 Cátia P. Frias
Cátia P. Frias Corette J. Wierenga
Corette J. Wierenga- Division of Cell Biology, Faculty of Science, Utrecht University, Utrecht, Netherlands
Synaptic connections in our brains change continuously and throughout our lifetime. Despite ongoing synaptic changes, a healthy balance between excitation and inhibition is maintained by various forms of homeostatic and activity-dependent adaptations, ensuring stable functioning of neuronal networks. In this review we summarize experimental evidence for activity-dependent changes occurring in inhibitory axons, in cultures as well as in vivo. Axons form many presynaptic terminals, which are dynamic structures sharing presynaptic material along the axonal shaft. We discuss how internal (e.g., vesicle sharing) and external factors (e.g., binding of cell adhesion molecules or secreted factors) may affect the formation and plasticity of inhibitory synapses.
Introduction
Our brain is a complex organ with tremendous self-organizing abilities. Its computational power is based in the adjustable synaptic connections between neurons. When new experiences and memories are established, specific synapses in specific brain regions are changed, both in strength and in number. To ensure proper global functioning despite changes in local connectivity, these synaptic changes must be coordinated within neurons, as well as within neuronal circuits. An important aspect is the coordination between changes in excitatory and inhibitory synapses to regulate and maintain an overall balance between excitation and inhibition. When this balance is disturbed, neurological diseases such as autism or schizophrenia can develop (Palop et al., 2007; Yizhar et al., 2011; Han et al., 2012; Bateup et al., 2013).
Homeostatic plasticity is a term that is used for plasticity mechanisms which ensure that overall neuronal spiking activity is maintained within neuronal networks. Many forms of homeostatic plasticity have been described in excitatory and inhibitory neurons (Turrigiano, 2008; Wenner, 2011; Pozo and Goda, 2010; Tyagarajan and Fritschy, 2010). In neuronal circuits in the brain, inhibitory neurons serve multiple functions, making connections to excitatory as well as inhibitory neurons, and providing feedforward inhibition to some neurons, while supplying feedback input to others. In such complicated networks, there are multiple ways to compensate for changes in network activity, which makes it hard, if not impossible, to classify synaptic changes in inhibitory axons as truly homeostatic. Therefore, we will discuss activity-dependent feedback signals in inhibitory axons in a more general context in this review. We will discuss experimental evidence showing that synaptic activity can affect the formation and plasticity of inhibitory synapses and we will speculate on possible mechanisms.
Activity-Dependent Adaptations of Inhibitory Synapses
When prolonged changes occur in network activity, homeostatic mechanisms come into play which adjust excitatory and inhibitory synapses to compensate and restore the activity level in the network (Turrigiano, 1999, 2011; Burrone and Murthy, 2003; Pozo and Goda, 2010; Wenner, 2011). Generally speaking, when the activity is too high, excitation must be downregulated, and inhibition should be increased to bring activity levels back to baseline. And opposite changes should occur during activity blockade. Homeostatic plasticity has been studied extensively in cultures, where neurons are randomly connected. Dissociated cultures provide superb access for experimental manipulations and therefore form an excellent system to study the cell biological mechanisms underlying homeostatic plasticity. However, in our brain neurons are embedded in multiple neuronal networks and make specific synaptic connections. Recurrent connections between neurons or groups of neurons are very common and different types of GABAergic interneurons are known to have high specificity, making inhibitory synapses onto specific target neurons, including inhibitory neurons (Pfeffer et al., 2013; Jiang et al., 2013). In such complex networks, it is not easy to determine rules of homeostatic plasticity. Adaptation to changes in the activity of the network will be strongly synapse-specific and likely depends on the precise function and location of the synapse in the network (Chen et al., 2011; Maffei et al., 2004; Maffei and Turrigiano, 2008). Here we briefly describe the experimental evidence for activity-dependent plasticity of inhibitory synapses from in vitro (i.e., in dissociated and organotypic cultures) and in vivo studies.
Primary cell cultures
Activity manipulations in cultures of dissociated hippocampal or neocortical neurons generally affect excitatory and inhibitory synapses in opposite directions. After a prolonged period of activity blockade, excitatory synapses get strengthened and inhibitory synapses are weakened, and synaptic changes are in opposite directions when activity is enhanced (Turrigiano et al., 1998; Kilman et al., 2002; Hartman et al., 2006; Swanwick et al., 2006). Therefore, changes in excitation and inhibition cooperate to compensate for the change in activity level. For inhibitory synapses, changes in mIPSC amplitude are most commonly reported, reflecting changes in synaptic strength. Sometimes they are accompanied by changes in mIPSC frequency, which could either reflect a change in the number of synapses or a change in release properties. Dissociated cultures provide excellent experimental access and are therefore well-suited for studying underlying mechanisms of homeostatic plasticity. However, the artificial environment in which neurons grow in culture may affect synaptic maturation (Wierenga et al., 2006; Rose et al., 2013) and consequently cellular or synaptic mechanisms of plasticity. Cellular mechanisms that were identified to mediate the changes in inhibitory synapses after activity manipulations include: changes in number of postsynaptic receptors (Kilman et al., 2002; Swanwick et al., 2006; Saliba et al., 2007; Peng et al., 2010; Rannals and Kapur, 2011) or scaffolding proteins (Vlachos et al., 2012; study in slice cultures) on the postsynaptic side, and changes in presynaptic release probability (Kim and Alger, 2010), presynaptic vesicle loading (De Gois et al., 2005; Hartman et al., 2006; Lau and Murthy, 2012), or GABA-producing enzymes (Peng et al., 2010; Rannals and Kapur, 2011) on the presynaptic side. Only in a few cases, changes in the number of inhibitory synapses were reported (Hartman et al., 2006; Peng et al., 2010). Homeostatic changes of inhibitory synapses could be induced in a cell autonomous fashion (Peng et al., 2010), or required a change in activity of the entire neuronal network (Hartman et al., 2006), emphasizing that there are multiple mechanisms of homeostatic plasticity at inhibitory synapses. In particular, distinct mechanisms could exist for activity-dependent downregulation and upregulation of inhibitory synapses.
Organotypic cultures
In contrast to dissociated cultures neurons in more intact tissue, such as acute slices or organotypic cultures, make more specific connections and form structured networks. This network configuration makes the interpretation of synaptic changes more complex. In slices that were submitted to activity manipulations, changes in inhibition have been observed opposite to (Marty et al., 2004; Karmarkar and Buonomano, 2006; Kim and Alger, 2010) as well as in conjunction with (Buckby et al., 2006; Echegoyen et al., 2007) changes in excitation. It was also shown that different types of homeostatic mechanisms have different time courses (Karmarkar and Buonomano, 2006) and that different subsets of inhibitory synapses can respond differently. For instance, the presence of cannabinoid receptors in a subset of inhibitory synapses renders them selectively receptive to changes in endocannabinoid levels induced by inactivity (Kim and Alger, 2010). In another example, inactivity differentially affected somatic and dendritic inhibitory inputs on CA1 pyramidal cells. Interestingly, both types of synapses showed reduction in the number of presynaptic boutons and upregulation of release probability, but the functional end-effect on inhibitory input to the postsynaptic cells was different (Chattopadhyaya et al., 2004; Bartley et al., 2008). This emphasizes that simple in vitro homeostatic rules for scaling inhibitory synapses get complicated in more complex networks. In addition, other factors such as different cell (glia) types or the extracellular environment in more intact tissue potentially influence homeostatic plasticity compared to dissociated cultured cells.
In vivo studies
Typically, when studying activity-dependent or homeostatic changes in vivo, sensory deprivation is used as experimental paradigm to lower activity levels in the primary sensory cortex (e.g., whisker trimming, monocular deprivation, or retinal lesion). While in vitro activity manipulations by pharmacological means affect the activity of all neurons in equal amounts, sensory deprivation in vivo will affect different neurons in the circuitry differentially. Therefore, in vivo responses of inhibitory synapses to changes in activity vary widely and strongly depend on the specific cell types, cortical layer, and specific circuitry (Maffei et al., 2004; Maffei and Turrigiano, 2008; Chen et al., 2011). Furthermore, it is well-known that inhibition in sensory cortex areas undergoes important developmental changes (Hensch, 2005), which means that the same deprivation paradigm can have different effects on inhibitory synapses depending on the postnatal period that is considered (Chattopadhyaya et al., 2004; Maffei et al., 2006; Maffei et al., 2010). An emerging theme from the in vivo studies is that inhibitory synapses can respond rapidly to sensory deprivation. It was shown that inhibitory axons in cortical layer 2/3 reduce the number of boutons within the first 24 h after a retinal lesion or monocular deprivation (Chen et al., 2011; Keck et al., 2011). Over longer periods, inhibitory axons in the barrel cortex were shown to sprout and form new axonal branches after whisker plucking (Marik et al., 2010). Interestingly, the reduction of inhibition was often found to precede adaptive changes of the excitatory circuitry (Marik et al., 2010; Keck et al., 2011). The rapid downregulation of inhibition might serve to render the local circuit more permissive for excitatory plasticity to occur (Ormond and Woodin, 2011; Gambino and Holtmaat, 2012). In two recent studies it was shown that inhibitory synapses that are located on spines (presumably next to an excitatory synapse) showed much higher turnover rates compared to inhibitory synapses on shaft after visual deprivation (Chen et al., 2012; vanVersendaal et al., 2012). It will be interesting to see whether direct cross talk of the two types of synapses exists.
In conclusion, there is a large amount of compelling evidence for activity-dependent adaptations in inhibitory synapses in vitro as well as in vivo. The precise expression mechanisms significantly vary between different preparations and experimental paradigms.
Axons
In this review we focus on possible feedback signals that occur in inhibitory axons in response to changes in network or synaptic activity and that induce changes in the number or properties of presynaptic terminals along the axon. The axon of a single neuron forms several thousands of presynaptic terminals (i.e., “boutons”) along its shaft and contacts many different postsynaptic neurons. Presynaptic boutons along an axon show a large variety in their volumes, in the number of synaptic vesicles and in the presence or absence of mitochondria (Shepherd and Harris, 1998). It is now well-established that neighboring boutons are not independent entities, but they continuously share and exchange molecular components of the release machinery and synaptic vesicles (Krueger et al., 2003; Darcy et al., 2006; Sabo et al., 2006; Staras, 2007; Yamada et al., 2013). Synaptic vesicles may not belong to a specific presynaptic terminal, but form a super pool of vesicles in the axonal shaft and are shared by multiple release sites (Staras et al., 2010).
The exchange of presynaptic proteins means that the exact composition of presynaptic terminals is continuously changing. These changes can occur in a correlated fashion with the postsynaptic site in some synapses, but can be uncoordinated in others (Fisher-Lavie et al., 2011; Fisher-Lavie and Ziv, 2013). Release properties and synaptic strength are highly variable between individual boutons along the same axon (Branco et al., 2008; Zhao et al., 2011; Rose et al., 2013). Therefore the demand for synaptic vesicles or other presynaptic proteins will vary between presynaptic boutons and neighboring boutons compete for available resources. Indeed, reduced availability of synaptic proteins within the axon has been shown to enhance competition between boutons (Yamada et al., 2013). In addition, vesicle exchange is regulated by neuronal activity through changes in axonal calcium levels (Kim and Ryan, 2013, 2010).
Synaptic vesicles are kept at the presynaptic terminal by interacting with a scaffolding meshwork of actin, β-catenin, synapsin, and many other proteins (Bamji et al., 2003; Takamori et al., 2006; Cingolani and Goda, 2008; Fernández-Busnadiego et al., 2010; Peng et al., 2012; Taylor et al., 2013). Synaptic vesicles can escape from the presynaptic terminal into the axon, while other vesicles that were traveling along the axonal shaft can be captured. Although the loss of a strict presynaptic compartmentalization may seem disadvantageous at first, the main advantage of sharing presynaptic material between boutons is flexibility. When presynaptic material is continuously being lost and gained at synapses, synapses can rapidly change their strength by adjusting the ratio of vesicle capture and release (Wu et al., 2013). In addition, synapses can be formed or disassembled within a few hours. It was shown that presynaptic proteins can be transported together in small packages in axons (Friedman et al., 2000; Zhai et al., 2001; Wu et al., 2013). Such multi-protein packages can be recruited to locations where new synapses are being formed and a few of these ready-to-go packages are enough to rapidly build a functional active zone and release site (Jin and Garner, 2008; Owald and Sigrist, 2009).
Live imaging of axons have shown that transient and mobile release sites exist (Krueger et al., 2003) and that transient boutons occur at predefined locations along the axon (Sabo et al., 2006; Ou and Shen, 2010; Bury and Sabo, 2011), presumably reflecting contact sites with potential postsynaptic targets (Wierenga et al., 2008; Schuemann et al., 2013). The transient nature of boutons in such locations suggest that presynaptic structures are immature or incomplete and may serve a role in “testing” a new synaptic location (Wierenga et al., 2008; Dobie and Craig, 2011; Fu et al., 2012; Schuemann et al., 2013). Transient boutons might therefore reflect failed attempts or intermediate stages of building new synapses, but they could also have a physiological function. Transient boutons, or orphan release sites, are likely capable of neurotransmitter release (Krueger et al., 2003; Coggan et al., 2005; Ratnayaka et al., 2011) and besides having a role in synapse formation, ectopic release of neurotransmitter by transient boutons could also serve to signal to nearby astrocytes or to regulate ambient neurotransmitter levels.
