

- 1School of Anatomy, Physiology and Human Biology, The University of Western Australia, Crawley, WA, Australia
- 2Netherlands Institute for Brain Research, Meibergdreef, Amsterdam, Netherlands
The Class 3 Semaphorins (Sema3s) are a sub-family of proteins whose known biological roles are varied and growing. The mechanism of action of the Sema3s requires binding to transmembrane receptors that comprise heteromeric complexes of Neuropilins, Plexins and cell adhesion molecules (CAMs). However, knowledge of the receptor components of the Sema3s remains incomplete, and there may be receptor components which are as yet undiscovered. The receptor complexes of the Sema3s share receptor components with each other, and it is the specific combination of these components within a heteromeric complex that is thought to give rise to selective binding and signalling for individual Sema3s. This crosstalk makes it experimentally difficult to define a single holoreceptor for each Sema3. Furthermore, the receptor composition for a given Sema3 may differ between cell types, and change as a function of developmental state or pathological situation. Nevertheless, there are at least some known differences in the constitutive structure of the receptors for the Sema3s. For example in neural cells, Sema3a and Sema3f signal through different Neuropilins (Nrp1 and Nrp2 respectively) and L1cam only appears important for Sema3a signaling, while Nrcam forms a complex with Nrp2. Further complexity arises from crosstalk of other families of ligands (e.g., VEGF) with Sema3 receptor components. Thus the Sema3s, which have been shown as antagonists for each other, can also act as antagonists for other families of molecules. This review compiles experimental evidence describing the receptor components for the Sema3s, detailing the current state of knowledge of which components are important for signaling of each Sema3 before going on to consider possible future directions for the field.
Introduction
The Class 3 Semaphorins (Sema3s) were first discovered as axon guidance molecules (Kolodkin et al., 1992; Luo et al., 1993), and in vertebrates are the only secreted members of the Semaphorin family (Semaphorin Nomenclature Committee, 1999). The known Sema3s consist of Sema3a through Sema3g (Kolodkin et al., 1993; Luo et al., 1993, 1995; Püschel et al., 1995; Roche et al., 1996; Sekido et al., 1996; Xiang et al., 1996; Feiner et al., 1997; Stevens and Halloran, 2005; Taniguchi et al., 2005), and their known physiological and pathological functions have expanded to include axon attraction and repulsion, apoptosis, cell migration, growth cone collapse, immune response, organogenesis, tumour suppression and promotion, and vasculature development (Yazdani and Terman, 2006; Roth et al., 2009; Takegahara and Kumanogoh, 2010; Staton, 2011; Sakurai et al., 2012; Takamatsu and Kumanogoh, 2012). Vital to our understanding of these functions of the Sema3s is our understanding of their receptors. However, our knowledge of the composition of the holoreceptors for the Sema3s is far from complete (Raper, 2000), and we lack a recent detailed and comprehensive review of what is known.
In this review we present the current state of knowledge on Sema3 receptors, deduced from bioassays and biochemical and in vivo analyses, with particular emphasis on neural cells. It is our hope that this review helps to shed light on those areas most in need of further research.
General Structure of a Class 3 Semaphorin Receptor
The receptors for the Sema3s are heterocomplexes of receptor subunits, with significant overlap between different Sema3 holoreceptors (Feiner et al., 1997; Takahashi et al., 1998; Rohm et al., 2000). The molecules first identified as receptors for the Sema3s were Neuropilin 1 and 2 (Nrp1, Nrp2), independently reported by two laboratories in 1997 (Chen et al., 1997; He and Tessier-Lavigne, 1997; Kolodkin et al., 1997). Both Nrp1 and Nrp2 were found to be essential for Sema3 signal transduction, but the specificity of Sema3 signaling could not be attributed to either Neuropilin alone (Chen et al., 1997; Takahashi et al., 1998). Indeed, even before the discovery that the Neuropilins were essential for Sema3 signaling, it was inferred from the structure of Nrp1 and its expression profile in the developing mouse nervous system that Nrp1 was likely heterophilic (Kawakami et al., 1996). Also, the cytoplasmic domains of Nrp1 were found to be not required for Sema3 signaling (Nakamura et al., 1998; Renzi et al., 1999), indicating that Neuropilins act in concert with other receptor co-receptors which transduce the extracellular signals to the intracellular signaling pathways.
Class A Plexins (PlexinAs) are the main co-receptors for the Sema3s, and were the first identified (Takahashi et al., 1999; Tamagnone et al., 1999). These initial studies found that PlexinAs associate with the Neuropilins and that this association is important for signal transduction of the Sema3s. Further studies discovered a number of other co-receptors for the Sema3s: L1cam (Castellani et al., 2000), Nrcam (Falk et al., 2005), Plxnb1 (Usui et al., 2003), and Plxnd1 (Gitler et al., 2004; Gu et al., 2005; Chauvet et al., 2007). A summary of known receptor components for each of the Sema3s is presented in Table 1.
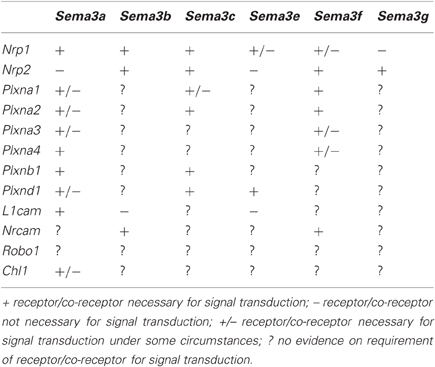
Table 1. Known receptor-ligand interactions.
Reviews of the general structure and signaling of the Sema3 holoreceptors have been published previously (Pasterkamp and Kolodkin, 2003; Geretti et al., 2008; Pellet-Many et al., 2008; Zhou et al., 2008; Yoshida, 2012), consequently these concepts are only briefly revisited here. With the exception of Sema3e, all Sema3 receptors require a Neuropilin to act as the binding site for the Sema3 ligand. The binding of the Sema3 ligands to the Neuropilins depends on their N-terminus Sema sequence, and a 70 amino acid stretch within that sequence determines specificity (Koppel et al., 1997). The receptors for the Sema3s are multimeric, with varying numbers of associated Plexins or cell adhesion molecules (CAMs) providing the intracellular signaling mechanics. Sema3e differs in that it is able to bind directly to Plxnd1 in the absence of a Neuropilin (Gu et al., 2005). The Plexins (Class A, with the exception of Plxnd1 for Sema3e) and CAMs (L1cam and Nrcam) cis-interact with Nrp1 and Nrp2 through their transmembrane, and extracellular domains (Tamagnone et al., 1999; Rohm et al., 2000; Takahashi and Strittmatter, 2001; Castellani et al., 2002; Roth et al., 2008).
Methodologies used to Investigate Receptor Compositions
A number of in vitro methods have been used to investigate the function of the Sema3s: axon repulsion/attraction assays, grown cone collapse assays, COS cell collapse, co-immunoprecipitation, and ligand binding assays. To aid the reader in understanding how deductions about the makeup of Sema3 receptor complexes have been made, an overview of the methods used to elucidate the makeup of the Sema3 receptor complexes is given in Figure 1, and a brief description of these assays is given below.
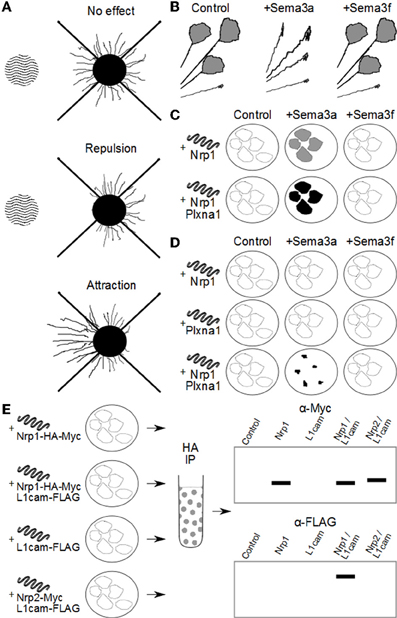
Figure 1. Methods used to investigate Sema3 receptor components. (A) Attraction/repulsion assay. The area of wavy lines represents aggregates of COS/HEK-293 cells over-expressing a Sema3. To the right is neuronal tissue with outgrowth of neurites. In this assay, the average or total lengths of neurites in the proximal and distal quadrants of the neuronal tissue (with respect to the COS/HEK-293 cell aggregates) are compared to quantify the amount of attraction or repulsion. (B) Neurites in vitro with active growth cones are exposed to exogenous Sema3 or vehicle. Changes in the percentage of active to collapsed growth cones are used to determine the biological activity of the Sema3s on different neuronal types. In this example, neurites are found to collapse in the presence of Sema3a but not Sema3f. (C) COS/HEK-293 cells are transfected to ectopically express one or more putative Sema3 receptor components. These cells are then exposed to exogenous recombinant Sema3 which includes an artificial conjugate, most often alkaline phosphatase. After incubation at 4°C the cultures are washed and stained either by immunocytochemistry, or direct application of a chromagen that interacts with the artificial conjugate (for example NBT/BCIP with alkaline phosphatase). The intensity of staining is proportional to the binding kinetics of the recombinant Sema3 to the putative receptor components and/or complexes, and the dissociation constant can be derived by Scatchard analysis. In this way researchers can compare the relative affinity for the Sema3s to putative receptor components and complexes. For example, in this diagram, Sema3a, but not Sema3f binds to cells expressing Nrp1, and Sema3a binds more to cells expressing both Nrp1 and Plxna1, than Nrp1 alone. (D) COS cells engineered to over-express one or more putative Sema3 receptor components collapse on exposure to exogenous Sema3, if those receptor components form a functional receptor for that Sema3. In the example here, Nrp1 or Plxna1 can transduce a Sema3a signal when expressed in concert, but not individually. Furthermore, Nrp1 and Plxna1 cannot transduce a Sema3f signal, either individually or together. (E) Co-immunoprecipitation experiments allow researchers to investigate whether two or more receptor components interact either in vivo, or in vitro. In this example HEK-293 cells are engineered to ectopically express combinations of either Nrp1 or Nrp2, and L1cam. The recombinant proteins are conjugated to artificial epitopes which allows for their selective immunoprecipitation, in this case by the HA tag on the Nrp1/Nrp2. Once selected for, the proteins are analysed by western blot for any other proteins of interest that were bound to Nrp1/Nrp2 in the cell. In this example L1cam is analysed by the immunoblot, and appears associated with only Nrp1, and not Nrp2. Importantly the controls reveal that the L1cam signal is not detected in cells expressing L1cam alone, confirming the positive signal in Nrp1/L1cam cultures as due to Nrp1 and L1cam interaction.
