 Inês Palmela1
Inês Palmela1 Hiroyuki Sasaki2,3 Filipa L. Cardoso1 Miguel Moutinho1 Kwang S. Kim4
Hiroyuki Sasaki2,3 Filipa L. Cardoso1 Miguel Moutinho1 Kwang S. Kim4 Dora Brites1,5
Dora Brites1,5 Maria A. Brito1,5*
Maria A. Brito1,5*- 1Faculty of Pharmacy, Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL), University of Lisbon, Lisbon, Portugal
- 2Division of Fine Morphology, Core Research Facilities, The Jikei University School of Medicine, Tokyo, Japan
- 3The Center for Advanced Medical Engineering and Informatics, Osaka University, Osaka, Japan
- 4Division of Infectious Diseases, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 5Faculty of Pharmacy, Department of Biochemistry and Human Biology, University of Lisbon, Lisbon, Portugal
In neonatal jaundice, high levels of unconjugated bilirubin (UCB) may induce neurological dysfunction (BIND). Recently, it was observed that UCB induces alterations on brain microvasculature, which may facilitate its entrance into the brain, but little is known about the steps involved. To evaluate if UCB damages the integrity of human brain microvascular endothelial cells (HBMECs), we used 50 or 100 μM UCB plus human serum albumin, to mimic the neuropathological conditions where levels of UCB free species correspond to moderate and severe neonatal jaundice, respectively. Our results point to a biphasic response of HBMEC to UCB depending on time of exposure. The early response includes increased number of caveolae and caveolin-1 expression, as well as upregulation of vascular endothelial growth factor (VEGF) and its receptor 2 (VEGFR-2) with no alterations of the paracellular permeability. In contrast, effects by sustained hyperbilirubinemia are the reduction in zonula occludens (ZO)-1 and β-catenin levels and thus of tight junctions (TJ) strands and cell-to-cell contacts. In addition, reduction of the transendothelial electrical resistance (TEER) and increased paracellular permeability are observed, revealing loss of the barrier properties. The 72 h of HBMEC exposure to UCB triggers a cell response to the stressful stimulus evidenced by increased autophagy. In this later condition, the UCB intracellular content and the detachment of both viable and non-viable cells are increased. These findings contribute to understand why the duration of hyperbilirubinemia is considered one of the risk factors of BIND. Indeed, facilitated brain entrance of the free UCB species will favor its parenchymal accumulation and neurological dysfunction.
Introduction
The blood-brain barrier (BBB) is a dynamic and complex interface between blood and the central nervous system (CNS) that strictly controls the exchanges between the blood and brain compartments, therefore playing a key role in brain homeostasis. The integrity and BBB barrier characteristics are mainly achieved through the presence of tight (TJ) and adherens junctions (AJ), which are able to restrict and regulate the passage of molecules through the paracellular pathway and are also involved in complex signaling mechanisms (Abbott et al., 2010; Cardoso et al., 2010). They are organized by networks of transmembrane proteins connected to the cytoskeleton by cytosolic proteins, such as the TJ proteins, occludin and the zonula occludens (ZO)-1, and the AJ proteins, vascular endothelial (VE)-cadherin, and β-catenin (Cardoso et al., 2010).
Neuroinflammation, acute hypoxia/ischemia, and several neurodegenerative diseases have been pointed to perturb BBB integrity (Wispelwey et al., 1988; Abbruscato and Davis, 1999; Davies, 2002). LPS revealed to lower the transendothelial electrical resistance (TEER), to increase the permeability to sodium fluorescein (NA-F) and to promote changes in the linear shape along the junctions between cells to a more zigzap shape morphology, altering the selective barrier function of TJ at the BBB (Sumi et al., 2010). Alterations in the subcellular localization of TJ proteins, including ZO-1 and occludin, leading to hyperpermeability of the BBB, were also observed by hydrogen peroxide (Lee et al., 2004; Fischer et al., 2005), as well as by reactive oxygen species (Schreibelt et al., 2007) and oxidative stress-induced by several compounds (Haorah et al., 2007). Occludin levels were also decreased, suggesting that vascular endothelial growth factor (VEGF) increases permeability by reducing occludin expression and disrupting ZO-1 and occludin organization, which leads to TJs disassembly. Accordingly, occludin and ZO-1 were suggested as downstream effectors of the VEGF signaling pathway (Petty and Lo, 2002). In addition, β-catenin evidenced to disappear subsequently to leukocyte extravasation (Petty and Lo, 2002). More recently, it was demonstrated that early life stress, both prenatal and postnatal, increase transvascular transport mediated by caveolae in brain endothelial cells (Gomez-Gonzalez and Escobar, 2009). Interestingly, increased expression of caveolin-1, an integral caveolar membrane protein, evidenced to be associated with early BBB breakdown and edema in brain injury (Nusrat et al., 2000; Song et al., 2007; Nag et al., 2009a). Caveolin-1 was also indicated to control signaling pathways that lead to the disruption of TJ proteins (Zhong et al., 2008). In particular, the interaction of caveolin-1 with VEGF and its receptor 2 (VEGFR-2) indicates that caveolin-1 plays multiple roles in the VEGF-induced signaling cascade and, consequently, on the endothelial permeability (Chen et al., 2002; Labrecque et al., 2003). Thus, caveolae and caveolin-1 have an important role in early BBB breakdown and are considered potential therapeutic targets.
Elevated levels of unconjugated bilirubin (UCB) during the neonatal period may be responsible for the occurrence of UCB-induced neurological dysfunction (BIND) associated to minor brain deficits, or bilirubin encephalopathy commonly known as kernicterus (Cohen et al., 2010; Shapiro, 2010). Therefore, the passage of UCB across the BBB into the CNS appears to be a crucial first step in the development of kernicterus (Sequeira et al., 2007). Autopsy findings in kernicterus cases include brain edema (Perlman et al., 1997), increased VEGF expression, and infiltration of erytrocytes and albumin into the brain parenchyma (Brito et al., 2011), features resulting from an enhanced permeability of vascular walls. Despite the indication that brain vascular system may be compromised during severe hyperbilirubinemia, entrance of UCB into the brain has been mainly attributed to simple diffusion of the UCB fraction not bound to albumin, usually designated as free UCB (Bf), through the endothelial cells (Diamond and Schmid, 1966; Wennberg, 2000). Toxicity is believed to occur when concentrations of Bf are too high, favoring its precipitation, and when risk factors such as prematurity and prolonged exposure are associated to the neonatal hyperbilirubinemia, thus enhancing the probability of BIND (Wennberg, 2000; Ahlfors, 2001).
In spite of the increased awareness of UCB effects to brain cells (Brites and Brito, 2012), knowledge about its passage across the endothelial monolayer, the rate in which this passage is achieved, and its effects on the integrity of the brain endothelial barrier is scarce (Akin et al., 2002; Sequeira et al., 2007). Our own previous study has shown that the endothelial lining is susceptible to UCB (Palmela et al., 2011), since high levels of the molecule increased endothelial cell death, compromised the cell antioxidant capacities, affected the expression of important endothelial proteins and, most notably, increased the production of mediators with great implications to the integrity and permeability of the BBB, such as nitric oxide and VEGF. More recently, our group has confirmed that UCB affects rat primary brain endothelial cells, inducing cell death, impairing P-glycoprotein function and activating metalloproteinases (Cardoso et al., 2012). Importantly, such study evaluated the integrity of the endothelial monolayers, either in mono or in co-cultures with astrocytes, and showed a decreased TEER and increased permeability after UCB exposure.
If the BBB is disturbed during hyperbilirubinemia, it is likely that the entrance of Bf or even albumin-bound UCB into the brain will be facilitated (Wennberg, 2000). Therefore, we decided to undertake a deeper analysis of the integrity of the endothelial lining upon UCB exposure to test the harmful effects of a short and sustained hyperbilirubinemia on BBB properties. To this end, we further explored the dynamics of UCB interaction with a human brain microvascular endothelial cell (HBMEC) monolayer, considered a simplified in vitro model of the human BBB, throughout incubation time. Our results indicate biphasic effects of UCB on the HBMEC that depend on the time of interaction. A short exposure to UCB increases the protein levels of caveolin-1, VEGF and VEGFR-2 and already elicits the release of small endothelial cell vesicles. In the late response UCB triggers a reduction of ZO-1 and β-catenin levels, loss of TJ strands and impairment of the cohesion between endothelial cells, with the consequent decrease in TEER and increase in permeability. Interestingly, in this last condition there is an increase in autophagy, thus indicating the activation of the cellular degradation system. Collectively, our findings support the hypothesis that a sustained and severe hyperbilirubinemia, by inducing BBB endothelial disruption, facilitates BIND, thus requiring a preventive clinical intervention.