Synapse assembly is a complicated process involving interactions of multiple proteins. It does not necessarily need to be a linear process, where one component necessarily recruits the next, but some of the interactions could occur in parallel and the sequence of protein recruitment may vary. Rapid self-assembly of presynaptic components may be an important element during synaptogenesis. This would mean that the formation of a presynaptic terminal merely needs an initial trigger to ascertain a specific axonal location or postsynaptic partner, but then the new presynaptic terminal “unfolds” automatically by spontaneous clustering of its components. It is likely that multiple triggers can induce self-assembly. Indeed, it was recently reported that synaptic material is actively prevented from aggregating and assembling new synapses during transport (Wu et al., 2013), supporting the self-assembly hypothesis. Without prevention of aggregation, presynaptic terminals were formed at locations where no postsynaptic targets were present and no postsynaptic specializations were recruited. Furthermore, the ectopic formation of presynaptic terminals on non-neuronal cells can be induced when these cells express “synaptogenic” cell adhesion molecules (Scheiffele et al., 2000; Graf et al., 2004; Takahashi et al., 2012), indicating that a single trans-synaptic trigger is enough to start the presynaptic cascade to assemble functional release sites.
A dynamic control of the strength and number of presynaptic terminals in axons implies that control of transport, capture, and release of synaptic material are essential processes regulating the formation, maintenance, and strength of presynaptic terminals. In a dynamic axon with competing presynaptic terminals, a general change in synaptic strength is expected to also have an effect on ongoing synapse formation within the same axon and vice versa (Figure 1). For instance, enhancement of synaptic strength by increasing vesicle capture or anchoring at presynaptic terminals would also result in lower amounts of “free” vesicles in the axonal shaft thereby reducing the chance that new synapses are formed at nascent sites (Yamada et al., 2013). However, a similar increase in synaptic strength could also be achieved by increasing vesicle clustering (Wu et al., 2013), but such a mechanism would actually promote synapse formation (Figure 1). This illustrates that presynaptic plasticity and synapse formation should be considered mutually dependent processes when neighboring presynaptic terminals are sharing synaptic proteins and vesicles.
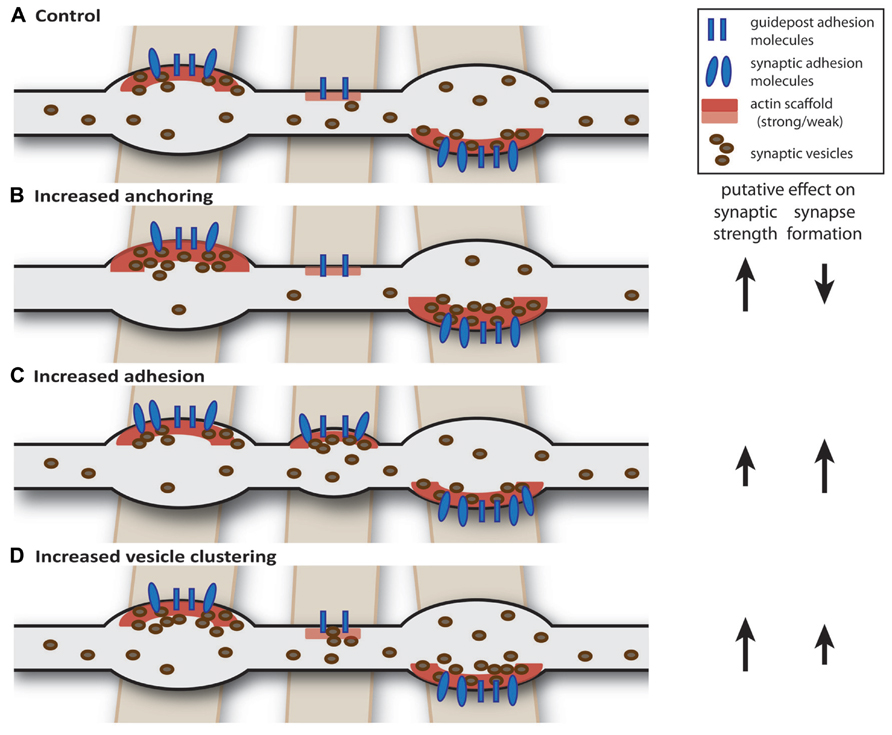
FIGURE 1. Intrinsic factors: axon-wide increase in synaptic strength or release properties may also affect synapse formation. (A) Schematic drawing of an axon (gray) forming two mature and one nascent bouton on crossing dendrites (brown). We hypothesize that axon-dendrite crossings are marked at potential synaptic locations and contain guidepost adhesion molecules (Shen and Bargmann, 2003; Shen et al., 2004) and weak actin scaffold (Chia et al., 2012). (B) Increasing anchoring of vesicles at presynaptic terminals could decrease the pool of “free” vesicles, thereby reducing the probability of forming new synapses (Yamada et al., 2013). (C) Increasing synaptic adhesion increases the number of synapses (Scheiffele et al., 2000; Takahashi et al., 2012; Kuzirian et al., 2013) and may also affect properties of existing synapses (Varoqueaux et al., 2006; Wittenmayer et al., 2009). (D) Overexpression of vesicle clustering factors induce changes in release properties, but may also promote synapse formation (Wentzel et al., 2013; Wu et al., 2013).
Inhibitory Axons
Most of the studies that were mentioned above were performed in excitatory axons and it is not entirely clear to what extent the results are also valid for inhibitory axons. Important observations have been made in live imaging studies of inhibitory axons. Presynaptic terminals in inhibitory axons were shown to be dynamic structures in vitro and in vivo. Inhibitory boutons can appear, disappear, and reappear over the course of several minutes to hours (Kuhlman and Huang, 2008; Marik et al., 2010; Keck et al., 2011; Fu et al., 2012; Schuemann et al., 2013), and the same has been shown for clusters of pre- or postsynaptic proteins at inhibitory synapses (Dobie and Craig, 2011; Chen et al., 2012; Kuriu et al., 2012; vanVersendaal et al., 2012). Bouton dynamics are comparable in vitro and in vivo and likely reflect physiological processes. Interestingly, these dynamic changes were shown to be affected by network activity and mediated, at least in part, by activation of GABA receptors (Fu et al., 2012; Kuriu et al., 2012; Schuemann et al., 2013). This could represent a mechanism by which the synaptic activity of inhibitory synapses may regulate their own stability using GABA as a feedback signal.
New inhibitory synapses can form rapidly by the appearance of a bouton at locations where the inhibitory axon is in close contact with a dendrite, without the involvement of dendritic protrusions (Wierenga et al., 2008; Dobie and Craig, 2011). This finding indicates an important contrast with the formation of excitatory synapses, in which new synapses are usually formed by the outgrowth of dendritic protrusions. It also emphasizes the important role of crosstalk between neighboring boutons within inhibitory axons for synapse formation. Nascent inhibitory synapses recruit release machinery proteins and synaptic vesicles on the presynaptic side and receptors and scaffolding molecules on the postsynaptic side within a few hours (Wierenga et al., 2008; Dobie and Craig, 2011; Kuriu et al., 2012; Schuemann et al., 2013). Interestingly, simultaneous translocations of pre- and postsynaptic proteins over several micrometers were observed in cultures (Dobie and Craig, 2011; Kuriu et al., 2012) and it will be interesting to see if such movement of inhibitory synapses can also occur in slices or in vivo. Together, these observations reveal the dynamic nature of inhibitory axons and strongly suggest that the exchange of presynaptic material between existing and emerging boutons within the axonal shaft plays an essential role in the activity-dependent formation, maintenance and plasticity of inhibitory synapses.
In general, it is not clear if molecular differences exist between excitatory and inhibitory axons, other than the neurotransmitter that is produced and loaded into synaptic vesicles. For instance, the extent or regulation of dynamic exchange between boutons could be different in these two types of axons. The protein composition of the release machinery at excitatory and inhibitory presynaptic terminals is surprisingly similar, although small difference have been reported (Gitler et al., 2004; Kerr et al., 2008; Kaeser et al., 2009; Grønborg et al., 2010; Zander et al., 2010; Boyken et al., 2013; Bragina et al., 2013). It is currently not known if some of these differences have consequences for plasticity or presynaptic dynamics within axons. Furthermore, it is not known if there are differences between axons of the various inhibitory cell types (Ascoli et al., 2008; Klausberger and Somogyi, 2008). However, there is a clear difference between excitatory and inhibitory axons in the expression of specific cell adhesion molecules at excitatory and inhibitory synapses.
Role of Cell-Adhesion Molecules in Synaptic Plasticity
The observation that inhibitory boutons appear at specific, predefined locations along the axon (Sabo et al., 2006; Wierenga et al., 2008; Schuemann et al., 2013), strongly suggests that something is marking these locations prior to bouton formation (Shen and Bargmann, 2003; Shen et al., 2004). Inhibitory axons are characterized by their tortuous and highly branched morphology and they are in close contacts with many nearby dendrites. In fact, it was shown that inhibitory axons have substantially larger overlap with the dendritic trees of their potential target neurons than expected from chance, whereas this is not the case for excitatory axons (Stepanyants et al., 2004). This suggests that inhibitory axons possibly search for or are attracted by dendrites during development. Contacts between dendrites and inhibitory axons could be maintained by guidepost cell-adhesion molecules, even without inhibitory synapses present (Shen and Bargmann, 2003; Shen et al., 2004). Their presence would mark the location of a postsynaptic dendrite and therefore a potential spot for an inhibitory synapse.
Cell adhesion molecules are transmembrane proteins, which play a role in recognition of synaptic partners during the initial contact and provide specificity of synaptic connections (Meijers et al., 2007; Wojtowicz et al., 2007). In addition, cell adhesion molecules have been shown to play a role in the process of synaptic maturation following the initial contact, in the recruitment of synaptic proteins as well as in maintaining proper synaptic function throughout the lifetime of the synapse (Dalva et al., 2007; Krueger et al., 2012; Thalhammer and Cingolani, 2013). Cell adhesion molecules can also play an active role in the process of synapse disassembly (O’Connor et al., 2009). In conclusion, cell adhesion molecules are an essential part of synapses and synaptic plasticity most likely involves regulation of cell-adhesion molecules. Here we discuss how synaptic adhesion could be regulated in an activity-dependent manner (Figure 2) and we summarize current knowledge of cell adhesion molecules that are specific for inhibitory synapses.
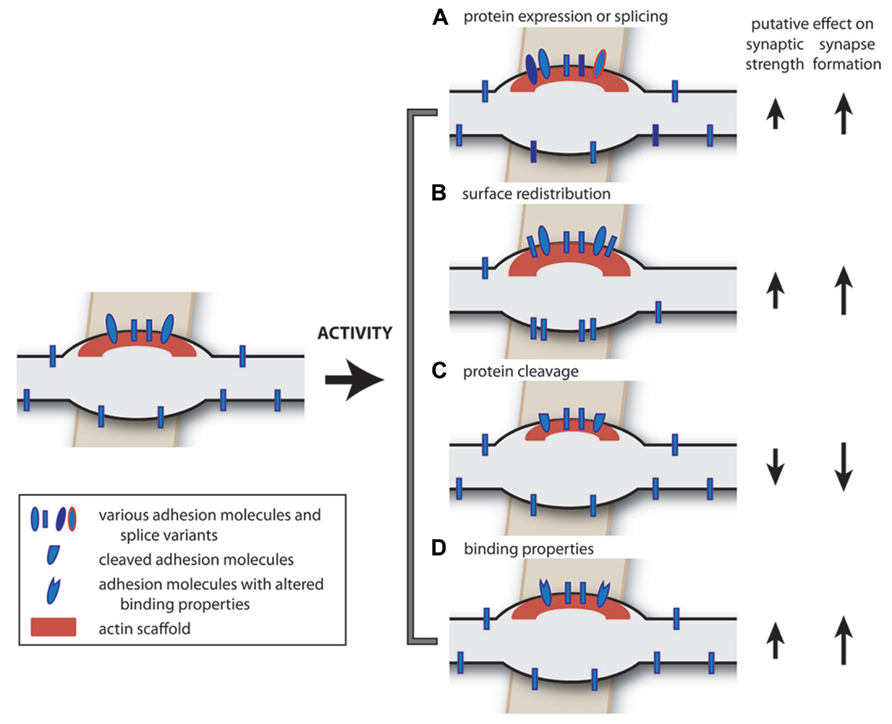
FIGURE 2. Extrinsic factors: possible activity-dependent changes in cell adhesion molecules. Neural activity may induce a number of changes in adhesion molecules. (A) The expression level of cell adhesion molecules (Cingolani et al., 2008), or their splice variants (Chih et al., 2006; Graf et al., 2006), can be regulated in an activity-dependent manner, potentially affecting synapse formation, and synapse specificity. (B) Activity-dependent redistribution of adhesion molecules over the axonal membrane can facilitate synapse formation (Fu and Huang, 2010). (C) Activity-dependent cleavage of synaptic adhesion molecules could induce synapse disassembly or plasticity (Matsumoto-Miyai et al., 2009; O’Connor et al., 2009; Peixoto et al., 2012; Suzuki et al., 2012). (D) Activity-dependent changes in binding properties of adhesion molecules (Kim et al., 2011a,b) could affect synaptic properties. In addition, the intracellular signaling pathways (not depicted) may also be regulated in an activity-dependent manner, affecting all of these processes.
