
- Division of Neurology and Translational Epilepsy Research Program, Department of Pediatrics, University of Colorado School of Medicine, Aurora, CO, USA
The term epileptogenesis refers to a dynamic alteration in neuronal excitability that promotes the appearance of spontaneous seizures. Temporal lobe epilepsy, the most common type of acquired epilepsy, often develops after an insult to the brain such as trauma, febrile seizures, encephalitis, or status epilepticus. During the pre-epileptic state (also referred as latent or silent period) there is a plethora of molecular, biochemical, and structural changes that lead to the generation of recurrent spontaneous seizures (or epilepsy). The specific contribution of these alterations to epilepsy development is unclear, but a loss of inhibition has been associated with the increased excitability detected in the latent period. A rapid increase in neuronal hyperexcitability could be due, at least in part, to a decline in the number of physiologically active GABAA receptors (GABAAR). Altered expression of scaffolding proteins involved in the trafficking and anchoring of GABAAR could directly impact the stability of GABAergic synapses and promote a deficiency in inhibitory neurotransmission. Uncovering the molecular mechanisms operating during epileptogenesis and its possible impact on the regulation of GABAAR and scaffolding proteins may offer new targets to prevent the development of epilepsy.
This review centers on the possible role of GABAA receptors (GABAAR) and the scaffolding protein gephyrin in epileptogenesis. The basic premise is that disruption of the network of proteins involved in the trafficking and anchoring of GABAAR might result in decreased inhibitory drive, which may promote the development of spontaneous seizures. A brief introduction to epilepsy and epileptogenesis is provided along with some of the fundamental aspects of the regulation of GABAAR by gephyrin. Finally an overview of the alterations in gephyrin and GABAAR function observed during epileptogenesis is included.
Epileptogenesis and Epilepsy
In the majority of patients, temporal lobe epilepsy (TLE) appears to be the result of an injury to the brain caused by trauma, febrile seizures, encephalitis, or status epilepticus (SE; Duncan et al., 2006; Sharma et al., 2007). The precise point in time when spontaneous seizures appear is unknown but it is suspected that following a brain injury there is a pre-epileptic state when many changes occur and transform a normal brain into an epileptic one. Since in many cases, after a brain injury, patients recover from a triggering injury without presenting overt spontaneous seizures, the period that precedes the appearance of recurrent epileptic seizures is known as latent or silent period (Sharma et al., 2007; O’Dell et al., 2012). During the early stages of the chronic period spontaneous seizures are more likely to be controlled with medication but as the disease progress seizures become less responsive to medication and might become intractable (Wieser, 2004). So far there is a consensus that acquired epilepsy results from a triggering insult to the brain and that the cellular and molecular alterations resulting from such insult play a key role in the development of epilepsy, but a direct link between the brain abnormalities detected in epileptic tissue and the generation of recurrent spontaneous seizures remains to be firmly established.
Due to the inherent or obvious limitations associated with the acquisition of human samples, the mechanisms responsible for the appearance of spontaneous seizures are being aggressively studied in experimental models of epilepsy. Post-SE models of chronic epilepsy closely mimic the clinical manifestations and tissue abnormalities observed in humans affected with TLE (Sharma et al., 2007; Curia et al., 2008; Loscher and Brandt, 2010). Induction of SE in rodents by systemic or local administration of a chemoconvulsant is usually followed by a silent (latent) period lasting days or weeks when no obvious seizure activity is observed. During the latent period brain abnormalities develop and inter-ictal activity becomes more frequent, and then suddenly with no apparent cause overt spontaneous seizures manifest (Curia et al., 2008; Scorza et al., 2009). To capitalize the methodological advantages provided by the experimental models, several laboratories have attempted to characterize and establish the duration of the latent (silent) period, unfortunately these attempts have yielded a wide range of measures (Williams et al., 2007). It appears that despite the advantages provided by a well-controlled experimental setting, the animals used in the experiments and the type and severity of injury being tested might impact the length of the latent period. More importantly, some methodological limitations (i.e., accuracy of seizure detection, continuous vs. intermittent monitoring, authentication of when the first seizure occurred, etc.) directly impact the accuracy of the measurements and need to be overcome in order to obtain a definitive characterization of the latent period (Williams et al., 2007).
The dynamic process characterized by progressive alterations in neuronal excitability that promotes appearance of spontaneous seizures and its associated structural lesions is known as epileptogenesis (Pitkanen and Lukasiuk, 2011). Currently, the terms epileptogenesis and latent (or silent) period are used interchangeably to describe the period that encompass the occurrence of an insult to the brain and the appearance epileptic seizures. However, recent findings in patients and experimental models suggest that the alterations resulting from an injury to the brain might progress beyond the appearance of the first spontaneous seizure. Accordingly, it has been suggested that during the natural evolution of epilepsy, each new episode of spontaneous seizures (an insult itself) produces new damage that compounds the damage produced by the original brain injury (Williams et al., 2007; Sloviter, 2008; Pitkanen and Lukasiuk, 2009; O’Dell et al., 2012). Many cellular and molecular alterations in both neuronal and non-neuronal cells have been observed during epileptogenesis. Neuronal alterations include neurodegeneration, neurogenesis, and axonal damage in addition to the architectural reorganization of neuronal processes that result from abnormal sprouting and altered dendritic plasticity. Astrocytes become hypertrophic, develop longer and thicker processes and increase the expression of glial fibrillary acidic protein all as part of a process known as reactive astrogliosis (Gibbons et al., 2012; Heinemann et al., 2012; Kovacs et al., 2012). During epileptogenesis there are many pathological processes that affect brain excitability including the breakdown of the blood–brain barrier (BBB) and inflammation. Ultrastructural analysis of epileptic tissue revealed significant abnormalities in the BBB components and focal opening of the BBB by direct application of albumin can lead to the generation of an epileptic focus (Ivens et al., 2007; Vezzani et al., 2011; Heinemann et al., 2012). Further, the magnitude of BBB leakage occurring during epileptogenesis has been shown to directly correlate with the seizure frequency detected during the chronic period (van Vliet et al., 2007; Vezzani et al., 2013). Inflammatory response due to activation of microglia and astrocytes is associated with brain damage and increased BBB permeability in the tissue adjacent to the region of injury (Vezzani et al., 2011; Marchi et al., 2012). SE-induced inflammation is observed during the epileptogenic period and appears to be reactivated by the occurrence of spontaneous seizures, suggesting that inflammation might have a role in epileptogenesis and promote epileptic activity during the chronic period due to the continuous presence of inflammation intermediaries (Vezzani et al., 2011, 2013).
