 Chrysanthi Samara
Chrysanthi Samara Olivier Poirot
Olivier Poirot Enric Domènech-Estévez
Enric Domènech-Estévez Roman Chrast
Roman Chrast- Department of Medical Genetics, University of Lausanne, Lausanne, Switzerland
The integrity and function of neurons depend on their continuous interactions with glial cells. In the peripheral nervous system glial functions are exerted by Schwann cells (SCs). SCs sense synaptic and extrasynaptic manifestations of action potential propagation and adapt their physiology to support neuronal activity. We review here existing literature data on extrasynaptic bidirectional axon-SC communication, focusing particularly on neuronal activity implications. To shed light on underlying mechanisms, we conduct a thorough analysis of microarray data from SC-rich mouse sciatic nerve at different developmental stages and in neuropathic models. We identify molecules that are potentially involved in SC detection of neuronal activity signals inducing subsequent glial responses. We further suggest that alterations in the activity-dependent axon-SC crosstalk impact on peripheral neuropathies. Together with previously reported data, these observations open new perspectives for deciphering glial mechanisms of neuronal function support.
Introduction
Neurons generate and propagate action potentials (APs) over long distances along their axons. Their functional and structural integrity depend on their partnership with adjacent glial cells. Glia confers trophic and metabolic support, regulates neuronal structure, insulates axons, controls the neuronal environment and has immunoprotective role. In the peripheral nervous system (PNS) the majority of these functions are exerted by Schwann cells (SCs) (Griffin and Thompson, 2008; Nave, 2010). Most SCs are aligned along peripheral axons of the sensory, motor, and autonomic nervous system, and are either myelinating (mSCs) or non-myelinating. The latter include immature SCs (iSCs) and mature non-myelinating SCs (nmSCs) in Remak bundles. Furthermore, the PNS contains perineuronal satellite cells enwrapping the neuronal soma, perisynaptic SCs in neuromuscular junctions (NMJs), and SCs of sensory transducers.
SCs were assumed to be passive in nature. However, experimental observations have radically challenged this concept. Converging evidence suggests that SCs are excitable, able to sense neuronal activity and generate appropriate feedback responses to support and control neuronal function. This dynamic reciprocal activity-dependent SC-neuron communication is the focus of our perspective. Although the majority of respective information has stemmed from studies on NMJs (Feng and Ko, 2007), we review here only the less well-studied extrasynaptic interactions between SCs and active axons under physiological and pathological conditions. We put into perspective the current literature with some of our recent data, and point to future directions in the field.
Detection of Axonal Activity by SCs
Intercellular interactions can be mediated through electrical fields generated in a cell and depolarizing neighboring cells bearing voltage sensors (ephaptic communication), via paracrine signaling, and by physical coupling, for instance through adhesion molecules or gap junctions (GJs). Indications exist for the utilization of all three means in activity-dependent interactions among PNS neurons and glia.
Signals Transmitted by Active Axons
APs are generated by activation of specific voltage–gated Na+ (NaV) and K+ (KV) channels, and propagate autoregeneratively along axons. In non-myelinated fibers APs travel successively through ion channels expressed all along the axons (Figure 1A1) (Debanne et al., 2011). In myelinated fibers, ion channels are mainly clustered in nodal (NaV1.6, KV7.2-3) and juxtaparanodal (JPN, KV1.1-2) regions, and conduction is saltatory (Figures 1A2,A3) (Debanne et al., 2011; Buttermore et al., 2013). Ion flows generate local currents in the periaxonal space, which can influence surrounding cells via ephaptic coupling (Debanne et al., 2011).
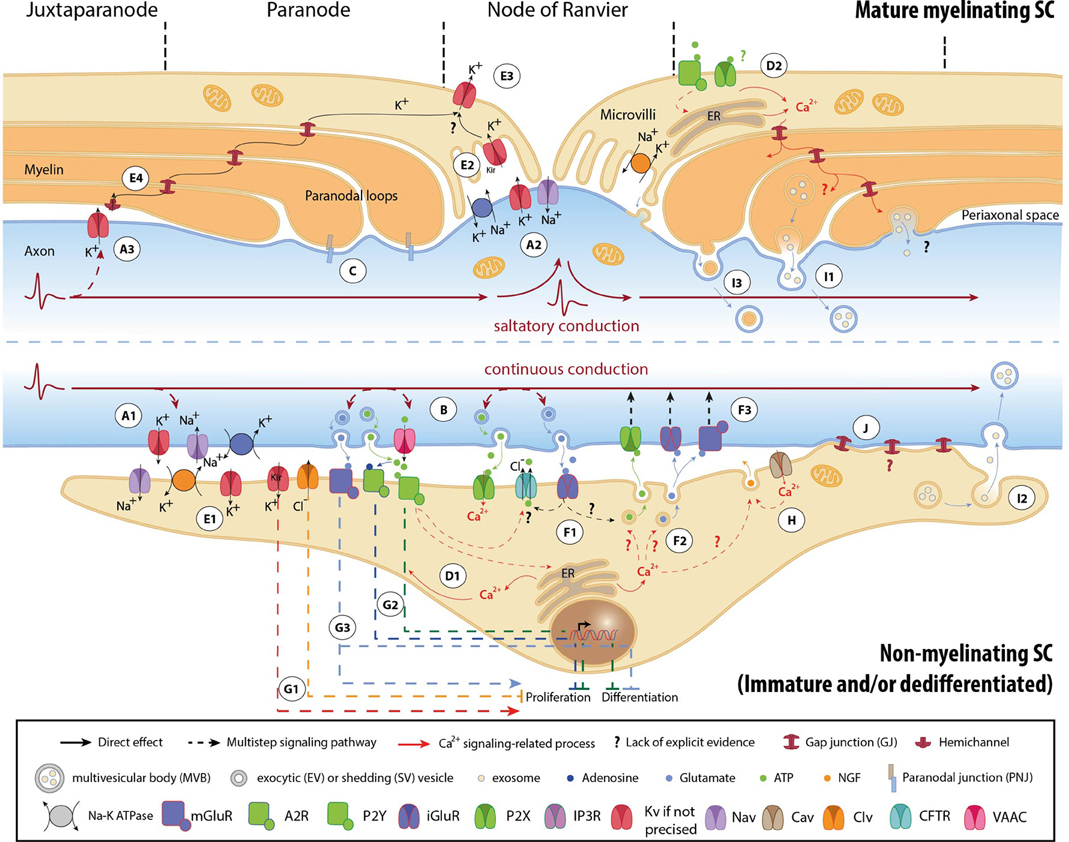
Figure 1. Mechanisms involved in activity-dependent axon-Schwann cell bilateral communication. Schematic representation of the different molecules and mechanisms described in myelinated (upper part) and non-myelinated (lower part) PNS fibers. (A) Ephaptic communication through ion flows across the plasmalemma of unmyelinated (A1) and myelinated axons (A2, A3). (B) Paracrine signaling from axons to SCs. (C) Physical coupling between axons and mSCs. (D) SC Ca2+ transients developing after neuronal stimulation. In nmSCs activation of purinergic receptors leads to increase of cytoplasmic Ca2+ due to influx from the extracellular space, or efflux from intracellular stores (D1) (Stevens et al., 1998; Stevens and Fields, 2000; Stevens et al., 2004). mSCs express both P2X and P2Y receptors, and also respond to ATP stimulation by Ca2+ increase (D2) (Mayer et al., 1998; Grafe et al., 1999). Indications suggest that Ca2+ transients expand in the whole paranodal region through GJs (Toews et al., 2007). The origin of ATP in mature myelinated fibers, however, is not clear. High ATP levels, sufficient to activate glial receptors, are probably generated only during high frequency activity or after injury. (E) K+ buffering and ion homeostasis. K+ uptake by nmSCs through the Na+/K+ pump and KV