 Nikolay Naumenko1†
Nikolay Naumenko1† Eveliina Pollari1† Antti Kurronen1† Raisa Giniatullina1 Anastasia Shakirzyanova1,2
Eveliina Pollari1† Antti Kurronen1† Raisa Giniatullina1 Anastasia Shakirzyanova1,2 Johanna Magga1
Johanna Magga1 Jari Koistinaho1,3
Jari Koistinaho1,3 Rashid Giniatullin1*
Rashid Giniatullin1*- 1 Department of Neurobiology, A. I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio, Finland
- 2 Kazan Institute of Biochemistry and Biophysics KSC RAS, Kazan, Russia
- 3 Department of Oncology, Kuopio University Hospital, Kuopio, Finland
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of motoneurons which progresses differentially in males and females for unknown reason. Here we measured gender differences in pre- and post-synaptic parameters of the neuromuscular transmission in a mutant G93A-SOD1 mouse model of ALS. Using intracellular microelectrode technique we recorded miniature and evoked end-plate potentials (MEPPs and EPPs) in the diaphragm muscle of G93A-SOD1 mice at early symptomatic stage. While no evident alterations in the amplitude of MEPPs was observed in male or female G93A-SOD1 mice, G93A-SOD1 mice displayed dramatically reduced probability of spontaneous acetylcholine release. In contrast, the EPPs evoked by single nerve stimulation had unchanged amplitude and quantal content. In males, but not females, this was accompanied by reduced readily releasable transmitter pool. Transmitter release in both sexes was sensitive to the inhibitory action of reactive oxygen species (ROS), but the production of ROS was increased in the spinal cords of male but not female G93A-SOD1 mice. Treatment with granulocyte colony stimulating factor (GCSF), which we previously found to be beneficial in males, attenuated the increased ROS production indicating involvement of the antioxidant mechanisms and improved ALS-induced synaptic dysfunctions only in males being ineffective in females. Consistent with our findings at synaptic level, GCSF did not change the survival rate or motor performance of female ALS mice. In summary, neuromuscular transmission in ALS mice is impaired at early symptomatic stage when a dramatic presynaptic decline of spontaneous release occurs. Beneficial effects of GCSF treatment on survival in males may be explained by GCSF-improved presynaptic functions in male G93A-SOD1 mice. Development of efficient treatment strategies for ALS may need to be directed in a gender-specific manner.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease leading to motoneuron death (Boillée et al., 2006; Rothstein, 2009). While 90% of the cases are sporadic, 20% of the remaining familial cases are caused by dominant mutations in the Cu/Zn superoxide dismutase (SOD1) gene (Valdmanis and Rouleau, 2008). Even though sporadic and familial forms of the disease display similar clinical features, a direct causative pathway to neuronal dysfunction and death has not yet been established. The central neuropathology of the disease, motoneuron degeneration, appears to be associated with accumulation of mutant SOD1 (Boillée et al., 2006). Such progressive neurodegeneration is observed in transgenic mice overexpressing mutant forms of human SOD1 (Shibata, 2001). Since the main function of the motoneuron is the transmission of motor commands to the muscle, it is important to characterize the electrophysiological parameters of neurotransmission in ALS. While numerous morphological studies indicate early distal axon degeneration (Fischer et al., 2004; David et al., 2007), only one electrophysiological study reported decreased amplitudes of miniature end-plate potentials (MEPPs) but unchanged average MEPP frequency along with reduced end-plate potentials (EPPs) quantal content in muscle biopsies taken from patients with clinical features of ALS (Maselli et al., 1993).
Epidemiological studies show that both the incidence and prevalence of ALS are greater in men than in women (McCombe and Henderson, 2010). Sex differences have also been reported in genetic mouse models overexpressing mutant SOD1. Thus, in female G93A-SOD1 mice, the onset is delayed compared to male G93A-SOD1 littermates. It has been shown that gender influences disease onset but not the progressive loss of motor units (Hegedus et al., 2009; Alves et al., 2011). However, the mechanisms underlying the role of gender in ALS and whether suggested treatments should take into account sexual dimorphism remain only partially understood.
Oxidative stress has often been implicated as a main reason for motoneuron death in ALS patients (Barber and Shaw, 2010; Cova et al., 2010). Morphological studies performed in G93A-SOD1 mice suggest that synapses are highly sensitive to ischemia/reperfusion injury (David et al., 2007), probably via free radicals. Although antioxidants have beneficial effects in some ALS animal models, no confirmation in human trials has been obtained (Barber and Shaw, 2010). This suggests that antioxidant therapy should probably be supplemented with additional neuroprotective treatments. One promising and clinically safe agent is granulocyte colony stimulating factor (GCSF), which prolonged survival in an animal model of ALS (Pitzer et al., 2008; Pollari et al., 2011). However, the cellular targets and the mechanisms of its beneficial effect remain largely unknown. In the present study, we tested the functional state of the neuromuscular junctions in G93A-SOD1 mice at early symptomatic stage of ALS and in animals treated with GCSF and investigated whether there are gender differences in neuromuscular junction function and in response to GCSF treatment.
Materials and Methods
Animals
Transgenic G93A-SOD1 (B6.Cg-Tg-(SOD1-G93A)1Gur/J) mice carrying a high copy number of human mutant G93A-SOD1 were obtained from Jackson laboratory and maintained on C57BL congenic background. The motor deficits appear at the age of 19 weeks in both the male and female mice reaching the end stage at the age of 24–26 weeks. The onset of the disease was determined with a wire-hang test as an inability to complete the 3 min observation period. The clinical end stage was determined as the inability of a mouse to right itself in a period of 30 s which was defined as a criterion for mouse sacrifice. The mice were used for electrophysiological studies at the early symptomatic stage at 20 weeks of age. For FOX1 assay spinal cords were collected at symptomatic stage at the age of 20–22 weeks. Animal experiments were conducted according to the national regulations of the usage and welfare of laboratory animals and approved by the Animal Experiment Committee in State Provincial Office of Southern Finland.
