 Jordan D. T. Engbers
Jordan D. T. Engbers Dustin Anderson
Dustin Anderson Gerald W. Zamponi
Gerald W. Zamponi Ray W. Turner
Ray W. Turner- 1Department of Cell Biology and Anatomy, Hotchkiss Brain Institute, University of Calgary, Calgary, Canada
- 2Department of Physiology and Pharmacology, Hotchkiss Brain Institute, University of Calgary, Calgary, Canada
T-type calcium channels of the Cav3 family are unique among voltage-gated calcium channels due to their low activation voltage, rapid inactivation, and small single channel conductance. These special properties allow Cav3 calcium channels to regulate neuronal processing in the subthreshold voltage range. Here, we review two different subthreshold ion channel interactions involving Cav3 channels and explore the ability of these interactions to expand the functional roles of Cav3 channels. In cerebellar Purkinje cells, Cav3 and intermediate conductance calcium-activated potassium (IKCa) channels form a novel complex which creates a low voltage-activated, transient outward current capable of suppressing temporal summation of excitatory postsynaptic potentials (EPSPs). In large diameter neurons of the deep cerebellar nuclei, Cav3-mediated calcium current (IT) and hyperpolarization-activated cation current (IH) are activated during trains of inhibitory postsynaptic potentials. These currents have distinct, and yet synergistic, roles in the subthreshold domain with IT generating a rebound burst and IH controlling first spike latency and rebound spike precision. However, by shortening the membrane time constant the membrane returns towards resting value at a faster rate, allowing IH to increase the efficacy of IT and increase the range of burst frequencies that can be generated. The net effect of Cav3 channels thus depends on the channels with which they are paired. When expressed in a complex with a KCa channel, Cav3 channels reduce excitability when processing excitatory inputs. If functionally coupled with an HCN channel, the depolarizing effect of Cav3 channels is accentuated, allowing for efficient inversion of inhibitory inputs to generate a rebound burst output. Therefore, signal processing relies not only on the activity of individual subtypes of channels but also on complex interactions between ion channels whether based on a physical complex or by indirect effects on membrane properties.
Introduction
Voltage- or calcium-gated ion channels can alter the output of a postsynaptic cell by modulating temporal or spatial summation of synaptic responses and thus the ability to attain spike threshold. Since the majority of a synaptic potential is subthreshold to spike generation, ion channels must be activated within a low voltage range or even during membrane hyperpolarizations. A limited subset of ion channels have the requisite properties to function in this manner, with low voltage-activated Cav3 T-type calcium channels directly activated during subthreshold postsynaptic potentials, and HCN channels activated within the negative voltage range traversed by inhibitory postsynaptic potentials (IPSPs; Magee and Johnston, 1995; Magee et al., 1995; Luthi and McCormick, 1998b; Swensen and Bean, 2003; Kole et al., 2006; Hildebrand et al., 2009; Engbers et al., 2011; Gastrein et al., 2011; Engbers et al., 2012). However, the ability for these channels to regulate synaptic responses can differ depending on direct or indirect interactions with other ion channels.
Interactions between calcium and potassium channels can be detected simply at a functional level, in which calcium influx is shown to drive potassium channel activation, or as an actual association at the molecular level based on either a direct interaction between alpha subunits or indirectly through an accessory protein. Cav3 channels have been shown to at least functionally couple to small conductance (SK, KCa2.x) calcium-dependent potassium channels (Wolfart et al., 2001; Wolfart and Roeper, 2002; Yanovsky et al., 2005; Cueni et al., 2008) or big conductance (BK, KCa1.1) potassium channels (Smith et al., 2002). More recently Cav3 channels have been found to associate at the molecular level with a host of potassium channels, including BK channels (Engbers et al., 2013; Rehak et al., 2013), intermediate conductance calcium-dependent potassium channels (IKCa, KCa3.1; Engbers et al., 2012), and the voltage-gated Kv4 family (Anderson et al., 2010a, b). Interactions between these channels allow Cav3-mediated calcium influx to modulate spike repolarization (Gittis and du Lac, 2007; Rehak et al., 2013), firing rate gain (Smith et al., 2002; Anderson et al., 2010a, 2013), and temporal summation of EPSPs (Engbers et al., 2012), all over a wider range of membrane voltage than would be possible with high voltage-activated (HVA) calcium channels. The roles for Cav3 calcium influx in modulating subthreshold responses can also reflect a synergistic interplay with other channels through their respective effects on membrane potential, such as the well-recognized case of Cav3 calcium and HCN channels that drive subthreshold oscillations in thalamic neurons (McCormick and Pape, 1990; McCormick and Bal, 1997; Kim et al., 2001; Cheong and Shin, 2013). HCN channels that give rise to IH are also recognized to modulate temporal summation of both excitatory (Magee, 1999; Angelo et al., 2007; Tsay et al., 2007) and inhibitory (McCormick and Pape, 1990; Atherton et al., 2010) synaptic responses.
Several reviews of the functional roles of Cav3 calcium channels and HCN channels have been published (Luthi and McCormick, 1998a; Perez-Reyes, 2003; Robinson and Siegelbaum, 2003; Yunker and McEnery, 2003; Biel et al., 2009; Cueni et al., 2009; Wahl-Schott and Biel, 2009; Cain and Snutch, 2010, 2013; Cheong and Shin, 2013; Weiss and Zamponi, 2013). In the current review, we contrast and compare the effects of a new direct molecular interaction between Cav3 and IKCa channels to that of a synergistic interaction between Cav3 and HCN channels on synaptic processing in cerebellar Purkinje cells and deep cerebellar nuclear (DCN) cells, the postsynaptic target of Purkinje cells. These two examples serve to emphasize how ion channel interactions involving Cav3 channels can result in a wide range of functional effects.
