 Jake Ormond
Jake Ormond Melanie A. Woodin*
Melanie A. Woodin*- Department of Cell and Systems Biology, University of Toronto, Toronto, ON, Canada
Paired pre- and postsynaptic activity in area CA1 of the hippocampus induces long-term inhibitory synaptic plasticity at GABAergic synapses. This pairing-induced GABAergic plasticity weakens synaptic inhibition due to a depolarization of the reversal potential for GABAA receptor-mediated currents (EGABA) through a decrease in the function of the neuron-specific K+–Cl− cotransporter KCC2. When pairing-induced GABAergic plasticity is induced at feed-forward inhibitory synapses in the CA1, the decrease in inhibition produces an increase in the amplitude of Schaffer collateral-mediated postsynaptic potentials in pyramidal neurons. This form of inhibitory synaptic plasticity is termed disinhibition-mediated long-term potentiation (LTP). In the present study, we investigated whether disinhibition-mediated LTP is synapse specific. We performed these experiments in hippocampal slices prepared from adult Sprague Dawley rats. We found that the underlying depolarization of EGABA is not restricted to the paired pathway, but rather is expressed to the same extent at unpaired control pathways. However, the overall strength of GABAergic transmission is maintained at the unpaired pathway by a heterosynaptic increase in GABAergic conductance. The pairing-induced depolarization of EGABA at the paired and unpaired pathways required Ca2+-influx through both the L-type voltage-gated Ca2+ channels and N-methyl-D-aspartic acid receptors. However, only Ca2+-influx through L-type channels was required for the increased conductance at the unpaired pathway. As a result of this increased GABAergic conductance, disinhibition-mediated LTP remains confined to the paired pathway and thus is synapse specific, suggesting it may be a novel mechanism for hippocampal-dependent learning and memory.
Introduction
Inhibitory synaptic transmission plays a central role in regulating the output of neurons and neuronal circuits throughout the nervous system (Pouille and Scanziani, 2001; Markram et al., 2004; Akerman and Cline, 2006). Inhibitory synaptic plasticity can be induced by repetitively pairing pre- and postsynaptic activity at mature GABAergic synapses in the hippocampus (Woodin et al., 2003; Fiumelli and Woodin, 2007; Balena and Woodin, 2008; Lamsa et al., 2010), which results in a decrease in the strength of inhibition. The mechanism underlying this form of inhibitory spike-timing dependent plasticity (STDP) is a Ca2+-dependent decrease in the function of KCC2 (Woodin et al., 2003; Balena et al., 2010), which is primarily responsible for maintaining neuronal Cl− gradients in the mature CNS (Rivera et al., 1999; Blaesse et al., 2009).
In hippocampal area CA1, the firing of the presynaptic Schaffer collaterals produces a mixed postsynaptic potential (PSP) composed of an excitatory PSP (EPSP) and an overlapping inhibitory PSP (IPSP). The IPSP is generated disynaptically by Schaffer collateral-driven firing of GABAergic basket cells (Glickfeld and Scanziani, 2006), and normally arrives prior to the preceding EPSP reaching its maximal amplitude (Pouille and Scanziani, 2001). The effect of this overlapping IPSP is to profoundly attenuate the firing of the postsynaptic CA1 pyramidals (Pouille and Scanziani, 2001), an effect which can be at least partly explained by the ability of feed-forward inhibition to limit the degree to which dendritic EPSPs are able to depolarize the soma of the pyramidal neuron (Ormond and Woodin, 2009). We recently demonstrated that a form of inhibitory synaptic plasticity, termed disinhibition-mediated long-term potentiation (LTP), regulates the activity of pyramidal neurons in the hippocampus (Saraga et al., 2008; Ormond and Woodin, 2009). Disinhibition-mediated LTP occurs when repetitive pairing of pre- and postsynaptic activity weakens inhibition at feed-forward inhibitory synapses (Woodin et al., 2003), thereby potentiating Schaffer collateral-mediated PSPs (Ormond and Woodin, 2009) and increasing pyramidal neuron spiking (Saraga et al., 2008). Thus, plasticity at feed-forward inhibitory synapses has the potential to regulate the efficacy of synaptic transmission between areas CA3 and CA1, much like classic LTP expressed at glutamatergic synapses.
An important requirement for cellular mechanisms of learning and memory is that they demonstrate synapse specificity (Hebb, 1949; Marr, 1969; Albus, 1971). For example, the expression of glutamatergic LTP, thought to play a central role in hippocampal-dependent memory formation (Tsien et al., 1996; Morris and Frey, 1997), is input specific, meaning that LTP is largely restricted to the pathway at which it was induced (Andersen et al., 1977; Engert and Bonhoeffer, 1997). Because the inhibitory plasticity underlying disinhibition-mediated LTP depends on a postsynaptic increase in intracellular chloride at feed-forward inhibitory synapses (Woodin et al., 2003; Ormond and Woodin, 2009), most of which are located on the soma (Glickfeld and Scanziani, 2006), we were doubtful that its expression would be confined to the set of synapses active during the induction. However, because the inhibitory effect of GABAergic transmission depends not just on the driving force for GABAergic currents, set primarily by chloride levels, but also on the size of the synaptic conductance, we investigated whether pairing might induce parallel changes in inhibitory synaptic conductance capable of confining disinhibition-mediated LTP to the pathway at which it was induced.
Materials and Methods
Brain Slice Preparation
Hippocampal brain slices were prepared from 50- to 75-day-old male Sprague Dawley rats as previously described (Ormond and Woodin, 2009). The Animal Care Ethics committee at the University of Toronto approved all experimental protocols. Briefly, rats were anesthetized with a mixture of xylazine and ketamine injected intraperitoneally (25 mg/kg), and the brain was perfused with chilled modified artificial cerebrospinal fluid (ACSF; saturated with 95% O2/5% CO2; pH 7.4, osmolarity ∼305 MΩ). Hippocampi were partially isolated by removing the midbrain and all cortex except that directly overlying the hippocampus, and 400 μm slices were cut with a Vibratome 1000 plus. Slices recovered for 1 h in 35–37°C ACSF saturated with 95% O2/5% CO2 (pH 7.4, osmolarity ∼305 mOsm).