Activity-dependent regulation of protein expression levels
Cell adhesion molecules often serve as recognition or identity signals to specify neuronal connectivity, and they can either promote or prevent synapse formation (Dalva et al., 2007; Bukalo and Dityatev, 2012). Neurons presumably express a combination of cell adhesion molecules and the specific combination (both the variety as well as relative levels) likely regulate the specificity and number of their synaptic contacts (Sassoè-Pognetto et al., 2011). Different cell adhesion molecules can cooperate to promote synapse formation, but the opposite is also possible: cis-interactions between different cell adhesion molecules within a neuron can preclude trans-interactions with cell adhesion molecules on neighboring neurons and thereby inhibit or prevent synapse formation (Taniguchi et al., 2007; Lee et al., 2013). Most importantly, the combination of cell adhesion molecules that a neuron expresses might not be static (Figure 2A). Indeed, for a number of cell adhesion molecules, activity-dependent changes in expression level have been observed (Pinkstaff et al., 1998; Cingolani et al., 2008; Pregno et al., 2013). Changes in expression level may be regulated by the activity level of the neuron itself or by extracellular signals from the environment, such as secreted factors from neighboring cells. For instance, TNFα, a glia-derived factor, which is secreted in an activity-dependent manner, regulates expression levels of β3 integrin and N-cadherin (Kubota et al., 2009; Thalhammer and Cingolani, 2013). In theory, local protein synthesis in the axon could also contribute to changes in expression level of cell adhesion proteins (Taylor et al., 2009, 2013; Zivraj et al., 2010), but direct experimental evidence is currently lacking.
Activity-dependent regulation of splicing
For many adhesion molecules different splice forms have been identified. Different splice variants often have different affinities for their binding partners and thereby differentially affect synapse formation or plasticity (Missler and Südhof, 1998; Hattori et al., 2008; Aoto et al., 2013). For instance, alternative splicing of neuroligins and neurexins affects specificity for excitatory or inhibitory synapses (Chih et al., 2006; Graf et al., 2006). Therefore, alternative splicing might be a way to enlarge the available set of adhesion molecules within a neuron and to enhance the range of molecular specificity of synaptic connections.
Activity-dependent regulation of cell surface distribution
To have their effect specifically at synapses, cell adhesion molecules should be enriched at synaptic membranes. There is experimental evidence that the distribution of cell adhesion molecules over the cellular surface can be regulated (Tai et al., 2007; Fu and Huang, 2010). For instance, while neurexin1α shows a diffuse pattern along the axonal membrane in inhibitory axons, neurexin1β is specifically enriched in the membrane at presynaptic terminals. Anchoring of neurexin1β at presynaptic boutons is regulated by presynaptic GABA release and subsequent GABAB receptor activation (Fu and Huang, 2010). Further investigation is needed to understand how such local changes are regulated by protein modifications or localized endo- or exocytosis and how they affect local synapse formation (Figure 2B).
Activity-dependent regulation of protein cleavage
Synaptic adhesion molecules execute their function by binding to a trans-synaptic partner at their extracellular domain. In some cases, the extracellular domain can be cleaved, with strong effects on local synaptic adhesion. For instance, activity-dependent cleavage of agrin was shown to mediate the formation of dendritic filopodia (Frischknecht et al., 2008; Matsumoto-Miyai et al., 2009) and cleavage of neuroligin-1 was shown to regulate synaptic strength of individual excitatory synapses in an activity-dependent manner (Peixoto et al., 2012; Suzuki et al., 2012). Many other adhesion molecules have known cleavage sites and it will be interesting to see whether this mechanism for activity-dependent regulation is also present at inhibitory synapses (Figure 2C).
Activity-dependent regulation of binding
For some cell adhesion molecules activity can regulate the binding properties of the proteins. For instance, interactions between cadherins are affected by extracellular calcium concentrations (Kim et al., 2011b) and integrins can switch between an active or inactive configuration by extra- or intracellular factors (Hynes, 2002). In this way, synaptic adhesion can be modulated in an activity-dependent manner without a change in synaptic composition (Figure 2D).
Activity-dependent regulation of interacting proteins
Upon binding to other cell adhesion molecules, cell adhesion molecules cluster at the cell membrane and signal through interactions with many intracellular proteins, whose levels may be regulated in an activity-dependent manner. Ultimately, signaling through synaptic adhesion molecules in the presynaptic terminal result in direct or indirect alterations of the actin cytoskeleton and vesicle recycling, affecting the number, function, and/or stability of synapses (Zhang et al., 2001; Tabuchi et al., 2002; Swiercz et al., 2008; Sun and Bamji, 2011; Takahashi and Craig, 2013). It will be crucial to identify the precise molecular pathways that are involved to fully understand how activity-dependent changes at inhibitory synapses occur.
Cell Adhesion Molecules at Inhibitory Synapses
The list of known synaptic adhesion molecules is rapidly growing, but our knowledge on the precise function of most of these proteins remains incomplete. Interestingly, several synaptic cell-adhesion molecules have been reported to be specifically involved in inhibitory, and not excitatory, synapses. These include sema4D (Paradis et al., 2007; Kuzirian et al., 2013), slitrk3 (Takahashi et al., 2012), and neuroligin-2 (Varoqueaux et al., 2004; Patrizi et al., 2008; Poulopoulos et al., 2009), and it is to be expected that new discoveries will be made in the near future. Here we briefly summarize what is known on the role of various cell adhesion molecules at inhibitory synapses.
Neuroligin-2
Postsynaptic neuroligins and their presynaptic partners neurexins are transmembrane cell adhesion molecules that have been established as important synaptic regulators (Südhof, 2008; Siddiqui and Craig, 2011; Krueger et al., 2012). When expressed in non-neuronal cells, neurexins as well as neuroligins can induce the formation of synapses in co-cultured neurons (Graf et al., 2004; Kang et al., 2008). This suggests that neurexins and neuroligins function in the initial assembly of synaptic connections. However, knock out studies showed that they are not strictly required for synaptogenesis, but they play a crucial role in the proper assembly and functional maturation of synapses (Varoqueaux et al., 2006). Neuroligin-2 localizes specifically to the postsynaptic membrane of inhibitory synapses (Varoqueaux et al., 2004; Chubykin et al., 2007) and has been shown to be a regulator of inhibitory synapse formation and function (Varoqueaux et al., 2006; Chubykin et al., 2007; Poulopoulos et al., 2009). Interestingly, a recent report suggested that the preferential localization of neuroligin-2 at inhibitory synapses can be contributed to the low abundance of β-neurexin1 in inhibitory axons (Futai et al., 2013), suggesting that the presynaptic axon determines specificity of cell adhesion molecules at inhibitory synapses. Mice lacking neuroligin-2 show impairments in inhibitory synaptic transmission and exhibit anxiety-like behavior and increased excitability (Blundell et al., 2009; Gibson et al., 2009; Jedlicka et al., 2011). Interestingly, although neuroligin-2 is present at all inhibitory synapses, only perisomatic synapses were affected in the absence of neuroligin-2 (Gibson et al., 2009). Recently, two adhesion molecules were found to show specific interactions with neuroligin-2 at inhibitory synapses. MDGA1 inhibits the interaction between neuroligin-2 and neurexins and therefore specifically suppresses the inhibitory synaptogenic activity of neuroligin-2 (Lee et al., 2013; Pettem et al., 2013). IgSF9 specifically localizes at inhibitory synapses on inhibitory neurons, where it binds to neuroligin-2 via the scaffolding protein S-SCAM (Woo et al., 2013). These findings raises the possibility that neuroligin-2 serves different functions at different inhibitory synapses, depending on its interactions with other cell adhesion molecules.
Slitrk3
Leucine-rich repeat (LRR) proteins have received considerable research attention recently. The members of the subfamily of Slitrk (Slit and Trk-like) proteins are involved in synapse formation and has been linked to several neurological disorders (Aruga and Mikoshiba, 2003; Takahashi and Craig, 2013). Slitrk3 has been shown to be present at the postsynaptic side of inhibitory synapses and it can induce the formation of inhibitory synapses through its interaction with the presynaptic tyrosine phosphatase receptor PTPδ (Takahashi et al., 2012; Yim et al., 2013). Here, the specificity for inhibitory synapses is dictated by the postsynaptic slitrk3, as it was shown that presynaptic PTPδ can interfere with other synaptic organizing molecules to promote formation of excitatory synapses (Yoshida et al., 2011, 2012). The slitrk3 knock out mouse has no gross defect in brain morphology, but shows decreased expression of inhibitory markers (Takahashi et al., 2012). Accordingly, these mice have an increased susceptibility for seizures and sometimes display spontaneous seizures. Interestingly, not all inhibitory synapses were equally affected by the loss of slitrk3. In the hippocampal CA1 region, specifically inhibitory synapses in the middle of the pyramidal layer were lost (Takahashi et al., 2012). It will be interesting to examine whether specificity of inhibitory synapses correlates with different subsets of pre- or postsynaptic neurons types or function.
Members of the closely related subfamily of leucine-rich transmembrane proteins (LRRTMs) have also been implicated in excitatory synapse formation and plasticity (Linhoff et al., 2009; Ko et al., 2011; de Wit et al., 2013; Siddiqui et al., 2013), but so far no LRRTM that is specific for inhibitory synapses has been identified.
Semaphorin-4D
Semaphorins are well-known as (repulsive) axon guidance molecules acting through rearrangements of the cytoskeleton in the growth cone. They play an important role in the early development of the brain (Pasterkamp, 2012). Some semaphorins are also expressed later in development and have been implicated in the formation and plasticity of neuronal connections (Sahay et al., 2005; Morita et al., 2006; Paradis et al., 2007; O’Connor et al., 2009; Ding et al., 2012; Mizumoto and Shen, 2013). Knocking down the membrane-bound semaphorin Sema4D was shown to specifically reduce the number of inhibitory synapses, while excitatory synapses were not affected (Paradis et al., 2007). Furthermore, application of soluble Sema4D was able to increase the density of GABAergic synapses within 30 min in rat hippocampal neurons (Kuzirian et al., 2013). These new inhibitory synapses became functional within 2 h and could restore normal levels of activity in an in vitro model for epilepsy (Kuzirian et al., 2013). The effect of sema4D on inhibitory synapses depends on the plexinB1 receptor (Kuzirian et al., 2013). It was earlier shown that activation of plexinB1 by sema4D can induce opposing responses on the cytoskeleton, depending on different interacting proteins (Basile et al., 2004; Swiercz et al., 2008; Tasaka et al., 2012), but the intracellular pathway used for inhibitory synapse formation is not known. Sema4D is a membrane-bound protein, but the protein can also be cleaved (Basile et al., 2007; Zhu et al., 2007). It was recently shown that extracellular cleavage of sema4D occurs in neurons, but does not interfere with its synaptogenic properties at inhibitory synapses (Raissi et al., 2013).
Other Cell Adhesion Molecules
There are many other cell adhesion molecule proteins and with continued research on inhibitory synapses, it is expected that more of them will be found to play a role at inhibitory synapses. Here we just mention a few that have been reported at inhibitory synapses.
Neural cell adhesion molecule (NCAM) has been reported to be important for the maturation of perisomatic inhibitory synapses in the cortex (Pillai-Nair et al., 2005; Brennaman and Maness, 2008; Chattopadhyaya et al., 2013). NCAM acts through activation of Fyn kinases and possibly recruits other adhesion molecules (Chattopadhyaya et al., 2013). Interestingly, it was recently reported that also members of the ephrin family, ephrinA5 and EphA3, can affect inhibitory synapses and they require NCAM for their action (Brennaman et al., 2013). In vivo, NCAM is polysialylated (NCAM-PSA) in an experience-dependent manner and developmental downregulation of NCAM-PSA was shown to coordinate maturation of perisomatic inhibitory synapses in the visual cortex (Di Cristo et al., 2007).
Several components of the dystrophin-associated glycoprotein complex (DGC), such as dystroglycan, dystrophin, and dystrobrevin, are also specifically located at a subset of inhibitory synapses (Knuesel et al., 1999; Brünig et al., 2002; Lévi et al., 2002; Grady et al., 2006), but the function of this complex at inhibitory synapses is not well understood. The DGC could be linked to postsynaptic neuroligin-2 via the scaffolding protein S-SCAM (Sumita et al., 2007) and to presynaptic neurexins (Sugita, 2001). Interestingly, a synaptic guanine exchange factor SynArfGEF has been identified that specifically co-localizes at inhibitory synapses, which could be involved in the downstream signaling pathway of the DGC (Fukaya et al., 2011), but its exact function remains to determined.