Treatment
In this study setup G93A-SOD1 mice were treated with pegfilgrastim, GCSF with sustained action (Neulasta, Amgen, Breda, The Netherlands) at the dosage of 300 μg/kg given subcutaneously once per week. Pegfilgrastim was diluted into 0.15 M sodium acetate right before use while sodium acetate injections were used as a vehicle control. The treatment was started at the age of 12 weeks at the presymptomatic stage and continued until the mouse was sacrificed. The littermates were randomly divided into treatment groups.
Electrophysiological Recordings
Electrical recordings were made from diaphragm muscle kept at room (∼21°C) temperature. In total 45 mice of both sexes (from 4 to 11 animals in a group) were used for electrophysiological recordings. Mice were decapitated under carbon dioxide anesthesia (100% CO2 for several seconds until the loss of reflexes) and diaphragms were immediately dissected. Preparations were placed in a small chamber continuously superfused with gassed (95% O2/5% CO2) Krebs solution containing (in mM): 120 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 11 glucose, 1 NaHPO4, 24 NaHCO3, pH was adjusted to 7.4. EPPs and MEPPs were recorded using a low-noise custom-made amplifier and standard microelectrodes technique (8–15 MΩ electrode resistance when filled with 3 M KCl) typically during 2 min (10–20 s in sucrose test). Each diaphragm was dissected into two to four muscle strips and 4–12 synapses were sampled in each strip. For recording evoked potentials the left phrenic nerves (14–16 mm length) were dissected together with the diaphragm. To evaluate the readily releasable pool (RRP) of transmitter in nerve terminals we used hypertonic sucrose solution (Kashani et al., 2001; Moulder and Mennerick, 2005; Virmani et al., 2005; Vanden Berghe and Klingauf, 2007). In the sucrose test recordings were made in the interval of 5–10 min in the sustain phase of stimulated release induced by bath application of 500 mM sucrose (Sara et al., 2002). In experiments with recording of spontaneous MEPPs and sucrose test 1 μM tetrodotoxin (TTX, Ascent scientific, UK) was present during recordings. To block muscle contractions and preserve the physiological level of transmitter release in the experiments with motor nerve stimulation we used transverse cutting of muscle fibers (Giniatullin et al., 1993). EPPs were induced by the suprathreshold stimulation of the phrenic nerve via a suction electrode connected to an isolated pulse stimulator Model 2100 (A-M Systems, USA). EPPs (recorded at the membrane potential ∼39–47 mV) and MEPPs (recorded at the membrane potential ∼69–72 mV) were digitized at 50 kHz and acquired to the PC using the data acquisition board NI PCI6221 (National Instruments, Austin, TX, USA) and visualized with the WinEDR V3.0.4 software (Strathclyde University, UK). Amplitudes of EPPs and MEPPs, interevent intervals of MEPPs were analyzed off-line with the WinEDR V3.0.4 and ClampFit V10.2.0.14 (Molecular Devices, Sunnyvale, CA, USA) software. Subsequent statistical analysis was made using the Origin graphic software (v.8.0, OriginLab Corp, Northampton, MA, USA).
FOX1 Assay
Concentration of hydroperoxides in the spinal cord samples were measured with the FOX1 assay of ferrous oxidation in xylenol orange (Wolff, 1994; Giniatullin et al., 2005; Morgan et al., 2008). In short, this method is based on oxidation of ferrous ions in an acidic medium containing the dye xylenol orange, which binds the resulting ferric ions to produce a complex with absorbance maximal at 560 nm. The FOX reagent was prepared as described by Wolff (1994) with slight modification: 1 part 25 mM heptahydrate ammonium ferrous sulfate, 2 parts H2O, and 2 parts 10 mM xylenol orange. After addition of cold (-20°C) acetone as a fixative the spinal cord samples were homogenized, sonicated, and centrifuged for 8 min at 12000 g. The supernatant for the assay was mixed with 50 μl of the FOX reagent. After completion of the reaction the tissue extracts were centrifuged for 3 min at 10000 g, and the supernatant was assayed spectrophotometrically (absorbance at 560 nm). The peroxide content of samples was determined with reference to a calibration curve obtained with known concentrations of H2O2 and expressed as micromoles of peroxide per milligram of tissue.
Statistical Analysis
Although we collected and averaged data from each synapse, as the basic statistical approach we compared averaged data from each mouse. Thus, all data are presented as the mean ± SEM with indication of number of animals used. Significance was assessed using Student’s t-test. Kaplan–Meier survival statistics with a log rank sum test was used for testing differences in survival. A P value of less than 0.05 was accepted as indicative of a statistically significant difference.
Results
Effect of GCSF on Survival of G93A-SOD1 Mice: The Gender Difference
We have shown, in the parallel study, that pegfilgrastim, GCSF with a sustained action, prolonged the survival of G93A-SOD1 male mice from 173 ± 7 to 185 ± 7 days (Pollari et al., 2011). When in the current project, we treated the female G93A-SOD1 mice with pegfilgrastim in the similar manner, surprisingly we did not detect any effect of GCSF on survival (P = 0.30, Figure 1). In addition, GCSF did not improve the motor performance in female G93A-SOD1 mice as assessed with wire-hang test (data not shown) as we previously detected in male G93A-SOD1 mice (Pollari et al., 2011). The life span was longer (by 15.0 ± 3.3 days, P = 0.001) in female G93A-SOD1 mice than in male G93A-SOD1 mice. The similar results were previously reported by Alves et al. (2011). This finding indicates that even though GCSF treatment significantly extends the lifespan of male ALS mice, it may be almost ineffective in female ALS mice. Therefore, in the current study we aimed to elucidate gender-dependent synaptic mechanisms of ALS development and cellular mechanisms of differential sensitivity to the therapeutic effectiveness of GCSF.
