



- 1Laboratorio Neurofisiología y Comportamiento, Facultad de Medicina de Ciudad Real, Universidad de Castilla-La Mancha, Ciudad Real, Spain
- 2Department of Fisiología y Farmacología, Universidad de Salamanca, Salamanca, Spain
Last evidences suggest that, in Alzheimer's disease (AD) early stage, Amyloid-β (Aβ) peptide induces an imbalance between excitatory and inhibitory neurotransmission systems resulting in the functional impairment of neural networks. Such alterations are particularly important in the septohippocampal system where learning and memory processes take place depending on accurate oscillatory activity tuned at fimbria-CA3 synapse. Here, the acute effects of Aβ on CA3 pyramidal neurons and their synaptic activation from septal part of the fimbria were studied in rats. A triphasic postsynaptic response defined by an excitatory potential (EPSP) followed by both early and late inhibitory potentials (IPSP) was evoked. The EPSP was glutamatergic acting on ionotropic receptors. The early IPSP was blocked by GABAA antagonists whereas the late IPSP was removed by GABAB antagonists. Aβ perfusion induced recorded cells to depolarize, increase their input resistance and decrease the late IPSP. Aβ action mechanism was localized at postsynaptic level and most likely linked to GABAB-related ion channels conductance decrease. In addition, it was found that the specific pharmacological modulation of the GABAB receptor effector, G-protein-coupled inward rectifier potassium (GirK) channels, mimicked all Aβ effects previously described. Thus, our findings suggest that Aβ altering GirK channels conductance in CA3 pyramidal neurons might have a key role in the septohippocampal activity dysfunction observed in AD.
Introduction
Being still lack of effective treatments for Alzheimer's disease (AD), current research efforts have focused on finding the relationships between amyloid-β peptide (Aβ) functions and toxic mechanisms to understand the development of AD (Huang and Mucke, 2012). Memory deficits and disorientation appear as the first symptoms of AD (McKhann et al., 1984; Swanberg et al., 2004) and, among the different regions early affected, damages found in septum and hippocampus could explain these cognitive deficits (Moreno et al., 2007; Palop et al., 2007; Villette et al., 2010; Rubio et al., 2012). Both structures are reciprocally interconnected through fimbria/fornix, and are functionally coupled to form the septohippocampal system (Bland and Colom, 1993), which is critical in generating certain oscillatory activity, such as theta rhythm, necessary for fundamental processes in learning and memory (Stewart and Fox, 1990; Bland and Oddie, 2001; Buzsaki, 2002; Sotty et al., 2003; Colom, 2006; Colom et al., 2010; Rubio et al., 2012). Theta oscillation coordinates septohippocampal network and depends on interconnections, which include well known cholinergic and GABAergic (Lynch et al., 1977; Kohler et al., 1984; Bland and Colom, 1993) as well as glutamatergic (Sotty et al., 2003; Huh et al., 2010) projections.
In animal models of AD, septohippocampal network dysfunction has extensively been reported (Colom, 2006; Palop and Mucke, 2010; Peña et al., 2010; Villette et al., 2010, 2012; Rubio et al., 2012; Verret et al., 2012). At the synaptic level, dysfunction induced by Aβ on inhibitory neurotransmission causes aberrant patterns of activity in its associated neural circuits, destabilizes neuronal networks and impairs oscillatory activity. This scenario, ultimately, seems to be responsible for the early alteration of the processes implicated in learning and memory tasks observed in AD patients (Palop and Mucke, 2010; Huang and Mucke, 2012). However, the specific mechanisms involving inhibitory neurotransmission at the molecular level, synaptic circuits or systems that consistently explain Aβ neurotoxic effects and associated neurological deficits remain unknown.
γ-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mammalian central nervous system and is involved in the regulation of many physiological processes. GABA mediates the inhibitory neurotransmission and accordingly, regulates excitatory activity preventing hyperexcitation, actions especially relevant to maintain neural network stability and oscillatory activity (Palop and Mucke, 2010). GABA metabotropic type receptors (GABAB) are coupled to intracellular signal transduction mechanisms via G proteins (Mott and Lewis, 1994; Kaupmann et al., 1998) and mediate slow and prolonged synaptic inhibition mainly by postsynaptic G protein-coupled activated inwardly-rectifying potassium (GirK) channels (Luscher et al., 1997; Kaupmann et al., 1998). Thus, GirK channels act as key players in the control of cellular and network excitability by modulating synaptic activity (Lujan and Ciruela, 2012).
In this study, we aimed to characterize Aβ effects on septohippocampal fimbria/CA3 synapsis. To address this question, we used an in vitro preparation taking advantage of the specific septo-hippocampal projection to CA3 pyramidal neurons, and evoked a characteristic complex synaptic response in CA3 recorded neurons by stimulating the septal part of the fimbria. For the first time, we provide evidence that Aβ decreased GABAB neurotransmission through altering GirK channel conductance.
Materials and Methods
Animals
Experiments were carried out on male and female rats (80–100 g) raised in the Salamanca University Animal House (Salamanca, Spain). All animal procedures were reviewed and approved by the Ethical Committee for Use of Laboratory Animals of the University of Salamanca and University of Castilla-La Mancha, and followed the European Communities Council (86/609/EEC).
Preparation of Slices
Animals were deeply anesthetized with halothane and decapitated. The brain was excised and rapidly immersed in oxygenated ice-cold (4–6°C) artificial cerebrospinal fluid (ACSF), with sucrose (234 mM) replacing the NaCl (117 mM) to maintain osmolarity. In order to preserve the optimal connectivity from fimbria fibers on CA3 pyramidal neurons (Gloveli et al., 2005; Bischofberger et al., 2006), horizontal slices containing the septal part of the fimbria, i.e., lateral fimbria (Alonso and Kohler, 1984; Amaral and Lavenex, 2007), and the hippocampus (350 μm-thick) were cut in cold oxygenated Ringer solution using a vibratome (Leica VT 1000S, Wetzlar, Germany) and placed in an incubation chamber, where they were maintained for at least 2 h at room temperature (22°C) before the recordings. Further details of this in vitro preparation have been described elsewhere (Yajeya et al., 2000).
Sharp Electrode Recordings
For recordings, a single septohippocampal slice was transferred to an interface recording chamber (BSC-HT and BSC-BU; Harvard Apparatus, Holliston, US) and perfused continuously with ACSF comprising (in mM) 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 glucose. The ACSF was bubbled with carbogen gas (95%O2–5%CO2) and maintained at room temperature during the recordings.
Intracellular sharp electrode recordings from CA3 pyramidal neurons were obtained with borosilicate glass microelectrodes (140–180 MΩ; WPI, Sarasota, US) filled with a potassium acetate solution (3 M) and connected to the headstage of an intracellular recording amplifier (Bio-logic VF180, Claix, France). Only data from neurons with both, stable resting membrane potential (RMP) with values ≤ −60 mV in the absence of direct current (DC) holding currents, and presenting overshooting action potentials, were collected for analysis. Spike amplitude, afterdepolarization and afterhyperpolarizing potentials were measured relative to threshold.
Excitatory and inhibitory postsynaptic potentials (EPSP and IPSP, respectively) were elicited orthodromically by stimulating the lateral fimbria where septal afferents to CA3 hippocampal neurons are mainly found (Alonso and Kohler, 1984; Amaral and Lavenex, 2007). For that purpose a monopolar stainless steel electrode (2 MΩ of effective resistance; WPI, Sarasota, US) and a programmable stimulator (MASTER-8, A8, A.M.P.I., Jerusalem, Israel) were used. Single, cathodal, square-wave pulses of 100–200 μs duration and 100–500 μA intensity were adjusted to subthreshold values for orthodromic spike generation. Postsynaptic potentials were characterized according to their amplitude (as a function of the RMP) and latency. Since horizontal slices were obtained at different level and angle, sometimes the location of the electrode along lateral fimbria had to be changed to evoke the characteristic triphasic response.
Identification of Stimulation and Recording Sites
Recorded neurons were identified following procedures described elsewhere (Navarro-Lopez et al., 2004). Briefly, selected neurons were stained by the intracellular injection of biocytin diluted in a 2 M potassium acetate solution, using positive current pulses of 0.2 nA for 6 min. Slices were fixed, and cut in sections (40 μm) using a freezing microtome (HM400R, Microm, Heidelberg, Germany). Sections were incubated with avidin-biotin-peroxidase complex (ABC, Vector Labs., Burlingame, US). 3,3′-Diaminobenzidine was used as chromogen for visualization of the biocytin complex. Sections were counterstained with cresyl violet. Neuron was reconstructed from serial sections using a graphic design software. Photographs were superimposed and orientated to obtain the best fit between the corresponding sectioned elements.
Drugs
All chemicals used in this study were purchased from Sigma (Poole, UK) and Tocris (Biogen Científica, Spain) and applied by superfusion in the ACSF. The chemicals used were amyloid-β peptides (Aβ25–35 and the reverse Aβ35–25), 6-cyano-nitroquinoxaline-2,3-dione (CNQX; a potent, competitive AMPA-kainate receptor antagonist), 2-amino-5-phosphonovalerate (APV; a specific blocker of NMDA receptors), Bicuculline Methiodide (specific blocker of GABAA receptors), (RS)-3-Amino-2-(4-chlorophenyl) propylphosphonic acid (Saclofen; blocker of GABAB receptors), (RS)-4-Amino-3-(4-chlorophenyl) butanoic acid (Baclofen; agonist of GABAB receptors), Tetrodotoxine (TTX; voltage dependent sodium channel blocker), Tertiapin-Q (selective blocker of GirK channels) and 2-methyl-2,4-pentanediol (MPD, agonist of GirK channels).
