 Dan Xu
Dan Xu Stephen D. Miller
Stephen D. Miller Sookyong Koh
Sookyong Koh- 1Department of Microbiology-Immunology and Interdepartmental Immunobiology, Feinberg School of Medicine, Northwestern University, Chicago IL, USA
- 2Department of Pediatrics, Division of Neurobiology, Children’s Research Center, Lurie Children’s Hospital of Chicago, Chicago IL, USA
- 3Department of Pediatrics, Feinberg School of Medicine, Northwestern University, Chicago IL, USA
Epilepsy is a chronic brain disorder that affects 1% of the human population worldwide. Immune responses are implicated in seizure induction and the development of epilepsy. Pre-clinical and clinical evidence have accumulated to suggest a positive feedback cycle between brain inflammation and epileptogenesis. Prolonged or recurrent seizures and brain injuries lead to upregulation of proinflammatory cytokines and activated immune responses to further increase seizure susceptibility, promote neuronal excitability, and induce blood–brain barrier breakdown. This review focuses on the potential role of innate and adaptive immune responses in the pathogenesis of epilepsy. Both human studies and animal models that help delineate the contributions of brain inflammation in epileptogenesis will be discussed. We highlight the critical role of brain-resident immune mediators and emphasize the contribution of brain-infiltrating peripheral leukocytes. Additionally, we propose possible immune mechanisms that underlie epileptogenesis. Several proinflammatory pathways are discussed, including the interleukin-1 receptor/toll-like receptor signaling cascade, the pathways activated by damage-associated molecular patterns, and the cyclooxygenase-2/prostaglandin pathway. Finally, development of better therapies that target the key constituents and processes identified in these mechanisms are considered, for instance, engineering antagonizing agents that effectively block these pathways in an antigen-specific manner.
Introduction
Epilepsy is a chronic neurological condition characterized by recurring seizures, and is often accompanied by cognitive deficits and mood disorder (Devinsky, 2004; Pellock, 2004; Jones et al., 2008). It affects approximately 1% of the world population, thus represents one of the most common brain disorders. Epilepsy arises from diverse etiologies including genetic, structural, metabolic, or in other instances, the cause is unknown. There is currently no medication available to effectively prevent epilepsy by targeting the mechanisms underlying the enduring predisposition to recurrent seizures, and nearly half of the patients with epilepsy fail to respond to anticonvulsants that only alleviate symptoms. Thus, there is a pressing need for the development of effective disease-modifying therapies that treat the underlying pathology. Such development can best be accomplished through an in depth understanding of the disease mechanisms.
Until a decade ago, epilepsy research focused on alterations of neuronal activities. Such neurocentric emphasis failed to address questions that arose in more complex models of epileptogenesis. A cumulative body of knowledge has suggested that the pathogenesis of epilepsy is associated with non-neuronal components, such as the glial cells that exceedingly outnumber neurons, brain vascular cells, and more importantly leukocytes from the periphery. Despite a long-held belief that the brain is an immunoprivileged site due to the vascular blood–brain barrier (BBB) that tightly regulates infiltration of blood constituents and the lack of a lymphatic drainage, mounting evidence has supported the critical role of immune responses in the initiation and maintenance of epilepsy (Vezzani and Granata, 2005; Vezzani and Baram, 2007; Choi et al., 2009; Riazi et al., 2010). Ongoing brain inflammation has the potential to lower seizure threshold, which in turn may promote neuronal excitability through modifications of neuronal channels, alterations of neurotransmitter uptake or release, and regulation of BBB permeability (Viviani et al., 2007; Wetherington et al., 2008; Friedman et al., 2009; Vezzani et al., 2011a).
Both innate and adaptive immune responses can be primed in the brain with the contribution of resident immune cells and mediators, as well as leukocytes infiltrating from the periphery (Ransohoff et al., 2003; Banks and Erickson, 2010). The innate arm of the response involves the activation of the IL-1 receptor/toll-like receptor (IL-1R/TLR) signaling pathways through ligation of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), activation of the cyclooxygenase-2 (COX-2) pathway, and initiation of the transforming growth factor-β/small mothers against decapentaplegic (TGF-β/Smad) signaling cascade. Inflammatory mediators produced by the innate immune system remodel the BBB by enhancing its permeability and upregulating leukocyte adhesion molecules on the endothelium, which acts to attract lymphocytes of the adaptive immune system leading to their infiltration into the CNS.
This review focuses on the roles of immune responses in the pathogenesis of epilepsy by summarizing the most recent findings generated from human studies and animal models that help delineate the contributions of brain inflammation in epileptogenesis. We will evaluate the causal relationship between inflammation and seizure activities and the positive feedback loop they establish by revisiting the experimental evidence from in vivo and in vitro models. Furthermore, we will provide mechanistic insights into the immunological cascade that precedes the establishment of epilepsy and assess the influences of immune mediators apart from the neurological aspect of seizure induction. Finally, we will propose potential mechanisms that underlie epileptogenesis and discuss development of therapies targeting the key constituents and processes identified in these mechanisms.
Pre-Clinical Data Underscoring the Relevance of Immune Responses in Epilepsy
Rodent models of epilepsy have provided ample evidence supporting the role of immune responses in the precipitation of epilepsy, modulation of seizure threshold, orchestration of seizure recurrence, regulation of brain cell survival or attrition, and rewiring of neuronal circuits that may lead to establishment of hyperexcitable neuronal networks (Dube et al., 2005; Kulkarni and Dhir, 2009; Riazi et al., 2010; Vezzani et al., 2011a,b). Adult and immature rats and mice are frequently used to elucidate the role of various immunological pathways that are potentially involved in seizure generation. Administration of proinflammatory or anti-inflammatory agents to rats and mice has been used to assess the influence of these immune mediators on latency to onset, frequency, duration, and phenotype of provoked seizures. Furthermore, the inflammatory pathways can be blocked pharmacologically in wildtype animals or manipulated in transgenic mice to evaluate their role in seizure severity (Kulkarni and Dhir, 2009; Maroso et al., 2010; Vezzani et al., 2011a). Additionally, the availability of genetically modified mice with impaired or constitutively hyperactive immunoregulatory pathways enables more detailed mechanistic studies of inflammation-related epileptogenesis (Campbell et al., 1993; Probert et al., 1997). Guinea pigs models, though less common, have also been used to elucidate the contribution of peripheral immune cells to seizure induction (Librizzi et al., 2010).