Cerebellar Synaptic Inputs
The cerebellum is a highly organized cortical structure that receives information from the periphery as well as input from descending cortical projections relayed through pontine nuclei in the form of excitatory synaptic inputs. The majority of input arrives as mossy fibers to granule cells that in turn project parallel fibers (PF) to the molecular layer, providing upwards of 150,000 inputs to individual Purkinje cells. Not all of these inputs are active, as long-term depression results in the silencing of up to 85% of the PF-PC synapses (Ekerot and Jorntell, 2001; Isope and Barbour, 2002). However, even accounting for silenced synapses, the Purkinje cell can still receive input from over 20,000 granule cells. Granule cell projections are highly organized in the context of sensory input but even a low rate of spontaneous activity (~1–4 Hz) can lead to an enormous level of background PF activity that Purkinje cells must discriminate from sensory-relevant bursts of input in a small subset of PFs (Chadderton et al., 2004; Rancz et al., 2007; Ekerot and Jorntell, 2008; D’Angelo and De Zeeuw, 2009). Purkinje cells that are excited by granule cells project inhibitory GABAergic output to cells in the DCN, a set of three bilateral nuclei at the base of cerebellum. The neural coding strategies used by DCN cells are still being determined, but extensive work in vitro has established that they have the capacity to respond to inhibition with a rebound increase in firing rate (Jahnsen, 1986a; Aizenman and Linden, 1999; Molineux et al., 2006, 2008; Tadayonnejad et al., 2009, 2010; Hoebeek et al., 2010; Sangrey and Jaeger, 2010; Engbers et al., 2011). The extent to which this occurs in response to physiological stimuli has recently been debated (Alvina et al., 2008; Person and Raman, 2012), although some consensus begins to emerge from both in vivo and in vitro work that DCN cells can exhibit a rebound increase in firing frequency given a significantly large inhibitory input that could arise in relation to the frequency, number, and in particular, synchronous input from Purkinje cells (Aizenman and Linden, 1999; Zhang et al., 2004; Wetmore et al., 2008; Tadayonnejad et al., 2009, 2010; Hoebeek et al., 2010; Bengtsson et al., 2011). Nevertheless, the ionic mechanisms that could facilitate a rebound response are still under investigation. The effects of PF input to Purkinje cells or Purkinje cell input onto DCN cells can be studied in an in vitro slice preparation either by directly stimulating synaptic inputs or by simulating synaptic potentials by injecting postsynaptic current waveforms at the soma (Figure 1). The current review compares the means by which Cav3 channels can interact with other ion channels to produce markedly different results between the PF-Purkinje cell synapse and the Purkinje-DCN cell synapse.
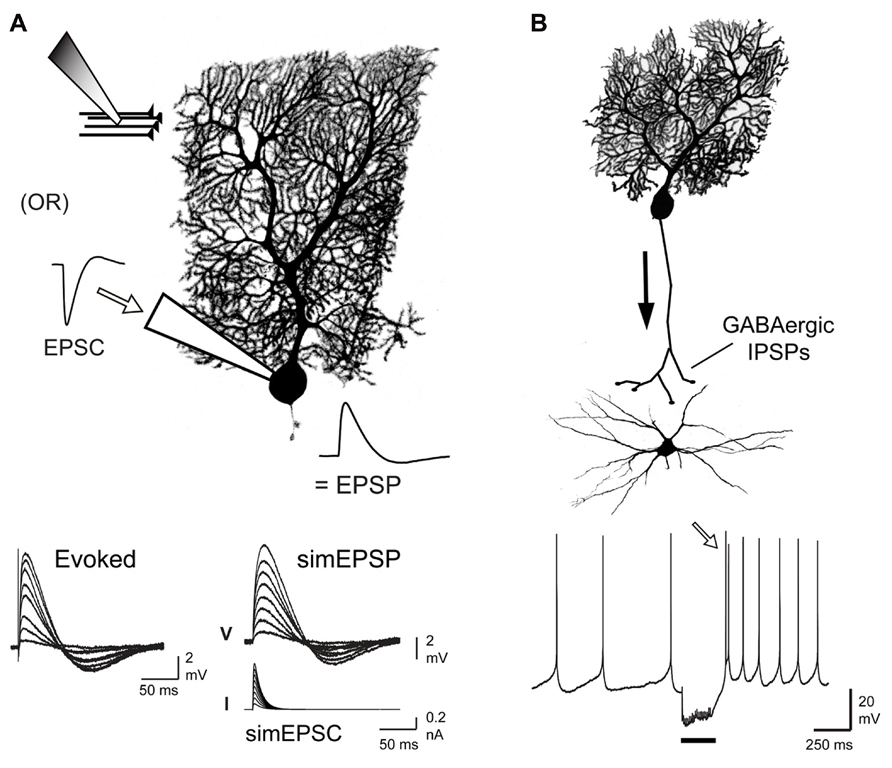
FIGURE 1. Comparison of postsynaptic responses modulated by Cav3 channel-mediated calcium influx. (A) Schematic illustrating means of evoking EPSPs through direct stimulation of parallel fiber input or by injecting simulated EPSCs through the electrode at the soma of a Purkinje cell. Traces below compare evoked and simEPSPs over a range of stimulus intensities. (B) Schematic of the inhibitory GABAergic input provided by Purkinje cells to DCN cells. Trace below shows a tonically firing DCN cell and a rebound firing response (open arrow) following a 100 Hz train of Purkinje cell input (20 pulses, black bar). All cells were biocytin filled during whole-cell recordings and labeled by streptavidin-Cy3. Modified from Engbers et al. (2012) and Tadayonnejad et al. (2010).