Materials and Methods
Chemicals
The basal medium Roswell Park Memorial Institute (RPMI) 1640, antibiotic-antimycotic solution, human serum albumin (HSA) (fraction V, fatty acid free), bovine serum albumin (BSA), Hoechst 33258 dye, sodium fluorescein, and UCB were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Non-essential amino acids (NEAA), sodium pyruvate, L-glutamine, fetal bovine serum (FBS), and minimum essential medium (MEM) vitamins were from Biochrom AG (Berlin, Germany). Nuserum IV and rat-tail collagen I were acquired from BD Biosciences (Erembodegem, Belgium). Rabbit anti-β-catenin, mouse anti-ZO-1, rabbit anti-tricellulin, Alexa Fluor 488 goat anti-rabbit IgG, and Alexa Fluor 594 goat anti-mouse IgG were purchased from Invitrogen (Carlsbad, CA, USA). Rabbit anti-caveolin-1, rabbit anti-microtubule-associated protein 1 light chain-3 (LC3), LumiGLO, and cell lysis buffer were acquired from Cell Signaling (Beverly, MA, USA). Horseradish peroxidase-labeled goat anti-rabbit IgG, mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH), rabbit anti-VEGF, and mouse anti-VEGFR-2 were acquired from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit anti-lamin B1 was from Abcam (Cambridge, UK). Pefabloc® SC AEBSF was obtained from Roche Applied Science (Mannheim, Germany). Horseradish peroxidase-labeled goat anti-mouse IgG, nitrocellulose membrane and Hyperfilm ECL were from Amersham Biosciences (Piscataway, NJ, USA). All other chemicals were of analytical grade and were purchased from Merck (Darmstadt, Germany).
Cell Culture and Treatment
We have previously used an HBMEC line as a simplified model of the human BBB to evaluate the effects of UCB on cell death, cytokine mRNA expression and release, as well as on nitrosative stress (Palmela et al., 2011). This cell line was derived from primary cultures of HBMEC transfected with SV40 large T antigen (Stins et al., 2001) and was cultured in RPMI medium supplemented with 10% FBS, 10% NuSerum IV, 1% NEAA, 1% MEM vitamins, 1 mM sodium pyruvate, 2 mM L-glutamine, and 1% antibiotic-antimycotic solution. For immunocytochemistry, electron microscopy and Western blot studies, cells were seeded at a density of 5 × 104 cell/mL in collagen I-coated coverslips and plates, respectively, and treated after 2 days in culture. For integrity studies, cells were seeded on collagen I-coated polyester transwell inserts (0.4 μM, Corning Costar Corp., USA) at a density of 8 × 104 cell/insert and treated after 8 days in culture. All experiments were performed at confluence. Endothelial cultures were maintained at 37°C in a humid atmosphere enriched with 5% CO2.
UCB was purified according to the method of McDonagh and Assisi (1972) and a 10 mM stock solution of UCB was prepared in 0.1 M NaOH and used immediately after preparation. The pH value was restored to 7.4 by addition of equal amounts of 0.1 M HCl, and all the procedures were performed under light protection to avoid photodegradation. Confluent monolayers of the HBMEC line were incubated with 50 or 100 μM UCB, or no addition (control), in the presence of 100 μM HSA, for 4, 24, 48, and 72 h. The incubation medium consisted in the regular medium without FBS and Nuserum to avoid disturbance of the final concentration of albumin in the incubation medium.
In the integrity experiments, endothelial cells were cultured on semipermeable filters with two well-defined compartments: the apical or upper compartment, which can be considered as the “blood-side” where UCB and HSA were added, and the basolateral or lower compartment, which is considered the “brain side.” At the end of each incubation period, and after TEER measurement, the cultured inserts were used for permeability studies and media from the upper and lower chambers were collected for Bf measurements. In separate experiments, the cells from the UCB-incubated inserts were collected for UCB extraction with chloroform and the medium of the upper chamber was collected for evaluation of the number of detached cells.
Determination of Bf Levels and UCB Cellular Content
Among the methods used to measure Bf, the peroxidase method is considered practical and reliable, and has been extensively employed (Roca et al., 2006; Ahlfors et al., 2009). Thus, in the present study, we used this method to evaluate Bf levels in our two UCB/HSA molar ratio conditions (Roca et al., 2006).
After determining the starting Bf levels in the two UCB conditions, we monitored Bf levels in the media of the upper and lower chambers of the treated inserts (4–72 h). In order to evaluate the intracellular content of UCB, a chloroform extraction was accomplished based on the study of Brito et al. (2000). Briefly, after lysis of the HBMEC monolayer, each sample was mixed with equal volumes of chloroform. After vortex shaking and centrifugation, UCB concentration in the chloroform extract was determined by direct measurement of the absorption at 454 nm. Results were expressed as nmol of UCB per mg of protein.
TEER Measurements
TEER evaluation was performed using an EndOhm™ chamber coupled to an EVOMX resistance meter (World Precision Instruments, Inc., USA). Readings were collected before UCB addition (time 0) and after 4, 24, 48, and 72 h of exposure. Results are shown as percentage of variation from average control readings, after deducting the empty insert values.
Permeability Studies
In order to evaluate the selective paracellular permeability of the HBMEC monolayer after UCB exposure, a permeability assay was conducted with sodium fluorescein (SF) (molecular weight: 376 Da). The permeability was determined as described by Veszelka et al. (2007). Briefly, cell culture inserts were transferred to 12-well plates containing Ringer–Hepes solution (118 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 5.5 mM D-glucose, 20 mM Hepes, pH 7.4) in the basolateral compartments. The SF solution (10 mg/mL SF in 1% BSA solution in Ringer–Hepes) was added to the upper chambers. The inserts were transferred to new wells at 20, 40, and 60 min. Lower chamber solutions were collected to determine fluorescein levels (Hitachi F-2000 fluorescence spectrophotometer, excitation: 440 nm and emission: 525 nm). Flux across cell-free inserts was also measured. The endothelial permeability coefficient Pe was calculated as described by Deli et al. (2005).
Evaluation of Detached Cells
At the end of the 72 h period of incubation, the number of viable and non-viable cells in suspension in the upper chamber of the UCB-treated inserts was measured. After media centrifugation, the cell pellet was recovered in RPMI medium and cells were counted with Trypan blue dye exclusion test. Results were shown as number of viable and non-viable cells per insert.
Immunocytochemistry
Immunostaining studies were performed to evaluate UCB effects on several relevant proteins that direct or indirectly affect the endothelial barrier functions. The proteins studied included the TJ protein ZO-1, the AJ protein β-catenin, the caveolae-mediated transcytosis protein caveolin-1, as well as VEGF and its receptor VEGFR-2. Detection of these proteins was performed using the primary antibodies mouse anti-ZO-1, rabbit anti-β-catenin, rabbit anti-caveolin-1, rabbit anti-VEGF and mouse anti-VEGFR-2 (all dilutions 1:100), and the secondary antibodies Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 594 goat anti-mouse (1:500). Fluorescence was visualized using a DFC 490 camera (Leica, Germany) adapted to an AxioScope.A1 microscope (Zeiss, Germany). For total fluorescence analysis, fluorescence of five random microscopic fields was acquired per sample. Evaluation of fluorescence intensity was determined using ImageJ 1.29x software (N.I.H., USA) and results were expressed as fold change (mean fluorescence per number of cells vs. control samples).
Western Blot Analysis
Total and region-specific β-catenin were further evaluated by Western blot. Subcellular fractionation was performed as described by Abcam (Dr. Richard Pattern). Briefly, cells were lysed with fractionation buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM Pefabloc) and passed through a 25 G needle. The nuclear pellet was collected after centrifugation and the supernatant (cytoplasmic and membrane fractions) was then ultracentrifuged (100,000 g) for 1 h. The cytoplasmic fraction (supernatant) was removed and the pellet (membrane fraction) was ressuspended in cell lysis buffer. The efficiency of fractionation was confirmed with the detection of GAPDH enrichment in the cytoplasmic fraction, Lamin B1 in the nuclear fraction and tricellulin in the membrane region. Results were normalized to total protein staining with Amido Black (Aldridge et al., 2008) due to UCB-induced changes in the expression of the membrane proteins. Therefore, in order to ensure consistency of results, we normalized the intensity of β-catenin in all the extracts to total protein staining.