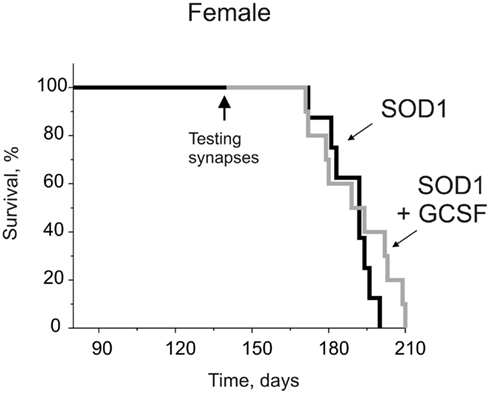
Figure 1. The time course of survival for female G93A-SOD1 mice with or without GCSF treatment (Kaplan–Meier survival graph). The female G93A-SOD1 mice were treated with GCSF starting at the age of 12 weeks. In contrary to the previous results we obtained from male G93A-SOD1 mice (Pollari et al., 2011), GCSF did not prolong the survival of female G93A-SOD1 mice (n = 8–10).
Low Probability of Transmitter Release in G93A-SOD1 Mice
In order to link disease related behavior, motor function changes, and sex-dependent effectiveness of the treatment with mechanisms of neuromuscular transmission, we recorded synaptic potentials in G93A-SOD1 mice at the symptomatic disease stage of 20 weeks when the pathological changes in behavior such as abnormal reflexes and decreased motor performance were evident. Independent from the sex or treatment all the mice reached the disease onset at age of 19 weeks as determined by the wire-hang test (data not shown). In the isolated diaphragm muscle taken from 20 week old G93A-SOD1 male mice only a trend toward the reduced amplitude of MEPP was observed. Thus, in males, MEPPs amplitude was 0.41 ± 0.06 mV (n = 8 mice) in the WT, 0.26 ± 0.02 mV in untreated G93A-SOD1 mice (n = 6 mice, P = 0.054 versus WT,) and 0.30 ± 0.04 mV (n = 5 mice, P = 0.42 versus G93A-SOD1) in the G93A-SOD1 mice treated with GCSF (Figures 2A–D). These data indicated that the functional state of the postsynaptic membrane in muscle fibers was not significantly affected, although P value was approaching the P < 0.05 limit, thus not excluding the possibility that with higher number of animals the difference between G93A-SOD1 and WT mice in the amplitude of postsynaptic responses could become significant.
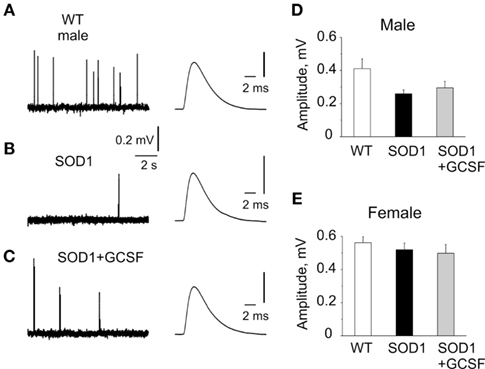
Figure 2. Spontaneous transmitter release at the neuromuscular junctions of WT, G93A-SOD1, and GCSF-treated G93A-SOD1 mice. (A–C) Examples of miniature end-plate potentials (MEPPs) in WT, G93A-SOD1, and GCSF-treated G93A-SOD1 mice, respectively (right, averaged MEPPs are shown in expanded scale). Note largely reduced frequency of MEPPs in G93A-SOD1 mice and improvement of this parameter in GCSF-treated animals. (D,E). Histograms showing MEPPs amplitude in WT, G93A-SOD1, and GCSF-treated G93A-SOD1 mice in males (D) and females (E) 80–140 MEPPs were averaged in every group.
In sharp contrast, the rate of spontaneous quantal acetylcholine (ACh) release from motor nerve endings (evaluated as the interevent intervals between single MEPPs, the longer interval indicated reduced frequency) was dramatically reduced in male G93A-SOD1 mice. Moreover, there was a huge variability in the rate of transmitter release in G93A-SOD1 mice most visible if we present data for each synapse (Figure 3A). Such large dispersion of MEPP frequency in G93A-SOD1 mice suggested that synapses were in different stages of disease progression. Comparing spontaneous quantal events, we found in symptomatic male G93A-SOD1 mice the interevent intervals as high as 9.45 ± 1.60 s (n = 6 mice) versus 1.59 ± 0.07 s in WT mice (n = 8 mice, P = 0.0001). GCSF reduced this parameter to 3.48 ± 0.21 s (n = 5 mice, P = 0.009, Figure 3A).
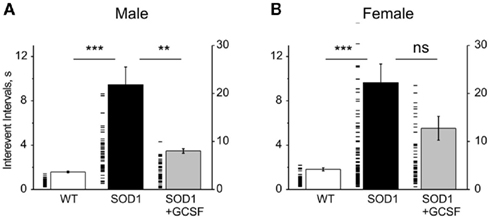
Figure 3. Interevent intervals of spontaneous MEPPs at the neuromuscular junctions of WT, G93A-SOD1, and GCSF-treated G93A-SOD1 mice. (A,B). Histograms showing mean interevent intervals of MEPPs (left axis) in the WT, G93A-SOD1, and GCSF-treated G93A-SOD1 male (A) and female (B) mice. Each dash line indicates individual synapse (right axis). Note huge variability of release probabilities between individual synapses. ** = P < 0.01, *** = P < 0.001.
In the WT females the amplitude of MEPPs was larger than in males (0.56 ± 0.04 mV, n = 10 mice, P < 0.0001) and was unchanged in G93A-SOD1 female mice without (0.52 ± 0.04 mV, n = 11 mice, P = 0.43) or with GCSF treatment (0.50 ± 0.05 mV, n = 5 mice, P = 0.77, Figure 2E).