The Role of Cav3 Channels in Suppressing Temporal Summation
Parallel fibers EPSPs are followed by an afterhyperpolarization (AHP) lasting up to ~200 ms, a striking feature given that these AHPs can be seen to follow even small amplitude EPSPs. Part of this AHP is due to deactivation of IH during the depolarizing phase of the EPSP, resulting in a hyperpolarizing voltage overshoot (Angelo et al., 2007). However, we recently showed that the remaining portion of the AHP is due to activation of IKCa channels via Cav3 channel-mediated calcium influx at hyperpolarized potentials (Engbers et al., 2012). This finding improves on our understanding of Cav3 ion channel interactions in three ways: (1) Cav3 channels can activate at much lower voltages than previously thought, (2) small amplitude Cav3-mediated current is sufficient to activate KCa channels, and (3) the functional role of Cav3 inward current can be inverted by association with a K+ channel.
Cav3 channels are known to be low voltage-activated, with the voltage for initial activation usually identified by the presence of measureable current on conductance plots derived from whole-cell voltage clamp recordings. However, it is important to recognize that activation of channels in the membrane may occur at voltage levels below those necessary to appear under whole-cell recording conditions. In this regard, voltage clamp analyses have established that Cav3 calcium channels exhibit overlap in the activation and inactivation profiles. This overlap establishes a window current that, while small near resting membrane potential (~2% of total available current), is constitutively active and sufficient to raise intracellular calcium concentration and affect neuronal firing (Figures 2A,B; Huguenard, 1996; Swensen and Bean, 2003; Perez-Reyes, 2006; Talavera and Nilius, 2006; Dreyfus et al., 2010; Engbers et al., 2012). Careful inspection of the degree of overlap of these curves recorded even under whole cell conditions reveals a Cav3 window current that extends from well below -65 mV all the way to ~-20 mV (Figure 2B). These values agree with previous work reporting that a significant proportion of calcium current activated during a sodium spike in Purkinje cells is mediated Cav3 channels (Chemin et al., 2002; Swensen and Bean, 2003). Therefore, LVA Cav3 channels can be a significant source of calcium at subthreshold and suprathreshold voltages, and provide a constitutive inward current even during rest.
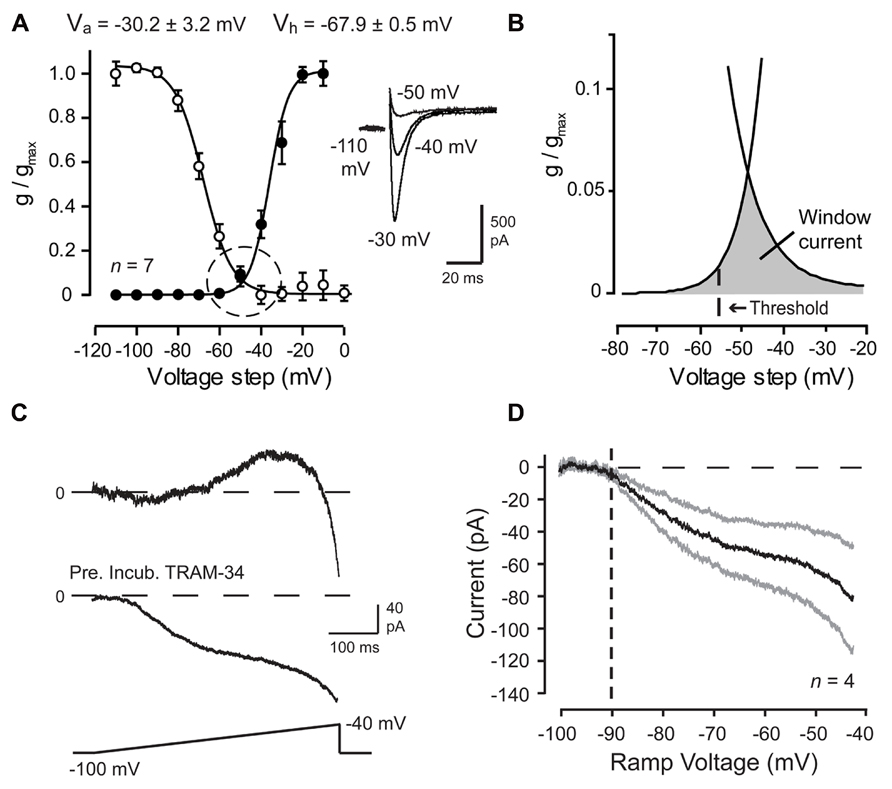
FIGURE 2. Cav3 T-type calcium current is evoked over a wide membrane voltage range in Purkinje cells. (A,B) Mean conductance and inactivation plots calculated for whole-cell Cav3 current recorded from P10–12 Purkinje cells. Inset shows Cav3 current evoked over relatively negative step commands. (B) Expanded image of the fits for activation and inactivation curves from plots in (A) (dashed circle) showing a wide voltage range for Cav3 window current (gray) in relation to spike threshold (dashed line). (C,D) Whole-cell recordings from mature (P18–30) Purkinje cells evoked by a ramp from -100 mV to -40 mV (500 ms) and Ni2+-sensitive currents extracted by applying 300 μM Ni2+. Shown are records of sequential inward-outward currents activated in one cell (top trace) and in another cell purely inward current when recorded in the presence of the potassium channel blocker TRAM-34 (100 nM) (bottom trace). (D) Current-voltage plot of the average Ni2+-sensitive Cav3 current evoked by a ramp command in the presence of TRAM-34. Inward current exhibits initial activation from a voltage of ~-90 mV (dashed line). Standard errors are shown by gray traces. Modified from Engbers et al. (2012).