Resident Immune Mediators of the Brain
It has long been appreciated that chemically induced or electrically stimulated prolonged seizures in rats and mice lead to induction of robust immune responses in the seizure-laden brain (Minami et al., 1991; Eriksson et al., 2000; De Simoni et al., 2000; Vezzani et al., 2000; Turrin and Rivest, 2004; Voutsinos-Porche et al., 2004; Gorter et al., 2006; Jung et al., 2006; Yoshikawa et al., 2006; Aronica et al., 2007; Dhote et al., 2007; Lee et al., 2007; Kulkarni and Dhir, 2009; Holtman et al., 2010; Polascheck et al., 2010). Immunohistochemical analyses performed on sections of affected brains have revealed that various cell type, including microglia, astrocytes, neurons, ependymal cells in the ventricles, and endothelial cells of the BBB, were involved in the waves of inflammation associated with seizure induction (Takao et al., 1990; Ban et al., 1991; Turrin and Rivest, 2004; Chakravarty and Herkenham, 2005; Ravizza and Vezzani, 2006). Activation of the IL-1R/TLR inflammatory pathway was among the first to be identified in these brain resident immune cells. Constitutive expression of IL-1R and TLR has been detected in the brain, though at a suboptimal level. Upon stimulation, such as viral or bacterial infections, cellular injuries, ischemia, and seizures, upregulation of these receptors is readily detectable (Ericsson et al., 1995; Allan et al., 2005; Peltier et al., 2010; Zurolo et al., 2011). Activation of TLRs, particularly TLR2 and TLR4, through systemic or cortical delivery of lipopolysaccharide (LPS) to rats, results in rapid changes in neuronal excitability, e.g., alteration in synaptic transmission and regulation of long-term potentiation (Bellinger et al., 1993; Plata-Salaman and Ffrench-Mullen, 1994; Schneider et al., 1998; Rodgers et al., 2009). The excitatory effect of IL-1β has also been reported to be associated with the reduction of gamma-aminobutyric acid (GABA) inhibition and reduced outward current of voltage-gated Ca2+ channels in the hippocampus (Zeise et al., 1997; Wang et al., 2000; Viviani et al., 2007; Schafers and Sorkin, 2008). The role of IL-1β in seizure sensitivity was further supported by an in vitro system that demonstrated that N-methyl-D-aspartate (NMDA)-mediated Ca2+ influx is enhanced by activation of Src-dependent NMDA receptor subtype 2B (NR2B) phosphorylation (Viviani et al., 2003). In addition to PAMPs, DAMPs have been recently shown to stimulate TLRs, especially TLR4, to exacerbate seizures. The endogenous signals of DAMPs, such as high mobility group box 1 (HMGB1), can be released by stressed neurons or activated microglia and astrocytes, suggesting that LPS may have mimicked the endogenous DAMP pathway to cause seizure (Maroso et al., 2010). Astrocytes can also enhance seizure induction through increased release of glutamate when the IL-1R/TLR pathway is activated (Wetherington et al., 2008; Friedman et al., 2009).
After the activation of the IL-1R/TLR pathway in the glial population, upregulation of COX-2 and prostaglandins in neurons often ensues (Yamagata et al., 1993; Rozovsky et al., 1994; Yoshikawa et al., 2006; Kulkarni and Dhir, 2009). Inhibition of COX-2 activation prior to seizure induction results in increased mortality and exacerbated seizures behaviors in mice (Baik et al., 1999; Toscano et al., 2008). In contrast, ablation of COX-2 production after seizure induction has been shown to be neuroprotective and resulted in decreased production of inflammatory cytokines by glia and prevented leakage of BBB in a conditional COX-2 knockout mouse strain (Serrano et al., 2011). However, constitutive inhibition of COX-2 failed to prevent the recurrent of unprovoked seizures (Holtman et al., 2010). Thus, it remains unclear whether induction of COX-2 in neurons leads to enhanced epileptogenesis.
After the induction of status epilepticus, serum albumin is detected in the brain suggesting that in BBB failure may contribute to the development of epilepsy (van Vliet et al., 2007). Extravasation of albumin into the cerebral cortex as a result of compromised BBB leads to activation of the TGF-β signaling pathway in astrocytes, and hence increases local inflammation. Such inflammatory responses in the brain parenchyma would most likely induce another wave of neuronal hyperactivation and attrition, which leads to excretion of danger signals, such as DAMPs that would further activate glia to boost inflammation. Thus, a positive feedback loop involves seizure, glia, neurons, and immune responses in the brain.
Infiltrating Immune Mediators of the Brain
It has long been appreciated that prolonged seizures lead to upregulation of adhesion molecules on brain endothelial cells to facilitate extravasation of circulating leukocytes. Expression of E-selectin, P-selectin, intracellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) are increased on the endothelial cells of the brain (Bell and Perry, 1995; Librizzi et al., 2007). The ligands of these molecules, integrins and mucins, are expressed by circulating leukocytes after seizure and facilitate rolling and tethering of granulocytes and lymphocytes (Fabene et al., 2008). Blockade of α4β1 integrins on leukocytes inhibits infiltration of this cell population into the brain, and therapeutic inhibition of α4 integrin activation prevents induction of seizure, and even development of epilepsy (Fabene et al., 2008).
Clinical Evidence of the Involvement of Immune Responses in Epilepsy
Anti-Inflammatory Therapies that are Effective at Treating Epilepsy
The efficacy of anti-inflammatory medications, such as corticosteroids and adrenocorticotrophic hormone (ACTH), in the treatment of some pediatric epilepsies that do not respond to conventional anticonvulsants was one of the first lines of clinical evidence that epilepsy has an immune inflammatory component (Hrachovy et al., 1983; Mackay et al., 2004). It has been shown that ACTH had superior efficacy in the cessation of spasms, improved developmental prognosis, and normalization of electroencephalography (EEG; Snead et al., 1983; Baram et al., 1996; Kivity et al., 2004). In the treatment of refractory epileptic encephalopathies, such as West syndrome, Ohtahara syndrome, Dravet syndrome, Lennox-Gastaut syndrome, Landau-Kleffner syndrome, epilepsy with continuous spike waves during slow-wave sleep and drug-resistant myoclonic atonic epilepsy, a significant proportion of ACTH- and steroid-treated pediatric patients were reported to be seizure-free for an extended period of time, albeit relapsed over time (Yamatogi et al., 1979; Snead et al., 1983; Donat, 1992; Engel, 2001).
In addition to, the corticosteroid therapies, intravenous gammaglobulin (IVIG) has been considered as another potential treatment for refractory epilepsy (Eibl and Wedgwood, 1989). The mechanisms of IVIG induced immunomodulation include suppression of proinflammatory cytokines, interference with antibody-dependent cytotoxicity through Fc receptor blockade, dampening innate immune responses by inhibition of phagocytosis by antigen presenting cells and complement uptake, as well as neutralization of autoantibodies. IgG has been reported to be readily detected in the cerebrospinal fluid (CSF) after a single dose of IVIG in neuromuscular disorders, suggesting that IVIG is capable of crossing the BBB (Cutler et al., 1970; Sekul et al., 1994; Dalakas, 1998). Furthermore, the compromised BBB in many types of epilepsy might further facilitate delivery of IVIG into the brain to exert local immuno- and neuro-modulating effects for seizure alleviation. In a double-blind clinical trial, seven doses of IVIG was administered over a time period of 6 weeks, and more than 50% of the patients in the treatment group had a significant reduction of seizures (van Rijckevorsel-Harmant et al., 1994). Similar effects of seizure reduction and temporary EEG normalization were observed in another trial using pediatric patients (Hart et al., 1994). The use of IVIG in intractable epilepsy and status epilepticus merits further investigation as consistent efficacious outcome has not been achieved and the dosing regimen remains to be optimized.