Integrins are receptors for extracellular matrix proteins, soluble factors, and counter-receptors on adjacent cells and they have an intracellular link to actin filaments via adaptor proteins (Hynes, 2002; Harburger and Calderwood, 2009). Integrins have been implicated in activity-dependent synaptic changes (Chavis and Westbrook, 2001; Chan et al., 2003) and in homeostatic scaling of excitatory synapses (Cingolani et al., 2008). At glycinergic inhibitory synapses in the spinal cord, postsynaptic β1 and β3 integrins have been reported to regulate glycine receptor stabilization at the postsynaptic membrane, with the two integrins acting in opposing directions (Charrier et al., 2010).
Finally, the cell adhesion molecule neurofascin has been shown to regulate the formation of a specific subset of inhibitory synapses on the axon initial segment of principal neurons (Ango et al., 2004; Burkarth et al., 2007; Kriebel et al., 2011).
Role of Secreted Factors and Retrograde Messengers at Inhibitory Synapses
Above we have described how cell adhesion molecules may provide signals to inhibitory axons from direct cell–cell contacts. However, inhibitory synapses may also be affected by signals from more distal origin. Nearby dendrites or surrounding cells can secrete trophic (or anti-trophic) factors, which may affect inhibitory synapse function and/or formation. Indeed, retrograde signals from the postsynaptic dendrite, such as endocannabinoids, nitric oxide (NO) or brain-derived neurotrophic factor (BDNF), or glutamate spillover from nearby excitatory synapses are known to regulate synaptic release at inhibitory synapses during many forms of short-term and long-term plasticity (Heifets and Castillo, 2009; Regehr et al., 2009; Castillo et al., 2011). Here we discuss secreted factors that have been linked to the formation of inhibitory synapses and that might play a role in activity-dependent regulation of the number of presynaptic terminals made by inhibitory axons.
Brain-Derived Neurotrophic Factor
Brain-derived neurotrophic factor (BDNF) is a secreted neurotrophin that has been shown in many different preparations to promote the formation and maturation of inhibitory synapses by presynaptic modifications (Vicario-Abejón et al., 1998; Huang et al., 1999; Marty et al., 2000; Yamada et al., 2002; Gottmann et al., 2009). Only excitatory neurons produce BDNF (Gottmann et al., 2009; Park and Poo, 2013) and BDNF is released from principal neurons in an activity-dependent manner (Kolarow et al., 2007; Kuczewski et al., 2008; Matsuda et al., 2009), which makes BDNF an attractive candidate molecule to regulate activity-dependent inhibitory synapse formation (Liu et al., 2007). Interestingly, the availability of postsynaptic BDNF signaling in individual neurons was shown to affect the number and strength of inhibitory synapses specifically onto the affected neurons (Ohba et al., 2005; Kohara et al., 2007; Peng et al., 2010). These cell-autonomous effects indicate the potential for BDNF in mediating changes in inhibitory synapses with high synaptic specificity. In excitatory axons, BDNF was shown to reduce the anchoring of synaptic vesicles at presynaptic terminals and thereby increase the exchange of vesicles between boutons (Bamji et al., 2006). It is currently not known if BDNF has a similar effect in inhibitory axons.
Neuregulin1
Neuregulin1 is a neurotrophic factor, which exists in various membrane-bound and diffusible isoforms. Mutations (both loss-of-functions and gain-of-function) in neuregulin1 have been linked to schizophrenia (Mei and Xiong, 2008). The main receptor for neuregulin1, ErbB4, is specifically expressed in interneurons (Vullhorst et al., 2009; Fazzari et al., 2010) and is located at postsynaptic densities of excitatory synapses in interneuron dendrites as well as at inhibitory axon terminals. An important role for neuregulin1 is the regulation of excitatory input onto interneurons through postsynaptic ErbB4 (Fazzari et al., 2010; Wen et al., 2010; Ting et al., 2011). Presynaptic ErbB4 can enhance GABA release from inhibitory synapses (Woo et al., 2007; Fazzari et al., 2010) and may affect the number of synapses made by inhibitory axons (delPino et al., 2013). In addition to ErbB4, neuregulin1 isoforms can also activate other receptors resulting in downregulation of postsynaptic GABAA receptors (Yin et al., 2013). This suggests that neuregulin1 has multiple actions on inhibitory synapses depending on the isoform and activated receptors.
FGF7
Fibroblast growth factors (FGFs) are secreted signaling glycoproteins, which exert their effect by binding to FGF receptor tyrosine kinases (FGFR). In the brain, FGF signaling is important for several developmental processes, including patterning of different brain structures and neurogenesis (Dono, 2003; Reuss and von Bohlen und Halbach, 2003). In addition, FGFs have been implicated as target-derived presynaptic organizers (Umemori et al., 2004). FGF7 is of particular interest, as it localizes specifically to inhibitory synapses in the hippocampal CA3 region, where it is secreted from the postsynaptic membrane and organizes presynaptic release properties (Terauchi et al., 2010). FGF receptors have been shown to directly interact with adenosine A2A receptors (Flajolet et al., 2008), which are important for GABA release (Cunha and Ribeiro, 2000) as well as for GABA uptake from the synaptic cleft (Cristóvão-Ferreira et al., 2009). In this way, FGFR and A2A receptors may act together to regulate GABAergic transmission in the hippocampus.
Factors From Glia Cells
Studies with neuronal and astrocyte co-cultures and astrocyte-conditioned medium have shown that astrocyte-released factors are crucial for synapse formation and plasticity (Elmariah et al., 2005; Christopherson et al., 2005a; Hughes et al., 2010; Crawford et al., 2012). For instance, thrombospondins, oligomeric proteins of the extracellular matrix produced by astrocytes (Christopherson et al., 2005b; Eroglu et al., 2009) are involved in the formation of glutamatergic synapses and the pro-inflammatory cytokine TNFα, coming from glia, (Stellwagen and Malenka, 2006) plays a role in homeostatic plasticity of these synapses. In addition, a different and so far unidentified, protein is secreted by astrocytes, which has been found to increase the density of inhibitory synapses in cultured neurons (Elmariah et al., 2005; Hughes et al., 2010).
GABA
A special secreted factor is the inhibitory neurotransmitter GABA itself. It is well-established that synapse formation does not depend on neurotransmitter release (Verhage, 2000; Harms and Craig, 2005; Schubert et al., 2013). However, the development and maturation of inhibitory synapses are influenced by their neurotransmitter GABA (Li et al., 2005; Huang and Scheiffele, 2008; Huang, 2009; Lau and Murthy, 2012). It was shown that individual axons of parvalbumin-positive basket cells are sensitive to their own GABA release (Chattopadhyaya et al., 2007; Wu et al., 2012) and that the amount of GABA release per vesicle can be regulated by activity (Hartman et al., 2006; Lau and Murthy, 2012). Inhibitory boutons are less dynamic in axons in which GABA release is impaired (Wu et al., 2012) or when GABA receptors are blocked (Schuemann et al., 2013), strongly suggesting that GABA is used as an important activity sensor for regulating activity-dependent presynaptic changes at inhibitory synapses. Both GABAA and GABAB receptors have been implicated in mediating this regulation (Fu et al., 2012; Schuemann et al., 2013), but the precise molecular mechanisms remain unknown.
Other Factors
In addition to cell adhesion molecules and secreted factors, there are many other factors that may affect activity-dependent plasticity of inhibitory axons. For instance, it is well-established that extracellular matrix molecules can play a role in the development and maturation of synapses in the central nervous system and specific interactions between cell adhesion molecules and the extracellular matrix have been revealed (Di Cristo et al., 2007; de Wit et al., 2013; Siddiqui et al., 2013). There are a few studies in which the absence or overexpression of extracellular matrix proteins affected inhibitory synapses specifically (Saghatelyan et al., 2001; Nikonenko et al., 2003; Brenneke et al., 2004; Pavlov et al., 2006; Su et al., 2010), but the underlying mechanisms remain largely unknown.
And finally, while it is clear that presynaptic components are continuously shared and exchanged between inhibitory boutons along the axons, it is not clear how exactly these proteins are dispersed along the axonal shaft. Presumably sharing occurs through passive diffusion of presynaptic proteins through the axonal shaft, but intracellular transport of synaptic cargo could also play a role. Axons contain extensive microtubule and actin networks and there are various motor proteins that deliver and ship transport vesicles, potentially affecting the amount of proteins available for exchange and synapse formation at boutons. For instance, it was shown that intra-axonal movement of mitochondria is enhanced when activity is blocked (Goldstein et al., 2008; Cai and Sheng, 2009; Obashi and Okabe, 2013), but it is not clear if this is due to enhanced motor protein activity or decreased anchoring at synapses. Further research on the possible activity-dependent regulation of intracellular transport of synaptic cargo (Guillaud et al., 2008; Maas et al., 2009; MacAskill et al., 2009) will be needed to address this issue in the future.
Conclusion
Research on activity-dependent adaptations in inhibitory axons continues to generate novel insight in the cellular processes of synapse formation and plasticity. Many open questions remain to be answered in the future and we listed a few of these in a small scheme (Figure 3). In this review we have painted a picture of the inhibitory axon as a dynamic structure that can quickly adjust to a changing environment, by responding to local signals from postsynaptic cells via adhesion molecules and to global signals from the local neuronal network. A highly dynamic inhibitory system might serve to quickly respond to changes to allow circuit rearrangements by excitatory connections. For a healthy brain changes at inhibitory and excitatory synapses need to be well-coordinated at all times as subtle defects in this coordination can cause defects in circuitry and may underlie psychiatric disorders. This means that the interplay between plasticity of excitatory and inhibitory synapses is an important factor for the stability of neuronal circuits. The precise response of the inhibitory axon will be determined by the combination of internal and external factors, such as the availability of synaptic proteins within the axon, or the combination of the extracellular factors and cell adhesion molecules that are present at the membrane. It will be an important challenge for future research to unravel the precise molecular and cellular mechanisms and to further uncover pathways that affect synapse formation and plasticity of inhibitory synapses.
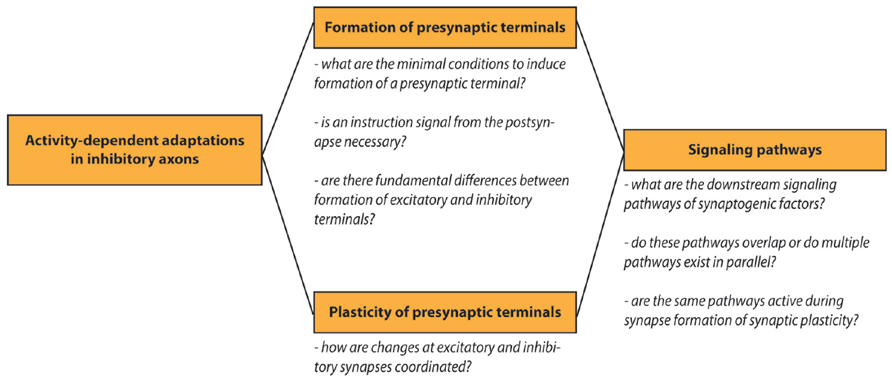
FIGURE 3. Outstanding research questions. Schematic overview of open research questions on activity-dependent adaptations in inhibitory axons.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors like to thank Anne Schuemann, Tara Keck, and Casper Hoogenraad for critical reading of the manuscript and helpful comments. Our research was supported by the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/ under REA grant agreement n 289581 (CPF), and a Marie Curie Reintegration Grant 256284 and Dutch NWO-VIDI grant (CJW).