Activation curves derived by voltage steps in whole-cell voltage clamp might suggest that no significant activation of Cav3 current will occur at potentials negative to -70 mV. However, in a study of HVA calcium channel-mediated transmitter release, the use of slow voltage ramps revealed Cav2.1 (P/Q-type) channel activation ~20 mV more negative than the activation threshold suggested by macroscopic activation curves (Awatramani et al., 2005). Experimental results had also suggested that Cav3 channels were capable of being activated at potentials as low as -75 mV (Engbers et al., 2012). Indeed, slow ramp commands (-100 to -40 mV) applied to Purkinje cells in whole-cell configuration established initial activation of a Ni2+-sensitive inward current at voltages as low as -90 mV (Figures 2C,D). These and other measures confirm that Cav3 channel window current can extend over a far wider range than previously anticipated, potentially providing a calcium current that can be activated by even small amplitude PF EPSPs. While this does not suggest that potentials of -90 mV will ever be reached under physiological conditions, it does indicate that Cav3 channels can act as a significant source of calcium influx over the entire subthreshold range and their availability as a functional current should not be discounted based solely on membrane potential in relation to the voltage for inactivation. In fact, Cav3 channels are known to contribute to Purkinje cell activity during tonic firing at a depolarized state (Swensen and Bean, 2003; Engbers et al., 2012). Furthermore, Cav3 activation properties are known to vary between cell types and even be regulated by their association with other structures, such as mGluR1 receptors (Hildebrand et al., 2009).
In addition to the Ni2+-sensitive inward current, ramp commands in Purkinje cells revealed a Ni2+-sensitive outward current that activated from ~-80 mV with a later onset than the initial Cav3 calcium current (Figure 2C), presumably the same current responsible for the PF-associated AHP. This finding indicated at least a functional coupling between Cav3 channels and KCa channel(s) of some identity. BK and SK2 channels are both expressed in Purkinje cells and are known to be activated by calcium influx. A functional coupling and molecular association between HVA calcium and BK channels is well-established and characterized (Raman and Bean, 1999; Swensen and Bean, 2003; Grunnet and Kaufmann, 2004; Womack et al., 2004; Berkefeld et al., 2006; Alvina and Khodakhah, 2008; Berkefeld and Fakler, 2008; Fakler and Adelman, 2008). A functional coupling between BK and Cav3 channels has also been demonstrated in some cells (Smith et al., 2002; Engbers et al., 2013; Rehak et al., 2013). A higher sensitivity of SK channels to intracellular calcium compared to BK channels allows the SK channel family to couple with a wide range of Ca2+ sources, including Cav1, Cav2.3, and Cav3 calcium channels, NMDA receptors, and Ca2+-permeable nicotinic acetylcholine receptors (Marrion and Tavalin, 1998; Oliver et al., 2000; Wolfart and Roeper, 2002; Ngo-Anh et al., 2005; Yanovsky et al., 2005; Bloodgood and Sabatini, 2007; Cueni et al., 2008; Fakler and Adelman, 2008; Faber, 2010).
Given the wide range of calcium sources capable of activating BK and SK channels, it was fully expected that one of these KCa channel subtypes would account for the PF-evoked AHP. However, a pharmacological assay established that the PF EPSP-AHP simulated by postsynaptic injection of a PF EPSC waveform (simEPSP) was fully insensitive to blockers of SK or BK channels as well as a range of HVA calcium channel blockers (Engbers et al., 2012). Instead the PF EPSP rate of decay and AHP were reduced by low Ni2+ and mibefradil, two established blockers of Cav3 current in Purkinje cells (McDonough and Bean, 1998; Isope and Murphy, 2005; Figure 3A). The rate of decay of the simEPSP and AHP were also blocked by charybdotoxin (ChTx) or TRAM-34, two blockers of IKCa channels (Figure 3A). This was surprising given that IKCa channels were only believed to be expressed in central regions in activated microglia and endothelial cells (Kaushal et al., 2007; Pedarzani and Stocker, 2008). However, applying TRAM-34 during ramp commands effectively blocked all Ni2+-sensitive outward current (Figures 2C,D). Moreover, a series of protein biochemical tests established that IKCa channels are expressed in Purkinje cells and exhibit an association with Cav3 channels at the molecular level (see Engbers et al., 2012). These results then established the existence of a Cav3-IKCa channel complex, adding to the number of complexes recognized between calcium and potassium channels.