In females, like in males, spontaneous release was strongly compromised in G93A-SOD1 mice. We found that the mean interevent interval was strongly increased from 1.82 ± 0.14 (n = 10 mice) to 9.63 ± 1.70 s (n = 11 mice, P = 0.0003 versus WT) in G93A-SOD1 females. However, we did not find significant improvement with GCSF treatment (5.53 ± 1.08 s, n = 5 mice, P = 0.14 versus G93A-SOD1 mice, Figure 3B).
To sum up, G93A-SOD1 mice of both sexes had significantly reduced probability of spontaneous transmitter release and this parameter was significantly restored in males but not in females by GCSF treatment.
Readily Releasable Pool of ACh and Ca2+-Dependent Evoked Release
In order to find reasons for reduced probability of spontaneous ACh release in ALS mice we measured the level of RRP of synaptic vesicles. The size of RRP is usually estimated by hypertonic treatments which trigger the intensive exocytosis from this transmitter pool (Kashani et al., 2001; Moulder and Mennerick, 2005; Virmani et al., 2005; Vanden Berghe and Klingauf, 2007). To this end, we applied hypertonic 500 mM sucrose solution which induced a large increase in the rate of quantal release (by shortening interevent intervals). Thus, in the WT males, interevent intervals after sucrose application were shortened to 0.076 ± 0.011 s (n = 6 mice) while in the G93A-SOD1 male mice they were shortened to 0.423 ± 0.170 s (n = 6 mice, P = 0.01 versus WT) indicating diminished RRP. The treatment with GCSF in males significantly restored the amount of synaptic vesicles in the RRP (0.091 ± 0.015 s, n = 5 mice, P = 0.02 versus G93A-SOD1, Figures 4A–D).
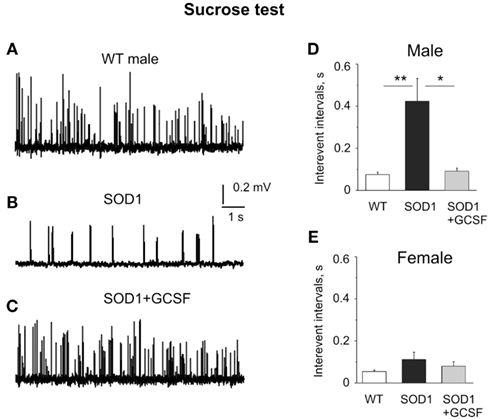
Figure 4. Evaluation of the readily releasable transmitter pool in the sucrose test. (A–C) Examples of sucrose-evoked quantal events in WT, G93A-SOD1, and GCSF-treated G93A-SOD1 male mice, respectively. (D,E) Histograms of mean interevent intervals in WT, G93A-SOD1, and GCSF-treated G93A-SOD1 male (D) and female (E) mice. Note the reduced RRP in G93A-SOD1 male mice and improvement of this parameter in GCSF-treated animals. *= P < 0.05, **= P < 0.01.
In females, interevent intervals in the WT mice after sucrose application were shortened to 0.055 ± 0.006 s (n = 5 mice) while in the G93A-SOD1 female mice they were shortened to 0.112 ± 0.034 s (n = 6 mice, P = 0.17 versus WT). GCSF did not change significantly sucrose-induced transmitter release (0.080 ± 0.021 s, n = 5 mice, P = 0.47 versus G93A-SOD1 mice, Figure 4E). Thus, unlike in males, RRP was not significantly compromised in female G93A-SOD1 mice.
To test potential changes in Ca2+-dependent quantal release in ALS we applied electrical stimulation to the motor nerve. Interestingly, unlike the dramatic drop in spontaneous release, Ca2+-dependent evoked release during single nerve stimulation was not changed in G93A-SOD1 mice of either sex. Thus, the amplitudes of EPPs were 13.2 ± 3.1 mV (n = 4 mice) in the WT, 13.4 ± 3.4 mV (n = 5 mice, P = 0.97 versus WT) in G93A-SOD1 male mice (Figures 5A,B). The quantal content of action potential-evoked transmitter release was 37.3 ± 4.7 quanta in the WT male mice (n = 4) and 34.8 ± 4.3 quanta in transgenic mice (n = 4, P = 0.71 versus WT). Likewise, in females, EPPs amplitudes were 13.2 ± 1.2 mV (n = 4 mice) in the WT and 13.6 ± 1.7 mV (n = 4 mice, P = 0.85 versus WT) in G93A-SOD1 mice, respectively (Figures 5C,D). The quantal content of transmitter release in females (26.9 ± 1.0 quanta) did not differ significantly from males (n = 4, P = 0.07) and was not changed in G93A-SOD1 females (n = 4, P = 0.53).
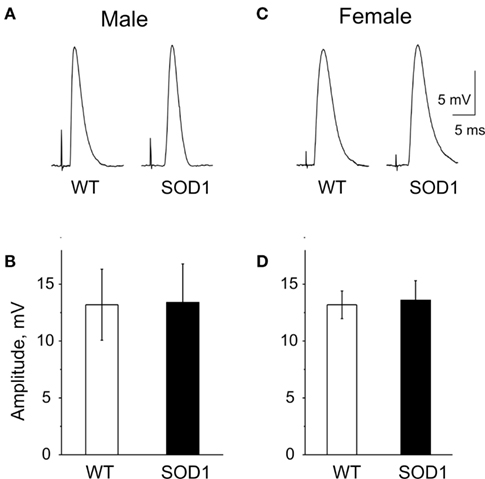
Figure 5. Evoked EPPs in the WT and G93A-SOD1 mice. (A,D) Examples of end-plate potentials (EPPs) in WT, G93A-SOD1 male, and female mice. (B,C). Histograms showing amplitudes of evoked EPPs in WT, G93A-SOD1 male, and female mice, respectively.