GABAA Receptors and its Associated Proteins
The majority of fast inhibitory neurotransmission in the mature brain is mediated by anion-selective GABAAR that are assembled as pentamers from an array of multiple subunit subtypes including α1–2, β1–3, γ1–3, δ, ε, π, θ, and σ1–3 (Fritschy, 2008; Jacob et al., 2008; Luscher et al., 2011; Brickley and Mody, 2012). The subunit composition of GABAAR governs the intrinsic properties of the channel such as affinity for GABA, receptor kinetics, conductance, and allosteric modulation. In addition, intracellular loops of each subunit have the potential to interact with scaffolding proteins and affect the cellular distribution and clustering (synaptic or extrasynaptic) of the channels (Jacob et al., 2008; Luscher et al., 2011; Brickley and Mody, 2012). GABAAR assembled by combining γ2 and α1–3 subunits (α1–3,βx,γ2) are more commonly located at synaptic sites and mostly responsible for phasic inhibition, whereas receptors located at perisynaptic or extrasynaptic sites are primarily composed of α4 or α6 subunits combined with δ subunits (α4/α6,βx,δ) and mediate most tonic inhibition (Fritschy, 2008; Jacob et al., 2008; Luscher et al., 2011; Hines et al., 2012). Notably, tonic currents in pyramidal neurons of the hippocampus can also be generated by receptors containing α5 subunits (α5βγ2) that are located at extrasynaptic locations (Brickley and Mody, 2012; Hines et al., 2012).
Typically, following synthesis in the endoplasmic reticulum, GABAAR are delivered to extrasynaptic compartments within the plasma membrane and then diffuse toward its final destination in either synaptic or extrasynaptic locations. GABAAR located at the plasma membrane also transit among different cellular compartments due to internalization and recycling events. Thus, the final number of receptors located at the cell surface is determined by continuous insertion of de novo synthesized and recycled receptors (Michels and Moss, 2007; Leidenheimer, 2008). There are a number of accessory proteins that facilitate the transit of GABAAR along the biosynthetic pathway and the different recycling compartments within the cell (Chen and Olsen, 2007; Jacob et al., 2008; Leidenheimer, 2008). Early on, during GABAAR oligomerization, accessory proteins like BIP (heavy chain binding protein), calnexin and BIG2 (Brefeldin-A-inhibited GDP/GTP exchange factor 2) form interactions with the nascent receptors within the membranous compartments of the endoplasmic reticulum and help with the translocation of the receptors into the Golgi apparatus. During the vesicular trafficking of receptors toward the plasma membrane, GABAAR containing γ subunits are linked to tubulin and the microtubules by GABARAP (GABAAR-associated protein) that acts as a bridge between vesicles containing GABAAR and the machinery that moves those vesicles toward the plasma membrane. GABARAP can also bind to NSF (N-ethylmaleimide-sensitive factor), PRIPs (phospholipase-C-related catalytically inactive proteins) and GRIF (GABAAR-interacting factors also known as TRAK), and together these proteins promote the interaction of intracellular vesicles containing GABAAR with the cytoskeleton and facilitate the motor-dependent transport of receptors toward the plasma membrane (Kneussel et al., 2000; Wang and Olsen, 2000; Charych et al., 2004). The final destination of GABAAR into synaptic or extrasynaptic sites is intrinsically determined by the subunits forming the channels and extrinsically by protein–protein interactions with scaffolding proteins (Luscher and Keller, 2004; Chen and Olsen, 2007; Jacob et al., 2008; Leidenheimer, 2008).
Gephyrin is the main structural scaffold that links proteins located at the subsynaptic compartment with the cytoskeleton and it is required for the organization and clustering of GABAAR at inhibitory synapses (Michels and Moss, 2007; Fritschy et al., 2008). During development, increased concentration of gephyrin precedes the accumulation of GABAAR at synaptic sites and facilitates the formation and stabilization of inhibitory synapses (Christie et al., 2002; Danglot et al., 2003; Swanwick et al., 2006; Fritschy et al., 2008). Removal of gephyrin by gene targeting or siRNA interference strongly affects GABAAR clustering and reduces inhibitory post-synaptic currents, a reciprocal effect on gephyrin clustering and GABAergic innervations has been observed following elimination of γ2 subunits (Essrich et al., 1998; Kneussel et al., 1999; Li et al., 2005; Yu et al., 2008). GABAAR clustering also occurs in neurons lacking gephyrin, but the clusters formed in these conditions have increased mobility and show less accumulation at inhibitory synapses, which reinforces the notion that gephyrin enhance anchoring of receptors at synaptic sites (Levi et al., 2004; Jacob et al., 2005; Yu et al., 2007). GABAAR containing α5 subunits are responsible for tonic inhibition and can also be found forming extrasynaptic clusters (Brunig et al., 2002; Loebrich et al., 2006). A crucial player in α5-containing GABAAR clustering is radixin. Both radixin antisense and genetic knockout causes a loss of α5 clusters in hippocampal neurons and hippocampal tissue (Loebrich et al., 2006; Kneussel and Loebrich, 2007). Radixin and gephyrin do not interact or colocalize with each other, suggesting that the mechanisms behind radixin and gephyrin-dependent clustering are independent (Loebrich et al., 2006; Kneussel and Loebrich, 2007).
