a postsynaptic response would depend both on pves and the probability that a given site (active zone) is available for release. Therefore, to obtain a full description of the developmental changes in the performance of a given type of synaptic connections binomial analysis or variance–mean analysis (Silver, 2003) need to be performed to determine Pr in addition to pves.
Apart from estimating the quantal parameters of synaptic transmission, the reliability or fatigability of a synaptic connection could be tested using the average eIPSC amplitude reached during the last 10 trials of a series of high-frequency pulses (50–100 Hz). This reveals the so called tetanic depression of synaptic transmission. If normalized to Q and RRP, this parameter is best suited to reflect the age-dependent changes in the release performance (Kirischuk et al., 2005). Tetanic eIPSC depression was strong at P3–6, but decreased at P11–P15. Although developmental acceleration of the RRP replenishment rate can not be excluded, the observed decrease of release probability accompanied with the increase of RRP size may underlie these changes. Interestingly, the decrease of tetanic depression coincides with the time of in-growth of cortical afferents and the massive up-regulation of glutamatergic synaptic transmission prior to eye opening at day P14–15 (see Aamodt and Constantine-Paton, 1999; Grantyn et al., 2004 for more). Experimentally, presynaptic GABA release can be modified by allowing cortical afferents to grow into tectal tissue (Henneberger et al., 2007) indicating that indeed changes in glutamatergic innervation shape the function of pre-existing GABAergic synapses.
Synapse Maturation is Characterized by a Dominance of Synchronous Over Asynchronous Release
A developmental study in the calyx of Held has reported a changing relationship between synchronous and asynchronous release (Chuhma et al., 2001; Yang and Xu-Friedman, 2010). Delayed release might even be present when evoked release is missing, and the two modes of release display a differential dependency on the presynaptic Ca2+ concentration in the vesicle area ([Ca2+]pre) (Kirischuk and Grantyn, 2003; Yang and Xu-Friedman, 2010). Asynchronous IPSCs (aIPSCs) were sampled during a train of high-frequency stimulation and a synchrony index of release was defined for the period of the last 10 intervals of the high-frequency train by dividing the charge transfer of stimulus-locked eIPSCs by the charge transfer of unlocked aIPSCs. It was found that this index increased with age, showing that the relative number of vesicles released in a stimulus-locked manner increases when neurons mature. As the delayed component of asynchronous release (charge of dIPSCs) displayed a developmental decrease as well, one can conclude that asynchronous release is a characteristic feature of immature synapses, being replaced by action potential-locked transmission at older age. Again, the changes were biggest around the time of eye opening (i.e., shortly after the time of massive in-growth of glutamatergic synapses).
While asynchronous release may be more pronounced in immature GABAergic connections, its persistence at more mature stages will depend on the type of interneuron activated and the Ca2+-binding proteins expressed (Daw et al., 2010).
In Immature Neurons GABA Acts as Depolarizing Transmitter
The pioneer role of GABA at initial stages of circuit formation in the brain has much to do with its depolarizing action (Cherubini et al., 1991; Ben Ari et al., 2007). The latter has long ago been discovered in the immature rat striatum (Misgeld et al., 1982) and the immature rabbit and rat hippocampus (Mueller et al., 1984; Cherubini et al., 1990). In the superior colliculus the depolarizing/excitatory action of GABA is prominent at P0–P1 (Juettner et al., 2001), but it already disappears by P3 (Grantyn et al., 2004). In the hippocampus, the change in the polarity of GABA action occurs around P5 (Ben Ari et al., 1989) and has been attributed to the developmental up-regulation of the expression and membrane targeting of KCC2, a Cl− exporter (Rivera et al., 1999; Ganguly et al., 2001; Hubner et al., 2001). High activity of KCC2 in relation to the activity of NKCC1, a Cl− importer, would ensure low intracellular Cl− concentrations and, consequently a hyperpolarizing GABA action (Blaesse et al., 2009).
However, a developmental switch from depolarizing to hyperpolarizing GABA is not observed in all developing neurons (Banke and McBain, 2006) and, where present, even parts of the somato-dendritic plasma membrane can differ with regard to their local chloride gradients (Gulledge and Stuart, 2003). Whether or not at the end shunt inhibition or excitatory GABA actions will dominate the overall effect of GABA on the neuronal output will, first of all, depend on the spatio-temporal relationships of the respective chloride channels with the glutamatergic inputs (see Bracci and Panzeri, 2006).
Synapse Maturation Implies IPSC Shortening due to an Increase in Alpha1 Subunit Expression
The developmental shortening of postsynaptic currents is a widely observed phenomenon in many brain areas, including the rodent hippocampus (Cohen et al., 2000; Hutcheon et al., 2004) and superior colliculus (Juettner et al., 2001; Henneberger et al., 2005a; Kirischuk et al., 2005). The slow decay kinetics of IPSCs shortly after birth has been associated with the high expression level of the alpha3 subunit of the GABA(A)R, while the subsequent shortening of IPSCs was attributed to an up-regulation of the alpha1/alpha3 ratio.