In order to study the HBMEC process of autophagy after UCB exposure, the conversion of LC3-I (free form) to LC3-II (phosphatidylethanolamine-conjugated form) was also evaluated by Western blot in total protein extracts. The Western blot assays were carried out as previously described (Fernandes et al., 2006). Briefly, total or region-specific protein extracts were separated on a 8 or 12% SDS-PAGE (for β-catenin and LC3 detection, respectively). Following electrophoretic transfer onto a nitrocellulose membrane and blocking with 5% milk solution, the blots were incubated with primary antibody overnight at 4°C [rabbit anti-LC3 (1:1000), rabbit anti-β-catenin (1:1000) or mouse anti-β-actin (1:5000)] and with horseradish peroxidase-labeled secondary antibody [anti-mouse (1:5000) or anti-rabbit (1:5000)] for 1 h at room temperature. Protein bands were detected by LumiGLO and visualized by autoradiography with Hyperfilm ECL.
Electron Microscopy (EM) and Freeze-Fracture
For scanning EM, cells and vesicles released into the culture medium were doubly fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (PB) and 1% osmium tetroxide in 0.1 M PB, and dehydrated with a graded series of ethanol. Samples were then critical-point dried, sputter coated, and observed by a JEOL JSM-5800LV scanning electron microscope (Tokyo, Japan) at an acceleration voltage of 15 kV. For transmission EM, cells were prepared as described above and then embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and observed with a Hitachi H-7500 transmission electron microscope (Tokyo, Japan) at an acceleration voltage of 80 kV. For freeze-fracture EM cells, fixed as described, were immersed overnight in 30% glycerol in 0.1 M PB and then frozen in liquid nitrogen. Frozen samples were fractured at −100°C and platinum-shadowed unidirectionally at an angle of 45° in BAL-TEC BAF060 Freeze Etching System. Samples were picked up on formovar-filmed grids and examined with a Hitachi H-7500 electron microscope at 100 kV.
Statistical Analysis
Results are expressed as means ± SEM from, at least, three separate experiments. Significant differences between groups were determined by the two-tailed t-test performed on the basis of equal and unequal variance as appropriate. Statistical significance was considered when P values were lower than 0.05.
Results
Early Interaction of UCB with the HBMEC Monolayer Triggers the Upregulation of Caveolin-1, VEGF and VEGFR-2, as well as the Release of Small Sized Vesicles
We began our studies by evaluating the effect of UCB on caveolin-1, VEGF and VEGFR-2 levels, since these proteins are important players in the mechanisms that regulate endothelial permeability.
As shown in Figure 1, the exposure of HBMEC to both UCB concentrations led to an initial increase in the immunostaining of caveolin-1 that returned to near control levels afterwards (Figure 1A). Maximum fluorescence was observed at 4 h, as represented in Figure 1B (increase of 56 and 91% for UCB 50 and 100 μM, respectively, P < 0.05). In accordance, transmission EM analysis of HBMEC also at 4 h (Figure 1C) evidenced an increased number of caveolae in the UCB-treated cells.
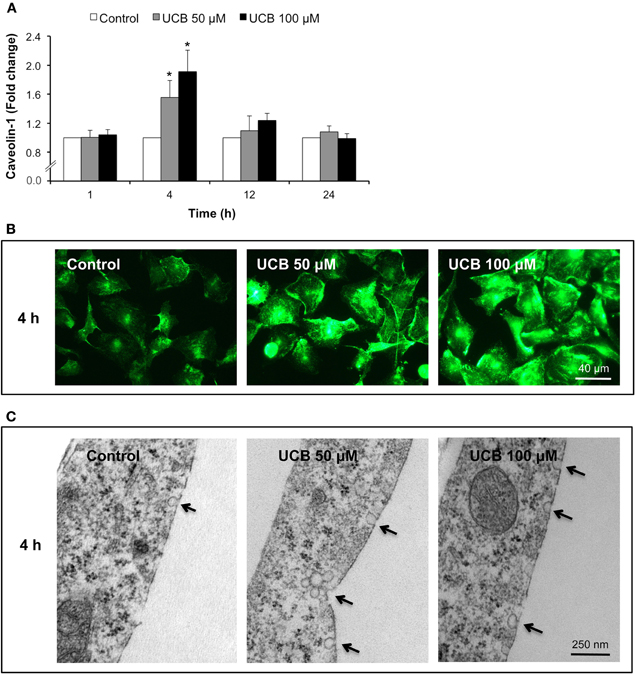
Figure 1. Early interaction of UCB with the HBMEC monolayer increases the expression of caveolin-1 and triggers the formation of caveolae. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA, for the indicated incubation periods. Caveolin-1 was analyzed by immunocytochemistry and protein levels were quantified by measurement of fluorescence intensity per number of cells. Results are shown as fold change from respective control (A). Representative immunocytochemistry results of the 4 h incubation point are shown (B). Ultrastructural analysis by transmission electron microscopy revealed an increased number of caveolae in UCB-treated cells (arrows) (C). Results are means ± SEM from at least three independent experiments performed in duplicate. *P < 0.05 vs. respective control.
Analysis of VEGF and VEGFR-2 fluorescence showed a surprising pattern of immunostainnings. The cytokine appeared in the perinuclear region, before secretion to the cultured medium as previously observed by us after prolonged UCB exposure (Palmela et al., 2011). On the other hand, receptor staining evidenced to not be restrained to the cell membrane but distributed in the entire cell, probably as a consequence of the interaction between VEGFR-2 and caveolae (Labrecque et al., 2003). Interestingly, the fluorescence showed a progressive increase from 4 to 24 h of exposure, decreasing to control levels afterwards (Figures 2A,B, respectively). Maximum immunostaining was observed at 24 h for both proteins (Figure 2C), correspondent to an increase of ∼16 and 12% in VEGF and VEGFR-2 levels, respectively (P < 0.01).
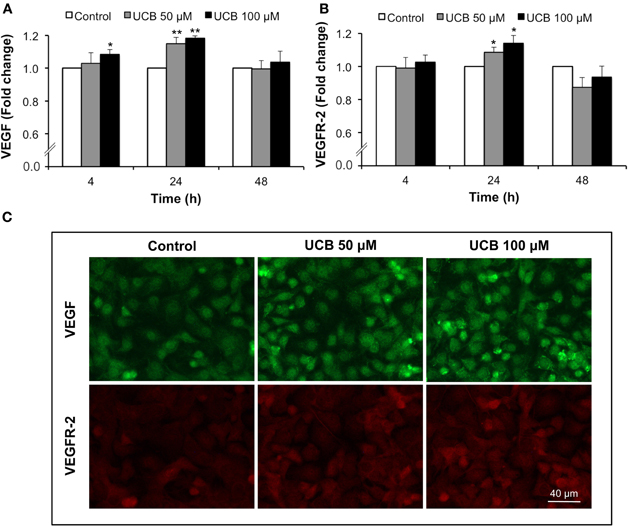
Figure 2. Early interaction of UCB with the HBMEC monolayer leads to the upregulation of VEGF and VEGFR-2. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA, for the indicated incubation periods. VEGF and VEGFR-2 levels were analyzed by immunocytochemistry and protein levels were quantified by measurement of fluorescence intensity per number of cells. Quantification of VEGF (A) and VEGFR-2 (B) levels is indicated as fold change from respective control. Representative results of the 24 h period of incubation are shown (C). Results are means ± SEM from at least five independent experiments performed in duplicate. *P < 0.05 and **P < 0.01 vs. respective control.
As shown in Figure 3, the scanning EM analysis revealed that during the early response to UCB (4 h incubation), a great number of vesicles with different sizes were released from the cells treated with both UCB concentrations. Interestingly, the highest concentration produced a very significant release of small vesicles, a distinct pattern from the one triggered by the lowest concentration of UCB. Despite this observation, no alterations were produced in the barrier properties of the monolayer or in the levels of β-catenin and ZO-1 at this time point (results not shown).
