 Vibeke S. Catts1,2,3
Vibeke S. Catts1,2,3 Samantha J. Fung1,2,3
Samantha J. Fung1,2,3 Leonora E. Long1,2,4
Leonora E. Long1,2,4 Dipesh Joshi1,2,3
Dipesh Joshi1,2,3 Ans Vercammen1,2,3,5
Ans Vercammen1,2,3,5 Katherine M. Allen1,2,3 Stu G. Fillman1,2,3 Debora A. Rothmond1,2 Duncan Sinclair1,2,3
Katherine M. Allen1,2,3 Stu G. Fillman1,2,3 Debora A. Rothmond1,2 Duncan Sinclair1,2,3 Yash Tiwari1,2,4 Shan-Yuan Tsai1,2,3 Thomas W. Weickert1,2,3
Yash Tiwari1,2,4 Shan-Yuan Tsai1,2,3 Thomas W. Weickert1,2,3 Cynthia Shannon Weickert1,2,3*
Cynthia Shannon Weickert1,2,3*- 1Schizophrenia Research Laboratory, Schizophrenia Research Institute, Sydney, NSW, Australia
- 2Neuroscience Research Australia, Sydney, NSW, Australia
- 3School of Psychiatry, University of New South Wales, Sydney, NSW, Australia
- 4School of Medical Sciences, University of New South Wales, Sydney, NSW, Australia
- 5School of Psychology, Australian Catholic University, Sydney, NSW, Australia
The schizophrenia brain is differentiated from the normal brain by subtle changes, with significant overlap in measures between normal and disease states. For the past 25 years, schizophrenia has increasingly been considered a neurodevelopmental disorder. This frame of reference challenges biological researchers to consider how pathological changes identified in adult brain tissue can be accounted for by aberrant developmental processes occurring during fetal, childhood, or adolescent periods. To place schizophrenia neuropathology in a neurodevelopmental context requires solid, scrutinized evidence of changes occurring during normal development of the human brain, particularly in the cortex; however, too often data on normative developmental change are selectively referenced. This paper focuses on the development of the prefrontal cortex and charts major molecular, cellular, and behavioral events on a similar time line. We first consider the time at which human cognitive abilities such as selective attention, working memory, and inhibitory control mature, emphasizing that attainment of full adult potential is a process requiring decades. We review the timing of neurogenesis, neuronal migration, white matter changes (myelination), and synapse development. We consider how molecular changes in neurotransmitter signaling pathways are altered throughout life and how they may be concomitant with cellular and cognitive changes. We end with a consideration of how the response to drugs of abuse changes with age. We conclude that the concepts around the timing of cortical neuronal migration, interneuron maturation, and synaptic regression in humans may need revision and include greater emphasis on the protracted and dynamic changes occurring in adolescence. Updating our current understanding of post-natal neurodevelopment should aid researchers in interpreting gray matter changes and derailed neurodevelopmental processes that could underlie emergence of psychosis.
Introduction
A recent perspective titled “Rethinking Schizophrenia” (Insel, 2010) forecast that if researchers and clinicians approached schizophrenia as a neurodevelopmental disorder, with several discernible disease stages, then prevention focusing on the pre-psychotic illness phase would be a real possibility by 2030. The perspective (Insel, 2010) relied on the popular neurodevelopmental theory that the consequences of genetic predisposition and early adverse events, such as mid-gestational insults, would be latent throughout the first two decades of life and become evident as psychosis in early adulthood as first proposed by Weinberger (1987). Weinberger emphasized that the normative maturational changes “unmasked” an earlier insult. While we agree that adolescence is a critical period and that pre- and perinatal development are potential vulnerable time periods when schizophrenia susceptibility genes and environments may contribute to the future onset of schizophrenia, current data indicate human neurodevelopment is not confined to the womb, but is a protracted process that continues in post-natal life well into adolescence and early adulthood. Thus, there is opportunity for perturbation of developmental processes beyond the fetal and perinatal period and, we suggest that developmental disruption well after birth may also contribute to onset of schizophrenia. Our emphasis on the importance of post-natal events is similar to what another schizophrenia neurodevelopmental theorist, Feinberg, originally proposed (Feinberg, 1983). Feinberg suggested that exuberant synaptic regression (“pruning”) in adolescence may underlie schizophrenia, a theory that has dominated our thinking about abnormal neurodevelopment in adolescence in schizophrenia for over 25 years. One of the attractions of Feinberg's hypothesis is that it seemingly fits with the emerging consensus that progressive brain changes in adolescence or early adulthood, thought to be related to tissue loss, are associated with schizophrenia, at least in some individuals (Borgwardt et al., 2009). While we suggest that Feinberg's over-exuberant synaptic pruning theory of schizophrenia is potentially flawed, as it relies on the timing of normative synaptic pruning with inaccurately extrapolated synaptic densities that do not appear consistent with more recent studies, we do submit that charting normal adolescent brain changes is essential if we are to understand the processes underlying schizophrenia pathology. Indeed, a better understanding of changes occurring in the normal adolescent brain may serve to unite protagonists of neurodevelopmental and of neurodegenerative pathologies of schizophrenia. Also, many more molecular studies on human cortical growth and development in the post-natal periods have appeared since the 1980s and the results of these studies, which show diverse and dynamic post-natal changes, deserve to also be incorporated into our working models of the physical substrates of brain maturation in humans and into our theories about abnormal neurodevelopment in schizophrenia.
Here we review current normative neurodevelopmental studies and place them in the context of what is known about schizophrenia neuropathology. We start by considering when emergent behavioral properties of the human prefrontal cortex, like executive function, mature in humans; as prefrontal cortex function is greatly impacted in schizophrenia. Next we consider the timing of cognitive decline in people with schizophrenia, as this can implicate distinct neurobiological events in the pathophysiology. Then we review the temporal development of the “hardware” of the brain; the genesis of new neurons, their journey to the cortex, myelination of axons, and growth of dendritic arbors. Changes in excitatory and inhibitory synapses are reviewed next. We then consider how changes in neurotransmitter receptors and psychopharmacological responses to drugs across development might provide clues to the developmental processes gone astray in schizophrenia. Although there are well-documented differences in the average age of onset of schizophrenia in males (earlier) as compared to females, our review does not cover gender differences in neurodevelopment because our main vantage point is transcriptional and gender differences account for relatively little (130 transcripts = 8%) of the variance of gene transcript levels in a large microarray study of a developmental brain collection and typically occur early in life, not during adolescence (Weickert et al., 2009; Kang et al., 2011).
Cognitive Development
Basic cognitive functions, such as selective attention and response selection are firmly established by early childhood. However, the development of complex executive functions are characterized by a protracted development well through adolescence (Levin et al., 1991; Anderson et al., 2001; Brocki and Bohlin, 2004). Executive functioning capabilities include keeping and manipulating information in short-term memory (also known as working memory), planning, ignoring irrelevant information (involving inhibition and cognitive control), solving problems, and applying existing information to novel situations to derive new solutions (also known as reasoning). Schizophrenia, a disease which typically has its onset during late adolescence or early adulthood, is coincident in time with cognitive maturation of the prefrontal and parietal cortices. This section will: (1) review the timing of the development of executive functions and relationship to measures of neural maturation; (2) place the prefrontal associated cognitive and neural dysfunction accompanying schizophrenia in a developmental perspective; and (3) consider the evidence regarding the timing of cognitive decline in schizophrenia.
Neurodevelopment of Executive Functioning
We will consider three aspects of executive processing (working memory, cognitive control, and reasoning), which are separate constructs with differing developmental trajectories and with both distinct and overlapping neural substrates (Brocki and Bohlin, 2004; Huizinga et al., 2006). During the attainment of increased working memory capacity as children get older the superior frontal sulcus (prefrontal cortex) and the intraparietal sulcus (parietal cortex) show a greater degree of task-related activation and have increased white matter connectivity (Klingberg et al., 2002; Kwon et al., 2002; Olesen et al., 2003; Nagy et al., 2004; Edin et al., 2007). When manipulation of information in working memory is required (such as repeating remembered information in reverse order), children perform more poorly and show less activation of the dorsolateral prefrontal cortex (DLPFC) and superior parietal cortices than adolescents and young adults during the same task (Crone et al., 2006). Conversely, other brain regions that are activated during working memory tasks in children, such as the ventromedial prefrontal cortex, show decreased activity in adolescence and early adulthood during working memory tests (Scherf et al., 2006). Thus, in some prefrontal (dorsal) and parietal regions children typically show less activity than adolescents and young adults during executive function/working memory tests; whereas in the ventral prefrontal cortex the pattern is reversed. This suggest that within the frontal lobes, there may be a preference to use more ventral areas to solve working memory problems initially, but this function may be taken over by more dorsal areas of the prefrontal cortex later in life.
Another aspect of executive function is cognitive control. Cognitive control relies on a broad group of mental abilities that work together in context to “allow information processing and behavior to vary adaptively from moment to moment depending on current goals, rather than remaining rigid and inflexible” and is thought to rely heavily on the prefrontal cortex (http://carterlab.ucdavis.edu/research/control.php, accessed 7 April 2013). Cognitive control increases as humans mature from childhood to adulthood and this increasing ability correlates with increases in myelination of white matter tracts between the frontal and parietal cortices (Fair et al., 2007; Spear, 2007). The Wisconsin Card Sorting test, a classic test of cognitive flexibility and control, which also relies on fronto-parietal function, shows a developmental pattern of gradual performance improvement interspersed with periods of accelerated improvement throughout childhood to young adulthood (Heaton et al., 1993; Figure 1). Other aspects of cognitive control involve performance monitoring, which is associated with dorsal anterior cingulate activity in adults. Similar to under activation of the dorsal frontal cortical regions in working memory performance, children and adolescents display less dorsal anterior cingulate activity during performance monitoring (Velanova et al., 2008). The protracted development of mature cognition has been linked to relatively late maturation of several association cortices, as compared to other cortical regions such as the sensorimotor and occipital lobes (Giedd et al., 1999; Casey et al., 2000; Gogtay et al., 2004; Blakemore and Choudhury, 2006). We suggest that while a developmental increase in task-related activity of the dorsal prefrontal cortex may be critical to executive function that is typical of adults, this dorsal prefrontal cortex functional maturation co-occurs with functional maturation of other associative regions.
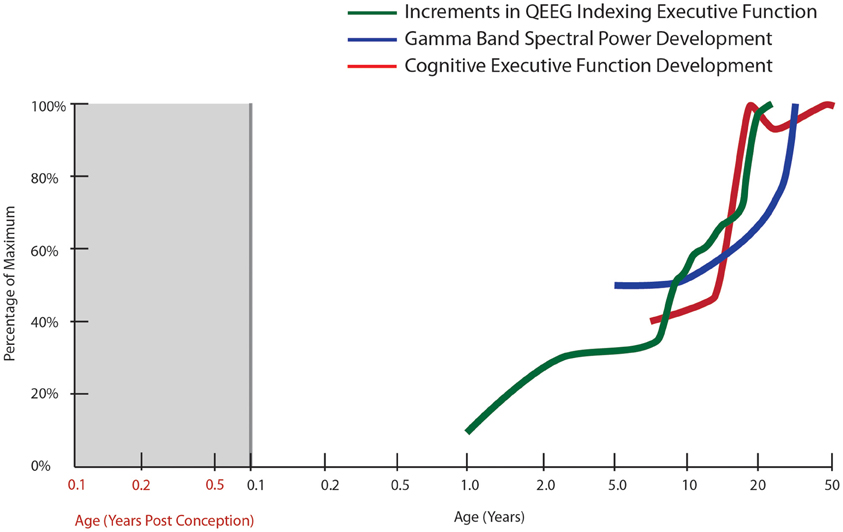
Figure 1. Normal developmental trajectory of cognitive executive function. The trajectory of cognitive executive functioning development is derived from developmental tables in the Wisconsin Card Sorting Manual [modified from Heaton et al. (1993)]. The other trajectories correspond to the spectral power in gamma bands during a face recognition task (30–75 Hz, all electrodes; Uhlhaas et al., 2009) as well as cumulative increments in computer quantified changes in EEG frequency spectrum (QEEG) between fronto-temporal cortices, derived from Hudspeth and Pribram (1990).
Reasoning (also referred to as a form of intelligence) is thought to rely to a large extent on “executive” processing again residing primarily in the frontal lobes. The two types of reasoning, crystallized and fluid, have somewhat different developmental trajectories. Crystallized intelligence refers to use of previously acquired knowledge to solve problems; whereas fluid intelligence refers to the ability to think logically and solve problems in novel situations independent of previously acquired knowledge. Fluid reasoning shows a rapid increase in ability from mid-childhood until early adulthood, whereas crystallized reasoning ability grows gradually, peaking in mid-life (Cattell, 1987; McArdle et al., 2002). The rostrolateral prefrontal cortex is consistently associated with logical reasoning ability (Prabhakaran et al., 1997; Christoff et al., 2001; Kroger et al., 2002), which also involves activation of other cortices that can vary depending on task demands (Bunge et al., 2005; Wright et al., 2008; Crone et al., 2009). While even toddlers (3-years old) can solve simple reasoning problems (Goswami, 1989), accurate performance on more difficult reasoning problems only occurs in later childhood to adolescence (Sternberg and Rifkin, 1979; Richland et al., 2006). During difficult reasoning problems rostral prefrontal cortical activity is delayed or fails to be sustained in children (Bunge et al., 2009; Crone et al., 2009). Thus, during difficult reasoning problems the timing and maintenance of activity increases as performance increases from early childhood to young adulthood. It is clear from these three examples of cognitive functional change during human development that most executive cognitive processes involve the ability to achieve and maintain activation of focal frontal regions in appropriate contexts. This suggests that molecular and cellular maturation in circuitry controlling the magnitude, extent and co-ordination of pyramidal neuron firing across association areas would be expected to increase during childhood and attain near adult levels during adolescence.
Mature ability to meet executive challenges of the adult are associated with more efficient prefrontal cortical activity (Casey et al., 1997; Bunge et al., 2002; Tamm et al., 2002; Durston et al., 2006). The increasing ability of the brain to transfer information more efficiently across widely distributed neural networks is an important part of adolescent development and likely involves faster and more synchronized axonal firing across long distances. In vivo brain imaging findings of macro-level increases in white matter (Snook et al., 2005; Liston et al., 2006; Eluvathingal et al., 2007; Giorgio et al., 2008) are thought to reflect progressive myelination at the micro-level (see Myelination section). However, while increased activity in frontal-parietal regions is a general rule throughout development from child to adulthood, for some tasks such as those requiring response inhibition, the lateral prefrontal cortex may show decreased activity (reflecting increased neural efficiency) as development progresses from children to young adults (Fair et al., 2007).
In general, based on its rich connections with other cortical and subcortical structures, the prefrontal cortex is also ideally suited to the task of coordinating activity within the neural network to facilitate increased neural efficiency and improved executive function. Electroencephalography (EEG) data show age-related changes in neural oscillations and synchrony that support enhanced temporal coordination of distributed cortical processes throughout development (Uhlhaas et al., 2009; Figure 1). Interestingly, this work also suggests a period of destabilization during adolescence, followed by reorganization during young adulthood (18–21 years of age), which is characterized by increases in gamma-band power, theta and beta band synchrony. In fact, EEG has detected several region-specific growth spurts (brief periods of accelerated neural development): the first typically occurring in toddlers, a second in early school age children, a third during puberty and early adolescence, and a final growth spurt in young adulthood (Hudspeth and Pribram, 1990, 1992; Figure 1). Thus, important changes in physiological and structural parameters may occur by gradual changes interspersed by occasional rapid increases, two distinct patterns of change that can also be detected with molecular markers, especially for inhibitory interneurons (see later sections of this review). These physiological changes parallel the patterns found for cognitive development where gradual change can be interspersed with brief periods of accelerated cognitive development (Thatcher, 1991, 1992, 1994). The other major point to consider is that while task-related activity of the prefrontal cortex increases in development, it appears that this activity must be integrated and coordinated with other regions and that both an increase in focal prefrontal activity and synchrony of this region with other association cortices may occur during adolescence. Thus, adolescence is a critical window for the organization and functional adjustment of cortical circuitry rendering this time of life particularly sensitive to disruptive effects. Given that the typical emergence of schizophrenia is during late adolescence or early adulthood, these later developmental changes, which may represent vulnerable periods, become especially relevant for the pathophysiology of schizophrenia, where abnormal patterns of oscillatory brain activity, especially in the gamma range, are observed in patients (Uhlhaas et al., 2008).
Executive Function and Prefrontal Cortex Development in Schizophrenia
One of the most debilitating problems for people with schizophrenia are the enduring cognitive deficits (Green, 1996), which are often unresponsive to antipsychotic medication (Heinrichs and Zakzanis, 1998; Goldberg et al., 2007). In terms of cognitive dysfunction, the most consistent findings are within the domains of executive function, working memory, inhibitory control, and reasoning (Weinberger et al., 1986; Goldman-Rakic, 1994; Weickert et al., 2000a; Silver et al., 2003; Ravizza et al., 2010). Almost three decades of functional and structural neuroimaging studies in schizophrenia provide converging evidence of localized abnormal activity and connectivity of the prefrontal cortex (Weinberger et al., 1986; Andreasen et al., 1997; Manoach et al., 1999, 2000; Barch et al., 2001; Meyer-Lindenberg et al., 2001; Perlstein et al., 2001, 2003; Callicott et al., 2003; Tan et al., 2006; Potkin et al., 2009). The concomitant refinement of cognitive executive processes, the physical maturation of neural circuitry underlying executive function, and the onset of schizophrenia in adolescence or early adulthood suggests that a failure in these maturational processes may play a critical role in the pathophysiology of schizophrenia.
One of the key questions for our field is when precisely do cognitive problems begin in people with schizophrenia? This is thought to be important as it may help point to the culprit neurodevelopmental event gone awry in the disease. Population and birth cohort studies show that some premorbid individuals displayed lower cognitive ability during childhood and adolescence. Significant deficits in premorbid IQ can be evident by the age of 16 years in people who subsequently develop schizophrenia (Dickson et al., 2012) and may precede the prodromal period (Woodberry et al., 2008; Khandaker et al., 2011). Overall group deficiencies in cognitive ability may be detectable before 8 years of age in people with schizophrenia (Jones et al., 1994; Cannon et al., 2002a; Seidman et al., 2013), and there is a relationship between the size of the IQ decrement and risk of subsequent schizophrenia (Khandaker et al., 2011; Schulz et al., 2012). However, not all people with schizophrenia start off with lower cognitive ability as a child; there is evidence showing that it is adolescent cognitive development in particular that is subject to perturbation in at least 40% of men and women later diagnosed with schizophrenia (Reichenberg et al., 2005). Additionally, there is a well-established 10 point drop from fairly normal pre-morbid IQ estimates in many adults with schizophrenia (50%), which demonstrates that many people with schizophrenia experience healthy childhood cognitive development, with only about 25% having a low premorbid IQ (Weickert et al., 2000a). The nature of the post-childhood schizophrenia-related cognitive declines affecting prefrontal-related verbal and executive abilities in particular (David et al., 1997; Bhojraj et al., 2010; Maccabe et al., 2013) suggests a deviation in the normal adolescent neurodevelopmental processes that typically support improvements in cognitive ability and efficiency with increasing age. We suggest that there may be at least two different patterns of cognitive decline in people who are destined to develop schizophrenia. One pattern where cognitive decline is found early in life and intellectual impairment is widespread, implying that early developmental processes occurring before 8 years of age may have gone awry (Weickert and Goldberg, 2000). However, the modal pattern of cognitive decline in people with schizophrenia is one of decline that is restricted to the second decade of life and may include more exaggerated loss of prefrontal associated cognitive functions (Weickert and Goldberg, 2000). Therefore, it may be informative to consider the underlying neurobiology in the context of alterations of developmental trajectories in later maturing executive functions in order to adequately address the risk factors for those people with schizophrenia demonstrating an adolescent IQ drop. There is some debate about whether premorbid cognitive changes represent liability- or disease-related processes. Whichever is the case, the disease-specific cognitive dysfunctions manifesting during adolescence (Heinrichs and Zakzanis, 1998) and prefrontal, temporal, and parietal cortical gray matter deficits associated with the prodrome (Wood et al., 2008) and schizophrenia itself (Cannon et al., 2002b) may be separate from or additional to the early childhood cognitive dysfunctions found in the subset of people who go onto develop schizophrenia.
The current literature suggests that brain changes in schizophrenia, if apparent premorbidly, are prominent in prefrontal and associative cortical regions and that they correlate with premorbid executive dysfunction. Structural brain changes present in adolescence or young adults during the early transition to illness provide further support for this view (Pantelis et al., 2003). The findings described above suggest a period of later development (e.g., adolescence) when neural perturbations may result in schizophrenia. Therefore, we suggest that schizophrenia should be considered in the context of alterations in the developmental trajectories of these executive functions and their underlying dynamic changes in neural substrates to adequately address the risk factors for aberrant neurodevelopment of schizophrenia. The remainder of this review will address the timing of normal prefrontal cortical development at the cellular and molecular level and its potential to inform us about the cause of neurobiological changes found in the cortex of people with schizophrenia.