Again, the most interesting questions concern the cause and the function of this phenomenon. We have tried to determine the timing of the developmental switch from slow to fast IPSCs in the superior colliculus and found that most of the change occurs during days P6 and 15, i.e., prior to eye opening (around P14–15 in mice, the day of birth being P0). It also coincided with the developmental peak of the NMDAR-mediated charge transfer in the glutamatergic synaptic currents and might be an activity-dependent phenomenon.
We therefore considered the possibility that glutamatergic/ NMDAR-mediated activity provided the drive for the switch in the GABA(A)R subunit composition. Respective culture experiments with chronic exposure to MK-801 confirmed this suggestion, while block of mGluR receptor activity with S-MCPG had no effect on IPSC decay kinetics (Henneberger et al., 2005a).
It is particularly telling that the decrease of IPSC duration also coincided with the increase of the synchrony index and the reduction of tetanic depression of the phasic stimulus-locked release. One may speculate that by the time of onset of patterned vision GABAergic synapses assume a new function enabling a better temporal resolution of inhibitory signals.
Extrasynaptic (Tonic) Conductances can Inhibit the Development of Synaptic (Phasic) GABA Actions
Tonic chloride conductances via extrasynaptic GABA(A) and/or glycine receptors represent a powerful means to adjust neuron excitability (see Farrant and Nusser, 2005), and is likely to happen as soon or even prior to the moment when a neuron becomes postmitotic. Indeed, non-synaptic responses to exogenous GABA can be detected much before neurons exhibit phasic synaptic activity, an example being the E14 rat retinal ganglion cells (Rörig and Grantyn, 1994).
Associated with changes in the subunit composition there might be a cell-type-specific up- or down-regulation of tonic GABA currents [I(Tonic)GABA]. In hippocampal granule cells (Holter et al., 2010) and in D1-expressing striatal output neurons (Santhakumar et al., 2010), I(Tonic)GABA displays a robust increase with age, while D2-expressing striatal output neurons showed a decrease, which possibly reflected the general tendency of GABA(A)R α5 down-regulation with age (Laurie et al., 1992). Extrasynaptic GABA(A)Rs typically contain α5 and/or δ subunits, which makes them very sensitive to low concentrations of ambient GABA.
Considering the idea that stability of GABAergic synaptic contacts requires local Ca2+ elevations on the background of low global (resting) Ca2+ levels in dendrites and somas (see Kirsch et al., 1993; Marty and Llano, 2005), one could expect an inverse relationship between tonic extrasynaptic and phasic synaptic Cl− conductances.
This question has recently been addressed by Meier and colleagues (Eichler et al., 2008) who transfected hippocampal neurons with a glycine receptor isoform that confers a particularly high agonist affinity to the extrasynaptic glycine receptors of hippocampal and collicular neurons (Meier et al., 2005). As expected, overexpression of the high affinity type of glycine receptor resulted in an increase of the tonic chloride conductance. Moreover, it also resulted in a massive decrease of GABAergic synapse numbers. As this effect could be reversed by transfecting hippocampal neurons with the chloride exporter KCC2, it was suggested that global changes of Ca2+ in response to depolarizing responses due to Cl− outflux through extrasynaptic Cl− channels and the resulting activation of voltage-gated Ca2+ channels in neurons with a high intracellular Cl− concentration could mask the signals required for stabilization of GABAergic contacts. In fact, Ca2+ was recently shown to impact on the stability of postsynaptic gephyrin and GABA(A)Rs (Förstera et al., 2010; Tyagarajan et al., 2011). Furthermore, if neurons were no longer able to generate action potentials (due to massive shunt inhibition provided by the extrasynaptic high affinity glycine receptors) they may be afflicted by neurodegeneration, as a worst-case scenario (Tao and Poo, 2005; Legendre et al., 2009).
More studies on the relationship between tonic extrasynaptic signals and phasic GABAergic synaptic transmission are needed, since it is known that a number of pathophysiological states, including epilepsy, depression, and neurodegenerative diseases, are associated with a return to depolarizing GABA actions (see Cherubini et al., 2011).
Neurotrophin Effects on Inhibitory Synapse Development
Effects of BDNF
Brain-derived neurotrophic factor (BDNF) is critically involved in the activity-dependent maturation of visual structures (see Frost, 2001 for review), and it affects a number of mechanisms underlying the maturation of synaptic inhibition (Huang et al., 1999; Frost, 2001). In the following we provide a short survey of BDNF-related findings from our studies on a range of model systems and approaches, such as hippocampal and collicular cultures and acute slices from wild-type and BDNF-deficient mice, acute and chronic BDNF treatment and overexpression of BDNF by transfection.