channels (E1) (Robert and Jirounek, 1994). In mSCs, inward rectifying KV channels (IRK1/Kir2.1 and IRK3/Kir2.3), and Na+/K+ ATPases are concentrated in microvilli (E2), where massive increase of K+ occurs during neuronal activity (Mi et al., 1996; Baker, 2002). Abaxonal KV1.5 channels in the nodal area may further assist to K+ removal (E3) (Mi et al., 1995; Baker, 2002). In juxtaparanodal and internodal regions, axonal KV1 channels may act in conjunction with closely apposed SC hemichannels and with GJs of the Schmidt-Lanterman incisures (SLIs) for the same purpose (E4, see also A3) (Altevogt et al., 2002; Mierzwa et al., 2010; Nualart-Marti et al., 2013). (F) Paracrine signaling from SCs to axons. Activation of P2Y and AMPA receptors acts in a positive feedback loop, triggering ATP release by nmSCs, through vesicular exocytosis or via ion transporters, such as CFTR (F1) (Liu and Bennett, 2003; Liu et al., 2005). Administration of ATP on proliferating SCs induces secretion of the excitatory amino acids Glu and aspartate, via intracellular Ca2+ store-dependent mechanisms (F2) (Jeftinija and Jeftinija, 1998). ATP and excitatory amino acids can reciprocally bind to ionotropic and metabotropic Glu-, and P2X-receptors on unmyelinated peripheral axons and influence their excitability (F3) (Agrawal and Evans, 1986; Kinkelin et al., 2000; Carlton et al., 2001; Irnich et al., 2001). (G) Regulation of SC fate by neuronal activity through activation of ion channels (G1) (Wilson and Chiu, 1993; Pappas and Ritchie, 1998; Sobko et al., 1998), purinergic metabotropic P2Y1 receptors and A2A GPCRs by ATP and its metabolite adenosine (G2) (Stevens and Fields, 2000; Stevens et al., 2004; Fields and Burnstock, 2006), and of mGluRs (G3) (Saitoh and Araki, 2010). (H) Neurotrophic axonal support by SCs. (I) Vesicular transfer of molecules from SCs to axons. Exosomes, which are enclosed in multivesicular bodies (MVB), move from mSCs to axons through cytoplasmic-rich regions like the SLIs and paranodal domains (I1), or can be released from dedifferentiated/iSCs close to neuronal growth cones after injury (I2) (Lopez-Verrilli and Court, 2012). Shedding vesicles (SVs) are directly generated from SC plasma membrane evaginations usually in microvilli and paranodal areas of mSCs, and can fuse or be endocytosed by axons (I3) (Court et al., 2008; Cocucci et al., 2009; Lopez-Verrilli and Court, 2012). (J) Potential direct transfer route of SC molecules via GJs. Abbreviations: CaV, voltage-gated Ca2+ channel; ClV, voltage-gated Cl− channel; KV, voltage-gated K+ channel; Kir, inwardly rectifying K+ channel, NaV, voltage-gated Na+ channel; CFTR, Cystic Fibrosis Transmembrane conductance Regulator; VAAC, Volume-Activated Anion Channel; A2R, adenosine receptor 2; P2X and P2Y, purinergic receptor; iGluR, ionotropic glutamate receptor; mGluR, metabotropic glutamate receptor; GPCR, G-protein coupled receptor; NGF, nerve growth factor; ER, Endoplasmic Reticulum.
Firing axons also release neurotransmitters (Figure 1B). Electrical or chemical stimulation in vitro induces extrasynaptic axonal ATP secretion through volume-activated anion channels (VAACs), via vesicular pathways (Verderio et al., 2006; Fields and Ni, 2010). Electrical stimulation (ES) evokes vesicular release of glutamate (Glu) along DRG axons, at least in cocultures with oligodendrocytes (Wake et al., 2011). Observations demonstrating exocytosis of large dense core vesicles by chemically depolarized axons of trigeminal ganglion neurons further support the concept of activity-induced extrasynaptic axonal secretion (Sobota et al., 2010).
In addition, axons are physically coupled to SCs via adhesive junctions, such as the paranodal junctions (PNJs) (Figure 1C) (Buttermore et al., 2013). The expression of specific axonal adhesion molecules is under regulation by ES in a pattern-specific manner (Itoh et al., 1997).
Detection of Axonal Signals by SC Activity Sensors
SC responses to neuronal activity were initially recorded on the squid giant axon by electrophysiology (Evans et al., 1991). ES of axons or perfusion of neurotransmitters induced SC membrane hyperpolarization (Evans et al., 1991). Similar responses have been also reported in vertebrates, mainly in the form of SC Ca2+ transients that develop subsequently to ES of myelinated and unmyelinated fibers (Figures 1D1,D2) (Brunet and Jirounek, 1994; Lev-Ram and Ellisman, 1995; Mayer et al., 1999).
mSCs and nmSCs express molecules, which allow them to respond to electrical or chemical axonal stimuli (Figure 1). SC “activity sensors,” including voltage- and ligand-gated ion channels, transporters, pumps, G-protein coupled receptors (GPCRs), connexins (Cx) of hemichannels and GJs, have been detected at mRNA and protein levels in vivo (animal tissues or human biopsies), ex vivo (nerve preparations) and/or in vitro (SC cultures), using biochemical and functional approaches (Dememes et al., 1995; Dezawa et al., 1998; Mayer et al., 1998; Verkhratsky and Steinhauser, 2000; Altevogt et al., 2002; Baker, 2002; Fields and Burnstock, 2006; Loreti et al., 2006; Magnaghi et al., 2006; Saitoh and Araki, 2010; Procacci et al., 2012; Nualart-Marti et al., 2013). A summary of the so far-identified SC receptors and ion channels is presented in Table 1.
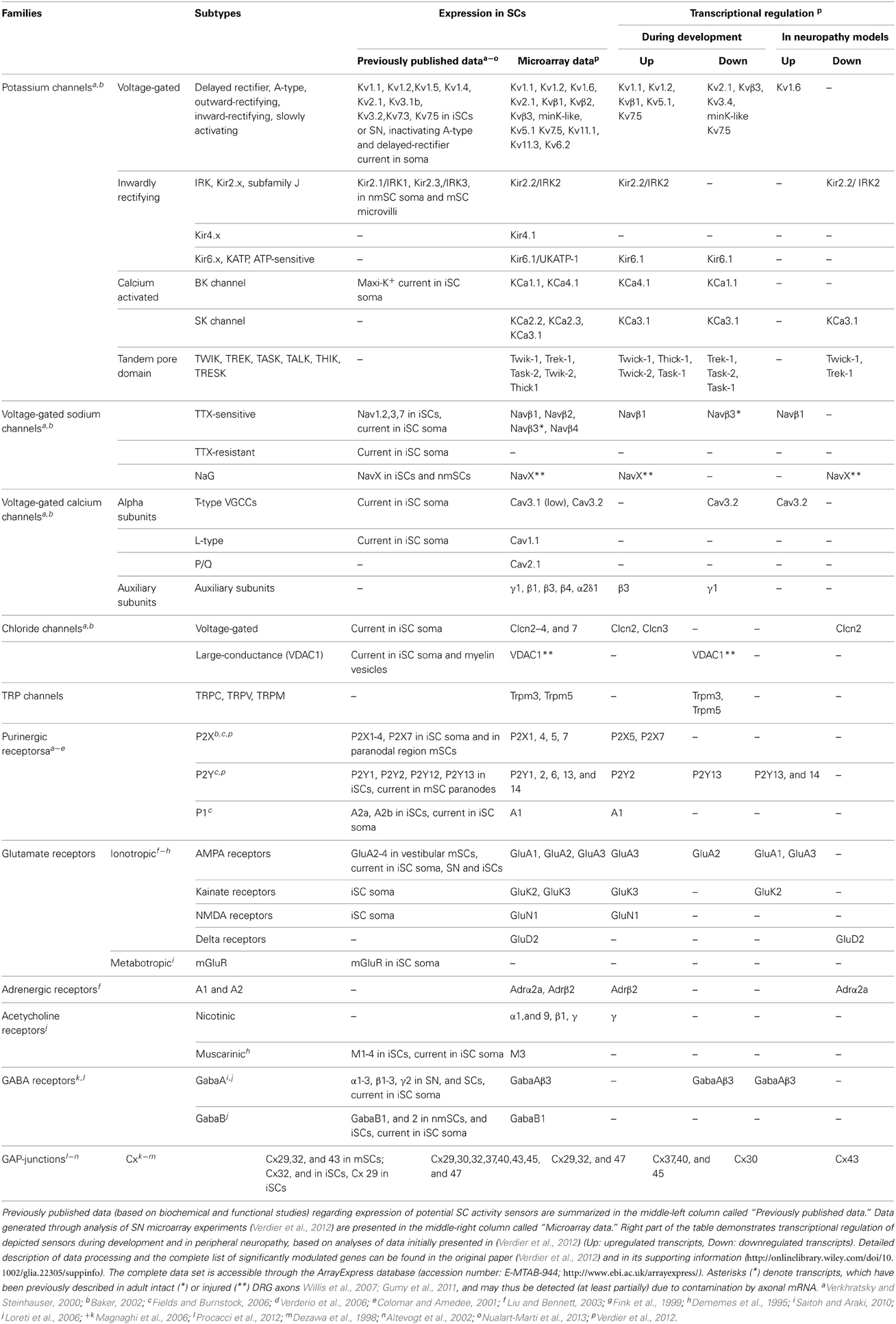
Table 1. Expression and regulation of potential SC activity sensors.