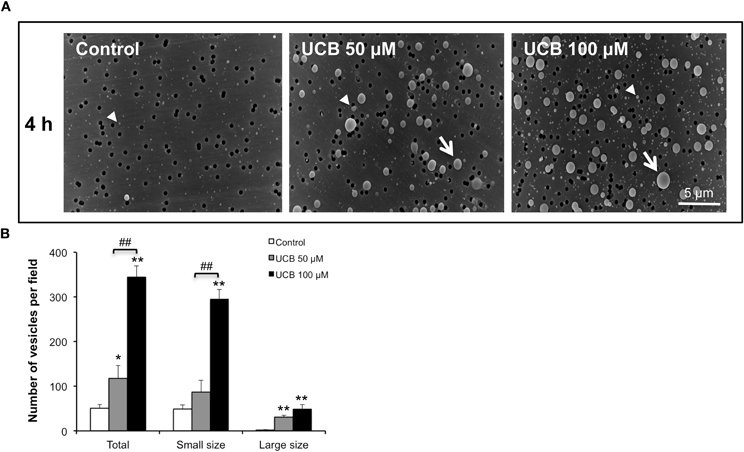
Figure 3. Early interaction of UCB with the HBMEC monolayer determines the release of vesicles. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA. Analysis of vesicles released into culture medium was performed by scanning electron microscopy and the results of the 4 h incubation period are shown (A). Quantification of small (arrowhead), large (arrow) and total number of vesicles is shown (B) and results are means ± SEM from three independent experiments performed in duplicate. *P < 0.05 and **P < 0.01 vs. control. ##P < 0.01 UCB 100 vs. 50 μM.
Sustained Interaction of UCB with the HBMEC Monolayer Determines the Down-Regulation of β-Catenin and ZO-1 Proteins
Next, we evaluated the effect of UCB in proteins from the TJ and AJ, ZO-1, and β-catenin, respectively, as they are key players in the maintenance of barrier properties of endothelial cells of the BBB.
Since β-catenin has also a crucial role as a transcription factor in the Wnt/β-catenin signaling pathway (Liebner and Plate, 2010), its location is not only relevant in the membrane but also in other subcellular regions. Therefore, we investigated if the cellular distribution of β-catenin was disturbed by UCB. To evaluate this, β-catenin levels were analyzed in three different subcellular locations: nucleus, cytoplasm and plasma membrane. As depicted in Figure 4A, the 24 h exposure to UCB caused a slight elevation of total β-catenin with increases of ∼5% for both UCB concentrations (P < 0.05). Interestingly, and specially for the highest UCB concentration, this minor elevation resulted from a balance between the upregulation of the nuclear fraction (increase of 11%, P < 0.01) and a down-regulation of the membrane content (decrease of 15%, P < 0.01). This pattern of results was also obtained for the 48 h period of incubation (Figure 4B), where the effects to the β-catenin membrane fraction were more marked between UCB concentrations (decrease of 7%, P < 0.05 and 18%, P < 0.01 for UCB 50, and 100 μM, respectively). After 72 h of exposure to the highest concentration of UCB the minor elevation of nuclear β-catenin, along with the sustained decrease in the membrane fraction, produced a significant decrease in total protein (11%, P < 0.05) (Figure 4C).
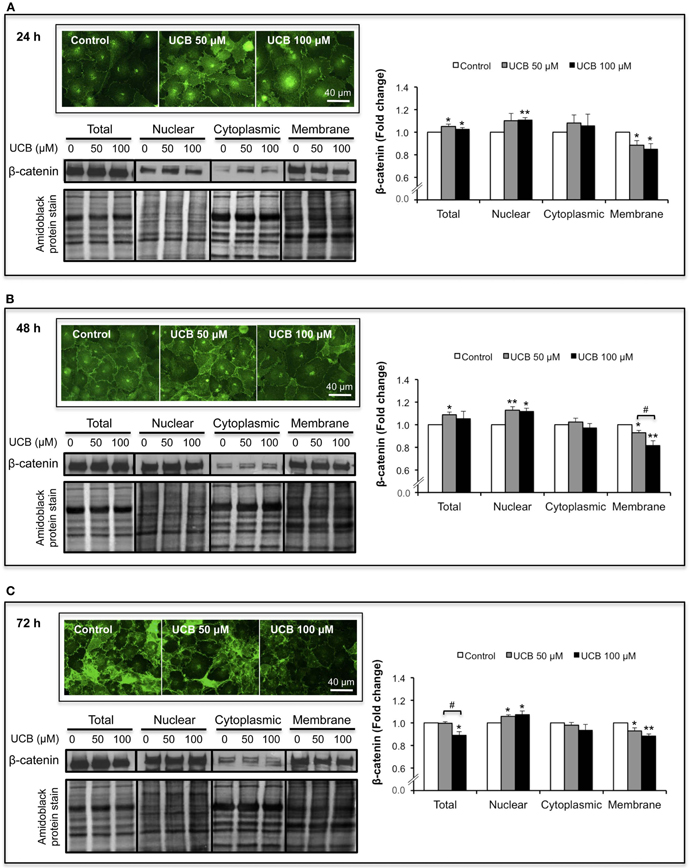
Figure 4. UCB interaction with the HBMEC monolayer determines an upregulation of nuclear β-catenin and a down-regulation in the membrane fraction. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA. β-catenin was evaluated by immunofluorescence and Western blot analysis of different subcellular locations (nucleus, cytoplasm, and membrane), after subcellular fractionation. Representative immunofluorescence photos, as well as representative Western blot results and respective quantification are shown for the incubation periods of 24 h (A), 48 h (B), and 72 h (C). Quantification of band intensity was done by scanning densitometry and standardized with respect to total protein stains. Results are means ± SEM from at least four independent experiments performed in duplicate. *P < 0.05 and **P < 0.01 vs. respective control; #P < 0.05 UCB 100 vs. 50 μM.
ZO-1 immunostaining analysis (Figure 5A) revealed also a decrease, although only evident after a 48 h interaction (Figure 5B). This 26% reduction for UCB 50 μM (P < 0.01) and 22% for UCB 100 μM (P < 0.05) suggests a weakness of the intercellular junctions. This alteration was corroborated either by freeze-fracture analysis evidencing loss of TJ strands by both UCB concentrations (Figure 5C), or scanning EM analysis also revealing increased number of retracted endothelial cells with deficient cell-to-cell contacts in the UCB-treated samples (Figure 5D).
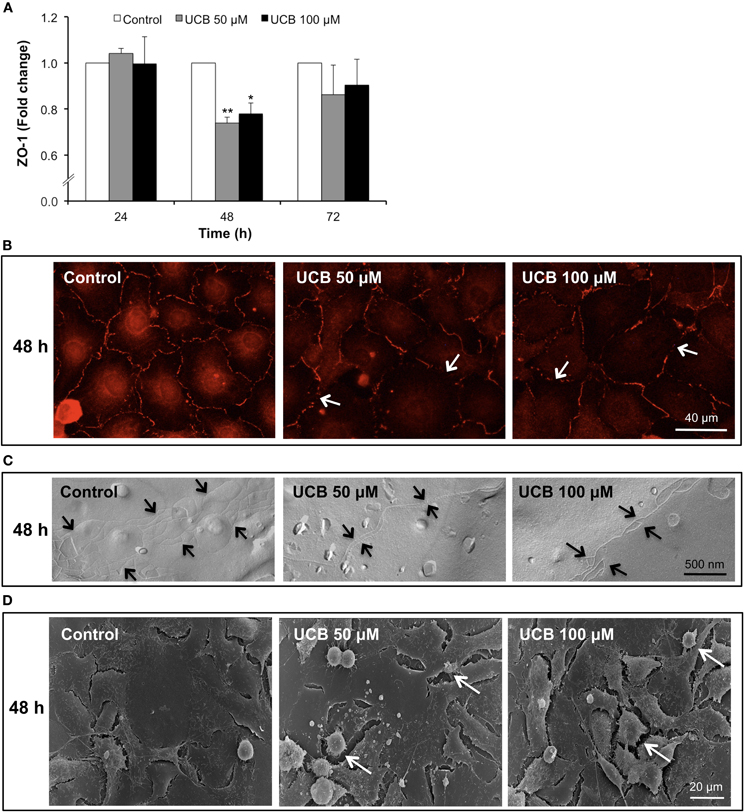
Figure 5. Interaction of UCB with the HBMEC monolayer determines a later down-regulation of ZO-1 protein. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA, for the indicated incubation periods. Total fluorescence of ZO-1 is shown as fold change from respective control (A). Representative results, showing loss of ZO-1 staining (arrows), after a 48 h period of incubation are shown (B). UCB exposure induces loss of tight junction strands (arrows) at 48 h, as observed by freeze-fracture (C). Scanning electron microscopy results evidence the effects of UCB on the intercellular contacts after 48 h, showing retracted endothelial cells (arrows) (D). Results are means ± SEM from at least three independent experiments performed in duplicate. *P < 0.05 and **P < 0.01 vs. respective control.