Early Events in the Formation of the Human Cortex
Appropriate genesis and migration of neurons are essential building blocks for a healthy brain. Relatively recent studies challenge dogma by suggesting that for some neuronal subtypes these processes may be ongoing throughout development and into adulthood (Gould et al., 1999; Weickert et al., 2000b; Bernier et al., 2002; Fung et al., 2010, 2011a; Wang et al., 2011). This has implications for adolescent-onset neurodevelopmental disorders, such as schizophrenia, as aberrations in these processes could be contributing to disease. Thus, while we begin with early events, we suggest that some of these early processes are not completely exhausted in post-natal life.
Neurogenesis and Neuronal Migration
When the nervous system begins, pluripotent cells from the outer embryonic layer, the ectoderm, produce neuroepithelial cells of the neural plate that invaginates to form the neural tube. The most rostral region of this tube swells to form the telencephalon with the lumen eventually becoming the lateral ventricles of the cerebral hemispheres. The telencephalon forms the cerebral cortex, basal ganglia, and hippocampus (Kandel et al., 2000). One of the most important divisions of the telencephalon relevant to schizophrenia neuropathology is the distinction between the dorsal (pallium) and ventral (subpallium) neurogenic zones as they by and large give rise to the two different neuronal types of the cortex. These zones are not only defined by their position, but by expression of distinct molecules [reviewed by Corbin and Butt (2011)]. Progenitors from the dorsal telencephalon spawn all the cortical excitatory pyramidal neurons that migrate radially to the cortical layers (Rakic, 1972). The ventral neurogenic zone gives rise to most of the cortical inhibitory neurons (Anderson et al., 1997; Tamamaki et al., 1997; Ayala et al., 2007) that migrate tangentially to reach the cortex (see below). Recent studies suggest that some inhibitory (GABAergic) interneurons in the human and non-human primate cortex, particularly many of those expressing calretinin, could also arise from progenitors within the dorsopallial proliferative zone (Letinic et al., 2002; Rakic and Zecevic, 2003; Fertuzinhos et al., 2009; Petanjek et al., 2009a,b; Zecevic et al., 2011), although it is unclear if these “dorsal” progenitors may be ventral in origin, having migrated to the dorsal neurogenic zone at an earlier developmental stage.
Cells in both germinal zones initially undergo proliferative, symmetric division, giving rise to two daughter cells that both retain their apical process and progenitor identity, resulting in expansion of the progenitor population (Kosodo et al., 2004; Kosodo and Huttner, 2009). The greatly expanded neuronal number and surface area of the human cerebral cortex depends on repeated rounds of symmetric cell division followed by later asymmetric division resulting in committed neuronal precursors. Given that there are not gross abnormalities in the size or shape of the human brain (in most cases of schizophrenia) it is likely that these very early embryonic events of division and early neuronal differentiation are intact in most people who later go on to develop schizophrenia. However, neuronal precursors residing in the ventral subventricular zone (vSVZ: Haubensak et al., 2004) retain the ability to divide and generate neurons well into post-natal human life and into adulthood (see below), and more selective disruption of the processes controlling vSVZ neurogenesis could lead to much more subtle changes impacting development of cortical interneurons. Therefore, disruption of the vSVZ neurogenesis process would be consistent with the pathology of mental illnesses involving GABAergic interneurons and with a post-natal age of onset, like what is found in schizophrenia.
Indeed, a deficit in GABAergic inhibition is one of the most robust findings in schizophrenia neuropathology, so the timing of cortical interneuron development warrants further consideration. In the human, GABAergic interneuron birth in the (vSVZ) ganglionic eminence starts as early as 5 gestational weeks (gw: Zecevic et al., 2011) and neurons are found in the nascent human cortex by 5.5–6 gw (Zecevic, 2004). Radial glial cells in the dorsal VZ form around 17 gw. Proliferation of the calretinin positive cell can be detected in the human dorsal SVZ at 20 gw (Zecevic et al., 2011). At 23 gw in the human, proliferation in dorsal VZ proliferation is no longer detectable suggesting that most of the cortical pyramidal neurons are born in embryonic life (Zecevic, 2004; Figure 2). Markers of immature neurons are, however, still abundant in the vSVZ (where cortical interneurons are born) during the first few post-natal years of life (Weickert et al., 2000b; Chong et al., 2008; Wang et al., 2011) and persist into adulthood in humans and monkeys (Fung et al., 2011a; Wang et al., 2011; Figure 2). Neurogenesis has been implicated in schizophrenia pathology by the involvement of disease-associated neurogenesis regulator genes such as DISC1 and neuregulin (Millar et al., 2000; Ghashghaei et al., 2006; Duan et al., 2007; Mao et al., 2009). Current studies using post-mortem material have reported reduced adult neurogenesis in the hippocampus (by PSA-NCAM and Ki67 immunoreactivity: Barbeau et al., 1995; Reif et al., 2006), however, neurogenesis in the vSVZ remains to be studied in schizophrenia.
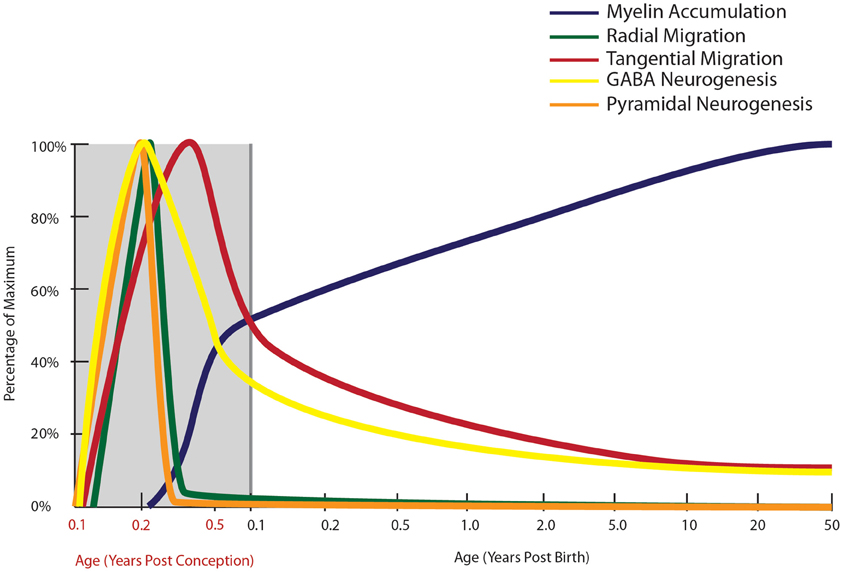
Figure 2. Normal developmental trajectory of neurogenesis, neuronal migration, and myelination in the human. Neurogenesis and the subsequent migration of neurons to the cortex begin within a few weeks of gestation in the human (Zecevic et al., 2011). Pyramidal neurogenesis and migration of these cells to the cortex occurs by radial migration and is completed by mid-gestation (Nadarajah and Parnavelas, 2002), while genesis and migration of GABAergic interneurons continues into early post-natal life, with emerging evidence based on the presence of molecular markers of immature neurons suggesting that this process continues into adulthood in primates (Gould et al., 1999, 2001; Bernier et al., 2002; Fung et al., 2011a; Wang et al., 2011). Prefrontal myelination occurs predominantly in early post-natal life, still increasing through adolescence before reaching adult levels (Kang et al., 2011).
Radial Migration of Neuroblasts From SVZ
After their birth, neurons travel either by radial (dorsal-born pyramidal neurons), or tangential (ventral-born GABAergic interneurons) migration to their final destination in the cortex. Radial migration, via radial glial cells, occurring around 20 gw in humans, is the main mode of migration for pyramidal neurons (Rakic, 1972; Nadarajah et al., 2001; Hatten, 2002; Nadarajah and Parnavelas, 2002; Nadarajah, 2003; Figure 2). Once radial migration of pyramidal neurons ceases, cortical maturation in the form of proper alignment of neuronal cells, synapse formation, and neurite outgrowth lasts from the sixteenth fetal week until the post-natal period (Sidman and Rakic, 1973; Lequin and Barkovich, 1999). One molecular regulator of radial migration and cortical layering, reelin, is an extracellular matrix protein produced by Cajal-Retzius cells in layer I of the cerebral cortex (Marin-Padilla, 1990; Del Rio et al., 1997). In mice lacking normal reelin function (reeler mice), inverted neocortical layers and abnormally dispersed cells are observed due to lack of neuronal ability to respond to migration signals in reeler mutants (Caviness, 1976; Goffinet, 1984; D'Arcangelo et al., 1995; Chai et al., 2009). Reelin is down-regulated in post-mortem brains from people with schizophrenia (Grayson et al., 2005), with significant (approximately 50%) reelin reductions in hippocampus, caudate nuclei, and cerebellum (Impagnatiello et al., 1998; Costa et al., 2002). However, the fact that cortical layering appears to be fairly intact in most cortical areas in schizophrenia, is not consistent with a major loss of function of the reelin gene early in development. Anomalous cortical development due to distorted distribution of neurons, especially in layer II of the entorhinal cortex from patients with schizophrenia has been reported (Jakob and Beckmann, 1986; Arnold et al., 1991, 1997; Falkai et al., 2000) at least in some cases, but not all (Jakob and Beckmann, 1986; Krimer et al., 1997; Bernstein et al., 1998), which could be consistent with more subtle alterations in reelin-mediated migration events than what is found in the reeler mouse.
Tangential Migration of Interneurons
In recent decades, there has been an increased understanding of the origins and migration of cortical interneurons (Anderson et al., 1997; Nadarajah and Parnavelas, 2002; Zecevic et al., 2011). During development of the mammalian telencephalon, interneurons migrate tangentially from the ventral ganglionic eminences (vSVZ) to the developing cortex (Corbin et al., 2001; Marin and Rubenstein, 2003; Takemura, 2005). Classically, the migration of cortical interneurons was thought to be completed during fetal life (Sidman and Rakic, 1973; Korr and Schmitz, 1999; Zecevic et al., 2005) and thus the number of interneurons was believed to remain stable throughout life (Spalding et al., 2005; Bhardwaj et al., 2006). While migration of interneurons occurs in the human embryo (Sidman and Rakic, 1973; O'Rahilly and Muller, 1994; Meyer, 2001; Bayatti et al., 2008) and may peak in the midgestational period in humans (Meyer, 2001), recent evidence suggests that it may also continue years after birth. Since we and others find a high density of nascent neurons in their birthplace, the SVZ, and a high neuronal density just below the cortex in human infants (Chong et al., 2008; Fung et al., 2011a; Wang et al., 2011), this suggest that addition of new cortical interneurons may contribute to the large growth of the human brain evidenced by a quadrupling of brain weight (Dekaban and Sadowsky, 1978; Beltaifa et al., 2005) and a large increase in cortical gray matter volume from birth to about 5 years of age (Iwasaki et al., 1997; Shankle et al., 1998a,b; Durston et al., 2001; Lenroot et al., 2007). So, while regressive events have received the bulk of the focus in considering cortical development in the context of schizophrenia there is in fact much larger cortical growth events, which are occurring within the first few years after birth.
Measurement of biochemical markers representing different interneuron subtypes suggests that parvalbumin and cholecystokinin mRNAs are increased over post-natal development in humans, most strikingly in the first 5 post-natal years (Figure 3). Parvalbumin immunoreactivity first appears in the DLPFC around 3–6 months of age and mRNA is increased 20-fold from neonatal to adult years, indicating that arrival and/or robust maturation of these interneurons occurs in post-natal primate life (Reynolds and Beasley, 2001; Erickson and Lewis, 2002; Cruz et al., 2003; Grateron et al., 2003; Fung et al., 2010). The cell density or mRNA expression of other interneuron markers, such as calbindin and vasoactive intestinal peptide increase similarly in the early post-natal years reaching a peak in children (~4–7 in human) prior to declining to adult levels in adolescence (Yan et al., 1995; Delalle et al., 1997; Fung et al., 2010; Figure 3). Post-natal expression of neuropeptide Y, somatostatin, and calretinin declines with age, indicating that these markers may contribute more to early brain development and become down-regulated with normal brain maturation (Figure 3). For example, density of dendrite-targeting somatostatin immunoreactive cells increases in the non-human primate cortex from E120 to E140, before declining through post-natal life and adulthood (Yamashita et al., 1989; Hayashi et al., 1990), with expression in human being dramatically reduced over the first 10 years of life (Fung et al., 2010). Taken together, these results suggest that recruitment, differentiation, growth, and refinement of cortical inhibitory interneurons is a major developmental event, if not the major developmental event, occurring as cognitive abilities like working memory and language are initially attained.
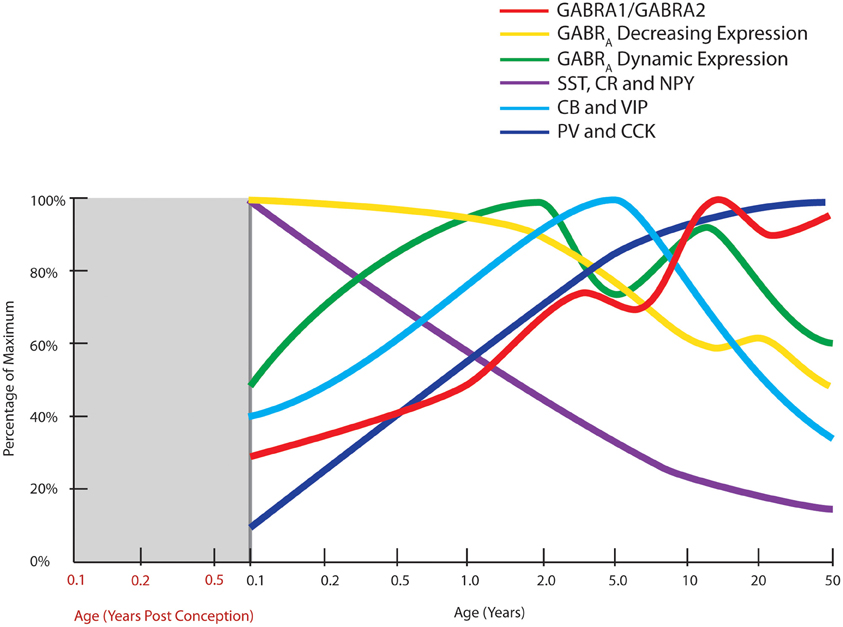
Figure 3. Normal developmental trajectories of expression of inhibitory system components in the human prefrontal cortex. Normal development of GABA receptor components of the inhibitory system is dynamic until adolescence where the trajectories reach a steady or declining state. GABAA (GABRA) subunits display two distinct patterns, one of decreasing expression following birth (α2, α5, β1, γ1, γ3 subunits) and a M pattern (dynamic expression) with peaks at toddler and teenage time periods (α1, α4, β2, β3, γ2 subunits; Fillman et al., 2010). The peak in the ratio of GABA α1 to α2 subunits (GABRA1/GABRA2) coincides with increased gamma band power in the prefrontal cortex (refer Figure 1). Expression of inhibitory neuron markers display either decline over post-natal life (SST, somatostatin; CR, calretinin; NPY, neuropeptide Y), initial up-regulation and then decline or plateau around school age (CB, calbindin; VIP, vasoactive intestinal peptide), or increased expression over post-natal life (PV, parvalbumin; CCK, cholecystokinin; Fung et al., 2010).
In addition to the developmental profiles of interneuron markers, high levels of neuronal migration markers such as DCX and poly-sialyated neuronal cell adhesion molecule (PSA-NCAM) in infants (Cox et al., 2009; Fung et al., 2011a; Kang et al., 2011; Xu et al., 2011) provide compelling evidence that cortical interneuron migration continues post-natally, particularly in the first few years of life (Figure 2). In the adult primate brain, new neurons are produced by two neurogenic regions, the sub-granular zone of the hippocampus (Eriksson et al., 1998) and the vSVZ (Curtis et al., 2003), producing granular cells in the dentate gyrus and interneurons thought to be destined for the olfactory bulb, respectively (Kornack and Rakic, 1999, 2001; Gould, 2007). In the rhesus macaque, we have found ~50,000–80,000 dividing cells/12 h along the SVZ of juvenile and adult macaques, using both BrdU and Ki67 positive cell markers of cell proliferation (unpublished; Shalaev et al., 2002). Other studies have demonstrated BrdU and NeuN/GAD67 co-labeled cells in the principal sulcus of adult macaques (Gould et al., 1999; Koketsu et al., 2003; Runyan et al., 2006; Ashrafi et al., 2007) suggesting that some of these dividing cells may contribute to an adult generated population of interneurons in the cortex. The abundance of DCX positive cells around the ventricle, and the presence of PSA-NCAM positive neurons in the white matter of the principal sulcus in non-human primate brain, further support post-natal generation of neurons from the vSVZ (Fung et al., 2011a). Additionally, the presence of GAD65/67+, somatostatin+ and neuropeptide Y+ neurons with morphology reminiscent of tangentially migrating cells (elongated cell body and leading and/or trailing processes parallel to the pial surface) in the post-natal white matter of primates may indicate the presence of post-natally migrating interneurons (Fung et al., 2011a; Yang et al., 2011; Joshi et al., 2012). A recent study by Alvarez-Buylla and colleagues (Sanai et al., 2011) provides further evidence of a major migratory pathway (DCX+, PSA-NCAM+ cells) in post-natal human brains originating from the SVZ and targeting prefrontal cortex. In primates, there are several reports of immature neurons in the amygdala, and in piriform, inferior temporal and prefrontal cortices in post-natal life (Gould et al., 1999, 2001; Bernier et al., 2002; Runyan et al., 2006; Ashrafi et al., 2007). The recent and emerging post-natal primate studies provide new insight into our still incomplete knowledge of the neurodevelopmental trajectory for tangential neuronal migration and we suggest that cortical interneurogenesis is protracted in humans and may continue at a low level throughout human life and we have incorporated this possibility on our graph (Figure 2).
Interestingly, most biochemical markers of interneurons are reduced in schizophrenia, supportive of an interneuron deficit in the disease. Specifically, there is strong evidence for reduced expression of the calcium binding protein parvalbumin (Beasley and Reynolds, 1997; Reynolds and Beasley, 2001; Reynolds et al., 2002; Hashimoto et al., 2008a,b; Morris et al., 2008; Sakai et al., 2008; Bitanihirwe et al., 2009; Mellios et al., 2009; Fung et al., 2010) and the neuromodulatory peptide, somatostatin (Hashimoto et al., 2008a,b; Morris et al., 2008; Fung et al., 2010). Given the developmental studies highlighting the protracted maturation of interneurons within the primate cortex over a decade or more (see Figure 3) the inhibitory neurons are implicated as a key substrate where brain development may be derailed in schizophrenia, with particular emphasis that a balance of cortical interneuron markers is normally achieved during adolescence (with stabilization of some markers, but down-regulation of others) and overlapping with timing of schizophrenia onset. This requires a new understanding of adolescence as a time of dynamic developmental change, not just as a time of regressive events, but one where the inhibitory system is still being rearranged and positioned for adult life.
There is new evidence that post-natal cortical interneuron migration is of relevance to the pathophysiology of schizophrenia, but this requires a re-interpretation of existing data. Ample evidence for increased interstitial white matter neuron (IWMN) densities in schizophrenia exists (Akbarian et al., 1993; Anderson et al., 1996; Eastwood and Harrison, 2003, 2005a; Kirkpatrick et al., 2003; Yang et al., 2011; Joshi et al., 2012). These observations were initially interpreted as increased remnants of early-generated cortical subplate neurons (a transient population of white matter neurons) derived from the dorsal pallium that failed to undergo programmed cell death (Kostovic and Rakic, 1980; Chun and Shatz, 1989). However, the observations of increased subcortical neurons found in schizophrenia can also be interpreted as being due to abnormal tangential migration of cortical inhibitory interneurons from the vSVZ (Yang et al., 2011; Joshi et al., 2012; Volk et al., 2012). The increase in IWMN density may be indicative of arrested migration of cortical interneurons during their journey to the cortex earlier in life or increased genesis and migration of new interneurons toward the cortex in response to possible cortical trauma, such as increased inflammation (Fillman et al., 2013). In either case, the recognition that cortical interneurons are generated from a unique birth place (vSVZ), require particular neurodevelopmental signals, can take years to establish themselves, and may have the ability to continually turn over, even at low levels, suggests that a greater understanding of the timing, the control and extent of interneurogenesis and migration may provide important clues as to the developmental origins of interneuron pathology in schizophrenia.