Blockade of cell-adhesion molecules involved in lymphocyte trafficking has also shown promise in ameliorating seizure severity in epilepsy. Natalizumab, an FDA approved humanized antibody specific to a homing molecule (α4β1 integrin) that directs lymphocyte migration to inflamed tissues, including the brain, has been shown to significantly reduce generalized seizures and status epilepticus in adult patients who also suffered from a autoimmune demyelinating disease, multiple sclerosis (Ley et al., 2007; Sotgiu et al., 2010; Fabene et al., 2013)
Epileptogenic Trigger with an Intrinsic Inflammatory Nature
Febrile status epilepticus is intrinsically associated with immune responses. Genetic susceptibility to inflammation, though not an obligatory factor, has been suggested to lower the seizure threshold, as nearly 30% of febrile seizure patients have such a family history. In addition, mutations in the IL-1β gene segment predispose patients to prolonged febrile convulsions (Millichap, 1959; Virta et al., 2002; Kanemoto et al., 2003). An elevation in a number of proinflammatory cytokines caused by neurotropic viral infections, for instance, human herpesvirus-6 and influenza viruses, is also commonly associated with febrile seizures in infants and young children (Hall et al., 1994; Chiu et al., 2001). Detection of viral DNA is more frequent in the CSF of patients with repetitive febrile seizures than in those patients with a single seizure (Kondo et al., 1993). Increased levels of Th1 and Th2 cytokines, such as interferon-γ (IFN-γ) and interleukin-6 (IL-6), have been reported in influenza-infected patients who later developed febrile seizures, when compared with the virally infected control subjects without seizures (Chiu et al., 2001; Masuyama et al., 2002; Kawada et al., 2003).
Rasmussen’s encephalitis, a prototypic childhood inflammatory epilepsy, is a progressive immune-mediated brain disorder characterized by focal recurrent seizures (epilepsia partialis continua), unilateral hemispheric atrophy, progressive neurological dysfunction and intractable epilepsy. Rasmussen encephalitis is associated with T-cell activation and production of proinflammatory cytokines by activated glia. Effector CD8+ cytotoxic T lymphocytes are proposed to induce astrocytic and neuronal apoptosis and degeneration, one of the hallmarks of Rasmussen’s encephalitis (Bauer et al., 2007). An autoantigen, a glutamate receptor, GluR3, has been detected in this disease. Removal of the GluR3-specific antibody from the circulation has been shown to ameliorate seizure severity, promote neurological functions, and improve the disease prognosis. However, anti-GluR3 antibody is not present in most cases and immune modulation including IVIG, steroid and plasmapheresis has limited efficacy (Rogers et al., 1994; He et al., 1998; Mantegazza et al., 2002; Watson et al., 2004). Hemispherectomy remains the only “cure” of disease progression.
In recent years, an increasing number of autoantibodies have been detected in serum and CSF of patients with new onset drug-resistant focal epilepsy. A definitive diagnosis and therapy have thus been possible for cases that would have been previously categorized as viral or idiopathic encephalitis/epilepsies. The California Encephalitis Project found that the frequency of autoimmune encephalitis was greater than any single viral etiology. Anti-N-methyl D-aspartate receptor (NMDAR) encephalitis was, in fact, the most frequent cause of immune-mediated encephalitis (Armangue et al., 2013). Autoimmune/inflammatory epilepsy is defined as immunologically mediated disorder where recurrent seizures are prominent feature and immune etiology is suggested by detection of neuronal antibodies, presence of inflammatory changes in CSF or upon MRI, or immunotherapy-responsive symptoms and exclusion of other etiologies. Autoimmune/inflammatory epilepsies include limbic encephalitis (both paraneoplastic and non-paraneoplastic), non-limbic encephalitis complicated by seizures, seizures in the context of autoimmune disease, and neural antibody-mediated CNS disorders where seizures are a significant feature. Disease causing autoantibodies have been detected against the following specific neuronal surface proteins: NMDAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), γ-aminobutyric acid beta receptor (GABA-BR), glycine receptor, voltage gated potassium channel leucine-rich glioma-inactivated 1 (VGKC LGI1), VGKC CASPR2 and P/Q or N-type voltage-gated calcium channel (VGCC; Armangue et al., 2012).
The paraneoplastic type is thought to be caused by self-reactive T lymphocytes, although antibodies specific for intracellular components are often detectable (Dalmau and Rosenfeld, 2008). The non-paraneoplastic category is consistently associated with seizure-inducing autoantibodies that target extracellular membrane components, including voltage-gated channels, NMDAR, and glutamic acid decarboxylase (Bien et al., 2007; Dalmau et al., 2008; Vincent and Bien, 2008; Dalmau, 2009; Vincent et al., 2010). These antibodies cause seizures by modifying neuronal excitability, which has been recapitulated in an in vitro model by measuring hippocampal neuronal firing frequency following autoantibody treatment (Vianello et al., 2008). Due to its autoimmune nature, limbic encephalitis responds to immunotherapies, including steroids and IVIG through the mechanisms of dampening T-cell immunity and neutralizing autoantibodies (Dalmau, 2009). Another type of autoimmune epilepsy that also responds to corticosteroid treatments is Hashimoto’s encephalopathy. Self-reactive antibodies in this syndrome target thyroid peroxidase or thyroglobulin, and are very potent in seizure induction (Castillo et al., 2006; Watemberg et al., 2006).
Detection of Immune Mediators in the Brain of Patients with Epilepsy
Leukocyte accumulation in the perivascular space, and occasionally the parenchyma, of epileptic brain in pediatric as well as adult patients provide another line of evidence that immune cell invasion of the central nervous system (CNS) may be critical in orchestrating epileptogenesis. CD3+ lymphocytes and myeloid-derived macrophages have been detected in resected brain samples of patients diagnosed with temporal lobe epilepsy (TLE; Ravizza et al., 2008). A subset of CD3+ cells, cytotoxic CD8+ T lymphocytes, has also been found in the gray and white matter of epileptic patients with tuberous sclerosis complex (Boer et al., 2008). Brain-infiltrating granulocytes and T cells have been additionally demonstrated in epilepsy of diverse etiologies (Fabene et al., 2008). In line with the above findings, our laboratory and others have detected a myriad of immune-related genes that are upregulated in surgical specimens from patients with intractable TLE using a global survey of changes in gene expression. These immune-related genes include chemokines, complement components, metalloproteinases and their inhibitors, adhesion molecules, immune receptors such as MHC molecules and Fc receptors, and heat shock proteins (Aronica and Gorter, 2007; van Gassen et al., 2008).