Electrophysiology
Whole-cell recordings were made in oxygenated ACSF at 35–37°C from CA1 pyramidal cells. Pyramidal neurons were identified by the presence of an after-depolarization following action potential firing, as well as action potential accommodation during prolonged action potential trains. Recording pipettes were pulled from thin-walled borosilicate (TW-150F; World Precision Industries, Sarasota, FL, USA) to resistances of 5–8 MΩ with a Sutter Instruments P-87 (Novato, CA, USA). Pipettes were filled with: 130 mM potassium gluconate, 10 mM KCl, 10 mM HEPES, 0.2 mM EGTA, 4 mM ATP, 0.3 mM GTP, 10 mM phosphocreatine (pH 7.25, osmolarity 275–285 mOsm). Signals were amplified using an Axon Instruments MultiClamp 700b and digitized using an Axon Instruments Digidata 1322a (Molecular Devices, Sunnyvale, CA, USA). The bridge was balanced upon going whole-cell, and then monitored and adjusted as necessary throughout the recording (to a maximum of 10 MΩ/experiment). Extracellular stimulation was applied through a whole-cell recording pipette containing a silver chloride wire and filled with ACSF at a baseline recording frequency of 0.03 Hz. CNQX was used to block glutamatergic transmission for recordings of isolated IPSPs. APV, BAPTA, nimodipine, and CNQX were purchased from Sigma-Aldrich (ON, Canada).
Plasticity induction
Plasticity was induced by pairing extracellular stimulation with postsynaptic current injection (1 nA for 10 ms); synaptic stimulation occurred 1 ms after the beginning of the 10-ms pulse. During induction the postsynaptic cell typically fired two action potentials per pairing, with the first action potential occurring 2–4 ms after extracellular stimulation, and the second occurring about 5 ms after the first. Three-hundred pairings were made at a frequency of 5 Hz. During recordings of pharmacologically isolated inhibition, plasticity was only induced at synapses where the initial EGABA value was between −70 and −80 mV.
Characterization of feed-forward inhibition, pathway independence, and stimulating electrode position
In experiments where EGABA and GABAergic conductance were recorded (Figures 1, 2, and 5), interneurons were stimulated directly by placing the stimulating electrodes in S. pyramidale, on either side of the recorded neuron to stimulate the paired and unpaired pathways (Figure 1A). The independence of pathways was demonstrated by the confinement of short-term plasticity within pathways (Figure A1 in Appendix). Four pulses at 20 Hz were applied to one pathway, followed by a pulse to other pathway; the protocol was then repeated with four pulses to the second pathway followed by one to the first. For each pathway, the ratio of the fourth to the first pulse was taken as the measure of short-term plasticity within the pathway, while the ratio of the single fifth pulse to the first pulse in the other recording gave the short-term plasticity between pathways.
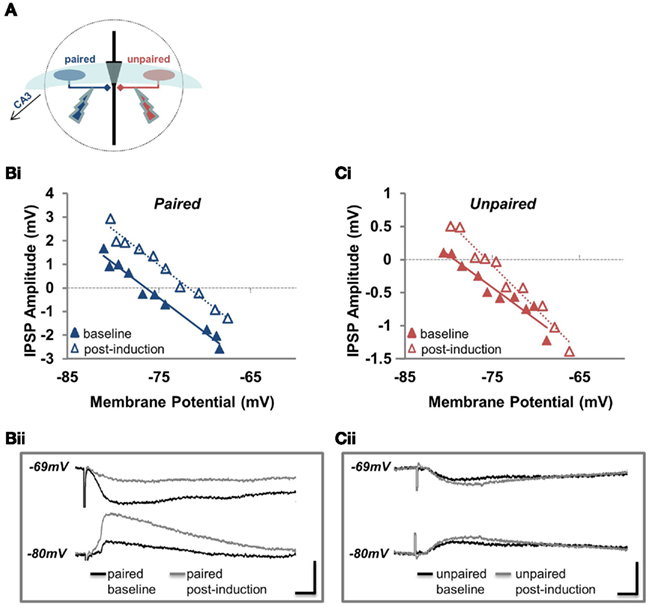
Figure 1. EGABA depolarizes and conductance increases at unpaired inhibitory synapses. (A) Position of the paired and unpaired stimulating electrodes for all experiments examining EGABA depolarization and GABA conductance. (Bi) Example of a recording used to determine EGABA at the paired pathway. IPSP amplitude plotted against Vm for the paired pathway before (baseline; solid blue triangles) and after pairing (post-induction; open blue triangles). (Bii) Sample traces from the experiment plotted in (Bi). Current waveforms are overlaid from the baseline period (black) and from the last post-induction time point (gray), for relatively hyperpolarized and depolarized potentials. Scale bars: 2 mV, 10 ms. (Ci) Example of a recording used to determine EGABA at the unpaired pathway. IPSP amplitude plotted against Vm for the unpaired pathway before (baseline; solid red triangles) and after pairing (post-induction; open red triangles). (Cii) Sample traces from the experiment plotted in (Ci). Current waveforms are overlaid from the baseline period (black) and from the last post-induction time point (gray), for relatively hyperpolarized and depolarized potentials. Scale bars: 2 mV, 10 ms.