The impact of BDNF on synapse function and development varies depending on the maturity of the synapses studied. In cultured neurons from the embryonic hippocampus, BDNF increased the number of glutamatergic synapses, but decreased the number of GABAergic, vGAT-positive, synaptic terminals (Singh et al., 2006). These changes were accompanied by an increase in the ratio between synaptic excitation and inhibition assessed by quantifying spontaneously occurring action potential-independent release events (mEPSC/mIPSC frequency; Singh et al., 2006). Our results suggest that in immature neuronal networks BDNF could promote glutamatergic synaptogenesis at the expense of GABAergic synapses.
Addition of exogenous BDNF to collicular cultures revealed two possible presynaptic targets in GABAergic synapses, vesicle loading, and asynchronous release. The former suggestion is based on the finding that BDNF treatment reduced the level of presynaptic vGAT, as judged by immunofluorescence (Henneberger et al., 2005b). No effect was found on the RRP and pves of evoked release, but it augmented asynchronous release (Figures 3E,G) – the characteristic feature of immature collicular synapses (see above, part 5). This could be a consequence of increased levels of the GABA-synthesizing enzyme GAD after BDNF treatment. Indeed, like hippocampal neurons (Aguado et al., 2003) collicular neurons reacted to BDNF with an increase in presynaptic GAD65 levels (Henneberger et al., 2005b), and the latter is known to facilitate asynchronous release (Tian et al., 1999). In some preparations, including hippocampal slice cultures, addition of exogenous BDNF was reported to produce a higher yield of GAD-labeled presynaptic terminals (Marty et al., 2000).
On the postsynaptic side (Figure 3H), BDNF treatment of cultured collicular neurons resulted in a suppression of GABAergic synaptic transmission (Tanaka et al., 1997; Brünig et al., 2001) which could be attributed to a reduction in the number of open channels contributing to the IPSCs without affecting their single channel conductance, RRP or pves (Henneberger et al., 2005b). In principle, the depressant effect of BDNF on the postsynaptic response to GABA could reflect reduced neurotransmitter loading into vesicles and thus a smaller number of open postsynaptic receptors/channels during transmission. However, we are not aware of any direct evidence supporting this interesting alternative hypothesis and would tentatively conclude that a reduction in postsynaptic receptor number accounts for most of the BDNF-mediated inhibition of GABAergic transmission elicited by single action potentials (Brünig et al., 2001).
Similar or related results were obtained in the intact superior colliculus of bdnf−/− mice, however only after P13/14, i.e., at a stage when glutamatergic inputs were available and active (Henneberger et al., 2002, 2005b). At a younger age (P1–5) evidence for a suppressive postsynaptic effect of BDNF was missing, although the chronic absence of BDNF delayed the switch from depolarizing to hyperpolarizing GABA action (Grantyn et al., 2004), in accord with similar findings in immature murine hippocampal slices (Aguado et al., 2003).
In summary, the actions of the neurotrophins BDNF vary with brain region and developmental stage. In the superior colliculus, BDNF is likely to accelerate the transition to hyperpolarizing GABA action and to facilitate asynchronous release during early postnatal development whereas at later stages it primarily inhibits GABAergic synaptic transmission.
Effects of NGF
That nerve growth factor (NGF) can be an influential player in the maturation and function of GABAergic synapses has only recently been recognized. Still very little is known on the possible role of NGF during in situ development of GABAergic synapses.
We have explored some acute effects of added NGF in hippocampal cultures and found that NGF promoted GABAergic synaptogenesis (Salama-Cohen et al., 2006). The effects of NGF included
(1) a TrkA-mediated up-regulation of vGAT expression
(2) an increase in the number of vGAT-immunopositive synaptic terminals in contact with hippocampal neurons
(3) a prominent reduction in the E/I ratio of contacting boutons.
The final outcome of exogenous NGF depended on its depressant action on the proneural gene neurogenin 3 (Ngn3), a nuclear transcription factor that is also under the control of Hes1/5.
Our results suggest that with regard to the glutamate/GABA or E/I balance of synaptic transmission and development BDNF and NGF might assume antagonistic roles. Therefore, to further explore the significance of NGF for the development and function of GABA synapses might be one of the most rewarding tasks in the near future.
Acute GABAergic Synaptogenesis after Lesion
Among the stimuli inducing inhibitory synaptogenesis are factors liberated from damaged tissue. Several labs have described “inhibitory sprouting” in the immediate environment (Mittmann and Eysel, 2001) or the terminal area (Deller et al., 1995) of lesioned cells.