Having first been discovered as repulsive axon guidance molecules, it was natural for investigators to use this function to dissect the receptor makeup for the Sema3s. Neural tissue explants [usually embryonic dorsal root glia (DRG)] are grown in a collagen matrix adjacent to cells (normally HEK-293 or COS cells) transfected to over-express a specific Sema3. The collagen matrix allows the formation of a gradient of Sema3s from the transfected cells toward the DRG explants. The most common method for quantifying the repulsion or attraction this bioassay is by measuring the length of neurites exiting the explant in the quadrants proximal and distal to the Sema3 source. Average neurite lengths are then either compared directly, or represented as a ratio of proximal/distal neurite lengths (Messersmith et al., 1995; Kolodkin et al., 1997).
Another bioassay enabling analysis of neuronal response to Sema3s is a growth cone collapse assay. Explants are grown in vitro and then exposed to exogenous Sema3 ligand for up to one hour before fixation and staining. The number of collapsed versus non-collapsed growth cones is then counted and compared by statistical means (Kapfhammer et al., 2007). It is important to note that this method captures only inhibitory effects on growth cones, and may miss any attractant or growth promoting effects (Campbell et al., 2001).
Another technology that has proved useful for investigating the Sema3 receptors is to engineer COS and HEK-293 cells to over express different combinations of Sema3 receptor components. When COS cells express a minimum functional Sema3 receptor, they collapse if presented with that Sema3 (Takahashi et al., 1999), thus elucidating necessary components for each Sema3 receptor. Further, COS and HEK-293 cells that over express combinations of receptor components have allowed researchers to study the interactions of these components with each other by co-immunoprecipitation. This technique is especially important, as until recently reliable commercial antibodies against specific Sema3s and their receptor components were scarce, making co-immunoprecipitation from animal tissue difficult at best. Ectopic expression of Sema3 receptor subunits also allows measurement of binding affinities of the Sema3s to different permutations of the general Sema3 receptor complex. For example, recombinant Sema3 ligands fused to a reporter sequence (for example FLAG/Myc/alkaline phosphatase) are exposed to engineered COS/HEK-293 cells, level of binding then visualized via the reporter sequence, and binding affinities calculated by Scatchard analysis (Chen et al., 1997; He and Tessier-Lavigne, 1997).
Recombinant Sema3 ligands conjugated to reporter sequences have also been used to map binding to different tissues both in vitro and ex vivo. Analysis of the expression of the different Sema3 receptor constituents in different tissues has then allowed deduction of the necessity of individual receptor components in each Sema3 holoreceptor.
The above methods have been combined with various models to study the makeup of the Sema3 receptors: genomic knock-out/knock-in, gene knock-downs, protein over expression, functional blocking antibodies, and tissues that are known to express different combinations of the Sema3 receptor components (for example sympathetic neurons versus sensory neurons). In addition to the above, researchers have also carefully analysed the in vivo phenotypes of single and double knock-out mutant animals.
Sema3a
Sema3a was the first Sema3 to be discovered (Kolodkin et al., 1992; Luo et al., 1993), and has been the most widely studied. As a result, knowledge of the Sema3a receptor is the most complete of all the Sema3s. Nonetheless, the make-up of the Sema3a receptor remains somewhat equivocal.
Neuropilins
Nrp1
Nrp1 was identified as a receptor for Sema3a by screening for Sema3a-AP binding of COS cells transfected with a cDNA expression library from E14 rat dorsal root ganglia (DRG, sensory neurons that bind recombinant Sema3a) (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997). The same studies confirmed Nrp1 as a candidate receptor protein for Sema3a by demonstrating that the proteins co-immunoprecipitate, and that Sema3a-AP binds to exogenously expressed recombinant Nrp1 in COS cells.
Additional evidence for Nrp1 as a receptor for Sema3a came from studies using functional blocking antibodies against Nrp1. Anti-Nrp1 antibodies ablate the axon repulsion and growth cone collapse effect of recombinant Sema3a on E14 rat DRGs (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997) and sympathetic neurons (Chen et al., 1998). Also, anti-Nrp1 antibodies abolish Sema3a mediated axon repulsion of mouse cortical neurons (Castellani et al., 2000). Similarly, Npr1Sema knock-out (a targeted deletion in the Sema region of Nrp1, removing Sema3 binding capability but leaving VEGF binding intact) in mice, causes Sema3a insensitivity in embryonic DRG neurons (Kitsukawa et al., 1997; Gu et al., 2003).
Corresponding evidence that Nrp1 is a receptor for Sema3a comes from over expression studies; Sema3a insensitive chick E8 retinal neurons become sensitive to Sema3a mediated growth cone collapse after they are transduced with recombinant Nrp1 (Nakamura et al., 1998). Furthermore, in Xenopus, retinal neurons become responsive to Sema3a induced growth cone collapse during development, concomitant with expression of Nrp1, and earlier sensitivity can be conferred using transfection to ectopically express Nrp1 (Campbell et al., 2001).
Nrp2
Initially it was shown that, unlike Nrp1, Nrp2 is unable to bind Sema3a in vitro (Chen et al., 1997; Takahashi et al., 1998). Also, soluble Nrp2-Fc abolishes Sema3c and Sema3f induced axon repulsion of embryonic rat sympathetic neurons in vitro, but has no effect on Sema3a induced repulsion in the same model (Chen et al., 1998). However, more recently Sema3a has been found to bind to COS cells expressing Nrp2 (albeit 1.3 fold less than Nrp1 expressing COS cells), and a functional blocking antibody against Nrp2 removes the chemorepulsive effect of Sema3a on at least one human glioma cell line in vitro (Nasarre et al., 2009). In vivo, Sema3a binding to tissue is abolished in areas such as the olfactory bulb in Nrp1Sema knockouts, but is still present in other tissues (Cariboni et al., 2011). Indeed, Sema3a binding is only completely absent in Nrp1Sema/Nrp2 double knockouts, and both Neuropilins appear to be partially redundant for one another in transducing Sema3a signals in mouse vomeronasal axons (Cariboni et al., 2011). Thus while Nrp1 appears to be an essential component of a functional Sema3a receptor complex, Nrp2 may or may not be necessary, depending on the cell type.
Class a Plexins
COS cells ectopically expressing both Nrp1 and Plxna1 undergo collapse, or rounding up, in the presence of Sema3a, but no such reaction occurs if only Nrp1 or Plxna1 is expressed (Takahashi et al., 1999; Takahashi and Strittmatter, 2001). Moreover, the presence of Plxna1 increases the affinity of Nrp1 for Sema3a in vitro, and Nrp1 and Plxna1 cluster in E7 chick DRG growth cones in response to exogenous Sema3a (Takahashi et al., 1999). In addition, expression of a dominant negative Plxna1 protein (lacking the intracellular domain) abolishes growth cone collapse in response to Sema3a in chick E7 DRG neurons (Takahashi et al., 1999), mouse E12.5 sensory ganglia (Rohm et al., 2000), and Xenopus laevis spinal neurons (Tamagnone et al., 1999). Intriguingly, it has been suggested that the presence of Plxna1 in the Sema3a holoreceptor acts as an adapter for the association of Plxnb1, increasing the possible signaling cascade complexity (Usui et al., 2003).
Similarly, embryonic mouse sensory ganglia lose their sensitivity to exogenous Sema3a in vitro when induced to express ectopic dominant negative Plxna2 protein (Rohm et al., 2000). Furthermore, COS cells expressing both Plxna2 and Nrp1 collapse in the presence of Sema3a, but not if expressing only one of Plxna2 or Nrp1 (Takahashi and Strittmatter, 2001). Finally, co-expression of both Plxna2 and Nrp1 in HEK-293 cells increases their affinity for Sema3a above those expressing Nrp1 alone (Rohm et al., 2000).
However, while both Plxna1 and Plxna2 are important for Sema3a signaling, neither Plxna1 nor Plxna2 appear to be essential components of the Sema3a holoreceptor. In mouse knockout models, the facial nerve of Plxna1 and Plxna2 knockouts does not exhibit the same level of abnormal phenotype as the Sema3a knockout (Schwarz et al., 2008). This indicates some degree of redundancy in the make-up of the full Sema3a receptor, with different components conveying overlapping signaling capabilities.
Such redundancy is particularly marked with Plxna3 and Plxna4 (Schwarz et al., 2008). Plxna3 knockouts have reduced sensitivity to Sema3a mediated axon repulsion and growth cone collapse in mouse embryonic DRG, hippocampal neurons, and embryonic sympathetic neurons (Cheng et al., 2001; Bagri et al., 2003; Yaron et al., 2005). Similarly, Plxna4 knockouts display a reduced sensitivity of mouse embryonic DRG and sympathetic neurons (Suto et al., 2005; Yaron et al., 2005). However, Plxna3/Plxna4 double-knockouts completely abolish sensitivity to Sema3a induced growth cone collapse and axon repulsion (Yaron et al., 2005), strongly suggesting that Plxna3 and Plxna4 are partially redundant for each other within the Sema3a holoreceptor. Further evidence for the reciprocal redundancy of Plxna3 and Plxna4 in the Sema3a receptor is found in the migration of sympathetic neurons, which is disrupted by double, but not single, Plxna3 and Plxna4 knockouts (Waimey et al., 2008). Finally, Plxna3 is required for Sema3a induced neuron cell death, but the absence of Plxna4 also reduces this effect of Sema3a by around 50% (Ben-Zvi et al., 2008).
It appears from the above evidence that Plxna3 and Plxna4 are part of the Sema3a holoreceptor, but partially mutually redundant, probably sharing overlapping signaling mechanisms. Interestingly, co-expression of Plxna3 and Nrp1 is not sufficient to confer sensitivity of COS cells to Sema3a mediated cell collapse, (Takahashi and Strittmatter, 2001), again indicating that whilse Plxna3 is part of the Sema3a holoreceptor, it is not essential. However it must be noted that all experimental avenues have as yet not been exhausted, as illustrated by a very recent report showing that Plxna4 is essential for Sema3a signaling in HUVECs, an endothelial cell line (Kigel et al., 2011).