Taken together these data indicated that EPPs evoked by a single action potential were unchanged. However, the fraction of vesicles included in the RRP was reduced in G93A-SOD1 male mice and remarkably preserved by the GCSF treatment.
Presynaptic Inhibition by ROS
Since oxidative stress has been implicated in ALS pathology we next evaluated the sensitivity of motor nerve terminals to reactive oxygen species (ROS). To this end we applied the diffusible ROS, H2O2, known as a potent inhibitor of transmitter release at the neuromuscular junction (Giniatullin and Giniatullin, 2003; Giniatullin et al., 2006) and measured MEPPs in the period of 15–20 min after H2O2 application. In line with previous observations, 300 μM H2O2 significantly reduced spontaneous quantal release of ACh in the WT males. Thus, the intervals between spontaneous events were lengthened from 1.59 ± 0.07 to 3.51 ± 0.80 s (n = 7 mice, P = 0.02, Figures 6A,B). In G93A-SOD1 male mice, despite the low basic level of spontaneous release, ROS application still reduced secretion since interevent intervals were lengthened from 9.44 ± 1.60 to 20.17 ± 5.23 s (n = 4 mice, P = 0.048). The depressant effect of H2O2 was also presented after GCSF treatment (not shown).
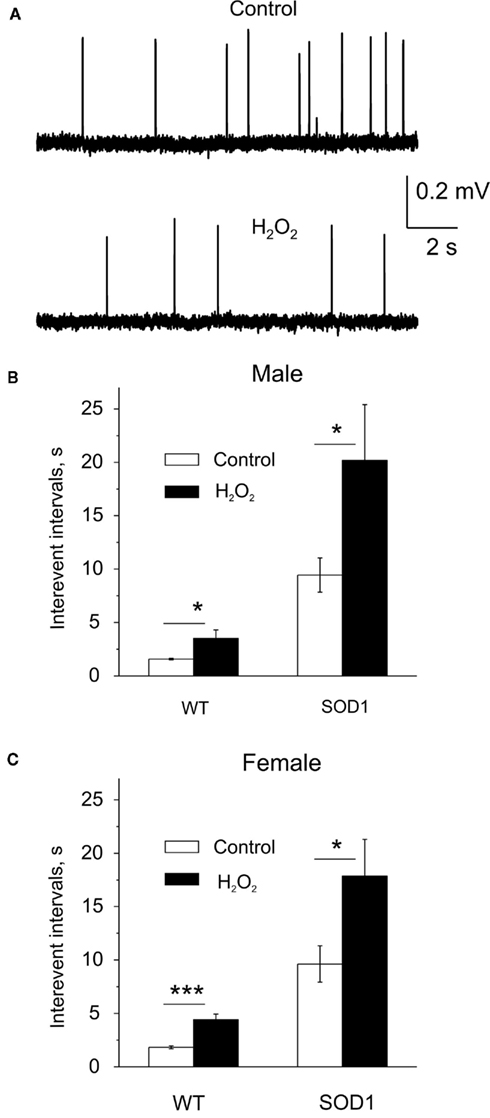
Figure 6. Reactive oxygen species-induced depression of quantal release by exogenous H2O2. (A) Examples of MEPPs in the WT mice before (control) and after application of 300 μM H2O2. (B,C) Histograms showing the mean interevent intervals of MEPPs before (control) and after application of 300 μM H2O2 to the neuromuscular junction in male (B) and female mice (C). Note greater depression in G93A-SOD1 males. *= P < 0.05, ***= P < 0.0001.
In the WT females, interevent intervals after ROS application were lengthened from 1.82 ± 0.14 to 4.41 ± 0.53 s (n = 8 mice, P < 0.0001, Figure 6C) indicating sex-independent inhibitory action of H2O2. Like in males, in G93A-SOD1 females H2O2 also increased interevent intervals (9.63 ± 1.70 s before and 17.85 ± 3.46 s after H2O2, n = 11 mice, P = 0.045).
Thus, after the onset of the disease G93A-SOD1 mice were still prone to oxidative stress-induced inhibition of quantal ACh release.
GCSF Diminishes Oxidative Stress in G93A-SOD1 Mice
As the relative proportion of motor nerve terminals to the total mass of the muscle is negligible, the level of oxidative stress in motoneurons of G93A-SOD1 and WT mice was evaluated from the spinal cord where the cell bodies of motoneurons are located. The level of endogenous peroxides in the spinal cord, as analyzed with the FOX1 assay, was elevated in G93A-SOD1 male mice (0.216 ± 0.020 μmol/mg in WT mice versus 0.301 ± 0.035 μmol/mg in G93A-SOD1 mice, n = 15 mice, P = 0.046, Figure 7A). However, in G93A-SOD1 females the level of peroxides (0.243 ± 0.020 μmol/mg) was undistinguishable from the WT (0.231 ± 0.026 μmol/mg, n = 9 mice, P = 0.73, Figure 7B). Interestingly, both G93A-SOD1 males and females responded to GCSF treatment by significantly reduced level of endogenous peroxides (0.181 ± 0.024 μmol/mg in males, n = 9, P < 0.024, and 0.173 ± 0.015 μmol/mg in females, n = 9, P < 0.013, Figures 7A,B) indicating that the therapeutic action of this agent includes an antioxidant mechanism, but in G93A-SOD1 females endogenous compensatory mechanisms are still able to keep the possible mutant G93A-SOD1-induced oxidative stress within normal range at the early symptomatic stage of the disease.