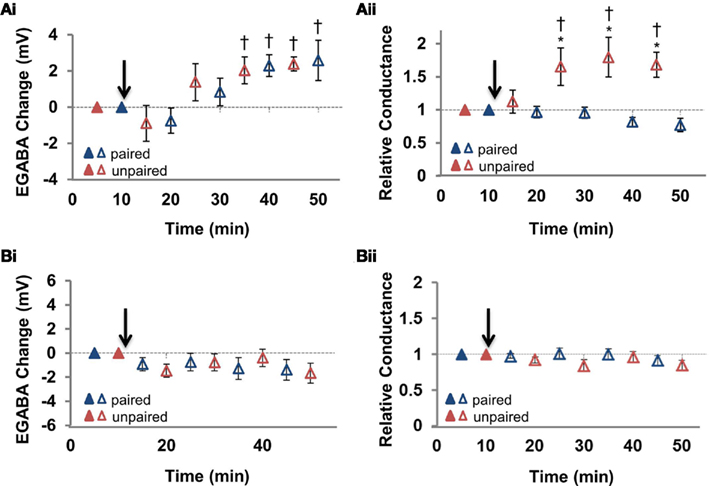
Figure 2. Summary of the changes in EGABA and conductance at the paired and unpaired pathways. (Ai) Summary of the change in EGABA at the paired and unpaired pathways (n = 6). (Aii) Summary of the change in relative GABAergic conductance at the paired and unpaired pathways [for the same cells in (Ai)]. Blue triangles denote paired pathways, red triangles denote unpaired pathways; filled triangles represent baseline recordings, open triangles represent recordings following plasticity induction. (Bi) Summary of the change in EGABA at the paired and unpaired pathways following stimulation of the postsynaptic neuron alone (n = 7). (Bii) Summary of the change in relative GABAergic conductance at the paired and unpaired pathways [for the same cells in (Ai)]. Blue triangles denote paired pathways, red triangles denote unpaired pathways; filled triangles represent baseline recordings, open triangles represent recordings following plasticity induction. IPSP–Vm curves were constructed at five time points, alternating between unpaired and paired pathways. Each of the five time points at the paired pathway were compared to the corresponding time point from the unpaired pathway.* Denotes statistical significance from unpaired pathway (p < 0.05). † Denotes statistical significance from baseline (p < 0.05). Paired activity denoted by arrow.
In experiments where disinhibition-mediated LTP was recorded (Figures 3, 4, and 6), interneurons were not stimulated directly. In these experiments the stimulating electrodes were placed in the CA3 (Figure 3Ai). To verify that the recorded inhibition was feed-forward (activated by Schaeffer collateral-mediated excitation of interneurons), and not due to direct stimulation of inhibitory fibers, CNQX was perfused at the end of all mixed PSP recordings (Figure 3Aii) and its effect on the slope of the PSP vs. membrane potential (Vm) relationship was assessed; because glutamatergic EPSP amplitude does not show voltage dependence at the membrane potentials examined (Ormond and Woodin, 2009), the reduction in slope after CNQX can be attributed to reduced feed-forward inhibition. Application of 10 μM CNQX reduced the mean slope of the PSP vs. membrane potential (Vm) relationship for all recordings by 89%.
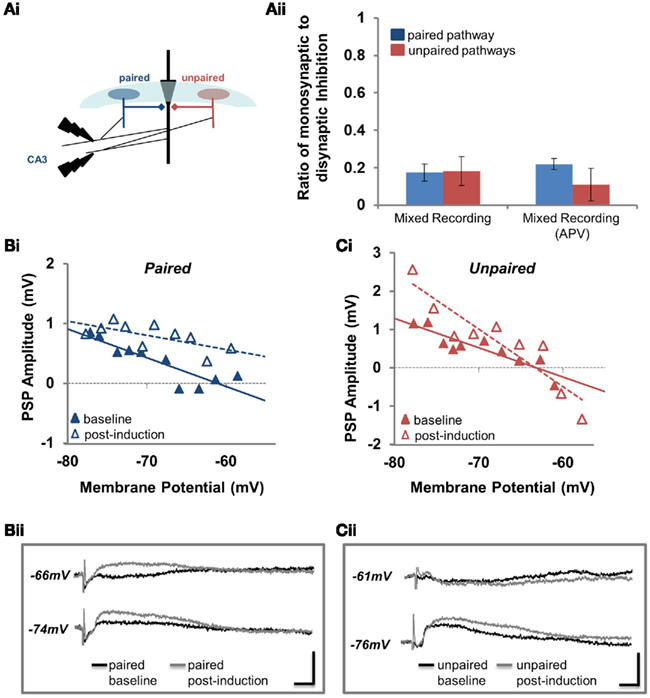
Figure 3. Disinhibition-mediated LTP is pathway specific. (Ai) Position of the paired and unpaired stimulating electrodes for all experiments where inhibition was stimulated disynaptically. (Aii) The ratio of monosynaptic to disynaptic inhibition (see Materials and Methods for details) at the paired (blue) and unpaired (red) pathways for disinhibition-mediated LTP (mixed recording) experiments performed in the absence and presence of APV. (Bi) PSP amplitude is plotted against Vm for the paired pathway before (baseline; solid blue triangles) and after pairing (post-induction; open blue triangles). (Bii) Sample traces from the experiment plotted in (Bi). Current waveforms are overlaid from the baseline period (black) and from the last post-induction time point (gray), for relatively hyperpolarized and depolarized potentials. Scale bars: 2 mV, 10 ms. (Ci) PSP amplitude is plotted against Vm for the unpaired pathway before (baseline; solid red triangles) and after pairing (post-induction; open red triangles). (Cii) Sample traces from the experiment plotted in (Ci). Current waveforms are overlaid from the baseline period (black) and from the last post-induction time point (gray), for relatively hyperpolarized and depolarized potentials. Scale bars: 2 mV, 10 ms.
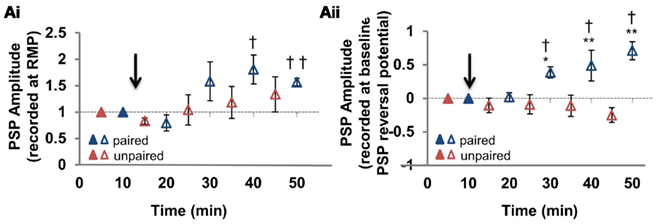
Figure 4. Summary of disinhibition-mediated LTP pathway specificity. (Ai) Summary of the change in mixed PSP amplitude measured from the resting membrane potential, at the paired and unpaired pathways (n = 6). (Aii) Summary of the change in mixed PSP amplitude measured from the initial PSP reversal potential [for the same cells as in (Ai)]. Blue triangles denote paired pathways, red triangles denote unpaired pathways; filled triangles represent baseline recordings, open triangles represent recordings following plasticity induction. PSP–Vm curves were constructed at five time points, alternating between unpaired and paired pathways. Each of the five time points at the paired pathway were compared to the corresponding time point from the unpaired pathway. * Denotes statistical significance from unpaired pathway (p < 0.05). † Denotes statistical significance from baseline (p < 0.05). Paired activity denoted by arrow.