Developmental Regulation of SC Activity Sensors
Responsiveness of SCs to neuronal activity is developmentally regulated. Downregulation of KV channel expression during early myelination, and clustering to microvilli in mature mSCs is a characteristic example (Figure 1) (Wilson and Chiu, 1990). However, scarce evidence exists regarding the developmental regulation of other SC activity sensors. To gain further insight, we analyzed microarray data previously published by our group (Verdier et al., 2012), on wild type (WT) mouse sciatic nerve (SN) at different developmental stages. Since the analyzed samples are highly enriched in SCs, we expect that the majority of the detected sensors represent SC molecules and do not derive from axon specific transcripts (Willis et al., 2007; Gumy et al., 2011), (see also Table 1). Our results -summarized in Table 1- corroborate and complete existing data, confirming the expression of specific voltage- (e.g., NaV, KV, voltage-gated Ca2+ channels; CaV, ClV), and ligand-gated (e.g., purinergic P2X and ionotropic glutamate receptors -iGluRs) ion channels, and of GPCRs (e.g., purinergic P2Y, muscarinic acetylcholine receptors, GABAB receptors) (Fink et al., 1999; Baker, 2002; Loreti et al., 2006; Magnaghi et al., 2006). In addition, they reveal previously non-described mammalian SC expression of nicotinic acetylcholine receptors and TRP channels. Apart from the known regulation of K+ channels, our data suggest that expression of Na+, Ca2+, Cl−, and TRP channels, purinergic receptors and iGluRs is also significantly regulated during development.
These transcriptional modulations could result as adaptations of SCs to different neuronal firing modes. The reduction and restriction of KV channels in mSC microvilli most likely corresponds to the need for K+ buffering mainly in nodal regions (see also paragraph “K+ uptake by SCs”) (Wilson and Chiu, 1990; Baker, 2002). In addition, nmSC inwardly rectifying K+ (Kir)-currents and T-type CaV depend on axonal firing (Konishi, 1994; Beaudu-Lange et al., 2000). Given that the firing patterns of nerve fibers change during maturation (Fitzgerald, 1987), we speculate that developmental regulation of SC activity sensors could be a direct glial response to axonal activity alterations. Alternatively, it may reflect mere phenotypic changes during SC maturation.
Further SC responses to neuronal activity will be the focus of the following paragraphs.
SC Responses to Axonal Activity Signals
Detection of axonal activity by glial sensors enables SCs to develop appropriate responses and -in a feedback loop- regulate the function of underlying axons. We will discuss the nature and the potential biological significance of those SC responses, focusing particularly on their direct (via ion balance regulation, neurotransmitter secretion and myelination) or indirect (by conferring metabolic support) impact on axonal activity.
Regulation of Axonal Excitability
K+ uptake by SCs
During prolonged neuronal activity, Na+ and K+ ions tend to accumulate in the axoplasm and in the periaxonal space respectively. Maintenance of neuronal excitability requires maintenance of ion homeostasis and fast restoration of the axonal resting potential. Both nmSC and mSCs contribute to it by buffering extracellular K+ ions, mainly through the activity of Na+/K+ pumps and KV channels (for more details see Figure 1E).
SC neurotransmitter secretion
Axonal firing leads to ATP and Glu release in the periaxonal space (Figure 1B, see also paragraph Signals transmitted by active axons) (Verderio et al., 2006; Fields and Ni, 2010; Wake et al., 2011). By activating P2Y and AMPA receptors on iSCs and nmSCs, these neurotransmitters reciprocally trigger secretion of ATP and the excitatory amino acids Glu and aspartate from SCs, via ion channels or vesicular mechanisms (Figures 1F1,F2) (Jeftinija and Jeftinija, 1998; Liu and Bennett, 2003; Liu et al., 2005). SCs may also secrete the inhibitory neurotransmitter GABA, known to modulate peripheral fiber excitability, but whether its secretion is induced by neuronal activity has not been determined (Morris et al., 1983; Carr et al., 2010; Magnaghi et al., 2010). SC-released neurotransmitters exert local effects on axonal excitability (Carlton et al., 2001; Irnich et al., 2001) (Figure 1F3). Moreover, they may initiate signals that propagate electrically or via retrograde axonal transport toward neuronal cell bodies, affecting soma signaling processes and gene expression (Itoh et al., 1997; Amir and Devor, 2003; Chen et al., 2012).
SC differentiation and myelination
Myelin production by SCs leads to the organization of enwrapped axons into distinct structural domains with highly specialized patterns of ion channel expression (Salzer, 2003; Buttermore et al., 2013). Internodes, electrically insulated by myelin layers with low electrical capacitance, alternate with ion-rich nodes of Ranvier, where APs are generated, so that fast and energy efficient saltatory stimulus propagation is achieved (Figures 1A–C). Hence, neuronal activity effects on SC differentiation can have significant consequences on axon excitability and AP conduction.
Early during development, firing of unmyelinated PNS fibers induces ionic imbalances and neurotransmitter secretion, which affect iSC maturation and myelin production. ClV and still unidentified K+ channels regulate iSC mitosis by modulating the SC membrane potential (Wilson and Chiu, 1993; Pappas and Ritchie, 1998; Sobko et al., 1998) (Figure 1G1). In vitro ES of embryonic DRG neurons, at low frequencies that mimic DRG spontaneous spiking at early developmental stages, leads to activation of purinergic signaling pathways and subsequent inhibition of both SC proliferation and differentiation (Figure 1G2) (Stevens and Fields, 2000; Stevens et al., 2004). Myelination reduction by low-frequency ES has been further attributed to downregulation of the axonal adhesion molecule L1 (Stevens et al., 1998). Glu and GABA also modulate SC maturation (Figure 1G3) (Magnaghi et al., 2006; Saitoh and Araki, 2010; Procacci et al., 2012). However, although GABA is known to be released by SCs (see paragraph “Neurotransmitter secretion”), its extrasynaptic secretion from PNS axons has not been demonstrated.
Few existing experimental data suggest that neuronal activity controls myelination also in the mature PNS. Subfunctional soleus nerve fibers in hindlimb-unloaded rats exhibit reduced myelin thickness (Canu et al., 2009). Administration of ATP modulates myelin lipid constitution in frog SN preparations (Kutuzov NP et al., 2013). Whether and how neuronal function is affected by these changes requires further investigation.
Trophic and Metabolic Support of Neurons
Neuronal activity depends on the maintenance of axonal integrity and energetic status. Both nmSCs and mSCs provide neurotropic and metabolic support to adjacent neurons (Griffin and Thompson, 2008; Nave, 2010). This support is under the control of axonal activity. In response to ES and ATP, cultured SCs secrete nerve growth factor (NGF) and brain-derived neurotropic factor (BDNF), respectively, promoting axonal growth (Figure 1H) (Verderio et al., 2006; Huang et al., 2010). In addition, chemical depolarization triggers vesicular transport of molecules from SCs to axons (Figure 1I) at least in invertebrates (Eyman et al., 2007). Reported molecular cargo of SC-to-axon transported vesicles includes ribosome-bound mRNA, cytoskeletal components and heat-shock proteins (Court et al., 2008; Cocucci et al., 2009; Lopez-Verrilli and Court, 2012). Their exact contributions to axonal function under physiological conditions are still unknown.