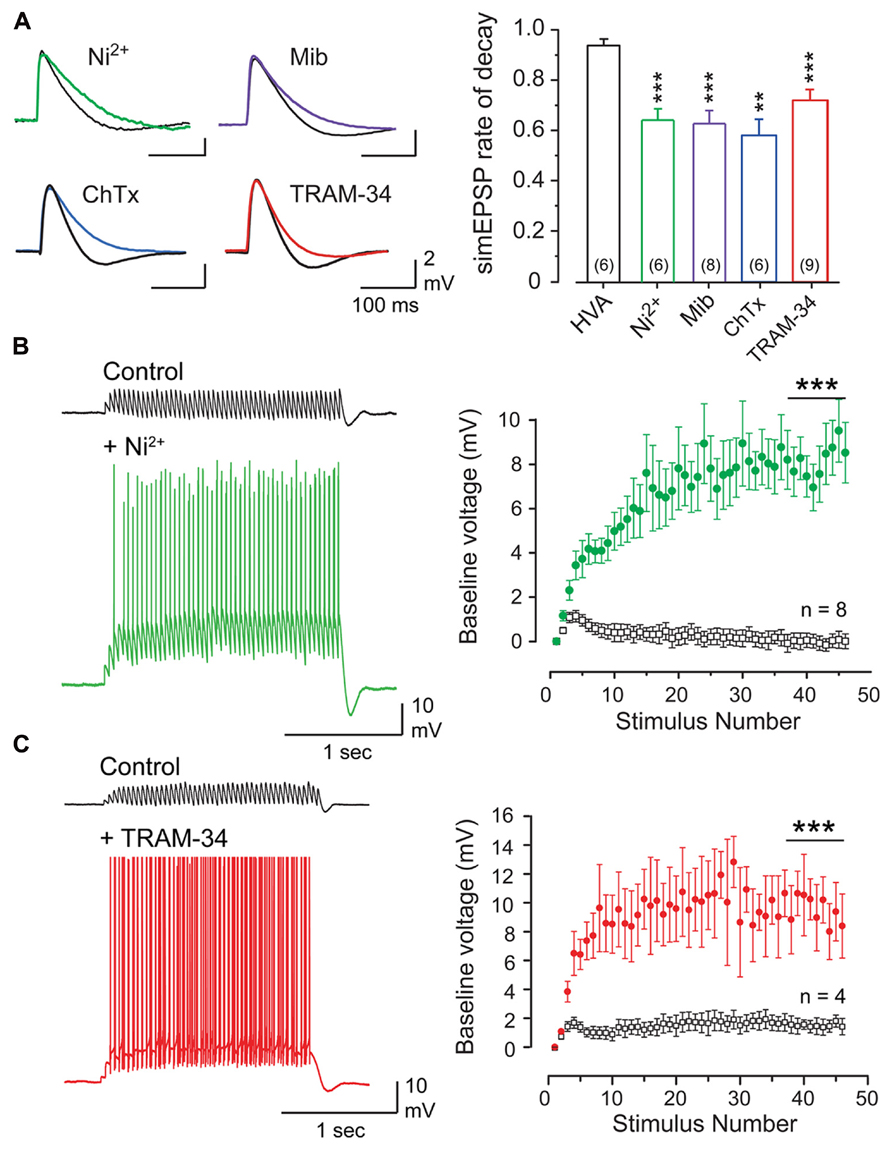
FIGURE 3. The parallel fiber EPSP activates a Cav3- and KCa3. 1-mediated AHP that suppresses temporal summation. (A) Superimposed records of simEPSPs in Purkinje cells before and after 100 μM Ni2+, 1 μM mibefradil (Mib), 100 nM ChTx, or 100 nM TRAM-34. Bar graphs show mean values of the reduction of simEPSP rate of decay. HVA refers to a cocktail of ω-conotoxin GVIA (1 μM), nifedipine (1 μM), and SNX-482 (200 nM). (B,C) Recordings and plots of the mean baseline voltage during 25 Hz trains of parallel fiber-evoked EPSPs before and after applying Ni2+ [(B), 100 μM)] or TRAM-34 [(C), 100 nM]. Recordings in (B,C) were conducted in 50 μM picrotoxin. Statistical significance tested for last 10 pulses of stimulus trains in (B,C) is denoted by bars. Sample values in (A) are shown in brackets within bar plots. **p < 0.01, ***p < 0.001. Modified from Engbers et al. (2012).
It is interesting to contemplate the pairing of Cav3 and IKCa channels. Cav3 channels have a small single channel conductance, low maximal open probability and exhibit rapid inactivation, resulting in small changes in intracellular calcium during subthreshold voltage changes. Therefore, any KCa channel depending on Cav3-mediated calcium influx would have to be exquisitely sensitive to calcium concentration. Interestingly, IKCa is the most sensitive of the KCa channels and exhibits a slow rate of deactivation once activated (Ishii et al., 1997; Joiner et al., 1997). Furthermore, the larger conductance of IKCa channels (20–90 pS) compared to SK channels (10–20 pS) allows small changes in open probability to have a greater effect on membrane voltage. The molecular association between Cav3 and IKCa channels is thus an ideal pairing of signal and transducer, with the KCa channel being tuned to sense the range of intracellular calcium concentration changes associated with Cav3 influx at the level of a nanodomain.
Given the factors that allow Cav3 calcium influx to activate IKCa channels, this ion channel complex should exhibit activation in the subthreshold region. Repetitive PF stimulation revealed an initial temporal summation of EPSPs over the first few stimuli, but then a marked suppression of summation for the remainder of the stimulus train (Figures 3B,C). This result was obtained in the presence or absence of picrotoxin to block GABAergic inhibition (not shown), revealing that suppression of temporal summation did not involve feedforward inhibition activated by PF inputs (Hausser and Clark, 1997; Mittmann et al., 2005). Rather, perfusion of either Ni2+ (100 μM) to block Cav3 calcium influx or TRAM-34 (100 nM) to block IKCa channels restored temporal summation to reveal a substantial increase in EPSP peak amplitude and repetitive spike output from Purkinje cells throughout the stimulus train (Figures 3B,C). Therefore, when coupled to IKCa channels, the Cav3 channel does not provide a net depolarizing effect, but has its influence effectively inverted by the IKCa channel, resulting in a net hyperpolarizing effect. The close association between these channels further allow a rapid response to subthreshold depolarizations and a pronounced effect on temporal summation of excitatory inputs.