One other Plexin that has been studied as a putative component of the Sema3a receptor is Plxnd1. Sema3a binding to Nrp1 is enhanced by the presence of Plxnd1, at least in transfected COS cells in vitro (Gitler et al., 2004), indicating a possible role for Plxnd1 in Sema3a signaling. However, there have been no functional studies performed to test this hypothesis.
L1cam
L1cam has been demonstrated to be important for signal transduction within the Sema3a receptor complex (Castellani et al., 2000, 2002, 2004). Mouse cortical and DRG neurites, normally repulsed by Sema3a in co-culture assays, are indifferent to Sema3a when L1cam is genetically knocked out (Castellani et al., 2000; Bechara et al., 2008). Importantly, COS cells over-expressing L1cam and Nrp1 collapse in the presence of Sema3a, indicating that L1cam can transduce the Sema3a signal (Castellani et al., 2004).
Interestingly, L1cam appears to be important for Nrp1, but not Nrp2, signaling. When co-expressed in COS cells, both Nrp1 and L1cam, but not Nrp2 and L1cam, naturally associate, independent of the presence of Sema3a (Castellani et al., 2000). Furthermore, Nrp1 and L1cam from postnatal mouse brain lysates also co-immunoprecipitate, with the extracellular portion of L1cam sufficient for binding of L1cam to Nrp1 (Castellani et al., 2000).
Nevertheless, the presence of L1cam is not necessary for the binding of Sema3a to Nrp1, at least in COS cells in vitro (Castellani et al., 2000). Furthermore, L1cam is not a necessary component of the Sema3a receptor, as shown under a number of circumstances. For example, neuronal apoptosis induced by Sema3a signaling is not affected by the lack of functional L1cam (Ben-Zvi et al., 2008). Therefore, as with other co-receptors for Sema3a, L1cam can transduce Sema3a signals but it is not essential for all Sema3a signaling under all circumstances.
Finally, the effect of the presence of L1cam in the Sema3a receptor is more nuanced than simply enabling/disabling Sema3a signaling. L1cam dependent, Sema3a induced neurite repulsion in vitro is converted into attraction by the addition of L1-Fc, the extracellular domain of L1cam fused with and Fc immunoglobulin fragment (Castellani et al., 2000), and similar extracellular soluble L1cam is present in vivo (Maretzky et al., 2005). Thus the presence of L1cam in the Sema3a receptor complex enables the cell to respond to the levels of both Sema3a and soluble L1cam.
Chl1
Chl1 (Close homologue of L1) associates with Nrp1 both in vivo and in vitro, and is necessary for Sema3a induced embryonic mouse thalamic and cortical neuron growth cone collapse (Wright et al., 2007; Schlatter et al., 2008). Interestingly, Chl1 mediated Sema3a signaling is dependent on a juxtamembrane region of Chl1's cytoplasmic domain, conserved with L1cam (Wright et al., 2007). However, the number of studies of Chl1 and Sema3 signaling are limited, and it is not possible to say whether Chl1 is an essential receptor component generally or only has a role in specific cell types.
Robo1
To date there has been one report of Robo1 acting as a co-receptor for Sema3a (Hernández-Miranda et al., 2011). Co-immunoprecipitation from embryonic mouse forebrain, and COS cells over-expressing Nrp1, Nrp2, Plxna1, Plxna4, and Robo1 demonstrated that Robo1 can form a complex with Nrp1, but not Nrp2, Plxna1, or Plxna4 individually. Further evidence was gathered from a covasphere aggregation assay [immunofluorescent beads (green or red) coated with relevant Fc proteins; heterophilic aggregates are yellow, homophilic aggregates are green or red], where Robo1 interacted with Nrp1, but not Nrp2, Plxna1, or Plxna4. Also, co-immunoprecipitation of Robo1 with Plxna1, and Plxna4 from embryonic mouse forebrain lysates indicates that it can bind to Nrp1 in complex with Plxna1 and Plxna4, a complex that would constitute a functional Sema3a receptor.
This binding of Robo1 to Nrp1 depends, at least in part, on the first two Ig domains in Robo1, but not the final three. Also, by using the same covasphere assay it was found that Robo1 cannot bind Sema3a directly, and Sema3a did not bind to COS cells ectopically expressing Robo1 alone. It is still unclear, however, whether Robo1 is part of the Sema3a receptor complex. The chemorepulsive effects of Sema3a and Sema3f on both mouse embryonic medial ganglionic eminence GN11 cells were reduced in Robo1 knockout animals, despite the fact that Robo1 only interacts with Nrp1 and not Nrp2, an essential component of the Sema3f receptor complex. Indeed it appears that the effect of Robo1 knockout on Sema3a and Sema3f induced chemorepulsion is the concomitant down-regulation of Nrp1 and Plxna1 expression. The mechanism by which Robo1 expression modulates Nrp1 and Plxna1 expression is as yet unknown, however, this genetic interaction is in itself an intriguing new facet of Sema3 receptor biology.
Sema3b
Unlike Sema3a, Sema3b binds to both Nrp1 and Nrp2 following ectopic expression in COS cells in vitro (Takahashi et al., 1998). Soluble Nrp2-Fc binds to mouse sub-ventricular zone (SVZ) regions expressing Sema3b, and this binding is markedly reduced or extinguished in Sema3b knockouts (Falk et al., 2005). Both anti-Nrp2 antibodies and exogenous soluble Nrp2 (Nrp2-Fc) abolished Sema3b induced attraction of mouse neonatal cortical neurons (Falk et al., 2005). Also, chick embryonic DRG neurons that are normally insensitive to Sema3b induced growth cone collapse in vitro become sensitized upon transduction with recombinant Nrp2 (Takahashi et al., 1998). While Sema3b can bind both Nrp1 and Nrp2, and it is known that Nrp2 is essential for Sema3b signal transduction, it remains unclear whether the presence of Nrp1 is necessary even though Sema3b can bind to Nrp1 in the absence of Nrp2, as demonstrated by its competitive antagonism to Sema3a in embryonic chick RGCs (which express Nrp1 but not Nrp2) (Takahashi et al., 1998).
Similarly, there is no evidence yet for the Plexins as constituents of the Sema3b receptor complex. However several IgCAMs have been investigated, with Nrcam but not Tag-1, Contactin, or L1cam able to bind Nrp2 when co-expressed in HEK-293 in vitro (Castellani et al., 2000; Falk et al., 2005). L1cam is not required for Sema3b signal transduction in neonatal mouse cortical neurons (Castellani et al., 2000), and Nrcam, either in its soluble or membrane bound form, cannot bind Sema3b directly (Falk et al., 2005) However, Nrcam is essential for neonatal mouse cortical neuron sensitivity to Sema3b (Falk et al., 2005). Further, functional blocking antibody against Nrcam abolishes Sema3b mediated growth cone collapse and axon attraction of neonatal mouse cortical neurons (Falk et al., 2005). Intriguingly, Nrcam can mediate neuronal sensitivity to Sema3b by reducing Calpain1 proteolytic activity on Plxna1 (Nawabi et al., 2010).
Lastly, there is no direct evidence for Robo1 as a constituent of the Sema3b holoreceptor. However, Robo1 does co-immunoprecipitate with Nrp1, Nrp2, Plxna1, and Plxna4, despite only being able to bind Nrp1 directly (Hernández-Miranda et al., 2011), This indicates that Robo1 is at least capable of forming a complex with a Nrp1/Nrp2 heterodimers, leaving open the possibility that Sema3b signals through a multimeric receptor with a composition that is as complex as the Sema3a receptor.
Sema3c
Similar to Sema3b, Sema3c binds to both Nrp1 and Nrp2 ectopically expressed in COS and HEK-293 cells in vitro (Chen et al., 1997; Feiner et al., 1997; Takahashi et al., 1998; Rohm et al., 2000). Expression of a dominant negative Nrp1 receptor (lacking extracellular domain) in embryonic chick sympathetic neurons abrogates Sema3c induced growth cone collapse (Renzi et al., 1999), and Sema3c acts as a competitive antagonist to Sema3a in chick embryonic retinal ganglion cells that express Nrp1 but not Npr2 (Takahashi et al., 1998). Also, COS cells ectopically expressing Plxna1/Plxna2/Plxna3 and either Nrp1 or Nrp2 do not collapse in the presence of exogenous Sema3c, but do collapse in the presence of either Sema3a or Sema3f (Takahashi et al., 1999; Takahashi and Strittmatter, 2001). Furthermore when embryonic chick DRG, which normally express Nrp1 but not Nrp2, are transduced to ectopically express Nrp2 they become responsive to Sema3c induced growth cone collapse (Takahashi et al., 1998). Thus the holoreceptor for Sema3c requires both Nrp1 and Nrp2 to be present.
There is little evidence that Plxna1 is essential for Sema3c signal transduction. Unlike for Sema3a and Sema3f, Plxna1 co-expression in COS cells with either Nrp1 or Nrp2 does not increase the binding affinity for Sema3c, and there is no collapse response (Takahashi et al., 1999; Rohm et al., 2000; Gitler et al., 2004). Conversely, there is some evidence that Plxna2 is important for Sema3c signaling; Plxna2 positive mouse cardiac neural crest cells show aberrant migration in Sema3c knockouts (Brown et al., 2001). However, the same line of evidence suggests that Nrp2 is not important for the migration of mouse cardiac neural crest cells (Chen et al., 1997), even though it has been shown that Nrp2 is essential for Sema3c signaling, at least in neurons (see above). Furthermore, Plxna2 co-expression with Nrp2 in COS cells does not increase the affinity of Sema3c binding above expression of Nrp2 alone (Rohm et al., 2000). It must be noted that binding analyses conducted so far have studied Plxna1 and Plxna2 in the presence of either Nrp1 or Nrp2, but not Nrp1 and Nrp2 together. It is possible that Plxna1 and Plxna2 can change the binding affinity of Sema3c to its receptor when both Nrp1 and Nrp2 are present, especially as both are essential for functional transduction of the Sema3c signal.