Consistent with the pioneer role of GABA at initial stages of synaptogenesis (see above, part 6), our investigation of synapse formation after slicing of late embryonic or neonatal rodent superior colliculi revealed that at this age lesion preferentially facilitates the formation of new GABAergic synapses (Meier et al., 2003). The up-regulation of GABAergic synapses was inferred from the increase of vGAT- or GAD65-positive terminals, mIPSC frequency, and postsynaptic GABA(A)R immunofluorescence. GABAergic synaptogenesis could be prevented/facilitated by blocking/enhancing, respectively, PKC activation, suggesting the involvement of phosphorylation-dependent mechanisms. Manipulations assisting the return of globally elevated intracellular Ca2+ levels to resting Ca2+ concentrations promoted this form of reactive synaptogenesis in the neonatal superior colliculus (J. Walter, C. Henneberger, J. C. Meier and R. Grantyn, unpublished observation).
GABAergic Synaptogenesis during Integration of Newly Generated Neurons in the Adult Brain
Adult neurogenesis represents an important response of the damaged as well as the learning brain (see Ma et al., 2010). Notably in the dentate gyrus newly formed neurons integrate into pre-existing networks, which requires the formation of new inhibitory synapses (Tozuka et al., 2005; Wang et al., 2005; Toni and Sultan, 2011). The functional properties of nascent GABAergic synapses and their subsequent development were shown to reproduce some essential features of immature synapses in the embryonic and neonatal brain: there was a GABA lead in the innervation of newborn granule cells, the initial action of GABA was depolarizing, the decay kinetic was slow, and synaptic currents were relatively insensitive to zolpidem (Overstreet-Wadiche et al., 2005; Ge et al., 2006; Karten et al., 2006). Preventing a depolarizing GABA action by knock-out of NKCC1 reduced and delayed GABAergic synaptogenesis (Ge et al., 2006).
These results validate major milestones, as defined above for the ontogeny of inhibitory synaptic connections in the brain, and suggest that the outlined developmental mechanisms may apply irrespective of the given local conditions of a neuronal network.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aamodt, S. M., and Constantine-Paton, M. (1999). The role of neural activity in synaptic development and its implications for adult brain function. Adv. Neurol. 79, 133–144.
Aguado, F., Carmona, M. A., Pozas, E., Aguilo, A., Martinez-Guijarro, F. J., Alcantara, S., Borrell, V., Yuste, R., Ibanez, C. F., and Soriano, E. (2003). BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K(+)/Cl(−) co-transporter KCC2. Development 130, 1267–1280.
Aizenman, C. D., and Cline, H. T. (2007). Enhanced visual activity in vivo forms nascent synapses in the developing retinotectal projection. J. Neurophysiol. 97, 2949–2957.
Banke, T. G., and McBain, C. J. (2006). GABAergic input onto CA3 hippocampal interneurons remains shunting throughout development. J. Neurosci. 26, 11720–11725.
Bellingham, M. C., and Walmsley, B. (1999). A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron 23, 159–170.
Ben Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325.
Ben Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.
Ben-Ari, Y., Khazipov, R., Leinekugel, X., Caillard, O., and Gaiarsa, J.-L. (1997). GABAA, NMDA and AMPA receptors: a developmentally regulated ‘mènage à trois’. Trends Neurosci. 20, 523–529.
Betz, W. J. (1970). Depression of transmitter release at the neuromuscular junction of the frog. J. Physiol. 206, 629–644.
Biro, A. A., Holderith, N. B., and Nusser, Z. (2006). Release probability-dependent scaling of the postsynaptic responses at single hippocampal GABAergic synapses. J. Neurosci. 26, 12487–12496.
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838.
Boehnke, S. E., and Munoz, D. P. (2008). On the importance of the transient visual response in the superior colliculus. Curr. Opin. Neurobiol. 18, 544–551.
Bolshakov, V., and Siegelbaum, S. A. (1995). Regulation of hippocampal transmitter release during development and long-term potentiation. Science 269, 1730–1734.
Borst, J. G., and Sakmann, B. (1999). Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J. Physiol. 521, 123–133.
Bracci, E., and Panzeri, S. (2006). Excitatory GABAergic effects in striatal projection neurons. J. Neurophysiol. 95, 1285–1290.
Brünig, I., Penschuck, S., Berninger, B., Benson, J., and Fritschy, J. M. (2001). BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur. J. Neurosci. 13, 1320–1328.
Chen, G., Harata, N. C., and Tsien, R. W. (2004). Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 101, 1063–1068.
Cherubini, E., Gaiarsa, J. L., and Ben Ari, Y. (1991). GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 14, 515–519.
Cherubini, E., Griguoli, M., Safiulina, V., and Lagostena, L. (2011). The depolarizing action of GABA controls early network activity in the developing hippocampus. Mol. Neurobiol. 43, 97–106.
Cherubini, E., Rovira, C., Gaiarsa, J.-L., Corradetti, R., and Ben-Ari, Y. (1990). GABA mediated excitation in immature rat CA3 hippocampal neurons. Int. J. Dev. Neurosci. 8, 481–490.
Chuhma, N., Koyano, K., and Ohmori, H. (2001). Synchronisation of neurotransmitter release during postnatal development in a calyceal presynaptic terminal of rat. J. Physiol. 530, 93–104.