Preparation of Aβ Peptides Solutions
Aβ25–35 and Aβ35–25 peptides were prepared as previously (Ashenafi et al., 2005; Santos-Torres et al., 2007). Briefly, the peptides were dissolved to 1 mM in bidistilled water and stored in aliquots at −20°C. Then aliquots were diluted in ACSF to required concentration and incubated for 24 h at 37°C before experiments were performed (Peña et al., 2010; Leao et al., 2012).
Data Storage and Statistical Analysis
Sharp electrode data were acquired online with the help of a CED 1401 interface (CED, Cambridge, UK), and stored on a personal computer (sample frequency 12.5 kHz). Analysis in both cases was performed using the MiniAnalysis Program, version 6.0.3 (Synaptosoft, Decatur, US). Unless otherwise indicated, the electrophysiological data are always expressed as mean ± standard error of the mean (SEM), and n represents the number of averaged neurons. Synaptic potentials were averaged (≥5) before quantitative analysis. Statistical analysis of collected data was performed using either Student's t-test or non-parametric test (Mann-Whitney U-test), accordingly with data distribution. When necessary, one-way ANOVA or equivalent non parametric test (Kruskal-Wallis test) and post-hoc analysis were performed. Statistical significance was determined at a level of p ≤ 0.05.
Results
Electrophysiological Characterization of Recorded Neurons and Their Synaptic Response to Fimbria Stimulation
This study comprises 110 intracellular recordings from pyramidal CA3 neurons (Figure 1), selected on the basis of their RMP (≤ −60 mV) and monosynaptic activation from the fimbria. Recorded neurons did not exhibit action potentials spontaneously at RMP values (−72.5 ± 1.8 mV). The input resistance (Ri) of the neurons was 113.4 ± 6.7 MΩ and the membrane time constant was 77.2 ± 25.4 ms. The direct activation of these neurons by depolarizing current injections (0.1–0.6 nA ; 300 ms) evoked a series of two to five spikes with marked spike frequency adaptation and decreased amplitude and longer duration of the second spike relative to the first one (Figure 1B). The spike amplitude was 101.1 ± 3.2 mV. These characteristics, together with neuronal morphology (Figure 1D) and other electrophysiological properties such as the presence of triphasic afterhyperpolarization (fAHP: 5.6 ± 0.8 mV; mAHP: 10.5 ± 1.5 mV; sAHP: 17.6 ± 1.3 mV) or afterdepolarization (ADP: 3.6 ± 0.5 mV), characterize the principal pyramidal-like neurons widely described in the hippocampus (Spruston and Johnston, 1992; Wittner et al., 2007). The location of selected neurons (n = 10) filled with biocytin is illustrated in Figure 1A. The morphology corresponds to pyramidal neurons in CA3 region of the hippocampus. The cell body is located into the stratum pyramidale and the visible basal dendrites on stratum oriens (Figure 1D).
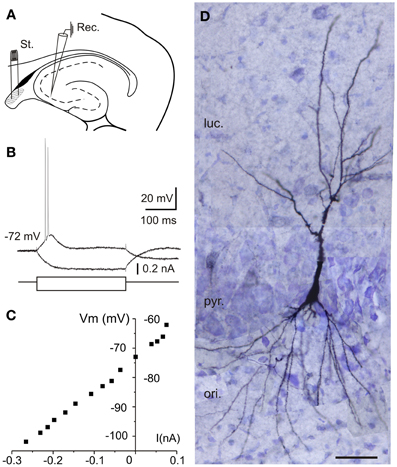
Figure 1. Location and electrophysiological characterization of the recorded neurons in hippocampal slices. (A) Experimental design. Diagram of stimulation and recording sites in an hippocampal horizontal section. Schematic location of recording (Rec.) and stimulating (St.) electrodes is shown. Stimuli were applied to the lateral part of the fimbria (shaded area). (B) Response of a CA3 neuron to depolarizing pulses consisted of two to five spikes with marked spike frequency adaptation with a depolarizing current pulse (100 pA, 300 ms) while hyperpolarizing current pulse injection (−160 pA, 300 ms) induced a hyperpolarizing response that allowed us monitoring the input resistance during the experiments. (C) Current-voltage (I-V) relationships for the pyramidal neuron recorded in (B). (D) Reconstruction of the CA3 neuron recorded in (B,C) labeled with biocytin after intracellular recording from 40-μm-thick serial sections. Note the pyramidal morphology of the injected hippocampal cell. ori, stratum oriens; pyr, stratum pyramidale; luc, stratum lucidum; Scale bar 50 μm.
Single subthreshold stimulation of the fimbria evoked stereotyped triphasic synaptic responses in CA3 pyramidal cells (Figure 2). The initial response was a fast EPSP, which occurred at a latency of 6.5 ± 0.6 ms following stimulus offset, suggesting the monosynaptic nature of the connection. The size of the EPSP was graded with the stimulus intensity and it increased in amplitude when elicited at progressively more negative membrane potentials (Figure 2A). The EPSP was followed by a rapidly developing hyperpolarization (early IPSP, Figures 2A,B) that reached its peak amplitude 30.4 ± 1.4 ms (n = 11) following fimbria stimulation. Finally a second hyperpolarization (late IPSP, Figures 2A,B) following the early IPSP, had a latency to peak amplitude of 247.4 ± 6.6 ms (n = 44). The amplitudes of both, IPSPs and EPSP, varied with membrane potential (Figures 2A,B), allowing us to determine the approximate reversal potential for both inhibitory components (early −61.1 mV and late −80.0 mV).
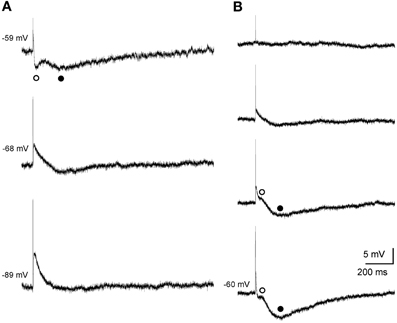
Figure 2. Postsynaptic septohippocampal response in CA3 pyramidal neurons. (A) Effect of membrane potential variations on the amplitude of the early and late IPSPs evoked by orthodromic activation in a pyramidal CA3 neuron. Stimulation of the fimbria elicited an EPSP followed by an early (open circles) and late (closed circles) IPSP. Traces shown are the average of five responses. (B) Effect of varying the intensity of fimbria stimulation on the complex postsynaptic response recorded in another neuron at a membrane potential of −60 mV. From top to bottom, traces represent synaptic responses which were evoked by progressive increments in fimbria stimulation. Stimulation of the fimbria at a low intensity evoked only EPSP followed by an early (◦, open circles) IPSP. Delivery of stimulation at higher intensities resulted in the elicitation of a subsequent late (•, closed circles) IPSP. The approximate reversal potential for both inhibitory components was early −61.1 mV and late −80.0 mV.
In order to determine the nature of the complex response, a pharmacological dissection of the postsynaptic potential components was performed (Figure 3). The early IPSP was blocked by bicuculline (10 μM, n = 6), a specific blocker of GABAA receptors (Figures 3A,C–E) whereas late IPSP was removed by saclofen (200 μM; n = 5), a specific blocker of GABAB receptors (Figures 3B,E). The excitatory component was increased by early IPSP block with bicuculline (n = 9; Figures 3A,C–E) and presented a glutamatergic nature acting mainly on non-NMDA (n = 5; Figure 3D) receptors, since although its complete elimination required CNQX (10 μM) and APV (50 μM) the cells were held at −75 mV (a membrane potential where NMDA receptor-mediated currents are null). However, when recorded cells were held at more positive values than RMP (n = 4; Figure 3C), NMDA channels were entirely functional and perfusion with APV decreased both bicuculline-enhanced responses, EPSP and late IPSP (Figure 3E), suggesting the participation of NMDA receptors in the response. As previously, complete blockage of EPSP also required CNQX (Figure 3C). In this regard, the pharmacological elimination of glutamatergic responses with CNQX plus APV also abolished the inhibitory response (Figures 3C,D) suggesting that IPSPs were produced by interneurons activation.
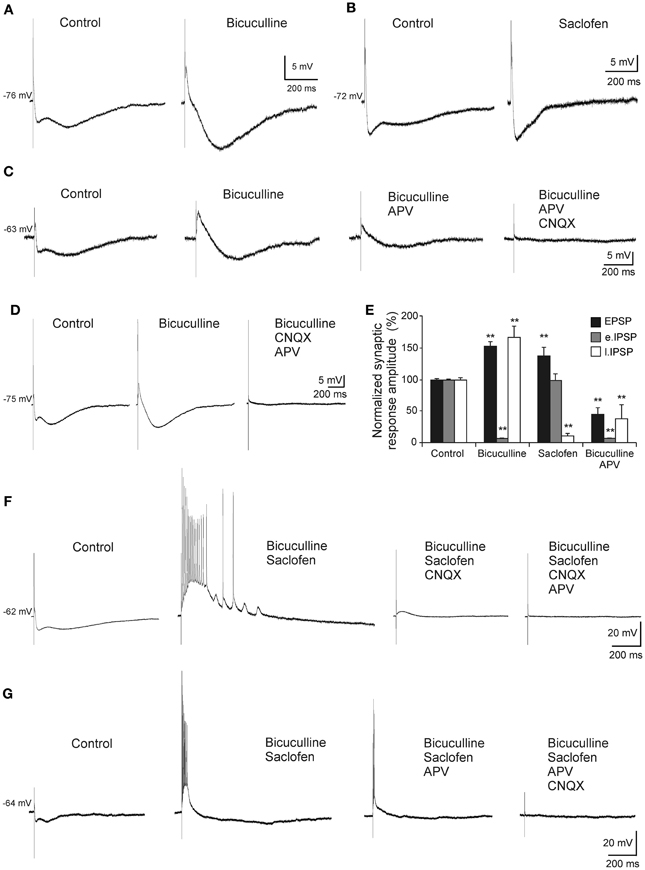
Figure 3. Pharmacological characterization of septohippocampal synaptic response. (A) Recordings from a pyramidal CA3 neuron illustrating a marked reduction of the early IPSP after perfusion with bicuculline (10 μM; specific blocker of GABAA receptors). (B) Blocking of the late IPSP after perfusion with saclofen (200 μM; blocker of GABAB receptor) was accompanied by a mild increase of the early IPSP and EPSP. (C) The increase in the amplitude of EPSP and late IPSP produced by perfusion of bicuculline was reduced by APV application (50 μM; specific blocker of NMDA receptor). Membrane potential was maintained at values more positives than resting membrane potential to assure NMDA receptors functionality. Synaptic response was completely removed with the addition of CNQX (10 μM; competitive non-NMDA receptor antagonist). (D) Blockade of the early IPSP with bicuculline (n = 4) was associated with a marked increase in amplitude of EPSP and late IPSP. Excitatory and inhibitory responses were blocked by CNQX and APV. The cells were held at −75 mV (a membrane potential where NMDA receptor-mediated currents are null). (E) Histograms with relative mean amplitude as percentage of control of the different components of the complex synaptic response (EPSP; early, e.IPSP; and late, l.IPSP) under pharmacological conditions described in (A–D) (**p < 0.001). (F) Both, early and late IPSPs were blocked by bicuculline and saclofen, respectively, while a large repetitive burst of action potential appeared (n = 4). At membrane potential values that assured NMDA activation, this epileptic-like activity could be removed by CNQX. Finally, residual excitatory NMDA response was eliminated by APV perfusion. (G) During the epileptic-like activity induced by both inhibitory components elimination, and at membrane potential values that led NMDA receptor activation, APV perfusion reduced the size of the epileptic response that had to be removed by addition of CNQX (n = 4).