Unlike Plxna1 and Plxna2, the interaction of Plxnd1 with either Nrp1 or Nrp2 does increase their binding affinity Sema3c (Gitler et al., 2004). However, Plxnd1 knockouts show defects in the cardiac outflow tract that are remarkably similar to those seen in Sema3c knockouts (Gitler et al., 2004). While, unlike Sema3e, Sema3c cannot bind directly to Plxnd1 in vitro (Gu et al., 2005), both Nrp1 and Nrp2 bind to Plxnd1 as demonstrated by co-immunoprecipitation (Gitler et al., 2004; Chauvet et al., 2007), and Plxnd1 may form a part of the Sema3c receptor complex via this interaction.
As with Sema3b, there is no direct evidence that Robo1 is a constituent of the Sema3b holoreceptor. However, because Robo1 co-immunoprecipitates with receptor components of the Sema3c receptor it is possible that Robo1 forms a complex within the Sema3c holoreceptor (Hernández-Miranda et al., 2011).
Sema3d
Little is known about the receptor for Sema3d. What is known is that Sema3d can bind to Nrp1 ectopically expressed by COS cells in vitro (Feiner et al., 1997). There is also some evidence in vivo that Nrp1 is important for Sema3d signaling in zebrafish, as Nrp1 knockdowns phenocopy Sema3d knockdowns; losing axon repulsion of axons from the nucleus of the medial longitudinal fasciculus (Wolman et al., 2004). The same study suggested that Nrp2 may be a constituent of the Sema3d receptor, as knock-down of Sema3d or Nrp2 showed a similar phenotype. It was also initially thought that Sema3d influenced fasciculation of axons in the nucleus of the medial longitudinal fasciculus through a receptor incorporating Nrp1, but it was later revealed that fasciculation is influenced by Sema3d modulating expression of L1cam on the axons (Wolman et al., 2007).
Sema3e
Unlike all other Sema3s, Sema3e can bind to a Plexin, Plxnd1, directly and independently of the Neuropilins. Also, exogenous Sema3e collapses COS cells expressing ectopic Plxnd1 (Gu et al., 2005). Further in vitro evidence implicating Plxnd1 as an essential component of the Sema3e receptor is that Sema3e mediated axon growth inhibition and growth cone collapse of embryonic mouse cortical neurons is abolished in both Plxnd1 knockout and knockdown models (Chauvet et al., 2007). Furthermore, Plxnd1 expression is essential for Sema3e mediated modelling of chick vasculature both in vivo, and in vitro (Gu et al., 2005), and observed metastatic activity of Sema3e is dependent on activation of Plxnd1 associated ErbB2/Neu oncogenic kinase (Casazza et al., 2010).
While Sema3e is able to exert a biological effect independent of Neuropilins, there is evidence that Nrp1 is able to modulate Sema3e signaling, because Sema3e induced growth cone collapse of embryonic mouse DRG is inhibited by the addition of anti-Nrp1 antibody (Miyazaki et al., 1999). Similarly, Sema3e induced neurite growth from subicular neurons in vitro is abolished by treatment with anti-Nrp1 functional blocking antibody, and by knockdown of Nrp1 (Chauvet et al., 2007). Interestingly, after Nrp1 gain of function, cortical neurons convert their response to Sema3e in vitro, from neurite inhibition to neurite extension, and this sensitivity is still able to be abolished by knockdown of Plxnd1 (Chauvet et al., 2007). The presence of Nrp1 in the Sema3e receptor complex may “gate” cellular response between attractive and repulsive, through interaction of the extracellular domains of Plxnd1 and Nrp2 (Chauvet et al., 2007).
Sema3f
Sema3f is one of the most studied of the Sema3s and its receptor complex is one of the best understood.
Neuropilins
Sema3f binds to COS cells ectopically expressing Nrp1, with a similar affinity as Sema3a binding to Nrp1 (Chen et al., 1997). In vitro, exogenous Sema3f inhibits cell attachment and spreading in the breast cancer cell line MCF7, a line that expresses Nrp1 but not Nrp2; this inhibition is blocked by the addition of anti-Nrp1 functional blocking antibody (Nasarre et al., 2003). However, exogenous Sema3f is unable to cause contraction of COS cells in vitro expressing Nrp1 or Nrp1 and one of Plxna1/Plxna2/Plxna3 (Takahashi and Strittmatter, 2001), raising the possibility that Sema3f signaling through Nrp1 requires the presence of an as yet unidentified co-receptor.
Intriguingly C100, a breast cancer cell line that expresses both Nrp1 and Nrp2, responds to exogenous Sema3f with inhibition of cell spreading, and this response is insensitive to the addition of anti-Nrp1 functional blocking antibody (Nasarre et al., 2003). Similarly in embryonic rat DRG, which also express both Nrp1 and Nrp2, Sema3f induced axon repulsion is unaffected by addition of anti-Nrp1 functional blocking antibody, even though the same antibody abolishes the repulsive effects of Sema3c and Sema3a (Chen et al., 1998). Perhaps, at least in some cell types, when both Nrp1 and Nrp2 are present, the Sema3f receptor is preferentially composed of Nrp2 over Nrp1. Indeed, the affinity for Sema3f for Nrp2 is around 10 fold greater than for Nrp1 (Chen et al., 1997).
It follows then that Nrp1 may not be an essential component of the Sema3f receptor complex, as long as Nrp2 is also present. This hypothesis is supported by the observation that embryonic chick sympathetic neurons, which normally express both Nrp1 and Nrp2, undergo growth cone collapse in the presence of exogenous Sema3f, and over expression of a dominant-negative Nrp1 receptor in these neurons does not remove their Sema3f mediated growth cone collapse, despite the same model abrogating Sema3a mediated growth cone collapse (Renzi et al., 1999). The obvious inference is that Nrp1 is an essential component of the Sema3a but not the Sema3f holoreceptor.
Sema3f binds to COS cells that ectopically express Nrp2 (Chen et al., 1997), and exogenous Sema3f induces collapse of COS cells when they express both Nrp2 and Plxna1 (Takahashi et al., 1999). Dopaminergic axons grown in vitro from the ventral tegmental area of mice are either repelled by, or attracted to, Sema3f depending on age, an effect which is dependent on Nrp2 (Kolk et al., 2009). Anti-Nrp2 functional blocking antibody also abolishes Sema3f induced growth cone collapse of embryonic rat sympathetic neurons, and axon repulsion in neonatal mouse cortical neurons (Chen et al., 1997; Falk et al., 2005).
Knockout models reinforce the importance of Nrp2 in Sema3f signaling. Embryonic mouse neural crest cells avoid focal Sema3f in vitro, but this effect is mollified in neural crest cells from Nrp2 knockout mice (Gammill et al., 2006). In vivo, Sema3f knockout models show aberrant growth in multiple Nrp2 expressing tracts in the mouse brain (Sahay et al., 2003). Similarly, Nrp2 and Sema3f knockouts demonstrate that migration of neural crest cells during mouse development is dependent on Sema3f signaling through Nrp2 (Gammill et al., 2006). Knockout models also demonstrate that Sema3f is important for the development of dopaminergic neurons in the mouse meso-diencephalon, and that this development is dependent on Nrp2 signaling (Kolk et al., 2009). In the olfactory bulb, Sema3f signaling in olfactory sensory neurons is dependent on their expression of Nrp2 (Takeuchi et al., 2010), and Sema3f and Nrp2 knockouts phenocopy each other (Cloutier et al., 2002, 2004).
Class A Plexins
There is also evidence supporting the role of Plxna1 in the Sema3f receptor; COS cells expressing both Nrp2 and Plxna1 bind Sema3f with greater affinity than those expressing Nrp2 alone (Plxna1 alone is unable to bind Sema3f) (Takahashi et al., 1999). Furthermore Nrp2 alone is insufficient to signal COS cell collapse in response to Sema3f, but Nrp2 and Plxna1 expression combined is able to transduce this signal (Takahashi et al., 1999; Takahashi and Strittmatter, 2001). Similarly, COS cells transfected with both Nrp2 and Plxna2, but not Nrp2 alone, collapse in the presence of exogenous Sema3f (Takahashi and Strittmatter, 2001). Consequently, at least in vitro, Plxna1 and Plxna2 are parts of a functioning Sema3f receptor complex.
Plxna3 also appears to be a part of the Sema3f holoreceptor. When Plxna3 is knocked out, mouse sympathetic and hippocampal neurons lose sensitivity to the repulsive effect of Sema3f in vitro (Cheng et al., 2001; Yaron et al., 2005; Waimey et al., 2008). Knockouts of Plxna3 and Sema3f are reported to phenocopy the defects of each other in the axon guidance of facial branchiomotor neurons (Schwarz et al., 2008), and in the olfactory bulb mosaic knockouts of Plxna3 disrupt Nrp2 and Sema3f dependent olfactory sensory neuron innervation (Takeuchi et al., 2010). In culture, CA1 pyramidal neurons respond to exogenous Sema3f with axon branch retraction, but this effect is absent in the same neurons from Plxna3 knockout mice (Bagri et al., 2003). However, Nrp2 and Plxna3 co-expression in COS cells is insufficient to generate a Sema3f mediated cell contraction response (Takahashi and Strittmatter, 2001), indicating that Plxna3 requires other co-receptors to form a functional Sema3f receptor.
Similar to the Sema3a receptor, there is evidence that both Plxna3 and Plxna4 are partially redundant constituents in the Sema3f holoreceptor. Embryonic mouse sympathetic neurons lose their migratory responsiveness to a gradient of Sema3f in vitro when both Plxna3 and Plxna4 are knocked out, but not if only one or the other is absent (Waimey et al., 2008). However, it appears that Plxna3 and Plxna4 do not coincide within the same Sema3f receptor complex, because co-immunoprecipitation studies demonstrate that they do not associate with one another, and this is unaffected by the presence of either Nrp1 or Nrp2 (Waimey et al., 2008). Thus Plxna3 and Plxna4 appear redundant for each other in Sema3f signaling, but as separate receptor complexes, and the preferential receptor complex is one that contains Plxna3 (Yaron et al., 2005; Schwarz et al., 2008). Indeed, some neurons may not form the Plxna4 constituting Sema3f receptor. For example mouse sympathetic neurons express both Plxna3 and Plxna4, however their Sema3f mediated growth cone collapse response is only abrogated by Plxna3, but not Plxna4, knockout (Cheng et al., 2001; Suto et al., 2005; Yaron et al., 2005).