The structural and molecular details underlying the regulation of GABAAR by gephyrin are starting to emerge. Gephyrin consist of three major domains, a 20-kDa N-terminal domain (G-domain), a 43-kDa C-terminal domain (E-domain), and an 18–21 kDa central domain (C-domain; Saiyed et al., 2007; Fritschy et al., 2008). Trimerization of the G-domain and dimerization of the E-domain appear to be the oligomerization pattern involved in the formation of the hexagonal lattice required for proper formation of inhibitory synapses (Sola et al., 2004; Saiyed et al., 2007; Fritschy et al., 2008; Tyagarajan and Fritschy, 2010; Herweg and Schwarz, 2012). In addition to its role in the formation of the gephyrin lattice, the E-domain is the binding site for GABAAR (Tyagarajan and Fritschy, 2010) whereas the C-domain binds to several accessory proteins like Pin1 (Zita et al., 2007), dynein light chain 1 and 2 (Fuhrmann et al., 2002; Maas et al., 2006) and collybistin (Kins et al., 2000). Experiments in recombinant expression systems have demonstrated that collybistin is essential for the distribution and stabilization of gephyrin at post-synaptic sites and that overexpression of selected collybistin domains increases the size and density of gephyrin clusters (Kins et al., 2000; Chiou et al., 2011; Tyagarajan et al., 2011), suggesting that collybistin aids with the proper clustering of GABAAR at synaptic sites by forming a partnership with gephyrin (Yu et al., 2008). The small GTPase Cdc42 also binds to the C-domain of gephyrin and works in collaboration with collybistin to help with the translocation of gephyrin toward the plasma membrane (Tyagarajan et al., 2011). Another protein that interacts with gephyrin and indirectly regulates the post-synaptic trafficking and/or accumulation of GABAAR is termed GRIP1 (glutamate receptor interacting protein 1). In vivo and in vitro evidence suggest that GRIP1 is located at GABAergic synapses and physically interacts with gephyrin to directly modulate gephyrin clustering and indirectly regulate the post-synaptic distribution of GABAAR (Yu et al., 2008).
Alterations in GABAAR and Scaffolding Proteins During Epileptogenesis
The belief that following a brain injury there is a quiescent, pre-epileptic state in which there is gradual changes at the molecular, cellular, and circuit levels that ultimately results in the manifestation of spontaneous seizures, has led to the search for mechanisms underlying epileptogenesis. Induction of SE using the chemoconvulsant pilocarpine produces a transient decrease in GABAergic drive readily detectable during the latent period. Abnormal electroencephalogram (EEG) patterns, such as large amplitude spikes and sharp waves can be detected as early as 3–5 days following SE, which overtime culminate with the appearance of full-blown electrographic seizures (El-Hassar et al., 2007). During SE there is a rapid increase in neuronal hyperexcitability due to a quick decline in the number of physiologically active GABAAR at the plasma membrane (Goodkin et al., 2005; Naylor et al., 2005). SE triggers a rapid loss of synaptic GABAAR containing β and γ subunits while extrasynaptic receptors containing α5 and δ subunits remain unaffected (Goodkin et al., 2008; Terunuma et al., 2008). A decrease in the phosphorylation of β3 subunits allows the interaction of β3-containing GABAAR with the clathrin-adaptor protein 2 and the recruitment of GABAAR into clathrin-coated pits promotes a faster removal of these receptors from the cell surface, suggesting that a decrease in the phosphorylation of β3 subunits may account for the selective loss of synaptic GABAAR observed following SE (Goodkin et al., 2008; Terunuma et al., 2008). These biochemical observations directly link the decrease in miniature inhibitory post-synaptic currents observed after induction of SE with the selective internalization of synaptic GABAAR containing β and γ subunits and explain why the currents mediated by extrasynaptic receptors are spared (Goodkin et al., 2005; Naylor et al., 2005).
The fate of internalized receptors following induction of SE is more likely to be determined by the cellular compartment where they are transiently stored (Figure 1). Receptors present in endosomal compartments can be reincorporated into the active pool of receptors at the plasma membrane or they can be relocated to the lysosomes for degradation (Chen et al., 2007; Wasterlain and Chen, 2008). Recent studies suggest that the network of proteins required for the proper trafficking and anchoring of GABAAR might be disrupted following SE. During the latent period there is a reduction in the total expression of gephyrin that appears to translate into a reduction in the number of gephyrin clusters (Knuesel et al., 2001; Thind et al., 2010; Fang et al., 2011; González et al., 2013). The pattern of gephyrin loss observed during the silent period parallels the changes in excitability previously observed, and suggests that the loss of scaffolding proteins directly impact the function of GABAAR during epileptogenesis. Intriguingly, during the chronic period there is an increase in both the total expression and the number of gephyrin clusters (Thind et al., 2010; Fang et al., 2011), but it is unclear if this rebound in gephyrin expression results in fully functional inhibitory synapses.