Sustained Interaction of UCB with the HBMEC Monolayer Reduces the TEER and Increases the Paracellular Permeability to SF
TEER and permeability are crucial parameters for the study of endothelial and epithelial barriers, reflecting the quality of the in vitro model and its paracellular barrier characteristics (Wilhelm et al., 2011). By measuring these parameters we were able to evaluate the consequences of UCB disturbance on an otherwise tight barrier.
Figure 6A shows that exposure of the HBMEC monolayer to 100 μM UCB led to a marked reduction of the TEER which, though initiated after 24 h of exposure, attained a maximum 34% decrease at 72 h incubation (P < 0.01). In accordance, the analysis of the HBMEC permeability to SF (Figure 6B) indicated a progressive increase of the flux of this paracellular tracer along the incubation time for the 100 μM UCB, doubling its values at 72 h (P < 0.05).
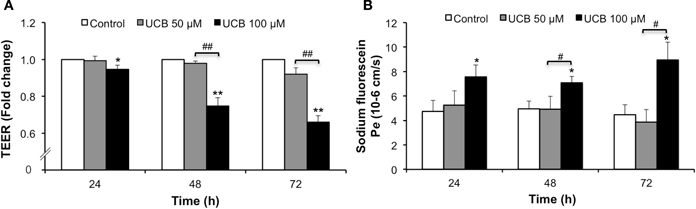
Figure 6. Prolonged interaction of UCB with the HBMEC monolayer decreases TEER and increases paracellular permeability to sodium fluorescein. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA, for the indicated incubation periods. TEER, expressed as fold change from control readings (A), and the transendothelial permeability (Pe) to sodium fluorescein (B) are shown. Results are means ± SEM from at least six independent experiments performed in duplicate. *P < 0.05 and **P < 0.01 vs. respective control. #P < 0.05 and ##P < 0.01 UCB 100 vs. 50 μM.
Sustained Interaction of UCB with the HBMEC Monolayer Activates Autophagy
Autophagy is a highly regulated cellular process that serves to remove damaged proteins and organelles from the cell (Kimmelman, 2011). This complex phenomenon contributes to the maintenance of cellular homeostasis, adaptation to adverse environments and activation of programmed cell death (Codogno and Meijer, 2005; Mizushima, 2007). The induced conversion of LC3-I to LC3-II, the protein associated with the autophagosome membrane, is widely used to monitor autophagy (Kabeya et al., 2000; Tanida et al., 2008). In order to understand if in HBMEC this process is stimulated by UCB treatment, we evaluated the protein levels of LC3 I and II. As depicted in Figure 7, prolonged exposure to UCB enhanced the expression of LC3-II, indicating a higher content of autophagosomes at 72 h (P < 0.01).
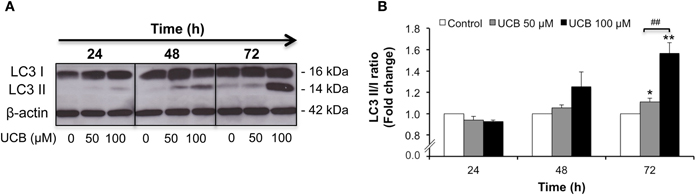
Figure 7. Prolonged interaction of UCB with the HBMEC monolayer activates autophagy. HBMEC line was incubated without (control) or with 50 or 100 μM UCB, in the presence of 100 μM HSA, for the indicated periods. Autophagy was evaluated by Western blot, with LC3 I (free form) and II (phosphatidylethanolamine-conjugated form) detection. Representative results of one experiment are shown (A). Quantification of LC3 I and II band intensity was done by scanning densitometry and the ratio between the membrane-bound and the cytosolic forms was calculated and standardized with respect to β-actin protein (B). Results are means ± SEM from at least four independent experiments performed in duplicate. *P < 0.05 and **P < 0.01 vs. control. ##P < 0.01 UCB 100 vs. 50 μM.
Sustained Interaction of UCB with the HBMEC Monolayer Causes Increased UCB Cellular Content, Detachment of Cells and Enhanced Passage across the Monolayer
Because Bf is believed to be the species crossing the BBB and its quantification is assumed to be a better predictor of BIND risk (Ahlfors, 2001; Ahlfors et al., 2009), we evaluated Bf levels in our current experimental model. The UCB concentrations of 50 and 100 μM (molar ratios of 0.5 and 1.0, respectively) yielded Bf levels of 13.2 and 23.6 nM, respectively.
Our prior results indicated that a prolonged exposure of the HBMEC monolayer to UCB increased the permeability, which could facilitate the passage of UCB across the monolayer. Therefore, we decided to measure the fraction of Bf that passed from the upper chamber of the inserts (where HSA and UCB were added, mimicking the systemic circulation) to the lower chamber (mimicking the brain parenchyma). Values of Bf became significantly detectable only after 72 h incubation, mainly for the highest UCB concentration used (Table 1).
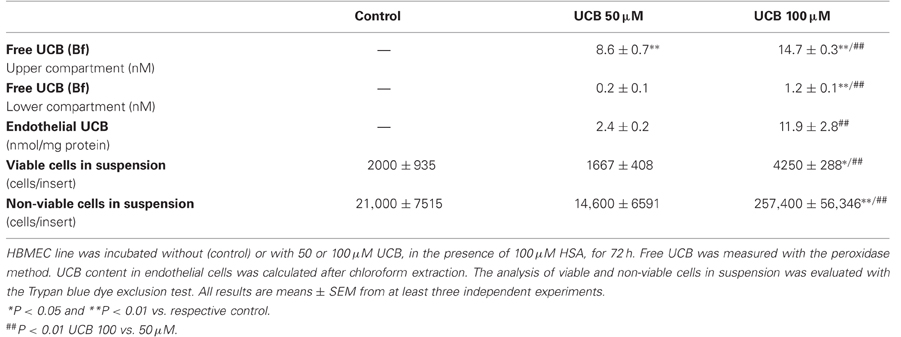
Table 1. Distribution of UCB across the HBMEC monolayer and consequent damage after 72 h of incubation.
Interestingly, a fraction of UCB remains in HBMEC as revealed after chloroform extraction. The content of UCB in endothelial cells showed a fivefold elevation in cells treated with 100 μM as compared with those exposed to 50 μM for 72 h (Table 1). We next sought to evaluate if the suggested UCB-induced fragility of the monolayer also included the detachment of cells and cellular fragments. As shown in Table 1, 100 μM had the greatest effect on the detachment of cells, increasing the number of cells in suspension, either viable (twofold) or non-viable (12-fold), from both the control condition and the lowest UCB concentration. These results are in agreement with those of TEER and permeability, as well as with those of Bf in the lower compartment and in HBMEC, and yet with the release of vesicles observed by scanning EM, and reflect a fragility of the monolayer that mainly occurs by exposure to the highest UCB concentration in sustained conditions.
Discussion
Though the role of the BBB in the progression, or even as a cause of CNS disorders, has often been dismissed, it is nowadays believed that BBB dysfunction plays an important role in a growing number of neurological pathologies, including cerebral ischemia, brain trauma, tumors, and neurodegenerative diseases (Zlokovic, 2008; Carvey et al., 2009; Weiss et al., 2009; Palmela et al., 2012). In these conditions, peripheral factors including toxins, monocytes, and complement more readily enter the brain, but also chemokines, metabolites, and neurotransmitters more freely leave the brain (Carvey et al., 2009). Therefore, a compromised BBB may facilitate the passage of UCB into the brain parenchyma (Wennberg, 2000), rendering the study of BBB permeability and integrity of utmost importance. Nevertheless, few studies have actually investigated how UCB passage across the endothelial lining is achieved (Diamond and Schmid, 1966; Sequeira et al., 2007) and the effects derived from the exposure of brain endothelial cells to UCB (Akin et al., 2002; Palmela et al., 2011; Cardoso et al., 2012). Interestingly, our data show that features of UCB interaction with the HBMEC depend on whether the exposure is short or prolonged. The early interaction of UCB affects the expression of caveolin-1, VEGF and VEGFR-2. Although not affecting junctions, TEER and permeability, a release of vesicles into the cultured media was observed for both UCB concentrations, which already suggests endothelial compromise. On the other hand, prolonged exposure to UCB has drastic effects in HBMEC transendothelial properties, altering the intercellular junction structure and protein levels, as well as inducing autophagy and UCB passage across the endothelial lining.