Nrcam
Finally, there is also evidence that Nrcam mediates Sema3f signaling. Mouse piriform cortical neuron growth cones collapse in the presence of Sema3f in vitro but this effect is abolished in the presence of soluble Nrcam-Fc or anti-Nrcam antibody (Falk et al., 2005). Nrcam associates with Nrp2, and when Nrcam is knocked out, thalamic neurons lose their sensitivity to Sema3f (Demyanenko et al., 2011).
Sema3g
Sema3g is the most recently discovered member of the Sema3s (Stevens and Halloran, 2005; Taniguchi et al., 2005). While there is growing evidence for the importance of Sema3g in cancer biology, cell migration, and axon guidance (Taniguchi et al., 2005; Bron et al., 2007; Karayan-Tapon et al., 2008; Kigel et al., 2008; Neufeld and Kessler, 2008), relatively little is known about its signaling mechanisms.
Taniguchi et al. (2005) reported that, Sema3g binds to COS cells expressing Nrp2, but not those expressing Nrp1. In the same study the researchers found that exogenous Sema3g repelled sympathetic axons which express Nrp2, but had no attractive/repulsive effect on dorsal root ganglion axons that don't express Nrp2. It can be inferred from these initial studies that Sema3g acts through Nrp2, but not Nrp1.
Discussion
Our current understanding of the receptors for the Sema3s has come a long way since the first discovery of Nrp1 and Nrp2. However, there is still a great deal that remains unclear. It is evident from the data reviewed above that there is not a single holoreceptor for each Sema3, and the make-up of a particular receptor complex depends on cell type, and perhaps also on the phenotypic status of the cell.
The methods used in the studies detailed in this review remain useful for further research into the specific receptor subunits that make up the receptor complexes for each Sema3. However, it is also important to understand the physical basis of the interactions between the Sema3 ligands, receptors, and co-receptors. This could contribute to our understanding of why different combinations of receptor components are necessary for binding and signaling of each Sema3. An example of how our knowledge of the physical interaction of different receptor subunits has led to a better understanding of how a Sema3 receptor complex functions is available from studies of the Sema3a/Nrp1/Plxna1 complex.
Takahashi and Strittmatter (2001) put forward a model where Plxna1 is constitutively inhibited by its Sema domain, and binding of both Nrp1 and Sema3a together causes a conformational change, removing the inhibition, and allowing downstream signaling. Also, within the Sema domain of the Sema3s, there is a 70 amino acid stretch that is responsible for the specificity in the Sema3s (Koppel et al., 1997). From this, and Takahashi and Strittmatter's model, it is probable that the specificity within that region involves both the ability of each Sema3 to bind specific Neuropilins, and co-receptors such as Plxna1 (Antipenko et al., 2003; Love et al., 2003; Liu et al., 2010). Interestingly, there is a conserved residue (K108) among all vertebrate Semaphorins that abuts the aforementioned 70 amino acid region, and when mutated in Sema3a and Sema3f, abrogates signaling without affecting binding to Nrp1 and Nrp2, respectively, (Merte et al., 2010). The same mutation in Sema3e does not affect that molecule's binding or signaling through Plxnd1, indicating that if this mutation is having an effect on Semaphorin-Plexin binding, it is only apparent in Semaphorin-Neuropilin-Plexin complexes. Indeed, the K108N Sema3a mutation did not affect binding to Nrp1, but did reduce binding to embryonic mouse DRG growth cones, indicating that even though the residue is strongly conserved, its effect is only observed in proximity to a Neuropilin. The above evidence supports Takahashi and Strittmatter's hypothesis, and also indicates that specific regions of the Sema3 ligands affect recruitment and/or activation of their co-receptors. Indeed, it appears that that Sema3 binding to Neuropilins, and Sema3 interaction with Plexins are on quite separate regions of the Sema3 protein, and targeting of these regions may allow specific interference with Sema3 signaling.
There are also several other issues relating to Sema3 receptors that are outside the purpose of this review, but merit comment. Firstly several of the receptor components of the Sema3 receptor also act as receptors for other classes of Semaphorins (Toyofuku et al., 2004; Suto et al., 2005; Yoshida et al., 2006; Suto et al., 2007; Matsuoka et al., 2011a,b; Taniguchi et al., 2011), and even other families of proteins such as the vascular endothelial growth factors (VEGFs) (Soker et al., 1998; Neufeld et al., 2002; Guttmann-Raviv et al., 2007; Geretti et al., 2008; Zachary et al., 2009), hepatocyte growth factor (HGF) (Sulpice et al., 2008; Zachary et al., 2009), and transforming growth factor β1 (TGFβ1) (Glinka and Prud'homme, 2008). Secondly, it remains to be determined how endogenous soluble forms of the Neuropilins and L1cam interact with Sema3 signaling (Gagnon et al., 2000; Rossignol et al., 2000; Castellani et al., 2002; Lu et al., 2009). Thirdly, the Sema3s interact with chondroitin sulphate proteoglycans (CSPGs) in the extracellular matrix (ECM), and this interaction appears to potentiate its repulsive activity (De Wit et al., 2005; Zimmer et al., 2010). Lastly, it is unclear how a particular cell or tissue regulates its expression of the different components of the Sema3 receptor complex.
If components of the Sema3 receptor, especially the binding subunits the Neuropilins, act as receptors for other molecules, does that mean the Sema3s can act as antagonists against those other molecules or vice versa? It is known that Sema3 signaling involves endocytosis of the receptor complex (Castellani et al., 2004; Tojima et al., 2010), as does VEGF signaling through Nrp1, albeit by a different mechanism (Narazaki and Tosato, 2006; Salikhova et al., 2008). Thus it is possible that by sequestering essential receptor subunits, both VEGFs and Sema3s can act as antagonists (Narazaki and Tosato, 2006; Narazaki et al., 2008). Indeed, antagonism between Sema3s and VEGFs has been observed (Nasarre et al., 2003; Geretti et al., 2008). Furthermore, competitive inhibition of Sema3s on VEGF binding of Nrp2 has been reported (Nasarre et al., 2005; Geretti et al., 2007), whereas it appears that Sema3s and VEGFs do not directly compete for binding to Nrp1 (Appleton et al., 2007). In other words, we should view the Sema3s not only as ligands for their receptors, but also as possible antagonists of molecules for which they share receptor components (Takahashi et al., 1998; Parker et al., 2010).
Soluble truncated Neuropilins and L1cam have been identified, and observed in vivo (Mechtersheimer et al., 2001; Lu et al., 2009). Soluble Neuropilins can bind VEGF165 (Gagnon et al., 2000), so it is possible then that they are also able to bind and sequester Sema3s from functional receptors. These soluble Neuropilins do not form part of the Sema3s's holoreceptors, but they are receptors for the Sema3s. Future studies into the effect of the Sema3s should be mindful of the presence of these soluble Neuropilins, and their buffering effect taken into account when considering ligand/receptor binding. Similar note should be made of L1cam expression, as its soluble form is known to modulate at least Sema3a signaling (Castellani et al., 2000, 2002, 2004). Another intriguing finding is that soluble amyloid precursor protein can bind Sema3a (Magdesian et al., 2011), and may act similar to soluble Neuropilins as an inhibitor to Sema3 signaling.
Based on the interaction of Sema3a with CSPGs (De Wit et al., 2005; Zimmer et al., 2010), Zimmer et al. (2010) speculate that amongst other possible mechanisms, CSPGs may interact directly with CAMs in the Sema3a receptor complex. CSPGs in the ECM could also act to stabilize Sema3a for presentation to Sema3a sensitive cells (De Wit et al., 2005). Interestingly, the presence of heparin and heparan sulphates, but not chondroitin sulphates, enhances binding and activity of Sema3a to Nrp1 expressing cells (De Wit et al., 2005), which may be related to the heparin binding site on Nrp1 (Vander Kooi et al., 2007). Also, via an as yet unexplained mechanism, Sema3c release from proteoglycans in the ECM in vitro increases its cell migration effect on MCF7 cells, despite no change in observed binding of Sema3c to the cell surface (Esselens et al., 2010). These studies show that in some respects, proteoglycans in the ECM can act as “helper” receptors for Sema3s, and may play an important role in Sema3 signaling in vivo.
Finally, factors that influence the regulation of expression of the Sema3 receptor components are still far from understood. It is known, for example, that there are changes in expression of the Neuropilins and PlexinAs during development, after injury, and in response to at least some growth factors (de Winter et al., 2002, 2004; Banerjee et al., 2006). Further factors that regulate expression of both Neuropilins are summarised by Bielenberg et al. (2006). VEGF regulates the expression of Plxnd1 in the endothelial cells in the developing mouse retina (Kim et al., 2011). In Xenopus laevis, fibroblast growth factor regulates expression of Sema3a (Atkinson-Leadbeater et al., 2010). It is also known that Sema3d can modulate expression of L1cam, (Wolman et al., 2004), and that Robo1 similarly affects expression of Nrp1 and Plxna1 (Hernández-Miranda et al., 2011). Interestingly, Nrcam sensitises commissural axons to Sema3b by inhibiting the proteolytic degradation of another receptor component, Plxna1 (Nawabi et al., 2010). Furthermore, Sema3a can induce protein synthesis at the growth cone via upregulation of translation (Campbell and Holt, 2001), and this could modulate expression of a wide range of proteins, including receptors. A deeper understanding of how the expression of Sema3 receptors is regulated will provide insight into the molecular mechanisms that cause aberrant expression or receptor components in pathological situations.
This review brings together the growing number of investigations into what receptor subunits are important for each Sema3 ligand. It is apparent that there is still much to learn about the Sema3 receptors, and that it is important we gain a fuller understanding of the Sema3 receptor complexes. When combined with greater knowledge of the signaling cascades involved with each receptor subunit, there is the tantalising possibility of designing therapies for the increasing number of pathological situations in which Sema3s and their receptors are implicated.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Antipenko, A., Himanen, J. P., Van Leyen, K., Nardi-Dei, V., Lesniak, J., Barton, W. A., Rajashankar, K. R., Lu, M., Hoemme, C., Püschel, A. W., and Nikolov, D. B. (2003). Structure of the semaphorin-3A receptor binding module. Neuron 39, 589–598.