Measurement of EGABA
EGABA was determined in current clamp mode by evoking IPSPs from progressively more depolarized membrane potentials. The protocol for each EGABA measurement consisted of 10 sweeps beginning with a negative current step (between −100 and −150 pA), with each subsequent step incremented by 25 pA; during each of these ten current steps an IPSP was evoked. These IPSPs were used to generate an IPSP–Vm curve (e.g., Figure 1B). IPSPs were stimulated with extracellular stimulating electrodes (described in Characterization of Feed-Forward Inhibition, Pathway Independence, and Stimulating Electrode Position) in the presence of 10 μM CNQX to block AMPA-mediated synaptic transmission. A linear regression of the IPSP amplitudes was then used to calculate the membrane potential dependence of IPSPs. The intercept of this line with the abscissa was taken as EGABA. The slope of the same line was taken as a measure of relative IPSP conductance, meaning slopes at different time points could be compared to determine whether conductance had changed. In some instances, when Vm was stepped to relatively depolarized potentials, the action potential threshold was reached and a spike was fired, which obscured the PSP. In these instances (such as Figure 3Ci), these data points were removed.
Data Analysis and Statistics
Data was acquired using Axon Instruments Clampex 9 software, and analyzed using Axon Instruments Clampfit (Molecular Devices, Sunnyvale, CA, USA) as previously described (Ormond and Woodin, 2009). Results are expressed as mean ± SEM. All statistical tests were performed in SigmaStat (Systat Software, San Jose, CA, USA). Significance (p < 0.05) was determined using a two-way repeated measures ANOVA with post hoc Tukey test.
Results
To investigate the synapse specificity of disinhibition-mediated LTP we first determined whether pairing-induced GABAergic plasticity was restricted to the stimulated pathway. To do this we made whole-cell patch-clamp recordings at 35–37°C from pyramidal neurons in hippocampal slices prepared from 50- to 75-day-old Sprague Dawley rats; GABAergic STDP is known to be expressed under whole-cell recording conditions (Woodin et al., 2003; Ormond and Woodin, 2009). We paired extracellular stimulation in S. pyramidale (see Characterization of Feed-Forward Inhibition, Pathway Independence, and Stimulating Electrode Position), the site of most interneurons providing feed-forward inhibition onto CA1 pyramidal neurons (Figure 1A; Pouille and Scanziani, 2001), with postsynaptic pyramidal neuron spiking evoked with intracellular current injection. We repeated this paired pre- and postsynaptic activity 300× at 5 Hz (see Plasticity Induction); from here forward we refer to this plasticity induction as “paired activity” (Woodin et al., 2003; Ormond and Woodin, 2009). We monitored EGABA and GABAergic conductance at 5 min intervals before and after plasticity induction, at both the paired pathway where we stimulated plasticity, and at an unpaired (control) pathway. The extracellular stimulating electrode for the unpaired pathway was also placed in the S. pyramidale, but on the opposite sides of the recorded neuron (Figure 1A). The independence of these pathways was demonstrated by the confinement of short-term plasticity within pathways (see Characterization of Feed-Forward Inhibition, Pathway Independence, and Stimulating Electrode Position for detailed explanation; Figure A1 in Appendix).
We found that paired activity induced a depolarization of EGABA at the paired pathway (Figures 1B and 2Ai; p < 0.05, n = 6): the magnitude of this depolarization is similar to that previously reported (Woodin et al., 2003; Ormond and Woodin, 2009). Likewise, EGABA depolarized to a similar magnitude and with a similar time course at the unpaired control pathway (Figures 1C and 2Ai; p < 0.05, n = 6), indicating that this form of synaptic plasticity is not pathway specific. However, synaptic conductance was significantly increased at the unpaired pathway (Figures 1C and 2Aii; p > 0.05, n = 6). The absolute values of EGABA, IPSP slope, and resting membrane potential (RMP) are listed in Table 1. Stimulation of only the postsynaptic neurons during the plasticity induction protocol failed to significantly alter EGABA or conductance at the paired or unpaired pathways (Figure 2B; p > 0.05, n = 7). We have previously demonstrated, under the same recording conditions and in the same preparation, that EGABA and GABAergic conductance does not significantly change during baseline recordings of the same duration as the recordings presented here (Ormond and Woodin, 2009).