Although information regarding glia-to-axon metabolic support in the PNS is scarce, inferences could be made from CNS data. Neuronal activity triggers exosome transfer of metabolic enzymes from oligodendrocytes to neurons (Fruhbeis et al., 2013), as well as release of lactate from astrocytes and uptake by neurons (Barros, 2013). Similar energy transfer processes may occur in the PNS. ES in SN increases O2 uptake and glucose consumption, and SCs seem to be the main metabolic SN niche (Heller and Hesse, 1961). Moreover, in vivo genetic disruption of mitochondria energy production in otherwise functional mouse SCs severely impairs the structure and function of peripheral fibers (Viader et al., 2011; Funfschilling et al., 2012), suggesting that there may be SC-to-neuron energy transfer also in the PNS. However, its characterization, and potential regulation by neuronal activity await further investigation.
Pathogenic Disruption of Activity-Dependent SC–Axon Communication
Significant insight into the physiological significance of the SC-axon cross-talk and its contribution to the maintenance of axonal excitability and function has been obtained by studies on PNS pathologies, such as inflammatory (e.g., chronic inflammatory demyelinating polyneuropathies), metabolic (e.g., diabetes) or genetic (e.g., Charcot-Marie Tooth, -CMT) diseases, and injury.
Dysregulation of SC Activity Sensors in Pathologies
Peripheral neuropathies have been linked to dysregulation of SC activity sensors. Overexpression of P2X7 receptors may have a causative role in CMT1A patient demyelination due to Ca2+ overload (Nobbio et al., 2009). Moreover, P2X7 activation induces BDNF secretion and activates K+ and Cl− conductances, through Big K+ channels and more likely via the cystic fibrosis transmembrane conductance regulator CFTR (Colomar and Amedee, 2001; Verderio et al., 2006). Interestingly, Cl− imbalance leads to axonal loss with primary or secondary dysmyelination in patients and animal models with dysfunctional CFTR or the K+-Cl− cotransporter KCC3 (Sun et al., 2010; Reznikov et al., 2013). Certain CMTX patients carry mutations in Cx32, which may lead to increased currents through the Cx32-hemichannel and to subsequent nerve damage (Abrams et al., 2002; Nualart-Marti et al., 2013). Dysregulation of SC sensors (e.g., upregulation of KV and NaV channels) also occurs after injury (Chiu, 1988).
To further investigate the contribution of SC activity sensor regulation to PNS dysfunctions, we checked for respective transcriptional modulations in our previously published microarray data on SN endoneuria from three mouse models of peripheral neuropathy: the Scap and Lpin1 conditional knockouts (KOs), which have defective lipid biosynthesis and exhibit PNS hypomyelination and progressive demyelination, respectively, and the Pmp22 total KO, which lacks the myelin protein PMP22 and is a model of Hereditary Neuropathy with Liability to Pressure Palsy (Table 1) (Adlkofer et al., 1995; Nadra et al., 2008; Verheijen et al., 2009; Verdier et al., 2012). With the exception of TRP channels and acetylcholine receptors, we are able to detect expression changes in all families of SC sensors. Their potential role in pathogenesis can be inferred from existing data. Upregulation of K+ channels may interfere with SC ability to buffer K+ ions or be associated with increased proliferation of dedifferentiated SCs (Wilson and Chiu, 1990, 1993) (Figures 1E2,G1). Upregulation of T-type CaV3.2 channels could trigger NGF release, in order to support underlying affected axons (Figure 1H) (Huang et al., 2010). A time-course analysis of the transcriptionally regulated genes during the progress of pathology, in conjunction with functional studies, would be necessary to delineate their potential destructive or protective roles in the development of neuropathy.
Disruption of Neuronal Activity Due to Myelin Defects
Myelin defects are a common feature of various peripheral neuropathies. Studies on animal models of demyelinating diseases (e.g., CMT1A, CMT1B, CMT1C, and CMTX) have demonstrated that myelin impairments affect neural influx conduction and axonal excitability through different mechanisms, including decreased electrical isolation of the axolemma, the exposure, redistribution or abnormal expression of voltage-gated ion channels, and the potential change from saltatory to continuous conduction (Brismar, 1981b, 1982; Rasminsky, 1982; Meiri et al., 1986; England et al., 1990, 1996; Schwarz et al., 1991; Rasband et al., 1998; Neuberg et al., 1999; Devaux and Scherer, 2005; Moldovan et al., 2011; Lee et al., 2013). Aberrant expression of nodal NaV channels and nodal or juxtaparanodal KV channels, has been confirmed in patients with CMT1A and CMT4C (Nodera et al., 2004; Arnaud et al., 2009). Computational simulations in combination with experimental observations correlate those demyelination-induced changes with alterations in axonal excitability and impulse propagation, leading to negative or positive clinical symptoms. Alteration in axonal domains can induce decreased excitability and even conduction failure underlying negative symptoms of peripheral neuropathies, such as muscle weakness (Brismar, 1981a,b; Cappelen-Smith et al., 2001; Nodera et al., 2004; Jani-Acsadi et al., 2008; Coggan et al., 2010; Moldovan et al., 2011). Alternatively, demyelination can lead to axonal hyperexcitability, spontaneous ectopic spiking and cross excitation of neighboring axons (by ephaptic coupling or crossed afterdischarge), leading to positive symptoms like neuropathic pain (Calvin et al., 1982; Rasminsky, 1982; Lisney and Pover, 1983; Lisney and Devor, 1987; Gillespie et al., 2000; Wallace et al., 2003; Gemignani et al., 2004; Coggan et al., 2010).
SC Support of Dysfunctional Axons
Axonal dysfunctions in pathologies and animal models with impaired SCs may also occur secondary to or without myelin abnormalities (Gabreels-Festen et al., 1992; Griffiths et al., 1998; Chen et al., 2003; Nave, 2010), indicating the implication of myelin-unrelated mechanisms. Failure of trophic or metabolic glia-to-neuron support may be one such mechanism. Glial support is particularly critical for neuropathic fibers, which have increased metabolic requirements, due to their decreased propagation efficiencies (Shrager and Rubinstein, 1990; De Waegh et al., 1992; Kirkpatrick and Brady, 1994; Moldovan et al., 2011). Glycogen stored in mSCs is utilized to provide neurons with lactate particularly during aglycemia (Brown et al., 2012). Likewise, exosome transport of metabolic enzymes from oligodendrocytes to axons is required to sustain neuronal survival and function under stress conditions (Fruhbeis et al., 2013), while vesicular transfer of ribosomes from mSCs is prominent in injured fibers, and promotes regeneration (Court et al., 2008, 2011; Lopez-Verrilli et al., 2013). Mutations affecting exosome-mediated intercellular communication have been recently described in CMT1C patients (Zhu et al., 2013). Direct transfer of SC molecules via GJs has been suggested in regenerating nerves (Figure 1J) (Dezawa et al., 1998). Apparently, under pathological conditions, SCs need to adjust their physiology in order to maintain the integrity and function of suffering axons.
To investigate whether glia-to-axon support mechanisms are affected in our Scap, Lpin1, and Pmp22 mouse models, we checked for transcriptional regulation of genes involved in cellular metabolism (excluding lipid metabolism, since its dysregulation is expected in the Scap and Lpin1 KOs) and vesicle trafficking, and for genes encoding potential SC exosome or other vesicular cargo (Lopez-Verrilli and Court, 2012; Fruhbeis et al., 2013). Results, depicted in Table S1, reveal changes in genes of all categories. Detailed analyses at both glial and neuronal levels are required to check the potential positive or negative impact of those alterations on the diseased phenotype, especially since some of the depicted transcripts are also present in axons (Willis et al., 2007; Gumy et al., 2011).