Myelination
Another developmental event that extends well into post-natal life is myelination (Figure 2). Brain axonal tracts are myelinated progressively across the lifespan in a region- and function-specific manner. Early human post-mortem work first described regional differences in the degree of myelination across the lifespan and highlighted the protracted development of a number of white matter tracts. Comparison of qualitative myelin staining in 200 cases from mid-gestation to 1-year old, from 1 to 30-years old, and numerous older cases yielded findings of earlier myelination of the hippocampus, primary motor, and primary sensory areas, and later myelination of frontal cortical white matter, extending into the 4th decade of life (Yakovlev and Lecours, 1967). A follow-up study (n = 12 brains) corroborated these findings in the primary motor and sensory cortices, but failed to confirm protracted myelination of the prefrontal cortex (Benes, 1989). Both studies were limited by critical age gaps in the cohort or very small sample sizes at certain stages. A comprehensive larger study graded myelination in 62 white matter sites within 162 post-mortem brains from newborn to 3-years of age (Brody et al., 1987; Kinney et al., 1988). This study confirmed that the major white matter tracts, subcortical regions, primary cortical areas, and hippocampus become myelinated sooner than association cortical areas, but estimated that even in association areas the majority of infants achieve substantial myelination by the age of 2. A more recent study investigated a marker for myelination, myelin basic protein (MBP), in the parietal cortex and found MBP expression commenced at around 3.5 months of age, and reached adult-like levels from 13 months of age (Haynes et al., 2005). Investigation of hippocampal myelination has also suggested the importance of early myelin development, with appearance of oligodendrocytes from gw 20 and of myelinated fibers from post-natal week 2 (Abraham et al., 2010). By 11 years of age, myelinated fiber density in the hippocampus was well developed but had still not reached the highest adult levels (Abraham et al., 2010). These findings suggest that most myelination and axonal growth occur within the first 2 years of early life, but may be ongoing in many telencephalic areas including the frontal cortex and hippocampus. Post-mortem microarray data from our laboratory (Harris et al., 2009; Weickert et al., 2009), have demonstrated dramatic increases in mRNA transcripts of myelin proteins in the prefrontal cortex across human post-natal life. We observed increasing mRNA expression of MBP, myelin-associated oligodendrocyte basic protein (MOBP) and proteolipid protein 1 (PLP1) during the first decade of life (all p < 0.001). From birth to 2 years of age, there was an approximately 100 times increase in MBP expression, a 20 times increase in MOBP expression and a 10 times increase in PLP1 expression, followed by a much more gradual increase of all three transcripts from early childhood into their peak in adolescence. These findings are supported by other recent microarray data, in which expression of genes associated with myelination reached approximately 50% of adult levels by birth and 95% of adult levels by 2–3 years of age in the neocortex (Kang et al., 2011). These post-mortem gene expression findings suggest that the most active myelination of the human prefrontal cortex occurs early in life, predominantly in the first 2 years of life, but continues throughout childhood and adolescence before full myelin maturation levels are reached in adulthood.
The molecular findings of developmental myelination trajectories in normal humans have been confirmed with brain imaging studies of growth of white matter tracts across human post-natal development. Throughout the brain, white matter, especially myelin, develops rapidly in the first 12 months of life (with most dramatic changes from 0 to 3 months) followed by slower change from 1 to 2 years and thereafter (Schneider et al., 2004; Dubois et al., 2006; Hermoye et al., 2006; Provenzale et al., 2007; Gao et al., 2009). After the first year of life, and throughout childhood and adolescence, changes in white matter measures (fractional anisotropy) are more subtle and are predominantly due to altered perpendicular diffusivity, reflecting increasing myelination (Giorgio et al., 2008; Lebel et al., 2008; Gao et al., 2009). However, global age-related white matter changes can still be observed in adolescents (Barnea-Goraly et al., 2005; Giorgio et al., 2008, 2010; Bava et al., 2010), while region-specific changes can continue in young adulthood (Giorgio et al., 2008). White matter maturation occurs in a region specific manner, with late maturation associated with the prefrontal cortex throughout the first 2 decades of life reaching full maturation at 18 years (Barnea-Goraly et al., 2005) and fronto-temporal tracts approaching full maturation after 25 years (Lebel et al., 2008).
Overall, these studies provide strong evidence that myelination throughout the brain takes place most rapidly early in life. However, in numerous regions, including prefrontal regions, the process continues throughout the second decade of life with full maturation only reached in late adolescence or young adulthood. In brain regions implicated in the pathogenesis of schizophrenia, such as the prefrontal cortex, full white matter maturation is mostly achieved prior to the period of life when schizophrenia emerges. However, improvements in working memory (which employs the prefrontal cortex) are associated with increased white matter maturation during adolescence (Nagy et al., 2004; Bava et al., 2010), while poorer performance is associated with less white matter maturation (Bava et al., 2010). Deficits of working memory are also consistently seen in schizophrenia (Goldman-Rakic, 1994; Meyer-Lindenberg et al., 2005), and structural abnormalities in white matter underlying the DLPFC have also been reported (Kyriakopoulos et al., 2008). Microarray studies of post-mortem brains from patients with schizophrenia have implicated myelination deficits in the pathology of schizophrenia (Hakak et al., 2001; Matthews et al., 2012). These observations have been validated by targeted investigations of cortex, which have revealed a decrease in mRNA (Matthews et al., 2012) and protein levels (Honer et al., 1999; Parlapani et al., 2009) of MBP in schizophrenia, though not consistently (Beasley et al., 2009). Thus, it seems likely that abnormal maturation of white matter could contribute to cognitive disturbances found in schizophrenia and that a better understanding of the molecular control of white matter maturation and how it could be disrupted in schizophrenia may reveal novel clues to neurodevelopmental origins of schizophrenia.
Excitatory Synapses
Probably the most popular theory for neurodevelopmental origins of schizophrenia is the theory that dendritic spines are over-pruned in adolescence. The formation and elimination of dendritic spines, subcellular structures specialized for receipt and integration of excitatory input to pyramidal neurons is important as spine numbers are thought to correlate with cognitive and learning ability (Elston, 2000).
Dendritic Arborization and Spine Densities
An early study (Huttenlocher, 1979) quantified synaptic density in the cerebral cortex across the human lifespan and suggested that synaptic regression occurred after the first two post-natal years; however, this study (Huttenlocher, 1979) had only one brain available in the age group between 7 and 25 years of age and one between 25 and 50 years of age, requiring a great deal of extrapolation from few data points. On the basis of the available data, it was concluded that synaptic regression occurs anytime between 2 and 16 years of age in human frontal cortex. It was a few years later that Feinberg interpreted these data as supporting the notion that a physical change, namely “synaptic pruning” could be occurring in the human brain during adolescence, and that emergence of schizophrenia symptomatology may be due to an overexuberance of synaptic elimination (Feinberg, 1983; Keshavan et al., 1994; Selemon and Goldman-Rakic, 1999). While this speculation has had enormous influence on neurodevelopmental models of schizophrenia and is found in most reviews on this topic, few investigators seem to carefully scrutinize the evidence upon which this theory is based, often interpreting a “dotted” line plotting supposed time of synaptic reduction as adolescent pruning, when in fact it is based on only 3 individuals and shows a putative reduction in synaptic density starting several years before adolescence. Further, as synapse densities change with brain volume changes and possible gain or loss of whole neurons, glia and blood vessels, it is not clear if changes in synapse densities across development are attributable to a net loss of number of synapses per neuron (pruning) or changes in these other factors (Huttenlocher, 1990). A subsequent report by Huttenlocher and Dabholkar (1997) with four adolescent cases could suggest in fact that there may indeed be no change or a small net increase in synapse density in the time period between infancy and adolescence in the human prefrontal cortex.
A more recent Golgi study of dendritic development in human cortex by Petanjek and colleagues (2008) spans a broad age range (newborn to age 91 years, n = 25) and includes 13 cases between the ages of 1 week and 10 years and studied the development of the layer III basilar dendritic tree from birth to the beginning of adolescence. There was an effect of age on all dendritic variables measured, except for the number of basal dendrites, which remained constant. The dendritic segment count reached adult levels by the age of 12 months. A 3-fold increase in the length of the dendritic tree of layer III pyramidal cells by 2.5 months of age with an additional 50% increase in the length of terminal dendritic segments between 16 and 30 months of age was observed. A plateau in intermediate dendritic segment length was observed during late adolescence, a period which was represented by three cases in this study (Figure 4). These data suggest that the major developmental event during early childhood is exuberant early growth of structures that are specialized to receive synapses and is not consistent with large-scale “pruning” of synapses or synaptic reductions that were believed to occur during post-natal life based on synaptic density counts (Huttenlocher, 1979).
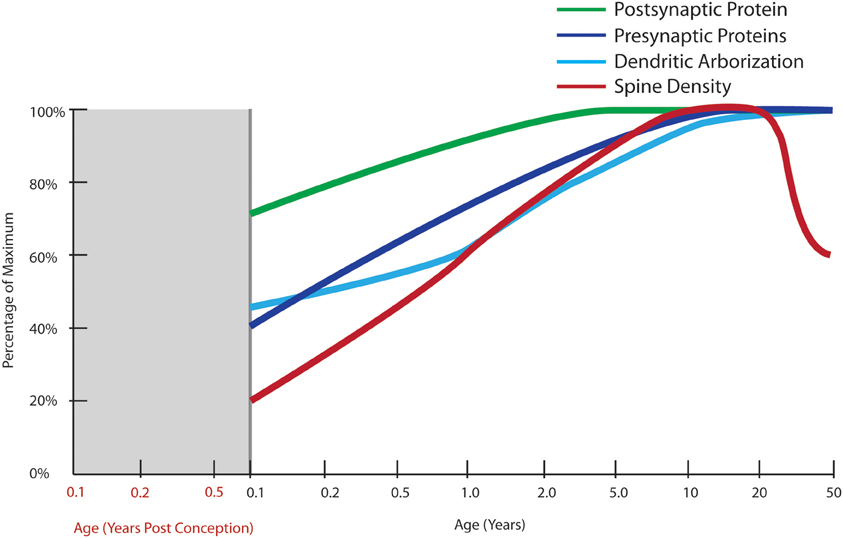
Figure 4. Normal developmental trajectories of excitatory system components in the human prefrontal cortex. Normal development of the excitatory system involves increasing expression of presynaptic SNARE complex proteins (SNAP-25, syntaxin 1A, VAMP1) into early adulthood (Webster et al., 2011) and a similar increase in protein expressions of the post-synaptic markers, PSD95 and spinophilin, though starting at a higher baseline at birth (Kang et al., 2011; Webster et al., 2011). Dendritic arborization indexed by average length of the total dendritic tree of layer III pyramidal neurons does not reach its maximum until adulthood with a slight decline after the age of 30 years (Petanjek et al., 2008). Quantification of Golgi-impregnated tissue suggests an increase in spine density from birth until early school age, followed by a period of gradually decreasing density starting in early adulthood which lasts until middle age (Petanjek et al., 2011).
In another study, basilar dendritic complexity appeared greater in younger individuals (14–40 years of age) than in older individuals (40–106 years of age; Jacobs et al., 1997). This was reflected in a statistically significant reduction of 9–12% in total dendritic length and a slight (4%), but statistically significant increase in the number of dendritic branches per cell in the older adult age group compared with the younger age group. The most substantial change observed was in dendritic spine measures, with a gradual 50% decrease in the number and density of spines from the mid-twenties to mid-adulthood (40 years: Jacobs et al., 1997). This suggests that the interpretation of earlier findings of changes in synaptic density reported by Huttenlocher (1979) could have been influenced by the many individuals in the 60–80 year range. However, Jacobs et al. (1997) found the number and density of spines between the ages of 40 to 106 years appeared stable in this study and the variability due to aging was relatively constricted to early adulthood. A subsequent study details changes in spine density in the DLPFC across the lifespan (Petanjek et al., 2011) using a post-mortem brain collection overlapping with that used in Petanjek's study described above. A peak in layer III pyramidal neuron spine densities in early life with higher level in school age as compared to the first year of life and an apparent stability from childhood through adolescence (5–20 years of age), and a change that begins after the second decade of life where spine density appears to gradually decline over the next 30 years (to age 50, Figure 4) and where this loss extends into aging (not graphed).
The studies reviewed above (Jacobs et al., 1997; Petanjek et al., 2008, 2011) had substantial inter-individual variability attributable to differences in the populations studied, and relatively small collection sizes in the context of broad age-ranges of the samples used. Taken together, these studies suggest that the timing of synaptic density change likely involves very early post-natal increases and that the timing and extent of synaptic density decreases and reductions in dendritic spines may be gradual and protracted in humans. The studies available suggest that synaptic pruning may not actually be prominent in human adolescence but may have its onset in toddlerhood until school age and/or begin after adolescence and extend into the 5th decade of life. Thus, the reviewed data suggest that the onset of synaptic regression is unlikely to be the trigger for the onset of schizophrenia. Another way to gather support for the timing of synaptic regression would be to examine quantitative measures of molecular makers of synapses during normal human cortical development as we and others have done and elaborate on next.
Molecular Markers of Excitatory Synapses
While development of the glutamatergic synapses begins prenatally with the majority of synapses formed during the neonatal period (Feldmeyer and Radnikow, 2009) other work suggests the levels of one protein often used as marker of synapse density, synaptophysin, peaks much later in life during school age years (6–10 years of age) while levels of a protein thought to be marker of dendritic spines, PSD-95, peaks even later, in early adolescence and then marginally declines (Glantz et al., 2007). Consistent with this, Salimi et al. (2008) found a similar pattern of a late peak in the protein expression of complexin 2, a marker of excitatory synapses. Importantly, the ratio of excitatory to inhibitory synapse markers (complexin 2/complexin 1) decreased in adolescence (Salimi et al., 2008), again pointing to the maturation of the inhibitory system as an important adolescent event. Our recent data from multiple molecular markers suggest that developmental increases in synaptic proteins are not followed by a significant decline either in childhood or in adolescence as would be expected with major synaptic regression (Webster et al., 2011). We have found that a variety of synaptic mRNAs and proteins increase steadily after birth during childhood years (Webster et al., 2011). This pattern of increasing expression was found for many presynaptic proteins and was particularly true of the SNARE complex proteins (syntaxin 1A, SNAP-25, and VAMP1) where syntaxin-1A levels peaked in young adulthood or even in adult age (Fung et al., 2011b; Webster et al., 2011; Figure 5). In terms of molecules localized to the postsynaptic elements, PSD-95 and spinophillin both had highest mRNA levels during infancy with protein levels increasing until school age and with no evidence of an adolescent decline in protein (Webster et al., 2011; Figure 5). Overall there is a major developmental increase in synaptic components, especially at the protein level, from neonates through school age and adolescence that remains relatively stable through adolescence and into adulthood. These observations are contradictory to the model proposed by earlier studies that suggested the major developmental event in adolescence is regression or loss of synaptic elements (Huttenlocher, 1979; Feinberg, 1983; Glantz et al., 2007). The work reviewed here provides evidence that the theory stating that exuberant excitatory synapse loss could be the basis for the aberrant adolescent brain development in schizophrenia may need to be reconsidered. This position gains additional support from the lack of consistent molecular changes in presynaptic mRNAs and proteins in schizophrenia brains (see below).
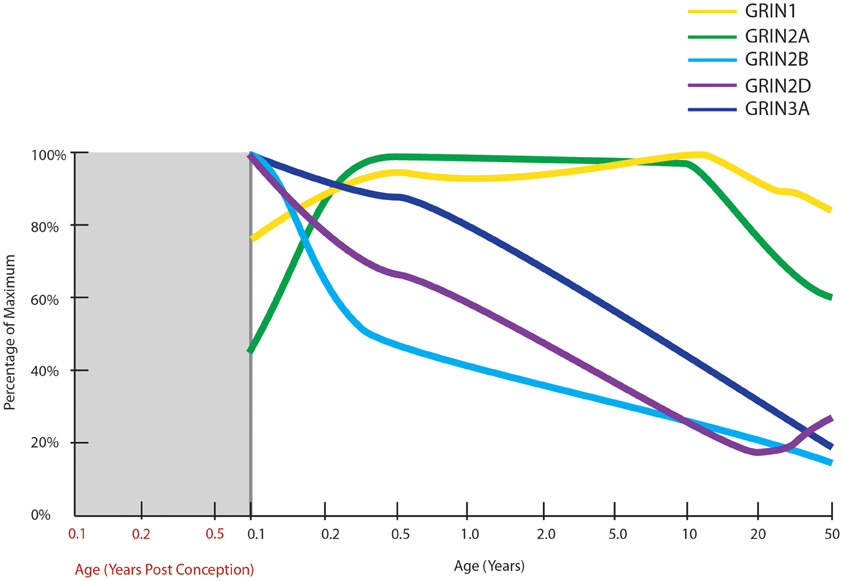
Figure 5. Normal developmental trajectories of NMDA receptor subunits in the human prefrontal cortex. Normal development of NMDA receptor subunits (Choi et al., 2009; Colantuoni et al., 2011; Kang et al., 2011). mRNA of the obligatory NMDA receptor subunit, GRIN1 (NR1), is expressed at fairly steady levels post-natally, at least until middle age. GRIN2A expression increases in infancy to attain fairly steady levels throughout childhood and then decreases through adolescence and into adulthood. GRIN2B, GRIN2D, and GRIN3A all have their highest expressions at birth with decreasing trajectories thereafter.
Studies of markers of excitatory synapses in neocortex of patients with schizophrenia primarily find no change (Eastwood and Harrison, 2001, 2005b; Sawada et al., 2002; Halim et al., 2003; Fung et al., 2011b) or slightly decreased levels (Eastwood and Harrison, 2005b). Similarly, measurement of excitatory spine markers has led to divergent results (Law et al., 2004; Weickert et al., 2004; Toro and Deakin, 2005; Kristiansen et al., 2006; Catts and Weickert, 2012), mainly suggesting there is no change or only slight decreased expression of spinophilin and PSD-95 in cortex of patients with schizophrenia. A molecular marker of dendrites, microtubule-associated protein 2, is also typically unaltered in level, but sometimes decreased in the cortex (Jones et al., 2002; Mukaetova-Ladinska et al., 2002; Somenarain and Jones, 2010). Even when slight decreases in molecular markers of excitatory synapses are found, they are typically identified in only a subset of cases (Faludi and Mirnics, 2011), not found in all anatomical regions (Webster et al., 2001; Law et al., 2004; Toro and Deakin, 2005), restricted to certain layers (layer III basilar dendrites only: Glantz and Lewis, 2000; Kolluri et al., 2005) and not consistent across cohorts (Halim et al., 2003; Eastwood and Harrison, 2005b; Castillo et al., 2010; Gray et al., 2010; Fung et al., 2011b). Taken together, schizophrenia, in general, does not appear to be characterized by a pervasive and consistent loss of excitatory synapse markers.
This raises the question of what physical elements could be responsible for cortical gray matter volume reductions found in schizophrenia? A recent and quite comprehensive consideration of the cellular components of cortical gray matter has yielded the following estimates for percentage of space occupied by each: neurons = ~64%, glial = ~12.5%, synapses = ~6%, capillaries = ~0.5%, and extracellular spaces = ~17% (Bennett, 2011). Based on available histological evidence from studies on people with schizophrenia, Bennett suggests that a deficit in spines (Glantz and Lewis, 2000; Kolluri et al., 2005) alone would be insufficient to explain the gray matter loss found in imaging studies which is typically on the order of a 8–10% reduction. The fact that the loss of spines is quite anatomically restricted, i.e., occurs in deep layer III, but not in superficial layer III, and is not found in layers V or layers VI in frontal cortex also supports that spine loss is likely only a partial explanation for cortical thinning (Glantz and Lewis, 2000; Kolluri et al., 2005). Bennett concludes that other elements must be involved and he suggests that loss of dendrites is the likely suspect (Bennett, 2011). However, the 30% loss of dendrites used to form this argument appears to only apply to basilar dendrites as apical dendrites and segment length are actually non-significantly increased in the cortex of people with schizophrenia (Kalus et al., 2000). Considering that basilar dendrites occupy an estimated 60% of the total dendritic length (Soloway et al., 2002), this reported loss of dendrites still may be only a partial explanation for cortical volume loss. What the anatomical substrate of the consistent reductions in gray matter volume found in patients with schizophrenia actually is, if it is not synaptic loss, remains an unanswered question. Clearly more quantitative anatomical studies of the brains of patients with schizophrenia are needed to determine the answer. We suggest that it is premature to consider all gray matter reduction in schizophrenia to be due solely to loss of synapses or to over-exuberant synaptic pruning.
Receptor Systems
Schizophrenia pathophysiology likely involves disruptions to three neurotransmitter systems, namely; excitatory glutamatergic, inhibitory GABAergic, and modulating dopaminergic, input [reviewed in Seshadri et al. (2013)], the developmental trajectories of some of the main receptors for each are reviewed below.