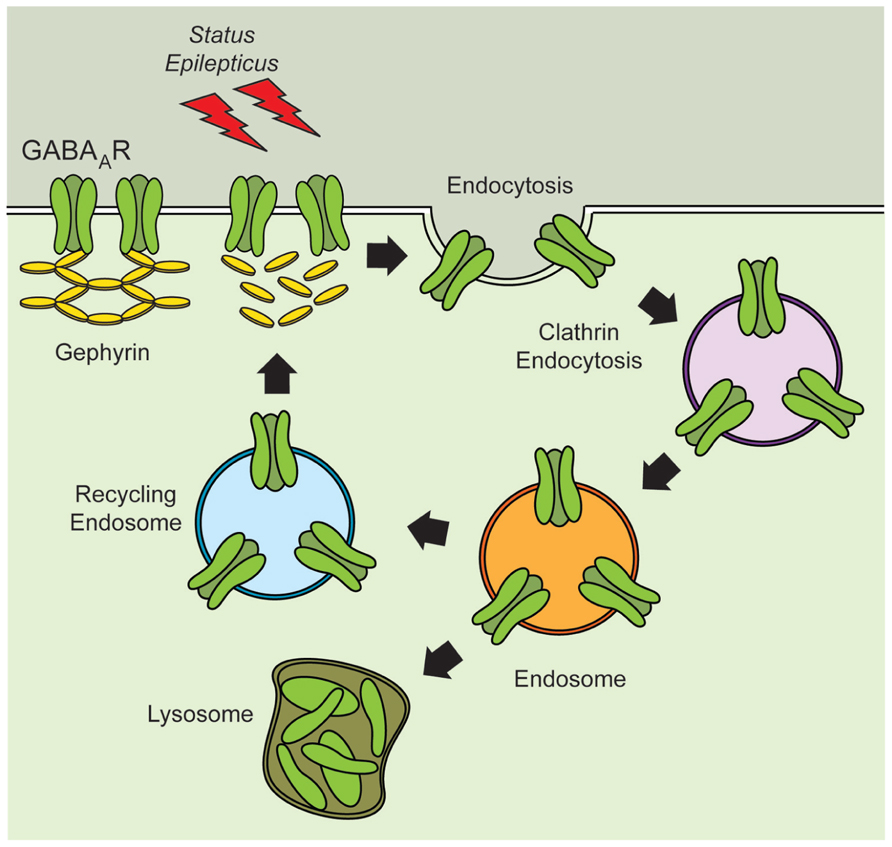
FIGURE 1. Altered stability of GABAAR and scaffolding proteins during epileptogenesis. Induction of status epilepticus produces alterations in the expression of gephyrin and might disrupt GABAAR anchoring. Decreased expression of gephyrin might compromise the recycling of GABAAR and reduce the stability of receptors present at the plasma membrane. Reduced expression of GABAAR at the plasma membrane may contribute to the increased excitability observed during epileptogenesis.
Alterations in the expression of GABAAR during the epileptogenic period include rapid down-regulation of α4, β2/3, γ2, and δ subunits (Schwarzer et al., 1997; Houser and Esclapez, 2003; Peng et al., 2004). Our recent characterization of the expression of several GABAAR subunits in microdissected CA1 also showed a reduction in the levels of α4, β2/3, and γ2 (but not α1) subunits as early as 4 days after SE (González et al., 2013). The loss of these subunits correlated with the down-regulation of gephyrin, suggesting that the loss of GABAAR might result from the lack of proper receptor anchoring and clustering (González et al., 2013). Accordingly, analysis of the cell surface levels of GABAAR revealed a time-dependent reduction in the plasma membrane levels of α4 and γ2 subunits that correlated with the down-regulation of gephyrin (González et al., 2013). These observations hint to the possibility that during the epileptogenic period, the stability of the GABAAR receptors that recycle back to the plasma membrane might be compromised because they cannot be properly anchored (González et al., 2013). They also suggest that the loss of inhibition and increased inter-ictal activity observed during the latent period might result from the persistent dysregulation of GABAAR trafficking and anchoring (Wasterlain and Chen, 2008). However, whether a loss of scaffolding proteins is a factor contributing to the hyperexcitability observed during the epileptogenic period remains to be fully characterized.
Additional support for hypothesis that a lack of GABAAR stability contributes to the hyperexcitability observed during the latent period comes from studies in Xenopus oocytes transplanted with cell membranes isolated from epileptic tissue (Palma et al., 2007; Mazzuferi et al., 2010). Repetitive stimulation of microtransplanted receptors induces a characteristic run-down of GABAAR-mediated currents that is independent of changes in receptor affinity or membrane potential (Palma et al., 2007). The run-down in GABAAR currents can be readily detected in receptors isolated following the manifestation of the first spontaneous seizure and its appearance has been associated with the transition from the latent to the chronic stage of epilepsy (Mazzuferi et al., 2010). Initial exploration of the molecular mechanisms behind this phenomenon revealed a switch in the composition of GABAAR and uncovered an increase in the ratio of α4/α1 subunits incorporated into receptors. More importantly, the switch in GABAAR assembly occurs at the same time that the current run-down appears, underscoring previous findings showing alterations in the expression of α4 and α1 subunits that affect the assembly, localization, and function of GABAAR and results in the impairment of tonic and phasic inhibition (Brooks-Kayal et al., 1998; Peng et al., 2004; Mazzuferi et al., 2010).