Conclusions and Perspectives
Neuronal activity plays a central role in the extrasynaptic communication between peripheral axons and SCs. SCs express proteins that allow them to detect signals produced by firing axons. Our microarray data indicate that the list of SC activity sensors may be more extensive than currently known, thus providing indications for novel axonal activity signals. Detection of those signals permits SCs to adjust their physiology, so as to sufficiently support and control neuronal activity. Although this reciprocal interaction is constantly required to sustain the PNS function, it becomes particularly critical in transitional periods, during development or under pathology-induced stress. By identifying SC activity sensor- and neuronal support-genes that are regulated during development and/or PNS disease, we attempt to shed light on mechanisms mobilized by SCs to cover the altered needs and increased requirements of the challenged nervous system. More questions, however, arise, especially regarding the potential contribution of neuronal activity signals to these regulations, their nature, the downstream signaling pathways mediating SC responses, and the role of the latter in the maintenance of neuronal integrity and the regulation of axonal function. Characterization of respective mechanisms can be facilitated by implementation of recently developed microfluidic compartmentalized cell culture technologies that enable cell-specific analyses and application of advanced microscopy techniques (Taylor et al., 2005). Combination with in vitro ES via conventional electrodes or microelectrode array platforms could be used to investigate the neuronal activity dependence and relevance of SC molecules and signaling pathways (Kanagasabapathi et al., 2011; Yang et al., 2012; Jokinen et al., 2013; Malone et al., 2013). Apart from revealing new modulators of myelination, we expect that such studies will also contribute to the understanding of myelin-independent mechanisms of SC-to-neuron crosstalk.
Author Contributions
Chrysanthi Samara and Olivier Poirot, concept and design, data analysis, and interpretation, manuscript writing; Enric Domènech-Estévez, manuscript writing; Roman Chrast, concept, and design, final approval of manuscript, financial support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the University of Lausanne, the EU Marie Curie fellowship (to Chrysanthi Samara) and the Swiss National Science Foundation (grant 31003A_135735/1 to Roman Chrast). We would like to thank Dr. Valerie Verdier for the generation of microarray data, and Dr. Fabien Pichon for his help in the design of Figure 1.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fncel.2013.00228/abstract
Table S1. Transcriptional regulation of genes encoding potential SC-to-neuron support molecules in mouse models of peripheral neuropathies. Re-analyzed microarray data were originally generated by characterization of endoneurial samples from adult, 56 days-old Scap, Lpin1, and Pmp22 knockout mice. The grouping in the categories of “Metabolism” and “Vesicle trafficking” was based on Gene Ontology, whereas grouping in the “Exosome-exocytic vesicle cargo” category was performed by manual annotation based on (Lopez-Verrilli and Court, 2012; Fruhbeis et al., 2013). For more information regarding the experiments and data analysis, see legend of Table 1 and (Verdier et al., 2012). Asterisk (*) indicates transcripts that have been previously described in axons of DRG neurons (Willis et al., 2007; Gumy et al., 2011).
References
Abrams, C. K., Bennett, M. V., Verselis, V. K., and Bargiello, T. A. (2002). Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc. Natl. Acad. Sci. U.S.A. 99, 3980–3984. doi: 10.1073/pnas.261713499
Adlkofer, K., Martini, R., Aguzzi, A., Zielasek, J., Toyka, K. V., and Suter, U. (1995). Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat. Genet. 11, 274–280. doi: 10.1038/ng1195-274
Agrawal, S. G., and Evans, R. H. (1986). The primary afferent depolarizing action of kainate in the rat. Br. J. Pharmacol. 87, 345–355. doi: 10.1111/j.1476-5381.1986.tb10823.x
Altevogt, B. M., Kleopa, K. A., Postma, F. R., Scherer, S. S., and Paul, D. L. (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22, 6458–6470.
Amir, R., and Devor, M. (2003). Electrical excitability of the soma of sensory neurons is required for spike invasion of the soma, but not for through-conduction. Biophys. J. 84, 2181–2191. doi: 10.1016/S0006-3495(03)75024-3
Arnaud, E., Zenker, J., De Preux Charles, A. S., Stendel, C., Roos, A., Medard, J. J., et al. (2009). SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc. Natl. Acad. Sci. U.S.A. 106, 17528–17533. doi: 10.1073/pnas.0905523106
Baker, M. D. (2002). Electrophysiology of mammalian Schwann cells. Prog. Biophys. Mol. Biol. 78, 83–103. doi: 10.1016/S0079-6107(02)00007-X
Barros, L. F. (2013). Metabolic signaling by lactate in the brain. Trends Neurosci. 36, 396–404. doi: 10.1016/j.tins.2013.04.002
Beaudu-Lange, C., Colomar, A., Israel, J. M., Coles, J. A., and Amedee, T. (2000). Spontaneous neuronal activity in organotypic cultures of mouse dorsal root ganglion leads to upregulation of calcium channel expression on remote Schwann cells. Glia 29, 281–287. doi: 10.1002/(SICI)1098-1136(20000201)29:3<281::AID-GLIA9>3.3.CO;2-X
Brismar, T. (1981a). Electrical properties of isolated demyelinated rat nerve fibres. Acta Physiol. Scand. 113, 161–166. doi: 10.1111/j.1748-1716.1981.tb06877.x
Brismar, T. (1981b). Specific permeability properties of demyelinated rat nerve fibres. Acta Physiol. Scand. 113, 167–176. doi: 10.1111/j.1748-1716.1981.tb06878.x
Brismar, T. (1982). Distribution of K-channels in the axolemma of myelinated fibres. Trends Neurosci. 5, 179. doi: 10.1016/0166-2236(82)90105-9
Brown, A. M., Evans, R. D., Black, J., and Ransom, B. R. (2012). Schwann cell glycogen selectively supports myelinated axon function. Ann. Neurol. 72, 406–418. doi: 10.1002/ana.23607
Brunet, P. C., and Jirounek, P. (1994). Long-range intercellular signalling in glial cells of the peripheral nerve. Neuroreport 5, 635–638. doi: 10.1097/00001756-199401000-00026
Buttermore, E. D., Thaxton, C. L., and Bhat, M. A. (2013). Organization and maintenance of molecular domains in myelinated axons. J. Neurosci. Res. 91, 603–622. doi: 10.1002/jnr.23197
Calvin, W. H., Devor, M., and Howe, J. F. (1982). Can neuralgias arise from minor demyelination? Spontaneous firing, mechanosensitivity, and afterdischarge from conducting axons. Exp. Neurol. 75, 755–763. doi: 10.1016/0014-4886(82)90040-1
Canu, M. H., Carnaud, M., Picquet, F., and Goutebroze, L. (2009). Activity-dependent regulation of myelin maintenance in the adult rat. Brain Res. 1252, 45–51. doi: 10.1016/j.brainres.2008.10.079
Cappelen-Smith, C., Kuwabara, S., Lin, C. S., Mogyoros, I., and Burke, D. (2001). Membrane properties in chronic inflammatory demyelinating polyneuropathy. Brain 124, 2439–2447. doi: 10.1093/brain/124.12.2439
Carlton, S. M., Hargett, G. L., and Coggeshall, R. E. (2001). Localization of metabotropic glutamate receptors 2/3 on primary afferent axons in the rat. Neuroscience 105, 957–969. doi: 10.1016/S0306-4522(01)00238-X
Carr, R. W., Sittl, R., Fleckenstein, J., and Grafe, P. (2010). GABA increases electrical excitability in a subset of human unmyelinated peripheral axons. PloS ONE 5:e8780. doi: 10.1371/journal.pone.0008780
Chen, S., Rio, C., Ji, R. R., Dikkes, P., Coggeshall, R. E., Woolf, C. J., et al. (2003). Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat. Neurosci. 6, 1186–1193. doi: 10.1038/nn1139
Chen, X. Q., Wang, B., Wu, C., Pan, J., Yuan, B., Su, Y. Y., et al. (2012). Endosome-mediated retrograde axonal transport of P2X3 receptor signals in primary sensory neurons. Cell Res. 22, 677–696. doi: 10.1038/cr.2011.197
Chiu, S. Y. (1988). Changes in excitable membrane properties in Schwann cells of adult rabbit sciatic nerves following nerve transection. J. Physiol. 396, 173–188.