On the other hand, when both inhibitory components were removed an epileptiform-like discharge was generated (Figures 3F,G). This response could reach a firing frequency of 90 Hz and showed a glutamatergic nature acting mainly on non-NMDA receptors, since CNQX completely abolished it (Figure 3F) whereas APV only could block it partially (Figure 3G).
These results indicate that the triphasic complex response involves excitatory and inhibitory neurotransmission mediated by glutamate and GABA receptors activation, suggesting that a precise tuning is required for information processing at this synapse.
Aβ25–35 Differential Effects on Membrane Properties
In all cases, the recordings were stabilized for at least 10 min. During this time, characterization of firing pattern, membrane potential, Ri and synaptic responses were performed. The specificity of the Aβ25–35 peptide action was confirmed by the use, as negative control, of the reverse sequence Aβ35–25 (1.5 μM), without any noticeable effect (Figures 4A,C). Then, slices were perfused with increasing concentrations of Aβ25–35 (0.5, 1.0, and 1.5 μM) for at least another 10 min at each concentration.
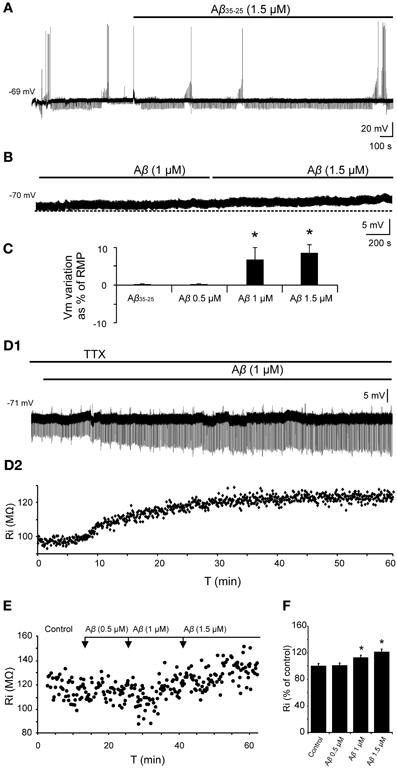
Figure 4. Effects of Aβ25–35 on membrane potential and Ri of CA3 neurons. (A) Recording of a CA3 pyramidal neuron during the perfusion of the reverse sequence of the peptide, Aβ35–25, used as negative control (n = 4). (B) Depolarization of membrane potential induced by perfusion of Aβ in a recorded pyramidal CA3 neuron (n = 10). (C) Plot of the membrane potential variations as a percentage of resting membrane potential (RMP). Perfusion of Aβ35–25 1 μM or Aβ25–35 0.5 μM did not induce any significant change in the membrane potential or Ri. Aβ25–35 higher concentrations induced the cells to depolarize (1 μM; 6.8 ± 3.2 %, n = 4; 1.5 μM, 8.6 ± 2.2 %, n = 10). (D1) Time course of Aβ effects on Ri in a CA3 pyramidal neuron after perfusion with TTX. The membrane potential was held at its RMP value by direct current (DC) holding current injection to cancel out the depolarization. (D2) For the same neuron, each point represents the Ri during the recording in (D1). Membrane potential was held at −71 mV. (E) Plot showing the time course of the effects of Aβ25–35 concentration increase on the Ri of a CA3 pyramidal neuron. Note that recordings last for a very long time. (F) Histogram with mean values in percentage for Ri (n = 10) at different Aβ25–35 concentration (*p < 0.05).
No significant differences were found in spike amplitude [F(3, 50) = 2.62, p = 0.062], threshold [F(3, 50) = 2.14, p = 0.108], ADP [F(3, 22) = 0.576, p = 0.638], fAHP [F(3, 31) = 1.22, p = 0.320], or sAHP [F(3, 33) = 1.625, p = 0.204] after perfusion with Aβ25–35 at increasing concentrations. However, as shown in Figures 4B,C, a significant depolarization was observed when Aβ25–35 was applied (1 μM; 4.3 ± 2.3 mV; n = 4; and 1.5 μM; 6.2 ± 1.6 mV; n = 10). The membrane potential of the recorded neurons was maintained at its RMP value by DC holding current injection to cancel out the depolarization induced by Aβ25–35 (Figure 4D). These variations in current injection were statistically significant at 1.0 μM (t = 2.557, p = 0.021) and 1.5 μM (t = 4.301, p < 0.001) concentrations and no difference was observed at 0.5 μM concentrations (t = 0.842, p = 0.412). Additionally, Aβ25–35 also produced a significant increase in the relative Ri (% = Ri recorded/Ri control *100; n = 16) (Figures 4D–F) at 1.0 μM concentration (t = −2.635, p = 0.018) and 1.5 μM (t = −3.236, p = 0.007) whereas no differences were found at 0.5 μM (Mann–Whitney U Statistic = 10.000, p = 0.690) (Figures 4E,F).
To determine the synaptic location of these Aβ25–35 effects, slices were perfused with TTX and any afferent synaptic activity was blocked. In these conditions, superfusion of the slice with Aβ25–35 was able to evoke both, depolarization and Ri increasing (Figure 4D) of intracellularly recorded CA3 pyramidal neurons, suggesting a postsynaptic location for the Aβ25–35 action mechanism (n = 5).
Differential Effects of Aβ25–35 on Fimbria-CA3 Synaptic Response
Since Aβ has widely shown to exert its effects through septohippocampal network impairing, we examined whether this peptide altered the fimbria-CA3 complex postsynaptic response (n = 20). In the experiment shown in Figure 5A, superfusion of Aβ25–35 produced a significant decrease of the late IPSP component (1 μM; t = 2.532, p = 0.030 and 1.5 μM; t = 2.519, p = 0.036) that was neither observed at 0.5 μM (t = 1.133, p = 0.295) nor on early IPSP (H = 2.578; p = 0.461). However, the late IPSP reduction was associated with an increase in the excitatory response observed at concentration of 1.5 μM (t = −2.503, p = 0.046) but not at lower concentrations (Figures 5B,C). These results indicate a possible mechanism to imbalance the particular excitatory/inhibitory tuning in the septohippocampal system, and therefore a differential Aβ25–35 effect, according to the specific neurotransmission system involved.
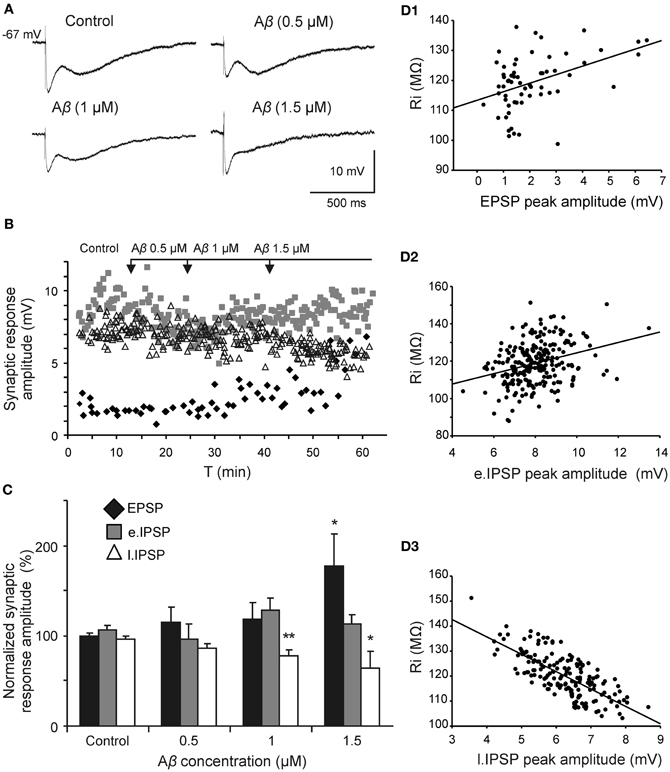
Figure 5. Selective effect of Aβ25–35 on different components of the complex postsynaptic response recorded in CA3 pyramidal neurons by fimbria stimulation. (A) Evoked responses obtained in a pyramidal CA3 neuron by fimbria orthodromic stimulation (control) and during perfusion of different concentrations of Aβ25–35 (0.5, 1, and 1.5 μM). The reduction of the late IPSP after perfusion with Aβ25–35 has been shown to be concentration-dependent. (B) Plot displaying the time course of Aβ25–35 perfusion effects on the amplitude (in mV) of the different components of the complex response (EPSP, black diamonds; early IPSP, gray squares; late IPSP, white triangles; see color code in C). (C) Histograms with relative mean amplitude (n = 20) of the different components of the complex synaptic response (EPSP; early, e.IPSP; and late, l.IPSP) 10 min after Aβ25–35 perfusion (0.5, 1, and 1.5 μM). Significant differences were found for EPSP at 1.5 μM Aβ25–35 and for late IPSP at 1–1.5 μM Aβ25–35. (D) Correlation analysis of Aβ25–35 perfusion on different components of the complex postsynaptic response vs. Ri values. Data showed a higher correlation between Aβ-induced Ri increase and late IPSP amplitude (R = −0.73, p < 0.001; D3) than Aβ-induced Ri increase and EPSP (R = 0.42, p < 0.001; D1) or early IPSP (R = 0.31, p < 0.01; D2)(*p < 0.05; **p < 0.01).