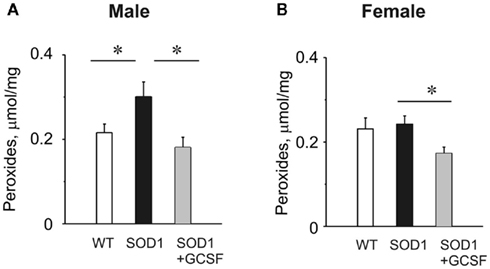
Figure 7. The level of endogenous peroxides in WT, G93A-SOD1 mice, and the antioxidant effect of GCSF. (A,B) Histograms showing the level of peroxides evaluated by FOX1 assay in spinal cord preparations obtained from male (A) or female (B) WT, G93A-SOD1, and GCSF-treated G93A-SOD1 mice. *= P < 0.05.
Discussion
Here, we have electrophysiologically characterized in G93A-SOD1 ALS mice a presynaptic decline in the neuromuscular junction, which was more severe in male than female mice. Importantly, we show that GCSF treatment reduced the oxidative stress in male mice to normal range and improved their compromised neurotransmission without having evident effect on the neurotransmission in female G93A-SOD1 mice. Our results are in agreement with the previously reported gender differences in the incidence and prevalence of ALS (McCombe and Henderson, 2010; Alves et al., 2011) and show that the function of neuromuscular junction can be improved in male but not female G93A-SOD1 mice with GCSF treatment. Importantly, the improved presynaptic function with GCSF treatment correlated with the gender-specific effects of GCSF on survival rate and motor performance, suggesting that the function of neuromuscular junction is a site of key pathology in ALS, reflecting the upcoming survival and the efficacy of neuroprotective treatment strategies in this disease.
Primary Presynaptic Dysfunction in ALS
Mice expressing human G93A mutant SOD1 are the most common animal model of ALS. Even though morphological data and the previous indirect functional approaches suggest that degeneration in ALS is initiated at the peripheral site (Frey et al., 2000; Fischer et al., 2004; David et al., 2007; Carrasco et al., 2010), the direct functional testing of the neuromuscular transmission in terms of synaptic EPPs in G93A-SOD1 ALS mice or other ALS animal models is not available. This is an important point since morphological changes could occur at later uncompensated stages of the disease, while subtle functional abnormalities could occur much earlier. Indeed, our study reveals that many motor terminals in the diaphragm muscle in the early disease stage were functional but exhibited a number of dysfunctional changes. Whereas some studies proposed that muscle fibers initiate the very early changes leading to ALS progression (Wong and Martin, 2010), our results indicate that presynaptic machinery is mostly affected. Thus, at the early symptomatic disease stage of 20 weeks in G93A-SOD1 mice, the amplitude of MEPPs was unchanged. These results are consistent with recent observations that expression of the mutant SOD1 in the muscle is not required for motor terminal degeneration (Nguyen et al., 2009; Carrasco et al., 2010). Unlike in the postsynaptic membrane, we observed a dramatically reduced probability of spontaneous release of ACh at the presynaptic level in both sexes. Interestingly, in biopsies taken from the anconeus muscle of ALS patients with uncharacterized etiology, the average frequency of MEPPs was not changed (Maselli et al., 1993). The discrepancy between our observation in G93A-SOD1 mice and the clinical findings by Maselli et al. (1993) may well be due to the difference in the disease progression between the arm (anconeus) and diaphragm muscles, etiology (a rare G93A-SOD1 mutation compared to common sporadic cases), and the large interindividual variability of the clinical study.
Alves et al. (2011) reported reduced values of amplitude and sciatic nerve conduction velocity and also an increased latency in ALS mice. Likewise, an increased latency of evoked EPPs was observed in our experiments (not shown) indicating, probably, a general trend to reduced conduction in various nerves. However, we did not see any significant differences in EPPs parameters of WT and G93A-SOD1 mice of either gender. Thus, it was shown in our study that spontaneous release was compromised much more strongly than evoked secretion underlying Ca2+-dependent EPPs. However, in the frame of oxidative mechanism of neurodegeneration in ALS this is a very consistent observation since spontaneous release was preferentially inhibited by the oxidative challenge (Giniatullin et al., 2006; Tsentsevitsky et al., 2011). Moreover, unlike earlier hypothesis, current evidence suggests the possibility of differential regulation of spontaneous and evoked neurotransmitter release (Ramirez and Kavalali, 2011).
In G93A-SOD1 mice of both sexes, the amplitude of EPPs induced by a single electrical stimulation of motor nerves and the quantal content of the evoked transmitter released was almost as large as in the WT mice. In contrast, the sucrose test, which is widely used for RRP measurements (Kashani et al., 2001; Moulder and Mennerick, 2005; Virmani et al., 2005; Vanden Berghe and Klingauf, 2007) revealed a significant decrease in the immediately available transmitter pool in nerve endings of males, which was likely one of the reasons for the dramatic drop in the spontaneous mediator release. Reduced RRP found in males could be a reason for the enhanced fatigability of ALS muscles at high rate firing (Schomburg et al., 2011).
Efficient Treatment with GCSF in Males
One important result of this study with clear clinical implication is that GCSF treatment started presymptomatically at the age of 12 weeks reduced the development of synaptic dysfunctions in male G93A-SOD1 mice. Recently, this agent showed beneficial effects in animal models of ALS (Pitzer et al., 2008; Pollari et al., 2011) and in a pilot human study (Zhang et al., 2009). Here we show beneficial effect of GCSF on the functional state of motor terminals which appear to be the most vulnerable structures in ALS (David et al., 2007). The protective effect of GCSF was illustrated here by increased level of spontaneous ACh release and larger amount of synaptic vesicles in the RRP in treated male mice. Thus, GCSF treatment prevented in males the early dysfunction of motor nerve terminals in the skeletal muscle. Remarkably, GCSF also showed strong antioxidant effect which should contribute to the complex neuroprotection in ALS. The protective action of GCSF to save nerve terminals might involve surrounding glial cells. In our previous study (Pollari et al., 2011) we demonstrated that long term GCSF treatment decreases inflammation, microgliosis, and astrogliosis. Astrocytes and microglia expressing mutant SOD1 have been shown to release soluble factors that are toxic to motoneurons (Nagai et al., 2007; Henkel et al., 2009) and thus GCSF could exert its protection to motor neurons by reducing astrocyte activation. Unlike in males, in females there was no positive effect of GCSF on survival tests or ex vivo synaptic recordings.