Our interest in studying caveolin-1 was due to the complex role of this protein in various signaling pathways that direct or indirectly affect endothelial permeability, since it is associated with proteins of the TJ and AJ (Nusrat et al., 2000; Song et al., 2007). Moreover, interaction of caveolin-1 with VEGFR-2 suggests a direct regulation of the receptor activity and a role in modulation of permeability (Labrecque et al., 2003; Tahir et al., 2009). Our results show that incubation with both UCB concentrations increase caveolin-1 expression after 4 h. In addition, transmission EM analysis reveals a higher number of caveolae in the UCB-treated cells. This is a very interesting fact since activation of caveolae-associated signaling proteins was indicated to have a role in the uptake of toxicants (Majkova et al., 2010) and upregulation of caveolin-1 has been associated with enhanced transcytosis (Xia et al., 2009; Wang et al., 2010). In accordance to Song et al. (2007) report that caveolin-1 acts to restrain processes that destabilize endothelial junctions, we may hypothesize that the upregulation at 4 h may translate an attempt to stabilize the HBMEC junctions and prevent their disorganization. Caveolin-1 is known to interact and inhibit VEGFR-2, although allowing VEGFR-2 to undergo VEGF-dependent activation (Labrecque et al., 2003). This evidence suggests a role of caveolin-1 in conferring ligand dependence to this receptor system and is in accordance to our results on VEGF and VEGFR-2 expression, evidencing that peak values shift from an increase of caveolin-1 at 4 h to maximal levels of VEGF and VEGFR-2 at 24 h. If after 4 h the reduced levels of VEGF and increased expression of caveolin-1 might prevent VEGFR-2 activation, at 24 h this is no longer present and VEGFR-2 increases. Stimulation of VEGFR-2 by VEGF is a key step in endothelial proliferation, migration and increased permeability, ultimately contributing to the angiogenic response (Holmes et al., 2007). Thus, our observed results with VEGFR-2 support the increase in paracellular permeability by UCB exposure, from 24 h onwards. Indeed, angiogenic sprouting with the presence of immature blood vessels and blood-borne components in brain parenchyma, suggesting an hyperpermeable BBB, was recently observed in a premature infant with kernicterus at autopsy (Brito et al., 2011).
Alterations produced by UCB in both TJ and AJ are only observed at prolonged conditions. Although TJ are the primary seal between endothelial cells, in BMEC, TJ and AJ are intermingled and AJ also contribute to the properties of the BBB endothelium (Cardoso et al., 2010). The AJ protein β-catenin plays an important role in maintaining barrier function by influencing TJ formation and directly activating signaling molecules and gene transcription (Lampugnani and Dejana, 2007). In our study, we questioned if UCB would alter β-catenin assembly, favoring any subcellular distribution and function. β-catenin peaked between 24 and 48 h for both UCB concentrations, similarly to our previous observations obtained by Western blot analysis (Palmela et al., 2011). Along the incubation period we observed an increase in the nuclear fraction of β-catenin, while the membrane content of this protein was decreased. This shift in the subcellular localization of β-catenin supports our results on the integrity parameters, reflecting a reduction of the protein at the junctional level and consequent fragility of the barrier. These effects were less marked for the 50 μM UCB, where total β-catenin remained unaltered or superior than in control samples, which supports the absence of effects on TEER and permeability with this concentration. When considering the time-dependent effects of 100 μM UCB in β-catenin, i.e., an upregulation followed by a great reduction of protein levels, we may assume that after an attempt to circumvent the UCB induced-damage, β-catenin may become vulnerable to UCB toxicity and further contribute to the decline of HBMEC integrity. Importantly, our results show a sustained β-catenin down-regulation in the membrane fraction, reflecting its reduction at the junction level. To note that VEGFR-2 stimulation by VEGF was shown to reduce the strength of VE-cadherin association to β-catenin and to actin cytoskeleton, contributing to an increased permeability (Liebner et al., 2006; Margariti et al., 2010). This corroborates our findings in that VEGF and VEGFR-2 increased prior to the reduction of β-catenin, and the alterations in TEER and permeability. It has been indicated that the reduced β-catenin in the cytoplasm is probably a consequence of protein degradation by polyubiquitination, as observed in other cell cultures (Zhang et al., 2011), and that is related with both BBB breakdown (Liebner et al., 2008) and cell death (Kim et al., 2010; Omori et al., 2011).
The decrease of ZO-1 protein expression at 48 h observed in this study is in line with the work of Raimondi et al. (2006) showing that UCB decreased occludin in Caco-2 cells, an intestinal epithelial cell line with barrier properties. The reduction we observed in ZO-1 was supported by freeze-fracture EM showing a decrease of TJ strands. In this regard, it was observed a correlation between increased organization of TJ cytoplasmic fibrils and decreased membrane permeability (Huber et al., 2001). Therefore, the poorer expression of ZO-1 and the less complex TJ in UCB-treated cells shall underline the decrease in TEER and the increase in the permeability to SF, supporting the lower capacity to maintain intercellular contacts as indicated by scanning EM analysis.
Curiously, in vasogenic cerebral edema, Nag et al. (2009b) reported that augmented levels of caveolin-1 at the onset of injury were followed by reduction and destabilization of TJ (late event), resulting in increased endothelial permeability and BBB breakdown (Zhong et al., 2008; Wang et al., 2011). We observed this sequence of events during the exposure of HBMEC to UCB, where prolonged incubation with the highest UCB concentration led to a reduction in TEER and an increase in permeability. This is not without precedent since it was previously shown in Caco-2 cells that Bf levels of 5–600 nM also triggered reduced TEER and increased permeability (Raimondi et al., 2006). Similar results were also recently obtained by us in rat brain endothelial cells, either in mono- and in co-culture with astrocytes (Cardoso et al., 2012).
The induction of autophagy occurs in response to extra or intracellular stress, and is associated with the activation of programmed cell death through a cell-autodigestive process (Levine and Klionsky, 2004; He and Klionsky, 2009). Since UCB-induced apoptotic cell death was not only observed in HBMEC (Palmela et al., 2011), but in other brain cells as well (Fernandes et al., 2004; Silva et al., 2010; Vaz et al., 2010), we hypothesized that UCB-mediated endothelial injury would include autophagy activation. We have observed that, in fact, UCB, mainly the highest concentration, induces the conversion of LC3-I (free form) to LC3-II (phosphatidylethanolamine-conjugated form), after 72 h of incubation. This provides the first demonstration that autophagy is among the mechanisms of UCB-induced cell death, at least in HBMEC. This finding indicates that UCB may induce HBMEC senescence since recent studies disclosed that autophagy facilitates the process of senescence (Young et al., 2009), an association already observed in stressed endothelial cells (Patschan et al., 2008). Curiously, the upregulation of genes suggestive of an induced autophagy in human neuroblastoma SH-SY5Y cells by exposure to UCB was previously indicated (Calligaris et al., 2009) and surely deserves to be explored in the future.
This study culminated in the evaluation of UCB passage across the HBMEC monolayer. It is generally assumed that albumin-bound UCB does not cross the intact BBB but that Bf readily does it (Wennberg, 2000; Hansen, 2002). Our results showed that the molar ratios of 0.5 and 1.0 used yielded levels of Bf comparable to those found in the serum of neonates with moderate or severe hyperbilirubinemia, respectively (Ahlfors et al., 2009). The maximum decrease in Bf in the upper compartment after 72 h of incubation, and the consequent increase in the lower compartment, reflects the passage of the pigment across the endothelial monolayer in the apical-basal direction, though not excluding the passage in the opposite direction, as observed by other studies with bovine brain microvascular endothelial cells (Sequeira et al., 2007). Additionally, and complementing our data, we observed that intracellular UCB levels were directly correlated with the concentration of UCB to which HBMEC were exposed to. This fraction of UCB in endothelial cells probably accounts for the HBMEC demise suggested by the autophagic activation, and impairment of barrier properties here noticed. Moreover, the observed cell detachment supported the notion of a fragile monolayer and perturbation of the endothelial integrity, particularly for 100 μM UCB, attesting the most harmful and acute effects produced by severe hyperbilirubinemia.