Appleton, B. A., Wu, P., Maloney, J., Yin, J., Liang, W. C., Stawicki, S., Mortara, K., Bowman, K. K., Elliott, J. M., Desmarais, W., Bazan, J. F., Bagri, A., Tessier-Lavigne, M., Koch, A. W., Wu, Y., Watts, R. J., and Wiesmann, C. (2007). Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. EMBO J. 26, 4902–4912.
Atkinson-Leadbeater, K., Bertolesi, G. E., Hehr, C. L., Webber, C. A., Cechmanek, P. B., and McFarlane, S. (2010). Dynamic expression of axon guidance cues required for optic tract development is controlled by fibroblast growth factor signaling. J. Neurosci. 30, 685–693.
Bagri, A., Cheng, H. J., Yaron, A., Pleasure, S. J., and Tessier-Lavigne, M. (2003). Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 113, 285–299.
Banerjee, S., Sengupta, K., Dhar, K., Mehta, S., D'Amore, P. A., Dhar, G., and Banerjee, S. K. (2006). Breast cancer cells secreted platelet-derived growth factor-induced motility of vascular smooth muscle cells is mediated through neuropilin-1. Mol. Carcinog. 45, 871–880.
Bechara, A., Nawabi, H., Moret, F., Yaron, A., Weaver, E., Bozon, M., Abouzid, K., Guan, J. L., Tessier-Lavigne, M., Lemmon, V., and Castellani, V. (2008). FAK-MAPK-dependent adhesion disassembly downstream of L1 contributes to semaphorin3A-induced collapse. EMBO J. 27, 1549–1562.
Ben-Zvi, A., Manor, O., Schachner, M., Yaron, A., Tessier-Lavigne, M., and Behar, O. (2008). The Semaphorin receptor PlexinA3 mediates neuronal apoptosis during dorsal root ganglia development. J. Neurosci. 28, 12427–12432.
Bielenberg, D. R., Pettaway, C. A., Takashima, S., and Klagsbrun, M. (2006). Neuropilins in neoplasms: expression, regulation, and function. Exp. Cell Res. 312, 584–593.
Bron, R., Vermeren, M., Kokot, N., Andrews, W., Little, G. E., Mitchell, K. J., and Cohen, J. (2007). Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev. 2, 21.
Brown, C. B., Feiner, L., Lu, M. M., Li, J., Ma, X., Webber, A. L., Jia, L., Raper, J. A., and Epstein, J. A. (2001). PlexinA2 and semaphorin signaling during cardiac neural crest development. Development 128, 3071–3080.
Campbell, D. S., and Holt, C. E. (2001). Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32, 1013–1026.
Campbell, D. S., Regan, A. G., Lopez, J. S., Tannahill, D., Harris, W. A., and Holt, C. E. (2001). Semaphorin 3A elicits stage-dependent collapse, turning, and branching in Xenopus retinal growth cones. J. Neurosci. 21, 8538–8547.
Cariboni, A., Davidson, K., Rakic, S., Maggi, R., Parnavelas, J. G., and Ruhrberg, C. (2011). Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum. Mol. Genet. 20, 336–344.
Casazza, A., Finisguerra, V., Capparuccia, L., Camperi, A., Swiercz, J. M., Rizzolio, S., Rolny, C., Christensen, C., Bertotti, A., Sarotto, I., Risio, M., Trusolino, L., Weitz, J., Schneider, M., Mazzone, M., Comoglio, P. M., and Tamagnone, L. (2010). Sema3E-Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J. Clin. Invest. 120, 2684–2698.
Castellani, V., Chedotal, A., Schachner, M., Faivre-Sarrailh, C., and Rougon, G. (2000). Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signaling pathways in axonal guidance. Neuron 27, 237–249.
Castellani, V., De Angelis, E., Kenwrick, S., and Rougon, G. (2002). Cis and trans interactions of L1 with neuropilin-1 control axonal responses to semaphorin 3A. EMBO J. 21, 6348–6357.
Castellani, V., Falk, J., and Rougon, G. (2004). Semaphorin3A-induced receptor endocytosis during axon guidance responses is mediated by L1 CAM. Mol. Cell. Neurosci. 26, 89–100.
Chauvet, S., Cohen, S., Yoshida, Y., Fekrane, L., Livet, J., Gayet, O., Segu, L., Buhot, M. C., Jessell, T. M., Henderson, C. E., and Mann, F. (2007). Gating of Sema3E/PlexinD1 signaling by neuropilin-1 switches axonal repulsion to attraction during brain development. Neuron 56, 807–822.
Chen, H., Chedotal, A., He, Z., Goodman, C. S., and Tessier-Lavigne, M. (1997). Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron 19, 547–559.
Chen, H., He, Z., Bagri, A., and Tessier-Lavigne, M. (1998). Semaphorin-neuropilin interactions underlying sympathetic axon responses to class III semaphorins. Neuron 21, 1283–1290.
Cheng, H. J., Bagri, A., Yaron, A., Stein, E., Pleasure, S. J., and Tessier-Lavigne, M. (2001). Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron 32, 249–263.
Cloutier, J. F., Giger, R. J., Koentges, G., Dulac, C., Kolodkin, A. L., and Ginty, D. D. (2002). Neuropilin-2 mediates axonal fasciculation, zonal segregation, but not axonal convergence, of primary accessory olfactory neurons. Neuron 33, 877–892.
Cloutier, J. F., Sahay, A., Chang, E. C., Tessier-Lavigne, M., Dulac, C., Kolodkin, A. L., and Ginty, D. D. (2004). Differential requirements for semaphorin 3F and Slit-1 in axonal targeting, fasciculation, and segregation of olfactory sensory neuron projections. J. Neurosci. 24, 9087–9096.
de Winter, F., Cui, Q., Symons, N., Verhaagen, J., and Harvey, A. R. (2004). Expression of class-3 semaphorins and their receptors in the neonatal and adult rat retina. Invest. Ophthalmol. Vis. Sci. 45, 4554–4562.
de Winter, F., Oudega, M., Lankhorst, A. J., Hamers, F. P., Blits, B., Ruitenberg, M. J., Pasterkamp, R. J., Gispen, W. H., and Verhaagen, J. (2002). Injury-induced class 3 semaphorin expression in the rat spinal cord. Exp. Neurol. 175, 61–75.
De Wit, J., De Winter, F., Klooster, J., and Verhaagen, J. (2005). Semaphorin 3A displays a punctate distribution on the surface of neuronal cells and interacts with proteoglycans in the extracellular matrix. Mol. Cell. Neurosci. 29, 40–55.
Demyanenko, G. P., Riday, T. T., Tran, T. S., Dalal, J., Darnell, E. P., Brennaman, L. H., Sakurai, T., Grumet, M., Philpot, B. D., and Maness, P. F. (2011). NrCAM deletion causes topographic mistargeting of thalamocortical axons to the visual cortex and disrupts visual acuity. J. Neurosci. 31, 1545–1558.
Esselens, C., Malapeira, J., Colome, N., Casal, C., Rodriguez-Manzaneque, J. C., Canals, F., and Arribas, J. (2010). The cleavage of semaphorin 3C induced by ADAMTS1 promotes cell migration. J. Biol. Chem. 285, 2463–2473.
Falk, J., Bechara, A., Fiore, R., Nawabi, H., Zhou, H., Hoyo-Becerra, C., Bozon, M., Rougon, G., Grumet, M., Püschel, A. W., Sanes, J. R., and Castellani, V. (2005). Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron 48, 63–75.
Feiner, L., Koppel, A. M., Kobayashi, H., and Raper, J. A. (1997). Secreted chick semaphorins bind recombinant neuropilin with similar affinities but bind different subsets of neurons in situ. Neuron 19, 539–545.
Gagnon, M. L., Bielenberg, D. R., Gechtman, Z., Miao, H. Q., Takashima, S., Soker, S., and Klagsbrun, M. (2000). Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: in vivo expression and antitumor activity. Proc. Natl. Acad. Sci. U.S.A. 97, 2573–2578.
Gammill, L. S., Gonzalez, C., Gu, C., and Bronner-Fraser, M. (2006). Guidance of trunk neural crest migration requires neuropilin 2/semaphorin 3F signaling. Development 133, 99–106.
Geretti, E., Shimizu, A., and Klagsbrun, M. (2008). Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. Angiogenesis 11, 31–39.
Geretti, E., Shimizu, A., Kurschat, P., and Klagsbrun, M. (2007). Site-directed mutagenesis in the B-neuropilin-2 domain selectively enhances its affinity to VEGF165, but not to semaphorin 3F. J. Biol. Chem. 282, 25698–25707.
Gitler, A. D., Lu, M. M., and Epstein, J. A. (2004). PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev. Cell 7, 107–116.
Glinka, Y., and Prud'homme, G. J. (2008). Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 84, 302–310.
Gu, C., Rodriguez, E. R., Reimert, D. V., Shu, T., Fritzsch, B., Richards, L. J., Kolodkin, A. L., and Ginty, D. D. (2003). Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 5, 45–57.
Gu, C., Yoshida, Y., Livet, J., Reimert, D. V., Mann, F., Merte, J., Henderson, C. E., Jessell, T. M., Kolodkin, A. L., and Ginty, D. D. (2005). Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science 307, 265–268.
Guttmann-Raviv, N., Shraga-Heled, N., Varshavsky, A., Guimaraes-Sternberg, C., Kessler, O., and Neufeld, G. (2007). Semaphorin-3A and semaphorin-3F work together to repel endothelial cells and to inhibit their survival by induction of apoptosis. J. Biol. Chem. 282, 26294–26305.
He, Z., and Tessier-Lavigne, M. (1997). Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 90, 739–751.
Hernández-Miranda, L. R., Cariboni, A., Faux, C., Ruhrberg, C., Cho, J. H., Cloutier, J. F., Eickholt, B. J., Parnavelas, J. G., and Andrews, W. D. (2011). Robo1 regulates semaphorin signaling to guide the migration of cortical interneurons through the ventral forebrain. J. Neurosci. 31, 6174–6187.
Kapfhammer, J. P., Xu, H., and Raper, J. A. (2007). The detection and quantification of growth cone collapsing activities. Nat. Protoc. 2, 2005–2011.
Karayan-Tapon, L., Wager, M., Guilhot, J., Levillain, P., Marquant, C., Clarhaut, J., Potiron, V., and Roche, J. (2008). Semaphorin, neuropilin and VEGF expression in glial tumours: SEMA3G, a prognostic marker? Br. J. Cancer 99, 1153–1160.