Due to this differential Aβ25–35 effect on both inhibitory components, the late component reduction may not be attributable to a decreased neurotransmitter release since GABAA component amplitude was maintained. This result pointed out to a selective postsynaptic action on GABAB complex.
Aβ25–35 Effects can be Explained by a Reduction in the Conductance of GirK Channels Coupled to GABAB Receptor
Depolarization caused by Aβ25–35 was associated to Ri increase and therefore, linked to a possible decrease in membrane conductance, which may very likely involve in ion channels closing. Given the correlation between reduction in GABAB component and Ri increase shown in Figure 5D, we investigated whether these Aβ25–35 effects could be mediated by conductance reduction of GABAB effector, GirK channel. We found that GirK blockade by its selective antagonist, tertiapin-Q (0.5 μM), not only removed the GABAB component (late IPSP; Figures 6A,B) of the synaptic response, but also induced a significant increase in Ri (Figures 6C,D; 122.5 ± 5.4%; t = −3.264; p = 0.022) as well as membrane depolarization (Figure 6E; 9.1 ± 2.6 mV; n = 5; t = 8.69, p < 0.001), therefore mimicking all Aβ25–35 effects previously described.
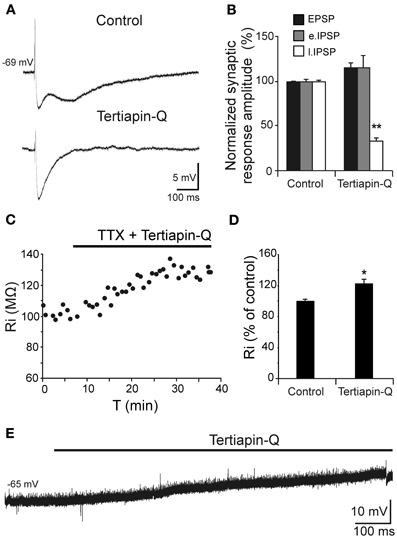
Figure 6. Effects of the selective GirK channel antagonist tertiapin-Q on CA3 pyramidal neurons complex response to fimbria stimulation. (A) Selective blockade by tertiapin-Q of the late IPSP recorded in CA3 pyramidal neurons after fimbria stimulation. Superfusion of the GirK antagonist tertiapin-Q (0.5 μM; selective blocker of GirK channels) selectively blocked the late IPSP but did not reduce the early IPSP that was elicited by fimbria stimulation. (B) Histograms with relative mean amplitude (n = 5) of the different components of the complex synaptic response (EPSP; early, e.IPSP; and late, l.IPSP) 40 min after tertiapin-Q perfusion. Significant differences were found for late IPSP. (C) Results of an experiment in another neuron designed to assess the postsynaptic effects of tertiapin-Q on Ri of CA3 pyramidal neurons. Perfusion of TTX (n = 5; 1 μM; voltage-dependent sodium channel blocker) blocked afferent neurotransmission and therefore any effect of tertiapin-Q took place at postsynaptic location. Note the significant Ri increase after 30 min. (D) Histogram with mean values in percentage for Ri (n = 6) after tertiapin-Q perfusion during 40 min (*p < 0.05). (E) Effect of tertiapin-Q on CA3 pyramidal neurons membrane potential (n = 5). Chart record shows that superfusion of tertiapin-Q (0.5 μM) produced a marked depolarization when applied at resting membrane potential (−65 mV). **p <0.01.
To determine the mechanism involved in the postsynaptic reduction of the late IPSP amplitude and Ri increase, pharmacological blockage of different receptors/channels was performed and effects of increasing Aβ25–35 concentrations on Ri were evaluated (Figure 7). The blockade of synaptic transmission by TTX did not produce significant changes in Ri (Mann-Whitney U, p = 0.700) compared to control (Figures 7A–C, Exp. 1–3). Then, Aβ25–35 perfusion produced a significant increase in Ri (n = 4) at high concentrations (Figure 7, Exp. 1; 1.0 μM, t = −7.424; p = 0.018 and 1.5 μM, t = −7.519; p = 0.002), which was not observed at 0.5 μM (Mann-Whitney U, p = 1.00). These results indicate a postsynaptic Aβ25–35 mechanism that quite likely involves a reduction in ion channels conductance.
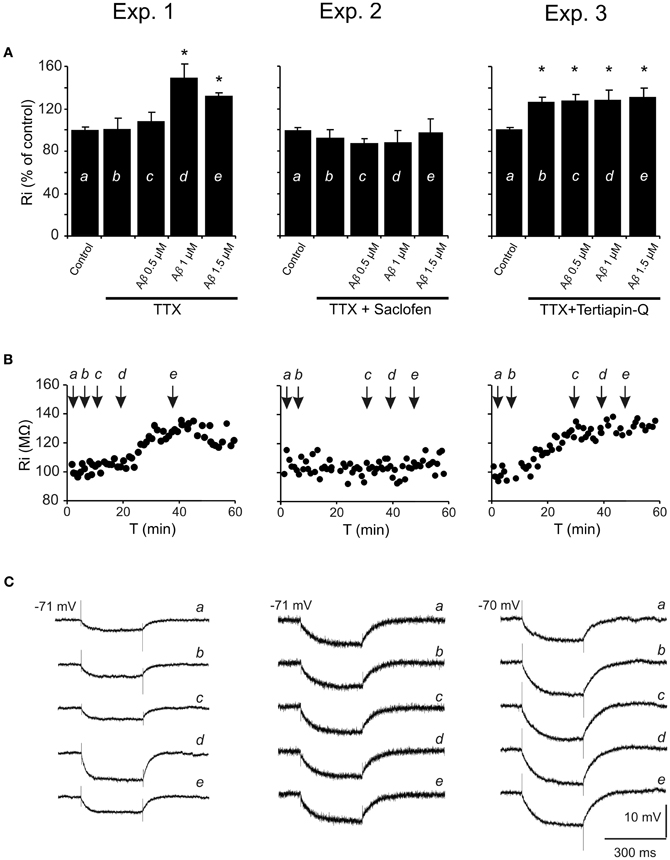
Figure 7. Histograms with Ri mean values in CA3 pyramidal neurons for increasing Aβ25–35 concentrations. (A) Normalized mean value of Ri as % of control after perfusion with three different Aβ25–35 concentrations in three different experimental conditions (Exp. 1–3). In all cases, TTX pre-treatment (1 μM; voltage-dependent sodium channel blocker) was unable to prevent Aβ25–35 effects on Ri, suggesting a postsynaptic location for Aβ25–35 action. Exp. 1: Significant increase on Ri was recorded after 1 and 1.5 μM Aβ25–35 perfusion (n = 16), even in the presence of TTX. Exp. 2: After perfusion of both TTX and saclofen (to block postsynaptic GABAB receptors), Aβ25–35 was unable to evoke any significant modification in Ri (n = 6). Exp. 3: As in the experiment showed in Figure 6B, TTX perfusion together with the selective antagonist of GirK channels, tertiapin-Q (0.5 μM), induced a significant increase in Ri compared to control values. However, when tertiapin-Q was perfused and GirK channels blocked, Aβ25–35 became unable to induce any additional Ri increase (n = 6). Control value for Ri was normalized to 100%. Data show mean ± SEM. *p < 0.05. Lower-case letters (a–e) indicate the five pharmacological conditions during each experimental treatment. (B) Representative examples for time course of Ri recorded in experimental conditions (Exp. 1–3). The arrows indicate the time point at which drugs were applied during the recordings. (C) Representative examples of recordings were expanded in time to show the changes in membrane potential during the presentation of hyperpolarizing pulses. This protocol allowed us to monitor Ri during the whole recording and to study the effect of different pharmacological treatments represented by lower-case letters (a–e).
On the other hand, since saclofen perfusion prevented Aβ25–35-induced changes on Ri (n = 6; H = 3.964, p = 0.411; Figure 7, Exp. 2), an interaction with GABAB receptors might be assumed. However, although saclofen also induced a reduction on late IPSP amplitude (see Figures 3B,E), in the presence of TTX it did not exhibit any noticeable effect on membrane potential (1.7 ± 2.8 mV; t = −0.541; p = 0.617; non illustrated) or Ri (Figure 7, Exp. 2; 94.5 ± 7.3%; t = 1.198; p = 0.285), in contrast to Aβ25–35. Altogether, these results suggest that Aβ25–35 exerts its effects acting preferentially on postsynaptic GirK channels, instead of GABAB receptors.
