 Jean-Luc Gaiarsa1,2,3*
Jean-Luc Gaiarsa1,2,3* Christophe Porcher1,2,3
Christophe Porcher1,2,3- 1Institut National de la Santé et de la Recherche Médicale U-901, Marseille, France
- 2Aix-Marseille Université, UMR S901, Marseille, France
- 3Institut de Neurobiologie de la Méditerranée, Marseille, France
The proper development of highly organized structures in the central nervous system is a complex process during which key events – neurogenesis, migration, growth, differentiation, and synaptogenesis – have to take place in an appropriate manner to create functional neuronal networks. It is now well established that GABA, the main inhibitory neurotransmitter in the adult mammalian brain, plays more than a classical inhibitory role and can function as an important developmental signal early in life. GABA binds to chloride-permeable ionotropic GABAA receptors and to G-protein-coupled GABAB receptors (GABAB-Rs). Although most of the trophic actions of GABA have been attributed to the activation of GABAA receptors, recent advances show that GABAB-Rs also regulate fundamental steps of network development. This review summarizes some of the recent progress about the neurotrophic role of GABAB-Rs to neuronal development.
Introduction
Gamma-aminobutyric acid (GABA) is the main inhibitory transmitter in the adult mammalian brain. GABAergic interneurons regulate neuronal excitability, synaptic integration, and the dynamic of network oscillations and as such are important for many cognitive functions. Recent advances, however, indicate that GABA can act beyond its classical role in synaptic communication and may function as an important developmental signal, being able to modulate nearly all key steps of neuronal network formation including cell survival and migration, neuritic growth and guidance, synapse formation and maturation (Owens and Kriegstein, 2002; Ben-Ari et al., 2007; Sernagor et al., 2010).
Most of the trophic actions of GABA have been attributed to the activation of the ionotropic, chloride permeable, GABAA receptors. Indeed, early in development, GABAA receptors activation induces a membrane depolarization and an increase in intracellular calcium concentration (Owens and Kriegstein, 2002; Ben-Ari et al., 2007). The depolarizing to hyperpolarizing shift of GABAA receptor-mediated response results from a developmental decrease in intracellular chloride concentration brought about by the increased contribution of the potassium/chloride co-transporter, KCC2, which extrudes chloride from the cells (Rivera et al., 1999). The role of GABAA receptors in neuronal development has been highlighted in studies in which the depolarizing/hyperpolarizing conversion of GABA responses is modified in vitro (Chudotvorova et al., 2005) and in vivo (Ge et al., 2006; Cancedda et al., 2007; Reynolds et al., 2008; Wang and Kriegstein, 2008). However, GABA also activates metabotropic GABAB receptors (GABAB-Rs) and accumulating evidence indicate that these receptors may promote cell migration, differentiation, and synaptogenesis. The goal of this review is to recapitulate the current knowledge about the overlooked role of GABAB-Rs in neuronal development.
A Short Introduction to the Basic Properties of GABAB-R Signaling
GABAB receptors are metabotropic receptors coupled to Gαi/o-guanosine triphosphate (GTP) type protein which inhibits adenylate cyclase and gates ion channels (Bowery, 1993; Bowery et al., 2002). Released GABA can feed back onto GABAB auto-receptors located on GABAergic terminals, and/or spillover to activate hetero-synaptic GABAB-Rs on neighboring glutamatergic terminals. Activation of the pre-synaptic GABAB-Rs inhibits neurotransmitter release through multiple targets including inactivation of voltage-dependent calcium channels (Mintz and Bean, 1993), gating of potassium conductance to shunt pre-synaptic action potentials (Thompson and Gahwiler, 1992a), reduction of vesicle priming (Sakaba and Neher, 2003), or interaction with the exocytosis machinery (Blackmer et al., 2005). Released GABA also signals onto post-synaptic GABAB-Rs located on dendritic shaft and spines (Kulik et al., 2003). Activation of these receptors generates slow (100-150 ms) inhibitory potentials via the opening of G-protein activated-inward rectifying potassium channels (G-protein-regulated inwardly rectifying K+ channels, GIRKs also known as inwardly rectifying potassium, Kir3 channels; Gähwiler and Brown, 1985).
The cloning of GABAB-Rs in the late 1990s has led to the identification of two GABAB gene products: the GABAB1 and GABAB2 subunits (Kaupmann et al., 1997). Recombinant experiments showed that heterodimerization of GABAB1 and GABAB2 subunits is mandatory for cell surface expression and coupling to G-protein (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998). Coiled–coil interactions in the C-terminal domain of the newly synthesized subunits in the endoplasmic reticulum masks a retention signal present on the C-terminal domain of the GABAB1 subunit so that only GABAB1 subunit assembled with GABAB2 subunit are trafficked to the cell surface. GABAB1/GABAB2 subunits assembly is also mandatory for agonist-induced signaling. In the heterodimeric GABAB-Rs, GABAB1 subunit is responsible for binding of GABA, whereas the GABAB2 subunit is necessary for G-protein coupling (Robbins et al., 2001). Transgenic mice lacking the GABAB1 subunit confirm that heterodimeric assembly is required to provide fully functional receptors in vivo since GABAB1-/- mice do not exhibit detectable electrophysiological, biochemical, or behavioral responses to GABAB-R agonists (Prosser et al., 2001; Schuler et al., 2001; Queva et al., 2003). Deletion of the GABAB2 subunit also abolished all known response to GABAB-R agonists (Gassmann et al., 2004). The GABAB2-/- mice, however, exhibit an atypical baclofen response, namely an inhibition of potassium channels, which is not observed in wild type (WT) mice (Gassmann et al., 2004). Thus GABAB1 subunits could assemble into functional receptor but such homomeric assembly may be a consequence of the knockout of the GABAB2 subunit (Gassmann et al., 2004).
The GABAB1 subunit further exists under two isoforms, named GABAB1a and GABAB1b, which differ by a pair of sushi domains on the N-terminal of the GABAB1a subunit (Kaupmann et al., 1997; Biermann et al., 2010). The two isoforms have similar pharmacological and physiological properties in heterologous expression systems precluding determination of the functional significance of this molecular diversity. The demonstration that the GABAB1a and GABAB1b isoforms contribute to distinct native GABAB-Rs and convey different functions was made possible by the generation of mice deficient in GABAB1a or GABAB1b isoform. Using this knocking down approach, it was shown that the GABAB1a isoform is preferentially targeted to the pre-synaptic glutamatergic terminals and assemble with GABAB2 subunit to form hetero-receptors whereas both GABAB1a and GABAB1b isoforms assemble with the GABAB2 subunit into auto-receptors at pre-synaptic GABAergic terminals (Vigot et al., 2006; Guetg et al., 2009). On the post-synaptic side, although both isoforms are present, GABAB1b isoform provides the majority of coupling with GIRK (Vigot et al., 2006) and inhibition of dendritic calcium spikes (Perez-Garci et al., 2006).
Ontogeny of GABAB Receptor-Mediated Responses
Information regarding the distribution pattern and subcellular distribution of the GABAB receptor subunits is crucial to gain insight into the contribution of these receptors to brain development. Several evidences indicate that both GABAB1 and GABAB2 subunits are expressed in the developing brain although some temporal and spatial differences exist, suggesting that the regulatory mechanisms for these subunits may differ. At the embryonic day (E) 11, GABAB1 transcripts are detected in the rat brain whereas the GABAB2 transcripts are below the level for detection (Kim et al., 2003; Lopez-Bendito et al., 2004; Martin et al., 2004). At E14, both transcripts are detected in most brain regions except in the olfactory bulb and striatum where GABAB2 transcripts remain barely detectable until E17 (Martin et al., 2004). At the protein levels, in a pioneer study, Turgeon and Albin (1994) performed quantitative receptor autoradiography with (3H)GABA to study the ontogeny of GABAB binding sites in the rat brain. They reported that GABAB binding levels is detected at early postnatal stages and peak at regionally specific times during the three first postnatal week of life. Western blot and immuno-histochemical analysis confirm that GABAB1 and GABAB2 subunits are present in the developing brain (Behar et al., 2001; Lopez-Bendito et al., 2002, 2004; Martin et al., 2004; Bianchi et al., 2005; Lujan and Shigemoto, 2006). These studies further show distinct expression patterns between GABAB1a and GABAB1b subunits (Fritschy et al., 1999; Fritschy et al., 2004). The GABAB1a subunit is highest during the first postnatal week of life, whereas GABAB1b subunit progressively increases to reach its maximum by P10 (Fritschy et al., 1999). Both subunits reach adult levels by the end of the third postnatal week of life. The expression of the GABAB1 and GABAB2 subunits overlaps in many regions but some laminar and cellular distinctions are found. For instance at birth, the GABAB1 labeling is uniform across all neocortical layers, while the GABAB2 labeling is most intense in the layers I and V–VI (Fritschy et al., 2004). Similarly, in the hippocampus, GABAB1 expression predominates in the pyramidal layer while GABAB2 is mostly expressed in the dendritic layers (Fritschy et al., 2004). In the gerbil medial superior olive (MSO), the GABAB1 expression changes from a predominantly dendritic to a somatic location during development (Hassfurth et al., 2010). Double immuno-labeling indicate that the GABAB1 and GABAB2 subunits co-localize in neurons of the marginal zone and subplate, while tangentially migrating neurons in the intermediate zone (iz) and Cajal–Retzus cells in the layer I only express the GABAB1 subunit (Lopez-Bendito et al., 2002). At the electron microscopic level, the GABAB1 and GABAB2 subunits are found at both pre- and post-synaptic levels in the developing cerebellum (Lujan and Shigemoto, 2006), neocortex (Lopez-Bendito et al., 2002), and hippocampus (Lopez-Bendito et al., 2004) at early postnatal development stages. At the post-synaptic levels, both subunits are present on dendritic shaft and spines at extra-synaptic and peri-synaptic sites. Of note is the transient expression of GABAB1 and GABAB2 subunits on glial cells in the hippocampus (Lopez-Bendito et al., 2004) and cerebellum (Lujan and Shigemoto, 2006). Activation of these receptors induce calcium transient in astrocytes in newborn rat hippocampal slices (Meier et al., 2008) that may in turn impact the development of neuronal networks.