NMDA Receptors
The majority of excitatory glutamate synapses, important for learning, are primarily found on postsynaptic dendritic spines (Sheng and Hoogenraad, 2007) with synapses equipped with both ionotropic and metabotropic glutamate receptors (Hollmann and Heinemann, 1994). The expression trajectories of some ionotropic glutamate receptor subunits, those of AMPA and kainite, do not change during post-natal life. The expression profiles are in fact quite similar to one another, with increasing expression during gestation reaching 100% of maximum around the time of birth and remaining steady thereafter (Kang et al., 2011). Another ionotropic glutamate receptor, N-methyl-D-aspartate receptor (NMDAR), is critically important for the calcium flow in and out of cells that is responsible for both long-term potentiation (LTP) and long-term depression (LTD: Li and Tsien, 2009) while metabotropic glutamate receptors can increase or decrease the excitability of the postsynaptic neuron and regulate postsynaptic protein synthesis through second messenger systems, in effect modulating synaptic plasticity (Hollmann and Heinemann, 1994). Given the interest in NMDARs in schizophrenia, due to the psychotomimetic effect if blocked (Catts and Catts, 2010) and for their role in cognitive ability and synaptic plasticity across development, we will focus on developmental change in NMDARs.
NMDARs are heterotetrameric receptors, comprised of two obligatory NR1 subunits together with two NR2 (A-D) or NR3 (A-B) subunits, with the different assembly of subunits determining the functional differences in receptor properties (Cull-Candy et al., 2001). One of the first studies to examine NR1 mRNA and protein levels across human development in the PFC (Henson et al., 2008) found expression significantly changed across age in a sample from 18 gw to 25-years old. NR1 protein levels were low prenatally, rose to a peak at 11–15 years then reduced slightly in young adults. The mRNA expression followed a similar pattern but with a less marked reduction in adulthood. Two recent transcriptome analyses have also charted NMDAR subunit mRNAs in human prefrontal cortex from early gestation to old age (Colantuoni et al., 2011; Kang et al., 2011; Figure 5). NR1 mRNA expression was generally consistent across post-natal life, confirming our unpublished findings, and not consistent with the putative peak in NMDAR1 early adolescent peak reported earlier (Henson et al., 2008).
In general, in rodents NR2B, 2D, and 3A predominate in early life and 2A, 2C, and 3B are expressed to a greater extent in adults as compared to neonates. Thus, in the developing rodent brain, there is a shift in the ratio of NR2A/NR2B containing receptors during post-natal development in several brain regions, with a higher proportion of NR2A-rich receptors in later life (Ritter et al., 2002; Turman et al., 2002). This ratio switch is also present in the human hippocampus (Law et al., 2003) and the visual cortex (Murphy et al., 2005). NR2B-rich receptors exhibit longer excitatory postsynaptic currents (EPSCs; Monyer et al., 1994; Flint et al., 1997), enabling the coincidence of pre- and postsynaptic events and thus facilitating experience dependent synaptic plasticity in the critical early post-natal period (Philpot et al., 2001). Thus, the decreasing NR2A/NR2B ratio across life may be responsible for the decrease in plasticity seen during maturation and ageing (Crair and Malenka, 1995). In the rat hippocampus, the NR2A/NR2B ratio is believed to mediate the process of synaptic remodeling (Gambrill and Barria, 2011). Higher NR2B expression increases the rate of addition and subtraction of spines, and higher NR2A expression reduces the number of synapses and their volume.
In two recent transcriptome analyses, the prenatal expression of all NMDAR subunit mRNAs were found to be low in both studies, with exception of NR2B mRNA which is high prenatally and peaks shortly before birth (Colantuoni et al., 2011; Kang et al., 2011). While NR2C mRNA expression was steady across most of the life span, it displayed a slight increase after 70 years of age (Colantuoni et al., 2011; Kang et al., 2011). In Henson and colleagues' study of the PFC, NR3A mRNA and protein were very low during prenatal development, at maximum levels in the first year after birth and then declined gradually from ages 1 to 25, to be at 30% of the maximum expression (Henson et al., 2008). We also studied NMDAR mRNA levels in individuals aged from 1 month to 49 years (Choi et al., 2009) and found NR3A (r = −0.933) and NR2D (r = −0.785) mRNA levels were high within the first few years of human life and were significantly and gradually down-regulated with advancing age. Adult NR3A mRNA expression decreased to 35% of neonatal levels, consistent with the findings of Henson and colleagues (2008). Adult NR2D mRNA decreased to 50% of neonatal levels in our microarray study (Choi et al., 2009). Unlike other NMDAR subunits, NR2D expression is thought to be absent from cell bodies (Thompson et al., 2002) and little is known of how this specific subunit influences the physiology of the receptor, however, there is evidence that recruitment of extrasynaptic NMDARs containing NR2D are important for the expression of LTP in the hippocampus in adult rodents (Harney et al., 2008). Similar to what we find in human cortical development, rodent NR3A expression peaks early in life during the period of synaptogenesis and is down-regulated prior to the critical period of plasticity. This suggests that the critical period for NMDAR mediated plasticity in humans may start around toddlerhood in the prefrontal cortex (Wong et al., 2002; Perez-Otano et al., 2006) at a time where language acquisition and utilization grows at a rapid pace. In rodents, deletion of NR3A increases spine density and leads to failure to maintain juvenile-type synapses (Das et al., 1998; Roberts et al., 2009). Taken together, these results suggest that the major post-natal change in human cortical NMDARs may not be the NR2B to NR2A switch, but rather may feature the down-regulation of NR2D and NR3A more prominently.
The extent to which NMDAR subunit mRNAs are altered in the brains of people with schizophrenia varies with subunit and brain region, however, mRNA changes remain controversial, with decreases (Humphries et al., 1996; Sokolov, 1998; Law and Deakin, 2001; Beneyto and Meador-Woodruff, 2008; Weickert et al., 2012), increases (Akbarian et al., 1996; Dracheva et al., 2001; Schmitt et al., 2010) and no change (Akbarian et al., 1996) observed in NR1. Direct studies of NMDAR protein expression in schizophrenia patients are rare. One study found no significant changes in NR1 or NR2A-D protein levels in schizophrenia patients, however, an alternatively spliced isoform of NR1, NR1C2, was increased in the anterior cingulate cortex (Kristiansen et al., 2006). Binding studies also show mixed results depending on brain region and binding site. In the frontal cortex, studies have shown no alteration in binding at the PCP (Dean et al., 1999, 2001; Scarr et al., 2005; Beneyto and Meador-Woodruff, 2008), glycine (Nudmamud and Reynolds, 2001, polyamine or glutamate sites (Beneyto and Meador-Woodruff, 2008) in schizophrenia, while one study found an increase in glycine site binding in the schizophrenia orbital frontal cortex (Simpson et al., 1991) and increases in glycine site binding have also been demonstrated in various other cortical areas including superior temporal cortex (Nudmamud and Reynolds, 2001), somesthetic, visual, and premotor areas (Ishimaru et al., 1992). One study found increased [3 H]MK-801 binding in the putamen in schizophrenia (Kornhuber et al., 1989), however, a subsequent study showed no change in binding in the major striatal structures (Noga et al., 1997). We recently demonstrated for the first time a robust decrease in NR1 protein in schizophrenia patients in prefrontal cortex (Weickert et al., 2012). Our study also described decreased expressions of the NR1 and NR2C mRNA subunits in schizophrenia patients (Weickert et al., 2012), which are consistent with an earlier study (Beneyto and Meador-Woodruff, 2008). The Meador–Woodruff group also reported a significant increase in NR3A mRNA (Mueller and Meador-Woodruff, 2004) which could indicate that the NMDARs are in a more immature state and that higher NR3A could interfere with age-appropriate cortical plasticity in schizophrenia. Elimination of interneuronal expression of mature NMDARs post-natally, but prior to onset of adolescence, leads to a schizophrenia-like phenotype in mice (Belforte et al., 2010), suggesting that either the lower levels of the obligatory subunit (NR1) or higher levels of the NR3A subunit could interfere with the maturational changes in NMDARs and contribute to schizophrenia.
Inhibitory Neurotransmitter Receptors
The major inhibitory neurotransmitter we will consider here is gamma-aminobutyric acid (GABA) which binds ligand gated ion channel (GABAA receptor) and G-protein coupled receptors (GABAB receptor; Aguayo et al., 2004; Olsen and Sieghart, 2008). GABA is synthesized by two enzymes, GAD65 (GAD2) and GAD67 (GAD1). GAD67 is responsible for 90% of the basal GABA synthesis and is produced at limiting levels in the brain (Asada et al., 1997; Kash et al., 1997). In the human, strong basal expression of GAD67 was shown from neonate through adult, with GAD65 increasing and peaking in the teenage years (Pinto et al., 2010). However, in the DLPFC, our unpublished microarray data as well as that from Kang et al. (2011) show the mRNA for GAD65 peaks around one year of age and stays consistently expressed from infants to adulthood with little or no drop-off. Our microarray data shows a similar pattern for GAD67 implying that mRNA for both constitutive and synaptic synthesis of GABA is fairly stable throughout post-natal life. This is interesting as the enzymes responsible for GABA production are expressed at a steady rate despite large changes in distribution, maturational state, and phenotype of the interneuron populations that occur in post-natal human life (see earlier section).
GABA receptors act to hyperpolarize adult neurons, but act to depolarize younger neurons (Flint et al., 1998; Ben-Ari et al., 2007). This reversal is due to the high chloride concentrations found in immature neurons due to high developmental expression of Na-K-Cl co-transporter 1 (NKCC1, also known as SLC12A2: Di Cristo, 2007). NKCC1 is highly expressed during early fetal human development and increases until reaching a plateau in early childhood in the DLPFC (Hyde et al., 2011). The other major potassium-chloride transporter member, KCC2 (SLC12A5), is also expressed in early fetal development, but at much lower levels and also increases before reaching its steady state during early childhood (Hyde et al., 2011). There is uncertainty in establishing an exact crossover point from which GABA switches from excitatory to inhibitory, but it could be as late as 10 years of age (Hyde et al., 2011). The uncertainty can be attributed to two main causes; firstly, the individual variability of the developmental expression of NKCC1 and KCC2, both in Hyde's study and our own unpublished microarray findings, is substantial and thus the point of crossover may occur anywhere from prenatal life to school age when examining the overall profiles. Secondly, it has been shown that individual cells may have differing chloride gradients due to variable subcellular distribution of the two chloride transporters even within the same neuron, particularly at the axon initial segment (Khirug et al., 2008). This would result in GABA having an inhibitory effect on the dendritic portions of a cell but perhaps an excitatory effect at the axon initial segment. In homogenate tissues of the DLPFC, this type of individual cell variation in chloride transporter expression would be diluted but may help to explain NKCC1's continued presence throughout life when GABA action is predominately inhibitory.
Since more comprehensive and validated developmental data are available for the GABAA receptor as compared to the GABAB receptor, we will consider the developmental changes for the GABAA receptor mRNAs here. The GABAA receptor is a pentameric ion channel that is made up from a number of possible subunits, namely 6α, 3β, 3γ, δ, ε, and π subunits (Szabadics et al., 2006; Olsen and Sieghart, 2008). In the DLPFC, the human post-natal developmental change in the GABAA receptor α/β/γ subunits can be divided into two groups (Figure 3). The first comprises the subunits with decreasing mRNA expression over the course of development (α2, α5, β1, γ1, γ3) and the second comprises those with a more dynamic expression pattern (α1, α4, β2, β3, γ2) with an M-shape peaking at the toddler and teenage time periods (Duncan et al., 2010; Fillman et al., 2010). The mature forms (predominant in adulthood) of the receptor subunits such as α1, β2, and γ2 typically have rapid channel opening/closing kinetics and greater sensitivity to GABA leading to greater temporal specificity of inhibitory action (Hevers and Lüddens, 1998; McClellan and Twyman, 1999).
The postsynaptic GABAA receptor α1 and α2 subunits show consistent change in post-natal life with α1 increasing in life with greatest rate of change soon after birth with more gradual increase until young adulthood and with quite dramatic down-regulation of α2 across post-natal life (Hashimoto et al., 2009; Duncan et al., 2010). This leads to an overall increase in the α1/α2 ratio in the prefrontal cortex, and it is interesting that the later peak in this ratio (in teenagers) aligns with the increase in gamma band power found in the prefrontal cortex in humans (Uhlhaas et al., 2009). However, changes in individual receptor subunit mRNAs for the β2, γ2 subunits do not achieve their peak of expression until the teenage/young adult years (Fillman et al., 2010) implying changes in the ratio of other GABAA receptor subunits also may contribute to increasing cognitive capacity of humans through development.
One of the most robust findings in schizophrenia neuropathology is deficits in cortical inhibitory interneurons across several cortical regions (Hashimoto et al., 2008a; Thompson et al., 2009), including DLPFC. Findings include consistent reductions in expression of GAD67, which have been recently reviewed (Gonzalez-Burgos et al., 2010). Given the reproducible changes in GAD67, one may expect to see correspondingly reproducible changes in the GABAA receptor. While there appears to be a replicable change toward an increase in GABA binding in the cortex of people with schizophrenia (Benes et al., 1992; Dean et al., 1999; Newell et al., 2007; Verdurand et al., 2013), there is less consensus as to changes occurring in the GABAA receptor mRNAs in schizophrenia. In DLPFC from schizophrenia patients, Volk et al. (2002) found an increase in GABAA α2 subunit labeled axon initial segments. However, as the axon initial segment lacks mature KCC2 transporters, the effect of this putative loss of GABA and increase in GABAA α2 is debated (Volk et al., 2002; Szabadics et al., 2006). It is noteworthy that the increased expression of GABAA α2 subunit has been independently replicated (Beneyto et al., 2011), but may not be found in all studies (Duncan et al., 2010). In contrast, decreased expression of the GABAA subunits α1 and α5 in the DLPFC from patients with schizophrenia has been more consistently observed (Hashimoto et al., 2008a; Duncan et al., 2010; Beneyto et al., 2011). The changes occurring in GABAA subunits in schizophrenia (an increase in α2 and a decrease α1) could be interpreted to reflect a cortex that may be held in a state of immaturity into adulthood. This concept is interesting as it fits with a number of other changes that would also indicate that the schizophrenia cortex could resemble a more immature cortical state, like increased NR3A, decreased parvalbumin and more calbindin with less cholecystokinin expression as just a few other notable examples consistent with this model.
Dopamine Receptors
Functions of the DLPFC, like working memory, cognitive control, and reasoning (Goldman-Rakic, 1996; Nieoullon, 2002; Glickstein et al., 2005; Mizoguchi et al., 2009) are impacted in schizophrenia and are sensitive to changes in cortical dopamine (Williams and Goldman-Rakic, 1995; Lewis, 1997; Romanides et al., 1999) As these cognitive functions change across development, it is of interest to consider how dopamine receptors may change as humans grow and mature. Cortical dopamine receptors fall into two distinct categories. DRD2-like receptors [DRD2 (S = short and L = long), DRD4] and DRD1-like receptors (DRD1, DRD5: Monsma et al., 1989; Missale et al., 1998). The expression of DRD2 (both forms), DRD4, DRD5 mRNAs all display a predominate pattern of developmental change with highest levels of expression in infants that then declines with age (Weickert et al., 2007; Rothmond et al., 2012; Figure 6). Interestingly, the opposite pattern of expression for both mRNA and protein is observed for DRD1 making DRD1 unique among the cortical dopamine receptors in terms of its developmental expression profile and suggests that there is an increased role of DRD1 as the human cortex matures (Weickert et al., 2007; Rothmond et al., 2012; Figure 6). By early adulthood DRD1 appears to be the most prevalent receptor in the human PFC (Lidow and Rakic, 1992; Meador-Woodruff et al., 1997) and DRD1 is critical to PFC cognitive functioning and in particular working memory (Arnsten et al., 1994). Thus, the gradual increase in DRD1 protein levels from pre-adolescence into young adulthood happens during a time in development when the cortex functionally matures. It coincides with improved performance on a cognitive test thought to tap into a core cognitive deficit in schizophrenia, the Wisconsin Card Sorting Test, which measures behavioral flexibility and working memory (see earlier section), suggesting that DRD1 function may have a particularly salient role in working memory. Indeed, DRD1 receptors in adolescent rodents were shown to interact with NMDARs on pyramidal neurons to increase the likelihood that they would be driven into “up-states” thought to be necessary for working memory function (Tseng et al., 2007).
Figure 6. Normal developmental trajectories of dopamine and cannabinoid expressions in the human prefrontal cortex. Expression of dopamine receptor, DRD1 suggests an inverted U-shape with increasing expression of receptors until the mid-twenties after which there is a slow decline (Rothmond et al., 2012). The DRD2 isoforms and DRD5 show an inverse pattern, peaking prenatally with a continuous decline in expression throughout life, excepting a small increase in the early twenties (Rothmond et al., 2012). The expression of DRD4 as determined by in situ hybridization is stable across the lifespan (Weickert et al., 2007). Measurement of this receptor by quantitative PCR has been quite variable and no statistical differences in expression across age groups have been detected (Rothmond et al., 2012). Cannabinoid receptor mRNA has its highest measured expression at the time of birth and shows a stepwise decline with age, reaching 30% of maximal expression in middle age (Long et al., 2012).
Electrophysiological studies show that in cortical pyramidal neurons, DRD1-like receptor agonism is excitatory (enhancing NMDA effects) and DRD2-like receptor agonism is inhibitory (attenuating AMPA and NMDA response: Wang and O'Donnell, 2001; Tseng and O'Donnell, 2004). In interneurons, DRD1-like receptors are excitatory throughout life (Tseng and O'Donnell, 2007), whereas DRD2-like receptors are weakly inhibitory in juvenile rodents but switch to being strongly excitatory in adult rodents, i.e., strongly increasing inhibitory firing of interneurons onto pyramidal neurons (Tseng and O'Donnell, 2004, 2007). In conditions of high dopamine, this leads to a greater contrast between pyramidal neurons firing due to strong excitatory input and the suppression of background activity of surrounding pyramidal cells (see O'Donnell, 2010 for a review). The cellular mechanisms underlying the switch from inhibitory to excitatory effect of DRD2-like receptors in interneurons are unclear (O'Donnell, 2010), but it yet again implicates peri-adolescent changes in cortical inhibitory circuits as critical foci of research for those seeking to understand the neurodevelopmental neuropathology of schizophrenia.
It is interesting to note that the other dopamine receptor linked to neuronal excitation, DRD5, has a developmental expression pattern opposite to that of DRD1 expression with the highest levels of expression within the first year of life, given that both receptors are nearly indistinguishable pharmacologically. DRD5 is consistently co-localized with DRD1 in the PFC (Bordelon-Glausier et al., 2008) and both are found on pyramidal neurons that are the neural substrates of working memory (Goldman-Rakic et al., 1989). Dopamine has a 10-fold higher affinity to DRD5 compared to DRD1 (Sunahara et al., 1991) suggesting DRD5 even when expressed at lower levels than DRD1 may be functionally important to cognitive processing of working memory (Amico et al., 2007). These results suggest that DRD5 may be the dominant receptor working in opposition to DRD2 early in life in infancy and toddlerhood, and that there may be a developmental switch that occurs such that DRD1 plays a more salient role in dopamine mediated excitation in neural processing during childhood and adolescence.
Schizophrenia studies investigating mRNA expression of all five dopamine receptors have found no change in DRD3, DRD4, and DRD5 in the DLPFC (Meador-Woodruff et al., 1997; Zhan et al., 2011). However, given the timing of late developmental up-regulation and the putative working model of an immature cortex found in schizophrenia, one may predict that DRD1 should be decreased in schizophrenia. While several in vivo binding studies support this (Okubo et al., 1997; Kosaka et al., 2010), only one post-mortem study has found a decrease in cortical DRD1 binding (Hess et al., 1987), which appears to be reflected in DRD1 mRNA expression levels (Meador-Woodruff et al., 1997; Zhan et al., 2011). There is, however, more support for the idea that DRD2 can be elevated in schizophrenia DLPFC (Hess et al., 1987; Kestler et al., 2001; Tallerico et al., 2001). Both DRD1 and DRD2 continue to be expressed throughout life and the interplay of the DRD1 and DRD2 receptors are integral to excitation and inhibition of adult cortical neurons (Onn et al., 2005; So et al., 2005; Cropley et al., 2006; Floresco and Magyar, 2006; Winter et al., 2009). Identifying the cellular localization of this up-regulation of DRD2 would be important for determining whether the increased DRD2 is perhaps counteracting DRD1 effects on pyramidal neurons or bolstering the inhibitory drive of interneurons in schizophrenia prefrontal cortex.