Proposed Immune Mechanisms of Epileptogenesis
Understanding the immune mechanisms underlying epileptogenesis provide insights into the development of more effective target-specific immunotherapies rather than general treatments that non-specifically suppress or regulate the immune responses. Ample experimental and clinical evidence has suggested that inflammation in the brain is likely to predispose, precipitate, and perpetuate epileptogenesis, however, protective immune responses that promote neuronal repair and restores homeostasis also exist in epilepsy as well as other immune-mediated neurological disorders, such as multiple sclerosis, Parkinson’s disease, and Alzheimer’s neurodegeneration. Thus, whether the immune response detected during the initiation and development of epilepsy is always deleterious to the survival of brain cells or perhaps may also mediate neuroprotective functions merits further in-depth investigation.
The current dominant view of immune-mediated epileptogenesis entails contributions from both brain-resident cells capable of innate immune responses as well as peripherally derived infiltrating innate and adaptive immune effector cells. The pathological triggering events that are initiated in the brain or the periphery for a variety of reasons, such as simple febrile seizures, trauma, stroke or infection, may lead to an inflammatory cascade. Activation of glia, neurons, and endothelial cells that constitute the BBB most likely result in the release proinflammatory cytokines, such as IL-1β and TNF-α, and danger signals, such as HMGB1.
These factors activate cognate pathways in neurons to cause an intracellular calcium ion surge, which results in modification of voltage-dependent ion channels. Dysregulated ion channels directly enhance the neuronal hyperexcitability and reduce seizure threshold. In addition, proinflammatory cytokines also stimulate chronic release of neuroexcitatory transmitters, inhibit uptake of these neurotransmitters by the glial population, and restrict the recycling of GABA receptors (Hu et al., 2000; Bezzi et al., 2001; Stellwagen et al., 2005; Ferguson et al., 2008). COX-2 and prostaglandin can also be involved in such a process that remodels the neuronal network by mobilizing intracellular calcium storage and an increase in cAMP production.
The inflammatory milieu and the neuronal hypersynochronization in the CNS are often accompanied by BBB leakage, which introduces tightly regulated blood components, such as albumin and potassium ion, into the brain (Seiffert et al., 2004; Oby and Janigro, 2006; Aronica et al., 2007; Ivens et al., 2007; Shlosberg et al., 2010). Increased leukocyte adhesion to the endothelial cells further modifies the BBB through cytoskeletal organization, which results in enhanced leukocyte infiltration into the brain (Greenwood et al., 2002). Upon entering the brain, activated peripheral immune cells are capable of generating free radicals, releasing additional chemokines, cytokines, nitric oxide, and cytotoxic enzymes to establish a self-amplifying cascade to further precipitate epileptogenesis.
Conclusion
Animal models and clinical evidence highlight the involvement of CNS resident and peripherally derived infiltrating immune mediators in seizure induction and epilepsy development (Vezzani et al., 2011a). Robust immune responses in the brain decrease seizure threshold, enhance neuronal excitability, induce BBB failure, promote synaptic reorganization, and regulate epileptogenesis (Figure 1). Despite the appreciation of the critical role of immunity in epileptogenesis and the advancements made in the recent years in understanding the immunological mechanisms underlying epilepsy, novel diagnostic measures and effective therapeutic treatments that targets immunological pathways are still lacking.
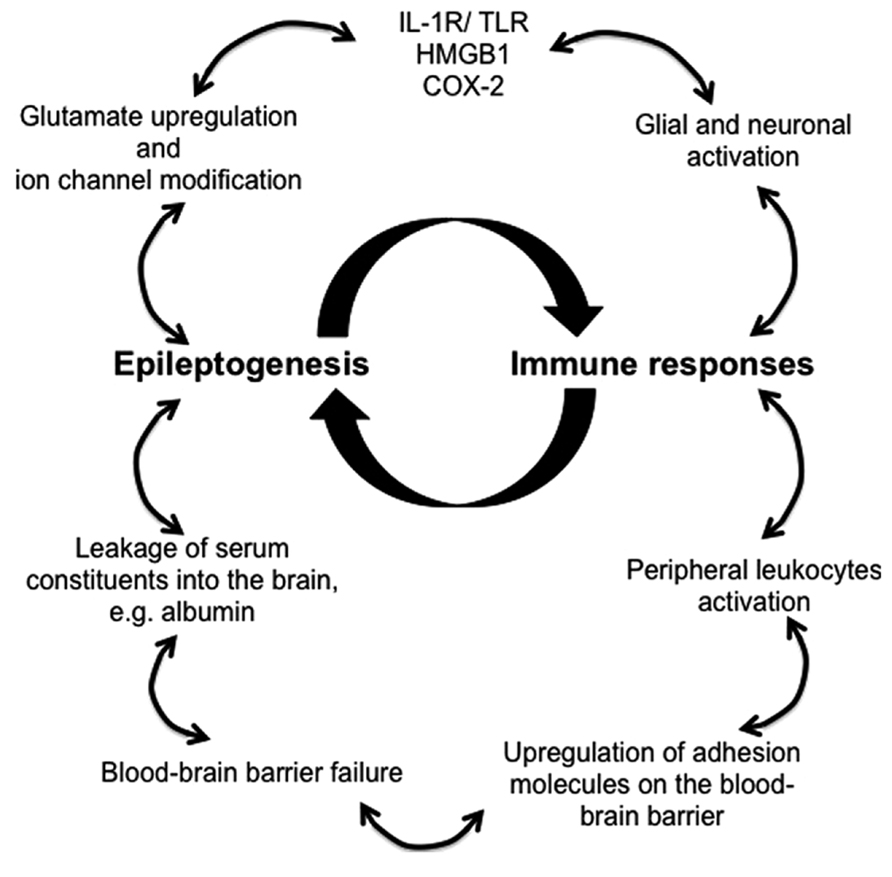
FIGURE 1. Proposed immune mechanisms in epileptogenesis. The relationship between the immune system and the development of epilepsy is non-linear, but rather better represented as an amplifying feedback loop. The immune response predisposes, precipitates and perpetuates epileptogenesis through activation of resident brain cells (glia and neurons) and facilitation of peripheral leukocyte infiltration. These immune mediators release proinflammatory cytokines and chemokines into the brain parenchyma and the blood, thereby activating downstream signaling cascades and compromising blood–brain barrier, which leads to pathophysiological outcomes, reoccurrence of seizures, and ultimately the development of epilepsy.