Studies investigating the ontogeny of GABAB-R mediated responses further indicate that GABAB-Rs are present and functional early in development albeit some regional distinctions in the signaling pathway activated. Post-synaptic GABAB-mediated opening of GIRK to baclofen applications are not recorded at birth in the hippocampus (Gaïarsa et al., 1995; Caillard et al., 1998; Nurse and Lacaille, 1999; Verheugen et al., 1999) and neocortex (Luhmann and Prince, 1991; Fukuda et al., 1993; Kirmse and Kirischuk, 2006), while pre-synaptic GABAB-R mediated inhibition is already functional and controls both GABA and glutamate release (Luhmann and Prince, 1991; Fukuda et al., 1993; McLean et al., 1996; Caillard et al., 1998). Post-synaptic GABAB-mediated responses increase until the middle of the second postnatal week and remain stable thereafter (Luhmann and Prince, 1991; Fukuda et al., 1993; Gaïarsa et al., 1995; Kirmse and Kirischuk, 2006). Hippocampal neuronal cultures from E18 rat embryos also lack of post-synaptic GABAB-R mediated currents until 11 days in vitro (Ehrengruber et al., 1997) although they express both GABAB1 and GABAB2 subunits (Martin et al., 2004). When transfected to overexpress GIRK, a typical GABAB-R mediated potassium outward current could be induced in these neurons (Ehrengruber et al., 1997), suggesting that the availability of the GIRK channels is the likely limiting factor for the appearance of functional post-synaptic GABAB-R mediated inhibition (but see Correa et al., 2004).
Other evidence confirm that the post-synaptic GABAB-Rs are present and interact with G-proteins at early developmental stages. For instance, GABAB-R agonists decrease forskolin-induced cyclic-adenosine monophosphate (cAMP) levels in E18 neocortical neuronal cultures (Martin et al., 2004; Bony et al., 2013) and increase intracellular calcium concentration in some dissociated embryonic cortical neurons (Behar et al., 1996). In the developing rat hippocampus, baclofen induces a rapid increase of protein kinase C (PKC) activity (Tremblay et al., 1995). Patch-clamp recordings performed on developing chick retina have shown that GABAB-Rs failed to activate potassium currents, while they inhibit calcium channels (Catsicas and Mobbs, 2001). Similarly, in the developing rat hypothalamus baclofen depresses the post-synaptic calcium rise induced by glutamate- and GABAA-R agonists (Obrietan and Van den Pol, 1998, 1999). A completely different situation has been observed in the gerbil MSO where GIRK currents activated by post-synaptic GABAB-Rs disappear during development (Hassfurth et al., 2010). How this developmental change in GABAB-R mediated responses is regulated in presently unknown.
Altogether, these observations show that post-synaptic GABAB-Rs are present and functional early in development, although they are not yet involved in the control of cell excitability via the opening of post-synaptic GIRK in most developing brain regions. At that stage, they are, however, coupled to signaling pathways that may instruct or modulate neuronal development.
The Conditions for the Activation of GABAB-RS are Gathered in the Developing Brain
To contribute to neuronal development, GABAB-Rs must be activated by ambient GABA. Early in development, GABA is released from growth cones (Taylor and Gordon-Weeks, 1991; Gao and Van den Pol, 2000) or in paracrine non-vesicular manner (Demarque et al., 2002) providing an endogenous and local source of GABA. Using a microchemotaxis assay, Behar et al. (2001) have shown that E18 cortical plate (cp) neurons release GABA and taurine, creating a gradient that direct cell migration via the GABAA-Rs and GABAB-Rs. When GABAergic synapses are established, released GABA must diffuse to activate extra-synaptic and/or peri-synaptic GABAB-Rs. Consequently, GABAB-R mediated responses are induced when a population of interneurons are synchronously activated or facilitated when the GABA transporters are blocked (Dutar and Nicoll, 1988; Thompson and Gahwiler, 1992b; Isaacson and Nicoll, 1993; Scanziani, 2000), both procedures promoting GABA spillover. One characteristic feature of developing neuronal networks is the presence of a primitive network-driven synaptic activity both in vivo and in vitro (reviewed in Khazipov and Luhmann, 2006; Ben-Ari et al., 2007). During this patterned activity, GABAergic interneurons fired synchronously (Khazipov et al., 1997; Khazipov and Luhmann, 2006), a condition that allows diffusion of GABA into the extracellular space. This diffusion is facilitated by the presence of a large extracellular space and a relative immaturity of the reuptake mechanisms (Caillard et al., 1998; Demarque et al., 2002). Combined with synchronous release of GABA, these immature features create the adequate conditions for GABA spillover and activation of GABAB-Rs in the developing brain.
Applications of specific antagonists have been used to reveal the activation of GABAB-Rs by ambient GABA. Application of GABAB-R antagonists reduces the incidence of calcium spikes in the embryonic Xenopus spinal cord (Root et al., 2008) and increases the basal level of cAMP in embryonic rodent cortex (Bony et al., 2013). In the developing rat hippocampus, the GABAB-R antagonists prolong the duration of the network-driven synaptic activity, termed giant depolarizing potentials (GDPs; Ben-Ari et al., 1989; McLean et al., 1996). A similar lengthening of GDPs was observed in newborn GABAB1-/- mice (Fiorentino et al., 2009) or after desensitization of GABAB-Rs following prolonged activation with baclofen (Tosetti et al., 2004, 2005). Interestingly, when the GDPs are blocked, the GABAB-R antagonist has no effect on GABAergic and glutamatergic synaptic activity (Colin-Le Brun et al., 2004; Fiorentino et al., 2009), supporting the idea that synchronous activation of GABAergic neurons (Khazipov et al., 1997), and subsequent GABA spillover, is required for GABAB-R activation. A similar GABAB-R dependent control of network-driven synaptic activity has been observed in developing retina (Catsicas and Mobbs, 2001), hypothalamus (Obrietan and Van den Pol, 1998, 1999), and cortex (Obrietan and Van den Pol, 1999; Kirmse and Kirischuk, 2006). In all these structures, the application of GABAB-R antagonists increase the frequency and/or duration of synaptic-driven intracellular calcium oscillations, showing that synaptically released GABA exerts a tonic inhibitory control of ongoing synaptic activity acting through GABABRs.
Altogether, these observations show that ambient GABA activates GABAB-Rs early in development, a prerequisite for a possible contribution of GABAB-Rs to neuronal development.