Psychopharmacological Responses to Drugs of Abuse
When considering schizophrenia from a developmental standpoint, it is of interest to consider that the time of onset of schizophrenia is also when humans are, for the first time, more likely to come in contact with mind-altering drugs. The adolescent brain seems to have unique responses to psychoactive drugs compared with children and adults. Adolescent mammals are less sensitive to the locomotor enhancing and cortisol-releasing effects of stimulants such as amphetamine and cocaine compared with adults (Laviola et al., 2002; Zombeck et al., 2010). Furthermore, the behavioral sensitization to stimulant drugs observed in adults can be absent in adolescent rodents (Laviola et al., 1995; Adriani et al., 1998). In contrast, effects of dopamine D2 receptor antagonism such as catalepsy, or prolonged fixed body posture, are more prominent in adolescents than adults (Spear et al., 1980; Campbell et al., 1988) perhaps due to the late up-regulation of D1 receptor and the changing D2R/D1R ratio, although these behaviors are thought to be more related to subcortical dopamine action than cortical dopamine action. Also, the ability of dopamine auto-receptors to suppress subcortical dopamine release matures during adolescence (Hedner and Lundborg, 1985), which might alter dopamine release following stimulant administration. Many people with schizophrenia show increased susceptibility to behavioral and dopamine-releasing effects of amphetamine (Lieberman et al., 1987; Laruelle et al., 1996), and hypersensitivity to typical psychostimulant effects such as locomotor hyperactivity is a benchmark of face validity of animal models of schizophrenia (Powell and Miyakawa, 2006).
Non-competitive NMDAR antagonists such as ketamine and phencyclidine produce psychotic and cognitive symptoms in healthy volunteers, exacerbate schizophrenia symptoms in patients and exert greater cognitive impairment in patients with schizophrenia as compared to healthy controls (Yago et al., 1981; Lahti et al., 1995, 2001; Malhotra et al., 1997). Ketamine and phencyclidine do not readily produce hallucinations in children, despite doing so in normal adults (Hirsch et al., 1997) perhaps due to post-natal difference in NMDAR subunit composition in children compared to adults (see earlier). Perinatal NMDAR antagonist administration induces impairments in attention, working memory, executive function, and social cognition in adulthood (Du Bois and Huang, 2007). In addition, these changes may be more pronounced in rodent pups treated perinatally than in animals exposed to NMDAR antagonists at later developmental stages (Mouri et al., 2007). This suggests that age of exposure to drugs may interact with normal developmental changes in NMDARs to bring about differential cortical effects and can result in distinct behavioral effects.
Compared with adults, adolescent rats find repeated administration of the active ingredient in cannabis, delta-9-tetrahydrocannabinol (THC), less aversive but are more likely to show residual deficits in social interaction and working memory after single or chronic THC exposure (Quinn et al., 2008). Similarly, adolescent rats show higher tolerance to the effects of alcohol (Swartzwelder et al., 1998) and are more likely to “binge-drink” than adults (Hargreaves et al., 2009a), but may also be more vulnerable to neurobiological change following alcohol exposure (Hargreaves et al., 2009b). Thus, while some drugs are more rewarding in younger individuals, this can lead to an increased potential for drug dependence (Quinn et al., 2008; Hargreaves et al., 2009a) and the likelihood of other detrimental effects of drugs of abuse. In humans, cannabinoid CB1 receptor mRNA is highly expressed in the DLPFC from birth until toddlerhood, when it begins to fall rapidly until adolescence and is reduced to a trough by adulthood (Long et al., 2012; Figure 6). Synthetic capacity for the major cortical endogenous cannabinoid neurotransmitter peaks at adolescence, but is followed by a reduction in adulthood (Long et al., 2012). This identifies significant turning points in the prominence of normative endogenous cannabinoid signaling and the subsequent developmental regulation of inhibitory neurotransmission, since the majority of CB1 receptors are localized to GABAergic terminals (Eggan et al., 2010a). Thus, the increased susceptibility to psychotomimetic effects of THC in people with schizophrenia (D'Souza et al., 2005) may be due to aberrant development of the endocannabinoid system during a critical period of change, and indeed numerous reports show that CB1 receptors are altered in schizophrenia (Zavitsanou et al., 2004; Newell et al., 2006; Eggan et al., 2008, 2010b; Dalton et al., 2011). While both increases and decreases in CB1 receptors are reported depending on brain region and if receptor binding, mRNA or protein is measured, it is possible that subtypes of schizophrenia may present with different alterations in the endogenous cannabinoid system (Dalton et al., 2011). In general, observations of the changes in the response to psychoactive drugs during development can be used together with developmental processes to understand not only detrimental effects of these drugs, such as psychotic reactions, but also the way in which development may have gone awry in schizophrenia.
Conclusion
The schizophrenia prodrome can occur during the adolescent years when the human prefrontal cortex is undergoing molecular and functional change (Cornblatt et al., 2003). Deficits in higher order cognitive abilities remain among the most important causes of persistent functional disability in schizophrenia. Therefore, closer examination of the normal developmental trajectories of these cognitive functions and their underlying neural substrates should be considered in the context of schizophrenia in order to adequately identify the key risk factors and determinants of abnormal cortical development in schizophrenia. To date, the predominant neurodevelopmental model used to underpin the emergence of schizophrenia during adolescence is one that posits synaptic regression as the dominant feature of adolescence, a theory based largely on work done over 25 years ago. We suggest that this view is an overly simplistic and a temporally inaccurate way to frame adolescent development of the human cortex. When considering schizophrenia in the context of neurodevelopment and giving appropriate emphasis on the more recently described post-natal developmental changes, a more modern, dynamic, and complex picture emerges. While many developmental changes are yet to be confirmed, many others have been replicated and these provide ample scope for changes in risk genes and environments to alter post-natal developmental trajectories. We also suggest that while some processes may be attenuated with human post-natal brain growth, many processes are accelerated and others are more accurately viewed as in an active growth phase and more dynamic state of change. Thus, it may be inaccurate to consider the major developmental event of the human cortex to be one of synaptic regression. Certainly, there is an overwhelming increase in brain size, in interneuron differentiation, in synaptic molecule expression, and in GABA and dopamine receptor mRNA levels that would indicate that gains in synaptic strength predominate in early life and continue during childhood. What appears to be the major developmental switch at adolescence is the slowing of the exuberant growth period typical of infants, toddlers, and children. We suggest that, when theorizing about the neurodevelopmental basis of schizophrenia, schizophrenia could be considered to result from a failure to reach the final state of cortical maturation resulting in retainment of an immature cortex (at least transiently) rather than resulting from excess of adolescent synaptic pruning. Further, it is clear that adolescence is a time of dynamic brain change and that schizophrenia could be viewed as resulting from a destabilization of a normal adolescence process. Since the adolescent brain is in a state of flux it may be possible to stabilize the adolescent brain of those at risk so that any disruption is only transitory and so that chronic schizophrenia does not emerge (McGorry, 2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Schizophrenia Research Institute (utilizing infrastructure funding from the NSW Ministry of Health and the Macquarie Group Foundation), the University of New South Wales, and Neuroscience Research Australia. Cynthia Shannon Weickert is supported by a Senior Research Fellowship from the National Health and Medical Research Council. The authors are grateful to members of the Schizophrenia Research Laboratory, including Professor Stanley Catts, Dr. Tertia Purves-Tyson, Mr. Heng Woon, and Ms. Alice Rothwell, for helpful discussions on the manuscript, and to Ms. Inara Bebris for editorial assistance.
References
Abraham, H., Vincze, A., Jewgenow, I., Veszpremi, B., Kravjak, A., Gomori, E., et al. (2010). Myelination in the human hippocampal formation from midgestation to adulthood. Int. J. Dev. Neurosci. 28, 401–410.
Adriani, W., Chiarotti, F., and Laviola, G. (1998). Elevated novelty seeking and peculiar d-amphetamine sensitization in periadolescent mice compared with adult mice. Behav. Neurosci. 112, 1152–1166.
Aguayo, L. G., van Zundert, B., Tapia, J. C., Carrasco, M. A., and Alvarez, F. J. (2004). Changes on the properties of glycine receptors during neuronal development. Brain Res. Brain Res. Rev. 47, 33–45.
Akbarian, S., Bunney, W. E., Potkin, S. G., Wigal, S. B., Hagman, J. O., Sandman, C. A., et al. (1993). Altered distribution of nicotinamide-adenine dinucleotide phosphate diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch. Gen. Psychiatry 50, 169–177.
Akbarian, S., Sucher, N. J., Bradley, D., Tafazzoli, A., Trinh, D., Hetrick, W. P., et al. (1996). Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 16, 19–30.
Amico, F., Spowart-Manning, L., Anwyl, R., and Rowan, M. J. (2007). Performance- and task-dependent effects of the dopamine D-1/D-5 receptor agonist SKF 38393 on learning and memory in the rat. Eur. J. Pharmacol. 577, 71–77.
Anderson, S., Eisenstat, D., Shi, L., and Rubenstein, J. (1997). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476.
Anderson, S. A., Volk, D. W., and Lewis, D. A. (1996). Increased density of microtubule associated protein 2-immunoreactive neurons in the prefrontal white matter of schizophrenic subjects. Schizophr. Res. 19, 111–119.
Anderson, V. A., Anderson, P., Northam, E., Jacobs, R., and Catroppa, C. (2001). Development of executive functions through late childhood and adolescence in an Australian sample. Dev. Neuropsychol. 20, 385–406.
Andreasen, N. C., O'Leary, D. S., Flaum, M., Nopoulos, P., Watkins, G. L., Boles Ponto, L. L., et al. (1997). Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naive patients. Lancet 349, 1730–1734.
Arnold, S. E., Hyman, B. T., Van Hoesen, G. W., and Damasio, A. R. (1991). Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Arch. Gen. Psychiatry 48, 625–632.
Arnold, S. E., Ruscheinsky, D. D., and Han, L. Y. (1997). Further evidence of abnormal cytoarchitecture of the entorhinal cortex in schizophrenia using spatial point pattern analyses. Biol. Psychiatry 42, 639–647.
Arnsten, A. F. T., Cai, J. X., Murphy, B. L., and Goldmanrakic, P. S. (1994). Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology (Berl.) 116, 143–151.
Asada, H., Kawamura, Y., Maruyama, K., Kume, H., Ding, R. G., Kanbara, N., et al. (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 94, 6496–6499.
Ashrafi, S., Runyan, C. A., Weickert, C. S., and Saunders, R. C. (2007). “Newly generated GABAergic cells in the adult primate prefrontal cortex,” in Society for Neuroscience (San Diego, CA, USA: Society for Neuroscience).
Ayala, R., Shu, T., and Tsai, L.-H. (2007). Trekking across the brain: the journey of neuronal migration. Cell 128, 29–43.
Barbeau, D., Laing, J., Robitalille, Y., Quirion, R., and Srivastava, L. (1995). Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc. Natl. Acad. Sci. U.S.A. 92, 2785–2789.
Barch, D. M., Carter, C. S., Braver, T. S., Sabb, F. W., MacDonald, A. 3rd, Noll, D. C., et al. (2001). Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch. Gen. Psychiatry 58, 280–288.
Barnea-Goraly, N., Menon, V., Eckert, M., Tamm, L., Bammer, R., Karchemskiy, A., et al. (2005). White matter development during childhood and adolescence: a cross-sectional diffusion tensor imaging study. Cereb. Cortex 15, 1848–1854.
Bava, S., Thayer, R., Jacobus, J., Ward, M., Jernigan, T. L., and Tapert, S. F. (2010). Longitudinal characterization of white matter maturation during adolescence. Brain Res. 1327, 38–46.
Bayatti, N., Moss, J. A., Sun, L., Ambrose, P., Ward, J. F., Lindsay, S., et al. (2008). A molecular neuroanatomical study of the developing human neocortex from 8 to 17 postconceptional weeks revealing the early differentiation of the subplate and subventricular zone. Cereb. Cortex 18, 1536–1548.
Beasley, C. L., Dwork, A. J., Rosoklija, G., Mann, J. J., Mancevski, B., Jakovski, Z., et al. (2009). Metabolic abnormalities in fronto-striatal-thalamic white matter tracts in schizophrenia. Schizophr. Res. 109, 159–166.
Beasley, C. L., and Reynolds, G. P. (1997). Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr. Res. 24, 349–355.
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z. H., Yu, G., Li, Y. Q., et al. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 13, 76–83.
Beltaifa, S., Webster, M. J., Ligons, D. L., Fatula, R. J., Herman, M. M., Kleinman, J. E., et al. (2005). Discordant changes in cortical TrkC mRNA and protein during the human lifespan. Eur. J. Neurosci. 21, 2433–2444.
Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.
Benes, F. M. (1989). Myelination of cortical-hippocampal relays during late adolescence. Schizophr. Bull. 15, 585–593.
Benes, F. M., Vincent, S. L., Alsterberg, G., Bird, E. D., and Sangiovanni, J. P. (1992). Increased GABA-A receptor binding in superficial layers of cingulate cortex in schizophrenics. J. Neurosci. 12, 924–929.
Beneyto, M., Abbott, A., Hashimoto, T., and Lewis, D. A. (2011). Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb. Cortex 21, 999–1011.
Beneyto, M., and Meador-Woodruff, J. H. (2008). Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 33, 2175–2186.
Bennett, M. R. (2011). Schizophrenia: susceptibility genes, dendritic-spine pathology and gray matter loss. Prog. Neurobiol. 95, 275–300.
Bernier, P. J., Bedard, A., Vinet, J., Levesque, M., and Parent, A. (2002). Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc. Natl. Acad. Sci. U.S.A. 99, 11464–11469.
Bernstein, H. G., Krell, D., Baumann, B., Danos, P., Falkai, P., Diekmann, S., et al. (1998). Morphometric studies of the entorhinal cortex in neuropsychiatric patients and controls: clusters of heterotopically displaced lamina II neurons are not indicative of schizophrenia. Schizophr. Res. 33, 125–132.
Bhardwaj, R. D., Curtis, M. A., Spalding, K. L., Buchholz, B. A., Fink, D., Bjork-Eriksson, T., et al. (2006). Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. U.S.A. 103, 12564–12568.
Bhojraj, T. S., Diwadkar, V. A., Sweeney, J. A., Prasad, K. M., Eack, S. M., Montrose, D. M., et al. (2010). Longitudinal alterations of executive function in non-psychotic adolescents at familial risk for schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 469–474.
Bitanihirwe, B. K. Y., Lim, M. P., Kelley, J. F., Kaneko, T., and Woo, T. U. W. (2009). Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 9:71. doi: 10.1186/1471-244X-9-71
Blakemore, S. J., and Choudhury, S. (2006). Development of the adolescent brain: implications for executive function and social cognition. J. Child Psychol. Psychiatry 47, 296–312.
Bordelon-Glausier, J. R., Khan, Z. U., and Muly, E. C. (2008). Quantification of D-1 and D-5 dopamine receptor localization in layers I, III, and V of Macaca mulatta prefrontal cortical area 9: coexpression in dendritic spines and axon terminals. J. Comp. Neurol. 508, 893–905.
Borgwardt, S. J., Dickey, C., Pol, H. H., Whitford, T. J., and Delisi, L. E. (2009). Workshop on defining the significance of progressive brain change in schizophrenia: December 12, 2008 American College of Neuropsychopharmacology (ACNP) all-day satellite, Scottsdale, Arizona The rapporteurs' report. Schizophr. Res. 112, 32–45.
Brocki, K. C., and Bohlin, G. (2004). Executive functions in children aged 6 to 13: a dimensional and developmental study. Dev. Neuropsychol. 26, 571–593.
Brody, B. A., Kinney, H. C., Kloman, A. S., and Gilles, F. H. (1987). Sequence of central nervous system myelination in human infancy. I. An autopsy study of myelination. J. Neuropathol. Exp. Neurol. 46, 283–301.
Bunge, S. A., Dudukovic, N. M., Thomason, M. E., Vaidya, C. J., and Gabrieli, J. D. E. (2002). Immature frontal lobe contributions to cognitive control in children: evidence from fMRI. Neuron 33, 301–311.
Bunge, S. A., Helskog, E. H., and Wendelken, C. (2009). Left, but not right, rostrolateral prefrontal cortex meets a stringent test of the relational integration hypothesis. Neuroimage 46, 338–342.
Bunge, S. A., Wendelken, C., Badre, D., and Wagner, A. D. (2005). Analogical reasoning and prefrontal cortex: evidence for separable retrieval and integration mechanisms. Cereb. Cortex 15, 239–249.
Callicott, J. H., Mattay, V. S., Verchinski, B. A., Marenco, S., Egan, M. F., and Weinberger, D. R. (2003). Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am. J. Psychiatry 160, 2209–2215.
Campbell, A., Baldessarini, R. J., and Teicher, M. H. (1988). Decreasing sensitivity to neuroleptic agents in developing rats; evidence for a pharmacodynamic factor. Psychopharmacology (Berl.) 94, 46–51.
Cannon, M., Caspi, A., Moffitt, T. E., Harrington, H., Taylor, A., Murray, R. M., et al. (2002a). Evidence for early-childhood, pan-developmental impairment specific to schizophreniform disorder – results from a longitudinal birth cohort. Arch. Gen. Psychiatry 59, 449–456.
Cannon, T. D., Thompson, P. M., van Erp, T. G. M., Toga, A. W., Poutanen, V. P., Huttunen, M., et al. (2002b). Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 99, 3228–3233.
Casey, B. J., Giedd, J. N., and Thomas, K. M. (2000). Structural and functional brain development and its relation to cognitive development. Biol. Psychol. 54, 241–257.
Casey, B. J., Trainor, R. J., Orendi, J. L., Schubert, A. B., Nystrom, L. E., Giedd, J. N., et al. (1997). A developmental functional MRI study of prefrontal activation during performance of a Go-No-Go task. J. Cogn. Neurosci. 9, 835–847.
Castillo, M. A., Ghose, S., Tamminga, C. A., and Ulery-Reynolds, P. G. (2010). Deficits in syntaxin 1 phosphorylation in schizophrenia prefrontal cortex. Biol. Psychiatry 67, 208–216.
Cattell, R. B. (1987). Intelligence: Its Structure, Growth, and Action. Amsterdam: North Holland: Elsevier.
Catts, V. S., and Catts, S. V. (2010). “The psychotomimetic effects of PCP, LSD and MDMA: Pharmacological models of schizophrenia?,” in Secondary Schizophrenia, eds. P. S. Sachdev and M. S. Keshavan (Cambridge: Cambridge Press), 141–168.
Catts, V. S., and Weickert, C. S. (2012). Gene expression analysis implicates a death receptor pathway in schizophrenia pathology. PLoS ONE 7:e35511. doi: 10.1371/journal.pone.0035511
Caviness, V. S. Jr. (1976). Patterns of cell and fiber distribution in the neocortex of the reeler mutant mouse. J. Comp. Neurol. 170, 435–447.
Chai, X., Forster, E., Zhao, S., Bock, H. H., and Frotscher, M. (2009). Reelin acts as a stop signal for radially migrating neurons by inducing phosphorylation of n-cofilin at the leading edge. Commun. Integr. Biol. 2, 375–377.
Choi, K. H., Zepp, M. E., Higgs, B. W., Weickert, C. S., and Webster, M. J. (2009). Expression profiles of schizophrenia susceptibility genes during human prefrontal cortical development. J. Psychiatry Neurosci. 34, 450–458.
Chong, V. Z., Webster, M. J., Rothmond, D., and Weickert, C. S. (2008). Specific developmental reductions in subventricular zone ErbB1 and ErbB4 mRNA in the human brain. Int. J. Dev. Neurosci. 26, 791–803.
Christoff, K., Prabhakaran, V., Dorfman, J., Zhao, Z., Kroger, J. K., Holyoak, K. J., et al. (2001). Rostrolateral prefrontal cortex involvement in relational integration during reasoning. Neuroimage 14, 1136–1149.
Chun, J. J. M., and Shatz, C. J. (1989). Interstitial cells of the adult neocortical white matter are the remnant of the early generated subplate neuron population. J. Comp. Neurol. 282, 555–569.
Colantuoni, C., Lipska, B. K., Ye, T., Hyde, T. M., Tao, R., Leek, J. T., et al. (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523.
Corbin, J., and Butt, S. (2011). Developmental mechanisms for the generation of telencephalic interneurons. Dev. Neurobiol. 71, 710–732.
Corbin, J. G., Nery, S., and Fishell, G. (2001). Telencephalic cells take a tangent: non-radial migration in the mammalian forebrain. Nat. Neurosci. 4, 1177–1182.
Cornblatt, B. A., Lencz, T., Smith, C. W., Correll, C. U., Auther, A. M., and Nakayama, E. (2003). The schizophrenia prodrome revisited: a neurodevelopmental perspective. Schizophr. Bull. 29, 633–651.