In accordance with this hypothesis, Aβ25–35 was found to be unable to generate additional increase on Ri after postsynaptic blockage of GirK channels by tertiapin-Q [Figure 7, Exp. 3; n = 6; F(3, 21) = 0.129, p = 0.941], suggesting that Aβ-induced Ri increase may be associated to a reduction in GirK channel conductance. However, because conductance depends, among others, on the number of channels, their open probability or membrane voltage, the methodology used in the present study has some limitations to determine the exact mechanism for Aβ-mediated Ri increase. In addition, in some experiments, it was necessary to inject DC to maintain a stable membrane potential and to prevent depolarization. Previous studies had reported that depolarization mechanisms induced by Aβ might involve activation of glutamatergic receptors (Blanchard et al., 2002a,b). In order to control these variables, tertiapin-Q was perfused together with glutamatergic antagonists CNQX (10 μM) and APV (50 μM). This protocol not only prevented the Aβ-mediated increase in Ri, but also abolished the dependence of DC injection to compensate membrane depolarization (n = 4; H = 3.709, p = 0.447; non illustrated). In contrast, perfusion with CNQX, APV and the GABAA blocker, bicuculline (10 μM), was not able to prevent the Ri increase induced by Aβ25–35 (n = 4; H = 21.681; p < 0.001; non illustrated) further suggesting an effect of Aβ25–35 on GirK channels.
Effects of Aβ25–35 on the Hyperpolarization Mediated by GABAB-GirK Activation
Given that, Ri changes may depend on multiple factors and might be affected by Aβ acting on different ion channels and receptors, we designed a protocol to specifically evaluate the effects of Aβ25–35 on GABAB response. To verify whether Aβ25–35 affects the postsynaptic response mediated by GABAB receptor activation, we used a drug cocktail including: TTX to block synaptic transmission, bicuculline to block GABAA receptors activation and baclofen to stimulate GABAB receptors (Figure 8). Cocktail application in the slice produced a postsynaptic membrane hyperpolarization in recorded CA3 pyramidal neurons (Figures 8A,D; −9.5 ± 2.8% of the RMP value). When membrane potential was stabilized, Aβ25–35 perfusion induced a pronounced depolarization (Figures 8A,D; 15.5 ± 4.3 % of the RMP value; n = 4), which confirms that Aβ25–35 reduces the postsynaptic GABAB response in a concentration and time dependent manner. But Aβ25–35 action on this GABAB response might also be explained by an effect on its final effector, GirK, which would also underlie the already described Aβ25–35 effects on Ri and membrane depolarization.
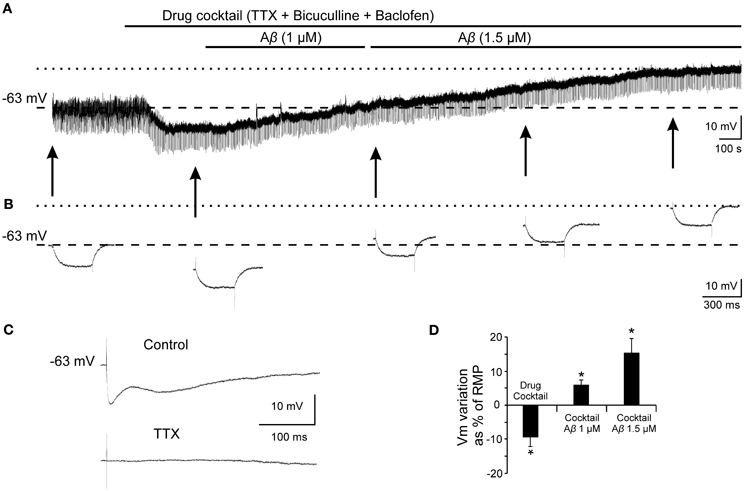
Figure 8. Effects of Aβ25–35 on postsynaptic hyperpolarization induced by activation of GABAB receptors. (A) Membrane potential recording from a CA3 neuron (top) after perfusion with TTX (1 μM; voltage-dependent sodium channels blocker), bicuculline (10 μM; specific blocker of GABAA receptor) and baclofen (15 μM; agonist of GABAB receptor). This treatment (n = 4) produced a membrane hyperpolarization depending on GABAB postsynaptic receptors activation. Perfusion with Aβ25–35 (1 and 1.5 μM) markedly induced the membrane to depolarize. (B) Intracellular hyperpolarizing current pulses. The arrows indicate the time points at which recordings were expanded in time to show the changes in the membrane potential during the presentation of hyperpolarizing pulses. This protocol allowed us to monitor Ri during the whole recording and check the viability of the neurons. Dashed lines in (A) and (B) indicate membrane resting potential. Maximum membrane potential evoked by Aβ25–35 superfusion is indicated by dotted lines. (C) In the same neuron, fimbria stimulation elicited the characteristic triphasic postsynaptic response before drugs cocktail perfusion. This complex postsynaptic potential was completely removed by TTX (1 μM; voltage-dependent sodium channels blocker) perfusion. (D) Plot of the membrane potential variations as a percentage of resting membrane potential (RMP) under pharmacological conditions presented in (A). TTX, bicuculline and baclofen (drug cocktail) induced pyramidal neurons to hyperpolarize while Aβ25–35 produced a noticeable depolarization (n = 4; 21.4 ± 5.9 mV). *p < 0.05.
In order to validate this hypothesis and evaluate the effect of Aβ25–35 on GirK response, we used a GirK channel agonist, MPD (Aryal et al., 2009). Bath application of the previous drugs cocktail together with MPD (50 mM) induced the cell to hyperpolarize by two postsynaptic mechanisms, GABAB receptor activation and direct increase in GirK conductance (Figure 9; −11.2 ± 3.8% of the RMP value; n = 15). Then, perfusion of Aβ25–35 (1–1.5 μM) removed the hyperpolarization mediated by GABAB-GirK stimulation (Figures 9A,E; 5.9 ± 2.7%; n = 3, Figures 9B,E; 4.2 ± 2.4%, n = 4), while this effect was not evident at 0.5 μM (Figures 9C,E; −7.2 ± 4.8% n = 4). In fact, the hyperpolarization could be eliminated when the cocktail was washed (Figure 9C). Finally, Aβ-induced depolarization was mimicked by tertiapin-Q, the specific antagonist of GirK channels (Figures 9D,E; 5.5 ± 1.5%; n = 4), indicating that Aβ25–35 directly affects GirK channels conductance.
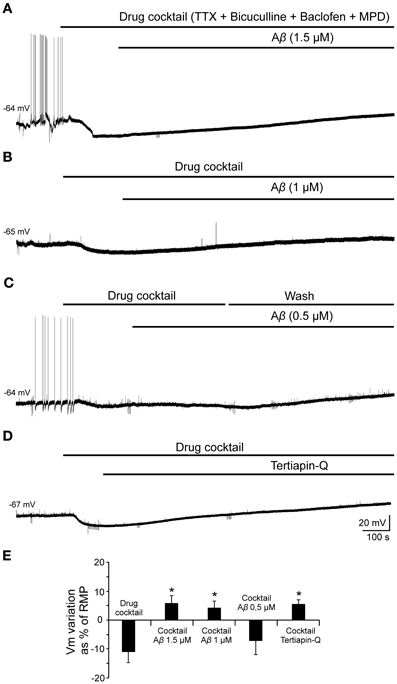
Figure 9. Effects of Aβ25–35 perfusion on the response evoked by pharmacological co-activation of GirK channels and GABAB receptors. (A,B) GirK/GABAB postsynaptic hyperpolarization was induced (n = 15) by perfusion of drugs cocktail including TTX (1 μM; voltage-dependent sodium channels blocker), bicuculline (10 μM; specific blocker of GABAA receptors), baclofen (15 μM; agonist of GABAB receptors), and MPD (50 mM; GirK channel agonist) perfusion. Aβ25–35 1.5 μM (n = 3) and 1.0 μM (n = 4)(A and B, respectively) evoked a postsynaptic depolarization. (C) Using the same protocol, Aβ25–35 0.5 μM was not able to produce this depolarization. The cocktail hyperpolarizing effect on membrane potential disappeared after cell washing with bathing solution (n = 4). (D) Tertiapin-Q (n = 4; 0.5 μM; selective blocker of GirK channels), the specific antagonist of GirK channels, induced a depolarization of the postsynaptic hyperpolarization mediated by GirK/GABAB activation, reproducing Aβ25–35 effects. (E) Plot showing the membrane potential changes as a percentage of resting membrane potential (RMP) under pharmacological conditions presented in (A–D). *p < 0.05.
Discussion
Despite the importance that rhythms as an emergent property of neural network seem to have, few studies have investigated how Aβ induces injury and how it may contribute to impair septohippocampal oscillatory activity, which in turn may underlie the early symptoms typically observed in AD patients (Colom, 2006; Palop and Mucke, 2010; Villette et al., 2010; Rubio et al., 2012; Verret et al., 2012). The present study identifies alterations in GirK channels conductance of fimbria-CA3 synapse as a putative mechanism of Aβ-induced synaptic dysfunction observed in the septohippocampal system activity.
Septohippocampal System and Aβ25–35 Neurotoxicity
Previously, it has been proposed that Aβ25–35 constitutes the biologically active fragment of Aβ (Millucci et al., 2010), and has been shown to induce major neuropathological signs related to early stages of AD in rats (Klementiev et al., 2007). In addition, Aβ25–35 is reported to be more soluble and presents toxic effects more rapidly than the parent peptide Aβ1–42 (Varadarajan et al., 2001), and has widely been used as a very useful tool to explore acutely the pathophysiological events related with neuronal dysfunction induced by soluble Aβ forms (Ashenafi et al., 2005; Santos-Torres et al., 2007; Peña et al., 2010; Leao et al., 2012). But the most important advantage for present work is that Aβ25–35 does not form ion-permeable pores in neuronal membrane (Jang et al., 2010; Chang et al., 2011; Leao et al., 2012) which could alter our protocols, especially input resistance measurements.