Neurotrophic Actions of GABAB-RS
When GABAB-RS Modulate Neuronal Migration
After their last division, immature postmitotic neurons migrate away from the germinal layers to reach their final position where they differentiate and establish appropriate synaptic connections. Among the many factors involved (Manent et al., 2011), GABAB-Rs have been reported to modulate migration or motility of embryonic spinal cord neurons (Behar et al., 1994, 1995), embryonic hypothalamic neurons (Davis et al., 2002; McClellan et al., 2008), and oligodendrocytes (Luyt et al., 2007). However, the best documented contributions of GABAB-Rs to neuronal migration have been obtained from the studies performed in the cortex. Two modes of migration coexist in the cortex: a radial migration of glutamatergic pyramidal cells from the ventricular/subventricular zone (VZ/SVZ) to the cp and a tangential migration of GABAergic neurons from the ganglionic eminences to the cp. The cortical VZ, cp, and tangentially migrating neurons express GABAB receptors (Behar et al., 2001; Lopez-Bendito et al., 2003; Bony et al., 2013) and both radial and tangential migration are modulated by GABAB-Rs (Figure 1). The first evidence that GABAB-Rs modulate cortical cell migration have been obtained on dissociated embryonic cortical neurons. Starting from E15, GABA induces both chemotaxis (directed migration along a chemical gradient) and chemokinesis (random motility; Behar et al., 1996, 1998). These effects are mimicked by GABAB-R agonists, prevented by GABAB-R antagonists and suppressed by pertussis toxin, an inhibitor of Go/i proteins (Behar et al., 1998). Interestingly, both migratory behaviors are eliminated when the cells are loaded with the calcium chelator BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)], indicating that cytosolic levels of calcium are important for GABA-induced cell motility. Accordingly, baclofen elevates intracellular calcium concentration in a subset of cells derived from E17 cortices (Behar et al., 1996). Using microdissection technique, Behar et al. (1998) have further shown that GABAB-Rs stimulates motility of glutamic acid decarboxylase (GAD)-positive cp neurons and directs a subset of GAD-negative VZ neurons to migrate, suggesting that these receptors may have a role in modulating radial and tangential modes of migration. Organotypic slices cultures and treatment with different GABA receptor antagonists were used to show that endogenous released GABA directs cortical cells to migrate. Thus, organotypic slice cultures treated with the GABAB-R antagonist CGP52342 display an accumulation in the VZ/SVZ of tangentially migrating neurons originating from the ganglionic eminences (Figure 1A; Lopez-Bendito et al., 2003). Similarly, treatment with the GABAB-R antagonist saclofen prevents most VZ BrdU-positive postmitotic neurons from entering into the cp (Behar et al., 2000). Pharmacological approaches show that GABA acts on both metabotropic and ionotropic GABA-Rs to direct neurons to their final position in the cp: GABAC-Rs signal cells to leave the VZ/SVZ and enter the iz; the GABAB-Rs direct cells to leave the iz to enter in the cp; and the GABAA-Rs provide a stop signal as the cells approach their target destination in the cp (Behar et al., 2000).

FIGURE 1. GABAB-Rs modulate radial and tangential migration. (A) E15 brain slices were cut at 300 μm and 4-chloromethyl benzoyl amino tetramethyl rhodamine (CMTMR)-coated particles were placed in the medial ganglionic eminence (MGE) of each hemisphere to label the tangentially migratory neurons. The slices were then incubated for 24 h in vitro in normal culture medium (a,c) or in the presence of 1 μM of the GABABR antagonist, CGP52432 (b,d). After the incubation period, the slices were fixed and counterstained with 0.002% bis-benzimide (blue) to reveal the different cortical compartments and brain structures. In control conditions more CMTMR-labeled cells located at the lower intermediate zone (LIZ) were observed (c) while in the presence of CGP52432 the majority of CMTMR-labeled cells were observed in the ventricular/subventricular zones (VZ/SVZ; d). Asterisks at (c,d) represent where the majority of the CMTMR-positive cells were localized. Abbreviations: ctx, cortex; LIZ, lower intermediate zone; mz, marginal zone; VZ, ventricular zone. Scale bars: a,b 200 μm, c,d 100 μm. Modified with permission from Lopez-Bendito et al. (2003). (B) Confocal images of tdTomato fluorescence in coronal sections of rat somatosensory cortices P16 after in utero transfection (at E17) with pRNAT-U6.3 (EGFP) siRNA empty vector together with pCAG-IRES-tdTomato (Tomato, Control), or functional GABAB2-siRNA (siRNA) together with Tomato construct. Dotted white lines delimitate the boundaries of the cortical layers. Note the presence of ectopic neurons that did not complete radial migration in the layers V and VI. Scale bar: 150 μm. LII–LIV, layer II–IV; WM, white matter. Modified with permission from Bony et al. (2013).
A recent study has addressed the role of GABAB-Rs on neuronal migration in vivo (Bony et al., 2013). To this aim, short interfering RNAs (siRNAs) targeting the GABAB2 subunit have been electroporated at E15 to silence GABAB-Rs mediated responses in cortical pyramidal neuron progenitors (Bony et al., 2013). Most of the GABAB2-silenced cells do not reach their target normal target position in the cortical layer II/III but remained in the deep layers at postnatal stages, indicating that ambient activation of GABAB-Rs modulate the radial migration in vivo (Figure 1B). The authors have gathered evidence identifying the cAMP/liver kinase B1 (LKB1) signaling as downstream effector of GABAB-Rs (Bony et al., 2013). Indeed, the silencing of the GABAB2 subunit increases the immuno-staining against the phosphorylated form of LKB1 at the protein kinase A (PKA) site (pLKB1). Moreover while the overexpression of both LKB1 and constitutively active pLKB1 results in similar migration defects, the overexpression GABAB-Rs only rescues the LKB1-induced defects, indicating that these receptors modulate LKB1 activity through the cAMP/PKA pathway. Finally, the silencing of LKB1 expression rescues the migration defects induced by GABAB2 down expression in vivo. Altogether these data indicate that GABAB-Rs affect neuronal migration by modulation of cAMP/LKB1 pathway in vivo.
If endogenous GABA directs the migration of cortical neurons, an endogenous source of GABA must exist. Because GABA exerts chemo-attractant effect and directs migration of cortical VZ neurons into the cp, a likely candidate for the endogenous source of GABA is the cp cells. To test this hypothesis, dissociated VZ cortical cells were placed in the upper half of a chemotaxis chamber opposite to cp neurons placed in the lower half (Behar et al., 2001). In this condition, the cp cells induce the VZ cells to migrate, an effect prevented by the GABAB-R antagonist saclofen. This observation suggests that cp cells release a diffusible signal that stimulates migration. In agreement with this hypothesis, high performance liquid chromatography (HPLC) analysis reveals that cp neurons release GABA and taurine, and that both molecules stimulate VZ migration in a GABAB-R dependent manner (Behar et al., 2001). More recent studies have identified the tangentially migrating interneurons as the possible source of released GABA (Manent et al., 2005; Bony et al., 2013).
When GABAB-RS Modulate Cell Differentiation
Proper neuronal specification and acquisition of correct neurotransmitter phenotype is crucial for the functioning of the nervous system. Knockdown of GAD, the GABA synthetizing enzyme, has been used in Xenopus embryos to investigate the contribution of GABA in this process (Root et al., 2008). The silencing of GAD results in a decreased incidence of GABA and glycine immuno-reactive cells and increased incidence of glutamate and acetylcholine (Ach) immuno-reactive cells in the Xenopus spinal cord. Agarose beads loaded with a GABAB-R antagonist and implanted in the neural tube pheno-copied the effects of GAD knocking down. With the observation that both the GABAB1 and GABAB2 subunits are detectable in the brain and spinal cord at the corresponding developing stage (Kaeser et al., 2011), these data show that ambient GABA may control neurotransmitter specification in the Xenopus embryos in vivo. The mechanisms involved are not entirely known but required the activation of PKA and PKC which in turn stimulate generation of calcium spikes in embryonic spinal neurons (Root et al., 2008).
When GABAB-RS Modulate Neuritic Outgrowth and Growth Cone Guidance
To ensure accurate targeting, neurons use different regulated mechanisms that provide growth and directional cues to advancing growth cones. Among the many factors, a large amount of evidence indicates that GABA can modulate neurite outgrowth in vitro in different brain regions and species (for review, see Sernagor et al., 2010). Although most studies have focused on the contribution of GABAA-Rs, there are some indications that GABAB-R activation also modulates neuritic growth and growth cone motility both in vitro and in vivo.
The first evidence that GABAB-Rs may affect neurite outgrowth was provided by Michler (1990). They have shown that the administration of the GABAB-R agonist baclofen to cultures obtained from chick tectum or rat cerebellum inhibits neurite outgrowth, while administration of the GABAB-R antagonist saclofen leads to opposite effect (Michler, 1990). Similar to the earlier study by Michler (1990), baclofen has been reported to inhibit the neurite outgrowth and growth cone motility of mice spinal cord (Bird and Owen, 1998) and olfactory bulb (Priest and Puche, 2004) neurons kept in cultures. GABAB-Rs also modulate the neurite outgrowth of cultured cortical neurons (Bony et al., 2013). Indeed, cortical neurons treated with baclofen exhibit longer and more branched dendrites associated with shorter and less branched axons. Conversely, treatment with the GABAB-R antagonist CGP55845 results in shorter dendrites and longer axons. Interestingly, siRNA transfection to silent the expression of the GABAB2 subunit also shortens dendritic length and promotes axonal length (Bony et al., 2013). With this approach, the post-synaptic GABAB-R mediated responses were silenced in a small population of neurons that grow in a normally developing network, hence ruling out impact on the overall network activity, in contrast to the pharmacological approach in which the pre- and post-synaptic responses were abolished in every cell. Thus, the neurotrophic action of GABAB-Rs is not an indirect consequence of altered synaptic activity, but rather results from the activation of post-synaptic downstream signaling pathways. GABAB-Rs not only modulate neuritic growth of some neuronal populations, they also modulate axon/dendrite polarity of cortical neuron in vitro. Baclofen inhibits axon initiation whereas CGP55845 or GABAB2 silencing promote axon initiation in vitro (Bony et al., 2013). The effects of baclofen and CGP55845 are abolished by, respectively, an activator or an inhibitor of the adenylate cyclase, indicating that GABAB-Rs modulate neuronal polarity through an inhibition of cAMP signaling. Gascon et al. (2006) have examined the effects of GABA signaling on dendritic development in vitro using purified neurons from the SVZ of newborn rats, intended to the olfactory bulb neurons. They found that although cells treated with the GABAA-R antagonist bicuculline exhibit reduced dendritic complexity, treatment with a GABAB-R antagonist has no significant effects. Similarly, blockade of GABAB-R does not affect the neuritic outgrowth of cultured cp neurons (Maric et al., 2001). These findings may suggest that either the modulation of neurite outgrowth exerted by the GABAB-Rs is cell type specific or that in these cultures the amount of GABA released do not reach the critical concentration allowing the activation of extra/peri-synaptic GABAB-Rs.