References
Ango, F., di Cristo, G., Higashiyama, H., Bennett, V., Wu, P., and Huang, Z. J. (2004). Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell 119, 257–272. doi: 10.1016/j.cell.2004.10.004
Aoto, J., Martinelli, D. C., Malenka, R. C., Tabuchi, K., and Südhof, T. C. (2013). Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 154, 75–88. doi: 10.1016/j.cell.2013.05.060
Aruga, J., and Mikoshiba, K. (2003). Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol. Cell. Neurosci. 24, 117–129. doi: 10.1016/S1044-7431(03)00129-5
Ascoli, G. A., Alonso-Nanclares, L., Anderson, S. A., Barrionuevo, G., Benavides-Piccione, R., Burkhalter, A., et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568. doi: 10.1038/nrn2402
Bamji, S. X., Rico, B., Kimes, N., and Reichardt, L. F. (2006). BDNF mobilizes synaptic vesicles and enhances synapse formation by disrupting cadherin-beta-catenin interactions. J. Cell Biol. 174, 289–299. doi: 10.1083/jcb.200601087
Bamji, S. X., Shimazu, K., Kimes, N., Huelsken, J., Birchmeier, W., Lu, B., et al. (2003). Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40, 719–731. doi: 10.1016/S0896-6273(03)00718-9
Bartley, A. F., Huang, Z. J., Huber, K. M., and Gibson, J. R. (2008). Differential activity-dependent, homeostatic plasticity of two neocortical inhibitory circuits. J. Neurophysiol. 100, 1983–1994. doi: 10.1152/jn.90635.2008
Basile, J. R., Barac, A., Zhu, T., Guan, K.-L., and Gutkind, J. S. (2004). Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Cancer Res. 64, 5212–5224. doi: 10.1158/0008-5472.CAN-04-0126
Basile, J. R., Holmbeck, K., Bugge, T. H., and Gutkind, J. S. (2007). MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J. Biol. Chem. 282, 6899–6905. doi: 10.1074/jbc.M609570200
Bateup, H. S., Johnson, C. A, Denefrio, C. L., Saulnier, J. L., Kornacker, K., and Sabatini, B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. doi: 10.1016/j.neuron.2013.03.017
Blundell, J., Tabuchi, K., Bolliger, M. F., Blaiss, C. A., Brose, N., Liu, X., et al. (2009). Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes. Brain. Behav. 8, 114–126. doi: 10.1111/j.1601-183X.2008. 00455.x
Boyken, J., Grønborg, M., Riedel, D., Urlaub, H., Jahn, R., and Chua, J. J. E. (2013). Molecular profiling of synaptic vesicle docking sites reveals novel proteins but few differences between glutamatergic and GABAergic synapses. Neuron 78, 285–297. doi: 10.1016/j.neuron.2013.02.027
Bragina, L., Fattorini, G., Giovedì, S., Bosco, F., Benfenati, F., and Conti, F. (2013). Heterogeneity of presynaptic proteins: do not forget isoforms. Front. Cell. Neurosci. 7:8. doi: 10.3389/fncel.2013.00008
Branco, T., Staras, K., Darcy, K. J., and Goda, Y. (2008). Local dendritic activity sets release probability at hippocampal synapses. Neuron 59, 475–485. doi: 10.1016/j.neuron.2008.07.006
Brennaman, L. H., and Maness, P. F. (2008). Developmental regulation of GABAergic interneuron branching and synaptic development in the prefrontal cortex by soluble neural cell adhesion molecule. Mol. Cell. Neurosci. 37, 781–793. doi: 10.1016/j.mcn.2008.01.006
Brennaman, L. H., Zhang, X., Guan, H., Triplett, J. W., Brown, A., Demyanenko, G. P., et al. (2013). Polysialylated NCAM and ephrinA/EphA regulate synaptic development of GABAergic interneurons in prefrontal cortex. Cereb. Cortex 23, 162–177. doi: 10.1093/cercor/bhr392
Brenneke, F., Bukalo, O., Dityatev, A., and Lie, A. A. (2004). Mice deficient for the extracellular matrix glycoprotein tenascin-r show physiological and structural hallmarks of increased hippocampal excitability, but no increased susceptibility to seizures in the pilocarpine model of epilepsy. Neuroscience 124, 841–855. doi: 10.1016/j.neuroscience.2003.11.037
Brünig, I., Suter, A., Knuesel, I., Lüscher, B., and Fritschy, J.-M. (2002). GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J. Neurosci. 22, 4805–4813.
Buckby, L. E., Jensen, T. P., Smith, P. J. E., and Empson, R. M. (2006). Network stability through homeostatic scaling of excitatory and inhibitory synapses following inactivity in CA3 of rat organotypic hippocampal slice cultures. Mol. Cell. Neurosci. 31, 805–816. doi: 10.1016/j.mcn.2006.01.009
Bukalo, O., and Dityatev, A. (2012). Synaptic cell adhesion molecules. Adv. Exp. Med. Biol. 970, 97–128. doi: 10.1007/978-3-7091-0932-8_5
Burkarth, N., Kriebel, M., Kranz, E. U., and Volkmer, H. (2007). Neurofascin regulates the formation of gephyrin clusters and their subsequent translocation to the axon hillock of hippocampal neurons. Mol. Cell. Neurosci. 36, 59–70. doi: 10.1016/j.mcn.2007.06.001
Burrone, J., and Murthy, V. N. (2003). Synaptic gain control and homeostasis. Curr. Opin. Neurobiol. 13, 560–567. doi: 10.1016/j.conb.2003.09.007
Bury, L. A., and Sabo, S. L. (2011). Coordinated trafficking of synaptic vesicle and active zone proteins prior to synapse formation. Neural Dev. 6, 24. doi: 10.1186/1749-8104-6-24
Cai, Q., and Sheng, Z.-H. (2009). Mitochondrial transport and docking in axons. Exp. Neurol. 218, 257–267. doi: 10.1016/j.expneurol.2009.03.024
Castillo, P. E., Chiu, C. Q., and Carroll, R. C. (2011). Long-term plasticity at inhibitory synapses. Curr. Opin. Neurobiol. 21, 328–338. doi: 10.1016/j.conb.2011.01.006
Chan, C.-S., Weeber, E. J., Kurup, S., Sweatt, J. D., and Davis, R. L. (2003). Integrin requirement for hippocampal synaptic plasticity and spatial memory. J. Neurosci. 23, 7107–7116.
Charrier, C., Machado, P., Tweedie-Cullen, R. Y., Rutishauser, D., Mansuy, I. M., and Triller, A. (2010). A crosstalk between β1 and β3 integrins controls glycine receptor and gephyrin trafficking at synapses. Nat. Neurosci. 13, 1388–1395. doi: 10.1038/nn.2645
Chattopadhyaya, B., Baho, E., Huang, Z. J., Schachner, M., and Di Cristo, G. (2013). Neural cell adhesion molecule-mediated Fyn activation promotes GABAergic synapse maturation in postnatal mouse cortex. J. Neurosci. 33, 5957–5968. doi: 10.1523/JNEUROSCI.1306-12.2013
Chattopadhyaya, B., Cristo, G. Di, Higashiyama, H., Knott, G. W., Kuhlman, S. J., Welker, E., et al. (2004). Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J. Neurosci. 24, 9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004
Chattopadhyaya, B., Di Cristo, G., Wu, C. Z., Knott, G., Kuhlman, S., Fu, Y., et al. (2007). GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron 54, 889–903. doi: 10.1016/j.neuron.2007.05.015
Chavis, P., and Westbrook, G. (2001). Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature 411, 317–321. doi: 10.1038/35077101
Chen, J. L., Lin, W. C., Cha, J. W., So, P. T., Kubota, Y., and Nedivi, E. (2011). Structural basis for the role of inhibition in facilitating adult brain plasticity. Nat. Neurosci. 14, 587–594. doi: 10.1038/nn.2799
Chen, J. L., Villa, K. L., Cha, J. W., So, P. T. C., Kubota, Y., and Nedivi, E. (2012). Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 74, 361–373. doi: 10.1016/j.neuron.2012.02.030
Chia, P. H., Patel, M. R., and Shen, K. (2012). NAB-1 instructs synapse assembly by linking adhesion molecules and F-actin to active zone proteins. Nat. Neurosci. 15, 234–242. doi: 10.1038/nn.2991
Chih, B., Gollan, L., and Scheiffele, P. (2006). Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron 51, 171–178. doi: 10.1016/j.neuron.2006.06.005
Christopherson, K. S., Stokes, C. C. A., Barres, B. A., Ullian, E. M., Mullowney, C. E., Hell, J. W., et al. (2005a). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. doi: 10.1016/j.cell.2004.12.020
Christopherson, K. S., Ullian, E. M., Stokes, C. C. A., Mullowney, C. E., Hell, J. W., Agah, A., et al. (2005b). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. doi: 10.1016/j.cell.2004.12.020
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Cingolani, L. A., and Goda, Y. (2008). Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 9, 344–356. doi: 10.1038/nrn2373
Cingolani, L. A., Thalhammer, A., Yu, L. M. Y., Catalano, M., Ramos, T., Colicos, M. A., et al. (2008). Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron 58, 749–762. doi: 10.1016/j.neuron.2008.04.011
Coggan, J. S., Bartol, T. M., Esquenazi, E., Stiles, J. R., Lamont, S., Martone, M. E., et al. (2005). Evidence for ectopic neurotransmission at a neuronal synapse. Science 309, 446–451. doi: 10.1126/science.1108239
Crawford, D. C., Jiang, X., Taylor, A., and Mennerick, S. (2012). Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J. Neurosci. 32, 13100–13110. doi: 10.1523/JNEUROSCI.2604-12.2012
Di Cristo, G., Chattopadhyaya, B., Kuhlman, S. J., Fu, Y., Bélanger, M.-C., Wu, C. Z., et al. (2007). Activity-dependent PSA expression regulates inhibitory maturation and onset of critical period plasticity. Nat. Neurosci. 10, 1569–1577. doi: 10.1038/nn2008
Cristóvão-Ferreira, S., Vaz, S. H., Ribeiro, J. A, and Sebastião, A. M. (2009). Adenosine A2A receptors enhance GABA transport into nerve terminals by restraining PKC inhibition of GAT-1. J. Neurochem. 109, 336–347. doi: 10.1111/j.1471-4159.2009.05963.x
Cunha, R. A, and Ribeiro, J. A. (2000). Purinergic modulation of [(3)H]GABA release from rat hippocampal nerve terminals. Neuropharmacology 39, 1156–1167. doi: 10.1016/S0028-3908(99)00237-3
Dalva, M. B., McClelland, A. C., and Kayser, M. S. (2007). Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220. doi: 10.1038/nrn2075
Darcy, K. J., Staras, K., Collinson, L. M., and Goda, Y. (2006). Constitutive sharing of recycling synaptic vesicles between presynaptic boutons. Nat. Neurosci. 9, 315–321. doi: 10.1038/nn1640
delPino, I., García-Frigola, C., Dehorter, N., Brotons-Mas, J. R., Alvarez-Salvado, E., Martínez de Lagrán, M., et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168. doi: 10.1016/j.neuron.2013.07.010
Ding, J. B., Oh, W.-J., Sabatini, B. L., and Gu, C. (2012). Semaphorin 3E-Plexin-D1 signaling controls pathway-specific synapse formation in the striatum. Nat. Neurosci. 15, 215–223. doi: 10.1038/nn.3003
Dobie, F. A., and Craig, A. M. (2011). Inhibitory synapse dynamics: coordinated presynaptic and postsynaptic mobility and the major contribution of recycled vesicles to new synapse formation. J. Neurosci. 31, 10481–10493. doi: 10.1523/JNEUROSCI.6023-10.2011
Dono, R. (2003). Fibroblast growth factors as regulators of central nervous system development and function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R867–R881. doi: 10.1152/ajpregu.00533.2002
Echegoyen, J., Neu, A., Graber, K. D., and Soltesz, I. (2007). Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS ONE 2:e700. doi:10.1371/journal.pone.0000700
Elmariah, S. B., Oh, E. J., Hughes, E. G., and Balice-Gordon, R. J. (2005). Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J. Neurosci. 25, 3638–3635. doi: 10.1523/JNEUROSCI.3980-04.2005
Eroglu, C., O’Rourke, N. A., Barres, B. A., Allen, N. J., Susman, M. W., Park, C. Y., et al. (2009). Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. doi: 10.1016/j.cell.2009.09.025
Fazzari, P., Paternain, A. V, Valiente, M., Pla, R., Luján, R., Lloyd, K., et al. (2010). Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380. doi: 10.1038/nature08928
Fernández-Busnadiego, R., Zuber, B., Maurer, U. E., Cyrklaff, M., Baumeister, W., and Lucic, V. (2010). Quantitative analysis of the native presynaptic cytomatrix by cryoelectron tomography. J. Cell Biol. 188, 145–156. doi: 10.1083/jcb.200908082
Fisher-Lavie, A., Zeidan, A., Stern, M., Garner, C. C., and Ziv, N. E. (2011). Use dependence of presynaptic tenacity. J. Neurosci. 31, 16770–16780. doi: 10.1523/JNEUROSCI.3384-11.2011
Fisher-Lavie, A., and Ziv, N. E. (2013). Matching dynamics of presynaptic and postsynaptic scaffolds. J. Neurosci. 33, 13094–13100. doi: 10.1523/JNEUROSCI.2144-13.2013
Flajolet, M., Wang, Z., Futter, M., Shen, W., Nuangchamnong, N., Bendor, J., et al. (2008). FGF acts as a co-transmitter through adenosine A(2A) receptor to regulate synaptic plasticity. Nat. Neurosci. 11, 1402–1409. doi: 10.1038/nn.2216
Friedman, H. V, Bresler, T., Garner, C. C., and Ziv, N. E. (2000). Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27, 57–69. doi: 10.1016/S0896-6273(00)00009-X
Frischknecht, R., Fejtova, A., Viesti, M., Stephan, A., and Sonderegger, P. (2008). Activity-induced synaptic capture and exocytosis of the neuronal serine protease neurotrypsin. Cultures 28, 1568–1579. doi: 10.1523/JNEUROSCI.3398-07.2008.