In conclusion, our data demonstrate that UCB impairs barrier function in an in vitro model of the human BBB. Interestingly, this interaction involves dual effects that depend on the time of incubation, with early transcytosis and late paracellular pathways facilitating UCB entrance into the brain (Figure 8). Collectively, these results underscore the need of prompt clinical intervention in cases of lasting hyperbilirubinemia to prevent BIND and related irreversible CNS damage.
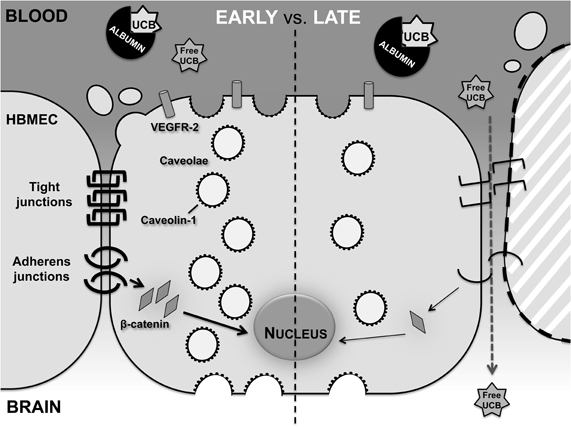
Figure 8. Effects of UCB on HBMEC differ between an early and late exposure. HBMEC early response includes increased number of caveolae and levels of caveolin-1 and VEGFR-2, while a delayed response presents alterations in the transendothelial properties of the HBMEC monolayer. This later response includes the disruption of the endothelial junctions, deficient cell-to-cell contacts and consequent detachment of cells, culminating in the passage of free UCB across the HBMEC monolayer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by PTDC/SAU-FCF/68819/2006 grant from Fundação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal (to Maria A. Brito) and PEst-OE/SAU/UI4013/2011 (to iMed.UL). Inês Palmela was recipient of a Ph.D. fellowship (SFRH/BD/61646/2009) from FCT.
References
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25.
Abbruscato, T. J., and Davis, T. P. (1999). Combination of hypoxia/aglycemia compromises in vitro blood-brain barrier integrity. J. Pharmacol. Exp. Ther. 289, 668–675.
Ahlfors, C. E., Wennberg, R. P., Ostrow, J. D., and Tiribelli, C. (2009). Unbound (free) bilirubin: improving the paradigm for evaluating neonatal jaundice. Clin. Chem. 55, 1288–1299.
Akin, E., Clower, B., Tibbs, R., Tang, J., and Zhang, J. (2002). Bilirubin produces apoptosis in cultured bovine brain endothelial cells. Brain Res. 931, 168–175.
Aldridge, G. M., Podrebarac, D. M., Greenough, W. T., and Weiler, I. J. (2008). The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 172, 250–254.
Brites, D., and Brito, M. A. (2012). “Bilirubin toxicity,” in Neonatal Jaundice, eds D. K. Stevenson, M. J. Maisels and J. F. Watchko (Mc-Graw-Hill Companies, Inc.), 115–143.
Brito, M. A., Silva, R. F. M., Tiribelli, C., and Brites, D. (2000). Assessment of bilirubin toxicity to erythrocytes. Implication in neonatal jaundice management. Eur. J. Clin. Invest. 30, 239–247.
Brito, M. A., Zurolo, E., Pereira, P., Barroso, C., Aronica, E., and Brites, D. (2011). Cerebellar axon/myelin loss, angiogenic sprouting, and neuronal increase of vascular endothelial growth factor in a preterm infant with kernicterus. J. Child Neurol. 27, 615–624.
Calligaris, R., Bellarosa, C., Foti, R., Roncaglia, P., Giraudi, P., Krmac, H., Tiribelli, C., and Gustincich, S. (2009). A transcriptome analysis identifies molecular effectors of unconjugated bilirubin in human neuroblastoma SH-SY5Y cells. BMC Genomics 10, 543.
Cardoso, F. L., Brites, D., and Brito, M. A. (2010). Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res. Rev. 64, 328–363.
Cardoso, F. L., Kittel, A., Veszelka, S., Palmela, I., Tóth, A., Brites, D., Deli, M. A., and Brito, M. A. (2012). Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells. PLoS ONE (in press).
Carvey, P. M., Hendey, B., and Monahan, A. J. (2009). The blood-brain barrier in neurodegenerative disease: a rhetorical perspective. J. Neurochem. 111, 291–314.
Chen, J., Braet, F., Brodsky, S., Weinstein, T., Romanov, V., Noiri, E., and Goligorsky, M. S. (2002). VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am. J. Physiol. Cell Physiol. 282, C1053–C1063.
Codogno, P., and Meijer, A. J. (2005). Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12, 1509–1518.
Cohen, R. S., Wong, R. J., and Stevenson, D. K. (2010). Understanding neonatal jaundice: a perspective on causation. Pediatr. Neonatol. 51, 143–148.
Davies, D. C. (2002). Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J. Anat. 200, 639–646.
Deli, M. A., Ábrahám, C. S., Kataoka, Y., and Niwa, M. (2005). Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell. Mol. Neurobiol. 25, 59–127.
Diamond, I., and Schmid, R. (1966). Experimental bilirubin encephalopathy. The mode of entry of bilirubin-14C into the central nervous system. J. Clin. Invest. 45, 678–689.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Gordo, A. C., Gama, M. J., Brito, M. A., and Brites, D. (2006). Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 96, 1667–1679.
Fernandes, A., Silva, R. F., Falcão, A. S., Brito, M. A., and Brites, D. (2004). Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J. Neuroimmunol. 153, 64–75.
Fischer, S., Wiesnet, M., Renz, D., and Schaper, W. (2005). H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur. J. Cell Biol. 84, 687–697.
Gomez-Gonzalez, B., and Escobar, A. (2009). Altered functional development of the blood-brain barrier after early life stress in the rat. Brain Res. Bull. 79, 376–387.
Hansen, T. W. R. (2002). Mechanisms of bilirubin toxicity: clinical implications. Clin. Perinatol. 29, 765–778.
Haorah, J., Ramirez, S. H., Schall, K., Smith, D., Pandya, R., and Persidsky, Y. (2007). Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 101, 566–576.
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93.
Holmes, K., Roberts, O. L., Thomas, A. M., and Cross, M. J. (2007). Vascular endothelial growth factor receptor-2, structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 19, 2003–2012.
Huber, J. D., Egleton, R. D., and Davis, T. P. (2001). Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 24, 719–725.
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728.
Kim, C. H., Bahn, J. H., Lee, S. H., Kim, G. Y., Jun, S. I., Lee, K., and Baek, S. J. (2010). Induction of cell growth arrest by atmospheric non-thermal plasma in colorectal cancer cells. J. Biotechnol. 150, 530–538.
Labrecque, L., Royal, I., Surprenant, D. S., Patterson, C., Gingras, D., and Béliveau, R. (2003). Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol. Biol. Cell 14, 334–347.
Lampugnani, M. G., and Dejana, E. (2007). The control of endothelial cell functions by adherens junctions. Novartis Found. Symp. 283, 4–13.
Lee, H. S., Namkoong, K., Kim, D. H., Kim, K. J., Cheong, Y. H., Kim, S. S., Lee, W. B., and Kim, K. Y. (2004). Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc. Res. 68, 231–238.
Levine, B., and Klionsky, D. J. (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477.
Liebner, S., and Plate, K. H. (2010). Differentiation of the brain vasculature: the answer came blowing by the Wnt. J. Angiogenes. Res. 2, 1.
Liebner, S., Cavallaro, U., and Dejana, E. (2006). The multiple languages of endothelial cell-to-cell communication. Arterioscler. Thromb. Vasc. Biol. 26, 1431–1438.
Liebner, S., Corada, M., Bangsow, T., Babbage, J., Taddei, A., Czupalla, C. J., Reis, M., Felici, A., Wolburg, H., Fruttiger, M., Taketo, M. M., von Melchner, H., Plate, K. H., Gerhardt, H., and Dejana, E. (2008). Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 183, 409–417.
Majkova, Z., Toborek, M., and Hennig, B. (2010). The role of caveolae in endothelial cell dysfunction with a focus on nutrition and environmental toxicants. J. Cell. Mol. Med. 14, 2359–2370.