Clements, J. D., and Silver, R. A. (2000). Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 23, 105–113.
Cohen, A. S., Lin, D. D., and Coulter, D. A. (2000). Protracted postnatal development of inhibitory synaptic transmission in rat hippocampal area CA1 neurons. J. Neurophysiol. 84, 2465–2476.
Daw, M. I., Pelkey, K. A., Chittajallu, R., and McBain, C. J. (2010). Presynaptic kainate receptor activation preserves asynchronous GABA release despite the reduction in synchronous release from hippocampal cholecystokinin interneurons. J. Neurosci. 30, 11202–11209.
Deller, T., Frotscher, M., and Nitsch, R. (1995). Morphological evidence for the sprouting of inhibitory commissural fibers in response to the lesion of the excitatory entorhinal input to the rat dentate gyrus. J. Neurosci. 15, 6868–6878.
Dumas, T. C., and Foster, T. C. (1995). Developmental increase in CA3-CA1 presynaptic function in the hippocampal slice. J. Neurophysiol. 73, 1821–1828.
Eichler, S. A., Kirischuk, S., Juttner, R., Legendre, P., Lehmann, T. N., Gloveli, T., Grantyn, R., and Meier, J. C. (2008). Glycinergic tonic inhibition of hippocampal neurons with depolarising GABAergic transmission elicits histopathological signs of temporal lobe epilepsy. J. Cell. Mol. Med. 12, 2848–2866.
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229.
Fedulova, S. A., and Veselovsky, N. S. (2002). Quantal GABA release in hippocampal synapses: role of local Ca2+ dynamics within the single terminals. Eur. J. Pharmacol. 447, 163–171.
Feldheim, D. A., and O’Leary, D. D. (2010). Visual map development: bidirectional signaling, bifunctional guidance molecules, and competition. Cold Spring Harb. Perspect. Biol. 2, a001768.
Förstera, B., Belaidi, A. A., Jüttner, R., Bernert, C., Tsokos, M., Lehmann, T. N., Horn, P., Dehnicke, C., Schwarz, G., and Meier, J. C. (2010). Irregular RNA splicing curtails postsynaptic gephyrin in the cornu ammonis of patients with epilepsy. Brain 133, 3778–3794.
Frost, D. O. (2001). BDNF/trkB signaling in the developmental sculpting of visual connections. Prog. Brain Res. 134, 35–49.
Ganguly, K., Schinder, A. F., Wong, S. T., and Poo, M. M. (2001). GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105, 521–532.
Ge, S., Goh, E. L., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593.
Gillespie, D. C., Kim, G., and Kandler, K. (2005). Inhibitory synapses in the developing auditory system are glutamatergic. Nat. Neurosci. 8, 332–338.
Gozlan, H., and Ben Ari, Y. (2003). Interneurons are the source and the targets of the first synapses formed in the rat developing hippocampal circuit. Cereb. Cortex 13, 684–692.
Grantyn, R., Jüttner, R., and Meier, J. (2004). “Development and use-dependent modification of synaptic connections in the visual layers of the rodent superior colliculus,” in The Superior Colliculus, eds. W. C. Hall and A. Moschovakis (Boca Raton: CRC Press), 173–210.
Gulledge, A. T., and Stuart, G. J. (2003). Excitatory actions of GABA in the cortex. Neuron 37, 299–309.
Gutierrez, R. (2003). The GABAergic phenotype of the “glutamatergic” granule cells of the dentate gyrus. Prog. Neurobiol. 71, 337–358.
Gutierrez, R., and Heinemann, U. (2001). Kindling induces transient fast inhibition in the dentate gyrus – CA3 projection. Eur. J. Neurosci. 13, 1371–1379.
Hanse, E., and Gustafsson, B. (2001). Vesicle release probability and pre-primed pool at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J. Physiol. 531, 481–493.
Henneberger, C., Jüttner, R., Rothe, T., and Grantyn, R. (2002). Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. J. Neurophysiol. 88, 595–603.
Henneberger, C., Jüttner, R., Schmidt, S., Walter, J. H., Meier, J., Rothe, T., and Grantyn, R. (2005a). GluR- and TrkB-mediated maturation of GABA(A) receptor function during the period of eye opening. Eur. J. Neurosci. 21, 431–440.
Henneberger, C., Kirischuk, S., and Grantyn, R. (2005b). Brain-derived neurotrophic factor modulates GABAergic synaptic transmission by enhancing presynaptic glutamic acid decarboxylase 65 levels, promoting asynchronous release and reducing the number of activated postsynaptic receptors. Neuroscience 135, 749–763.
Henneberger, C., Redman, S. J., and Grantyn, R. (2007). Cortical efferent control of subcortical sensory neurons by synaptic disinhibition. Cereb. Cortex 17, 2039–2049.