Kawakami, A., Kitsukawa, T., Takagi, S., and Fujisawa, H. (1996). Developmentally regulated expression of a cell surface protein, neuropilin, in the mouse nervous system. J. Neurobiol. 29, 1–17.
Kigel, B., Rabinowicz, N., Varshavsky, A., Kessler, O., and Neufeld, G. (2011). Plexin-A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 118, 4285–4296.
Kigel, B., Varshavsky, A., Kessler, O., and Neufeld, G. (2008). Successful inhibition of tumor development by specific class-3 semaphorins is associated with expression of appropriate semaphorin receptors by tumor cells. PLoS ONE 3:e3287. doi: 10.1371/journal.pone.0003287
Kim, J., Oh, W. J., Gaiano, N., Yoshida, Y., and Gu, C. (2011). Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 25, 1399–1411.
Kitsukawa, T., Shimizu, M., Sanbo, M., Hirata, T., Taniguchi, M., Bekku, Y., Yagi, T., and Fujisawa, H. (1997). Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 19, 995–1005.
Kolk, S. M., Gunput, R. A., Tran, T. S., Van Den Heuvel, D. M., Prasad, A. A., Hellemons, A. J., Adolfs, Y., Ginty, D. D., Kolodkin, A. L., Burbach, J. P., Smidt, M. P., and Pasterkamp, R. J. (2009). Semaphorin 3F is a bifunctional guidance cue for dopaminergic axons and controls their fasciculation, channeling, rostral growth, and intracortical targeting. J. Neurosci. 29, 12542–12557.
Kolodkin, A. L., Levengood, D. V., Rowe, E. G., Tai, Y. T., Giger, R. J., and Ginty, D. D. (1997). Neuropilin is a semaphorin III receptor. Cell 90, 753–762.
Kolodkin, A. L., Matthes, D. J., and Goodman, C. S. (1993). The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 75, 1389.
Kolodkin, A. L., Matthes, D. J., O'Connor, T. P., Patel, N. H., Admon, A., Bentley, D., and Goodman, C. S. (1992). Fasciclin IV: sequence, expression, and function during growth cone guidance in the grasshopper embryo. Neuron 9, 831–845.
Koppel, A. M., Feiner, L., Kobayashi, H., and Raper, J. A. (1997). A 70 amino acid region within the semaphorin domain activates specific cellular response of semaphorin family members. Neuron 19, 531–537.
Liu, H., Juo, Z. S., Shim, A. H., Focia, P. J., Chen, X., Garcia, K. C., and He, X. (2010). Structural basis of semaphorin-plexin recognition and viral mimicry from Sema7A and A39R complexes with PlexinC1. Cell 142, 749–761.
Love, C. A., Harlos, K., Mavaddat, N., Davis, S. J., Stuart, D. I., Jones, E. Y., and Esnouf, R. M. (2003). The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat. Struct. Biol. 10, 843–848.
Lu, Y., Xiang, H., Liu, P., Tong, R. R., Watts, R. J., Koch, A. W., Sandoval, W. N., Damico, L. A., Wong, W. L., and Meng, Y. G. (2009). Identification of circulating neuropilin-1 and dose-dependent elevation following anti-neuropilin-1 antibody administration. MAbs 1, 364–369.
Luo, Y., Raible, D., and Raper, J. A. (1993). Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75, 217–227.
Luo, Y., Shepherd, I., Li, J., Renzi, M. J., Chang, S., and Raper, J. A. (1995). A family of molecules related to collapsin in the embryonic chick nervous system. Neuron 14, 1131–1140.
Magdesian, M. H., Gralle, M., Guerreiro, L. H., Beltrao, P. J. I., Carvalho, M., Santos, L. E. D., De Mello, F. G., Reis, R. A. M., and Ferreira, S. T. (2011). Secreted human amyloid precursor protein binds semaphorin 3a and prevents semaphorin-induced growth cone collapse. PLoS ONE 6:e22857. doi: 10.1371/journal.pone.0022857
Maretzky, T., Schulte, M., Ludwig, A., Rose-John, S., Blobel, C., Hartmann, D., Altevogt, P., Saftig, P., and Reiss, K. (2005). L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol. Cell. Biol. 25, 9040–9053.
Matsuoka, R. L., Chivatakarn, O., Badea, T. C., Samuels, I. S., Cahill, H., Katayama, K., Kumar, S. R., Suto, F., Chedotal, A., Peachey, N. S., Nathans, J., Yoshida, Y., Giger, R. J., and Kolodkin, A. L. (2011a). Class 5 transmembrane semaphorins control selective Mammalian retinal lamination and function. Neuron 71, 460–473.
Matsuoka, R. L., Nguyen-Ba-Charvet, K. T., Parray, A., Badea, T. C., Chedotal, A., and Kolodkin, A. L. (2011b). Transmembrane semaphorin signalling controls laminar stratification in the mammalian retina. Nature 470, 259–263.
Mechtersheimer, S., Gutwein, P., Agmon-Levin, N., Stoeck, A., Oleszewski, M., Riedle, S., Postina, R., Fahrenholz, F., Fogel, M., Lemmon, V., and Altevogt, P. (2001). Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J. Cell Biol. 155, 661–673.
Merte, J., Wang, Q., Vander Kooi, C. W., Sarsfield, S., Leahy, D. J., Kolodkin, A. L., and Ginty, D. D. (2010). A forward genetic screen in mice identifies Sema3A(K108N), which binds to neuropilin-1 but cannot signal. J. Neurosci. 30, 5767–5775.
Messersmith, E. K., Leonardo, E. D., Shatz, C. J., Tessier-Lavigne, M., Goodman, C. S., and Kolodkin, A. L. (1995). Semaphorin III can function as a selective chemorepellent to pattern sensory projections in the spinal cord. Neuron 14, 949.
Miyazaki, N., Furuyama, T., Sakai, T., Fujioka, S., Mori, T., Ohoka, Y., Takeda, N., Kubo, T., and Inagaki, S. (1999). Developmental localization of semaphorin H messenger RNA acting as a collapsing factor on sensory axons in the mouse brain. Neuroscience 93, 401–408.
Nakamura, F., Tanaka, M., Takahashi, T., Kalb, R. G., and Strittmatter, S. M. (1998). Neuropilin-1 extracellular domains mediate semaphorin D/III-induced growth cone collapse. Neuron 21, 1093–1100.
Narazaki, M., Segarra, M., and Tosato, G. (2008). Sulfated polysaccharides identified as inducers of neuropilin-1 internalization and functional inhibition of VEGF165 and semaphorin3A. Blood 111, 4126–4136.
Narazaki, M., and Tosato, G. (2006). Ligand-induced internalization selects use of common receptor neuropilin-1 by VEGF165 and semaphorin3A. Blood 107, 3892–3901.
Nasarre, C., Koncina, E., Labourdette, G., Cremel, G., Roussel, G., Aunis, D., and Bagnard, D. (2009). Neuropilin-2 acts as a modulator of Sema3A-dependent glioma cell migration. Cell Adh. Migr. 3, 383–389.
Nasarre, P., Constantin, B., Rouhaud, L., Harnois, T., Raymond, G., Drabkin, H. A., Bourmeyster, N., and Roche, J. (2003). Semaphorin SEMA3F and VEGF have opposing effects on cell attachment and spreading. Neoplasia 5, 83–92.
Nasarre, P., Kusy, S., Constantin, B., Castellani, V., Drabkin, H. A., Bagnard, D., and Roche, J. (2005). Semaphorin SEMA3F has a repulsing activity on breast cancer cells and inhibits E-cadherin-mediated cell adhesion. Neoplasia 7, 180–189.
Nawabi, H., Briancon-Marjollet, A., Clark, C., Sanyas, I., Takamatsu, H., Okuno, T., Kumanogoh, A., Bozon, M., Takeshima, K., Yoshida, Y., Moret, F., Abouzid, K., and Castellani, V. (2010). A midline switch of receptor processing regulates commissural axon guidance in vertebrates. Genes Dev. 24, 396–410.
Neufeld, G., Cohen, T., Shraga, N., Lange, T., Kessler, O., and Herzog, Y. (2002). The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc. Med. 12, 13–19.
Neufeld, G., and Kessler, O. (2008). The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 8, 632–645.
Parker, M. W., Hellman, L. M., Xu, P., Fried, M. G., and Vander Kooi, C. W. (2010). Furin processing of semaphorin 3F determines its anti-angiogenic activity by regulating direct binding and competition for neuropilin. Biochemistry 49, 4068–4075.
Pasterkamp, R. J., and Kolodkin, A. L. (2003). Semaphorin junction: making tracks toward neural connectivity. Curr. Opin. Neurobiol. 13, 79–89.
Pellet-Many, C., Frankel, P., Jia, H., and Zachary, I. (2008). Neuropilins: structure, function and role in disease. Biochem. J. 411, 211–226.
Püschel, A. W., Adams, R. H., and Betz, H. (1995). Murine semaphorin D/collapsin is a member of a diverse gene family and creates domains inhibitory for axonal extension. Neuron 14, 941–948.
Raper, J. A. (2000). Semaphorins and their receptors in vertebrates and invertebrates. Curr. Opin. Neurobiol. 10, 88–94.
Renzi, M. J., Feiner, L., Koppel, A. M., and Raper, J. A. (1999). A dominant negative receptor for specific secreted semaphorins is generated by deleting an extracellular domain from neuropilin-1. J. Neurosci. 19, 7870–7880.
Roche, J., Boldog, F., Robinson, M., Robinson, L., Varella-Garcia, M., Swanton, M., Waggoner, B., Fishel, R., Franklin, W., Gemmill, R., and Drabkin, H. (1996). Distinct 3p21.3 deletions in lung cancer and identification of a new human semaphorin. Oncogene 12, 1289–1297.
Rohm, B., Ottemeyer, A., Lohrum, M., and Püschel, A. W. (2000). Plexin/neuropilin complexes mediate repulsion by the axonal guidance signal semaphorin 3A. Mech. Dev. 93, 95–104.
Rossignol, M., Gagnon, M. L., and Klagsbrun, M. (2000). Genomic organization of human neuropilin-1 and neuropilin-2 genes: identification and distribution of splice variants and soluble isoforms. Genomics 70, 211–222.