Margariti, A., Zampetaki, A., Xiao, Q., Zhou, B., Karamariti, E., Martin, D., Yin, X., Mayr, M., Li, H., Zhang, Z., de Falco, E., Hu, Y., Cockerill, G., Xu, Q., and Zeng, L. (2010). Histone deacetylase 7 controls endothelial cell growth through modulation of β-catenin. Circ. Res. 106, 1202–1211.
McDonagh, A. F., and Assisi, F. (1972). The ready isomerization of bilirubin IX-α in aqueous solution. Biochem. J. 129, 797–800.
Nag, S., Manias, J. L., and Stewart, D. J. (2009a). Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol. Appl. Neurobiol. 35, 417–426.
Nag, S., Manias, J. L., and Stewart, D. J. (2009b). Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 118, 197–217.
Nusrat, A., Parkos, C. A., Verkade, P., Foley, C. S., Liang, T. W., Innis-Whitehouse, W., Eastburn, K. K., and Madara, J. L. (2000). Tight junctions are membrane microdomains. J. Cell Sci. 113, 1771–1781.
Omori, E., Matsumoto, K., and Ninomiya-Tsuji, J. (2011). Non-canonical β-catenin degradation mediates reactive oxygen species-induced epidermal cell death. Oncogene 30, 3336–3344.
Palmela, I., Brites, D., and Brito, M. A. (2012). “Blood-brain barrier in health and disease,” in The Blood-Brain Barrier: New Research, eds P. A. Montenegro S. M. and Juárez (Nova Science Publishers), (in press).
Palmela, I., Cardoso, F. L., Bernas, M., Correia, L., Vaz, A. R., Silva, R. F. M., Fernandes, A., Kim, K. S., Brites, D., and Brito, M. A. (2011). Elevated levels of bilirubin and long-term exposure impair human brain microvascular endothelial cell integrity. Curr. Neurovasc. Res. 8, 153–169.
Patschan, S., Chen, J., Polotskaia, A., Mendelev, N., Cheng, J., Patschan, D., and Goligorsky, M. S. (2008). Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 294, H1119–H1129.
Perlman, J. M., Rogers, B. B., and Burns, D. (1997). Kernicteric findings at autopsy in two sick near term infants. Pediatrics 99, 612–615.
Petty, M. A., and Lo, E. H. (2002). Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog. Neurobiol. 68, 311–323.
Raimondi, F., Crivaro, V., Capasso, L., Maiuri, L., Santoro, P., Tucci, M., Barone, M. V., Pappacoda, S., and Paludetto, R. (2006). Unconjugated bilirubin modulates the intestinal epithelial barrier function in a human-derived in vitro model. Pediatr. Res. 60, 30–33.
Roca, L., Calligaris, S., Wennberg, R. P., Ahlfors, C. E., Malik, S. G., Ostrow, J. D., and Tiribelli, C. (2006). Factors affecting the binding of bilirubin to serum albumins: validation and application of the peroxidase method. Pediatr. Res. 60, 724–728.
Schreibelt, G., Kooij, G., Reijerkerk, A., van Doorn, R., Gringhuis, S. I., van der Pol, S., Weksler, B. B., Romero, I. A., Couraud, P. O., Piontek, J., Blasig, I. E., Dijkstra, C. D., Ronken, E., and de Vries, H. E. (2007). Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 21, 3666–3676.
Sequeira, D., Watchko, J. F., Daood, M. J., O'Day, T. L., and Mahmood, B. (2007). Unconjugated bilirubin efflux by bovine brain microvascular endothelial cells in vitro. Pediatr. Crit. Care Med. 8, 570–575.
Shapiro, S. M. (2010). Chronic bilirubin encephalopathy: diagnosis and outcome. Semin. Fetal Neonatal Med. 15, 157–163.
Silva, S. L., Vaz, A. R., Barateiro, A., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M., and Brites, D. (2010). Features of bilirubin-induced reactive microglia: from phagocytosis to inflammation. Neurobiol. Dis. 40, 663–675.
Song, L., Ge, S., and Pachter, J. S. (2007). Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood 109, 1515–1523.
Stins, M. F., Badger, J., and Kim, K. S. (2001). Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 30, 19–28.
Sumi, N., Nishioku, T., Takata, F., Matsumoto, J., Watanabe, T., Shuto, H., Yamauchi, A., Dohgu, S., and Kataoka, Y. (2010). Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell. Mol. Neurobiol. 30, 247–253.
Tahir, S. A., Park, S., and Thompson, T. C. (2009). Caveolin-1 regulates VEGF-stimulated angiogenic activities in prostate cancer and endothelial cells. Cancer Biol. Ther. 8, 2286–2296.
Vaz, A. R., Delgado-Esteban, M., Brito, M. A., Bolaños, J. P., Brites, D., and Almeida, A. (2010). Bilirubin selectively inhibits cytochrome c oxidase activity and induces apoptosis in immature cortical neurons: assessment of the protective effects of glycoursodeoxycholic acid. J. Neurochem. 112, 56–65.
Veszelka, S., Pásztói, M., Farkas, A. E., Krizbai, I., Ngo, T. K., Niwa, M., Ábrahám, C. S., and Deli, M. A. (2007). Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem. Int. 50, 219–228.
Wang, P., Liu, Y., Shang, X., and Xue, Y. (2011). CRM197-induced blood-brain barrier permeability increase is mediated by upregulation of caveolin-1 protein. J. Mol. Neurosci. 43, 485–492.
Wang, P., Xue, Y., Shang, X., and Liu, Y. (2010). Diphtheria toxin mutant CRM197-mediated transcytosis across blood-brain barrier in vitro. Cell. Mol. Neurobiol. 30, 717–725.
Weiss, N., Miller, F., Cazaubon, S., and Couraud, P. O. (2009). The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 1788, 842–857.
Wennberg, R. P. (2000). The blood-brain barrier and bilirubin encephalopathy. Cell. Mol. Neurobiol. 20, 97–109.
Wilhelm, I., Fazakas, C., and Krizbai, I. A. (2011). In vitro models of the blood-brain barrier. Acta Neurobiol. Exp. 71, 113–128.
Wispelwey, B., Lesse, A. J., Hansen, E. J., and Scheld, W. M. (1988). Haemophilus influenzae lipopolysaccharide-induced blood brain barrier permeability during experimental meningitis in the rat. J. Clin. Invest. 82, 1339–1346.
Xia, C. Y., Zhang, Z., Xue, Y. X., Wang, P., and Liu, Y. H. (2009). Mechanisms of the increase in the permeability of the blood-tumor barrier obtained by combining low-frequency ultrasound irradiation with small-dose bradykinin. J. Neurooncol. 94, 41–50.
Young, A. R., Narita, M., Ferreira, M., Kirschner, K., Sadaie, M., Darot, J. F., Tavare, S., Arakawa, S., Shimizu, S., and Watt, F. M. (2009). Autophagy mediates the mitotic senescence transition. Genes Dev. 23, 798–803.
Zhang, L., Zhou, F., van Laar, T., Zhang, J., van Dam, H., and Ten Dijke, P. (2011). Fas-associated factor 1 antagonizes Wnt signaling by promoting β-catenin degradation. Mol. Biol. Cell 22, 1617–1624.
Zhong, Y., Smart, E. J., Weksler, B., Couraud, P. O., Hennig, B., and Toborek, M. (2008). Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J. Neurosci. 28, 7788–7796.
Keywords: blood-brain barrier, caveolae, endothelial cells, integrity, intercellular junctions, unconjugated bilirubin, VEGF, VEGFR-2
Citation: Palmela I, Sasaki H, Cardoso FL, Moutinho M, Kim KS, Brites D and Brito MA (2012) Time-dependent dual effects of high levels of unconjugated bilirubin on the human blood-brain barrier lining. Front. Cell. Neurosci. 6:22. doi: 10.3389/fncel.2012.00022
Received: 13 January 2012; Accepted: 23 April 2012;
Published online: 10 May 2012.
Edited by:
Joana A. Palha, Universidade do Minho - Escola de Ciências da Saúde (ECS/UM), PortugalReviewed by:
Stefan H. Heinemann, Friedrich-Schiller-Universität, GermanySilvia Gazzin, Italian Liver Foundation (Fondazione Italiana Fegato), Italy
Copyright: © 2012 Palmela, Sasaki, Cardoso, Moutinho, Kim, Brites and Brito. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Maria A. Brito, Faculdade de Farmácia da Universidade de Lisboa, Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Avenida Professor Gama Pinto, Lisbon 1649-003, Portugal. e-mail:YWJyaXRvQGZmLnVsLnB0