Despite a large body of data indicating a role for GABAB-Rs in neurite outgrowth in vitro, in vivo evidence have been provided only recently. In the Xenopus laevis, the axons of retinal ganglion cells (RGCs) extend through the brain toward their major target, the optic tectum. The axon growth cones of RGCs express GABAB-Rs, and application of baclofen stimulate RGC neurite outgrowth in cultures (Ferguson and McFarlane, 2002). To determine the contribution of the GABAB-Rs in vivo, Ferguson and McFarlane (2002) have applied the GABAB-R antagonist CGP54626 and found that it causes a shortening of the optic projections, indicating that ambient activation of GABAB-Rs controls the growth of RGC axons in vivo. To investigate whether GABAB-Rs may also modulate the neurite outgrowth of cortical pyramidal neurons in vivo, siRNA were electroporated in utero to downregulate the expression of the GABAB2 subunit to silence post-synaptic GABAB-R mediated responses (Bony et al., 2013). Morphological analysis revealed that the specific downregulation of endogenous GABAB2 subunit in vivo leads to reduced apical dendritic length and branch number, and increased axonal growth of cortical neurons. In contrast, the dendritic length and complexity of hippocampal CA3 pyramidal neurons is not altered in GABAB1-/- knockout mice (Fiorentino et al., 2009). This observation may suggest that GABAB receptors regulate neurite outgrowth in some, but not all, brain regions or, that compensatory mechanism take place in the knockout mice (see below).
If GABAB-Rs can modulate neurite outgrowth, an obvious question was to determine whether they could also serve as guidance signal for axon pathfinding. To answer that question, Xiang et al. (2002) applied a gradient of baclofen to Xenopus spinal cord neurons in cultures. They found that the growth cones turn away from the baclofen gradient. Interestingly, repulsion is converted to attraction in the presence of PKC inhibitors. Pharmacological investigations show that baclofen exerts a bi-directional control of growth cone guidance through the activation of different signaling pathways: the activation of the PLC–PKC pathway leading to growth cone repulsion and the PLC-IP3 pathway that attracts the growth cone toward the baclofen source (Xiang et al., 2002). Whether and which of these signaling pathways contribute to axon guidance in vivo remains to be elucidated.
When GABAB-RS Modulate Synaptogenesis
The above gathered studies indicate that activation of GABAB-Rs could serve as a growth and guidance signal, thereby participating in neuronal network construction. Recent evidence indicates that GABAB-Rs may also regulate synaptogenesis. Chattopadhyaya et al. (2007) demonstrated in organotypic cortical slices that endogenous GABA regulates the axonal branching of basket-cell interneurons through the activation of GABAA and GABAB receptors. The authors found that knocking down GAD67, the major GABA synthetizing enzyme, in single basket cells results in a cell autonomous decrease in the number of peri-somatic GABAergic synapses formed by these interneurons on pyramidal cells. The beauty of this approach is that a single cell manipulation minimally impacts the overall level of synaptic activity in contrast to pharmacological manipulations. The deficit induced by the down expression of GAD67 is partially rescued by adding inhibitors of GABA uptake or GABAA as well as GABAB receptor agonists. These findings therefore indicate that ambient GABA, acting on both GABAA and GABAB receptors, contributes to the development of peri-somatic cortical inhibition. In line with GAD silencing, knockout of the GABAB1 subunit also results in altered development of GABAergic synaptic transmission in the mice hippocampus (Fiorentino et al., 2009). Whole cell recordings performed on acute hippocampal slices obtained from newborn GABAB1-/- mice reveals a lower level of miniature GABAergic synaptic activity in the mutant CA3 pyramidal neurons compared to the WT. This deficit is reproduced in vitro in newborn WT intact hippocampi incubated overnight with the GABAB-R antagonist CGP55845 or with tetrodotoxin (TTX) to block action potential-dependent synaptic activity. Importantly, the deficit induced by TTX treatment is rescued by baclofen, indicating that synaptic activation of GABAB-Rs by ambient GABA contributes to the functional development of hippocampal GABAergic synapses. The mechanism by which GABAB-Rs promote the development of GABAergic synapses likely involved regulated secretion of the brain-derived neurotrophic factor (BDNF) and subsequent activation of the tropomyosin receptor kinase (TrkB) pathway (Figure 2). Indeed, the deficit in GABAergic activity observed in WT intact hippocampi treated overnight with CGP55845 is rescued by BDNF treatment and occluded by the absence of BDNF (i.e., in intact hippocampi obtained from BDNF-/- mice or treated with the BDNF/NT4 scavenger TrkB–immunoglobulin G, IgG; Fiorentino et al., 2009). Furthermore, time lapse fluorescence imaging of BDNF–GFP expressing neurons and immuno-histochemical studies show that the activation of GABAB-Rs triggers a calcium-dependent secretion of BDNF in vitro through the activation of the phospholipase C (PLC)–PKC pathway and opening of L-type voltage-dependent calcium channels (Fiorentino et al., 2009; Kuczewski et al., 2011). A previous study has shown that the phosphorylation of the α-CamKII, a critical step in BDNF secretion (Kolarow et al., 2007), is enhanced by GABAB-R activation in the developing rat hippocampus (Xu et al., 2008). Therefore, post-synaptic elevation of calcium and phosphorylation of α-CamKII may underlie the GABAB-R induced secretion of BDNF and functional maturation of GABAergic synaptic transmission in the developing hippocampus.
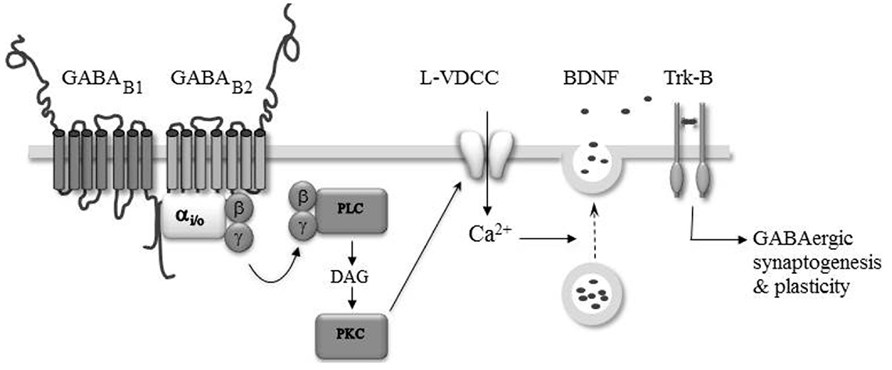
FIGURE 2. GABAB-Rs modulate GABAergic synaptogenesis and plasticity. The activation of GABAB-Rs induces the formation of diacylglycerol (DAG), activation of protein kinase C (PKC), and the opening of L-type voltage-dependent Ca2+ channels (L-VDCC). The intracellular Ca2+ rise triggers the secretion of BDNF which acting on TrkB-Rs modulate the formation and efficacy of developing GABAergic synapses.
Brain-derived neurotrophic factor and tropomyosin receptor kinase signaling is necessary for the full development of inhibitory circuitry (Gottmann et al., 2009). Moreover, endogenous activation of GABAB-Rs (McLean et al., 1996; Obrietan and Van den Pol, 1998, 1999; Catsicas and Mobbs, 2001; Kirmse and Kirischuk, 2006) and GABAB-R dependent modulation of intracellular calcium levels have been reported in several developing brain structures (Behar et al., 1996; Hirono et al., 2001; New et al., 2006; Root et al., 2008; Kuczewski et al., 2011). Therefore, the GABAB-R induced secretion of BDNF may be important for the maturation of synaptic connections throughout the nervous system. Accordingly, a GABAB-R and BDNF–TrkB dependent plasticity of developing inhibitory transmission has been observed in the auditory system (Kotak and Sanes, 2000, 2002; Chang et al., 2003) and visual cortex (Komatsu and Iwakiri, 1993; Inagaki et al., 2008). In these structures, the plasticity is induced during a restricted period of development suggesting that it may have a developmental function in the refinement of initially coarse patterns of synaptic connections (Katz and Shatz, 1996). How GABAB-R and BDNF are linked is presently not fully understood. However, based on the observation that bath application of BDNF rescues the inhibitory plasticity in neurons loaded with the calcium chelator BAPTA, it was proposed that the induction of the inhibitory plasticity relies on the endogenous activation of GABAB-Rs leading to a calcium-dependent secretion of BDNF from the target neurons, which through the TrkB pathway triggered synaptic plasticity (Inagaki et al., 2008; Figure 2).