Fu, Y., and Huang, Z. J. (2010). Differential dynamics and activity-dependent regulation of α- and β-neurexins at developing GABAergic synapses. Proc. Natl. Acad. Sci. U.S.A. 107, 22699–22704. doi: 10.1073/pnas.1011233108
Fu, Y., Wu, X., Lu, J., and Huang, Z. J. (2012). Presynaptic GABAB receptor regulates activity-dependent maturation and patterning of inhibitory synapses through dynamic allocation of synaptic vesicles. Front. Cell. Neurosci. 6:57. doi: 10.3389/fncel.2012.00057
Fukaya, M., Kamata, A., Hara, Y., Tamaki, H., Katsumata, O., Ito, N., et al. (2011). SynArfGEF is a guanine nucleotide exchange factor for Arf6 and localizes preferentially at post-synaptic specializations of inhibitory synapses. J. Neurochem. 116, 1122–1137. doi: 10.1111/j.1471-4159.2010.07167.x
Futai, K., Doty, C. D., Baek, B., Ryu, J., and Sheng, M. (2013). Specific trans-synaptic interaction with inhibitory interneuronal neurexin underlies differential ability of neuroligins to induce functional inhibitory synapses. J. Neurosci. 33, 3612–3623. doi: 10.1523/JNEUROSCI.1811-12.2013
Gambino, F., and Holtmaat, A. (2012). Spike-timing-dependent potentiation of sensory surround in the somatosensory cortex is facilitated by deprivation-mediated disinhibition. Neuron 75, 490–502. doi: 10.1016/j.neuron.2012.05.020
Gibson, J. R., Huber, K. M., and Südhof, T. C. (2009). Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. Cell 29, 13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009.
Gitler, D., Takagishi, Y., Feng, J., Ren, Y., Rodriguiz, R. M., Wetsel, W. C., et al. (2004). Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J. Neurosci. 24, 11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004
De Gois, S., Schäfer, M. K.-H., Defamie, N., Chen, C., Ricci, A., Weihe, E., et al. (2005). Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J. Neurosci. 25, 7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005
Goldstein, A. Y. N., Wang, X., and Schwarz, T. L. (2008). Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 18, 495–503. doi: 10.1016/j.conb.2008.10.003
Gottmann, K., Lessmann, V., and Mittmann, T. (2009). BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 199, 203–234. doi: 10.1007/s00221-009-1994-z
Grady, R. M., Wozniak, D. F., Ohlemiller, K. K., and Sanes, J. R. (2006). Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J. Neurosci. 26, 2841–2851. doi: 10.1523/JNEUROSCI.4823-05.2006
Graf, E. R., Kang, Y., Hauner, A. M., and Craig, A. M. (2006). Structure function and splice site analysis of the synaptogenic activity of the neurexin-1 beta LNS domain. J. Neurosci. 26, 4256–4265. doi: 10.1523/JNEUROSCI.1253-05.2006
Graf, E. R., Zhang, X., Jin, S. X., Linhoff, M. W., and Craig, A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026. doi: 10.1016/j.cell.2004.11.035
Grønborg, M., Pavlos, N. J., Brunk, I., Chua, J. J. E., Münster-Wandowski, A., Riedel, D., et al. (2010). Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J. Neurosci. 30, 2–12. doi: 10.1523/JNEUROSCI.4074-09.2010
Guillaud, L., Wong, R., and Hirokawa, N. (2008). Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat. Cell Biol. 10, 19–29. doi: 10.1038/ncb1665
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a± mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Harburger, D. S., and Calderwood, D. A. (2009). Integrin signalling at a glance. J. Cell Sci. 122, 159–163. doi: 10.1242/jcs.018093
Harms, K. J., and Craig, A. M. (2005). Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J. Comp. Neurol. 490, 72–84. doi: 10.1002/cne.20635
Hartman, K. N., Pal, S. K., Burrone, J., and Murthy, V. N. (2006). Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat. Neurosci. 9, 642–649. doi: 10.1038/nn1677
Hattori, D., Millard, S. S., Wojtowicz, W. M., and Zipursky, S. L. (2008). Dscam-mediated cell recognition regulates neural circuit formation. Annu. Rev. Cell Dev. Biol. 24, 597–620. doi: 10.1146/annurev.cellbio.24.110707.175250
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306. doi: 10.1146/annurev.physiol.010908.163149
Hensch, T. K. (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888. doi: 10.1038/nrn1787
Huang, Z. J. (2009). Activity-dependent development of inhibitory synapses and innervation pattern: role of GABA signalling and beyond. J. Physiol. 587, 1881–1888. doi: 10.1113/jphysiol.2008.168211
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., et al. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. doi: 10.1016/S0092-8674(00)81509-3
Huang, Z. J., and Scheiffele, P. (2008). GABA and neuroligin signaling: linking synaptic activity and adhesion in inhibitory synapse development. Curr. Opin. Neurobiol. 18, 77–83. doi: 10.1016/j.conb.2008.05.008
Hughes, E. G., Elmariah, S. B., and Balice-Gordon, R. J. (2010). Astrocyte secreted proteins selectively increase hippocampal GABAergic axon length, branching, and synaptogenesis. Mol. Cell. Neurosci. 43, 136–145. doi: 10.1016/j.mcn.2009.10.004
Hynes, R. O. (2002). Integrins?: bidirectional, allosteric signaling machines. Cell 110, 673–687. doi: 10.1016/S0092-8674(02)00971-6
Jedlicka, P., Hoon, M., Papadopoulos, T., Vlachos, A., Winkels, R., Poulopoulos, A., et al. (2011). Increased dentate gyrus excitability in neuroligin-2-deficient mice in vivo. Cereb. Cortex 21, 357–367. doi: 10.1093/cercor/bhq100
Jiang, X., Wang, G., Lee, A. J., Stornetta, R. L., and Zhu, J. J. (2013). The organization of two new cortical interneuronal circuits. Nat. Neurosci. 16, 210–220. doi: 10.1038/nn.3305
Jin, Y., and Garner, C. C. (2008). Molecular mechanisms of presynaptic differentiation. Annu. Rev. Cell Dev. Biol. 24, 237–262. doi: 10.1146/annurev.cellbio.23.090506.123417
Kaeser, P. S., Deng, L., Chávez, A. E., Liu, X., Castillo, P. E., and Südhof, T. C. (2009). ELKS2alpha/CAST deletion selectively increases neurotransmitter release at inhibitory synapses. Neuron 64, 227–239. doi: 10.1016/j.neuron.2009.09.019
Kang, Y., Zhang, X., Dobie, F., Wu, H., and Craig, A. M. (2008). Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J. Biol. Chem. 283, 2323–2334. doi: 10.1074/jbc.M703957200
Karmarkar, U. R., and Buonomano, D. V (2006). Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur. J. Neurosci. 23, 1575–1584. doi: 10.1111/j.1460-9568.2006.04692.x
Keck, T., Scheuss, V., Jacobsen, R. I. I., Wierenga, C. J. J., Eysel, U. T. T., Bonhoeffer, T., et al. (2011). Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron 71, 869–882. doi: 10.1016/j.neuron.2011.06.034
Kerr, A. M., Reisinger, E., and Jonas, P. (2008). Differential dependence of phasic transmitter release on synaptotagmin 1 at GABAergic and glutamatergic hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 105, 15581–15586. doi: 10.1073/pnas.0800621105
Kilman, V., Van Rossum, M. C., and Turrigiano, G. G. (2002). Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA. J. Neurosci. 22, 1328–1337.
Kim, C., Ye, F., and Ginsberg, M. H. (2011a). Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27, 321–345. doi: 10.1146/annurev-cellbio-100109-104104
Kim, J., and Alger, B. E. (2010). Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat. Neurosci. 13, 592–600. doi: 10.1038/nn.2517
Kim, S. A., Tai, C.-Y., Mok, L.-P., Mosser, E. A., and Schuman, E. M. (2011b). Calcium-dependent dynamics of cadherin interactions at cell-cell junctions. Proc. Natl. Acad. Sci. U.S.A. 108, 9857–9862. doi: 10.1073/pnas.1019003108
Kim, S. H., and Ryan, T. A. (2013). Balance of calcineurin A and CDK5 activities sets release probability at nerve terminals. J. Neurosci. 33, 8937–8950. doi: 10.1523/JNEUROSCI.4288-12.2013
Kim, S. H., and Ryan, T. A. (2010). CDK5 serves as a major control point in neurotransmitter release. Neuron 67, 797–809. doi: 10.1016/j.neuron.2010.08.003
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. doi: 10.1126/science.1149381
Knuesel, I., Mastrocola, M., Zuellig, R. A., Bornhauser, B., Schaub, M. C., and Fritschy, J. (1999). Altered synaptic clustering of GABA A receptors in mice lacking dystrophin (mdx mice). Eur. J. Neurosci. 11, 4457–4462. doi: 10.1046/j.1460-9568.1999.00887.x
Ko, J., Soler-Llavina, G. J., Fuccillo, M. V, Malenka, R. C., and Südhof, T. C. (2011). Neuroligins/LRRTMs prevent activity- and Ca2+/calmodulin-dependent synapse elimination in cultured neurons. J. Cell Biol. 194, 323–334. doi: 10.1083/jcb.201101072
Kohara, K., Yasuda, H., Huang, Y., Adachi, N., Sohya, K., and Tsumoto, T. (2007). A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J. Neurosci. 27, 7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007
Kolarow, R., Brigadski, T., and Lessmann, V. (2007). Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J. Neurosci. 27, 10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007
Kriebel, M., Metzger, J., Trinks, S., Chugh, D., Harvey, R. J., Harvey, K., et al. (2011). The cell adhesion molecule neurofascin stabilizes axo-axonic GABAergic terminals at the axon initial segment. J. Biol. Chem. 286, 24385–24393. doi: 10.1074/jbc.M110.212191
Krueger, D. D., Tuffy, L. P., Papadopoulos, T., and Brose, N. (2012). The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol. 22, 412–422. doi: 10.1016/j.conb.2012.02.012
Krueger, S. R., Kolar, A., and Fitzsimonds, R. M. (2003). The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron 40, 945–957. doi: 10.1016/S0896-6273(03)00729-3
Kubota, K., Inoue, K., Hashimoto, R., Kumamoto, N., Kosuga, A., Tatsumi, M., et al. (2009). Tumor necrosis factor receptor-associated protein 1 regulates cell adhesion and synaptic morphology via modulation of N-cadherin expression. J. Neurochem. 110, 496–508. doi: 10.1111/j.1471-4159.2009.06099.x
Kuczewski, N., Porcher, C., Ferrand, N., Pellegrino, C., Kolarow, R., Lessmann, V., et al. (2008). Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J. Neurosci. 28, 7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008
Kuhlman, S. J., and Huang, Z. J. (2008). High-resolution labeling and functional manipulation of specific neuron types in mouse brain by Cre-activated viral gene expression. PLoS ONE 3:e2005. doi: 10.1371/journal.pone.0002005
Kuriu, T., Yanagawa, Y., and Konishi, S. (2012). Activity-dependent coordinated mobility of hippocampal inhibitory synapses visualized with presynaptic and postsynaptic tagged-molecular markers. Mol. Cell. Neurosci. 49, 184–195. doi: 10.1016/j.mcn.2011.11.002
Kuzirian, M. S., Moore, A. R., Staudenmaier, E. K., Friedel, R. H., and Paradis, S. (2013). The class 4 semaphorin sema4D promotes the rapid assembly of GABaergic synapses in rodent hippocampus. J. Neurosci. 33, 8961–8973. doi: 10.1523/JNEUROSCI.0989-13.2013
Lau, C. G., and Murthy, V. N. (2012). Activity-dependent regulation of inhibition via GAD67. J. Neurosci. 32, 8521–8531. doi: 10.1523/JNEUROSCI.1245-12.2012
Lee, K., Kim, Y., Lee, S.-J., Qiang, Y., Lee, D., Lee, H. W., et al. (2013). MDGAs interact selectively with neuroligin-2 but not other neuroligins to regulate inhibitory synapse development. Proc. Natl. Acad. Sci. U.S.A. 110, 336–341. doi: 10.1073/pnas.1219987110
Lévi, S., Grady, R. M., Henry, M. D., Campbell, K. P., Sanes, J. R., and Craig, A. M. (2002). Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J. Neurosci. 22, 4274–4285.
Li, R.-W., Yu, W., Christie, S., Miralles, C. P., Bai, J., Loturco, J. J., et al. (2005). Disruption of postsynaptic GABA receptor clusters leads to decreased GABAergic innervation of pyramidal neurons. J. Neurochem. 95, 756–770. doi: 10.1111/j.1471-4159.2005.03426.x
Linhoff, M. W., Laurén, J., Cassidy, R. M., Dobie, F. A., Takahashi, H., Nygaard, H. B., et al. (2009). An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron 61, 734–749. doi: 10.1016/j.neuron.2009.01.017
Liu, Y., Zhang, L. I., and Tao, H. W. (2007). Heterosynaptic scaling of developing GABAergic synapses: dependence on glutamatergic input and developmental stage. J. Neurosci. 27, 5301–5312. doi: 10.1523/JNEUROSCI.0376-07.2007
Maas, C., Belgardt, D., Lee, H. K., Heisler, F. F., Lappe-Siefke, C., Magiera, M. M., et al. (2009). Synaptic activation modifies microtubules underlying transport of postsynaptic cargo. Proc. Natl. Acad. Sci. U.S.A. 106, 8731–8736. doi: 10.1073/pnas.0812391106
MacAskill, A. F., Rinholm, J. E., Twelvetrees, A. E., Arancibia-Carcamo, I. L., Muir, J., Fransson, A., et al. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. doi: 10.1016/j.neuron.2009.01.030
Maffei, A., Lambo, M. E., and Turrigiano, G. G. (2010). Critical period for inhibitory plasticity in rodent binocular V1. J. Neurosci. 30, 3304–3309. doi: 10.1523/JNEUROSCI.5340-09.2010
Maffei, A., Nataraj, K., Nelson, S. B., and Turrigiano, G. G. (2006). Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84. doi: 10.1038/nature05079
Maffei, A., Nelson, S. B., and Turrigiano, G. G. (2004). Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat. Neurosci. 7, 1353–1359. doi: 10.1038/nn1351
Maffei, A., and Turrigiano, G. G. (2008). Multiple modes of network homeostasis in visual cortical layer 2/3. J. Neurosci. 28, 4377–4384. doi: 10.1523/JNEUROSCI.5298-07.2008
Marik, S. A, Yamahachi, H., McManus, J. N. J., Szabo, G., and Gilbert, C. D. (2010). Axonal dynamics of excitatory and inhibitory neurons in somatosensory cortex. PLoS Biol. 8:e1000395. doi: 10.1371/journal.pbio.1000395
Marty, S., Wehrlé, R., Fritschy, J., and Sotelo, C. (2004). Quantitative effects produced by modifications of neuronal activity on the size of GABA A receptor clusters in hippocampal slice cultures. Eur. J. Neurosci. 20, 427–440. doi: 10.1111/j.1460-9568.2004.03491.x
Marty, S., Wehrle, R., and Sotelo, C. (2000). Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J. Neurosci. 20, 8087–8095.