Why are Female G93A-SOD1 Mice Insensitive to GCSF?
Although ALS develops also in females, the progression of this disease is faster in males. Such puzzling sexual dimorphism is presented also in G93A-SOD1 transgenic rats (Suzuki et al., 2007) and was confirmed in our survival tests in G93A-SOD1 mice. Thus, the treatment with GCSF was successful in males (Pollari et al., 2011) but almost not effective in females (current study, Figure 1). Interestingly, in our ex vivo tests at the age of 20 weeks the resting spontaneous quantal release of ACh was similarly dropped in both sexes. Nevertheless, also in ex vivo tests, GCSF was little effective in females. There could be several reasons why females were resistant to the therapy with GCSF. First, GCSF in a motoneuronal cell line had better protective effect under conditions of oxidative stress (Tanaka et al., 2006) suggesting that the prior oxidative stress is required for GCSF-induced neuroprotection. Notably, expression of GCSF and its receptor was induced by ischemia (Schneider et al., 2005) which is usually associated with the oxidative stress. Thus, the gender discrepancy in the effectiveness of GCSF could be related to higher level of oxidative stress in males. Indeed, the increased levels of ROS were observed in male G93A-SOD1 spinal cords but not in females suggesting that in the latter, the redox balance between pro- and anti-oxidant mechanisms was already shifted toward neuroprotection.
Second, in our recent study we showed that GCSF stimulated several pathways for the neuroprotection including mobilization of monocytes (Pollari et al., 2011). Given that GCSF has multiple targets, several mechanisms of the neuroprotection could coexist and determine the higher effectiveness of GCSF in males. Of course, data obtained in a mouse model cannot be directly extrapolated to ALS patients but they elucidate cellular and molecular mechanisms of sexual dimorphism and identify a need for new gender-specific treatments. Our study implies that the measurement of a simple parameter such as the frequency of MEPPs, could serve as a highly sensitive biomarker to test novel therapeutic molecules in ALS.
Taken together our data provide for the first time detailed electrophysiological characterization of compromised synaptic function in an animal model of ALS and demonstrate gender differences in the decline of the presynaptic machinery at the early symptomatic stage of the disease. The therapeutic efficacy of GCSF only in male ALS mice emphasizes the complex action of this compound involving essential antioxidant component.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the Academy of Finland (122910 and 135112 to Jari Koistinaho, 135179 to Rashid Giniatullin); Sigrid Juselius Foundation (1064/02.05.01/2011 to Jari Koistinaho); CIMO (to Nikolay Naumenko and Anastasia Shakirzyanova); and the Finnish Funding Agency for Technology and Innovation (2816/31/09. to Jari Koistinaho).
Abbreviations
Ach, acetylcholine; ALS, amyotrophic lateral sclerosis; EPPs, evoked end-plate potentials; GCSF, granulocyte colony stimulating factor; MEPPs, miniature end-plate potentials; ROS, reactive oxygen species; RRP, readily releasable pool; SOD1, Cu/Zn superoxide dismutase.
References
Alves, C. J., de Santana, L. P., Santos, A. J., de Oliveira, G. P., Duobles, T., Scorisa, J. M., Martins, R. S., Maximino, J. R., and Chadi, G. (2011). Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 1394, 90–104.
Barber, S. C., and Shaw, P. J. (2010). Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 48, 629–641.
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their non neuronal neighbors. Neuron 52, 39–59.
Carrasco, D. I., Bichler, E. K., Seburn, K. L., and Pinter, M. J. (2010). Nerve terminal degeneration is independent of muscle fiber genotype in SOD1 mice. PLoS ONE 5, e9802. doi:10.1371/journal.pone.0009802
Cova, E., Bongioanni, P., Cereda, C., Metelli, M. R., Salvaneschi, L., Bernuzzi, S., Guareschi, S., Rossi, B., and Ceroni, M. (2010). Time course of oxidant markers and antioxidant defenses in subgroups of amyotrophic lateral sclerosis patients. Neurochem. Int. 56, 687–693.
David, G., Nguyen, K., and Barrett, E. F. (2007). Early vulnerability to ischemia/reperfusion injury in motor terminals innervating fast muscles of SOD1-G93A mice. Exp. Neurol. 204, 411–420.
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., Khan, J., Polak, M. A., and Glass, J. D. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240.
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542.
Giniatullin, A. R., Darios, F., Shakirzyanova, A., Davletov, B., and Giniatullin, R. (2006). SNAP25 is a pre-synaptic target for the depressant action of reactive oxygen species on transmitter release. J. Neurochem. 98, 1789–1797.
Giniatullin, A. R., and Giniatullin, R. A. (2003). Dual action of hydrogen peroxide on synaptic transmission at the frog neuromuscular junction. J. Physiol. 552, 283–293.
Giniatullin, A. R., Grishin, S. N., Sharifullina, E. R., Petrov, A. M., Zefirov, A. L., and Giniatullin, R. A. (2005). Reactive oxygen species contribute to the presynaptic action of extracellular ATP at the frog neuromuscular junction. J. Physiol. 565, 229–242.
Giniatullin, R. A., Khazipov, R. N., and Vyskocil, F. (1993). A correlation between quantal content and decay time of endplate currents in frog muscles with intact cholinesterase. J. Physiol. 466, 95–103.
Hegedus, J., Putman, C. T., and Gordon, T. (2009). Progressive motor unit loss in the G93A mouse model of amyotrophic lateral sclerosis is unaffected by gender. Muscle Nerve 39, 318–327.