These studies were important in demonstrating that Cav3 calcium current has the capacity to activate at very negative membrane voltages, providing an inward current that could potentially accentuate temporal summation and other membrane depolarizations. However, by forming a complex with IKCa channels, Cav3 calcium influx instead provides a net inhibitory influence over temporal summation. These and other experiments help support the hypothesis that the purpose of a Cav3-IKCa mediated inhibition is to suppress the background level of PF input expected to be transmitted to Purkinje cells (Engbers et al., 2012). In this way Purkinje cells will be able to respond to the high frequency bursts of PF input that convey sensory-relevant information.
Synergistic Interplay between Cav3 and HCN Channels Boosts a Rebound Response
A different interaction between Cav3 and HCN channels becomes apparent in the integration of inhibitory synaptic input to DCN cells. Recordings in vitro have primarily focused on large diameter cells that exhibit a characteristic rebound increase in firing rate following a period of membrane hyperpolarization or train of IPSPs evoked by stimulation of Purkinje cell input (Molineux et al., 2008; Tadayonnejad et al., 2009, 2010; Pedroarena, 2010; Sangrey and Jaeger, 2010; Engbers et al., 2011). Transient calcium current through Cav3 channels (IT) and the inward tail current associated with hyperpolarization-activated HCN channels (IH) have been proposed to contribute to rebound responses from some of the earliest recordings (Jahnsen, 1986b; McCormick and Pape, 1990; Muri and Knopfel, 1994; Huguenard, 1996). We also found a correlation between the expression of the Cav3.1 calcium channel isoform in DCN cells and the generation of a high frequency Transient Burst phenotype, and for Cav3.3 channels to a form of Weak Burst response (Molineux et al., 2006). For the purpose of this review we will restrict examples to Transient Burst DCN cells.
IT recorded under steady-state whole-cell voltage clamp conditions is shown in Figure 4, where a step from a negative holding potential to -50 mV (a value just below spike threshold) reveals a large amplitude calcium current. The correspondence of this current to Cav3 channels is supported by an insensitivity to the general HVA calcium channel blocker Cd2+ (50 μM) but full block by 300 μM Ni2+ (Figures 4A,B). IH recorded under whole cell conditions and isolated as Cs+-sensitive current is evident as a slowly activating and non-inactivating inward current that activates at physiological temperatures negative to -50 mV (Figures 4C,D). Of most apparent relevance to a rebound response is a prolonged inward IH tail current (~500 ms) that follows the end of a negative command pulse (Engbers et al., 2011). This is important in that a prolonged IH deactivation will be expected to generate a depolarization with potential influence on the rebound response (Aizenman and Linden, 1999; Engbers et al., 2011; Steuber et al., 2011). Indeed, perfusion of either a low concentration of Ni2+ (300 μM) or Cs+ (2 mM) under current clamp conditions reduced the frequency of the rebound response (Figure 4E), results consistent with a depolarizing influence for both IT and IH. In fact, the ability for calcium channel blockers to reduce the rebound response has been reported using voltage clamp step commands (Molineux et al., 2006, 2008), calcium imaging experiments (Muri and Knopfel, 1994; Zhang et al., 2004; Zheng and Raman, 2009; Schneider et al., 2013), and recently more selective IT blockers (Alvina et al., 2009; Boehme et al., 2011). Blocking IH with external Cs+ uncovered an additional influence of IH in controlling the first spike latency (FSL) to the burst response, an effect that was graded with the stimulus (Figure 4F; Sangrey and Jaeger, 2010; Tadayonnejad et al., 2010). Thus, the greater the number of stimuli (and the longer the hyperpolarization) the shorter the FSL when expressed as a ratio to the control interspike interval (ISI). Similar results on FSL/ISI were not obtained by perfusing Cav3 channel blockers (not shown; Engbers et al., 2011).

FIGURE 4. DCN cells express both IT and IH. (A), IT recorded from a Transient Burst DCN cell under whole-cell conditions at physiological temperature for a single step to -50 mV. (B) IT recorded in a separate cell reveals no effects of applying Cd2+ but nearly full block by Ni2+. (C) IH recorded from a DCN cell at physiological temperature and identified as Cs+-sensitive current. An inward tail current is evident following the end of step commands (inset, open arrow). (D) Mean conductance plot for Cs+-sensitive IH. Dashed line and box denotes the physiological range of membrane voltage traversed by IPSPs. (E) Recordings and mean bar plots of rebound responses in DCN cells evoked by a hyperpolarizing current pulse reveal a decrease in burst frequency after blocking either Cav3 (Ni2+) or HCN (Cs+) channels. (F) Blocking IH with Cs+ reveals an increase in FSL during the rebound response evoked by a train of Purkinje cell IPSPs that increases in a graded fashion with the number of stimuli. *p < 0.05, **p < 0.01. Modified from Molineux et al. (2008) and Engbers et al. (2011).
The most direct interpretation of these results is that both Cav3 and HCN channels contribute a membrane depolarization that directly increases rebound spike frequency and reduces the FSL. To more carefully compare the roles for IT and IH in either aspect of a rebound, we used available data on these currents (Figures 2 and 4) to construct a two compartment reduced model based on Hodgkin–Huxley formalism (for details, see Engbers et al., 2011). Importantly, this model proved capable of reproducing the stimulus-dependent decrease in FSL/ISI and increase in spike frequency of the rebound response (Figure 5A). We note that the frequency response here was measured over only the first 100 ms of the rebound, and does not account for the longer duration depolarizations that reflect contributions by HVA calcium channels or INaP (Zheng and Raman, 2009; Sangrey and Jaeger, 2010; Steuber et al., 2011). Comparing the output of the model upon removing IT or IH was highly instructive in first showing that if only IH was present the model preserved the inverse relationship between FSL/ISI and hyperpolarization strength (greater depth and longer duration) but also exhibited a significant decrease in rebound frequency (Figure 5B). If only IT was present, the FSL/ISI relationship was reversed, with longer hyperpolarizations resulting in a longer FSL/ISI ratio (Figure 5C). However, the rebound response was only present when the model contained IT (Figures 5A,C). Removing both IT and IH caused the FSL/ISI relationship to become directly proportional to the depth and duration of hyperpolarization, as would be expected without active currents in the subthreshold range, and abolished the rebound response (Figure 5D).