Hollrigel, G. S., and Soltesz, I. (1997). Slow kinetics of miniature IPSCs during early postnatal development in granule cells of the dentate gyrus. J. Neurosci. 17, 5119–5128.
Holter, N. I., Zylla, M. M., Zuber, N., Bruehl, C., and Draguhn, A. (2010). Tonic GABAergic control of mouse dentate granule cells during postnatal development. Eur. J. Neurosci. 32, 1300–1309.
Hsu, S. F., Augustine, G. J., and Jackson, M. B. (1996). Adaptation of Ca2+-triggered exocytosis in presynaptic terminals. Neuron 17, 501–512.
Huang, C. H., Bao, J., and Sakaba, T. (2010). Multivesicular release differentiates the reliability of synaptic transmission between the visual cortex and the somatosensory cortex. J. Neurosci. 30, 11994–12004.
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., Maffei, L., and Tonegawa, S. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755.
Hubner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524.
Hutcheon, B., Fritschy, J. M., and Poulter, M. O. (2004). Organization of GABA receptor alpha-subunit clustering in the developing rat neocortex and hippocampus. Eur. J. Neurosci. 19, 2475–2487.
Jensen, K., Lambert, J. D. C., and Jensen, M. S. (1999). Activity-dependent depression of GABAergic IPSCs in cultured hippocampal neurons. J. Neurophysiol. 82, 42–49.
Juettner, R., Meier, J., and Grantyn, R. (2001). Slow IPSC kinetics, low levels of alpha1 subunit expression and paired-pulse depression are distinct properties of neonatal inhibitory GABAergic synaptic connections in the mouse superior colliculus. Eur. J. Neurosci. 13, 2088–2098.
Juettner, R., Moré, M. I., Das, D., Babich, A., Meier, J., Henning, M., Erdmann, B., Müller, E. C., Otto, A., Grantyn, R., and Rathjen, F. G. (2005). Impaired synapse function during postnatal development in the absence of CALEB, an EGF-like protein processed by neuronal activity. Neuron 46, 233–245.
Karten, Y. J., Jones, M. A., Jeurling, S. I., and Cameron, H. A. (2006). GABAergic signaling in young granule cells in the adult rat and mouse dentate gyrus. Hippocampus 16, 312–320.
Kirischuk, S., Clements, J. D., and Grantyn, R. (2002). Presynaptic and postsynaptic mechanisms underlie paired pulse depression at single GABAergic boutons. J. Physiol. 543, 99–116.
Kirischuk, S., and Grantyn, R. (2000). A readily releasable pool of single inhibitory boutons in culture. Neuroreport 11, 3709–3713.
Kirischuk, S., and Grantyn, R. (2002). Inter-bouton variability of synaptic strength correlates with heterogeneity of presynaptic Ca(2+) signals. J. Neurophysiol. 88, 2172–2176.
Kirischuk, S., and Grantyn, R. (2003). Intraterminal Ca2+ concentration and asynchronous transmitter release at single GABAergic boutons in rat collicular cultures. J. Physiol. 548, 753–764.
Kirischuk, S., Juettner, R., and Grantyn, R. (2005). Time-matched pre- and postsynaptic changes of GABAergic synaptic transmission in the developing mouse superior colliculus. J. Physiol. 563, 795–807.
Kirischuk, S., Veselovsky, N., and Grantyn, R. (1999). Relationship between presynaptic calcium transients and postsynaptic currents at single gamma-aminobutyric acid (GABA)ergic boutons. Proc. Natl. Acad. Sci. U.S.A. 96, 7520–7525.
Kirmse, K., and Kirischuk, S. (2006). N-ethylmaleimide increases release probability at GABAergic synapses in layer I of the mouse visual cortex. Eur. J. Neurosci. 24, 2741–2748.
Kirsch, J., Wolters, I., Triller, A., and Betz, H. (1993). Gephyrin antisense oligonucleotides prevent glycine receptor clustering in spinal neurons. Nature 366, 745–748.
Korn, H., and Faber, D. S. (1987). “Regulation and significance of probabilistic release mechanisms at central synapses,” in Synaptic Function, eds G. M. Edelman, W. E. Gall, and W. M. Cowan (New York: Wiley), 57–108.
Laurie, D. J., Wisden, W., and Seeburg, P. H. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172.
Legendre, P., Forstera, B., Juttner, R., and Meier, J. C. (2009). Glycine receptors caught between genome and proteome – functional implications of RNA editing and splicing. Front. Mol. Neurosci. 2:23. doi: 10.3389/neuro.02.023.2009
Llano, I., Gonzalez, J., Caputo, C., Lai, F. A., Blayney, L. M., Tan, Y. P., and Marty, A. (2000a). Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat. Neurosci. 3, 1256–1265.