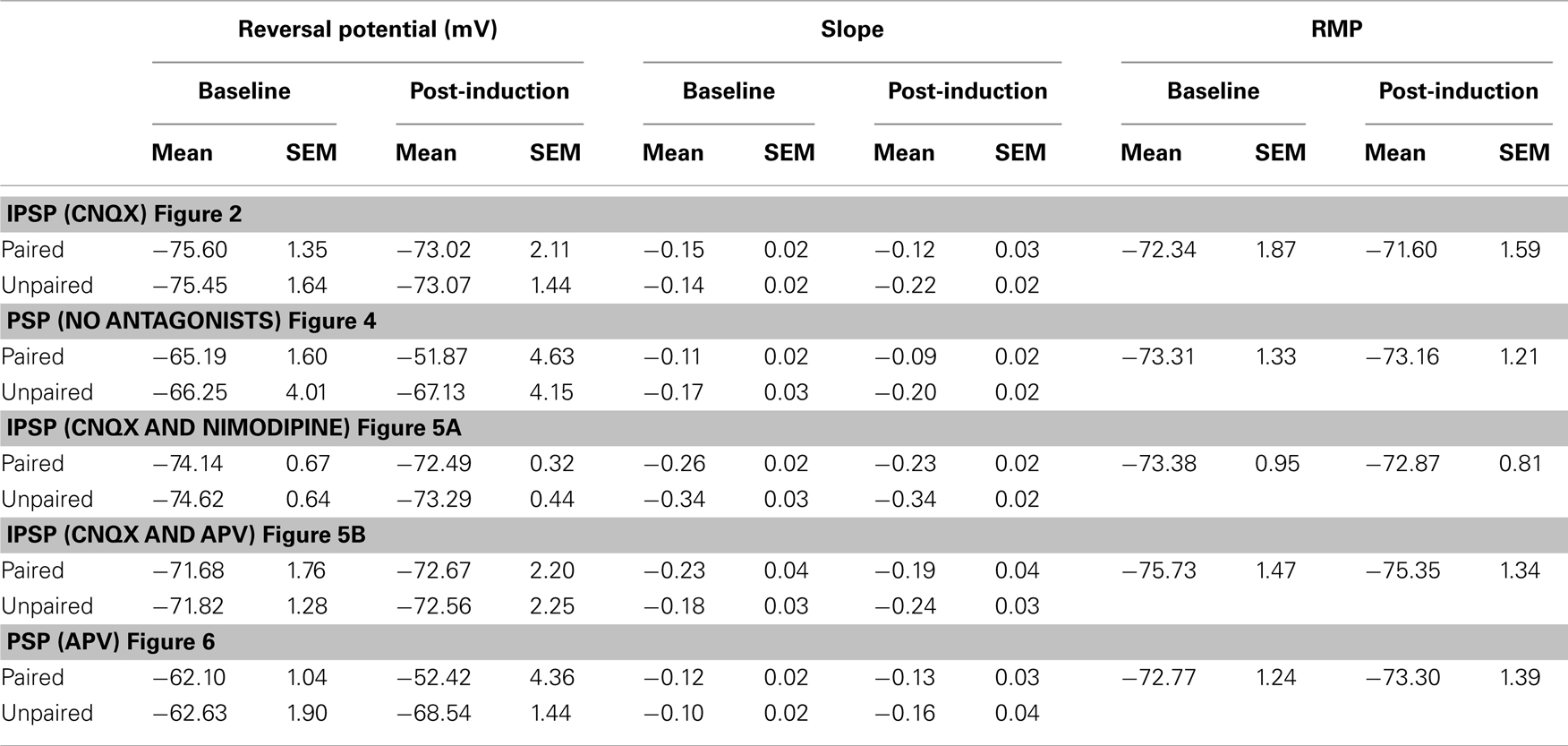
Table 1. Electrophysiological properties of CA1 pyramidal neurons. Reversal potential, PSP slope, and RMP are reported for baseline and post-induction recordings made during plasticity recordings at both paired and unpaired inputs.
We had initially hypothesized that if pairing-induced GABAergic plasticity was not synapse specific, then disinhibition-mediated LTP would also not be synapse specific. However our results demonstrating an increase in conductance accompanying the depolarization of EGABA at unpaired pathways suggested our hypothesis might be false. To determine whether disinhibition-mediated LTP was in fact synapse specific we paired extracellular stimulation of the Schaffer collaterals in CA3 with postsynaptic pyramidal spiking (we were careful to place the stimulating electrode in the CA3 to avoid eliciting inhibition directly; Figure 3Ai; see Characterization of Feed-Forward Inhibition, Pathway Independence, and Stimulating Electrode Position for details). The absence of monosynaptic inhibition was confirmed at the end of recordings with CNQX, demonstrating that the majority of inhibition in these recordings was disynaptic and feed-forward (Figure 3Aii; see Characterization of Feed-Forward Inhibition, Pathway Independence, and Stimulating Electrode Position). This configuration allowed us to record mixed glutamatergic and GABAergic PSPs from pyramidal neurons; we refer to this recording configuration as “mixed recording.” Following paired activity, we observed how the PSP amplitude changed relative to the membrane potential. In the example shown in Figure 3B, there was an increase in PSP amplitude at the paired pathway; this increase in PSP amplitude occurred at all membrane potentials from which PSPs were elicited (from −58 to −78 mV). However, the situation was different at the unpaired pathway; in the example shown in Figure 3C, there is an increase in PSP amplitude at −70 mV, but a decrease in amplitude at −60 mV. This suggests that the effect of inhibitory synaptic plasticity at the unpaired pathway, which is comprised of both EGABA depolarization and increased conductance, depends on the membrane potential.
To examine this relationship more fully we used our PSP–Vm curves for each recording to calculate PSP amplitude at two membrane potentials: (1) the RMP (Figure 4Ai); and (2) the reversal potential of mixed PSPs recorded during the pre-induction baseline period (which was more depolarized than RMP; Figure 4Aii). Regardless of which PSP values we used (PSP amplitude at RMP or at initial PSP reversal potential), the paired pathway always demonstrated disinhibition-mediated LTP (Figure 4; n = 6; p < 0.001). However, at the unpaired pathway we did not observe disinhibition-mediated LTP. When RMP values were used (Figure 4Ai), there was neither a significant change from baseline (p = 0.173) nor from the potentiated pathway (p = 0.345). However, when measured from the baseline PSP reversal, our analysis showed that PSPs became slightly hyperpolarizing after pairing, making the paired and unpaired pathways significantly different (Figure 4Aii; p < 0.001). Because there is very little voltage dependence of EPSP amplitude in the range of membrane potentials we recorded (Ormond and Woodin, 2009), the voltage dependence of the change in PSP amplitude at the unpaired pathway strongly suggested the involvement of the GABAergic plasticity described above (Figures 1 and 2). Taken together, our recordings revealed that at the paired pathway the depolarization of EGABA produced disinhibition-mediated LTP, while at the unpaired pathway the depolarization of EGABA was counteracted by the increase in GABAergic conductance to prevent disinhibition-mediated LTP (Figure 2Cii). This counteracting effect increases when the membrane potential is depolarized.