From neuroanatomical and electrophysiological points of view, hippocampus receives different projections from the medial septum diagonal broca band mainly through lateral fimbria (Wyss et al., 1980; Alonso and Kohler, 1984; Colom, 2006; Amaral and Lavenex, 2007). Cholinergic neurons innervate pyramidal neurons and interneurons (Widmer et al., 2006), GABAergic fibers project onto interneurons (Freund and Antal, 1988; Chamberland et al., 2010) and glutamatergic projections contact with CA3 pyramidal cells (Huh et al., 2010). These septo-hippocampal circuits are involved in both, generating hippocampal theta rhythm, as well as in learning and memory processes. To maintain these high-level functions, a precise septohippocampal network activity requires of finely regulated excitatory and inhibitory neurotransmission (Buzsaki, 2002; Sotty et al., 2003; Borhegyi et al., 2004; Colom, 2006) whose alteration could lead to impairments that would be consistent with the deficits described for AD initial stages (Bland and Colom, 1993; Palop and Mucke, 2010). In this sense, it has been reported that in hippocampus, Aβ mainly induces aberrant inhibitory septohippocampal network activity (Palop et al., 2007; Villette et al., 2010, 2012). Therefore, although an Aβ effect on inhibitory neurotransmission might be expected, the putative mechanism to detune the network coordination of this system remains unclear.
Aβ25–35 was found not to affect the active properties of CA3 pyramidal recorded neurons suggesting that Aβ25–35 does not modify the conductances that mediate active membrane properties such as sodium or potassium voltage-gated channels for spike amplitude (Hille, 2001), BK/SK channels for AHPs (Sah and Faber, 2002) or R-type calcium channels for ADP (Metz et al., 2005). Similar results have also been shown in other AD related regions as amygdala (Ashenafi et al., 2005), septum (Santos-Torres et al., 2007), or cortex (Wang et al., 2009).
On the other hand, Aβ25–35 induced an Ri increase associated with membrane depolarization. We have previously showed that Aβ25–35 exerts variable effects on membrane potential and Ri in amygdalar pyramidal neurons, possibly due to a presynaptic mechanism (Ashenafi et al., 2005) and in septal neurons, depending on pre- and postsynaptic actions (Santos-Torres et al., 2007). In the present study, TTX was not able to prevent Aβ25–35 effects suggesting a direct effect of Aβ25–35 on CA3 pyramidal neurons membrane.
Previous research has shown that neurotransmission in the septohippocampal system is affected by Aβ through altering the theta oscillatory activity (Colom et al., 2010; Rubio et al., 2012), but studies at the synaptic level on the mechanisms underlying this alteration have not been exhaustively performed. Thus, the possibility of studying in a single preparation and in a particular synapse, excitatory and inhibitory neurotransmission, presents fimbria-CA3 synapse preparation as an excellent model for dissecting the possible mechanisms involved in Aβ action on different septohippocampal neurotransmission systems.
A biphasic (glutamatergic, non-NMDA, and GABAergic, GABAA) response in CA3 region after fimbria stimulation (Schneiderman et al., 1992) has previously been reported. However, we found a complex synaptic response comprising three phases: an ionotropic glutamatergic EPSP (Huh et al., 2010) followed by two IPSPs, early (GABAA), and late (GABAB). This complex response has also been described in pyramidal neurons of basolateral amygdaloid nucleus (Washburn and Moises, 1992a), in CA3 pyramidal cells after hilus and mossy fibers stimulation (Malouf et al., 1990; Scanziani et al., 1991). In our study, the inhibitory feedback seems to be mediated by activation of GABAergic interneurons and depends on glutamatergic activation, since the block of EPSP also eliminated both IPSPs. The loss of GABAA inhibition led to an increase in EPSP and late IPSP amplitudes whereas complete GABAergic component block induced an epileptic-like response mainly mediated by non-NMDA receptors, although NMDA antagonist reduced the response partially. Very similar results have been shown in neurons from CA3 hippocampal region (Scanziani et al., 1991) or frontal cortex (Sutor and Luhmann, 1998) possibly caused by an increased excitation of interneurons arising from disinhibited excitatory neurons, and disinhibition of interneurons due to block of GABAA receptors on the inhibitory cell. This machinery has been suggested as a self-protective mechanism for the control of recurrent activity when an imbalance of the system occurs (Scanziani et al., 1991; Sutor and Luhmann, 1998).
On the other hand, although it has been reported that cholinergic septal neurons innervate pyramidal CA3 neurons and interneurons (Widmer et al., 2006), it is also known that cholinergic axons must be activated by train stimulation (>30 Hz) (Washburn and Moises, 1992b; Faber and Sah, 2002; Navarro-Lopez et al., 2004). It can therefore be suggested that cholinergic axons projecting onto CA3 neurons were not activated during single stimulation of the fimbria.
Aβ25–35 Effects on the Complex Fimbria-CA3 Synaptic Response
In the present study, pharmacological characterization of the complex septohippocampal synaptic response revealed the glutamatergic nature of the EPSP. Glutamatergic septohippocampal neurons (Colom et al., 2005; Huh et al., 2010) have shown spontaneous firing at theta frequencies and Aβ increases this frequency (Leao et al., 2012), suggesting Aβ to be likely to impair septohippocampal excitatory and network activity through more than one mechanism.
We found Aβ25–35 to increase the EPSP. This result may be explained because excitatory response is strongly braked by the GABAB activity (Otmakhova and Lisman, 2004; Chen and Johnston, 2005), and Aβ25–35 diminished late IPSP. Since late IPSP was generated by GABAB receptors stimulation, an Aβ25–35 effect on such receptors, on its intracellular signaling mechanism, or on its final effector, GirK channels, could be hypothesized. Assuming that the early GABAA component is not affected by Aβ25–35, the reduction of GABAB component would not be due to an inhibition of GABA release or other presynaptic mechanism. Together with Ri increase and membrane depolarization, our results lead to a reduction of the conductance of potassium channels coupled to GABAB receptor.
Binding studies in postmortem AD patients have shown a reduction in GABA receptors density in the hippocampus (Chu et al., 1987). More recently, the 17A polymerase has been described to be responsible for generating alternative splicing of GABAB2 subunit in AD patients (Massone et al., 2011). This modification affects intracellular signaling pathway and activation of GirK channels, and is also associated with an increased secretion of Aβ, suggesting a relationship between the metabolism of APP protein and dysfunction in the GABAB receptor signaling.
Aβ25–35 Action on GirK Channels
When fimbria is stimulated, GABAergic interneurons activate GABAB receptors of CA3 pyramidal cells; Aβ25–35 behaves as a selective antagonist and reduces late IPSP. However, the GABAB system has a very low tonic activity in basal conditions, and only when the system is activated pharmacologically or by afferent stimulation, the antagonist effect becomes obvious (Bowery and Smart, 2006) as occurs, for example, in chronic pain (Malcangio and Bowery, 1994). For this reason, blocking GABAB receptors induces little effects on membrane properties, i.e., membrane potential or Ri (Lambert et al., 1989; Emri et al., 1996), so GABAB receptor antagonism would not explain all the effects induced by Aβ25–35.
Another aspect to consider is that both, GABAB receptors and GirK channels are coupled and co-expressed in the postsynaptic membrane of CA3 pyramidal neurons (Luscher et al., 1997; Kulik et al., 2003; Lujan et al., 2009) conforming an oligomeric stable molecular complex (Lujan et al., 2009; Ciruela et al., 2010). Therefore, Aβ25–35 action on the membrane should include the effector coupled to GABAB receptor. GirK channels exhibit a tonic basal activity, even without receptor signaling, due to their direct binding to the Gα subunit of G proteins (Lujan et al., 2009). Hence, administration of GirK channel selective antagonist (tertiapin-Q) simulates all the effects of Aβ25–35 on the postsynaptic membrane, i.e., late IPSP reduction, Ri increase and membrane depolarization, even in the presence of GirK and GABAB agonists. Furthermore, Aβ25–35 was found to be unable to generate an additional significant increase in Ri after pharmacological blocking of GirK channels. These results not only suggest that GirK channels are functional in the basal state, but also that Aβ25–35 action seems to be more evident when these GirK channels are activated.
GirK channels activity alteration may have multiple implications for synaptic activity and neuronal network function. Numerous studies have emphasized its role in several pathological processes in the nervous system such as epilepsy, pain, addiction, Parkinson or Down syndrome (Luscher and Slesinger, 2010). Deletion studies of GirK channels have revealed their role in learning and memory processes. GIRK4 knock-out mice exhibited impaired performance in spatial learning and memory test (Wickman et al., 2000). Moreover, mutations in GIRK2 subunit reduced LTP and increased LTD in hippocampus (Sago et al., 1998; Siarey et al., 1999; Luscher and Slesinger, 2010) and it is especially relevant in Down syndrome, where cerebral Aβ accumulation is greatly accelerated and leads to invariant early-onset AD neuropathology (Lott and Head, 2005; Moncaster et al., 2010; Cooper et al., 2012).
The present study proposes a putative synaptic mechanism for neural network hyperactivity, which is considered as an early event in AD pathogenesis and is associated with early Aβ deposition in non-demented humans with or without mild cognitive impairment (Sperling et al., 2009). An alteration in GirK channel conductance of pyramidal CA3 neurons might underlie this hyperactivity and the impaired inhibition which has been related to network dysfunction and alteration of rhythm generation required for information processing and memory storage in the septohippocampal system (Palop et al., 2007; Palop and Mucke, 2010; Villette et al., 2010, 2012; Rubio et al., 2012; Verret et al., 2012). Our data could be in accordance to the notion that reducing network hyperactivity would have beneficial effects on cognitive functions (Palop and Mucke, 2010; Verret et al., 2012). Subsequently, the present work shows GirK channel as a new target to study Aβ pathophysiology in early and mild cognitive impairment in AD. Since cholinergic or glutamatergic treatments in AD have shown limited success, therapies combining modulators of different neurotransmission systems seem to be a more promising tool for the treatment, and overall prevention, of this dementia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Spanish MINECO Grants BFU2009-07341 (Javier Yajeya) and SAF2010-14878, BFU2011-22740 (Juan D. Navarro-Lopez). The research leading to these results also received funding (Juan D. Navarro-Lopez) from Fundación Eugenio Rodríguez Pascual. Juan D. Navarro-Lopez held a Ramón y Cajal Research Fellow (RYC-2009-03827). We thank Drs. A. Munera and D. Soto for their enlightening comments about this work, N. Gonzalez for her excellent technical assistance and Eureka-science for its help in manuscript editing.