In addition to, the glial populations and neurons, which are the major brain-resident immune mediators in epilepsy, peripheral leukocytes that infiltrate the brain are also being investigated for their contribution to epileptogenesis as a result of a compromised BBB, for instance, macrophages, monocytes, dendritic cells, αβ T lymphocytes, γδ T lymphocytes, and regulatory cells. Several inflammatory signaling pathways have been identified which initiate immune responses involving the aforementioned immune mediators. Activation of the IL-1R/TLR pathway may be due to brain injury or infection, but could also be caused by DAMPs, such as HMGB1. Furthermore, COX-2 induced production of prostaglandins is capable of triggering brain inflammation (Fabene et al., 2008; Serrano et al., 2011). Therefore, pharmacological blockade of these signaling pathways and inhibitors that antagonizing the main immune mediators have the potential of becoming the next generation of effective anti-epileptic treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by NIH/NINDS R01NS073768 to Sookyong Koh and Stephen D. Miller, as well as a post-doctoral fellowship 262243 from the Epilepsy Foundation of America to Dan Xu.
Abbreviations
ACTH, adrenocorticotrophic hormone; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BBB, blood–brain barrier; CNS, central nervous system; COX-2, cyclooxygenase-2; CSF, cerebrospinal fluid; DAMPs, damage-associated molecular patterns; EEG, electroencephalography; FDA, U.S. food and drug administration; GABA, gamma-aminobutyric acid; GABA-BR, gamma-aminobutyric acid beta receptor; GLuR3, glutamate receptor 3; HMGB1, high mobility group box 1; ICAM-1, intracellular adhesion molecule-1; IFN-γ, interferon-γ; IgG, immunoglobulin G; IL-1β, interleukin-1β; IL-1R/TLR, interleukin-1 receptor/toll-like receptor; IL-6, interleukin-6; IVIG, intravenous gammaglobulin; LPS, lipopolysaccharide; NMDA, N-methyl-D-aspartate; NMDAR, N-methyl D-aspartate receptor; NR2B, NMDA receptor subtype 2B; PAMPs, pathogen-associated molecular patterns; Smad, small mothers against decapentaplegic; TGF-β, transforming growth factor-β; TLE, temporal lobe epilepsy; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1; VGCC, N-type voltage-gated calcium channel; VGKC LGI1, glycine receptor, voltage gated potassium channel leucine-rich glioma-inactivated 1.
References
Allan, S. M., Tyrrell, P. J., and Rothwell, N. J. (2005). Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5, 629–640. doi: 10.1038/nri1664
Armangue, T., Petit-Pedrol, M., and Dalmau, J. (2012). Autoimmune encephalitis in children. J. Child Neurol. 27, 1460–1469. doi: 10.1177/0883073812448838
Armangue, T., Titulaer, M. J., Malaga, I., Bataller, L., Gabilondo, I., Graus, F., et al. (2013). Pediatric anti-N-methyl-D-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J. Pediatr. 162, 850–856. doi: 10.1016/j.jpeds.2012.10.011
Aronica, E., Boer, K., van Vliet, E. A., Redeker, S., Baayen, J. C., Spliet, W. G., et al. (2007). Complement activation in experimental and human temporal lobe epilepsy. Neurobiol. Dis. 26, 497–511. doi: 10.1016/j.nbd.2007.01.015
Aronica, E., and Gorter, J. A. (2007). Gene expression profile in temporal lobe epilepsy. Neuroscientist 13, 100–108. doi: 10.1177/1073858406295832
Baik, E. J., Kim, E. J., Lee, S. H., and Moon, C. (1999). Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res. 843, 118–129. doi: 10.1016/S0006-8993(99)01797-7
Ban, E., Milon, G., Prudhomme, N., Fillion, G., and Haour, F. (1991). Receptors for interleukin-1 (alpha and beta) in mouse brain: mapping and neuronal localization in hippocampus. Neuroscience 43, 21–30. doi: 10.1016/0306-4522(91)90412-H
Banks, W. A., and Erickson, M. A. (2010). The blood–brain barrier and immune function and dysfunction. Neurobiol. Dis. 37, 26–32. doi: 10.1016/j.nbd.2009.07.031
Baram, T. Z., Mitchell, W. G., Tournay, A., Snead, O. C., Hanson, R. A., and Horton, E. J. (1996). High-dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics 97, 375–379.
Bauer, J., Elger, C. E., Hans, V. H., Schramm, J., Urbach, H., Lassmann, H., et al. (2007). Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann. Neurol. 62, 67–80. doi: 10.1002/ana.21148
Bell, M. D., and Perry, V. H. (1995). Adhesion molecule expression on murine cerebral endothelium following the injection of a proinflammagen or during acute neuronal degeneration. J. Neurocytol. 24, 695–710. doi: 10.1007/BF01179819
Bellinger, F. P., Madamba, S., and Siggins, G. R. (1993). Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 628, 227–234. doi: 10.1016/0006-8993(93)90959-Q
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq, E., et al. (2001). CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710. doi: 10.1038/89490
Bien, C. G., Urbach, H., Schramm, J., Soeder, B. M., Becker, A. J., Voltz, R., et al. (2007). Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology 69, 1236–1244. doi: 10.1212/01.wnl.0000276946.08412.ef
Boer, K., Jansen, F., Nellist, M., Redeker, S., van den Ouweland, A. M., Spliet, W. G., et al. (2008). Aronica, Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res. 78, 7–21. doi: 10.1016/j.eplepsyres.2007.10.002
Campbell, I. L., Abraham, C. R., Masliah, E., Kemper, P., Inglis, J. D., Oldstone, M. B., et al. (1993). Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. U.S.A. 90, 10061–10065. doi: 10.1073/pnas.90.21.10061
Castillo, P., Woodruff, B., Caselli, R., Vernino, S., Lucchinetti, C., Swanson, J., et al. (2006). Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch. Neurol. 63, 197–202. doi: 10.1001/archneur.63.2.197
Chakravarty, S., and Herkenham, M. (2005). Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J. Neurosci. 25, 1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005
Chiu, S. S., Tse, C. Y., Lau, Y. L., and Peiris, M. (2001). Influenza A infection is an important cause of febrile seizures. Pediatrics 108, E63. doi: 10.1542/peds.108.4.e63
Choi, J., Nordli, D. R. Jr., Alden, T. D., DiPatri, A. Jr., Laux, L., Kelley, K., et al. (2009). Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J. Neuroinflammation 6, 38. doi: 10.1186/1742-2094-6-38
Cutler, R. W., Watters, G. V., and Hammerstad, J. P. (1970). The origin and turnover rates of cerebrospinal fluid albumin and gamma-globulin in man. J. Neurol. Sci. 10, 259–268. doi: 10.1016/0022-510X(70)90154-1
Dalakas, M. C. (1998). Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology 51, S2–S8. doi: 10.1212/WNL.51.6_Suppl_5.S2
Dalmau, J. (2009). Status epilepticus due to paraneoplastic and nonparaneoplastic encephalitides. Epilepsia 50(Suppl. 12), S58–S60. doi: 10.1111/j.1528-1167.2009.02352.x
Dalmau, J., Gleichman, A. J., Hughes, E. G., Rossi, J. E., Peng, X., Lai, M., et al. (2008). Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 7, 1091–1098. doi: 10.1016/S1474-4422(08)70224-2
Dalmau, J., and Rosenfeld, M. R. (2008). Paraneoplastic syndromes of the CNS. Lancet Neurol. 7, 327–340. doi: 10.1016/S1474-4422(08)70060-7
De Simoni, M. G., Perego, C., Ravizza, T., Moneta, D., Conti, M., Marchesi, F., et al. (2000). Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 12, 2623–2633. doi: 10.1046/j.1460-9568.2000.00140.x
Devinsky, O. (2004). Effects of seizures on autonomic and cardiovascular function. Epilepsy Curr. 4, 43–46. doi: 10.1111/j.1535-7597.2004.42001.x
Dhote, F., Peinnequin, A., Carpentier, P., Baille, V., Delacour, C., Foquin, A., et al. (2007). Prolonged inflammatory gene response following soman-induced seizures in mice. Toxicology 238, 166–176. doi: 10.1016/j.tox.2007.05.032
Donat, J. F. (1992). The age-dependent epileptic encephalopathies. J. Child Neurol. 7, 7–21. doi: 10.1177/088307389200700102
Dube, C., Vezzani, A., Behrens, M., Bartfai, T., and Baram, T. Z. (2005). Interleukin-1beta contributes to the generation of experimental febrile seizures. Ann. Neurol. 57, 152–155. doi: 10.1002/ana.20358
Eibl, M. M., and Wedgwood, R. J. (1989). Intravenous immunoglobulin: a review. Immunodefic. Rev. 1(Suppl.), S1–S42.