To confirm that the increased GABAergic conductance at the unpaired pathway was in fact responsible for the synapse specificity of disinhibition-mediated LTP, we wanted to further examine the effect in pharmacological isolation from glutamatergic LTP and EGABA depolarization. Because both glutamatergic LTP and pairing-induced EGABA depolarization require an elevation in intracellular Ca2+ concentration, blocking this elevation seemed a logical approach for the first of these experiments. When postsynaptic Ca2+ was chelated by including 30 mM BAPTA in the pipette, both EGABA and the increased conductance at the unpaired pathway were blocked (Figures 5Ai,ii); at the paired pathway, conductance increased slightly, and was significantly different from the unpaired pathway (n = 6; p = 0.02) but not baseline. Having failed to spare the increased conductance at the unpaired pathway, we next tried to block EGABA depolarization using the L-type Ca2+ channel blocker nimodipine, as it has previously been reported to block the depolarization of EGABA induced by pairing in hippocampal cell culture (Woodin et al., 2003). With nimodipine (15 μM) in the bath, pairing failed to produce a significant EGABA depolarization at either pathway (Figure 5Bi; Table 1; n = 6; paired pathway p = 0.162, unpaired pathway p = 0.137). This suggests that EGABA depolarization in hippocampal slices from adults also depends on Ca2+ entry through L-type Ca2+ channels. However, nimodipine also completely blocked the increase in GABAergic conductance at the unpaired pathway (Figure 5Bii; Table 1; n = 6; p = 0.551), so it was of no use as a tool to isolate the increase in GABAergic conductance.
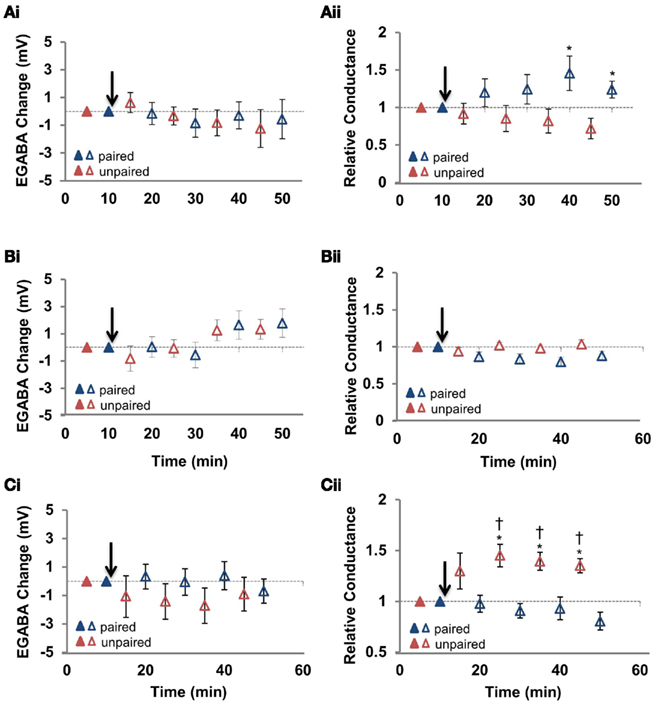
Figure 5. N-methyl-D-aspartic acid receptor blockade with APV spares the increased GABAergic conductance at the unpaired pathway. (Ai) Summary of the change in EGABA at the paired and unpaired pathways induced in the presence of cell-loaded BAPTA to chelate Ca2+ (n = 5). (Aii) Summary of the change in relative GABAergic conductance at the paired and unpaired pathways for all cells in (Ai). (Bi) Summary of the change in EGABA at the paired and unpaired pathways induced in the presence of bath applied nimodipine to block L-type Ca2+ channels (15 μM applied for the duration of the recording; n = 5). (Bii) Summary of the change in relative GABAergic conductance at the paired and unpaired pathways for all cells in (Bi). (Ci) Summary of the change in EGABA at the paired and unpaired pathways in the presence of bath applied APV to block NMDARs (25 μM applied for the duration of the recording; n = 6). (Cii) Summary of the change in relative GABAergic conductance at the paired and unpaired pathways for all cells in (Ci). Blue triangles denote paired pathways, red triangles denote unpaired pathways; filled triangles represent baseline recordings, open triangles represent recordings following plasticity induction. IPSP–Vm curves were constructed at five time points, alternating between unpaired and paired pathways. Each of the five time points at the paired pathway were compared to the corresponding time point from the unpaired pathway. * Denotes statistical significance from unpaired pathway (p < 0.05). † Denotes statistical significance from baseline (p < 0.05). Paired activity denoted by arrow.
We next attempted to isolate the pairing-induced increase in conductance at the unpaired pathway with the N-methyl-D-aspartic acid receptor (NMDAR) antagonist APV. Previously, we found that pairing-induced depolarization of EGABA is dependent on NMDARs (Ormond and Woodin, 2009), confirming the importance of postsynaptic Ca2+ influx for GABAergic plasticity demonstrated in hippocampal cell cultures and slices from juvenile animals (Woodin et al., 2003; Balena et al., 2010). NMDAR inhibition (with 25 μM APV) prevented EGABA depolarization at both pathways (Figure 5Ci; Table 1; n = 8; paired pathway p = 0.434, unpaired pathway p = 0.525), but did not prevent the increase in conductance at the unpaired pathway (Figure 5Cii; Table 1; n = 8; p < 0.05), allowing us to test the ability of increased GABAergic conductance at the unpaired pathway to differentiate the gain of Schaffer collateral inputs relative to the paired pathway.
Our prediction was that this increase in conductance at unpaired synapses would depress PSP amplitude at depolarized membrane potentials in the absence of EGABA depolarization. We returned to recording mixed PSPs (combined EPSPs and IPSPs; with the recording configuration in Figure 3A) and found that our prediction was correct. When PSPs were recorded at the RMP, there was no significant change seen at either pathway compared to baseline (Figures 6A,B,Ci; Table 1; n = 6; paired pathway p = 0.147, unpaired pathway p = 0.617) or between the two pathways (p = 0.092), but as the membrane was depolarized toward action potential threshold, the effect at the unpaired pathway became apparent. Specifically, when we calculated PSP amplitude at initial PSP reversal potential from our PSP–Vm plots (as in Figure 4Aii), we found PSPs became hyperpolarizing, indicating that the strength of inhibition was considerably increased relative to excitation (Figures 6B,Cii; Table 1; p < 0.001). These recordings confirm that increased synaptic conductance at the GABAergic synapses of the unpaired pathway underlies the synapse specificity of disinhibition-mediated LTP.