The Inconsistency of GABAB-R Knockout Mice
Although in vitro pharmacological studies indicate that GABAB-Rs can function as a development signal, studies of knockout mice lacking functional GABAB-Rs could lead to opposite conclusions. Indeed, the complete silencing of GABAB-Rs in mice causes behavioral alterations such as epilepsy, impaired memory, hyperalgesia, hyperthermia, and hyperactivity, highlighting the importance of GABAB-Rs in the appropriate functioning of the nervous system (Prosser et al., 2001; Queva et al., 2003; Haller et al., 2004; Wu et al., 2007). Yet, knockout of either GABAB1 or GABAB2 subunit does not reveal significant alteration in cortical layer organization, although a role of GABAB-Rs in cortical migration has been identified in vitro. Similarly, morphological analysis of hippocampal pyramidal neurons reveals no significant alteration of GABAB1-/- mice (Fiorentino et al., 2009), although endogenous GABAB-Rs have been reported to modulate neurite growth in vitro (Michler, 1990; Priest and Puche, 2004; Bony et al., 2013). The same discrepancy is observed in mice knockout for the GABA-synthetizing enzyme GAD. Indeed, although GABA is scarcely detected in double knockout GAD 65 and 67, no disorders of histogenesis are observed (Ji et al., 1999). It should be mentioned, however, that a developmental phenotype has been observed in GABAB1-/- mice, in which whole cell recordings of hippocampal slices reveal a delayed functional maturation of GABAergic synapses in the mutant mice (Fiorentino et al., 2009). Therefore, a more detailed determination of the morphological–functional properties of the neuronal networks in GABAB-/- mice is essential to ensure that the mutation has developmental consequences. The discrepancy between in vitro pharmacological studies and genetically modified mice in vivo may be explained by compensatory mechanisms frequently experienced by knockout mice. For instance, in the knockout GAD mice, taurine can bind GABAB-Rs and could potentially compensate for the absence of GABA (Behar et al., 2001). Moreover, some of the downstream signaling pathways activated by the GABAB-Rs are also the targets of other receptors, and activation of these receptors may substitute for the absence of functional GABAB-Rs. A recent study shows that silencing GABAB-Rs in a small population of excitatory neurons leads to ectopic positions and morphological alterations of these neurons (Bony et al., 2013), hence highlighting the contribution of GABAB-Rs to neuronal development in vivo.
Conclusion
A large body of studies has documented the prominent role of GABAA-Rs to neuronal development. For several years, the possible contribution of GABAB-Rs has lagged behind. The studies gathered in this review indicate that GABAB-Rs do play a role in crucial steps of neuronal network formation, including migration, neurite growth, synapse formation and plasticity, both in vitro and in vivo (Table 1). The neurotrophic action of GABAB-Rs raises several immediate questions. Do they play an instructive or a permissive role? What are the downstream signaling pathways involved? Does the use pharmacological drugs during pregnancy lead to GABAB-dependent alterations in brain construction?
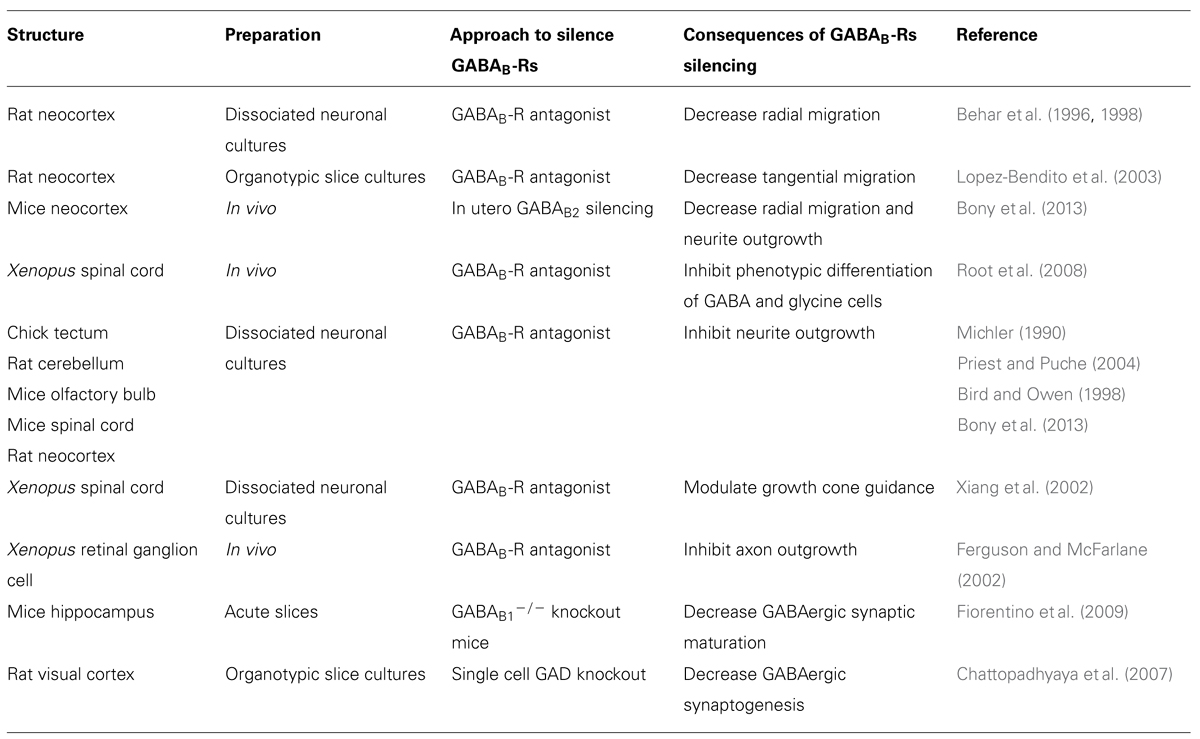
TABLE 1. Neurotrophic action of GABAB.
The use of antagonists has been extremely fruitful to unmask the endogenous activation of GABAB-Rs and reveal their neurotrophic actions. However, because GABAB-Rs control the level of synaptic activity, it is often difficult with this approach to distinguish between indirect actions on synaptic activity from direct consequences of GABAB-R activation. However, the functional silencing of GABAB-Rs in a small population of neurons that minimally impact the level of synaptic activity offers an interesting alternative, and confirms that endogenous GABAB-Rs exert a neurotrophic action on neuronal development in vivo. Some signaling pathways have been identified, involving changes in calcium and cAMP levels, but much remained to be done. In vitro studies have shown that the stimulation of GABAB-Rs leads to a translocation of the transcriptor factor 4 (ATF4; White et al., 2000; Vernon et al., 2001) and to the phosphorylation cAMP-response element binding (CREB). The interactions with ATF4 and/or CREB may be important to regulate gene expression and may underlie some of the neurotrophic actions of the GABAB-Rs. Accordingly, the direct activation of GABAB-Rs, in the presence of TTX to block spontaneous synaptic activity, leads to an up-regulation of BDNF expression, a key modulator of neuronal network wiring (Gottmann et al., 2009).
A recent study has revealed that patients with autoimmune encephalitis associated with antibodies to GABAB1 subunits show seizures, confusion and memory deficit (Lancaster et al., 2010). Moreover, some GABAB-R polymorphisms confer a highly increased susceptibility to temporal-lobe epilepsy in human beings (Gambardella et al., 2003; Wang et al., 2008). Thus, defective GABAB-Rs functions in the brain could underlie neurological and psychiatric diseases. It is therefore tempting to speculate that children exposed to drugs that affect GABA levels or act on GABAB-Rs during their fetal or postnatal life may develop GABAB-R related developmental disorders. In future studies, it will be essential to consider the emerging role of GABAB-Rs in network formation to fully understand the neurotrophic role of GABA and pathological consequences of altered GABAergic signaling.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We wish to thank actual and former members of our laboratory that contribute to original research.
References
Behar, T. N., Li, Y. X., Tran, H. T., Ma, W., Dunlap, V., Scott, C., et al. (1996). GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J. Neurosci. 16, 1808–1818.
Behar, T. N., Schaffner, A. E., Colton, C. A., Somogyi, R., Olah, Z., Lehel, C., et al. (1994). GABA-induced chemokinesis and NGF-induced chemotaxis of embryonic spinal cord neurons. J. Neurosci. 14, 29–38.
Behar, T. N., Schaffner, A. E., Scott, C. A., Greene, C. L., and Barker, J. L. (2000). GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb. Cortex 10, 899–909. doi: 10.1093/cercor/10.9.899
Behar, T. N., Schaffner, A. E., Scott, C. A., O'Connell, C., and Barker, J. L. (1998). Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J. Neurosci. 18, 6378–6387.