Engel, J. Jr. (2001). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 42, 796–803. doi: 10.1046/j.1528-1157.2001.10401.x
Ericsson, A., Liu, C., Hart, R. P., and Sawchenko, P. E. (1995). Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J. Comp. Neurol. 361, 681–698. doi: 10.1002/cne.903610410
Eriksson, C., Tehranian, R., Iverfeldt, K., Winblad, B., and Schultzberg, M. (2000). Increased expression of mRNA encoding interleukin-1beta and caspase-1, and the secreted isoform of interleukin-1 receptor antagonist in the rat brain following systemic kainic acid administration. J. Neurosci. Res. 60, 266–279. doi: 10.1002/(SICI)1097-4547(20000415)60:2<266::AID-JNR16>3.0.CO;2-P
Fabene, P. F., Laudanna, C., and Constantin, G. (2013). Leukocyte trafficking mechanisms in epilepsy. Mol. Immunol. 55, 100–104. doi: 10.1016/j.molimm.2012.12.009
Fabene, P. F., Navarro Mora, G., Martinello, M., Rossi, B., Merigo, F., Ottoboni, L., et al. (2008). A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 14, 1377–1383. doi: 10.1038/nm.1878
Ferguson, A. R., Christensen, R. N., Gensel, J. C., Miller, B. A., Sun, F., Beattie, E. C., et al. (2008). Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J. Neurosci. 28, 11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008
Friedman, A., Kaufer, D., and Heinemann, U. (2009). Blood–brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 85, 142–149. doi: 10.1016/j.eplepsyres.2009.03.005
Gorter, J. A., van Vliet, E. A., Aronica, E., Breit, T., Rauwerda, H., and Lopes da Silva, F. H. (2006). Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J. Neurosci. 26, 11083–11110. doi: 10.1523/JNEUROSCI.2766-06.2006
Greenwood, J., Etienne-Manneville, S., Adamson, P., and Couraud, P. O. (2002). Lymphocyte migration into the central nervous system: implication of ICAM-1 signalling at the blood–brain barrier. Vascul. Pharmacol. 38, 315–322. doi: 10.1016/S1537-1891(02)00199-4
Hall, C. B., Long, C. E., Schnabel, K. C., Caserta, M. T., McIntyre, K. M., Costanzo, M. A., et al. (1994). Human herpesvirus-6 infection in children. A prospective study of complications and reactivation. N. Engl. J. Med. 331, 432–438. doi: 10.1056/NEJM199408183310703
Hart, Y. M., Cortez, M., Andermann, F., Hwang, P., Fish, D. R., Dulac, O., et al. (1994). Medical treatment of Rasmussen’s syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology 44, 1030–1036. doi: 10.1212/WNL.44.6.1030
He, X. P., Patel, M., Whitney, K. D., Janumpalli, S., Tenner, A., and McNamara, J. O. (1998). Glutamate receptor GluR3 antibodies and death of cortical cells. Neuron 20, 153–163. doi: 10.1016/S0896-6273(00)80443-2
Holtman, L., van Vliet, E. A., Edelbroek, P. M., Aronica, E., and Gorter, J. A. (2010). Cox-2 inhibition can lead to adverse effects in a rat model for temporal lobe epilepsy. Epilepsy Res. 91, 49–56. doi: 10.1016/j.eplepsyres.2010.06.011
Hrachovy, R. A., Frost, J. D. Jr., Kellaway, P., and Zion, T. E. (1983). Double-blind study of ACTH vs prednisone therapy in infantile spasms. J. Pediatr. 103, 641–645. doi: 10.1016/S0022-3476(83)80606-4
Hu, S., Sheng, W. S., Ehrlich, L. C., Peterson, P. K., and Chao, C. C. (2000). Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7, 153–159. doi: 10.1159/000026433
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., et al. (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547. doi: 10.1093/brain/awl317
Jones, J. E., Austin, J. K., Caplan, R., Dunn, D., Plioplys, S., and Salpekar, J. A. (2008). Psychiatric disorders in children and adolescents who have epilepsy. Pediatr. Rev. 29, e9–e14. doi: 10.1542/pir.29-2-e9
Jung, K. H., Chu, K., Lee, S. T., Kim, J., Sinn, D. I., Kim, J. M., et al. (2006). Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol. Dis. 23, 237–246. doi: 10.1016/j.nbd.2006.02.016
Kanemoto, K., Kawasaki, J., Yuasa, S., Kumaki, T., Tomohiro, O., Kaji, R., et al. (2003). Increased frequency of interleukin-1beta-511T allele in patients with temporal lobe epilepsy, hippocampal sclerosis, and prolonged febrile convulsion. Epilepsia 44, 796–799. doi: 10.1046/j.1528-1157.2003.43302.x
Kawada, J., Kimura, H., Ito, Y., Hara, S., Iriyama, M., Yoshikawa, T., et al. (2003). Systemic cytokine responses in patients with influenza-associated encephalopathy. J. Infect. Dis. 188, 690–698. doi: 10.1086/377101
Kivity, S., Lerman, P., Ariel, R., Danziger, Y., Mimouni, M., and Shinnar, S. (2004). Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia 45, 255–262. doi: 10.1111/j.0013-9580.2004.30503.x
Kondo, K., Nagafuji, H., Hata, A., Tomomori, C., and Yamanishi, K. (1993). Association of human herpesvirus 6 infection of the central nervous system with recurrence of febrile convulsions. J. Infect. Dis. 167, 1197–1200. doi: 10.1093/infdis/167.5.1197
Kulkarni, S. K., and Dhir, A. (2009). Cyclooxygenase in epilepsy: from perception to application. Drugs Today (Barc.) 45, 135–154. doi: 10.1358/dot.2009.45.2.1322481
Lee, B., Dziema, H., Lee, K. H., Choi, Y. S., and Obrietan, K. (2007). CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiol. Dis. 25, 80–91. doi: 10.1016/j.nbd.2006.08.015
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. doi: 10.1038/nri2156
Librizzi, L., Ravizza, T., Vezzani, A., and de Curtis, M. (2010). Expression of IL-1 beta induced by epileptiform activity in the isolated guinea pig brain in vitro. Epilepsia 51(Suppl. 4), 18.