Matsuda, N., Lu, H., Fukata, Y., Noritake, J., Gao, H., Mukherjee, S., et al. (2009). Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J. Neurosci. 29, 14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009
Matsumoto-Miyai, K., Sokolowska, E., Zurlinden, A., Gee, C. E., Lüscher, D., Hettwer, S., et al. (2009). Coincident pre- and postsynaptic activation induces dendritic filopodia via neurotrypsin-dependent agrin cleavage. Cell 136, 1161–1171. doi: 10.1016/j.cell.2009.02.034
Mei, L., and Xiong, W.-C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452. doi: 10.1038/nrn2392
Meijers, R., Puettmann-Holgado, R., Skiniotis, G., Liu, J., Walz, T., Wang, J., et al. (2007). Structural basis of Dscam isoform specificity. Nature 449, 487–491. doi: 10.1038/nature06147
Missler, M., and Südhof, T. C. (1998). Neurexins: three genes and 1001 products. Trends Genet. 14, 20–26. doi: 10.1016/S0168-9525(97)01324-3
Mizumoto, K., and Shen, K. (2013). Interaxonal interaction defines tiled presynaptic innervation in C. elegans. Neuron 77, 655–666. doi: 10.1016/j.neuron.2012.12.031
Morita, A., Yamashita, N., Sasaki, Y., Uchida, Y., Nakajima, O., Nakamura, F., et al. (2006). Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J. Neurosci. 26, 2971–2980. doi: 10.1523/JNEUROSCI.5453-05.2006
Nikonenko, A., Schmidt, S., Skibo, G., Brückner, G., and Schachner, M. (2003). Tenascin-R-deficient mice show structural alterations of symmetric perisomatic synapses in the CA1 region of the hippocampus. J. Comp. Neurol. 456, 338–349. doi: 10.1002/cne.10537
O’Connor, T. P., Cockburn, K., Wang, W., Tapia, L., Currie, E., and Bamji, S. X. (2009). Semaphorin 5B mediates synapse elimination in hippocampal neurons. Neural Dev. 4, 18. doi: 10.1186/1749-8104-4-18
Obashi, K., and Okabe, S. (2013). Regulation of mitochondrial dynamics and distribution by synapse position and neuronal activity in the axon. Eur. J. Neurosci. 38, 2350–2363. doi: 10.1111/ejn.12263
Ohba, S., Ikeda, T., Ikegaya, Y., Nishiyama, N., Matsuki, N., and Yamada, M. K. (2005). BDNF locally potentiates GABAergic presynaptic machineries: target-selective circuit inhibition. Cereb. Cortex 15, 291–298. doi: 10.1093/cercor/bhh130
Ormond, J., and Woodin, M. A. (2011). Disinhibition-mediated LTP in the hippocampus is synapse specific. Front. Cell. Neurosci. 5:17. doi: 10.3389/fncel.2011.00017
Ou, C.-Y., and Shen, K. (2010). Setting up presynaptic structures at specific positions. Curr. Opin. Neurobiol. 20, 489–493. doi: 10.1016/j.conb.2010.04.011
Owald, D., and Sigrist, S. J. (2009). Assembling the presynaptic active zone. Curr. Opin. Neurobiol. 19, 311–318. doi: 10.1016/j.conb.2009.03.003
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Paradis, S., Harrar, D. B., Lin, Y., Koon, A. C., Hauser, J. L., Griffith, E. C., et al. (2007). An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron 53, 217–232. doi: 10.1016/j.neuron.2006.12.012
Park, H., and Poo, M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23. doi: 10.1038/nrn3379
Pasterkamp, R. J. (2012). Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 13, 605–618. doi: 10.1038/nrn3302
Patrizi, A., Scelfo, B., Viltono, L., Briatore, F., Fukaya, M., Watanabe, M., et al. (2008). Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 13151–13156. doi: 10.1073/pnas.0802390105
Pavlov, I., Rauvala, H., and Taira, T. (2006). Enhanced hippocampal GABAergic inhibition in mice overexpressing heparin-binding growth-associated molecule. Neuroscience 139, 505–511. doi: 10.1016/j.neuroscience.2005.11.070
Peixoto, R. T., Kunz, P. A., Kwon, H., Mabb, A. M., Sabatini, B. L., Philpot, B. D., et al. (2012). Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 76, 396–409. doi: 10.1016/j.neuron.2012.07.006
Peng, A., Rotman, Z., Deng, P.-Y., and Klyachko, V. A. (2012). Differential motion dynamics of synaptic vesicles undergoing spontaneous and activity-evoked endocytosis. Neuron 73, 1108–1115. doi: 10.1016/j.neuron.2012.01.023
Peng, Y.-R., Zeng, S.-Y., Song, H.-L., Li, M.-Y., Yamada, M. K., and Yu, X. (2010). Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J. Neurosci. 30, 16220–16231. doi: 10.1523/JNEUROSCI.3085-10.2010
Pettem, K. L., Yokomaku, D., Takahashi, H., Ge, Y., and Craig, A. M. (2013). Interaction between autism-linked MDGAs and neuroligins suppresses inhibitory synapse development. J. Cell Biol. 200, 321–326. doi: 10.1083/jcb.201206028
Pfeffer, C. K., Xue, M., He, M., Huang, Z. J., and Scanziani, M. (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci. 16, 1068–1076. doi: 10.1038/nn.3446
Pillai-Nair, N., Panicker, A. K., Rodriguiz, R. M., Gilmore, K. L., Demyanenko, G. P., Huang, J. Z., et al. (2005). Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J. Neurosci. 25, 4659–4657. doi: 10.1523/JNEUROSCI.0565-05.2005
Pinkstaff, J. K., Lynch, G., and Gall, C. M. (1998). Localization and seizure-regulation of integrin beta 1 mRNA in adult rat brain. Brain Res. Mol. Brain Res. 55, 265–276. doi: 10.1016/S0169-328X(98)00007-2
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Harvey, K., et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. doi: 10.1016/j.neuron.2009.08.023
Pozo, K., and Goda, Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. doi: 10.1016/j.neuron.2010.04.028
Pregno, G., Frola, E., Graziano, S., Patrizi, A., Bussolino, F., Arese, M., et al. (2013). Differential regulation of neurexin at glutamatergic and GABAergic synapses. Front. Cell. Neurosci. 7:35. doi: 10.3389/fncel.2013.00035
Raissi, A. J., Staudenmaier, E. K., David, S., Hu, L., and Paradis, S. (2013). Sema4D localizes to synapses and regulates GABAergic synapse development as a membrane-bound molecule in the mammalian hippocampus. Mol. Cell. Neurosci. 57C, 23–32. doi: 10.1016/j.mcn.2013.08.004
Rannals, M. D., and Kapur, J. (2011). Homeostatic strengthening of inhibitory synapses is mediated by the accumulation of GABAA receptors. J. Neurosci. 31, 17701–17712. doi: 10.1523/JNEUROSCI.4476-11.2011
Ratnayaka, A., Marra, V., Branco, T., and Staras, K. (2011). Extrasynaptic vesicle recycling in mature hippocampal neurons. Nat. Commun. 2, 531. doi: 10.1038/ncomms1534
Regehr, W. G., Carey, M. R., and Best, A. R. (2009). Activity-dependent regulation of synapses by retrograde messengers. Neuron 63, 154–170. doi: 10.1016/j.neuron.2009.06.021
Reuss, B., and von Bohlen und Halbach, O. (2003). Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 313, 139–157. doi: 10.1007/s00441-003-0756-7
Rose, T., Schoenenberger, P., Jezek, K., and Oertner, T. G. (2013). Developmental refinement of vesicle cycling at Schaffer collateral synapses. Neuron 77, 1109–1121. doi: 10.1016/j.neuron.2013.01.021
Sabo, S. L., Gomes, R. A., and McAllister, A. K. (2006). Formation of presynaptic terminals at predefined sites along axons. J. Neurosci. 26, 10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006
Saghatelyan, A. K., Dityatev, A., Schmidt, S., Schuster, T., Bartsch, U., and Schachner, M. (2001). Reduced perisomatic inhibition, increased excitatory transmission, and impaired long-term potentiation in mice deficient for the extracellular matrix glycoprotein tenascin-R. Mol. Cell. Neurosci. 17, 226–240. doi: 10.1006/mcne.2000.0922
Sahay, A., Kim, C.-H., Sepkuty, J. P., Cho, E., Huganir, R. L., Ginty, D. D., et al. (2005). Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J. Neurosci. 25, 3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005
Saliba, R. S., Michels, G., Jacob, T. C., Pangalos, M. N., and Moss, S. J. (2007). Activity-dependent ubiquitination of GABA(A) receptors regulates their accumulation at synaptic sites. J. Neurosci. 27, 13341–13351. doi: 10.1523/JNEUROSCI.3277-07.2007
Sassoè-Pognetto, M., Frola, E., Pregno, G., Briatore, F., and Patrizi, A. (2011). Understanding the molecular diversity of GABAergic synapses. Front. Cell. Neurosci. 5:4. doi: 10.3389/fncel.2011.00004
Scheiffele, P., Fan, J., Choih, J., Fetter, R., and Serafini, T. (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. doi: 10.1016/S0092-8674(00)80877-6
Schubert, T., Hoon, M., Euler, T., Lukasiewicz, P. D., and Wong, R. O. L. (2013). Developmental regulation and activity-dependent maintenance of GABAergic presynaptic inhibition onto rod bipolar cell axonal terminals. Neuron 78, 124–137. doi: 10.1016/j.neuron.2013.01.037
Schuemann, A., Klawiter, A., Bonhoeffer, T., Wierenga, C. J., Huang, J., and Harbor, C. S. (2013). Structural plasticity of GABAergic axons is regulated by network activity and GABA A receptor activation. Front. Neural Circuits 7:113. doi: 10.3389/fncir.2013.00113
Shen, K., and Bargmann, C. I. (2003). The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans. Cell 112, 619–630. doi: 10.1016/S0092-8674(03)00113-2
Shen, K., Fetter, R. D., and Bargmann, C. I. (2004). Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell 116, 869–881. doi: 10.1016/S0092-8674(04)00251-X
Shepherd, G. M., and Harris, K. M. (1998). Three-dimensional structure and composition of CA3 – >CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J. Neurosci. 18, 8300–8310.
Siddiqui, T. J., and Craig, A. M. (2011). Synaptic organizing complexes. Curr. Opin. Neurobiol. 21, 132–143. doi: 10.1016/j.conb.2010.08.016
Siddiqui, T. J., Tari, P. K., Connor, S. A., Zhang, P., Dobie, F. A., She, K., et al. (2013). An LRRTM4-HSPG complex mediates excitatory synapse development on dentate gyrus granule cells. Neuron 79, 680–695. doi: 10.1016/j.neuron.2013.06.029
Staras, K. (2007). Share and share alike: trading of presynaptic elements between central synapses. Trends Neurosci. 30, 292–298. doi: 10.1016/j.tins.2007.04.005
Staras, K., Branco, T., Burden, J. J., Pozo, K., Darcy, K., Marra, V., et al. (2010). A vesicle superpool spans multiple presynaptic terminals in hippocampal neurons. Neuron 66, 37–44. doi: 10.1016/j.neuron.2010.03.020
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. doi: 10.1038/nature04671
Stepanyants, A., Tamás, G., and Chklovskii, D. B. (2004). Class-specific features of neuronal wiring. Neuron 43, 251–259. doi: 10.1016/j.neuron.2004.06.013
Su, J., Gorse, K., Ramirez, F., and Fox, M. A. (2010). Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J. Comp. Neurol. 518, 229–253. doi: 10.1002/cne.22228
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Sugita, S. (2001). A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 154, 435–446. doi: 10.1083/jcb.200105003
Sumita, K., Sato, Y., Iida, J., Kawata, A., Hamano, M., Hirabayashi, S., et al. (2007). Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J. Neurochem. 100, 154–166. doi: 10.1111/j.1471-4159.2006.04170.x
Sun, Y., and Bamji, S. X. (2011). β-Pix modulates actin-mediated recruitment of synaptic vesicles to synapses. J. Neurosci. 31, 17123–17133. doi: 10.1523/JNEUROSCI.2359-11.2011
Suzuki, K., Hayashi, Y., Nakahara, S., Kumazaki, H., Prox, J., Horiuchi, K., et al. (2012). Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76, 410–422. doi: 10.1016/j.neuron.2012.10.003
Swanwick, C. C., Murthy, N. R., and Kapur, J. (2006). Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol. Cell. Neurosci. 31, 481–492. doi: 10.1016/j.mcn.2005.11.002
Swiercz, J. M., Worzfeld, T., and Offermanns, S. (2008). ErbB-2 and met reciprocally regulate cellular signaling via plexin-B1. J. Biol. Chem. 283, 1893–1901. doi: 10.1074/jbc.M706822200
Tabuchi, K., Biederer, T., Butz, S., and Sudhof, T. C. (2002). CASK participates in alternative tripartite complexes in which Mint 1 competes for binding with caskin 1, a novel CASK-binding protein. J. Neurosci. 22, 4264–4273.