Llano, I., Gonzalez, J., Caputo, C., Lai, F. A., Blayney, L. M., Tan, Y. P., and Marty, A. (2000b). Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat. Neurosci. 3, 1256–1265.
Lund, R. D., and Lund, J. S. (1972). Development of synaptic patterns in the superior colliculus of the rat. Brain Res. 42, 1–20.
Ma, D. K., Marchetto, M. C., Guo, J. U., Ming, G. L., Gage, F. H., and Song, H. (2010). Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat. Neurosci. 13, 1338–1344.
Marty, A., and Llano, I. (2005). Excitatory effects of GABA in established brain networks. Trends Neurosci. 28, 284–289.
Marty, S., Wehrlé, R., and Sotelo, C. (2000). Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J. Neurosci. 20, 8087–8095.
McBain, C. J., and Kauer, J. A. (2009). Presynaptic plasticity: targeted control of inhibitory networks. Curr. Opin. Neurobiol. 19, 254–262.
Meier, J., Akyeli, J., Kirischuk, S., and Grantyn, R. (2003). GABA(A) receptor activity and PKC control inhibitory synaptogenesis in CNS tissue slices. Mol. Cell. Neurosci. 23, 600–613.
Meier, J. C., Henneberger, C., Melnick, I., Racca, C., Harvey, R. J., Heinemann, U., Schmieden, V., and Grantyn, R. (2005). RNA editing produces glycine receptor alpha3(P185L), resulting in high agonist potency. Nat. Neurosci. 8, 736–744.
Misgeld, U., Wagner, A., and Ohno, T. (1982). Depolarizing IPSPs and Depolarization by GABA of rat neostriatum cells in vitro. Exp. Brain Res. 45, 108–114.
Mittmann, T., and Eysel, U. T. (2001). Increased synaptic plasticity in the surround of visual cortex lesions in rats. Neuroreport 12, 3341–3347.
Mozhayeva, M. G., Sara, Y., Liu, X., and Kavalali, E. T. (2002). Development of vesicle pools during maturation of hippocampal synapses. J. Neurosci. 22, 654–665.
Mueller, A. L., Taube, J. S., and Schwartzkroin, P. A. (1984). Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J. Neurosci. 4, 860–867.
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872.
Oertner, T. G., Sabatini, B. L., Nimchinsky, E. A., and Svoboda, K. (2002). Facilitation at single synapses probed with optical quantal analysis. Nat. Neurosci. 5, 657–664.
Overstreet-Wadiche, L., Bromberg, D. A., Bensen, A. L., and Westbrook, G. L. (2005). GABAergic signaling to newborn neurons in dentate gyrus. J. Neurophysiol. 94, 4528–4532.
Pan, B., and Zucker, R. S. (2009). A general model of synaptic transmission and short-term plasticity. Neuron 62, 539–554.
Pang, Z. P., and Sudhof, T. C. (2010). Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol. 22, 496–505.
Poncer, J. C., McKinney, R. A., Gahwiler, B. H., and Thompson, S. M. (2000). Differential control of GABA release at synapses from distinct interneurons in rat hippocampus. J. Physiol. 528(Pt 1), 123–130.
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., Pirvola, U., Saarma, M., and Kaila, K. (1999). The K+/Cl− cotransporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255.
Rörig, B., and Grantyn, R. (1994). Ligand- and voltage-gated ion channels are expressed by embryonic mouse retinal neurons. Neuroreport 5, 1197–1200.
Safiulina, V. F., Caiati, M. D., Sivakumaran, S., Bisson, G., Migliore, M., and Cherubini, E. (2010). Control of GABA release at mossy fiber-CA3 connections in the developing hippocampus. Front. Synaptic Neurosci. 2:1. doi: 10.3389/neuro.19.001.2010
Safiulina, V. F., Fattorini, G., Conti, F., and Cherubini, E. (2006). GABAergic signaling at mossy fiber synapses in neonatal rat hippocampus. J. Neurosci. 26, 597–608.
Sakaba, T., Schneggenburger, R., and Neher, E. (2002). Estimation of quantal parameters at the calyx of held synapse. Neurosci. Res. 44, 343–356.
Salama-Cohen, P., Arevalo, M. A., Grantyn, R., and Rodríguez-Tébar, A. (2006). Notch and NGF/p75NTR control dendrite morphology and the balance of excitatory/inhibitory synaptic input to hippocampal neurons through Neurogenin 3. J. Neurochem. 97, 1269–1278.
Santhakumar, V., Jones, R. T., and Mody, I. (2010). Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience 167, 644–655.
Scheuss, V., and Neher, E. (2001). Estimating synaptic parameters from mean, variance, and covariance in trains of synaptic responses. Biophys. J. 81, 1970–1989.
Schneggenburger, R., Meyer, A. C., and Neher, E. (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23, 399–409.
Senn, W., Segev, I., and Tsodyks, M. (1998). Reading neuronal synchrony with depressing synapses. Neural. Comput. 10, 815–819.