Librizzi, L., Regondi, M. C., Pastori, C., Frigerio, S., Frassoni, C., and de Curtis, M. (2007). Expression of adhesion factors induced by epileptiform activity in the endothelium of the isolated guinea pig brain in vitro. Epilepsia 48, 743–751. doi: 10.1111/j.1528-1167.2007.01047.x
Mackay, M. T., Weiss, S. K., Adams-Webber, T., Ashwal, S., Stephens, D., Ballaban-Gill, K., et al. (2004). Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology 62, 1668–1681. doi: 10.1212/01.WNL.0000127773.72699.C8
Mantegazza, R., Bernasconi, P., Baggi, F., Spreafico, R., Ragona, F., Antozzi, C., et al. (2002). Antibodies against GluR3 peptides are not specific for Rasmussen’s encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J. Neuroimmunol. 131, 179–185. doi: 10.1016/S0165-5728(02)00261-8
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., et al. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419. doi: 10.1038/nm.2127
Masuyama, T., Matsuo, M., Ichimaru, T., Ishii, K., Tsuchiya, K., and Hamasaki, Y. (2002). Possible contribution of interferon-alpha to febrile seizures in influenza. Pediatr. Neurol. 27, 289–292. doi: 10.1016/S0887-8994(02)00452-6
Millichap, J. G. (1959). Studies in febrile seizures. I. Height of body temperature as a measure of the febrile-seizure threshold. Pediatrics 23, 76–85.
Minami, M., Kuraishi, Y., and Satoh, M. (1991). Effects of kainic acid on messenger RNA levels of IL-1 beta, IL-6, TNF alpha and LIF in the rat brain. Biochem. Biophys. Res. Commun. 176, 593–598. doi: 10.1016/S0006-291X(05)80225-6
Oby, E., and Janigro, D. (2006). The blood–brain barrier and epilepsy. Epilepsia 47, 1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x
Pellock, J. M. (2004). Understanding co-morbidities affecting children with epilepsy. Neurology 62, S17–S23. doi: 10.1212/WNL.62.5_suppl_2.S17
Peltier, D. C., Simms, A., Farmer, J. R., and Miller, D. J. (2010). Human neuronal cells possess functional cytoplasmic and TLR-mediated innate immune pathways influenced by phosphatidylinositol-3 kinase signaling. J. Immunol. 184, 7010–7021. doi: 10.4049/jimmunol.0904133
Plata-Salaman, C. R., and Ffrench-Mullen, J. M. (1994). Interleukin-1 beta inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur. J. Pharmacol. 266, 1–10. doi: 10.1016/0922-4106(94)90202-X
Polascheck, N., Bankstahl, M., and Loscher, W. (2010). The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 224, 219–233. doi: 10.1016/j.expneurol.2010.03.014
Probert, L., Akassoglou, K., Kassiotis, G., Pasparakis, M., Alexopoulou, L., and Kollias, G. (1997). TNF-alpha transgenic and knockout models of CNS inflammation and degeneration. J. Neuroimmunol. 72, 137–141. doi: 10.1016/S0165-5728(96)00184-1
Ransohoff, R. M., Kivisakk, P., and Kidd, G. (2003). Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 3, 569–581. doi: 10.1038/nri1130
Ravizza, T., Gagliardi, B., Noe, F., Boer, K., Aronica, E., and Vezzani, A. (2008). Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 29, 142–160. doi: 10.1016/j.nbd.2007.08.012
Ravizza, T., and Vezzani, A. (2006). Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 137, 301–308. doi: 10.1016/j.neuroscience.2005.07.063
Riazi, K., Galic, M. A., and Pittman, Q. J. (2010). Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 89, 34–42. doi: 10.1016/j.eplepsyres.2009.09.004
Rodgers, K. M., Hutchinson, M. R., Northcutt, A., Maier, S. F., Watkins, L. R., and Barth, D. S. (2009). The cortical innate immune response increases local neuronal excitability leading to seizures. Brain 132, 2478–2486. doi: 10.1093/brain/awp177
Rogers, S. W., Andrews, P. I., Gahring, L. C., Whisenand, T., Cauley, K., Crain, B., et al. (1994). Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 265, 648–651. doi: 10.1126/science.8036512
Rozovsky, I., Morgan, T. E., Willoughby, D. A., Dugichi-Djordjevich, M. M., Pasinetti, G. M., Johnson, S. A., et al. (1994). Selective expression of clusterin (SGP-2) and complement C1qB and C4 during responses to neurotoxins in vivo and in vitro. Neuroscience 62, 741–758. doi: 10.1016/0306-4522(94)90473-1
Schafers, M., and Sorkin, L. (2008). Effect of cytokines on neuronal excitability. Neurosci. Lett. 437, 188–193. doi: 10.1016/j.neulet.2008.03.052
Schneider, H., Pitossi, F., Balschun, D., Wagner, A., del Rey, A., and Besedovsky, H. O. (1998). A neuromodulatory role of interleukin-1beta in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 95, 7778–7783. doi: 10.1073/pnas.95.13.7778
Seiffert, E., Dreier, J. P., Ivens, S., Bechmann, I., Tomkins, O., Heinemann, U., et al. (2004). Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci. 24, 7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004
Sekul, E. A., Cupler, E. J., and Dalakas, M. C. (1994). Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy: frequency and risk factors. Ann. Intern. Med. 121, 259–262. doi: 10.7326/0003-4819-121-4-199408150-00004
Serrano, G. E., Lelutiu, N., Rojas, A., Cochi, S., Shaw, R., Makinson, C. D., et al. (2011). Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J. Neurosci. 31, 14850–14860. doi: 10.1523/JNEUROSCI.3922-11.2011
Shlosberg, D., Benifla, M., Kaufer, D., and Friedman, A. (2010). Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 6, 393–403. doi: 10.1038/nrneurol.2010.74
Snead, O. C. III, Benton, J. W., and Myers, G. J. (1983). ACTH and prednisone in childhood seizure disorders. Neurology 33, 966–970. doi: 10.1212/WNL.33.8.966
Sotgiu, S., Murrighile, M. R., and Constantin, G. (2010). Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report. BMC Neurol. 10:84. doi: 10.1186/1471-2377-10-84
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Takao, T., Tracey, D. E., Mitchell, W. M., and De Souza, E. B. (1990). Interleukin-1 receptors in mouse brain: characterization and neuronal localization. Endocrinology 127, 3070–3078. doi: 10.1210/endo-127-6-3070