The specific mechanisms involved in the regulation of gephyrin during epileptogenesis and epilepsy remain to be fully characterized, but some clues are starting to emerge. Analysis of samples obtained from epileptic patients show a reduction in gephyrin expression, which correlates with the appearance of protein fragments probably resulting from gephyrin degradation (Forstera et al., 2010; Fang et al., 2011). The process involved in the generation of gephyrin fragments remains unclear but a favored hypothesis is that cellular stress (alkalosis and hyperthermia) might be sufficient to induce the skipping of exons in gephyrin messenger RNA resulting in the production of abnormally spliced variants of gephyrin. These abnormal variants may then interact with normal gephyrin molecules and act as dominant-negative mutants and promote the accumulation of gephyrin in ubiquitin positive inclusions (Forstera et al., 2010). The impact abnormally spliced variants of gephyrin has been associated with oligomerization deficits and aberrant clustering of GABAAR containing α2 subunits (Forstera et al., 2010). Induction of mild seizures or inflammatory events triggers the generation of adult-born hippocampal neurons and increases the expression of gephyrin, mostly in the newly generated neurons (Jakubs et al., 2006, 2008; Jackson et al., 2012). Electrophysiological recordings revealed that newborn neurons produced after these injuries have reduced excitatory and increased inhibitory drive partially associated with the increase in gephyrin expression. Together, these observations point out to the possibility that newly generated cells might increase gephyrin expression as a compensatory mechanism to mitigate the hippocampal hyperexcitability observed after an insult to the brain (Jakubs et al., 2006; Jackson et al., 2012).
Another line of evidence implicating gephyrin dysfunction as a key element in the generation of epileptic seizures includes recent genetic evidence found in individuals affected by pathologies associated with a seizure phenotype. Rare hemizygous microdeletions in the chromosome 14q23.3 that encompass exons 3–5 in the coding region the G-domain were found in individuals from six unrelated families presenting autism, schizophrenia, and seizures (Lionel et al., 2013). Other mutations that interfere with collybistin function have also been associated with abnormalities observed in patients with epilepsy and mental retardation (Harvey et al., 2004; Marco et al., 2008; Kalscheuer et al., 2009; Lesca et al., 2011). These mutations found in the coding sequence of collybistin interfere with the somatic and synaptic localization of gephyrin via dominant-negative mechanisms that indirectly affect the distribution and synaptic clustering of GABAAR (Harvey et al., 2004; Kalscheuer et al., 2009).
The impact that disruption of gephyrin and other scaffolding proteins might have on GABAAR function during epileptogenesis remains to be elucidated. If a loss of gephyrin directly impacts the number and function of GABAAR at inhibitory synapses, interventions to promote the stability of gephyrin and GABAAR might ameliorate the deleterious changes in excitability observed during epileptogenesis and epilepsy. Altered expression of gephyrin has been observed in several pathologies presenting symptomatic seizures, but it is unclear if changes in gephyrin are beneficial or pathologic (Jakubs et al., 2008; Thind et al., 2010; Jackson et al., 2012). Understanding the molecular mechanism(s) behind the dysregulation of scaffolding proteins involved in the regulation of GABAAR might provide new insights into the pathologic events that contribute to the generation of spontaneous seizures and might offer new targets to disrupt epileptogenesis and prevent epilepsy.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Marco I. González is supported by the National Institutes of Health Grant K01-NS069583.
References
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Brooks-Kayal, A. R., Shumate, M. D., Jin, H., Rikhter, T. Y., and Coulter, D. A. (1998). Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat. Med. 4, 1166–1172. doi: 10.1038/2661
Brunig, I., Scotti, E., Sidler, C., and Fritschy, J. M. (2002). Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J. Comp. Neurol. 443, 43–55. doi: 10.1002/cne.10102
Charych, E. I., Yu, W., Miralles, C. P., Serwanski, D. R., Li, X., Rubio, M., et al. (2004). The brefeldin A-inhibited GDP/GTP exchange factor 2, a protein involved in vesicular trafficking, interacts with the beta subunits of the GABA receptors. J. Neurochem. 90, 173–189. doi: 10.1111/j.1471-4159.2004.02481.x
Chen, J. W., Naylor, D. E., and Wasterlain, C. G. (2007). Advances in the pathophysiology of status epilepticus. Acta Neurol. Scand. Suppl. 186, 7–15. doi: 10.1111/j.1600-0404.2007.00803.x
Chen, Z. W., and Olsen, R. W. (2007). GABAA receptor associated proteins: a key factor regulating GABAA receptor function. J. Neurochem. 100, 279–294. doi: 10.1111/j.1471-4159.2006.04206.x
Chiou, T. T., Bonhomme, B., Jin, H., Miralles, C. P., Xiao, H., Fu, Z., et al. (2011). Differential regulation of the postsynaptic clustering of gamma-aminobutyric acid type A (GABAA) receptors by collybistin isoforms. J. Biol. Chem. 286, 22456–22468. doi: 10.1074/jbc.M111.236190
Christie, S. B., Miralles, C. P., and De Blas, A. L. (2002). GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J. Neurosci. 22, 684–697.