Sernagor, E., Chabrol, F., Bony, G., and Cancedda, L. (2010). GABAergic control of neurite outgrowth and remodeling during development and adult neurogenesis: general rules and differences in diverse systems. Front. Cell. Neurosci. 4:11. doi: 10.3389/fncel.2010.00011
Silver, R. A. (2003). Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J. Neurosci. Methods 130, 127–141.
Singh, B., Henneberger, C., Betances, D., Arevalo, M. A., Rodriguez-Tebar, A., Meier, J. C., and Grantyn, R. (2006). Altered balance of glutamatergic/GABAergic synaptic input and associated changes in dendrite morphology after BDNF expression in BDNF-deficient hippocampal neurons. J. Neurosci. 26, 7189–7200.
Stein, B. E., Perrault, T. J. Jr., Stanford, T. R., and Rowland, B. A. (2009). Postnatal experiences influence how the brain integrates information from different senses. Front Integr. Neurosci. 3:21. doi: 10.3389/neuro.07.021.2009
Stricker, C., Field, A. C., and Redman, S. J. (1996). Statistical analysis of amplitude fluctuations in EPSCs evoked in rat CA1 pyramidal neurones in vitro. J. Physiol. 490, 419–441.
Tanaka, T., Saito, H., and Matsuki, N. (1997). Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J. Neurosci. 17, 2959–2966.
Tao, H. W., and Poo, M. M. (2005). Activity-dependent matching of excitatory and inhibitory inputs during refinement of visual receptive fields. Neuron 45, 829–836.
Taschenberger, H., Scheuss, V., and Neher, E. (2005). Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J. Physiol. 568, 513–537.
Tian, N., Petersen, C., Kash, S., Baekkeskov, S., Copenhagen, D., and Nicoll, R. (1999). The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc. Natl. Acad. Sci. U.S.A. 96, 12911–12916.
Toni, N., and Sultan, S. (2011). Synapse formation on adult-born hippocampal neurons. Eur. J. Neurosci. 33, 1062–1068.
Tozuka, Y., Fukuda, S., Namba, T., Seki, T., and Hisatsune, T. (2005). GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 47, 803–815.
Tyagarajan, S.K., Ghosh, H., Yevenes, G.E., Nikonenko, I., Ebeling, C., Schwerdel, C., Sidler, C., Zeilhofer, H.U., Gerrits, B., Muller, D., and Fritschy, J.M. (2011). Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. U. S. A 108, 379–384.
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Aniksztejn, L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19, 10372–10382.
Unsoeld, T., Stradomska, A. M., Wang, R., Rathjen, F. G., and Juttner, R. (2008). Early maturation of GABAergic synapses in mouse retinal ganglion cells. Int. J. Dev. Neurosci. 26, 233–238.
Waldeck, R. F., Pereda, A., and Faber, D. S. (2000). Properties and plasticity of paired-pulse depression at a central synapse. J. Neurosci. 20, 5312–5320.
Wang, L. P., Kempermann, G., and Kettenmann, H. (2005). A subpopulation of precursor cells in the mouse dentate gyrus receives synaptic GABAergic input. Mol. Cell. Neurosci. 29, 181–189.
Wasling, P., Hanse, E., and Gustafsson, B. (2004). Developmental changes in release properties of the CA3-CA1 glutamate synapse in rat hippocampus. J. Neurophysiol. 92, 2714–2724.
Wilcox, K. S., and Dichter, M. A. (1994). Paired pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J. Neurosci. 14, 1775–1788.
Yang, H., and Xu-Friedman, M. A. (2010). Developmental mechanisms for suppressing the effects of delayed release at the endbulb of held. J. Neurosci. 30, 11466–11475.
Keywords: GABAergic synaptic transmission, quantal analysis, presynaptic function, tonic inhibition, synapse development, BDNF, NGF, excitatory–inhibitory balance
Citation: Grantyn R, Henneberger C, Jüttner R, Meier JC and Kirischuk S (2011) Functional hallmarks of GABAergic synapse maturation and the diverse roles of neurotrophins. Front. Cell. Neurosci. 5:13. doi: 10.3389/fncel.2011.00013
Received: 01 April 2011;
Paper pending published: 23 April 2011;
Accepted: 17 June 2011;
Published online: 04 July 2011.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Rafael Gutierrez, Centro de Investigación y Estudios Avanzados del Instituto Politécnico Nacional, MexicoDonald S. Faber, Albert Einstein College of Medicine, USA
Copyright: © 2011 Grantyn, Henneberger, Jüttner, Meier and Kirischuk. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Rosemarie Grantyn, Experimental Neurology and Cluster of Excellence Neurocure, University Medicine Charité, Robert-Koch-Platz 4, D-10115 Berlin, Germany. e-mail:cm9zZW1hcmllLmdyYW50eW5AY2hhcml0ZS5kZQ==