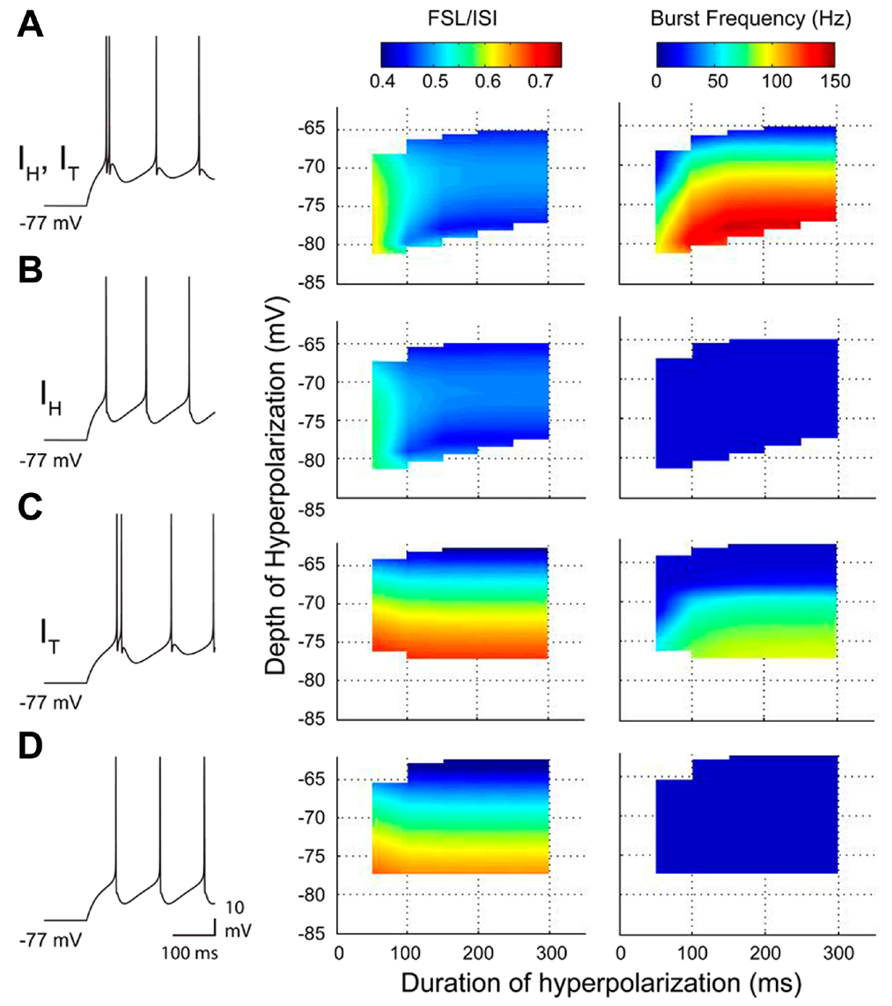
FIGURE 5. A reduced model of the roles for IT and IH in rebound response frequency and FSL. (A–D), Output from a two compartment model containing combinations of IT and/or IH, with representative spike output following a 300 ms hyperpolarizing step to -77 mV from a resting potential of ~-60 mV (10 Hz baseline firing) (left). Changes in frequency and FSL/ISI are plotted in three dimensions in relation to the frequency and ISI present at a resting membrane potential of ~-60 mV. Note a decrease in FSL/ISI and increase in frequency with longer durations or larger amplitude hyperpolarizations when both IT and IH are present (A). Removing IT dramatically abolishes all rebound responses (B), while removing IH reverses the FSL/ISI relationship and reduces rebound frequency (C). Removing both IT and IH blocks the shifts in both FSL/ISI and spike frequency found when both are present (D).
These results emphasize that IH is the primary determinant of the FSL of a rebound response, while IT by itself failed to reproduce the normal FSL response. In addition, the large reduction in rebound frequency when IH was removed revealed that IH has a key role in controlling the frequency of a rebound response. The most straightforward explanation for this would be that IH increases rebound frequency by way of a direct depolarization of the membrane following hyperpolarization. However, further examination revealed that the contribution by IH to rebound frequency could instead be accounted for by a synergistic effect of IH on IT, revealing an indirect interaction between HCN and Cav3 channels.