Costa, E., Chen, Y., Davis, J., Dong, E., Noh, J. S., Tremolizzo, L., et al. (2002). REELIN and schizophrenia: a disease at the interface of the genome and the epigenome. Mol. Interv. 2, 47–57.
Cox, E. T., Brennaman, L. H., Gable, K. L., Hamer, R. M., Glantz, L. A., Lamantia, A. S., et al. (2009). Developmental regulation of neural cell adhesion molecule in human prefrontal cortex. Neuroscience 162, 96–105.
Crair, M. C., and Malenka, R. C. (1995). A critical period for long-term potentiation at thalamocortical synapses. Nature 375, 325–328.
Crone, E. A., Wendelken, C., Donohue, S., van Leijenhorst, L., and Bunge, S. A. (2006). Neurocognitive development of the ability to manipulate information in working memory. Proc. Natl. Acad. Sci. U.S.A. 103, 9315–9320.
Crone, E. A., Wendelken, C., van Leijenhorst, L., Honomichl, R. D., Christoff, K., and Bunge, S. A. (2009). Neurocognitive development of relational reasoning. Dev. Sci. 12, 55–66.
Cropley, V. L., Fujita, M., Innis, R. B., and Nathan, P. J. (2006). Molecular imaging of the dopaminergic system and its association with human cognitive function. Biol. Psychiatry 59, 898–907.
Cruz, D. A., Eggan, S. M., and Lewis, D. A. (2003). Postnatal development of pre- and postsynaptic GABA markers at chandelier cell connections with pyramidal neurons in monkey prefrontal cortex. J. Comp. Neurol. 465, 385–400.
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335.
Curtis, M. A., Penney, E. B., Pearson, A. G., van Roon-Mom, W. M., Butterworth, N. J., Dragunow, M., et al. (2003). Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc. Natl. Acad. Sci. U.S.A. 100, 9023–9027.
D'Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I., and Curran, T. (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723.
D'Souza, D. C., Abi-Saab, W. M., Madonick, S., Forselius-Bielen, K., Doersch, A., Braley, G., et al. (2005). Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol. Psychiatry 57, 594–608.
Dalton, V. S., Long, L. E., Weickert, C. S., and Zavitsanou, K. (2011). Paranoid schizophrenia is characterized by increased CB1 receptor binding in the dorsolateral prefrontal cortex. Neuropsychopharmacology 36, 1620–1630.
Das, S., Sasaki, Y. F., Rothe, T., Premkumar, L. S., Takasu, M., Crandall, J. E., et al. (1998). Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature 393, 377–381.
David, A. S., Malmberg, A., Brandt, L., Allebeck, P., and Lewis, G. (1997). IQ and risk for schizophrenia: a population-based cohort study. Psychol. Med. 27, 1311–1323.
Dean, B., Hussain, T., Hayes, W., Scarr, E., Kitsoulis, S., Hill, C., et al. (1999). Changes in serotonin(2A) and GABA(A) receptors in schizophrenia: studies on the human dorsolateral prefrontal cortex. J. Neurochem. 72, 1593–1599.
Dean, B., Pavey, G., McLeod, M., Opeskin, K., Keks, N., and Copolov, D. (2001). A change in the density of [(3)H]flumazenil, but not [(3)H]muscimol binding, in Brodmann's Area 9 from subjects with bipolar disorder. J. Affect. Disord. 66, 147–158.
Dekaban, A. S., and Sadowsky, D. (1978). Changes in brain weights during span of human life – relation of brain weights to body heights and body weights. Ann. Neurol. 4, 345–356.
Del Rio, J. A., Heimrich, B., Borrell, V., Forster, E., Drakew, A., Alcantara, S., et al. (1997). A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature 385, 70–74.
Delalle, I., Evers, P., Kostovic, I., and Uylings, H. B. (1997). Laminar distribution of neuropeptide Y-immunoreactive neurons in human prefrontal cortex during development. J. Comp. Neurol. 379, 515–522.
Di Cristo, G. (2007). Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin. Genet. 72, 1–8.
Dickson, H., Laurens, K. R., Cullen, A. E., and Hodgins, S. (2012). Meta-analyses of cognitive and motor function in youth aged 16 years and younger who subsequently develop schizophrenia. Psychol. Med. 42, 743–755.
Dracheva, S., Marras, S. A., Kramer, F. R., Davis, K. L., and Haroutunian, V. (2001). N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am. J. Psychiatry 158, 2107.
Du Bois, T. M., and Huang, X. F. (2007). Early brain development disruption from NMDA receptor hypofunction: relevance to schizophrenia. Brain Res. Rev. 53, 260–270.
Duan, X., Chang, J. H., Ge, S., Faulkner, R. L., Kim, J. Y., Kitabatake, Y., et al. (2007). Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130, 1146–1158.
Dubois, J., Hertz-Pannier, L., Dehaene-Lambertz, G., Cointepas, Y., and Le Bihan, D. (2006). Assessment of the early organization and maturation of infants' cerebral white matter fiber bundles: a feasibility study using quantitative diffusion tensor imaging and tractography. Neuroimage 30, 1121–1132.
Duncan, C., Webster, M., Rothmond, D. A., Bahn, S., Elashoff, M., and Shannon Weickert, C. (2010). Prefrontal GABA-A receptor α subunit expression in normal postnatal human development and schizophrenia. J. Psychiatr. Res. 44, 673–681.
Durston, S., Davidson, M. C., Tottenham, N., Galvan, A., Spicer, J., Fossella, J. A., et al. (2006). A shift from diffuse to focal cortical activity with development. Dev. Sci. 9, 1–8.
Durston, S., Pol, H. E. H., Casey, B. J., Giedd, J. N., Buitelaar, J. K., and van Engeland, H. (2001). Anatomical MRI of the developing human brain: what have we learned? J. Am. Acad. Child Adolesc. Psychiatry 40, 1012–1020.
Eastwood, S. L., and Harrison, P. J. (2001). Synaptic pathology in the anterior cingulate cortex in schizophrenia and mood disorders. A review and a Western blot study of synaptophysin, GAP-43 and the complexins. Brain Res. Bull. 55, 569–578.
Eastwood, S. L., and Harrison, P. J. (2003). Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol. Psychiatry 8, 821–831.
Eastwood, S. L., and Harrison, P. J. (2005a). Interstitial white matter neuron density in the dorsolateral prefrontal cortex and parahippocampal gyrus in schizophrenia. Schizophr. Res. 79, 181–188.
Eastwood, S. L., and Harrison, P. J. (2005b). Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr. Res. 73, 159–172.
Edin, F., MacOveanu, J., Olesen, P., Tegner, J., and Klingberg, T. (2007). Stronger synaptic connectivity as a mechanism behind development of working memory-related brain activity during childhood. J. Cogn. Neurosci. 19, 750–760.
Eggan, S. M., Hashimoto, T., and Lewis, D. A. (2008). Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch. Gen. Psychiatry 65, 772–784.
Eggan, S. M., Melchitzky, D. S., Sesack, S. R., Fish, K. N., and Lewis, D. A. (2010a). Relationship of cannabinoid CB1 receptor and cholecystokinin immunoreactivity in monkey dorsolateral prefrontal cortex. Neuroscience 169, 1651–1661.
Eggan, S. M., Stoyak, S. R., Verrico, C. D., and Lewis, D. A. (2010b). Cannabinoid CB1 receptor immunoreactivity in the prefrontal cortex: comparison of schizophrenia and major depressive disorder. Neuropsychopharmacology 35, 2060–2071.
Elston, G. N. (2000). Pyramidal cells of the frontal lobe: all the more spinous to think with. J. Neurosci. 20, RC95.
Eluvathingal, T. J., Hasan, K. M., Kramer, L., Fletcher, J. M., and Ewing-Cobbs, L. (2007). Quantitative diffusion tensor tractography of association and projection fibers in normally developing children and adolescents. Cereb. Cortex 17, 2760–2768.
Erickson, S. L., and Lewis, D. A. (2002). Postnatal development of parvalbumin- and GABA transporter-immunoreactive axon terminals in monkey prefrontal cortex. J. Comp. Neurol. 448, 186–202.
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A., et al. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Fair, D. A., Dosenbach, N. U. F., Church, J. A., Cohen, A. L., Brahmbhatt, S., Miezin, F. M., et al. (2007). Development of distinct control networks through segregation and integration. Proc. Natl. Acad. Sci. U.S.A. 104, 13507–13512.
Falkai, P., Schneider-Axmann, T., and Honer, W. G. (2000). Entorhinal cortex pre-alpha cell clusters in schizophrenia: quantitative evidence of a developmental abnormality. Biol. Psychiatry 47, 937–943.
Faludi, G., and Mirnics, K. (2011). Synaptic changes in the brain of subjects with schizophrenia. Int. J. Dev. Neurosci. 29, 305–309.
Feinberg, I. (1983). Schizophrenia – caused by a fault in programmed synaptic elimination during adolescence. J. Psychiatr. Res. 17, 319–334.
Feldmeyer, D., and Radnikow, G. (2009). Developmental alterations in the functional properties of excitatory neocortical synapses. J. Physiol. 587, 1889–1896.
Fertuzinhos, S., Krsnik, Z., Kawasawa, Y., Rasin, M., Kwan, K., Chen, J., et al. (2009). Selective depletion of molecularly defined cortical interneurons in human holoprosencephaly with severe striatal hypoplasia. Cereb. Cortex 19, 2196–2207.
Fillman, S. G., Cloonan, N., Catts, V. S., Miller, L. C., Wong, J., McCrossin, T., et al. (2013). Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 18, 206–214.
Fillman, S. G., Duncan, C. E., Webster, M. J., Elashoff, M., and Weickert, C. S. (2010). Developmental co-regulation of the beta and gamma GABAA receptor subunits with distinct alpha subunits in the human dorsolateral prefrontal cortex. Int. J. Dev. Neurosci. 28, 513–519.
Flint, A. C., Liu, X., and Kriegstein, A. R. (1998). Nonsynaptic glycine receptor activation during early neocortical development. Neuron 20, 43–53.
Flint, A. C., Maisch, U. S., Weishaupt, J. H., Kriegstein, A. R., and Monyer, H. (1997). NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J. Neurosci. 17, 2469–2476.
Floresco, S. B., and Magyar, O. (2006). Mesocortical dopamine modulation of executive functions: beyond working memory. Psychopharmacology (Berl.) 188, 567–585.
Fung, S., Joshi, D., Allen, K., Sivagnanasundaram, S., Rothmond, D., Saunders, R., et al. (2011a). Developmental patterns of doublecortin expression and white matter neuron density in the postnatal primate prefrontal cortex and schizophrenia. PLoS ONE 6:e25194. doi: 10.1371/journal.pone.0025194
Fung, S. J., Sivagnanasundaram, S., and Weickert, C. S. (2011b). Lack of change in markers of presynaptic terminal abundance alongside subtle reductions in markers of presynaptic terminal plasticity in prefrontal cortex of schizophrenia patients. Biol. Psychiatry 69, 71–79.
Fung, S. J., Webster, M. J., Sivagnanasundaram, S., Duncan, C., Elashoff, M., and Weickert, C. S. (2010). Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 167, 1479–1488.
Gambrill, A. C., and Barria, A. (2011). NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. U.S.A. 108, 5855–5860.
Gao, W., Lin, W., Chen, Y., Gerig, G., Smith, J. K., Jewells, V., et al. (2009). Temporal and spatial development of axonal maturation and myelination of white matter in the developing brain. Am. J. Neuroradiol. 30, 290–296.
Ghashghaei, H. T., Weber, J., Pevny, L., Schmid, R., Schwab, M. H., Lloyd, K. C., et al. (2006). The role of neuregulin-ErbB4 interactions on the proliferation and organization of cells in the subventricular zone. Proc. Natl. Acad. Sci. U.S.A. 103, 1930–1935.
Giedd, J. N., Blumenthal, J., Jeffries, N. O., Castellanos, F. X., Liu, H., Zijdenbos, A., et al. (1999). Brain development during childhood and adolescence: a longitudinal MRI study. Nat. Neurosci. 2, 861–863.
Giorgio, A., Watkins, K. E., Chadwick, M., James, S., Winmill, L., Douaud, G., et al. (2010). Longitudinal changes in grey and white matter during adolescence. Neuroimage 49, 94–103.
Giorgio, A., Watkins, K. E., Douaud, G., James, A. C., James, S., De Stefano, N., et al. (2008). Changes in white matter microstructure during adolescence. Neuroimage 39, 52–61.
Glantz, L. A., Gilmore, J. H., Hamer, R. M., Lieberman, J. A., and Jarskog, L. F. (2007). Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience 149, 582–591.
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73.
Glickstein, S. B., Desteno, D. A., Hof, P. R., and Schmauss, C. (2005). Mice lacking dopamine D2 and D3 receptors exhibit differential activation of prefrontal cortical neurons during tasks requiring attention. Cereb. Cortex 15, 1016–1024.
Goffinet, A. M. (1984). Events governing organization of postmigratory neurons: studies on brain development in normal and reeler mice. Brain Res. 319, 261–296.
Gogtay, N., Giedd, J. N., Lusk, L., Hayashi, K. M., Greenstein, D., Vaituzis, A. C., et al. (2004). Dynamic mapping of human cortical development during childhood through early adulthood. Proc. Natl. Acad. Sci. U.S.A. 101, 8174–8179.
Goldberg, T. E., Goldman, R. S., Burdick, K. E., Malhotra, A. K., Lencz, T., Patel, R. C., et al. (2007). Cognitive improvement after treatment with second-generation antipsychotic medications in first-episode schizophrenia: is it a practice effect? Arch. Gen. Psychiatry 64, 1115–1122.
Goldman-Rakic, P. S. (1994). Working memory dysfunction in schizophrenia. J. Neuropsychiatry Clin. Neurosci. 6, 348–357.
Goldman-Rakic, P. S. (1996). Regional and cellular fractionation of working memory. Proc. Natl. Acad. Sci. U.S.A. 93, 13473–13480.
Goldman-Rakic, P. S., Leranth, C., Williams, S. M., Mons, N., and Geffard, M. (1989). Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 86, 9015–9019.
Gonzalez-Burgos, G., Hashimoto, T., and Lewis, D. A. (2010). Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr. Psychiatry Rep. 12, 335–344.
Goswami, U. (1989). Relational complexity and the development of analogical reasoning. Cogn. Dev. 4, 251–268.
Gould, E., Reeves, A. J., Graziano, M. S., and Gross, C. G. (1999). Neurogenesis in the neocortex of adult primates. Science 286, 548–552.
Gould, E., Vail, N., Wagers, M., and Gross, C. G. (2001). Adult-generated hippocampal and neocortical neurons in macaques have a transient existence. Proc. Natl. Acad. Sci. U.S.A. 98, 10910–10917.
Grateron, L., Cebada-Sanchez, S., Marcos, P., Mohedano-Moriano, A., Insausti, A. M., Munoz, M., et al. (2003). Postnatal development of calcium-binding proteins immunoreactivity (parvalbumin, calbindin, calretinin) in the human entorhinal cortex. J. Chem. Neuroanat. 26, 311–316.
Gray, L. J., Dean, B., Kronsbein, H. C., Robinson, P. J., and Scarr, E. (2010). Region and diagnosis-specific changes in synaptic proteins in schizophrenia and bipolar I disorder. Psychiatry Res. 178, 374–380.
Grayson, D. R., Jia, X., Chen, Y., Sharma, R. P., Mitchell, C. P., Guidotti, A., et al. (2005). Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 102, 9341–9346.
Green, M. F. (1996). What are the functional consequences of neurocognitive deficits in schizophrenia? Am. J. Psychiatry 153, 321–330.
Hakak, Y., Walker, J. R., Li, C., Wong, W. H., Davis, K. L., Buxbaum, J. D., et al. (2001). Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 98, 4746–4751.
Halim, N. D., Weickert, C. S., McClintock, B. W., Hyde, T. M., Weinberger, D. R., Kleinman, J. E., et al. (2003). Presynaptic proteins in the prefrontal cortex of patients with schizophrenia and rats with abnormal prefrontal development. Mol. Psychiatry 8, 797–810.
Hargreaves, G. A., Monds, L., Gunasekaran, N., Dawson, B., and McGregor, I. S. (2009a). Intermittent access to beer promotes binge-like drinking in adolescent but not adult Wistar rats. Alcohol 43, 305–314.
Hargreaves, G. A., Quinn, H., Kashem, M. A., Matsumoto, I., and McGregor, I. S. (2009b). Proteomic analysis demonstrates adolescent vulnerability to lasting hippocampal changes following chronic alcohol consumption. Alcohol. Clin. Exp. Res. 33, 86–94.
Harney, S. C., Jane, D. E., and Anwyl, R. (2008). Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J. Neurosci. 28, 11685–11694.
Harris, L. W., Lockstone, H. E., Khaitovich, P., Weickert, C. S., Webster, M. J., and Bahn, S. (2009). Gene expression in the prefrontal cortex during adolescence: implications for the onset of schizophrenia. BMC Med. Genomics 2:28. doi: 10.1186/1755-8794-2-28
Hashimoto, T., Arion, D., Unger, T., Maldonado-Aviles, J. G., Morris, H. M., Volk, D. W., et al. (2008a). Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 13, 147–161.
Hashimoto, T., Bazmi, H. H., Mirnics, K., Wu, Q., Sampson, A. R., and Lewis, D. A. (2008b). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165, 479–489.
Hashimoto, T., Nguyen, Q. L., Rotaru, D., Keenan, T., Arion, D., Beneyto, M., et al. (2009). Protracted developmental trajectories of GABA(A) receptor alpha 1 and alpha 2 subunit expression in primate prefrontal cortex. Biol. Psychiatry 65, 1015–1023.
Haubensak, W., Attardo, A., Denk, W., and Huttner, W. B. (2004). Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 3196–3201.
Hayashi, M., Yamashita, A., Shimizu, K., Sogawa, K., and Fujii, Y. (1990). Somatostatin gene expression in the developing monkey frontal and cerebellar cortices. Brain Res. Dev. Brain Res. 57, 37–41.
Haynes, R. L., Borenstein, N. S., Desilva, T. M., Folkerth, R. D., Liu, L. G., Volpe, J. J., et al. (2005). Axonal development in the cerebral white matter of the human fetus and infant. J. Comp. Neurol. 484, 156–167.
Heaton, R. K., Chelune, G. J., Talley, T. L., Kay, G. G., and Curtiss, G. (1993). Wisconsin Card Sorting Test Manual. Odessa, FL: Psychological Assessment Resources.
Hedner, T., and Lundborg, P. (1985). Development of dopamine autoreceptors in the postnatal rat brain. J. Neural Transm. 62, 53–63.
Heinrichs, R. W., and Zakzanis, K. K. (1998). Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology 12, 426–445.
Henson, M. A., Roberts, A. C., Salimi, K., Vadlamudi, S., Hamer, R. M., Gilmore, J. H., et al. (2008). Developmental regulation of the NMDA receptor subunits, NR3A and NR1, in human prefrontal cortex. Cereb. Cortex 18, 2560–2573.
Hermoye, L., Saint-Martin, C., Cosnard, G., Lee, S.-K., Kim, J., Nassogne, M.-C., et al. (2006). Pediatric diffusion tensor imaging: normal database and observation of the white matter maturation in early childhood. Neuroimage 29, 493–504.
Hess, E. J., Bracha, H. S., Kleinman, J. E., and Creese, I. (1987). Dopamine receptor subtype imbalance in schizophrenia. Life Sci. 40, 1487–1497.
Hevers, W., and Lüddens, H. (1998). The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol. Neurobiol. 18, 35–86.
Hirsch, S. R., Das, I., Garey, L. J., and De Belleroche, J. (1997). A pivotal role for glutamate in the pathogenesis of schizophrenia, and its cognitive dysfunction. Pharmacol. Biochem. Behav. 56, 797–802.
Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108.
Honer, W. G., Falkal, P., Chen, C., Arango, V., Mann, J. J., and Dwork, A. J. (1999). Synaptic and plasticity-associated proteins in anterior frontal cortex in severe mental illness. Neuroscience 91, 1247–1255.
Hudspeth, W. J., and Pribram, K. H. (1990). Stages of brain and cognitive maturation. J. Educ. Psychol. 82, 881–884.
Hudspeth, W. J., and Pribram, K. H. (1992). Psychophysiological indexes of cerebral maturation. Int. J. Psychophysiol. 12, 19–29.
Huizinga, M., Dolan, C. V., and van der Molen, M. W. (2006). Age-related change in executive function: developmental trends and a latent variable analysis. Neuropsychologia 44, 2017–2036.
Humphries, C., Mortimer, A., Hirsch, S., and Debelleroche, J. (1996). NMDA receptor mRNA correlation with antemortem cognitive impairment in schizophrenia. Neuroreport 7, 2051–2055.