References
Alonso, A., and Kohler, C. (1984). A study of the reciprocal connections between the septum and the entorhinal area using anterograde and retrograde axonal transport methods in the rat brain. J. Comp. Neurol. 225, 327–343. doi: 10.1002/cne.902250303
Amaral, D. G., and Lavenex, P. (2007). “Hippocampal neuroanatomy,” in The Hippocampus Book, eds P. Andersen, R. Morris, D. G. Amaral, T. Bliss, and J. O'Keefe (New York, NY: Oxford University Press), 37–114.
Aryal, P., Dvir, H., Choe, S., and Slesinger, P. A. (2009). A discrete alcohol pocket involved in GIRK channel activation. Nat. Neurosci. 12, 988–995. doi: 10.1038/nn.2358
Ashenafi, S., Fuente, A., Criado, J. M., Riolobos, A. S., Heredia, M., and Yajeya, J. (2005). Beta-Amyloid peptide 25–35 depresses excitatory synaptic transmission in the rat basolateral amygdala “in vitro”. Neurobiol. Aging 26, 419–428. doi: 10.1016/j.neurobiolaging.2004.05.008
Bischofberger, J., Engel, D., Li, L., Geiger, J. R., and Jonas, P. (2006). Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat. Protoc. 1, 2075–2081. doi: 10.1038/nprot.2006.312
Blanchard, B. J., Stockwell, B. R., and Ingram, V. M. (2002a). Eliminating membrane depolarization caused by the Alzheimer peptide Abeta(1-42, aggr.). Biochem. Biophys. Res. Commun. 293, 1204–1208. doi: 10.1016/S0006-291X(02)00290-5
Blanchard, B. J., Thomas, V. L., and Ingram, V. M. (2002b). Mechanism of membrane depolarization caused by the Alzheimer Abeta1-42 peptide. Biochem. Biophys. Res. Commun. 293, 1197–1203. doi: 10.1016/S0006-291X(02)00346-7
Bland, B. H., and Colom, L. V. (1993). Extrinsic and intrinsic properties underlying oscillation and synchrony in limbic cortex. Prog. Neurobiol. 41, 157–208. doi: 10.1016/0301-0082(93)90007-F
Bland, B. H., and Oddie, S. D. (2001). Theta band oscillation and synchrony in the hippocampal formation and associated structures: the case for its role in sensorimotor integration. Behav. Brain Res. 127, 119–136. doi: 10.1016/S0166-4328(01)00358-8
Borhegyi, Z., Varga, V., Szilagyi, N., Fabo, D., and Freund, T. F. (2004). Phase segregation of medial septal GABAergic neurons during hippocampal theta activity. J. Neurosci. 24, 8470–8479. doi: 10.1523/JNEUROSCI.1413-04.2004
Bowery, N. G., and Smart, T. G. (2006). GABA and glycine as neurotransmitters: a brief history. Br. J. Pharmacol. 147 (Suppl. 1), S109–S119. doi: 10.1038/sj.bjp.0706443
Buzsaki, G. (2002). Theta oscillations in the hippocampus. Neuron 33, 325–340. doi: 10.1016/S0896-6273(02)00586-X
Chamberland, S., Salesse, C., Topolnik, D., and Topolnik, L. (2010). Synapse-specific inhibitory control of hippocampal feedback inhibitory circuit. Front. Cell. Neurosci. 4:130. doi: 10.3389/fncel.2010.00130
Chang, Z., Luo, Y., Zhang, Y., and Wei, G. (2011). Interactions of Abeta25–35 beta-barrel-like oligomers with anionic lipid bilayer and resulting membrane leakage: an all-atom molecular dynamics study. J. Phys. Chem. B 115, 1165–1174. doi: 10.1021/jp107558e
Chen, X., and Johnston, D. (2005). Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J. Neurosci. 25, 3787–3792. doi: 10.1523/JNEUROSCI.5312-04.2005
Chu, D. C., Penney, J. B. Jr., and Young, A. B. (1987). Quantitative autoradiography of hippocampal GABAB and GABAA receptor changes in Alzheimer's disease. Neurosci. Lett. 82, 246–252. doi: 10.1016/0304-3940(87)90264-3
Ciruela, F., Fernandez-Duenas, V., Sahlholm, K., Fernandez-Alacid, L., Nicolau, J. C., Watanabe, M., et al. (2010). Evidence for oligomerization between GABAB receptors and GIRK channels containing the GIRK1 and GIRK3 subunits. Eur. J Neurosci. 32, 1265–1277. doi: 10.1111/j.1460-9568.2010.07356.x
Colom, L. V. (2006). Septal networks: relevance to theta rhythm, epilepsy and Alzheimer's disease. J. Neurochem. 96, 609–623. doi: 10.1111/j.1471-4159.2005.03630.x
Colom, L. V., Castaneda, M. T., Banuelos, C., Puras, G., Garcia-Hernandez, A., Hernandez, S., et al. (2010). Medial septal beta-amyloid 1-40 injections alter septo-hippocampal anatomy and function. Neurobiol. Aging 31, 46–57. doi: 10.1016/j.neurobiolaging.2008.05.006
Colom, L. V., Castaneda, M. T., Reyna, T., Hernandez, S., and Garrido-Sanabria, E. (2005). Characterization of medial septal glutamatergic neurons and their projection to the hippocampus. Synapse 58, 151–164. doi: 10.1002/syn.20184
Cooper, A., Grigoryan, G., Guy-David, L., Tsoory, M. M., Chen, A., and Reuveny, E. (2012). Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 2642–2647. doi: 10.1073/pnas.1109099109
Emri, Z., Turner, J. P., and Crunelli, V. (1996). Tonic activation of presynaptic GABA(B) receptors on thalamic sensory afferents. Neuroscience 72, 689–698. doi: 10.1016/0306-4522(95)00590-0
Faber, E. S., and Sah, P. (2002). Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J. Neurosci. 22, 1618–1628.
Freund, T. F., and Antal, M. (1988). GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature 336, 170–173. doi: 10.1038/336170a0
Gloveli, T., Dugladze, T., Rotstein, H. G., Traub, R. D., Monyer, H., Heinemann, U., et al. (2005). Orthogonal arrangement of rhythm-generating microcircuits in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 102, 13295–13300. doi: 10.1073/pnas.0506259102
Huang, Y., and Mucke, L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. doi: 10.1016/j.cell.2012.02.040
Huh, C. Y., Goutagny, R., and Williams, S. (2010). Glutamatergic neurons of the mouse medial septum and diagonal band of Broca synaptically drive hippocampal pyramidal cells: relevance for hippocampal theta rhythm. J. Neurosci. 30, 15951–15961. doi: 10.1523/JNEUROSCI.3663-10.2010
Jang, H., Arce, F. T., Ramachandran, S., Capone, R., Azimova, R., Kagan, B. L., et al. (2010). Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer's Disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 6538–6543. doi: 10.1073/pnas.0914251107
Kaupmann, K., Schuler, V., Mosbacher, J., Bischoff, S., Bittiger, H., Heid, J., et al. (1998). Human gamma-aminobutyric acid type B receptors are differentially expressed and regulate inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U.S.A. 95, 14991–14996. doi: 10.1073/pnas.95.25.14991
Klementiev, B., Novikova, T., Novitskaya, V., Walmod, P. S., Dmytriyeva, O., Pakkenberg, B., et al. (2007). A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by Abeta25–35. Neuroscience 145, 209–224. doi: 10.1016/j.neuroscience.2006.11.060
Kohler, C., Chan-Palay, V., and Wu, J. Y. (1984). Septal neurons containing glutamic acid decarboxylase immunoreactivity project to the hippocampal region in the rat brain. Anat. Embryol. (Berl.) 169, 41–44. doi: 10.1007/BF00300585
Kulik, A., Vida, I., Lujan, R., Haas, C. A., Lopez-Bendito, G., Shigemoto, R., et al. (2003). Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J. Neurosci. 23, 11026–11035.
Lambert, N. A., Harrison, N. L., Kerr, D. I., Ong, J., Prager, R. H., and Teyler, T. J. (1989). Blockade of the late IPSP in rat CA1 hippocampal neurons by 2-hydroxy-saclofen. Neurosci. Lett. 107, 125–128. doi: 10.1016/0304-3940(89)90803-3
Leao, R. N., Colom, L. V., Borgius, L., Kiehn, O., and Fisahn, A. (2012). Medial septal dysfunction by Abeta-induced KCNQ channel-block in glutamatergic neurons. Neurobiol. Aging 33, 2046–2061. doi: 10.1016/j.neurobiolaging.2011.07.013
Lott, I. T., and Head, E. (2005). Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol. Aging 26, 383–389. doi: 10.1016/j.neurobiolaging.2004.08.005
Lujan, R., and Ciruela, F. (2012). GABAB receptors-associated proteins: potential drug targets in neurological disorders? Curr. Drug Targets 13, 129–144. doi: 10.2174/138945012798868425
Lujan, R., Maylie, J., and Adelman, J. P. (2009). New sites of action for GIRK and SK channels. Nat. Rev. Neurosci. 10, 475–480. doi: 10.1038/nrn2668
Luscher, C., Jan, L. Y., Stoffel, M., Malenka, R. C., and Nicoll, R. A. (1997). G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695. doi: 10.1016/S0896-6273(00)80381-5
Luscher, C., and Slesinger, P. A. (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315. doi: 10.1038/nrn2834
Lynch, G., Rose, G., and Gall, C. (1977). Anatomical and functional aspects of the septo-hippocampal projections. Ciba Found. Symp. 58, 5–24.