Cocucci, E., Racchetti, G., and Meldolesi, J. (2009). Shedding microvesicles: artefacts no more. Trends Cell Biol. 19, 43–51. doi: 10.1016/j.tcb.2008.11.003
Coggan, J. S., Prescott, S. A., Bartol, T. M., and Sejnowski, T. J. (2010). Imbalance of ionic conductances contributes to diverse symptoms of demyelination. Proc. Natl. Acad. Sci. U.S.A. 107, 20602–20609. doi: 10.1073/pnas.1013798107
Colomar, A., and Amedee, T. (2001). ATP stimulation of P2X(7) receptors activates three different ionic conductances on cultured mouse Schwann cells. Eur. J. Neurosci. 14, 927–936. doi: 10.1046/j.0953-816x.2001.01714.x
Court, F. A., Hendriks, W. T., Macgillavry, H. D., Alvarez, J., and Van Minnen, J. (2008). Schwann cell to axon transfer of ribosomes: toward a novel understanding of the role of glia in the nervous system. J. Neurosci. 28, 11024–11029. doi: 10.1523/JNEUROSCI.2429-08.2008
Court, F. A., Midha, R., Cisterna, B. A., Grochmal, J., Shakhbazau, A., Hendriks, W. T., et al. (2011). Morphological evidence for a transport of ribosomes from Schwann cells to regenerating axons. Glia 59, 1529–1539. doi: 10.1002/glia.21196
Debanne, D., Campanac, E., Bialowas, A., Carlier, E., and Alcaraz, G. (2011). Axon physiology. Physiol. Rev. 91, 555–602. doi: 10.1152/physrev.00048.2009
Dememes, D., Lleixa, A., and Dechesne, C. J. (1995). Cellular and subcellular localization of AMPA-selective glutamate receptors in the mammalian peripheral vestibular system. Brain Res. 671, 83–94. doi: 10.1016/0006-8993(94)01322-9
Devaux, J. J., and Scherer, S. S. (2005). Altered ion channels in an animal model of Charcot-Marie-Tooth disease type IA. J. Neurosci. 25, 1470–1480. doi: 10.1523/JNEUROSCI.3328-04.2005
De Waegh, S. M., Lee, V. M., and Brady, S. T. (1992). Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 68, 451–463. doi: 10.1016/0092-8674(92)90183-D
Dezawa, M., Mutoh, T., Dezawa, A., and Adachi-Usami, E. (1998). Putative gap junctional communication between axon and regenerating Schwann cells during mammalian peripheral nerve regeneration. Neuroscience 85, 663–667. doi: 10.1016/S0306-4522(98)00051-7
England, J. D., Gamboni, F., Levinson, S. R., and Finger, T. E. (1990). Changed distribution of sodium channels along demyelinated axons. Proc. Natl. Acad. Sci. U.S.A. 87, 6777–6780. doi: 10.1073/pnas.87.17.6777
England, J. D., Levinson, S. R., and Shrager, P. (1996). Immunocytochemical investigations of sodium channels along nodal and internodal portions of demyelinated axons. Microsc. Res. Tech. 34, 445–451. doi: 10.1002/(SICI)1097-0029(19960801)34:5<445::AID-JEMT4>3.0.CO;2-L
Evans, P. D., Reale, V., Merzon, R. M., and Villegas, J. (1991). Mechanisms of axon-Schwann cell signaling in the squid nerve fiber. Ann. N.Y. Acad. Sci. 633, 434–447. doi: 10.1111/j.1749-6632.1991.tb15634.x
Eyman, M., Cefaliello, C., Ferrara, E., De Stefano, R., Lavina, Z. S., Crispino, M., et al. (2007). Local synthesis of axonal and presynaptic RNA in squid model systems. Eur. J. Neurosci. 25, 341–350. doi: 10.1111/j.1460-9568.2007.05304.x
Feng, Z., and Ko, C. P. (2007). Neuronal glia interactions at the vertebrate neuromuscular junction. Curr. Opin. Pharmacol. 7, 316–324. doi: 10.1016/j.coph.2006.12.003
Fields, R. D., and Burnstock, G. (2006). Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 7, 423–436. doi: 10.1038/nrn1928
Fields, R. D., and Ni, Y. (2010). Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci. Signal. 3, ra73. doi: 10.1126/scisignal.2001128
Fink, T., Davey, D. F., and Ansselin, A. D. (1999). Glutaminergic and adrenergic receptors expressed on adult guinea pig Schwann cells in vitro. Can. J. Physiol. Pharmacol. 77, 204–210. doi: 10.1139/y99-008
Fitzgerald, M. (1987). Spontaneous and evoked activity of fetal primary afferents in vivo. Nature 326, 603–605. doi: 10.1038/326603a0
Fruhbeis, C., Frohlich, D., Kuo, W. P., Amphornrat, J., Thilemann, S., Saab, A. S., et al. (2013). Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11:e1001604. doi: 10.1371/journal.pbio.1001604
Funfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. doi: 10.1038/nature11007
Gabreels-Festen, A. A., Gabreels, F. J., Jennekens, F. G., Joosten, E. M., and Janssen-Van Kempen, T. W. (1992). Autosomal recessive form of hereditary motor and sensory neuropathy type I. Neurology 42, 1755–1761. doi: 10.1212/WNL.42.9.1755
Gemignani, F., Melli, G., Alfieri, S., Inglese, C., and Marbini, A. (2004). Sensory manifestations in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 9, 7–14. doi: 10.1111/j.1085-9489.2004.09103.x
Gillespie, C. S., Sherman, D. L., Fleetwood-Walker, S. M., Cottrell, D. F., Tait, S., Garry, E. M., et al. (2000). Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 26, 523–531. doi: 10.1016/S0896-6273(00)81184-8
Grafe, P., Mayer, C., Takigawa, T., Kamleiter, M., and Sanchez-Brandelik, R. (1999). Confocal calcium imaging reveals an ionotropic P2 nucleotide receptor in the paranodal membrane of rat Schwann cells. J. Physiol. 515(Pt 2), 377–383. doi: 10.1111/j.1469-7793.1999.377ac.x
Griffin, J. W., and Thompson, W. J. (2008). Biology and pathology of nonmyelinating Schwann cells. Glia 56, 1518–1531. doi: 10.1002/glia.20778
Griffiths, I., Klugmann, M., Anderson, T., Yool, D., Thomson, C., Schwab, M. H., et al. (1998). Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610–1613. doi: 10.1126/science.280.5369.1610
Gumy, L. F., Yeo, G. S., Tung, Y. C., Zivraj, K. H., Willis, D., Coppola, G., et al. (2011). Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17, 85–98. doi: 10.1261/rna.2386111
Heller, I. H., and Hesse, S. (1961). Substance in peripheral nerve which influences oxygen uptake. Science 133, 1708–1709. doi: 10.1126/science.133.3465.1708
Huang, J., Ye, Z., Hu, X., Lu, L., and Luo, Z. (2010). Electrical stimulation induces calcium-dependent release of NGF from cultured Schwann cells. Glia 58, 622–631. doi: 10.1002/glia.20951
Irnich, D., Burgstahler, R., Bostock, H., and Grafe, P. (2001). ATP affects both axons and Schwann cells of unmyelinated C fibres. Pain 92, 343–350. doi: 10.1016/S0304-3959(01)00277-9
Itoh, K., Ozaki, M., Stevens, B., and Fields, R. D. (1997). Activity-dependent regulation of N-cadherin in DRG neurons: differential regulation of N-cadherin, NCAM, and L1 by distinct patterns of action potentials. J. Neurobiol. 33, 735–748. doi: 10.1002/(SICI)1097-4695(19971120)33:6<735::AID-NEU3>3.0.CO;2-A
Jani-Acsadi, A., Krajewski, K., and Shy, M. E. (2008). Charcot-Marie-Tooth neuropathies: diagnosis and management. Semin. Neurol. 28, 185–194. doi: 10.1055/s-2008-1062264
Jeftinija, S. D., and Jeftinija, K. V. (1998). ATP stimulates release of excitatory amino acids from cultured Schwann cells. Neuroscience 82, 927–934. doi: 10.1016/S0306-4522(97)00310-2
Jokinen, V., Sakha, P., Suvanto, P., Rivera, C., Franssila, S., Lauri, S. E., et al. (2013). A microfluidic chip for axonal isolation and electrophysiological measurements. J. Neurosci. Methods 212, 276–282. doi: 10.1016/j.jneumeth.2012.10.013
Kanagasabapathi, T. T., Ciliberti, D., Martinoia, S., Wadman, W. J., and Decre, M. M. (2011). Dual-compartment neurofluidic system for electrophysiological measurements in physically segregated and functionally connected neuronal cell culture. Front. Neuroeng. 4:13. doi: 10.3389/fneng.2011.00013
Kinkelin, I., Brocker, E. B., Koltzenburg, M., and Carlton, S. M. (2000). Localization of ionotropic glutamate receptors in peripheral axons of human skin. Neurosci. Lett. 283, 149–152. doi: 10.1016/S0304-3940(00)00944-7
Kirkpatrick, L. L., and Brady, S. T. (1994). Modulation of the axonal microtubule cytoskeleton by myelinating Schwann cells. J. Neurosci. 14, 7440–7450.