Toscano, C. D., Kingsley, P. J., Marnett, L. J., and Bosetti, F. (2008). NMDA-induced seizure intensity is enhanced in COX-2 deficient mice. Neurotoxicology 29, 1114–1120. doi: 10.1016/j.neuro.2008.08.008
Turrin, N. P., and Rivest, S. (2004). Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol. Dis. 16, 321–334. doi: 10.1016/j.nbd.2004.03.010
van Gassen, K. L., de Wit, M., Koerkamp, M. J., Rensen, M. G., van Rijen, P. C., Holstege, F. C., et al. (2008). Possible role of the innate immunity in temporal lobe epilepsy. Epilepsia 49, 1055–1065. doi: 10.1111/j.1528-1167.2007.01470.x
van Rijckevorsel-Harmant, K., Delire, M., Schmitz-Moorman, W., and Wieser, H. G. (1994). Treatment of refractory epilepsy with intravenous immunoglobulins. Results of the first double-blind/dose finding clinical study. Int. J. Clin. Lab. Res. 24, 162–126. doi: 10.1007/BF02592447
van Vliet, E. A., da Costa Araujo, S., Redeker, S., van Schaik, R., Aronica, E., and Gorter, J. A. (2007). Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130, 521–534. doi: 10.1093/brain/awl318
Vezzani, A., and Baram, T. Z. (2007). New roles for interleukin-1 beta in the mechanisms of epilepsy. Epilepsy Curr. 7, 45–50. doi: 10.1111/j.1535-7511.2007.00165.x
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011a). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Vezzani, A., Maroso, M., Balosso, S., Sanchez, M. A., and Bartfai, T. (2011b). IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 25, 1281–1289. doi: 10.1016/j.bbi.2011.03.018
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x
Vezzani, A., Moneta, D., Conti, M., Richichi, C., Ravizza, T., De Luigi, A., et al. (2000). Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 11534–11539. doi: 10.1073/pnas.190206797
Vianello, M., Bisson, G., Dal Maschio, M., Vassanelli, S., Girardi, S., Mucignat, C., et al. (2008). Increased spontaneous activity of a network of hippocampal neurons in culture caused by suppression of inhibitory potentials mediated by anti-gad antibodies. Autoimmunity 41, 66–73. doi: 10.1080/08916930701619565
Vincent, A., and Bien, C. G. (2008). Anti-NMDA-receptor encephalitis: a cause of psychiatric, seizure, and movement disorders in young adults. Lancet Neurol. 7, 1074–1075. doi: 10.1016/S1474-4422(08)70225-4
Vincent, A., Irani, S. R., and Lang, B. (2010). The growing recognition of immunotherapy-responsive seizure disorders with autoantibodies to specific neuronal proteins. Curr. Opin. Neurol. 23, 144–150. doi: 10.1097/WCO.0b013e32833735fe
Virta, M., Hurme, M., and Helminen, M. (2002). Increased frequency of interleukin-1beta (-511) allele 2 in febrile seizures. Pediatr. Neurol. 26, 192–195. doi: 10.1016/S0887-8994(01)00380-0
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700.
Viviani, B., Gardoni, F., and Marinovich, M. (2007). Cytokines and neuronal ion channels in health and disease. Int. Rev. Neurobiol. 82, 247–263. doi: 10.1016/S0074-7742(07)82013-7
Voutsinos-Porche, B., Koning, E., Kaplan, H., Ferrandon, A., Guenounou, M., Nehlig, A., et al. (2004). Temporal patterns of the cerebral inflammatory response in the rat lithium-pilocarpine model of temporal lobe epilepsy. Neurobiol. Dis. 17, 385–402. doi: 10.1016/j.nbd.2004.07.023
Wang, S., Cheng, Q., Malik, S., and Yang, J. (2000). Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J. Pharmacol. Exp. Ther. 292, 497–504.
Watemberg, N., Greenstein, D., and Levine, A. (2006). Encephalopathy associated with Hashimoto thyroiditis: pediatric perspective. J. Child Neurol. 21, 1–5. doi: 10.1177/08830738060210010201
Watson, R., Jiang, Y., Bermudez, I., Houlihan, L., Clover, L., McKnight, K., et al. (2004). Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology 63, 43–50. doi: 10.1212/01.WNL.0000132651.66689.0F
Wetherington, J., Serrano, G., and Dingledine, R. (2008). Astrocytes in the epileptic brain. Neuron 58, 168–178. doi: 10.1016/j.neuron.2008.04.002
Yamagata, K., Andreasson, K. I., Kaufmann, W. E., Barnes, C. A., and Worley, P. F. (1993). Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron 11, 371–386. doi: 10.1016/0896-6273(93)90192-T
Yamatogi, Y., Ohtsuka, Y., Ishida, T., Ichiba, N., Ishida, S., Miyake, S., et al. (1979). Treatment of the Lennox syndrome with ACTH: a clinical and electroencephalographic study. Brain Dev. 1, 267–276. doi: 10.1016/S0387-7604(79)80041-8
Yoshikawa, K., Kita, Y., Kishimoto, K., and Shimizu, T. (2006). Profiling of eicosanoid production in the rat hippocampus during kainic acid-induced seizure: dual phase regulation and differential involvement of COX-1 and COX-2. J. Biol. Chem. 281, 14663–14669. doi: 10.1074/jbc.M511089200
Zeise, M. L., Espinoza, J., Morales, P., and Nalli, A. (1997). Interleukin-1beta does not increase synaptic inhibition in hippocampal CA3 pyramidal and dentate gyrus granule cells of the rat in vitro. Brain Res. 768, 341–344. doi: 10.1016/S0006-8993(97)00787-7
Keywords: seizure, epilepsy, epileptogenesis, immune response, inflammation, microglia, astrocytes, T lymphocytes
Citation: Xu D, Miller SD and Koh S (2013) Immune mechanisms in epileptogenesis. Front. Cell. Neurosci. 7:195. doi: 10.3389/fncel.2013.00195
Received: 11 June 2013; Accepted: 08 October 2013;
Published online: 08 November 2013.
Edited by:
Roberto Di Maio, University of Pittsburgh, USAReviewed by:
Michela Matteoli, University of Milano, ItalyRoberto Di Maio, University of Pittsburgh, USA
Copyright © 2013 Xu, Miller and Koh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sookyong Koh, Department of Pediatrics, Division of Neurobiology, Children’s Research Center, Lurie Children’s Hospital of Chicago, 225 E. Chicago Avenue, Epilepsy Box 29, Chicago, IL 60611, USA e-mail:c2tvaEBsdXJpZWNoaWxkcmVucy5vcmc=;
Stephen D. Miller, Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, 303 E. Chicago Avenue, Chicago, IL 60611, USA e-mail:cy1kLW1pbGxlckBub3J0aHdlc3Rlcm4uZWR1