Behar, T. N., Schaffner, A. E., Tran, H. T., and Barker, J. L. (1995). GABA-induced motility of spinal neuroblasts develops along a ventrodorsal gradient and can be mimicked by agonists of GABAA and GABAB receptors. J. Neurosci. Res. 42, 97–108. doi: 10.1002/jnr.490420111
Behar, T. N., Smith, S. V., Kennedy, R. T., McKenzie, J. M., Maric, I., and Barker, J. L. (2001). GABA(B) receptors mediate motility signals for migrating embryonic cortical cells. Cereb. Cortex 11, 744–753. doi: 10.1093/cercor/11.8.744
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaïarsa, J.-L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. (Lond.) 416, 303–325.
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Bianchi, M. S., Lux-Lantos, V. A., Bettler, B., and Libertun, C. (2005). Expression of gamma-aminobutyric acid B receptor subunits in hypothalamus of male and female developing rats. Brain Res. Dev. Brain Res. 160, 124–129. doi: 10.1016/j.devbrainres.2005.06.017
Biermann, B., Ivankova-Susankova, K., Bradaia, A., Abdel, A. S., Besseyrias, V., Kapfhammer, J. P., et al. (2010). The sushi domains of GABAB receptors function as axonal targeting signals. J. Neurosci. 30, 1385–1394. doi: 10.1523/JNEUROSCI.3172-09.2010
Bird, M., and Owen, A. (1998). Neurite outgrowth-regulating properties of GABA and the effect of serum on mouse spinal cord neurons in culture. J. Anat. 193(Pt 4), 503–508. doi: 10.1046/j.1469-7580.1998.19340503.x
Blackmer, T., Larsen, E. C., Bartleson, C., Kowalchyk, J. A., Yoon, E. J., Preininger, A. M., et al. (2005). G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat. Neurosci. 8, 421–425.
Bony, G., Szczurkowska, J., Tamagno, I., Shelly, M., Contestabile, A., and Cancedda, L. (2013). Non-hyperpolarizing GABAB receptor activation regulates neuronal migration and neurite growth and specification by cAMP/LKB1. Nat. Commun. 4, 1800. doi: 10.1038/ncomms2820
Bowery, N. G. (1993). GABAB receptor pharmacology. Annu. Rev. Pharmacol. Toxicol. 33, 109–147. doi: 10.1146/annurev.pa.33.040193.000545
Bowery, N. G., Bettler, B., Froestl, W., Gallagher, J. P., Marshall, F., Raiteri, M., et al. (2002). International union of pharmacology. XXXIII. Mammalian gamma-aminobutyric acid (B) receptors: structure and function. Pharmacol. Rev. 54, 247–264. doi: 10.1124/pr.54.2.247
Caillard, O., McLean, H. A., Ben-Ari, Y., and Gaïarsa, J.-L. (1998). Ontogenesis of presynaptic GABAB receptor-mediated inhibition in the CA3 region of the rat hippocampus. J. Neurophysiol. 79, 1341–1348.
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007
Catsicas, S., and Mobbs, P. (2001). GABAB receptors regulate chick retinal calcium waves. J. Neurosci. 21, 897–910.
Chang, E. H., Kotak, V. C., and Sanes, D. H. (2003). Long-term depression of synaptic inhibition is expressed postsynaptically in the developing auditory system. J. Neurophysiol. 90, 1479–1488. doi: 10.1152/jn.00386.2003
Chattopadhyaya, B., Di Cristo, G., Wu, C. Z., Knott, G., Kuhlman, S., Fu, Y., et al. (2007). GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron 54, 889–903. doi: 10.1016/j.neuron.2007.05.015
Chudotvorova, I., Ivanov, A., Rama, S., Hubner, C. A., Pellegrino, C., Ben-Ari, Y., et al. (2005). Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 566, 671–679. doi: 10.1113/jphysiol.2005.089821
Colin-Le Brun, I., Ferrand, N., Caillard, O., Tosetti, P., Ben-Ari, Y., and Gaïarsa, J. L. (2004). Spontaneous synaptic activity is required for the formation of functional GABAergic synapses in the developing rat hippocampus. J. Physiol. 559, 129–139. doi: 10.1113/jphysiol.2004.065060
Correa, S. A., Munton, R., Nishimune, A., Fitzjohn, S., and Henley, J. M. (2004). Development of GABAB subunits and functional GABAB receptors in rat cultured hippocampal neurons. Neuropharmacology 47, 475–484. doi: 10.1016/j.neuropharm.2004.04.021
Davis, A. M., Henion, T. R., and Tobet, S. A. (2002). Gamma-aminobutyric acid B receptors and the development of the ventromedial nucleus of the hypothalamus. J. Comp. Neurol. 449, 270–280. doi: 10.1002/cne.10293
Demarque, M., Represa, A., Becq, H., Khalilov, I., Ben-Ari, Y., and Aniksztejn, L. (2002). Paracrine intercellular communication by a Ca(2+)- and SNARE-independent release of GABA and glutamate prior to synapse formation. Neuron 36, 1051–1061. doi: 10.1016/S0896-6273(02)01053-X
Dutar, P., and Nicoll, R. A. (1988). A physiological role for GABAB receptors in the central nervous system. Nature 332, 156–158. doi: 10.1038/332156a0
Ehrengruber, M. U., Doupnik, C. A., Xu, Y., Garvey, J., Jasek, M. C., Lester, H. A., et al. (1997). Activation of heteromeric G protein-gated inward rectifier K+ channels overexpressed by adenovirus gene transfer inhibits the excitability of hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 7070–7075. doi: 10.1073/pnas.94.13.7070
Ferguson, S. C., and McFarlane, S. (2002). GABA and development of the Xenopus optic projection. J. Neurobiol. 51, 272–284. doi: 10.1002/neu.10061
Fiorentino, H., Kuczewski, N., Diabira, D., Ferrand, N., Pangalos, M. N., Porcher, C., et al. (2009). GABA(B) receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses. J. Neurosci. 29, 11650–11661. doi: 10.1523/JNEUROSCI.3587-09.2009
Fritschy, J. M., Meskenaite, V., Weinmann, O., Honer, M., Benke, D., and Mohler, H. (1999). GABAB-receptor splice variants GB1a and GB1b in rat brain: developmental regulation, cellular distribution and extrasynaptic localization. Eur. J. Neurosci. 11, 761–768. doi: 10.1046/j.1460-9568.1999.00481.x
Fritschy, J. M., Sidler, C., Parpan, F., Gassmann, M., Kaupmann, K., Bettler, B., et al. (2004). Independent maturation of the GABA(B) receptor subunits GABA(B1) and GABA(B2) during postnatal development in rodent brain. J. Comp. Neurol. 477, 235–252. doi: 10.1002/cne.20188
Fukuda, A., Mody, I., and Prince, D. A. (1993). Differential ontogenesis of presynaptic and postsynaptic GABAB inhibition in the rat somatosensory cortex. J. Neurophysiol. 70, 448–452.
Gähwiler, B. H., and Brown, D. A. (1985). GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proc. Natl. Acad. Sci. U.S.A. 82, 1558–1562. doi: 10.1073/pnas.82.5.1558
Gaïarsa, J.-L., Tseeb, V., and Ben-Ari, Y. (1995). Postnatal development of pre- and postsynaptic GABAB-mediated inhibitions in the CA3 hippocampal region of the rat. J. Neurophysiol. 73, 246–255.