Huttenlocher, P. (1979). Synaptic density in human frontal cortex – developmental changes and effects of aging. Brain Res. 163, 195–205.
Huttenlocher, P. R. (1990). Morphometric study of human cerebral cortex development. Neuropsychologia 28, 517–527.
Huttenlocher, P. R., and Dabholkar, A. S. (1997). Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 387, 167–178.
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095.
Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95, 15718–15723.
Ishimaru, M., Kurumaji, A., and Toru, M. (1992). NMDA-associated glycine binding site increases in schizophrenic brains. Biol. Psychiatry 32, 379–381.
Iwasaki, N., Hamano, K., Okada, Y., Horigome, Y., Nakayama, J., Takeya, T., et al. (1997). Volumetric quantification of brain development using MRI. Neuroradiology 39, 841–846.
Jacobs, B., Driscoll, L., and Schall, M. (1997). Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J. Comp. Neurol. 386, 661–680.
Jakob, H., and Beckmann, H. (1986). Prenatal developmental disturbances in the limbic allocortex in schizophrenics. J. Neural Transm. 65, 303–326.
Jones, L. B., Johnson, N., and Byne, W. (2002). Alterations in MAP2 immunocytochemistry in areas 9 and 32 of schizophrenic prefrontal cortex. Psychiatry Res. 114, 137–148.
Jones, P., Rodgers, B., Murray, R., and Marmot, M. (1994). Child developmental risk-factors for adult schizophrenia in the British 1946 birth cohort. Lancet 344, 1398–1402.
Joshi, D., Fung, S. J., Rothwell, A., and Weickert, C. S. (2012). Higher gamma-aminobutyric acid neuron density in the white matter of orbital frontal cortex in schizophrenia. Biol. Psychiatry 72, 725–733.
Kalus, P., Muller, T. J., Zuschratter, W., and Senitz, D. (2000). The dendritic architecture of prefrontal pyramidal neurons in schizophrenic patients. Neuroreport 11, 3621–3625.
Kandel, E., Schwartz, J., and Jessell, T. (2000). Principles of Neural Science. New York, NY: McGraw-Hill.
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489.
Kash, S. F., Johnson, R. S., Tecott, L. H., Noebels, J. L., Mayfield, R. D., Hanahan, D., et al. (1997). Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 94, 14060–14065.
Keshavan, M. S., Anderson, S., and Pettegrew, J. W. (1994). Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex – the Feinberg hypothesis revisisted. J. Psychiatr. Res. 28, 239–265.
Kestler, L. P., Walker, E., and Vega, E. M. (2001). Dopamine receptors in the brains of schizophrenia patients: a meta-analysis of the findings. Behav. Pharmacol. 12, 355–371.
Khandaker, G. M., Barnett, J. H., White, I. R., and Jones, P. B. (2011). A quantitative meta-analysis of population-based studies of premorbid intelligence and schizophrenia. Schizophr. Res. 132, 220–227.
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639.
Kinney, H. C., Brody, B. A., Kloman, A. S., and Gilles, F. H. (1988). Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J. Neuropathol. Exp. Neurol. 47, 217–234.
Kirkpatrick, B., Messias, N. C., Conley, R. R., and Roberts, R. C. (2003). Interstitial cells of the white matter in the dorsolateral prefrontal cortex in deficit and nondeficit schizophrenia. J. Nerv. Ment. Dis. 191, 563–567.
Klingberg, T., Forssberg, H., and Westerberg, H. (2002). Increased brain activity in frontal and parietal cortex underlies the development of visuospatial working memory capacity during childhood. J. Cogn. Neurosci. 14, 1–10.
Koketsu, D., Mikami, A., Miyamoto, Y., and Hisatsune, T. (2003). Nonrenewal of neurons in the cerebral neocortex of adult macaque monkeys. J. Neurosci. 23, 937–942.
Kolluri, N., Sun, Z. X., Sampson, A. R., and Lewis, D. A. (2005). Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am. J. Psychiatry 162, 1200–1202.
Kornack, D. R., and Rakic, P. (1999). Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc. Natl. Acad. Sci. U.S.A. 96, 5768–5773.
Kornack, D. R., and Rakic, P. (2001). The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc. Natl. Acad. Sci. U.S.A. 98, 4752–4757.
Kornhuber, J., Mack-Burkhardt, F., Riederer, P., Hebenstreit, G. F., Reynolds, G. P., Andrews, H. B., et al. (1989). [3H]MK-801 binding sites in postmortem brain regions of schizophrenic patients. J. Neural Transm. 77, 231–236.
Korr, H., and Schmitz, C. (1999). Facts and fictions regarding post-natal neurogenesis in the developing human cerebral cortex. J. Theor. Biol. 200, 291–297.
Kosaka, J., Takahashi, H., Ito, H., Takano, A., Fujimura, Y., Matsumoto, R., et al. (2010). Decreased binding of C-11 NNC112 and C-11 SCH23390 in patients with chronic schizophrenia. Life Sci. 86, 814–818.
Kosodo, Y., and Huttner, W. B. (2009). Basal process and cell divisions of neural progenitors in the developing brain. Dev. Growth Differ. 51, 251–261.
Kosodo, Y., Roper, K., Haubensak, W., Marzesco, A. M., Corbeil, D., and Huttner, W. B. (2004). Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 23, 2314–2324.
Kostovic, I., and Rakic, P. (1980). Cytology and time of origin of interstitial neurons in the white matter in infant and adult human and monkey telencephalon. J. Neurocytol. 9, 219–242.
Krimer, L. S., Herman, M. M., Saunders, R. C., Boyd, J. C., Hyde, T. M., Carter, J. M., et al. (1997). A qualitative and quantitative analysis of the entorhinal cortex in schizophrenia. Cereb. Cortex 7, 732–739.
Kristiansen, L. V., Beneyto, M., Haroutunian, V., and Meador-Woodruff, J. H. (2006). Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 11, 737–747.
Kroger, J. K., Sabb, F. W., Fales, C. L., Bookheimer, S. Y., Cohen, M. S., and Holyoak, K. J. (2002). Recruitment of anterior dorsolateral prefrontal cortex in human reasoning: a parametric study of relational complexity. Cereb. Cortex 12, 477–485.
Kwon, H., Reiss, A. L., and Menon, V. (2002). Neural basis of protracted developmental changes in visuo-spatial working memory. Proc. Natl. Acad. Sci. U.S.A. 99, 13336–13341.
Kyriakopoulos, M., Bargiotas, T., Barker, G. J., and Frangou, S. (2008). Diffusion tensor imaging in schizophrenia. Biol. Psychiatry 23, 255–273.
Lahti, A. C., Koffel, B., Laporte, D., and Tamminga, C. A. (1995). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13, 9–19.
Lahti, A. C., Weiler, M. A., Tamara Michaelidis, B. A., Parwani, A., and Tamminga, C. A. (2001). Effects of ketamine in normal and schizophrenic volunteers. Neuro-psychopharmacology 25, 455–467.
Laruelle, M., Abi-Dargham, A., van Dyck, C. H., Gil, R., D'Souza, C. D., Erdos, J., et al. (1996). Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. U.S.A. 93, 9235–9240.
Laviola, G., Adriani, W., Morley-Fletcher, S., and Terranova, M. L. (2002). Peculiar response of adolescent mice to acute and chronic stress and to amphetamine: evidence of sex differences. Behav. Brain Res. 130, 117–125.
Laviola, G., Wood, R. D., Kuhn, C., Francis, R., and Spear, L. P. (1995). Cocaine sensitization in periadolescent and adult rats. J. Pharmacol. Exp. Ther. 275, 345–357.
Law, A. J., and Deakin, J. F. W. (2001). Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport 12, 2971–2974.
Law, A. J., Weickert, C. S., Hyde, T. M., Kleinman, J. E., and Harrison, P. J. (2004). Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am. J. Psychiatry 161, 1848–1855.
Law, A. J., Weickert, C. S., Webster, M. J., Herman, M. M., Kleinman, J. E., and Harrison, P. J. (2003). Changes in NMDA receptor subunit mRNAs and cyclophilin mRNA during development of the human hippocampus. Ann. N.Y. Acad. Sci. 1003, 426–430.
Lebel, C., Walker, L., Leemans, A., Phillips, L., and Beaulieu, C. (2008). Microstructural maturation of the human brain from childhood to adulthood. Neuroimage 40, 1044–1055.
Lenroot, R. K., Gogtay, N., Greenstein, D. K., Wells, E. M., Wallace, G. L., Clasen, L. S., et al. (2007). Sexual dimorphism of brain developmental trajectories during childhood and adolescence. Neuroimage 36, 1065–1073.
Lequin, M. H., and Barkovich, A. J. (1999). Current concepts of cerebral malformation syndromes. Curr. Opin. Pediatr. 11, 492–496.
Letinic, K., Zoncu, R., and Rakic, P. (2002). Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649.
Levin, H. S., Culhane, K. A., Hartmann, J., Evankovich, K., Mattson, A. J., Harward, H., et al. (1991). Developmental changes in performance on tests of purported frontal lobe functioning. Dev. Neuropsychol. 7, 377–395.
Lewis, D. A. (1997). Development of the prefrontal cortex during adolescence: insights into vulnerable neural circuits in schizophrenia. Neuropsychopharmacology 16, 385–398.
Lidow, M. S., and Rakic, P. (1992). Scheduling of monoaminergic neurotransmitter receptor expression in the primate neocortex during postnatal development. Cereb. Cortex 2, 401–416.
Lieberman, J. A., Kane, J. M., and Alvir, J. (1987). Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl.) 91, 415–433.
Liston, C., Watts, R., Tottenham, N., Davidson, M. C., Niogi, S., Ulug, A. M., et al. (2006). Frontostriatal microstructure modulates efficient recruitment of cognitive control. Cereb. Cortex 16, 553–560.
Long, L. E., Lind, J., Webster, M., and Weickert, C. S. (2012). Developmental trajectory of the endocannabinoid system in human dorsolateral prefrontal cortex. BMC Neurosci. 13:87. doi: 10.1186/1471-2202-13-87
Maccabe, J. H., Wicks, S., Lofving, S., David, A. S., Berndtsson, A., Gustafsson, J. E., et al. (2013). Decline in cognitive performance between ages 13 and 18 years and the risk for psychosis in adulthood. A Swedish longitudinal cohort study in males. JAMA Psychiatry. 70, 261–270.
Malhotra, A. K., Pinals, D. A., Adler, C. M., Elman, I., Clifton, A., Pickar, D., et al. (1997). Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17, 141–150.
Manoach, D. S., Gollub, R. L., Benson, E. S., Searl, M. M., Goff, D. C., Halpern, E., et al. (2000). Schizophrenic subjects show aberrant fMRI activation of dorsolateral prefrontal cortex and basal ganglia during working memory performance. Biol. Psychiatry 48, 99–109.
Manoach, D. S., Press, D. Z., Thangaraj, V., Searl, M. M., Goff, D. C., Halpern, E., et al. (1999). Schizophrenic subjects activate dorsolateral prefrontal cortex during a working memory task, as measured by fMRI. Biol. Psychiatry 45, 1128–1137.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., et al. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Marin-Padilla, M. (1990). Three-dimensional structural organization of layer I of the human cerebral cortex: a Golgi study. J. Comp. Neurol. 299, 89–105.
Marin, O., and Rubenstein, J. L. (2003). Cell migration in the forebrain. Annu. Rev. Neurosci. 26, 441–483.
Matthews, P. R., Eastwood, S. L., and Harrison, P. J. (2012). Reduced myelin basic protein and actin-related gene expression in visual cortex in schizophrenia. PLoS ONE 7:e38211. doi: 10.1371/journal.pone.0038211
McArdle, J. J., Ferrer-Caja, E., Hamagami, F., and Woodcock, R. W. (2002). Comparative longitudinal structural analyses of the growth and decline of multiple intellectual abilities over the life span. Dev. Psychol. 38, 115–142.
McClellan, A. M., and Twyman, R. E. (1999). Receptor system response kinetics reveal functional subtypes of native murine and recombinant human GABAA receptors. J. Physiol. 515, 711–727.
McGorry, P. (2011). Transition to adulthood: the critical period for pre-emptive, disease-modifying care for schizophrenia and related disorders. Schizophr. Bull. 37, 524–530.
Meador-Woodruff, J. H., Haroutunian, V., Powchik, P., Davidson, M., Davis, K. L., and Watson, S. J. (1997). Dopamine receptor transcript expression in striatum and prefrontal and occipital cortex – focal abnormalities in orbitofrontal cortex in schizophrenia. Arch. Gen. Psychiatry 54, 1089–1095.
Mellios, N., Huang, H. S., Baker, S. P., Gaidzicka, M., Ginns, E., and Akbarian, S. (2009). Molecular determinants of dysregulated GABAergic gene expression in the prefrontal cortex of subjects with schizophrenia. Biol. Psychiatry 65, 1006–1014.
Meyer-Lindenberg, A., Poline, J. B., Kohn, P. D., Holt, J. L., Egan, M. F., Weinberger, D. R., et al. (2001). Evidence for abnormal cortical functional connectivity during working memory in schizophrenia. Am. J. Psychiatry 158, 1809–1817.
Meyer-Lindenberg, A. S., Olsen, R. K., Kohn, P. D., Brown, T., Egan, M. F., Weinberger, D. R., et al. (2005). Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Arch. Gen. Psychiatry 62, 379–386.
Meyer, G. (2001). Human neocortical development: the importance of embryonic and early fetal events. Neuroscientist 7, 303–314.
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423.
Missale, C., Nash, S. R., Robinson, S. W., Jaber, M., and Caron, M. G. (1998). Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225.
Mizoguchi, K., Shoji, H., Tanaka, Y., Maruyama, W., and Tabira, T. (2009). Age-related spatial working memory impairment is caused by prefrontal cortical dopaminergic dysfunction in rats. Neuroscience 162, 1192–1201.
Monsma, F. J., McVittie, L. D., Gerfen, C. R., Mahan, L. C., and Sibley, D. R. (1989). Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature 342, 926–929.
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540.
Morris, H. M., Hashimoto, T., and Lewis, D. A. (2008). Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb. Cortex 18, 1575–1587.
Mouri, A., Noda, Y., Enomoto, T., and Nabeshima, T. (2007). Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem. Int. 51, 173–184.
Mueller, H. T., and Meador-Woodruff, J. H. (2004). NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophr. Res. 71, 361–370.
Mukaetova-Ladinska, E. B., Hurt, J., Honer, W. G., Harrington, C. R., and Wischik, C. M. (2002). Loss of synaptic but not cytoskeletal proteins in the cerebellum of chronic schizophrenics. Neurosci. Lett. 317, 161–165.
Murphy, K. M., Beston, B. R., Boley, P. M., and Jones, D. G. (2005). Development of human visual cortex: a balance between excitatory and inhibitory plasticity mechanisms. Dev. Psychobiol. 46, 209–221.
Nadarajah, B. (2003). Radial glia and somal translocation of radial neurons in the developing cerebral cortex. Glia 43, 33–36.
Nadarajah, B., Brunstrom, J. E., Grutzendler, J., Wong, R. O., and Pearlman, A. L. (2001). Two modes of radial migration in early development of the cerebral cortex. Nat. Neurosci. 4, 143–150.
Nadarajah, B., and Parnavelas, J. G. (2002). Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 3, 423–432.
Nagy, Z., Westerberg, H., and Klingberg, T. (2004). Maturation of white matter is associated with the development of cognitive functions during childhood. J. Cogn. Neurosci. 16, 1227–1233.
Newell, K. A., Deng, C., and Huang, X. F. (2006). Increased cannabinoid receptor density in the posterior cingulate cortex in schizophrenia. Exp. Brain Res. 172, 556–560.
Newell, K. A., Zavitsanou, K., Jew, S. K., and Huang, X. F. (2007). Alterations of muscarinic and GABA receptor binding in the posterior cingulate cortex in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 225–233.
Nieoullon, A. (2002). Dopamine and the regulation of cognition and attention. Prog. Neurobiol. 67, 53–83.
Noga, J. T., Hyde, T. M., Herman, M. M., Spurney, C. F., Bigelow, L. B., Weinberger, D. R., et al. (1997). Glutamate receptors in the postmortem striatum of schizophrenic, suicide, and control brains. Synapse 27, 168–176.
Nudmamud, S., and Reynolds, G. P. (2001). Increased density of glutamate/N-methyl-D-aspartate receptors in superior temporal cortex in schizophrenia. Neurosci. Lett. 304, 9–12.
O'Rahilly, R., and Muller, F. (1994). “Neurulation in the normal human embryo,” in Neural Tube Effects, eds G. Bock and J. Marsh (Chichester: Wiley), 70–82.
Okubo, Y., Suhara, T., Suzuki, K., Kobayashi, K., Inoue, O., Terasaki, O., et al. (1997). Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 385, 634–636.
Olesen, P. J., Nagy, Z., Westerberg, H., and Klingberg, T. (2003). Combined analysis of DTI and fMRI data reveals a joint maturation of white and grey matter in a fronto-parietal network. Cogn. Brain Res. 18, 48–57.
Olsen, R. W., and Sieghart, W. (2008). International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260.
Onn, S. P., Wang, X. B., Lin, M., and Grace, A. A. (2005). Dopamine D1 and D4 receptor subtypes differentially modulate recurrent excitatory synapses in prefrontal cortical pyramidal neurons. Neuropsychopharmacology 31, 318–338.
Pantelis, C., Yucel, M., Wood, S. J., McGorry, P. D., and Velakoulis, D. (2003). Early and late neurodevelopmental disturbances in schizophrenia and their functional consequences. Aust. N.Z. J. Psychiatry 37, 399–406.
Parlapani, E., Schmitt, A., Erdmann, A., Bernstein, H. G., Breunig, B., Gruber, O., et al. (2009). Association between myelin basic protein expression and left entorhinal cortex pre-alpha cell layer disorganization in schizophrenia. Brain Res. 1301, 126–134.
Perez-Otano, I., Lujan, R., Tavalin, S. J., Plomann, M., Modregger, J., Liu, X. B., et al. (2006). Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat. Neurosci. 9, 611–621.
Perlstein, W. M., Carter, C. S., Noll, D. C., and Cohen, J. D. (2001). Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am. J. Psychiatry 158, 1105–1113.
Perlstein, W. M., Dixit, N. K., Carter, C. S., Noll, D. C., and Cohen, J. D. (2003). Prefrontal cortex dysfunction mediates deficits in working memory and prepotent responding in schizophrenia. Biol. Psychiatry 53, 25–38.
Petanjek, Z., Berger, B., and Esclapez, M. (2009a). Origins of cortical GABAergic neurons in the cynomolgus monkey. Cereb. Cortex 19, 249–262.
Petanjek, Z., Kostovic, I., and Esclapez, M. (2009b). Primate-specific origins and migration of cortical GABAergic neurons. Front. Neuroanat. 3:26. doi: 10.3389/neuro.05.026.2009
Petanjek, Z., Judas, M., Kostovic, I., and Uylings, H. B. M. (2008). Lifespan alterations of basal dendritic trees of pyramidal neurons in the human prefrontal cortex: a layer-specific pattern. Cereb. Cortex 18, 915–929.
Petanjek, Z., Judas, M., Simic, G., Rasin, M. R., Uylings, H. B. M., Rakic, P., et al. (2011). Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 108, 13281–13286.
Philpot, B. D., Sekhar, A. K., Shouval, H. Z., and Bear, M. F. (2001). Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29, 157–169.
Pinto, J. G. A., Hornby, K. R., Jones, D. G., and Murphy, K. M. (2010). Developmental changes in GABAergic mechanisms in human visual cortex across the lifespan. Front. Cell. Neurosci. 4:16. doi: 10.3389/fncel.2010.00016
Potkin, S. G., Turner, J. A., Brown, G. G., McCarthy, G., Greve, D. N., Glover, G. H., et al. (2009). Working memory and DLPFC inefficiency in schizophrenia: the FBIRN study. Schizophr. Bull. 35, 19–31.
Powell, C. M., and Miyakawa, T. (2006). Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol. Psychiatry 59, 1198–1207.
Prabhakaran, V., Smith, J. A. L., Desmond, J. E., Glover, G. H., and Gabrieli, J. D. E. (1997). Neural substrates of fluid reasoning: an fMRI study of neocortical activation during performance of the Raven's Progressive Matrices Test. Cogn. Psychol. 33, 43–63.
Provenzale, J. M., Liang, L., Delong, D., and White, L. E. (2007). Diffusion tensor imaging assessment of brain white matter maturation during the first postnatal year. Am. J. Roentgenol. 189, 476–486.
Quinn, H. R., Matsumoto, I., Callaghan, P. D., Long, L. E., Arnold, J. C., Gunasekaran, N., et al. (2008). Adolescent rats find repeated Delta(9)-THC less aversive than adult rats but display greater residual cognitive deficits and changes in hippocampal protein expression following exposure. Neuropsychopharmacology 33, 1113–1126.