Curia, G., Longo, D., Biagini, G., Jones, R. S., and Avoli, M. (2008). The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 172, 143–157. doi: 10.1016/j.jneumeth.2008.04.019
Danglot, L., Triller, A., and Bessis, A. (2003). Association of gephyrin with synaptic and extrasynaptic GABAA receptors varies during development in cultured hippocampal neurons. Mol. Cell. Neurosci. 23, 264–278. doi: 10.1016/S1044-7431(03)00069-1
Duncan, J. S., Sander, J. W., Sisodiya, S. M., and Walker, M. C. (2006). Adult epilepsy. Lancet 367, 1087–1100. doi: 10.1016/S0140-6736(06)68477-8
El-Hassar, L., Milh, M., Wendling, F., Ferrand, N., Esclapez, M., and Bernard, C. (2007). Cell domain-dependent changes in the glutamatergic and GABAergic drives during epileptogenesis in the rat CA1 region. J. Physiol. 578, 193–211. doi: 10.1113/jphysiol.2006.119297
Essrich, C., Lorez, M., Benson, J. A., Fritschy, J. M., and Luscher, B. (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1, 563–571. doi: 10.1038/2798
Fang, M., Shen, L., Yin, H., Pan, Y. M., Wang, L., Chen, D., et al. (2011). Downregulation of gephyrin in temporal lobe epilepsy neurons in humans and a rat model. Synapse 65, 1006–1014. doi: 10.1002/syn.20928
Forstera, B., Belaidi, A. A., Juttner, R., Bernert, C., Tsokos, M., Lehmann, T. N., et al. (2010). Irregular RNA splicing curtails postsynaptic gephyrin in the cornu ammonis of patients with epilepsy. Brain 133, 3778–3794. doi: 10.1093/brain/awq298
Fritschy, J. M. (2008). Epilepsy, E/I balance and GABA(A) receptor plasticity. Front. Mol. Neurosci. 1:5. doi: 10.3389/neuro.02.005.2008
Fritschy, J. M., Harvey, R. J., and Schwarz, G. (2008). Gephyrin: where do we stand, where do we go? Trends Neurosci. 31, 257–264. doi: 10.1016/j.tins.2008.02.006
Fuhrmann, J. C., Kins, S., Rostaing, P., El Far, O., Kirsch, J., Sheng, M., et al. (2002). Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J. Neurosci. 22, 5393–5402.
Gibbons, M. B., Smeal, R. M., Takahashi, D. K., Vargas, J. R., and Wilcox, K. S. (2012). Contributions of astrocytes to epileptogenesis following status epilepticus: opportunities for preventive therapy? Neurochem. Int. doi: 10.1016/j.neuint.2012.12.008 [Epub ahead of print].
González, M. I., Cruz Del Angel, Y., and Brooks-Kayal, A. (2013). Down-regulation of gephyrin and GABA(A) receptor subunits during epileptogenesis in the CA1 region of hippocampus. Epilepsia 54, 616–624. doi: 10.1111/epi.12063
Goodkin, H. P., Joshi, S., Mtchedlishvili, Z., Brar, J., and Kapur, J. (2008). Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 28, 2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008
Goodkin, H. P., Yeh, J. L., and Kapur, J. (2005). Status epilepticus increases the intracellular accumulation of GABAA receptors. J. Neurosci. 25, 5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005
Harvey, K., Duguid, I. C., Alldred, M. J., Beatty, S. E., Ward, H., Keep, N. H., et al. (2004). The GDP–GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826. doi: 10.1523/JNEUROSCI.1184-04.2004
Heinemann, U., Kaufer, D., and Friedman, A. (2012). Blood–brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 60, 1251–1257. doi: 10.1002/glia.22311
Herweg, J., and Schwarz, G. (2012). Splice-specific glycine receptor binding, folding, and phosphorylation of the scaffolding protein gephyrin. J. Biol. Chem. 287, 12645–12656. doi: 10.1074/jbc.M112.341826
Hines, R. M., Davies, P. A., Moss, S. J., and Maguire, J. (2012). Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 22, 552–558. doi: 10.1016/j.conb.2011.10.007
Houser, C. R., and Esclapez, M. (2003). Downregulation of the alpha5 subunit of the GABA(A) receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus 13, 633–645. doi: 10.1002/hipo.10108
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., et al. (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547. doi: 10.1093/brain/awl317
Jackson, J., Chugh, D., Nilsson, P., Wood, J., Carlstrom, K., Lindvall, O., et al. (2012). Altered synaptic properties during integration of adult-born hippocampal neurons following a seizure insult. PLoS ONE 7:e35557. doi: 10.1371/journal.pone.0035557
Jacob, T. C., Bogdanov, Y. D., Magnus, C., Saliba, R. S., Kittler, J. T., Haydon, P. G., et al. (2005). Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J. Neurosci. 25, 10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005
Jacob, T. C., Moss, S. J., and Jurd, R. (2008). GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343. doi: 10.1038/nrn2370
Jakubs, K., Bonde, S., Iosif, R. E., Ekdahl, C. T., Kokaia, Z., Kokaia, M., et al. (2008). Inflammation regulates functional integration of neurons born in adult brain. J. Neurosci. 28, 12477–12488. doi: 10.1523/JNEUROSCI.3240-08.2008
Jakubs, K., Nanobashvili, A., Bonde, S., Ekdahl, C. T., Kokaia, Z., Kokaia, M., et al. (2006). Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron 52, 1047–1059. doi: 10.1016/j.neuron.2006.11.004
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., et al. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68. doi: 10.1002/humu.20814
Kins, S., Betz, H., and Kirsch, J. (2000). Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat. Neurosci. 3, 22–29. doi: 10.1038/71096
Kneussel, M., Brandstatter, J. H., Laube, B., Stahl, S., Muller, U., and Betz, H. (1999). Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J. Neurosci. 19, 9289–9297.