Tai, C.-Y., Mysore, S. P., Chiu, C., and Schuman, E. M. (2007). Activity-regulated N-cadherin endocytosis. Neuron 54, 771–785. doi: 10.1016/j.neuron.2007.05.013
Takahashi, H., and Craig, A. M. (2013). Protein tyrosine phosphatases PTPδ, PTPσ, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 36, 522–534. doi: 10.1016/j.tins.2013.06.002
Takahashi, H., Katayama, K., Sohya, K., Miyamoto, H., Prasad, T., Matsumoto, Y., et al. (2012). Selective control of inhibitory synapse development by Slitrk3-PTPδ trans-synaptic interaction. Nat. Neurosci. 15, 389–398. doi: 10.1038/nn.3040
Takamori, S., Holt, M., Stenius, K., Lemke, E. A., Grønborg, M., Riedel, D., Urlaub, H., et al. (2006). Molecular anatomy of a trafficking organelle. Cell 127, 831–846. doi: 10.1016/j.cell.2006.10.030
Taniguchi, H., Gollan, L., Scholl, F. G., Mahadomrongkul, V., Dobler, E., Limthong, N., et al. (2007). Silencing of neuroligin function by postsynaptic neurexins. J. Neurosci. 27, 2815–2824. doi: 10.1523/JNEUROSCI.0032-07.2007
Tasaka, G.-I., Negishi, M., and Oinuma, I. (2012). Semaphorin 4D/plexin-B1-mediated M-Ras GAP activity regulates actin-based dendrite remodeling through Lamellipodin. J. Neurosci. 32, 8293–8305. doi: 10.1523/JNEUROSCI.0799-12.2012
Taylor, A. M., Berchtold, N. C., Perreau, V. M., Tu, C. H., Li Jeon, N., and Cotman, C. W. (2009). Axonal mRNA in uninjured and regenerating cortical mammalian axons. J. Neurosci. 29, 4697–4707. doi: 10.1523/JNEUROSCI.6130-08.2009
Taylor, A. M., Wu, J., Tai, H.-C., and Schuman, E. M. (2013). Axonal translation of β-catenin regulates synaptic vesicle dynamics. J. Neurosci. 33, 5584–5589. doi: 10.1523/JNEUROSCI.2944-12.2013
Terauchi, A., Johnson-Venkatesh, E. M., Toth, A. B., Javed, D., Sutton, M. A., and Umemori, H. (2010). Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465, 783–787. doi: 10.1038/nature09041
Thalhammer, A., and Cingolani, L. A. (2013). Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology pii: S0028-3908(13)00111-1. doi: 10.1016/j.neuropharm.2013.03.015
Ting, A. K., Chen, Y., Wen, L., Yin, D.-M., Shen, C., Tao, Y., et al. (2011). Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons. J. Neurosci. 31, 15–25. doi: 10.1523/JNEUROSCI.2538-10.2011
Turrigiano, G. (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 89–103. doi: 10.1146/annurev-neuro-060909-153238
Turrigiano, G. G. (1999). Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 22, 221–227. doi: 10.1016/S0166-2236(98)01341-1
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. doi: 10.1016/j.cell.2008.10.008
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. doi: 10.1038/36103
Tyagarajan, S. K., and Fritschy, J.-M. (2010). GABA(A) receptors, gephyrin and homeostatic synaptic plasticity. J. Physiol. 588, 101–106. doi: 10.1113/jphysiol.2009.178517
Umemori, H., Linhoff, M. W., Ornitz, D. M., and Sanes, J. R. (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257–270. doi: 10.1016/j.cell.2004.06.025
vanVersendaal, D., Rajendran, R., Saiepour, M. H., Klooster, J., Smit-Rigter, L., Sommeijer, J.-P., et al. (2012). Elimination of inhibitory synapses is a major component of adult ocular dominance plasticity. Neuron 74, 374–383. doi: 10.1016/j.neuron.2012.03.015
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., et al. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. doi: 10.1016/j.neuron.2006.09.003
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 83, 449–456. doi: 10.1078/0171-9335-00410
Verhage, M. (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287, 864–869. doi: 10.1126/science.287.5454.864
Vicario-Abejón, C., Collin, C., Mckay, R. D. G., and Segal, M. (1998). Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J. Neurosci. 18, 7256–7271.
Vlachos, A., Reddy-Alla, S., Papadopoulos, T., Deller, T., and Betz, H. (2012). Homeostatic regulation of gephyrin scaffolds and synaptic strength at mature hippocampal GABAergic postsynapses. Cereb. Cortex 23, 2700–2711. doi: 10.1093/cercor/bhs260
Vullhorst, D., Neddens, J., Karavanova, I., Tricoire, L., Petralia, R. S., McBain, C. J., et al. (2009). Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J. Neurosci. 29, 12255–12264. doi: 10.1523/JNEUROSCI.2454-09.2009
Wen, L., Lu, Y.-S., Zhu, X.-H., Li, X.-M., Woo, R.-S., Chen, Y.-J., et al. (2010). Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. U.S.A. 107, 1211–1216. doi: 10.1073/pnas.0910302107
Wenner, P. (2011). Mechanisms of GABAergic homeostatic plasticity. Neural Plast. 2011, 489470. doi: 10.1155/2011/489470
Wentzel, C., Sommer, J. E., Nair, R., Stiefvater, A., Sibarita, J.-B., and Scheiffele, P. (2013). mSYD1A, a mammalian synapse-defective-1 protein, regulates synaptogenic signaling and vesicle docking. Neuron 78, 1012–1023. doi: 10.1016/j.neuron.2013.05.010
Wierenga, C. J., Becker, N., and Bonhoeffer, T. (2008). GABAergic synapses are formed without the involvement of dendritic protrusions. Nat. Neurosci. 11, 1044–1052. doi: 10.1038/nn.2180
Wierenga, C. J., Walsh, M. F., and Turrigiano, G. G. (2006). Temporal regulation of the expression locus of homeostatic plasticity. J. Neurophysiol. 96, 2127–33. doi: 10.1152/jn.00107.2006
de Wit, J., O’Sullivan, M. L., Savas, J. N., Condomitti, G., Caccese, M. C., Vennekens, K. M., et al. (2013). Unbiased discovery of glypican as a receptor for LRRTM4 in regulating excitatory synapse development. Neuron 79, 696–711. doi: 10.1016/j.neuron.2013.06.049
Wittenmayer, N., Körber, C., Liu, H., Kremer, T., Varoqueaux, F., Chapman, E. R., et al. (2009). Postsynaptic neuroligin1 regulates presynaptic maturation. Proc. Natl. Acad. Sci. U.S.A. 106, 13564–13569. doi: 10.1073/pnas.0905819106
Wojtowicz, W. M., Wu, W., Andre, I., Qian, B., Baker, D., and Zipursky, S. L. (2007). A vast repertoire of Dscam binding specificities arises from modular interactions of variable Ig domains. Cell 130, 1134–1145. doi: 10.1016/j.cell.2007.08.026
Woo, J., Kwon, S.-K., Nam, J., Choi, S., Takahashi, H., Krueger, D., et al. (2013). The adhesion protein IgSF9b is coupled to neuroligin 2 via S-SCAM to promote inhibitory synapse development. J. Cell Biol. 201, 929–944. doi: 10.1083/jcb.201209132
Woo, R.-S., Li, X.-M., Tao, Y., Carpenter-Hyland, E., Huang, Y. Z., Weber, J., et al. (2007). Neuregulin-1 enhances depolarization-induced GABA release. Neuron 54, 599–610. doi: 10.1016/j.neuron.2007.04.009
Wu, X., Fu, Y., Knott, G., Lu, J., Di Cristo, G., and Huang, J. (2012). GABA signaling promotes synapse elimination and axon pruning in developing cortical inhibitory interneurons. J. Neurosci. 32, 331–343. doi: 10.1523/JNEUROSCI.3189-11.2012
Wu, Y. E., Huo, L., Maeder, C. I., Feng, W., and Shen, K. (2013). The balance between capture and dissociation of presynaptic proteins controls the spatial distribution of synapses. Neuron 78, 994–1011. doi: 10.1016/j.neuron.2013.04.035
Yamada, M. K., Nakanishi, K., Ohba, S., Nakamura, T., Ikegaya, Y., Nishiyama, N., et al. (2002). Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J. Neurosci. 22, 7580–7585.
Yamada, T., Yang, Y., Huang, J., Coppola, G., Geschwind, D. H., and Bonni, A. (2013). Sumoylated MEF2A coordinately eliminates orphan presynaptic sites and promotes maturation of presynaptic boutons. J. Neurosci. 33, 4726–4740. doi: 10.1523/JNEUROSCI.4191-12.2013
Yim, Y. S., Kwon, Y., Nam, J., In, H., Lee, K., Goo, D., et al. (2013). Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 110, 4057–4062. doi: 10.1073/pnas.1209881110
Yin, D.-M., Chen, Y.-J., Lu, Y.-S., Bean, J. C., Sathyamurthy, A., Shen, C., et al. (2013). Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron 78, 644–657. doi: 10.1016/j.neuron.2013.03.028
Yizhar, O., Fenno, L. E., Prigge, M., Schneider, F., Davidson, T. J., O’Shea, D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. doi: 10.1038/nature10360
Yoshida, T., Shiroshima, T., Lee, S.-J., Yasumura, M., Uemura, T., Chen, X., et al. (2012). Interleukin-1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J. Neurosci. 32, 2588–2600. doi: 10.1523/JNEUROSCI.4637-11.2012
Yoshida, T., Yasumura, M., Uemura, T., Lee, S.-J., Ra, M., Taguchi, R., et al. (2011). IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase δ. J. Neurosci. 31, 13485–13499. doi: 10.1523/JNEUROSCI.2136-11.2011
Zander, J.-F., Münster-Wandowski, A., Brunk, I., Pahner, I., Gómez-Lira, G., Heinemann, U., et al. (2010). Synaptic and vesicular coexistence of VGLUT and VGAT in selected excitatory and inhibitory synapses. J. Neurosci. 30, 7634–7645. doi: 10.1523/JNEUROSCI.0141-10.2010
Zhai, R. G., Vardinon-Friedman, H., Cases-Langhoff, C., Becker, B., Gundelfinger, E. D., Ziv, N. E., et al. (2001). Assembling the presynaptic active zone: a characterization of an active zone precursor vesicle. Neuron 29, 131–143. doi: 10.1016/S0896-6273(01)00185-4
Zhang, Y., Luan, Z., Liu, A., and Hu, G. (2001). The scaffolding protein CASK mediates the interaction between rabphilin3a and beta-neurexins. FEBS Lett. 497, 99–102. doi: 10.1016/S0014-5793(01)02450-4
Zhao, C., Dreosti, E., and Lagnado, L. (2011). Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J. Neurosci. 31, 7492–7496. doi: 10.1523/JNEUROSCI.6636-10.2011
Zhu, L., Bergmeier, W., Wu, J., Jiang, H., Stalker, T. J., Cieslak, M., et al. (2007). Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc. Natl. Acad. Sci. U.S.A. 104, 1621–1626. doi: 10.1073/pnas.0606344104
Keywords: GABAergic synapses, interneurons, homeostatic plasticity, axons, cell adhesion molecules
Citation: Frias CP and Wierenga CJ (2013) Activity-dependent adaptations in inhibitory axons. Front. Cell. Neurosci. 7:219. doi: 10.3389/fncel.2013.00219
Received: 27 September 2013; Accepted: 30 October 2013;
Published online: 21 November 2013.
Edited by:
Jaichandar Subramanian, Massachusetts Institute of Technology, USAReviewed by:
Arianna Maffei, Suny Stony Brook, USAAndreas Vlachos, Goethe University Frankfurt, Germany
Copyright © 2013 Frias and Wierenga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Corette J. Wierenga, Division of Cell Biology, Faculty of Science, Utrecht University, Padualaan 8, 3584 CH Utrecht, Netherlands e-mail:Yy5qLndpZXJlbmdhQHV1Lm5s