Rakic, P. (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83.
Rakic, S., and Zecevic, N. (2003). Emerging complexity of layer I in human cerebral cortex. Cereb. Cortex 13, 1072–1083.
Ravizza, S. M., Moua, K. C. K., Long, D., and Carter, C. S. (2010). The impact of context processing deficits on task-switching performance in schizophrenia. Schizophr. Res. 116, 274–279.
Reichenberg, A., Weiser, M., Rapp, M. A., Rabinowitz, J., Caspi, A., Schmeidler, J., et al. (2005). Elaboration on premorbid intellectual performance in schizophrenia – premorbid intellectual decline and risk for schizophrenia. Arch. Gen. Psychiatry 62, 1297–1304.
Reif, A., Fritzen, S., Finger, M., Strobel, A., Lauer, M., Schmitt, A., et al. (2006). Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol. Psychiatry 11, 514–522.
Reynolds, G. P., and Beasley, C. L. (2001). GABAergic neuronal subtypes in the human frontal cortex – development and deficits in schizophrenia. J. Chem. Neuroanat. 22, 95–100.
Reynolds, G. P., Beasley, C. L., and Zhang, Z. J. (2002). Understanding the neurotransmitter pathology of schizophrenia: selective deficits of subtypes of cortical GABAergic neurons. J. Neural Transm. 109, 881–889.
Richland, L. E., Morrison, R. G., and Holyoak, K. J. (2006). Children's development of analogical reasoning: insights from scene analogy problems. J. Exp. Child Psychol. 94, 249–273.
Ritter, L. M., Vazquez, D. M., and Meador-Woodruff, J. H. (2002). Ontogeny of ionotropic glutamate receptor subunit expression in the rat hippocampus. Brain Res. Dev. Brain Res. 139, 227–236.
Roberts, A. C., Diez-Garcia, J., Rodriguiz, R. M., Lopez, I. P., Lujan, R., Martinez-Turrillas, R., et al. (2009). Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron 63, 342–356.
Romanides, A. J., Duffy, P., and Kalivas, P. W. (1999). Glutamatergic and dopaminergic afferents to the prefrontal cortex regulate spatial working memory in rats. Neuroscience 92, 97–106.
Rothmond, D. A., Weickert, C. S., and Webster, M. J. (2012). Developmental changes in human dopamine neurotransmission: cortical receptors and terminators. BMC Neurosci. 13:18. doi: 10.1186/1471-2202-13-18
Runyan, C. A., Weickert, C. S., and Saunders, R. C. (2006). “Adult neurogenesis and immediate early gene response to working memory stimulation in the primate prefrontal cortex,” in Society for Neuroscience (Atlanta, GA, USA: Society for Neuroscience).
Sakai, T., Oshima, A., Nozaki, Y., Ida, I., Haga, C., Akiyama, H., et al. (2008). Changes in density of calcium-binding-protein-immunoreactive GABAergic neurons in prefrontal cortex in schizophrenia and bipolar disorder. Neuropathology 28, 143–150.
Salimi, K., Glantz, L. A., Hamer, R. M., German, T. T., Gilmore, J. H., and Jarskog, L. F. (2008). Regulation of complexin 1 and complexin 2 in the developing human prefrontal cortex. Synapse 62, 273–282.
Sanai, N., Nguyen, T., Ihrie, R. A., Mirzadeh, Z., Tsai, H. H., Wong, M., et al. (2011). Corridors of migrating neurons in the human brain and their decline during infancy. Nature 478, 382–386.
Sawada, K., Young, C. E., Barr, A. M., Longworth, K., Takahashi, S., Arango, V., et al. (2002). Altered immunoreactivity of complexin protein in prefrontal cortex in severe mental illness. Mol. Psychiatry 7, 484–492.
Scarr, E., Beneyto, M., Meador-Woodruff, J. H., and Dean, B. (2005). Cortical glutamatergic markers in schizophrenia. Neuropsychopharmacology 30, 1521–1531.
Scherf, K. S., Sweeney, J. A., and Luna, B. (2006). Brain basis of developmental change in visuospatial working memory. J. Cogn. Neurosci. 18, 1045–1058.
Schmitt, A., Koschel, J., Zink, M., Bauer, M., Sommer, C., Frank, J., et al. (2010). Gene expression of NMDA receptor subunits in the cerebellum of elderly patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 260, 101–111.
Schneider, J. F., Il'yasov, K. A., Hennig, J., and Martin, E. (2004). Fast quantitative diffusion-tensor imaging of cerebral white matter from the neonatal period to adolescence. Neuroradiology 46, 258–266.
Schulz, J., Sundin, J., Leask, S., and Done, D. J. (2012). Risk of adult schizophrenia and its relationship to childhood IQ in the 1958 British Birth Cohort. Schizophr. Bull. doi: 10.1093/schbul/sbs157. [Epub ahead of print].
Seidman, L. J., Cherkerzian, S., Goldstein, J. M., Agnew-Blais, J., Tsuang, M. T., and Buka, S. L. (2013). Neuropsychological performance and family history in children at age 7 who develop adult schizophrenia or bipolar psychosis in the New England Family Studies. Psychol. Med. 43, 119–131.
Selemon, L. D., and Goldman-Rakic, P. S. (1999). The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol. Psychiatry 45, 17–25.
Seshadri, S., Zeledon, M., and Sawa, A. (2013). Synapse-specific contributions in the cortical pathology of schizophrenia. Neurobiol. Dis. 53, 26–35.
Shalaev, I., Weickert, C. S., Webster, M. J., and Saunders, R. C. (2002). “Neurogenesis in the dentate gurys and subventricular zone of the macaque,” in Society for Neuroscience (Orlando, FL, USA: Society for Neuroscience).
Shankle, W. R., Landing, B. H., Rafii, M. S., Schiano, A., Chen, J. M., and Hara, J. (1998a). Evidence for a postnatal doubling of neuron number in the developing human cerebral cortex between 15 months and 6 years. J. Theor. Biol. 191, 115–140.
Shankle, W. R., Romney, A. K., Landing, B. H., and Hara, J. (1998b). Developmental patterns in the cytoarchitecture of the human cerebral cortex from birth to 6 years examined by correspondence analysis. Proc. Natl. Acad. Sci. U.S.A. 95, 4023–4028.
Sheng, M., and Hoogenraad, C. C. (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76, 823–847.
Sidman, R. L., and Rakic, P. (1973). Neuronal migration, with special reference to developing human brain – review. Brain Res. 62, 1–35.
Silver, H., Feldman, P., Bilker, W., and Gur, R. C. (2003). Working memory deficit as a core neuropsychological dysfunction in schizophrenia. Am. J. Psychiatry 160, 1809–1816.
Simpson, M. D., Slater, P., Royston, M. C., and Deakin, J. F. (1991). Alterations in phencyclidine and sigma binding sites in schizophrenic brains. Effects of disease process and neuroleptic medication. Schizophr. Res. 6, 41–48.
Snook, L., Paulson, L. A., Roy, D., Phillips, L., and Beaulieu, C. (2005). Diffusion tensor imaging of neuro development in children and young adults. Neuroimage 26, 1164–1173.
So, C. H., Varghese, G., Curley, K. J., Kong, M. M. C., Alijaniaram, M., Ji, X. D., et al. (2005). D1 and D2 dopamine receptors form heterooligomers and cointernalize after selective activation of either receptor. Mol. Pharmacol. 68, 568–578.
Sokolov, B. P. (1998). Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 71, 2454–2464.
Soloway, A. S., Pucak, M. L., Melchitzky, D. S., and Lewis, D. A. (2002). Dendritic morphology of callosal and ipsilateral projection neurons in monkey prefrontal cortex. Neuroscience 109, 461–471.
Somenarain, L., and Jones, L. B. (2010). A comparative study of MAP2 immunostaining in areas 9 and 17 in schizophrenia and Huntington chorea. J. Psychiatr. Res. 44, 694–699.
Spalding, K. L., Bhardwaj, R. D., Buchholz, B. A., Druid, H., and Frisen, J. (2005). Retrospective birth dating of cells in humans. Cell 122, 133–143.
Spear, L. P. (2007). Adolescent Psychopathology and the Developing Brain: Integrating Brain and Prevention Science. New York, NY: Oxford University Press.
Spear, L. P., Shalaby, I. A., and Brick, J. (1980). Chronic administration of haloperidol during development: behavioral and psychopharmacological effects. Psychopharmacology (Berl.) 70, 47–58.
Sternberg, R. J., and Rifkin, B. (1979). Development of analogical reasoning processes. J. Exp. Child Psychol. 27, 195–232.
Sunahara, R. K., Guan, H. C., Odowd, B. F., Seeman, P., Laurier, L. G., Ng, G., et al. (1991). Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 350, 614–619.
Swartzwelder, H. S., Richardson, R. C., Markwiese-Foerch, B., Wilson, W. A., and Little, P. J. (1998). Developmental differences in the acquisition of tolerance to ethanol. Alcohol 15, 311–314.
Szabadics, J., Varga, C., Molnar, G., Olah, S., Barzo, P., and Tamas, G. (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235.
Takemura, N. U. (2005). Evidence for neurogenesis within the white matter beneath the temporal neocortex of the adult rat brain. Neuroscience 134, 121–132.
Tallerico, T., Novak, G., Liu, I. S. C., Ulpian, C., and Seeman, P. (2001). Schizophrenia: elevated mRNA for dopamine D2(Longer) receptors in frontal cortex. Mol. Brain Res. 87, 160–165.
Tamamaki, N., Fujimori, K., and Takauji, R. (1997). Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J. Neurosci. 17, 8313–8323.
Tamm, L., Menon, V., and Reiss, A. L. (2002). Maturation of brain function associated with response inhibition. J. Am. Acad. Child Adolesc. Psychiatry 41, 1231–1238.
Tan, H. Y., Sust, S., Buckholtz, J. W., Mattay, V. S., Meyer-Lindenberg, A., Egan, M. F., et al. (2006). Dysfunctional prefrontal regional specialization and compensation in schizophrenia. Am. J. Psychiatry 163, 1969–1977.
Thatcher, R. W. (1991). Maturation of the human frontal lobes – physiological evidence for staging. Dev. Neuropsychol. 7, 397–419.
Thatcher, R. W. (1992). Cyclic cortical reorganization during early childhood. Brain Cogn. 20, 24–50.
Thatcher, R. W. (1994). Psychopathology for early frontal lobe damage – dependence on cycles of development. Dev. Psychopathol. 6, 565–596.
Thompson, C. L., Drewery, D. L., Atkins, H. D., Stephenson, F. A., and Chazot, P. L. (2002). Immunohistochemical localization of N-methyl-D-aspartate receptor subunits in the adult murine hippocampal formation: evidence for a unique role of the NR2D subunit. Brain Res. Mol. Brain Res. 102, 55–61.
Thompson, M., Weickert, C. S., Wyatt, E., and Webster, M. J. (2009). Decreased glutamic acid decarboxylase(67) mRNA expression in multiple brain areas of patients with schizophrenia and mood disorders. J. Psychiatr. Res. 43, 970–977.
Toro, C., and Deakin, J. F. W. (2005). NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr. Res. 80, 323–330.
Tseng, K. Y., Lewis, B. L., Lipska, B. K., and O'Donnell, P. (2007). Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol. Psychiatry 62, 730–738.
Tseng, K. Y., and O'Donnell, P. (2004). Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J. Neurosci. 24, 5131–5139.
Tseng, K. Y., and O'Donnell, P. (2007). D-2 dopamine receptors recruit a GABA component for their attenuation of excitatory synaptic transmission in the adult rat prefrontal cortex. Synapse 61, 843–850.
Turman, J. E. Jr., Lee, O. K., and Chandler, S. H. (2002). Differential NR2A and NR2B expression between trigeminal neurons during early postnatal development. Synapse 44, 76–85.
Uhlhaas, P. J., Haenschel, C., Nikolic, D., and Singer, W. (2008). The role of oscillations and synchrony in cortical networks and their putative relevance for the pathophysiology of schizophrenia. Schizophr. Bull. 34, 927–943.
Uhlhaas, P. J., Roux, F., Singer, W., Haenschel, C., Sireteanu, R., and Rodriguez, E. (2009). The development of neural synchrony reflects late maturation and restructuring of functional networks in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 9866–9871.
Velanova, K., Wheeler, M. E., and Luna, B. (2008). Maturational changes in anterior cingulate and frontoparietal recruitment support the development of error processing and inhibitory control. Cereb. Cortex 18, 2505–2522.
Verdurand, M., Fillman, S. G., Weickert, C. S., and Zavitsanou, K. (2013). Increases in [3H]muscimol and [3H]flumazenil binding in the dorsolateral prefrontal cortex in schizophrenia are linked to alpha4 and gamma2s mRNA levels respectively. PLoS ONE 8:e52724. doi: 10.1371/journal.pone.0052724
Volk, D. W., Matsubara, T., Li, S. Y., Sengupta, E. J., Georgiev, D., Minabe, Y., et al. (2012). Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am. J. Psychiatry 169, 1082–1091.
Volk, D. W., Pierri, J. N., Fritschy, J. M., Auh, S., Sampson, A. R., and Lewis, D. A. (2002). Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 12, 1063–1070.
Wang, C., Liu, F., Liu, Y. Y., Zhao, C. H., You, Y., Wang, L., et al. (2011). Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res. 21, 1534–1550.
Wang, J., and O'Donnell, P. (2001). D-1 dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cereb. Cortex 11, 452–462.
Webster, M. J., Elashoff, M., and Weickert, C. S. (2011). Molecular evidence that cortical synaptic growth predominates during the first decade of life in humans. Int. J. Dev. Neurosci. 29, 225–236.
Webster, M. J., Weickert, C. S., Herman, M. M., Hyde, T. M., and Kleinman, J. E. (2001). Synaptophysin and GAP-43 mRNA levels in the hippocampus of subjects with schizophrenia. Schizophr. Res. 49, 89–98.
Weickert, C. S., Elashoff, M., Richards, A. B., Sinclair, D., Bahn, S., Paabo, S., et al. (2009). Transcriptome analysis of male-female differences in prefrontal cortical development. Mol. Psychiatry 14, 558–561.
Weickert, C. S., Fung, S. J., Catts, V. S., Schofield, P. R., Allen, K. M., Moore, L. T., et al. (2012). Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry. doi: 10.1038/mp.2012.137. [Epub ahead of print].
Weickert, C. S., Straub, R. E., McClintock, B. W., Matsumoto, M., Hashimoto, R., Hyde, T. M., et al. (2004). Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch. Gen. Psychiatry 61, 544–555.
Weickert, T. W., Goldberg, T. E., Gold, J. M., Bigelow, L. B., Egan, M. F., and Weinberger, D. R. (2000a). Cognitive impairments in patients with schizophrenia displaying preserved and compromised intellect. Arch. Gen. Psychiatry 57, 907–913.
Weickert, C. S., Webster, M. J., Colvin, S. M., Herman, M. M., Hyde, T. M., Weinberger, D. R., et al. (2000b). Localization of epidermal growth factor receptors and putative neuroblasts in human subependymal zone. J. Comp. Neurol. 423, 359–372.
Weickert, C. S., Webster, M. J., Gondipalli, P., Rothmond, D., Fatula, R. J., Herman, M. M., et al. (2007). Postnatal alterations in dopaminergic markers in the human prefrontal cortex. Neuroscience 144, 1109–1119.
Weickert, T. W., and Goldberg, T. E. (2000). “The course of cognitive impairments in patients with schizophrenia,” in Cognition in Schizophrenia: Impairments, Importance and Treatment Strategies, eds T. Sharma and P. Harvey (New York, NY: Oxford University Press), 3–15.
Weinberger, D. R. (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669.
Weinberger, D. R., Berman, K. F., and Zec, R. F. (1986). Physiological dysfunction of dorsolateral prefrontal cortex in schizophrenia. 1. Regional cerebral blood flow evidence. Arch. Gen. Psychiatry 43, 114–124.
Williams, G. V., and Goldman-Rakic, P. S. (1995). Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature 376, 572–575.
Winter, S., Dieckmann, M., and Schwabe, K. (2009). Dopamine in the prefrontal cortex regulates rats behavioral flexibility to changing reward value. Behav. Brain Res. 198, 206–213.
Wong, H. K., Liu, X. B., Matos, M. F., Chan, S. F., Perez-Otano, I., Boysen, M., et al. (2002). Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J. Comp. Neurol. 450, 303–317.
Wood, S. J., Pantelis, C., Velakoulis, D., Yucel, M., Fornito, A., and McGorry, P. D. (2008). Progressive changes in the development toward schizophrenia: studies in subjects at increased symptomatic risk. Schizophr. Bull. 34, 322–329.
Woodberry, K. A., Giuliano, A. J., and Seidman, L. J. (2008). Premorbid IQ in schizophrenia: a meta-analytic review. Am. J. Psychiatry 165, 579–587.
Wright, S. B., Matlen, B. J., Baym, C. L., Ferrer, E., and Bunge, S. A. (2008). Neural correlates of fluid reasoning in children and adults. Front. Hum. Neurosci. 1:8. doi: 10.3389/neuro.09.008.2007
Xu, G., Broadbelt, K. G., Haynes, R. L., Folkerth, R. D., Borenstein, N. S., Belliveau, R. A., et al. (2011). Late development of the GABAergic system in the human cerebral cortex and white matter. J. Neuropathol. Exp. Neurol. 70, 841–858.
Yago, K. B., Pitts, F. N. Jr., Burgoyne, R. W., Aniline, O., Yago, L. S., and Pitts, A. F. (1981). The urban epidemic of phencyclidine (PCP) use: clinical and laboratory evidence from a public psychiatric hospital emergency service. J. Clin. Psychiatry 42, 193–196.
Yakovlev, P. I., and Lecours, A. (1967). “The myelogenetic cycles of regional maturation of the brain,” in Regional Development of the Brain Early in Life, ed A. Minkowski (Oxford: Blackwell Scientific Publications Inc.).
Yamashita, A., Hayashi, M., Shimizu, K., and Oshima, K. (1989). Ontogeny of somatostatin in cerebral cortex of macaque monkey: an immunohistochemical study. Brain Res. Dev. Brain Res. 45, 103–111.
Yan, Y. H., Van Brederode, J. F., and Hendrickson, A. E. (1995). Transient co-localization of calretinin, parvalbumin, and calbindin-D28K in developing visual cortex of monkey. J. Neurocytol. 24, 825–837.
Yang, Y., Fung, S. J., Rothwell, A., Tianmei, S., and Weickert, C. S. (2011). Increased interstitial white matter neuron density in the dorsolateral prefrontal cortex of people with schizophrenia. Biol. Psychiatry 69, 63–70.
Zavitsanou, K., Garrick, T., and Huang, X. F. (2004). Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 355–360.
Zecevic, N. (2004). Specific characteristic of radial glia in the human fetal telencephalon. Glia 48, 27–35.
Zecevic, N., Chen, Y., and Filipovic, R. (2005). Contributions of cortical subventricular zone to the development of the human cerebral cortex. J. Comp. Neurol. 491, 109–122.
Zecevic, N., Hu, F., and Jakovcevski, I. (2011). Interneurons in the developing human neocortex. Dev. Neurobiol. 71, 18–33.
Zhan, L., Kerr, J. R., Lafuente, M. J., Maclean, A., Chibalina, M. V., Liu, B., et al. (2011). Altered expression and coregulation of dopamine signalling genes in schizophrenia and bipolar disorder. Neuropathol. Appl. Neurobiol. 37, 206–219.
Keywords: cognition, neurogenesis, myelination, excitatory synapses, neural migration, NMDA receptor, GABA receptor, dopamine receptor
Citation: Catts VS, Fung SJ, Long LE, Joshi D, Vercammen A, Allen KM, Fillman SG, Rothmond DA, Sinclair D, Tiwari Y, Tsai S-Y, Weickert TW and Shannon Weickert C (2013) Rethinking schizophrenia in the context of normal neurodevelopment. Front. Cell. Neurosci. 7:60. doi: 10.3389/fncel.2013.00060
Received: 13 February 2013; Accepted: 16 April 2013;
Published online: 15 May 2013.
Edited by:
Brian Dean, Mental Health Research Institute, AustraliaReviewed by:
Elizabeth A. Thomas, The Scripps Research Institute, USAAndrew S. Gibbons, The University of Melbourne, Australia
Copyright © 2013 Catts, Fung, Long, Joshi, Vercammen, Allen, Fillman, Rothmond, Sinclair, Tiwari, Tsai, Weickert and Shannon Weickert. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Cynthia Shannon Weickert, Schizophrenia Research Laboratory, Neuroscience Research Australia, PO Box 1165, Randwick, NSW 2031, Australia. e-mail:Yy53ZWlja2VydEBuZXVyYS5lZHUuYXU=