Kneussel, M., Haverkamp, S., Fuhrmann, J. C., Wang, H., Wassle, H., Olsen, R. W., et al. (2000). The gamma-aminobutyric acid type A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc. Natl. Acad. Sci. U.S.A. 97, 8594–8599. doi: 10.1073/pnas.97.15.8594
Kneussel, M., and Loebrich, S. (2007). Trafficking and synaptic anchoring of ionotropic inhibitory neurotransmitter receptors. Biol. Cell 99, 297–309. doi: 10.1042/BC20060120
Knuesel, I., Zuellig, R. A., Schaub, M. C., and Fritschy, J. M. (2001). Alterations in dystrophin and utrophin expression parallel the reorganization of GABAergic synapses in a mouse model of temporal lobe epilepsy. Eur. J. Neurosci. 13, 1113–1124. doi: 10.1046/j.0953-816x.2001.01476.x
Kovacs, R., Heinemann, U., and Steinhauser, C. (2012). Mechanisms underlying blood–brain barrier dysfunction in brain pathology and epileptogenesis: role of astroglia. Epilepsia 53(Suppl. 6), 53–59. doi: 10.1111/j.1528-1167.2012.03703.x
Leidenheimer, N. J. (2008). Regulation of excitation by GABA(A) receptor internalization. Results Probl. Cell Differ. 44, 1–28. doi: 10.1007/400_2007_039
Lesca, G., Till, M., Labalme, A., Vallee, D., Hugonenq, C., Philip, N., et al. (2011). De novo Xq11.11 microdeletion including ARHGEF9 in a boy with mental retardation, epilepsy, macrosomia, and dysmorphic features. Am. J. Med. Genet. A 155A, 1706–1711. doi: 10.1002/ajmg.a.34004
Levi, S., Logan, S. M., Tovar, K. R., and Craig, A. M. (2004). Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J. Neurosci. 24, 207–217. doi: 10.1523/JNEUROSCI.1661-03.2004
Li, R. W., Yu, W., Christie, S., Miralles, C. P., Bai, J., Loturco, J. J., et al. (2005). Disruption of postsynaptic GABA receptor clusters leads to decreased GABAergic innervation of pyramidal neurons. J. Neurochem. 95, 756–770. doi: 10.1111/j.1471-4159.2005.03426.x
Lionel, A. C., Vaags, A. K., Sato, D., Gazzellone, M. J., Mitchell, E. B., Chen, H. Y., et al. (2013). Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 22, 2055–2066. doi: 10.1093/hmg/ddt056
Loebrich, S., Bahring, R., Katsuno, T., Tsukita, S., and Kneussel, M. (2006). Activated radixin is essential for GABAA receptor alpha5 subunit anchoring at the actin cytoskeleton. EMBO J. 25, 987–999. doi: 10.1038/sj.emboj.7600995
Loscher, W., and Brandt, C. (2010). Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol. Rev. 62, 668–700. doi: 10.1124/pr.110.003046
Luscher, B., Fuchs, T., and Kilpatrick, C. L. (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409. doi: 10.1016/j.neuron.2011.03.024
Luscher, B., and Keller, C. A. (2004). Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol. Ther. 102, 195–221. doi: 10.1016/j.pharmthera.2004.04.003
Maas, C., Tagnaouti, N., Loebrich, S., Behrend, B., Lappe-Siefke, C., and Kneussel, M. (2006). Neuronal cotransport of glycine receptor and the scaffold protein gephyrin. J. Cell Biol. 172, 441–451. doi: 10.1083/jcb.200506066
Marchi, N., Granata, T., Ghosh, C., and Janigro, D. (2012). Blood–brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia 53, 1877–1886. doi: 10.1111/j.1528-1167.2012.03637.x
Marco, E. J., Abidi, F. E., Bristow, J., Dean, W. B., Cotter, P., Jeremy, R. J., et al. (2008). ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J. Med. Genet. 45, 100–105. doi: 10.1136/jmg.2007.052324
Mazzuferi, M., Palma, E., Martinello, K., Maiolino, F., Roseti, C., Fucile, S., et al. (2010). Enhancement of GABA(A)-current run-down in the hippocampus occurs at the first spontaneous seizure in a model of temporal lobe epilepsy. Proc. Natl. Acad. Sci. U.S.A. 107, 3180–3185. doi: 10.1073/pnas.0914710107
Michels, G., and Moss, S. J. (2007). GABAA receptors: properties and trafficking. Crit. Rev. Biochem. Mol. Biol. 42, 3–14. doi: 10.1080/10409230601146219
Naylor, D. E., Liu, H., and Wasterlain, C. G. (2005). Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 25, 7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005
O’Dell, C. M., Das, A., Wallace, G. T., Ray, S. K., and Banik, N. L. (2012). Understanding the basic mechanisms underlying seizures in mesial temporal lobe epilepsy and possible therapeutic targets: a review. J. Neurosci. Res. 90, 913–924. doi: 10.1002/jnr.22829
Palma, E., Roseti, C., Maiolino, F., Fucile, S., Martinello, K., Mazzuferi, M., et al. (2007). GABA(A)-current rundown of temporal lobe epilepsy is associated with repetitive activation of GABA(A) “phasic” receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 20944–20948. doi: 10.1073/pnas.0710522105
Peng, Z., Huang, C. S., Stell, B. M., Mody, I., and Houser, C. R. (2004). Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J. Neurosci. 24, 8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004
Pitkanen, A., and Lukasiuk, K. (2009). Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 14(Suppl. 1), 16–25. doi: 10.1016/j.yebeh.2008.09.023
Pitkanen, A., and Lukasiuk, K. (2011). Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10, 173–186. doi: 10.1016/S1474-4422(10)70310-0