Konishi, T. (1994). Activity-dependent regulation of inwardly rectifying potassium currents in non-myelinating Schwann cells in mice. J. Physiol. 474, 193–202.
Kutuzov, N. P. B. A., Yusipovich, A. I., Maksimov, G. V., Dracheva, O. E., Lyaskovskiy, V. l., Bulygin, F. V., et al. (2013). ATP-induced lipid membrane reordering in the myelinated nerve fiber identified using Raman spectroscopy. Laser Phys. Lett. 10, 5. doi: 10.1088/1612-2011/10/7/075606
Lee, S. M., Sha, D., Mohammed, A. A., Asress, S., Glass, J. D., Chin, L.-S., et al. (2013). Motor and sensory neuropathy due to myelin infolding and paranodal damage in a transgenic mouse model of Charcot–Marie–Tooth disease type 1C. Hum. Mol. Genet. 22, 1755–1770. doi: 10.1093/hmg/ddt022
Lev-Ram, V., and Ellisman, M. H. (1995). Axonal activation-induced calcium transients in myelinating Schwann cells, sources, and mechanisms. J. Neurosci. 15, 2628–2637.
Lisney, S. J., and Devor, M. (1987). Afterdischarge and interactions among fibers in damaged peripheral nerve in the rat. Brain Res. 415, 122–136. doi: 10.1016/0006-8993(87)90275-7
Lisney, S. J., and Pover, C. M. (1983). Coupling between fibres involved in sensory nerve neuromata in cats. J. Neurol. Sci. 59, 255–264. doi: 10.1016/0022-510X(83)90043-6
Liu, G. J., and Bennett, M. R. (2003). ATP secretion from nerve trunks and Schwann cells mediated by glutamate. Neuroreport 14, 2079–2083. doi: 10.1097/00001756-200311140-00014
Liu, G. J., Werry, E. L., and Bennett, M. R. (2005). Secretion of ATP from Schwann cells in response to uridine triphosphate. Eur. J. Neurosci. 21, 151–160. doi: 10.1111/j.1460-9568.2004.03831.x
Lopez-Verrilli, M. A., and Court, F. A. (2012). Transfer of vesicles from schwann cells to axons: a novel mechanism of communication in the peripheral nervous system. Front. Physiol. 3:205. doi: 10.3389/fphys.2012.00205
Lopez-Verrilli, M. A., Picou, F., and Court, F. A. (2013). Schwann cell-derived exosomes enhance axonal regeneration in the peripheral nervous system. Glia 61, 1795–1806. doi: 10.1002/glia.22558
Loreti, S., Vilaro, M. T., Visentin, S., Rees, H., Levey, A. I., and Tata, A. M. (2006). Rat Schwann cells express M1-M4 muscarinic receptor subtypes. J. Neurosci. Res. 84, 97–105. doi: 10.1002/jnr.20874
Magnaghi, V., Ballabio, M., Consoli, A., Lambert, J. J., Roglio, I., and Melcangi, R. C. (2006). GABA receptor-mediated effects in the peripheral nervous system: a cross-interaction with neuroactive steroids. J. Mol. Neurosci. 28, 89–102. doi: 10.1385/JMN:28:1:89
Magnaghi, V., Parducz, A., Frasca, A., Ballabio, M., Procacci, P., Racagni, G., et al. (2010). GABA synthesis in Schwann cells is induced by the neuroactive steroid allopregnanolone. J. Neurochem. 112, 980–990. doi: 10.1111/j.1471-4159.2009.06512.x
Malone, M., Gary, D., Yang, I. H., Miglioretti, A., Houdayer, T., Thakor, N., et al. (2013). Neuronal activity promotes myelination via a cAMP pathway. Glia 61, 843–854. doi: 10.1002/glia.22476
Mayer, C., Quasthoff, S., and Grafe, P. (1998). Differences in the sensitivity to purinergic stimulation of myelinating and non-myelinating Schwann cells in peripheral human and rat nerve. Glia 23, 374–382. doi: 10.1002/(SICI)1098-1136(199808)23:4<374::AID-GLIA9>3.0.CO;2-2
Mayer, C., Quasthoff, S., and Grafe, P. (1999). Confocal imaging reveals activity-dependent intracellular Ca2+ transients in nociceptive human C fibres. Pain 81, 317–322. doi: 10.1016/S0304-3959(99)00015-9
Meiri, H., Steinberg, R., and Medalion, B. (1986). Detection of sodium channel distribution in rat sciatic nerve following lysophosphatidylcholine-induced demyelination. J. Membr. Biol. 92, 47–56. doi: 10.1007/BF01869015
Mi, H., Deerinck, T. J., Ellisman, M. H., and Schwarz, T. L. (1995). Differential distribution of closely related potassium channels in rat Schwann cells. J. Neurosci. 15, 3761–3774.
Mi, H., Deerinck, T. J., Jones, M., Ellisman, M. H., and Schwarz, T. L. (1996). Inwardly rectifying K+ channels that may participate in K+ buffering are localized in microvilli of Schwann cells. J. Neurosci. 16, 2421–2429.
Mierzwa, A., Shroff, S., and Rosenbluth, J. (2010). Permeability of the paranodal junction of myelinated nerve fibers. J. Neurosci. 30, 15962–15968. doi: 10.1523/JNEUROSCI.4047-10.2010
Moldovan, M., Alvarez, S., Pinchenko, V., Klein, D., Nielsen, F. C., Wood, J. N., et al. (2011). Na(v)1.8 channelopathy in mutant mice deficient for myelin protein zero is detrimental to motor axons. Brain 134, 585–601. doi: 10.1093/brain/awq336
Morris, M. E., Di Costanzo, G. A., Fox, S., and Werman, R. (1983). Depolarizing action of GABA (gamma-aminobutyric acid) on myelinated fibers of peripheral nerves. Brain Res. 278, 117–126. doi: 10.1016/0006-8993(83)90230-5
Nadra, K., De Preux Charles, A. S., Medard, J. J., Hendriks, W. T., Han, G. S., Gres, S., et al. (2008). Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev. 22, 1647–1661. doi: 10.1101/gad.1638008
Nave, K. A. (2010). Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 11, 275–283. doi: 10.1038/nrn2797
Neuberg, D. H., Sancho, S., and Suter, U. (1999). Altered molecular architecture of peripheral nerves in mice lacking the peripheral myelin protein 22 or connexin32. J. Neurosci. Res. 58, 612–623. doi: 10.10002/(SICI)1097-4547(19991201)58:5<612::AID-JNR2>3.0.CO;2-x
Nobbio, L., Sturla, L., Fiorese, F., Usai, C., Basile, G., Moreschi, I., et al. (2009). P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 284, 23146–23158. doi: 10.1074/jbc.M109.027128
Nodera, H., Bostock, H., Kuwabara, S., Sakamoto, T., Asanuma, K., Jia-Ying, S., et al. (2004). Nerve excitability properties in Charcot-Marie-Tooth disease type 1A. Brain 127, 203–211. doi: 10.1093/brain/awh020
Nualart-Marti, A., Solsona, C., and Fields, R. D. (2013). Gap junction communication in myelinating glia. Biochim. Biophys. Acta 1828, 69–78. doi: 10.1016/j.bbamem.2012.01.024
Pappas, C. A., and Ritchie, J. M. (1998). Effect of specific ion channel blockers on cultured Schwann cell proliferation. Glia 22, 113–120. doi: 10.1002/(SICI)1098-1136(199802)22:2<113::AID-GLIA2>3.3.CO;2-Q
Procacci, P., Ballabio, M., Castelnovo, L. F., Mantovani, C., and Magnaghi, V. (2012). GABA-B receptors in the PNS have a role in Schwann cells differentiation? Front. Cell. Neurosci. 6:68. doi: 10.3389/fncel.2012.00068
Rasband, M. N., Trimmer, J. S., Schwarz, T. L., Levinson, S. R., Ellisman, M. H., Schachner, M., et al. (1998). Potassium channel distribution, clustering, and function in remyelinating rat axons. J. Neurosci. 18, 36–47.