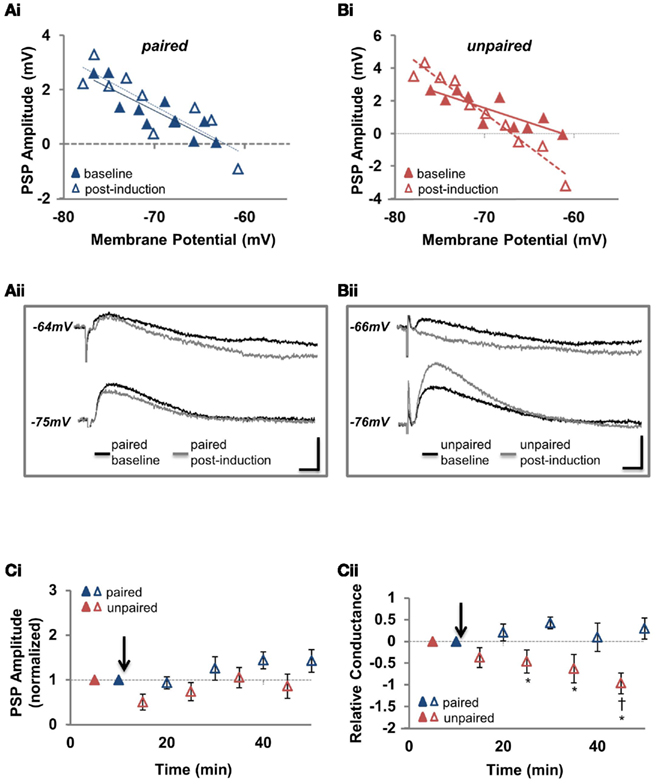
Figure 6. N-methyl-D-aspartic acid receptor blockade reveals that increased GABAergic conductance at the unpaired pathway underlies the pathway specificity of disinhibition-mediated LTP. (Ai) Example recording of the PSP amplitude plotted against Vm for the paired pathway before and after pairing. (Aii) Sample traces before and after pairing for the same cell as in (Ai). (Bi) Example recording of the PSP amplitude plotted against Vm for the unpaired pathway before and after pairing. (Bii) Sample traces before and after pairing for the same cell as in (Bi). (Ci) Summary of the change in mixed PSP amplitude, measured from the resting membrane potential, at the paired and unpaired pathways with NMDARs antagonized (n = 6). (Cii) Summary of the change in mixed PSP amplitude, measured from the initial PSP reversal potential, at the paired and unpaired pathways with NMDARs antagonized [for the same cells as in (Cii)]. Blue triangles denote paired pathways, red triangles denote unpaired pathways; filled triangles represent baseline recordings, open triangles represent recordings following plasticity induction. PSP–Vm curves were constructed at five time points, alternating between unpaired and paired pathways. Each of the five time points at the paired pathway were compared to the corresponding time point from the unpaired pathway. * Denotes statistical significance from unpaired pathway (p < 0.05). † Denotes statistical significance from baseline (p < 0.05). Paired activity denoted by arrow.
Discussion
To our knowledge, this is the first report of a synapse specific enhancement of excitatory transmission by inhibitory plasticity. In our LTP recordings, we took great care to ensure we elicited inhibition physiologically in order to avoid overestimating its impact on disinhibition-mediated LTP. First, we confirmed inhibition was activated disynaptically to maintain the appropriate EPSP/IPSP delay, and to prevent the recruitment of additional non-feed-forward interneurons. Second, the chloride concentration (10 mM) used in our intracellular pipette maintains the driving force at the level measured when recording in the gramicidin perforated-patch configuration, which leaves intracellular chloride unperturbed (Ormond and Woodin, 2009); this is particularly important, as the effect of feed-forward inhibition on excitatory transmission can be enhanced artificially by increasing the driving force for Cl− by lowering its concentration in the intracellular solution to levels below the physiological norm. Last, we used slices from adult rats in all experiments, as ongoing neural development, particularly as it relates to chloride regulation (Rivera et al., 1999), complicates the interpretation of data from younger animals. Highlighting this fact, we have previously shown using gramicidin perforated-patch recording that EGABA hyperpolarizes a further 15 mV between 3 and 7 weeks of age (Ormond and Woodin, 2009), indicating that the developmental changes involved in strengthening inhibition last much longer than the time it takes KCC2 expression to peak (Rivera et al., 1999).
While our data suggests that the pairing-induced depolarization of EGABA is not synapse specific, it is entirely possible that the depolarization of EGABA was confined to the soma if we were primarily stimulating somatic inhibitory synapses. Such a compartmentalized Cl− regulation has been demonstrated in pyramidal cells of the cerebral cortex. In these cells, GABAergic inhibition is strongly depolarizing at the axon-initial segment, but not in the soma or dendrites, due to the absence of KCC2 expression (Szabadics et al., 2006) and the presence of the Na+–K+–2Cl− cotransporter NKCC1 in the axon (Khirug et al., 2008). In contrast, EGABA is hyperpolarizing at the soma, and becomes progressively more negative with distance into the dendrites (Khirug et al., 2008). It will be interesting to examine in further studies whether the same phenomenon exists at inhibitory synapses made onto the distal dendrites of CA1 pyramidals, such as those made by the O-LM interneurons.
It is interesting to note that despite our results showing that the pairing-induced depolarization of EGABA is not confined to the synapses at which it is induced, they also suggest a certain amount of synapse specific EGABA regulation. This is because the value of EGABA was never exactly the same at the paired and unpaired pathways (e.g., Figures 1Bi,Ci; unpublished observations). A recent study has shown that EGABA can in fact be regulated locally at individual basket-cell synapses on the soma of CA1 pyramidals (Földy et al., 2010), so our observation is perhaps not so surprising. Nevertheless, current data suggests both global and local regulation of EGABA at feed-forward inhibitory synapses in CA1. Thus, it should not be assumed that the pairing-induced EGABA depolarization reported in the present study is expressed uniformly throughout the soma.