Roth, L., Koncina, E., Satkauskas, S., Cremel, G., Aunis, D., and Bagnard, D. (2009). The many faces of semaphorins: from development to pathology. Cell. Mol. Life Sci. 66, 649–666.
Roth, L., Nasarre, C., Dirrig-Grosch, S., Aunis, D., Cremel, G., Hubert, P., and Bagnard, D. (2008). Transmembrane domain interactions control biological functions of neuropilin-1. Mol. Biol. Cell 19, 646–654.
Sahay, A., Molliver, M. E., Ginty, D. D., and Kolodkin, A. L. (2003). Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J. Neurosci. 23, 6671–6680.
Sakurai, A., Doci, C. L., and Gutkind, J. S. (2012). Semaphorin signaling in angiogenesis, lymphangiogenesis and cancer. Cell Res. 22, 23–32.
Salikhova, A., Wang, L., Lanahan, A. A., Liu, M., Simons, M., Leenders, W. P., Mukhopadhyay, D., and Horowitz, A. (2008). Vascular endothelial growth factor and semaphorin induce neuropilin-1 endocytosis via separate pathways. Circ. Res. 103, e71–e79.
Schlatter, M. C., Buhusi, M., Wright, A. G., and Maness, P. F. (2008). CHL1 promotes Sema3A-induced growth cone collapse and neurite elaboration through a motif required for recruitment of ERM proteins to the plasma membrane. J. Neurochem. 104, 731–744.
Schwarz, Q., Waimey, K. E., Golding, M., Takamatsu, H., Kumanogoh, A., Fujisawa, H., Cheng, H. J., and Ruhrberg, C. (2008). Plexin A3 and plexin A4 convey semaphorin signals during facial nerve development. Dev. Biol. 324, 1–9.
Sekido, Y., Bader, S., Latif, F., Chen, J.-Y., Duh, F.-M., Wei, M.-H., Albanesi, J. P., Lee, C.-C., Lerman, M. I., and Minna, J. D. (1996). Human semaphorins A(V) and IV reside in the 3p21.3 small cell lung cancer deletion region and demonstrate distinct expression patterns. Proc. Natl. Acad. Sci. U.S.A. 93, 4120–4125.
Semaphorin Nomenclature Committee. (1999). Unified nomenclature for the semaphorins/collapsins. Cell 97, 551–552.
Soker, S., Takashima, S., Miao, H. Q., Neufeld, G., and Klagsbrun, M. (1998). Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92, 735–745.
Staton, C. A. (2011). Class 3 semaphorins and their receptors in physiological and pathological angiogenesis. Biochem. Soc. Trans. 39, 1565–1570.
Stevens, C. B., and Halloran, M. C. (2005). Developmental expression of sema3G, a novel zebrafish semaphorin. Gene Expr. Patterns 5, 647–653.
Sulpice, E., Plouet, J., Berge, M., Allanic, D., Tobelem, G., and Merkulova-Rainon, T. (2008). Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood 111, 2036–2045.
Suto, F., Ito, K., Uemura, M., Shimizu, M., Shinkawa, Y., Sanbo, M., Shinoda, T., Tsuboi, M., Takashima, S., Yagi, T., and Fujisawa, H. (2005). Plexin-a4 mediates axon-repulsive activities of both secreted and transmembrane semaphorins and plays roles in nerve fiber guidance. J. Neurosci. 25, 3628–3637.
Suto, F., Tsuboi, M., Kamiya, H., Mizuno, H., Kiyama, Y., Komai, S., Shimizu, M., Sanbo, M., Yagi, T., Hiromi, Y., Chedotal, A., Mitchell, K. J., Manabe, T., and Fujisawa, H. (2007). Interactions between plexin-A2, plexin-A4, and semaphorin 6A control lamina-restricted projection of hippocampal mossy fibers. Neuron 53, 535–547.
Takahashi, T., Fournier, A., Nakamura, F., Wang, L. H., Murakami, Y., Kalb, R. G., Fujisawa, H., and Strittmatter, S. M. (1999). Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell 99, 59–69.
Takahashi, T., Nakamura, F., Jin, Z., Kalb, R. G., and Strittmatter, S. M. (1998). Semaphorins A and E act as antagonists of neuropilin-1 and agonists of neuropilin-2 receptors. Nat. Neurosci. 1, 487–493.
Takahashi, T., and Strittmatter, S. M. (2001). Plexina1 autoinhibition by the plexin sema domain. Neuron 29, 429–439.
Takamatsu, H., and Kumanogoh, A. (2012). Diverse roles for semaphorin-plexin signaling in the immune system. Trends Immunol. 33, 127–135.
Takeuchi, H., Inokuchi, K., Aoki, M., Suto, F., Tsuboi, A., Matsuda, I., Suzuki, M., Aiba, A., Serizawa, S., Yoshihara, Y., Fujisawa, H., and Sakano, H. (2010). Sequential arrival and graded secretion of Sema3F by olfactory neuron axons specify map topography at the bulb. Cell 141, 1056–1067.
Takegahara, N., and Kumanogoh, A. (2010). Involvement of semaphorins and their receptors in neurological diseases. Clin. Exp. Neuroimmunol. 1, 33–45.
Tamagnone, L., Artigiani, S., Chen, H., He, Z., Ming, G. I., Song, H., Chedotal, A., Winberg, M. L., Goodman, C. S., Poo, M., Tessier-Lavigne, M., and Comoglio, P. M. (1999). Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99, 71–80.
Taniguchi, M., Masuda, T., Fukaya, M., Kataoka, H., Mishina, M., Yaginuma, H., Watanabe, M., and Shimizu, T. (2005). Identification and characterization of a novel member of murine semaphorin family. Genes Cells 10, 785–792.
Taniguchi, M., Masuda, T., Mikami, Y., Kimura, M., Yoshida, T., Mishina, M., and Shimizu, T. (2011). Identification and characterization of a novel zebrafish semaphorin. Neurosci. Lett. 488, 215–220.
Tojima, T., Itofusa, R., and Kamiguchi, H. (2010). Asymmetric clathrin-mediated endocytosis drives repulsive growth cone guidance. Neuron 66, 370–377.
Toyofuku, T., Zhang, H., Kumanogoh, A., Takegahara, N., Suto, F., Kamei, J., Aoki, K., Yabuki, M., Hori, M., Fujisawa, H., and Kikutani, H. (2004). Dual roles of Sema6D in cardiac morphogenesis through region-specific association of its receptor, Plexin-A1, with off-track and vascular endothelial growth factor receptor type 2. Genes Dev. 18, 435–447.
Usui, H., Taniguchi, M., Yokomizo, T., and Shimizu, T. (2003). Plexin-A1 and plexin-B1 specifically interact at their cytoplasmic domains. Biochem. Biophys. Res. Commun. 300, 927–931.
Vander Kooi, C. W., Jusino, M. A., Perman, B., Neau, D. B., Bellamy, H. D., and Leahy, D. J. (2007). Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. U.S.A. 104, 6152–6157.
Waimey, K. E., Huang, P. H., Chen, M., and Cheng, H. J. (2008). Plexin-A3 and plexin-A4 restrict the migration of sympathetic neurons but not their neural crest precursors. Dev. Biol. 315, 448–458.
Wolman, M. A., Liu, Y., Tawarayama, H., Shoji, W., and Halloran, M. C. (2004). Repulsion and attraction of axons by semaphorin3D are mediated by different neuropilins in vivo. J. Neurosci. 24, 8428–8435.
Wolman, M. A., Regnery, A. M., Becker, T., Becker, C. G., and Halloran, M. C. (2007). Semaphorin3D regulates axon axon interactions by modulating levels of L1 cell adhesion molecule. J. Neurosci. 27, 9653–9663.
Wright, A. G., Demyanenko, G. P., Powell, A., Schachner, M., Enriquez-Barreto, L., Tran, T. S., Polleux, F., and Maness, P. F. (2007). Close homolog of L1 and neuropilin 1 mediate guidance of thalamocortical axons at the ventral telencephalon. J. Neurosci. 27, 13667–13679.
Xiang, R. H., Hensel, C. H., Garcia, D. K., Carlson, H. C., Kok, K., Daly, M. C., Kerbacher, K., Van den Berg, A., Veldhuis, P., Buys, C. H., and Naylor, S. L. (1996). Isolation of the human semaphorin III/F gene (SEMA3F) at chromosome 3p21, a region deleted in lung cancer. Genomics 32, 39–48.
Yaron, A., Huang, P. H., Cheng, H. J., and Tessier-Lavigne, M. (2005). Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 semaphorins. Neuron 45, 513–523.
Yoshida, Y., Han, B., Mendelsohn, M., and Jessell, T. M. (2006). PlexinA1 signaling directs the segregation of proprioceptive sensory axons in the developing spinal cord. Neuron 52, 775–788.
Yoshida, Y. (2012). Semaphorin signaling in vertebrate neural circuit assembly. Front. Mol. Neurosci. 5:71. doi: 10.3389/fnmol.2012.00071
Zachary, I. C., Frankel, P., Evans, I. M., and Pellet-Many, C. (2009). The role of neuropilins in cell signalling. Biochem. Soc. Trans. 37, 1171–1178.
Zhou, Y., Gunput, R. A., and Pasterkamp, R. J. (2008). Semaphorin signaling: progress made and promises ahead. Trends Biochem. Sci. 33, 161–170.
Keywords: adhesion molecules, axon guidance, axon repulsion, Neuropilins, Plexins, Robo, Semaphorins
Citation: Sharma A, Verhaagen J and Harvey AR (2012) Receptor complexes for each of the Class 3 Semaphorins. Front. Cell. Neurosci. 6:28. doi: 10.3389/fncel.2012.00028
Received: 10 May 2012; Paper pending published: 11 June 2012;
Accepted: 20 June 2012; Published online: 05 July 2012.
Edited by:
Dieter Wicher, Max Planck Institute for Chemical Ecology, GermanyReviewed by:
Corette Wierenga, Utrecht University, NetherlandsYuanquan Song, University of California, San Francisco, USA
Copyright © 2012 Sharma, Verhaagen and Harvey. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Alan R. Harvey, School of Anatomy, Physiology and Human Biology, Mailbag M309, The University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia. e-mail:YWxhbi5oYXJ2ZXlAdXdhLmVkdS5hdQ==