Rasminsky, M. (1982). Physiological properties of dystrophic mouse spinal root axons. Electroencephalogr. Clin. Neurophysiol. Suppl. 36, 99–105.
Reznikov, L. R., Dong, Q., Chen, J. H., Moninger, T. O., Park, J. M., Zhang, Y., et al. (2013). CFTR-deficient pigs display peripheral nervous system defects at birth. Proc. Natl. Acad. Sci. U.S.A. 110, 3083–3088. doi: 10.1073/pnas.1222729110
Robert, A., and Jirounek, P. (1994). Uptake of potassium by nonmyelinating Schwann cells induced by axonal activity. J. Neurophysiol. 72, 2570–2579.
Saitoh, F., and Araki, T. (2010). Proteasomal degradation of glutamine synthetase regulates schwann cell differentiation. J. Neurosci. 30, 1204–1212. doi: 10.1523/JNEUROSCI.3591-09.2010
Salzer, J. L. (2003). Polarized domains of myelinated axons. Neuron 40, 297–318. doi: 10.1016/S0896-6273(03)00628-7
Schwarz, J. R., Corrette, B. J., Mann, K., and Wietholter, H. (1991). Changes of ionic channel distribution in myelinated nerve fibres from rats with experimental allergic neuritis. Neurosci. Lett. 122, 205–209. doi: 10.1016/0304-3940(91)90859-R
Shrager, P., and Rubinstein, C. T. (1990). Optical measurement of conduction in single demyelinated axons. J. Gen. Physiol. 95, 867–889. doi: 10.1085/jgp.95.5.867
Sobko, A., Peretz, A., Shirihai, O., Etkin, S., Cherepanova, V., Dagan, D., et al. (1998). Heteromultimeric delayed-rectifier K+ channels in schwann cells: developmental expression and role in cell proliferation. J. Neurosci. 18, 10398–10408.
Sobota, J. A., Mohler, W. A., Cowan, A. E., Eipper, B. A., and Mains, R. E. (2010). Dynamics of peptidergic secretory granule transport are regulated by neuronal stimulation. BMC Neurosci. 11:32. doi: 10.1186/1471-2202-11-32
Stevens, B., and Fields, R. D. (2000). Response of Schwann cells to action potentials in development. Science 287, 2267–2271. doi: 10.1126/science.287.5461.2267
Stevens, B., Ishibashi, T., Chen, J. F., and Fields, R. D. (2004). Adenosine: an activity-dependent axonal signal regulating MAP kinase and proliferation in developing Schwann cells. Neuron Glia Biol. 1, 23–34. doi: 10.1017/S1740925X04000055
Stevens, B., Tanner, S., and Fields, R. D. (1998). Control of myelination by specific patterns of neural impulses. J. Neurosci. 18, 9303–9311.
Sun, Y. T., Lin, T. S., Tzeng, S. F., Delpire, E., and Shen, M. R. (2010). Deficiency of electroneutral K+-Cl- cotransporter 3 causes a disruption in impulse propagation along peripheral nerves. Glia 58, 1544–1552. doi: 10.1002/glia.21028
Taylor, A. M., Blurton-Jones, M., Rhee, S. W., Cribbs, D. H., Cotman, C. W., and Jeon, N. L. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605. doi: 10.1038/nmeth777
Toews, J. C., Schram, V., Weerth, S. H., Mignery, G. A., and Russell, J. T. (2007). Signaling proteins in the axoglial apparatus of sciatic nerve nodes of Ranvier. Glia 55, 202–213. doi: 10.1002/glia.20448
Verderio, C., Bianco, F., Blanchard, M. P., Bergami, M., Canossa, M., Scarfone, E., et al. (2006). Cross talk between vestibular neurons and Schwann cells mediates BDNF release and neuronal regeneration. Brain Cell Biol. 35, 187–201. doi: 10.1007/s11068-007-9011-6
Verdier, V., Csardi, G., De Preux-Charles, A. S., Medard, J. J., Smit, A. B., Verheijen, M. H., et al. (2012). Aging of myelinating glial cells predominantly affects lipid metabolism and immune response pathways. Glia 60, 751–760. doi: 10.1002/glia.22305
Verheijen, M. H., Camargo, N., Verdier, V., Nadra, K., De Preux Charles, A. S., Medard, J. J., et al. (2009). SCAP is required for timely and proper myelin membrane synthesis. Proc. Natl. Acad. Sci. U.S.A. 106, 21383–21388. doi: 10.1073/pnas.0905633106
Verkhratsky, A., and Steinhauser, C. (2000). Ion channels in glial cells. Brain Res. Brain Res. Rev. 32, 380–412. doi: 10.1016/S0165-0173(99)00093-4
Viader, A., Golden, J. P., Baloh, R. H., Schmidt, R. E., Hunter, D. A., and Milbrandt, J. (2011). Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J. Neurosci. 31, 10128–10140. doi: 10.1523/JNEUROSCI.0884-11.2011
Wake, H., Lee, P. R., and Fields, R. D. (2011). Control of local protein synthesis and initial events in myelination by action potentials. Science 333, 1647–1651. doi: 10.1126/science.1206998
Wallace, V. C., Cottrell, D. F., Brophy, P. J., and Fleetwood-Walker, S. M. (2003). Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J. Neurosci. 23, 3221–3233. doi: 10.1083/jcb.200703209
Willis, D. E., Van Niekerk, E. A., Sasaki, Y., Mesngon, M., Merianda, T. T., Williams, G. G., et al. (2007). Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J. Cell Biol. 178, 965–980. doi: 10.1083/jcb.200703209
Wilson, G. F., and Chiu, S. Y. (1990). Potassium channel regulation in Schwann cells during early developmental myelinogenesis. J. Neurosci. 10, 1615–1625.
Wilson, G. F., and Chiu, S. Y. (1993). Mitogenic factors regulate ion channels in Schwann cells cultured from newborn rat sciatic nerve. J. Physiol. 470, 501–520.
Yang, I. H., Gary, D., Malone, M., Dria, S., Houdayer, T., Belegu, V., et al. (2012). Axon myelination and electrical stimulation in a microfluidic, compartmentalized cell culture platform. Neuromolecular Med. 14, 112–118. doi: 10.1007/s12017-012-8170-5
Keywords: peripheral nervous system, Schwann cell, axon-glia interaction, neuronal activity, microarray, neuronal support
Citation: Samara C, Poirot O, Domènech-Estévez E and Chrast R (2013) Neuronal activity in the hub of extrasynaptic Schwann cell-axon interactions. Front. Cell. Neurosci. 7:228. doi: 10.3389/fncel.2013.00228
Received: 29 August 2013; Accepted: 05 November 2013;
Published online: 25 November 2013.
Edited by:
Martin Stangel, Hannover Medical School, GermanyReviewed by:
Felipe A. Court, Pontificia Universidad Catolica de Chile, ChileMark Verheijen, VU University, Netherlands
Copyright © 2013 Samara, Poirot, Domènech-Estévez and Chrast. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chrysanthi Samara and Roman Chrast, Department of Medical Genetics, University of Lausanne, Rue du Bugnon 27, Lausanne, CH-1005, Switzerland e-mail:Q2hyeXNhbnRoaS5TYW1hcmFAdW5pbC5jaA==;Um9tYW4uQ2hyYXN0QHVuaWwuY2g=
†These authors have contributed equally to this work.