Malcangio, M., and Bowery, N. G. (1994). Spinal cord SP release and hyperalgesia in monoarthritic rats: involvement of the GABAB receptor system. Br. J. Pharmacol. 113, 1561–1566. doi: 10.1111/j.1476-5381.1994.tb17174.x
Malouf, A. T., Robbins, C. A., and Schwartzkroin, P. A. (1990). Phaclofen inhibition of the slow inhibitory postsynaptic potential in hippocampal slice cultures: a possible role for the GABAB-mediated inhibitory postsynaptic potential. Neuroscience 35, 53–61. doi: 10.1016/0306-4522(90)90119-O
Massone, S., Vassallo, I., Fiorino, G., Castelnuovo, M., Barbieri, F., Borghi, R., et al. (2011). 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 41, 308–317. doi: 10.1016/j.nbd.2010.09.019
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34, 939–944. doi: 10.1212/WNL.34.7.939
Metz, A. E., Jarsky, T., Martina, M., and Spruston, N. (2005). R-type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J. Neurosci. 25, 5763–5773. doi: 10.1523/JNEUROSCI.0624-05.2005
Millucci, L., Ghezzi, L., Bernardini, G., and Santucci, A. (2010). Conformations and biological activities of amyloid beta peptide 25–35. Curr. Protein Pept. Sci. 11, 54–67. doi: 10.2174/138920310790274626
Moncaster, J. A., Pineda, R., Moir, R. D., Lu, S., Burton, M. A., Ghosh, J. G., et al. (2010). Alzheimer's disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS ONE 5:e10659. doi: 10.1371/journal.pone.0010659
Moreno, H., Wu, W. E., Lee, T., Brickman, A., Mayeux, R., Brown, T. R., et al. (2007). Imaging the Abeta-related neurotoxicity of Alzheimer disease. Arch. Neurol. 64, 1467–1477. doi: 10.1001/archneur.64.10.1467
Mott, D. D., and Lewis, D. V. (1994). The pharmacology and function of central GABAB receptors. Int. Rev. Neurobiol. 36, 97–223. doi: 10.1016/S0074-7742(08)60304-9
Navarro-Lopez, J. D., Alvarado, J. C., Marquez-Ruiz, J., Escudero, M., Delgado-Garcia, J. M., and Yajeya, J. (2004). A cholinergic synaptically triggered event participates in the generation of persistent activity necessary for eye fixation. J. Neurosci. 24, 5109–5118. doi: 10.1523/JNEUROSCI.0235-04.2004
Otmakhova, N. A., and Lisman, J. E. (2004). Contribution of Ih and GABAB to synaptically induced afterhyperpolarizations in CA1: a brake on the NMDA response. J. Neurophysiol. 92, 2027–2039. doi: 10.1152/jn.00427.2004
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Peña, F., Ordaz, B., Balleza-Tapia, H., Bernal-Pedraza, R., Marquez-Ramos, A., Carmona-Aparicio, L., et al. (2010). Beta-amyloid protein (25–35) disrupts hippocampal network activity: role of Fyn-kinase. Hippocampus 20, 78–96.
Rubio, S. E., Vega-Flores, G., Martinez, A., Bosch, C., Perez-Mediavilla, A., Del, R. J., et al. (2012). Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer's disease. FASEB J. 26, 4458–4467. doi: 10.1096/fj.12-208413
Sago, H., Carlson, E. J., Smith, D. J., Kilbridge, J., Rubin, E. M., Mobley, W. C., et al. (1998). Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. U.S.A. 95, 6256–6261. doi: 10.1073/pnas.95.11.6256
Sah, P., and Faber, E. S. (2002). Channels underlying neuronal calcium-activated potassium currents. Prog. Neurobiol. 66, 345–353. doi: 10.1016/S0301-0082(02)00004-7
Santos-Torres, J., Fuente, A., Criado, J. M., Riolobos, A. S., Heredia, M., and Yajeya, J. (2007). Glutamatergic synaptic depression by synthetic amyloid beta-peptide in the medial septum. J. Neurosci. Res. 85, 634–648. doi: 10.1002/jnr.21150
Scanziani, M., Gahwiler, B. H., and Thompson, S. M. (1991). Paroxysmal inhibitory potentials mediated by GABAB receptors in partially disinhibited rat hippocampal slice cultures. J. Physiol. 444, 375–396.
Schneiderman, J. H., Cairns, A., and Sterling, C. A. (1992). Evoked CA3 field potentials corresponding to both EPSPs and IPSPs in hippocampal slice. Brain Res. 569, 287–294. doi: 10.1016/0006-8993(92)90641-L
Siarey, R. J., Carlson, E. J., Epstein, C. J., Balbo, A., Rapoport, S. I., and Galdzicki, Z. (1999). Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38, 1917–1920. doi: 10.1016/S0028-3908(99)00083-0
Sotty, F., Danik, M., Manseau, F., Laplante, F., Quirion, R., and Williams, S. (2003). Distinct electrophysiological properties of glutamatergic, cholinergic and GABAergic rat septohippocampal neurons: novel implications for hippocampal rhythmicity. J. Physiol. 551, 927–943. doi: 10.1113/jphysiol.2003.046847
Sperling, R. A., LaViolette, P. S., O'Keefe, K., O'Brien, J., Rentz, D. M., Pihlajamaki, M., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. doi: 10.1016/j.neuron.2009.07.003
Spruston, N., and Johnston, D. (1992). Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J. Neurophysiol. 67, 508–529.
Stewart, M., and Fox, S. E. (1990). Do septal neurons pace the hippocampal theta rhythm? Trends Neurosci. 13, 163–168. doi: 10.1016/0166-2236(90)90040-H
Sutor, B., and Luhmann, H. J. (1998). Involvement of GABA(B) receptors in convulsant-induced epileptiform activity in rat neocortex in vitro. Eur. J Neurosci. 10, 3417–3427. doi: 10.1046/j.1460-9568.1998.00351.x
Swanberg, M. M., Tractenberg, R. E., Mohs, R., Thal, L. J., and Cummings, J. L. (2004). Executive dysfunction in Alzheimer disease. Arch. Neurol. 61, 556–560. doi: 10.1001/archneur.61.4.556
Varadarajan, S., Kanski, J., Aksenova, M., Lauderback, C., and Butterfield, D. A. (2001). Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's A beta(1–42) and A beta(25–35). J. Am. Chem. Soc. 123, 5625–5631. doi: 10.1021/ja010452r
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Villette, V., Poindessous-Jazat, F., Bellessort, B., Roullot, E., Peterschmitt, Y., Epelbaum, J., et al. (2012). A new neuronal target for beta-amyloid peptide in the rat hippocampus. Neurobiol. Aging 33, 1126–1114. doi: 10.1016/j.neurobiolaging.2011.11.024
Villette, V., Poindessous-Jazat, F., Simon, A., Lena, C., Roullot, E., Bellessort, B., et al. (2010). Decreased rhythmic GABAergic septal activity and memory-associated theta oscillations after hippocampal amyloid-beta pathology in the rat. J. Neurosci. 30, 10991–11003. doi: 10.1523/JNEUROSCI.6284-09.2010
Wang, Y., Zhang, G., Zhou, H., Barakat, A., and Querfurth, H. (2009). Opposite effects of low and high doses of Abeta42 on electrical network and neuronal excitability in the rat prefrontal cortex. PLoS ONE 4:e8366. doi: 10.1371/journal.pone.0008366
Washburn, M. S., and Moises, H. C. (1992a). Inhibitory responses of rat basolateral amygdaloid neurons recorded in vitro. Neuroscience 50, 811–830. doi: 10.1016/0306-4522(92)90206-H
Washburn, M. S., and Moises, H. C. (1992b). Muscarinic responses of rat basolateral amygdaloid neurons recorded in vitro. J. Physiol. 449, 121–154.
Wickman, K., Karschin, C., Karschin, A., Picciotto, M. R., and Clapham, D. E. (2000). Brain localization and behavioral impact of the G-protein-gated K+ channel subunit GIRK4. J. Neurosci. 20, 5608–5615.
Widmer, H., Ferrigan, L., Davies, C. H., and Cobb, S. R. (2006). Evoked slow muscarinic acetylcholinergic synaptic potentials in rat hippocampal interneurons. Hippocampus 16, 617–628. doi: 10.1002/hipo.20191
Wittner, L., Henze, D. A., Zaborszky, L., and Buzsaki, G. (2007). Three-dimensional reconstruction of the axon arbor of a CA3 pyramidal cell recorded and filled in vivo. Brain Struct. Funct. 212, 75–83. doi: 10.1007/s00429-007-0148-y
Wyss, J. M., Swanson, L. W., and Cowan, W. M. (1980). The organization of the fimbria, dorsal fornix and ventral hippocampal commissure in the rat. Anat. Embryol. (Berl.) 158, 303–316. doi: 10.1007/BF00301819
Keywords: septohippocampal system, fimbria-CA3 synapse, amyloid-β25–35 peptide, GABAB, GirK channels, Alzheimer's disease, brain slices, intracellular recordings
Citation: Nava-Mesa MO, Jiménez-Díaz L, Yajeya J and Navarro-Lopez JD (2013) Amyloid-β induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria-CA3 hippocampal synapse. Front. Cell. Neurosci. 7:117. doi: 10.3389/fncel.2013.00117
Received: 19 April 2013; Accepted: 05 July 2013;
Published online: 25 July 2013.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Lisa Topolnik, Laval University, CanadaClaudia Lodovichi, Venetian Institute of Molecular Medicine, Italy
Copyright © 2013 Nava-Mesa, Jiménez-Díaz, Yajeya and Navarro-Lopez. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Juan D. Navarro-López, Neurophysiology and Bahavior Lab, School of Medicine of Ciudad Real, University of Castilla-La Mancha, Paseo Moledores s/n, 13071-Ciudad Real, Spain e-mail:anVhbi5uYXZhcnJvQHVjbG0uZXM=