Gambardella, A., Manna, I., Labate, A., Chifari, R., La, R. A., Serra, P., et al. (2003). GABA(B) receptor 1 polymorphism (G1465A) is associated with temporal lobe epilepsy. Neurology 60, 560–563. doi: 10.1212/01.WNL.0000046520.79877.D8
Gao, X. B., and Van den Pol, A. N. (2000). GABA release from mouse axonal growth cones. J. Physiol. 523(Pt 3), 629–637. doi: 10.1111/j.1469-7793.2000.t01-1-00629.x
Gascon, E., Dayer, A. G., Sauvain, M. O., Potter, G., Jenny, B., De, R. M., et al. (2006). GABA regulates dendritic growth by stabilizing lamellipodia in newly generated interneurons of the olfactory bulb. J. Neurosci. 26, 12956–12966. doi: 10.1523/JNEUROSCI.4508-06.2006
Gassmann, M., Shaban, H., Vigot, R., Sansig, G., Haller, C., Barbieri, S., et al. (2004). Redistribution of GABAB(1) protein and atypical GABAB responses in GABAB(2)-deficient mice. J. Neurosci. 24, 6086–6097. doi: 10.1523/JNEUROSCI.5635-03.2004
Ge, S., Goh, E. L., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593. doi: 10.1038/nature04404
Gottmann, K., Mittmann, T., and Lessmann, V. (2009). BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 199, 203–234. doi: 10.1007/s00221-009-1994-z
Guetg, N., Seddik, R., Vigot, R., Turecek, R., Gassmann, M., Vogt, K. E., et al. (2009). The GABAB1a isoform mediates heterosynaptic depression at hippocampal mossy fiber synapses. J. Neurosci. 29, 1414–1423. doi: 10.1523/JNEUROSCI.3697-08.2009
Haller, C., Casanova, E., Muller, M., Vacher, C. M., Vigot, R., Doll, T., et al. (2004). Floxed allele for conditional inactivation of the GABAB(1) gene. Genesis 40, 125–130. doi: 10.1002/gene.20073
Hassfurth, B., Grothe, B., and Koch, U. (2010). The mammalian interaural time difference detection circuit is differentially controlled by GABAB receptors during development. J. Neurosci. 30, 9715–9727. doi: 10.1523/JNEUROSCI.1552-10.2010
Hirono, M., Yoshioka, T., and Konishi, S. (2001). GABA(B) receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat. Neurosci. 4, 1207–1216. doi: 10.1038/nn764
Inagaki, T., Begum, T., Reza, F., Horibe, S., Inaba, M., Yoshimura, Y., et al. (2008). Brain-derived neurotrophic factor-mediated retrograde signaling required for the induction of long-term potentiation at inhibitory synapses of visual cortical pyramidal neurons. Neurosci. Res. 61, 192–200. doi: 10.1016/j.neures.2008.02.006
Isaacson, J. S., and Nicoll, R. A. (1993). Local and diffuse synaptic actions of GABA in the hippocampus. Neuron 10, 165–175. doi: 10.1016/0896-6273(93)90308-E
Ji, F., Kanbara, N., and Obata, K. (1999). GABA and histogenesis in fetal and neonatal mouse brain lacking both the isoforms of glutamic acid decarboxylase. Neurosci. Res. 33, 187–194. doi: 10.1016/S0168-0102(99)00011-5
Jones, K. A., Borowsky, B., Tamm, J. A., Craig, D. A., Durkin, M. M., Dai, M., et al. (1998). GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 396, 674–679. doi: 10.1038/25348
Kaeser, G. E., Rabe, B. A., and Saha, M. S. (2011). Cloning and characterization of GABAA alpha subunits and GABAB subunits in Xenopus laevis during development. Dev. Dyn. 240, 862–873. doi: 10.1002/dvdy.22580
Katz, L. C., and Shatz, C. J. (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. doi: 10.1126/science.274.5290.1133
Kaupmann, K., Huggel, K., Heid, J., Flor, P. J., Bischoff, S., Mickel, S. J., et al. (1997). Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature 386, 239–246. doi: 10.1038/386239a0
Kaupmann, K., Malitschek, B., Schuler, V., Heid, J., Froestl, W., Beck, P., et al. (1998). GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 396, 683–687. doi: 10.1038/25360
Khazipov, R., Leinekugel, X., Khalilov, I., Gaïarsa, J.-L., and Ben-Ari, Y. (1997). Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J. Physiol. (Lond.) 498, 763–772.
Khazipov, R., and Luhmann, H. J. (2006). Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 29, 414–418. doi: 10.1016/j.tins.2006.05.007
Kim, M. O., Li, S., Park, M. S., and Hornung, J. P. (2003). Early fetal expression of GABA(B1) and GABA(B2) receptor mRNAs on the development of the rat central nervous system. Brain Res. Dev. Brain Res. 143, 47–55. doi: 10.1016/S0165-3806(03)00099-3
Kirmse, K., and Kirischuk, S. (2006). Ambient GABA constrains the strength of GABAergic synapses at Cajal-Retzius cells in the developing visual cortex. J. Neurosci. 26, 4216–4227. doi: 10.1523/JNEUROSCI.0589-06.2006
Kolarow, R., Brigadski, T., and Lessmann, V. (2007). Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J. Neurosci. 27, 10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007
Komatsu, Y., and Iwakiri, M. (1993). Long-term modification of inhibitory synaptic transmission in developing visual cortex. Neuroreport 4, 907–910. doi: 10.1097/00001756-199307000-00017
Kotak, V. C., and Sanes, D. H. (2000). Long-lasting inhibitory synaptic depression is age- and calcium-dependent. J. Neurosci. 20, 5820–5826.
Kotak, V. C., and Sanes, D. H. (2002). Postsynaptic kinase signaling underlies inhibitory synaptic plasticity in the lateral superior olive. J. Neurobiol. 53, 36–43. doi: 10.1002/neu.10107
Kuczewski, N., Fuchs, C., Ferrand, N., Jovanovic, J. N., Gaiarsa, J. L., and Porcher, C. (2011). Mechanism of GABA(B) receptor-induced BDNF secretion and promotion of GABA(A) receptor membrane expression. J. Neurochem. 118, 533–545. doi: 10.1111/j.1471-4159.2011.07192.x
Kulik, A., Vida, I., Lujan, R., Haas, C. A., Lopez-Bendito, G., Shigemoto, R., et al. (2003). Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J. Neurosci. 23, 11026–11035.
Lancaster, E., Lai, M., Peng, X., Hughes, E., Constantinescu, R., Raizer, J., et al. (2010). Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 9, 67–76. doi: 10.1016/S1474-4422(09)70324-2
Lopez-Bendito, G., Lujan, R., Shigemoto, R., Ganter, P., Paulsen, O., and Molnar, Z. (2003). Blockade of GABA(B) receptors alters the tangential migration of cortical neurons. Cereb. Cortex 13, 932–942. doi: 10.1093/cercor/13.9.932
Lopez-Bendito, G., Shigemoto, R., Kulik, A., Paulsen, O., Fairen, A., and Lujan, R. (2002). Expression and distribution of metabotropic GABA receptor subtypes GABABR1 and GABABR2 during rat neocortical development. Eur. J. Neurosci. 15, 1766–1778. doi: 10.1046/j.1460-9568.2002.02032.x
Lopez-Bendito, G., Shigemoto, R., Kulik, A., Vida, I., Fairen, A., and Lujan, R. (2004). Distribution of metabotropic GABA receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus during prenatal and postnatal development. Hippocampus 14, 836–848. doi: 10.1002/hipo.10221
Luhmann, H. J., and Prince, D. A. (1991). Postnatal maturation of the GABAergic system in rat neocortex. J. Neurophysiol. 247–263.
Lujan, R., and Shigemoto, R. (2006). Localization of metabotropic GABA receptor subunits GABAB1 and GABAB2 relative to synaptic sites in the rat developing cerebellum. Eur. J. Neurosci. 23, 1479–1490. doi: 10.1111/j.1460-9568.2006.04669.x
Luyt, K., Slade, T. P., Dorward, J. J., Durant, C. F., Wu, Y., Shigemoto, R., et al. (2007). Developing oligodendrocytes express functional GABA(B) receptors that stimulate cell proliferation and migration. J. Neurochem. 100, 822–840. doi: 10.1111/j.1471-4159.2006.04255.x
Manent, J. B., Beguin, S., Ganay, T., and Represa, A. (2011). Cell-autonomous and cell-to-cell signalling events in normal and altered neuronal migration. Eur. J. Neurosci. 34, 1595–1608. doi: 10.1111/j.1460-9568.2011.07867.x
Manent, J. B., Demarque, M., Jorquera, I., Pellegrino, C., Ben-Ari, Y., Aniksztejn, L., et al. (2005). A noncanonical release of GABA and glutamate modulates neuronal migration. J. Neurosci. 25, 4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005
Maric, D., Liu, Q. Y., Maric, I., Chaudry, S., Chang, Y. H., Smith, S. V., et al. (2001). GABA expression dominates neuronal lineage progression in the embryonic rat neocortex and facilitates neurite outgrowth via GABA(A) autoreceptor/Cl-channels. J. Neurosci. 21, 2343–2360.
Martin, S. C., Steiger, J. L., Gravielle, M. C., Lyons, H. R., Russek, S. J., and Farb, D. H. (2004). Differential expression of gamma-aminobutyric acid type B receptor subunit mRNAs in the developing nervous system and receptor coupling to adenylyl cyclase in embryonic neurons. J. Comp. Neurol. 473, 16–29. doi: 10.1002/cne.20094
McClellan, K. M., Calver, A. R., and Tobet, S. A. (2008). GABAB receptors role in cell migration and positioning within the ventromedial nucleus of the hypothalamus. Neuroscience 151, 1119–1131. doi: 10.1016/j.neuroscience.2007.11.048
McLean, H. A., Caillard, O., Khazipov, R., Ben-Ari, Y., and Gaïarsa, J.-L. (1996). Spontaneous release of GABA activates GABAB receptors and controls network activity in the neonatal rat hippocampus. J. Neurophysiol. 76, 1036–1046.