Henkel, J. S., Beers, D. R., Zhao, W., and Appel, S. H. (2009). Microglia in ALS: the good, the bad, and the resting. J. Neuroimmune Pharmacol. 4, 389–398.
Kashani, A. H., Chen, B. M., and Grinnell, A. D. (2001). Hypertonic enhancement of transmitter release from frog motor nerve terminals: Ca2+ independence and role of integrins. J. Physiol. 530, 243–252.
Maselli, R. A., Wollman, R. L., Leung, C., Distad, B., Palombi, S., Richman, D. P., Salazar-Grueso, E. F., and Roos, R. P. (1993). Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve 16, 1193–1203.
McCombe, P. A., and Henderson, R. D. (2010). Effects of gender in amyotrophic lateral sclerosis. Gend. Med. 7, 557–570.
Morgan, P. E., Pattison, D. I., Hawkins, C. L., and Davies, M. J. (2008). Separation, detection, and quantification of hydroperoxides formed at side-chain and backbone sites on amino acids, peptides, and proteins. Free Radic. Biol. Med. 45, 1279–1289.
Moulder, K. L., and Mennerick, S. (2005). Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J. Neurosci. 25, 3842–3850.
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., and Przedborski, S. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622.
Nguyen, K. T., García-Chacón, L. E., Barrett, J. N., Barrett, E. F., and David, G. (2009). The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc. Natl. Acad. Sci. U.S.A. 106, 2007–2011.
Pitzer, C., Krüger, C., Plaas, C., Kirsch, F., Dittgen, T., Müller, R., Laage, R., Kastner, S., Suess, S., Spoelgen, R., Henriques, A., Ehrenreich, H., Schäbitz, W. R., Bach, A., and Schneider, A. (2008). Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 131, 3335–3347.
Pollari, E., Savchenko, E., Jaronen, M., Kanninen, K., Malm, T., Wojciechowski, S., Ahtoniemi, T., Goldsteins, G., Giniatullina, R., Giniatullin, R., Koistinaho, J., and Magga, J. (2011). Granulocyte colony stimulating factor attenuates inflammation in a mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 8, 74.
Ramirez, D. M., and Kavalali, E. T. (2011). Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr. Opin. Neurobiol. 21, 275–282.
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl. 1), S3–S9.
Sara, Y., Mozhaeva, M. G., Liu, X., and Kavalali, E. T. (2002). Fast vesicle recycling supports neurotransmission during sustained stimulation at hippocampal synapses. J. Neurosci. 22, 1608–1617.
Schneider, A., Krüger, C., Steigleder, T., Weber, D., Pitzer, C., Laage, R., Aronowski, J., Maurer, M. H., Gassler, N., Mier, W., Hasselblatt, M., Kollmar, R., Schwab, S., Sommer, C., Bach, A., Kuhn, H. G., and Schäbitz, W. R. (2005). The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Invest. 115, 2083–2098.
Schomburg, E. D., Steffens, H., Zschüntzsch, J., Dibaj, P., and Keller, B. U. (2011). Fatigability of spinal reflex transmission in a mouse model [SOD1(G93A)] of amyotrophic lateral sclerosis. Muscle Nerve 43, 230–236.
Shibata, N. (2001). Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology 21, 82–92.
Suzuki, M., Tork, C., Shelley, B., McHugh, J., Wallace, K., Klein, S. M., Lindstrom, M. J., and Svendsen, C. N. (2007). Sexual dimorphism in disease onset and progression of a rat model of ALS. Amyotroph. Lateral Scler. 8, 20–25.
Tanaka, M., Kikuchi, H., Ishizu, T., Minohara, M., Osoegawa, M., Motomura, K., Tateishi, T., Ohyagi, Y., and Kira, J. (2006). Intrathecal upregulation of granulocyte colony stimulating factor and its neuroprotective actions on motor neurons in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 65, 816–825.
Tsentsevitsky, A., Nikolsky, E., Giniatullin, R., and Bukharaeva, E. (2011). Opposite modulation of time course of quantal release in two parts of the same synapse by reactive oxygen species. Neuroscience 189, 93–99.
Valdmanis, P. N., and Rouleau, G. A. (2008). Genetics of familial amyotrophic lateral sclerosis. Neurology 70, 144–152.
Vanden Berghe, P., and Klingauf, J. (2007). Spatial organization and dynamic properties of neurotransmitter release sites in the enteric nervous system. Neuroscience 145, 88–99.
Virmani, T., Ertunc, M., Sara, Y., Mozhayeva, M., and Kavalali, E. T. (2005). Phorbol esters target the activity-dependent recycling and spare spontaneous vesicle recycling. J. Neurosci. 25, 10922–10929.
Wolff, S. P. (1994). Ferrous oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Meth. Enzymol. 233, 182–189.
Wong, M., and Martin, L. J. (2010). Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 19, 2284–2302.
Keywords: synaptic transmission, transmitter release, amyotrophic lateral sclerosis, neuromuscular junction, GCSF
Citation: Naumenko N, Pollari E, Kurronen A, Giniatullina R, Shakirzyanova A, Magga J, Koistinaho J and Giniatullin R (2011) Gender-specific mechanism of synaptic impairment and its prevention by GCSF in a mouse model of ALS. Front. Cell. Neurosci. 5:26. doi: 10.3389/fncel.2011.00026
Received: 21 October 2011;
Accepted: 25 November 2011;
Published online: 12 December 2011.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyCopyright: © 2011 Naumenko, Pollari, Kurronen, Giniatullina, Shakirzyanova, Magga, Koistinaho and Giniatullin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Rashid Giniatullin, Department of Neurobiology, A. I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, P.O. Box 1627/Neulaniementie 2, Kuopio 70211, Finland. e-mail:cmFzaGlkLmdpbmlhdHVsbGluQHVlZi5maQ==
†Nikolay Naumenko and Eveliina Pollari and Antti Kurronen have contributed equally to this work.