Saiyed, T., Paarmann, I., Schmitt, B., Haeger, S., Sola, M., Schmalzing, G., et al. (2007). Molecular basis of gephyrin clustering at inhibitory synapses: role of G- and E-domain interactions. J. Biol. Chem. 282, 5625–5632. doi: 10.1074/jbc.M610290200
Schwarzer, C., Tsunashima, K., Wanzenbock, C., Fuchs, K., Sieghart, W., and Sperk, G. (1997). GABA(A) receptor subunits in the rat hippocampus II: altered distribution in kainic acid-induced temporal lobe epilepsy. Neuroscience 80, 1001–1017. doi: 10.1016/S0306-4522(97)00145-0
Scorza, F. A., Arida, R. M., Naffah-Mazzacoratti Mda, G., Scerni, D. A., Calderazzo, L., and Cavalheiro, E. A. (2009). The pilocarpine model of epilepsy: what have we learned? An. Acad. Bras. Cienc. 81, 345–365. doi: 10.1590/S0001-37652009000300003
Sharma, A. K., Reams, R. Y., Jordan, W. H., Miller, M. A., Thacker, H. L., and Snyder, P. W. (2007). Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol. Pathol. 35, 984–999. doi: 10.1080/01926230701748305
Sloviter, R. S. (2008). Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the “latent period” and other concepts. Epilepsia 49(Suppl. 9), 85–92. doi: 10.1111/j.1528-1167.2008.01931.x
Sola, M., Bavro, V. N., Timmins, J., Franz, T., Ricard-Blum, S., Schoehn, G., et al. (2004). Structural basis of dynamic glycine receptor clustering by gephyrin. EMBO J. 23, 2510–2519. doi: 10.1038/sj.emboj.7600256
Swanwick, C. C., Murthy, N. R., Mtchedlishvili, Z., Sieghart, W., and Kapur, J. (2006). Development of gamma-aminobutyric acidergic synapses in cultured hippocampal neurons. J. Comp. Neurol. 495, 497–510. doi: 10.1002/cne.20897
Terunuma, M., Xu, J., Vithlani, M., Sieghart, W., Kittler, J., Pangalos, M., et al. (2008). Deficits in phosphorylation of GABA(A) receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J. Neurosci. 28, 376–384. doi: 10.1523/JNEUROSCI.4346-07.2008
Thind, K. K., Yamawaki, R., Phanwar, I., Zhang, G., Wen, X., and Buckmaster, P. S. (2010). Initial loss but later excess of GABAergic synapses with dentate granule cells in a rat model of temporal lobe epilepsy. J. Comp. Neurol. 518, 647–667. doi: 10.1002/cne.22235
Tyagarajan, S. K., and Fritschy, J. M. (2010). GABA(A) receptors, gephyrin and homeostatic synaptic plasticity. J. Physiol. 588, 101–106. doi: 10.1113/jphysiol.2009.178517
Tyagarajan, S. K., Ghosh, H., Harvey, K., and Fritschy, J. M. (2011). Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J. Cell Sci. 124, 2786–2796. doi: 10.1242/jcs.086199
van Vliet, E. A., Da Costa Araujo, S., Redeker, S., Van Schaik, R., Aronica, E., and Gorter, J. A. (2007). Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130, 521–534. doi: 10.1093/brain/awl318
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Vezzani, A., Friedman, A., and Dingledine, R. J. (2013). The role of inflammation in epileptogenesis. Neuropharmacology 69, 16–24. doi: 10.1016/j.neuropharm.2012.04.004
Wang, H., and Olsen, R. W. (2000). Binding of the GABA(A) receptor-associated protein (GABARAP) to microtubules and microfilaments suggests involvement of the cytoskeleton in GABARAPGABA(A) receptor interaction. J. Neurochem. 75, 644–655. doi: 10.1046/j.1471-4159.2000.0750644.x
Wasterlain, C. G., and Chen, J. W. (2008). Mechanistic and pharmacologic aspects of status epilepticus and its treatment with new antiepileptic drugs. Epilepsia 49(Suppl. 9), 63–73. doi: 10.1111/j.1528-1167.2008.01928.x
Wieser, H. G. (2004). ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 45, 695–714. doi: 10.1111/j.0013-9580.2004.09004.x
Williams, P. A., Hellier, J. L., White, A. M., Staley, K. J., and Dudek, F. E. (2007). Development of spontaneous seizures after experimental status epilepticus: implications for understanding epileptogenesis. Epilepsia 48(Suppl. 5), 157–163. doi: 10.1111/j.1528-1167.2007.01304.x
Yu, W., Charych, E. I., Serwanski, D. R., Li, R. W., Ali, R., Bahr, B. A., et al. (2008). Gephyrin interacts with the glutamate receptor interacting protein 1 isoforms at GABAergic synapses. J. Neurochem. 105, 2300–2314. doi: 10.1111/j.1471-4159.2008.05311.x
Yu, W., Jiang, M., Miralles, C. P., Li, R. W., Chen, G., and De Blas, A. L. (2007). Gephyrin clustering is required for the stability of GABAergic synapses. Mol. Cell. Neurosci. 36, 484–500. doi: 10.1016/j.mcn.2007.08.008
Keywords: epilepsy, epileptogenesis, GABA receptors, gephyrin, status epilepticus
Citation: González MI (2013) The possible role of GABAA receptors and gephyrin in epileptogenesis. Front. Cell. Neurosci. 7:113. doi: 10.3389/fncel.2013.00113
Received: 23 April 2013; Accepted: 26 June 2013;
Published online: 22 July 2013.
Edited by:
Roberto Di Maio, University of Pittsburgh, USAReviewed by:
Enrico Cherubini, International School for Advanced Studies, ItalyHermona Soreq, The Hebrew University of Jerusalem, Israel
Copyright: © 2013 González. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Marco I. González, Division of Neurology and Translational Epilepsy Research Program, Department of Pediatrics, University of Colorado School of Medicine, Room 3111, Pharmacy Building (V20), Mail Stop 8605, 12850 East Montview Boulevard, Aurora, CO 80045, USA e-mail:bWFyY28uZ29uemFsZXpAdWNkZW52ZXIuZWR1