While many of the mechanisms underlying disinhibition-mediated LTP are likely quite different from those underlying classic LTP, there is one striking similarity. Classic LTP expression is accompanied by heterosynaptic plasticity expressed as a depression of glutamatergic transmission at control pathways which further differentiates the gain of test and control pathways (Lynch et al., 1977; Abraham and Goddard, 1983; Scanziani et al., 1996). This is not unlike the situation presented here, where the heterosynaptic GABAergic plasticity not only maintains inhibitory strength but actually strengthens it as the membrane potential is depolarized toward action potential threshold, enhancing the contrast between pathways.
Our results demonstrate that disinhibition-mediated LTP in the adult rodent hippocampus is synapse specific. The specificity arises from an increase in GABAergic conductance at the unpaired pathway that counteracts the non-specific increase in internal Cl− concentration. We propose that disinhibition-mediated LTP may be a novel form of plasticity underlying learning and memory.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a Natural Sciences and Engineering Research Council (NSERC) of Canada Graduate Scholarship to Jake Ormond and an NSERC Discovery Grant to Melanie A. Woodin.
References
Abraham, W. C., and Goddard, G. V. (1983). Asymmetric relationships between homosynaptic long-term potentiation and heterosynaptic long-term depression. Nature 305, 717–719.
Akerman, C. J., and Cline, H. T. (2006). Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26, 5117–5130.
Andersen, P., Sundberg, S. H., Sveen, O., and Wigstrom, H. (1977). Specific long-lasting potentiation of synaptic transmission in hippocampal slices. Nature 266, 736–737.
Balena, T., Acton, B. A., and Woodin, M. A. (2010). GABAergic synaptic transmission regulates calcium influx during spike-timing dependent plasticity. Front. Synaptic Neurosci. 2:16.
Balena, T., and Woodin, M. A. (2008). Coincident pre- and postsynaptic activity downregulates NKCC1 to hyperpolarize E(Cl) during development. Eur. J. Neurosci. 27, 2402–2412.
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838.
Engert, F., and Bonhoeffer, T. (1997). Synapse specificity of long-term potentiation breaks down at short distances. Nature 388, 279–284.
Fiumelli, H., and Woodin, M. A. (2007). Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr. Opin. Neurobiol. 17, 81–86.
Földy, C., Lee, S. H., Morgan, R. J., and Soltesz, I. (2010) Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat. Neurosci. 13, 1047–1049.
Glickfeld, L. L., and Scanziani, M. (2006). Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat. Neurosci. 9, 807–815.
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639.
Lamsa, K., Kullmann, D. M., and Woodin, M. A. (2010). Inhibitory circuit plasticity. Front. Synaptic Neurosci. 2:8.
Lynch, G. S., Dunwiddie, T., and Gribkoff, V. (1977). Heterosynaptic depression: a postsynaptic correlate of long-term potentiation. Nature 266, 737–739.
Markram, H., Toledo-Rodriguez, M., Wang, Y., Gupta, A., Silberberg, G., and Wu, C. (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807.
Morris, R. G., and Frey, U. (1997). Hippocampal synaptic plasticity: role in spatial learning or the automatic recording of attended experience? Philos. Trans. R. Soc. Lond. B Biol. Sci. 352, 1489–1503.
Ormond, J., and Woodin, M. A. (2009). Disinhibition mediates a form of hippocampal long-term potentiation in area CA1. PLoS ONE 4, e7224.
Pouille, F., and Scanziani, M. (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293, 1159–1163.
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., Pirvola, U., Saarma, M., and Kaila, K. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255.
Saraga, F., Balena, T., Wolansky, T., Dickson, C. T., and Woodin, M. A. (2008). Inhibitory synaptic plasticity regulates pyramidal neuron spiking in the rodent hippocampus. Neuroscience 155, 64–75.
Scanziani, M., Nicoll, R. A., and Malenka, R. C. (1996). Heterosynaptic long-term depression in the hippocampus. J. Physiol. Paris 90, 165–166.
Szabadics, J., Varga, C., Molnar, G., Olah, S., Barzo, P., and Tamas, G. (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235.
Tsien, J. Z., Huerta, P. T., and Tonegawa, S. (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338.
Woodin, M. A., Ganguly, K., and Poo, M. M. (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron 39, 807–820.
Appendix
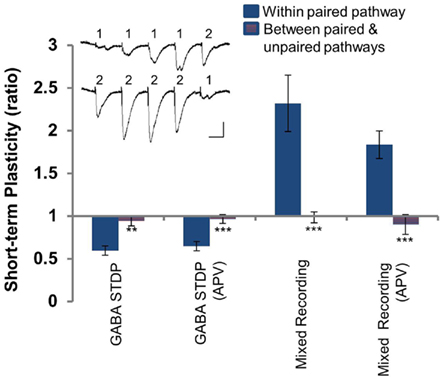
Figure A1. Paired and unpaired pathways are independent. Short-term plasticity within the paired pathway (solid blue) and between the paired and unpaired pathways (blue and red hatched) for experimental conditions [GABA STDP, Figures 1 and 2; GABA STDP (APV), Figure 5C; Mixed LTP, Figures 3 and 4; Mixed LTP (APV), Figure 6]. Inset: sample recording demonstrating the protocol used. Scale bars: 50 pA, 25 ms. *p < 0.05; **p < 0.005; ***p < 0.001.
Keywords: GABA, synaptic plasticity, spike-timing dependent plasticity, KCC2, chloride (Cl−), LTP
Citation: Ormond J and Woodin MA (2011) Disinhibition-mediated LTP in the hippocampus is synapse specific. Front. Cell. Neurosci. 5:17. doi: 10.3389/fncel.2011.00017
Received: 15 June 2011;
Accepted: 27 August 2011;
Published online: 19 September 2011.
Edited by:
Yehezkel Ben-Ari, Institut National de la Santé et de la Recherche Médicale, FranceReviewed by:
Gianmaria Maccaferri, Northwestern University, USARustem Khazipov, Institut National de la Santé et de la Recherche Médicale, France
Shaoyu Ge, SUNY Stony Brook, USA
Copyright: © 2011 Ormond and Woodin. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Melanie A. Woodin, Department of Cell and Systems Biology, University of Toronto, 25 Harbord Street, Toronto, ON, Canada M5S 3G5. e-mail:bS53b29kaW5AdXRvcm9udG8uY2E=