Meier, S. D., Kafitz, K. W., and Rose, C. R. (2008). Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia 56, 1127–1137. doi: 10.1002/glia.20684
Michler, A. (1990). Involvement of GABA receptors in the regulation of neurite growth in cultured embryonic chick tectum. Int. J. Dev. Neurosci. 8, 463–472. doi: 10.1016/0736-5748(90)90078-G
Mintz, I. M., and Bean, B. P. (1993). GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron 10, 889–898. doi: 10.1016/0896-6273(93)90204-5
New, D. C., An, H., Ip, N. Y., and Wong, Y. H. (2006). GABAB heterodimeric receptors promote Ca2+ influx via store-operated channels in rat cortical neurons and transfected Chinese hamster ovary cells. Neuroscience 137, 1347–1358. doi: 10.1016/j.neuroscience.2005.10.033
Nurse, S., and Lacaille, J. C. (1999). Late maturation of GABA(B) synaptic transmission in area CA1 of the rat hippocampus. Neuropharmacology 38, 1733–1742. doi: 10.1016/S0028-3908(99)00122-7
Obrietan, K., and Van den Pol, A. N. (1998). GABAB receptor-mediated inhibition of GABAA receptor calcium elevations in developing hypothalamic neurons. J. Neurophysiol. 79, 1360–1370.
Obrietan, K., and Van den Pol, A. N. (1999). GABAB receptor-mediated regulation of glutamate-activated calcium transients in hypothalamic and cortial neuron development. J. Neurophysiol. 81, 94–102.
Owens, D. F., and Kriegstein, A. R. (2002). Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 3, 715–727. doi: 10.1038/nrn919
Perez-Garci, E., Gassmann, M., Bettler, B., and Larkum, M. E. (2006). The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron 50, 603–616. doi: 10.1016/j.neuron.2006.04.019
Priest, C. A., and Puche, A. C. (2004). GABAB receptor expression and function in olfactory receptor neuron axon growth. J. Neurobiol. 60, 154–165. doi: 10.1002/neu.20011
Prosser, H. M., Gill, C. H., Hirst, W. D., Grau, E., Robbins, M., Calver, A., et al. (2001). Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol. Cell. Neurosci. 17, 1059–1070. doi: 10.1006/mcne.2001.0995
Queva, C., Bremner-Danielsen, M., Edlund, A., Ekstrand, A. J., Elg, S., Erickson, S., et al. (2003). Effects of GABA agonists on body temperature regulation in GABA(B(1))-/-mice. Br. J. Pharmacol. 140, 315–322. doi: 10.1038/sj.bjp.0705447
Reynolds, A., Brustein, E., Liao, M., Mercado, A., Babilonia, E., Mount, D. B., et al. (2008). Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J. Neurosci. 28, 1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Robbins, M. J., Calver, A. R., Filippov, A. K., Hirst, W. D., Russell, R. B., Wood, M. D., et al. (2001). GABA(B2) is essential for G-protein coupling of the GABA(B) receptor heterodimer. J. Neurosci. 21, 8043–8052.
Root, C. M., Velazquez-Ulloa, N. A., Monsalve, G. C., Minakova, E., and Spitzer, N. C. (2008). Embryonically expressed GABA and glutamate drive electrical activity regulating neurotransmitter specification. J. Neurosci. 28, 4777–4784. doi: 10.1523/JNEUROSCI.4873-07.2008
Sakaba, T., and Neher, E. (2003). Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature 424, 775–778. doi: 10.1038/nature01859
Scanziani, M. (2000). GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron 25, 673–681. doi: 10.1016/S0896-6273(00)81069-7
Schuler, V., Luscher, C., Blanchet, C., Klix, N., Sansig, G., Klebs, K., et al. (2001). Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA(B(1)). Neuron 31, 47–58. doi: 10.1016/S0896-6273(01)00345-2
Sernagor, E., Chabrol, F., Bony, G., and Cancedda, L. (2010). GABAergic control of neurite outgrowth and remodeling during development and adult neurogenesis: general rules and differences in diverse systems. Front. Cell. Neurosci. 4:11. doi: 10.3389/fncel.2010.00011
Taylor, J., and Gordon-Weeks, P. R. (1991). Calcium-indenpendent g-aminobutyric acid release from growth cones: role of g-aminobutyric acid transport. J. Neurochem. 56, 273–280. doi: 10.1111/j.1471-4159.1991.tb02592.x
Thompson, S. M., and Gahwiler, B. H. (1992a). Comparison of the actions of baclofen at pre- and postsynaptic receptors in the rat hippocampus in vitro. J. Physiol. 451, 329–345.
Thompson, S. M., and Gahwiler, B. H. (1992b). Effects of the GABA uptake inhibitor tiagabine on inhibitory synaptic potentials in rat hippocampal slice cultures. J. Neurophysiol. 67, 1698–1701.
Tosetti, P., Bakels, R., Colin-Le Brun, I., Ferrand, N., Gaïarsa, J. L., and Caillard, O. (2004). Acute desensitization of presynaptic GABAB-mediated inhibition and induction of epileptiform discharges in the neonatal rat hippocampus. Eur. J. Neurosci. 19, 3227–3234. doi: 10.1111/j.0953-816X.2004.03413.x
Tosetti, P., Ferrand, N., Brun, I. C., and Gaiarsa, J. L. (2005). Epileptiform activity triggers long-term plasticity of GABAB receptor signalling in the developing rat hippocampus. J. Physiol. 568, 951–966. doi: 10.1113/jphysiol.2005.094631
Tremblay, E., Ben-Ari, Y., and Roisin, M. P. (1995). Different GABAB-mediated effects on protein kinase C activity and immunoreactivity in neonatal and adult rat hippocampal slices. J. Neurochem. 65, 863–870. doi: 10.1046/j.1471-4159.1995.65020863.x
Turgeon, S. M., and Albin, R. L. (1994). Postnatal ontogeny of GABAB binding in rat brain. Neuroscience 62, 601–613. doi: 10.1016/0306-4522(94)90392-1
Verheugen, J. A., Fricker, D., and Miles, R. (1999). Noninvasive measurements of the membrane potential and GABAergic action in hippocampal interneurons. J. Neurosci. 19, 2546–2555.
Vernon, E., Meyer, G., Pickard, L., Dev, K., Molnar, E., Collingridge, G. L., et al. (2001). GABA(B) receptors couple directly to the transcription factor ATF4. Mol. Cell. Neurosci. 17, 637–645. doi: 10.1006/mcne.2000.0960
Vigot, R., Barbieri, S., Brauner-Osborne, H., Turecek, R., Shigemoto, R., Zhang, Y. P., et al. (2006). Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron 50, 589–601. doi: 10.1016/j.neuron.2006.04.014
Wang, D. D., and Kriegstein, A. R. (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28, 5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008
Wang, X., Sun, W., Zhu, X., Li, L., Wu, X., Lin, H., et al. (2008). Association between the gamma-aminobutyric acid type B receptor 1 and 2 gene polymorphisms and mesial temporal lobe epilepsy in a Han Chinese population. Epilepsy Res. 81, 198–203. doi: 10.1016/j.eplepsyres.2008.06.001
White, J. H., McIllhinney, R. A., Wise, A., Ciruela, F., Chan, W. Y., Emson, P. C., et al. (2000). The GABAB receptor interacts directly with the related transcription factors CREB2 and ATFx. Proc. Natl. Acad. Sci. U.S.A. 97, 13967–13972. doi: 10.1073/pnas.240452197
White, J. H., Wise, A., Main, M. J., Green, A., Fraser, N. J., Disney, G. H., et al. (1998). Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396, 679–682. doi: 10.1038/25354
Wu, Y., Chan, K. F., Eubanks, J. H., Guin Ting, W. C., Cortez, M. A., Shen, L., et al. (2007). Transgenic mice over-expressing GABA(B)R1a receptors acquire an atypical absence epilepsy-like phenotype. Neurobiol. Dis. 26, 439–451. doi: 10.1016/j.nbd.2007.01.013
Xiang, Y., Li, Y., Zhang, Z., Cui, K., Wang, S., Yuan, X. B., et al. (2002). Nerve growth cone guidance mediated by G protein-coupled receptors. Nat. Neurosci. 5, 843–848. doi: 10.1038/nn899
Keywords: GABAB receptors, GABA, cortical development, synaptogenesis, migration
Citation: Gaiarsa J-L and Porcher C (2013) Emerging neurotrophic role of GABAB receptors in neuronal circuit development. Front. Cell. Neurosci. 7:206. doi: 10.3389/fncel.2013.00206
Received: 19 September 2013; Accepted: 18 October 2013;
Published online: 12 November 2013.
Edited by:
Laura Cancedda, Istituto Italiano di Tecnologia, ItalyReviewed by:
Melanie A. Woodin, University of Toronto, CanadaVictoria Adela Lux-Lantos, Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina
Copyright © 2013 Gaiarsa and Porcher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Luc Gaiarsa, Institut de Neurobiologie de la Méditerranée, Parc Scientifique de Luminy, BP 13, 13273 Marseille Cedex 09, France e-mail:amVhbi1sdWMuZ2FpYXJzYUBpbnNlcm0uZnI=