Recognition of an IT–IH interaction was gained when considering a well-established role for HCN channels in regulating resting membrane conductance. In hippocampal cells, IH has been shown to account for a large percentage of the resting conductance and acts to normalize dendritic EPSPs (Magee, 1999). In Purkinje cells, basal activation of IH at resting membrane potentials shortens the width of PF EPSPs (Angelo et al., 2007) and reduces the bistable range (Williams et al., 2002; Fernandez et al., 2007). To determine if IH exerted its role through a direct depolarization or by modifying the membrane conductance, we compared charging curves of the three models for IT alone, IT–IH, and when IT was expressed alone and membrane capacitance adjusted in the somatic compartment (IT-C; Figure 6A). This test showed that the model of IT alone had a slower rate of charging compared to IT–IH or IT-C, but that a model of IT could be adjusted to the same time constant when model capacitance was increased as a substitute for IH (Figure 6A). When compared in current clamp, it was apparent that spikes discharged at a shorter latency for either the IT–IH model or IT-C than for IT alone (Figure 6B). These results were borne out when comparing three dimensional plots of the rebound burst frequency for different levels and durations of hyperpolarization, where the profile of burst frequency for the IT-C strongly resembled that of IT–IH (Figure 6C). In contrast, adjusting the capacitance in a model with just IT could not replicate the FSL/ISI relationship generated by a model of IT–IH (Figure 6D), indicating that the voltage dependence and kinetics of IH determine the FSL/ISI relationship. Finally, a comparison of the degree of IT inactivation for different depths of hyperpolarization revealed that without IH the membrane potential returned to rest at the end of the stimulus slowly enough to induce greater IT inactivation, and thus less ability to contribute to a rebound response (Figure 6E). Thus, a normally slow rate of return of membrane potential at the end of a stimulus is rescued by virtue of the effects of IH on the membrane time constant, an effect that can be mimicked by lowering cell capacitance.
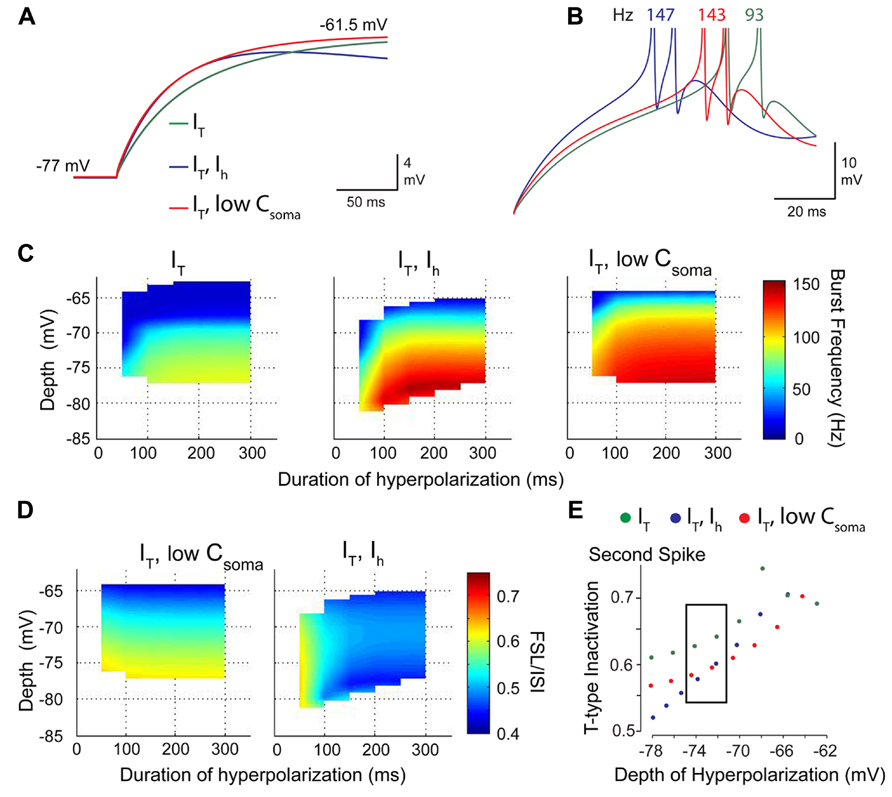
FIGURE 6. IH acts to increase the role of IT in determining rebound burst frequency by modifying the membrane time constant. (A) Expanded image of membrane charging profiles during repolarization from -77 mV. Superimposed traces are for models containing IT and IH (IT–IH), IT alone, and a model with only IT in which membrane capacitance was lowered (red trace) to match the time constant for a model containing IH (purple trace). (B) In the absence of IH, a decrease in capacitance increases burst frequency and decreases FSL (red trace) when compared to a model with IT alone (green trace). (C) Three dimensional colour plots comparing the response of models containing IT, IT–IH, and IT with a low somatic membrane capacitance to simulate the effect of IH. Note that the IT-LowC model reaches the same peak frequency of 150 Hz as the IT–IH model, indicating that a reduction in time constant increases burst frequency. (D) A comparison of models of IT-LowC to IT–IH indicate that the voltage-dependence and kinetics of IH determine the normal voltage-FSL relationship. (E) A comparison of the amount of IT inactivation during the rebound between the three models shows that incorporating low C or IH reduces the extent of IT inactivation compared to a model of IT alone. Modified from Engbers et al. (2011).
A second important parameter of FSL is the degree of precision of the first spike, as measured by the standard deviation of the latency of jitter in a population of spike discharges. To test the contribution of IH and IT to this parameter, we injected the models with stochastic excitatory and inhibitory synaptic conductances and injected current to hyperpolarize the model to -75 mV for 300 ms (Figures 7A,B). The standard deviation of the latency of discharge of the first spike for each of the different models was used as a measure of spike precision. In a model of IT–IH the latency to first spike displayed a high degree of precision or minimal standard deviation (Figures 7A,B). By comparison, the model with IT alone presented a much greater degree of variation in FSL, the majority of which were at longer latencies than for the model of IT–IH (Figures 7A,B). Here, a model of IT alone with an adjustment of membrane capacitance could not restore spike precision despite a decrease in latency (Figures 7A,B). This suggests that the direct depolarizing effect of IH following hyperpolarization affects the precision of the first spike. These data were important in showing that IH modifies the actions of IT both by decreasing latency and increasing precision of first spike discharge, but through different mechanisms.
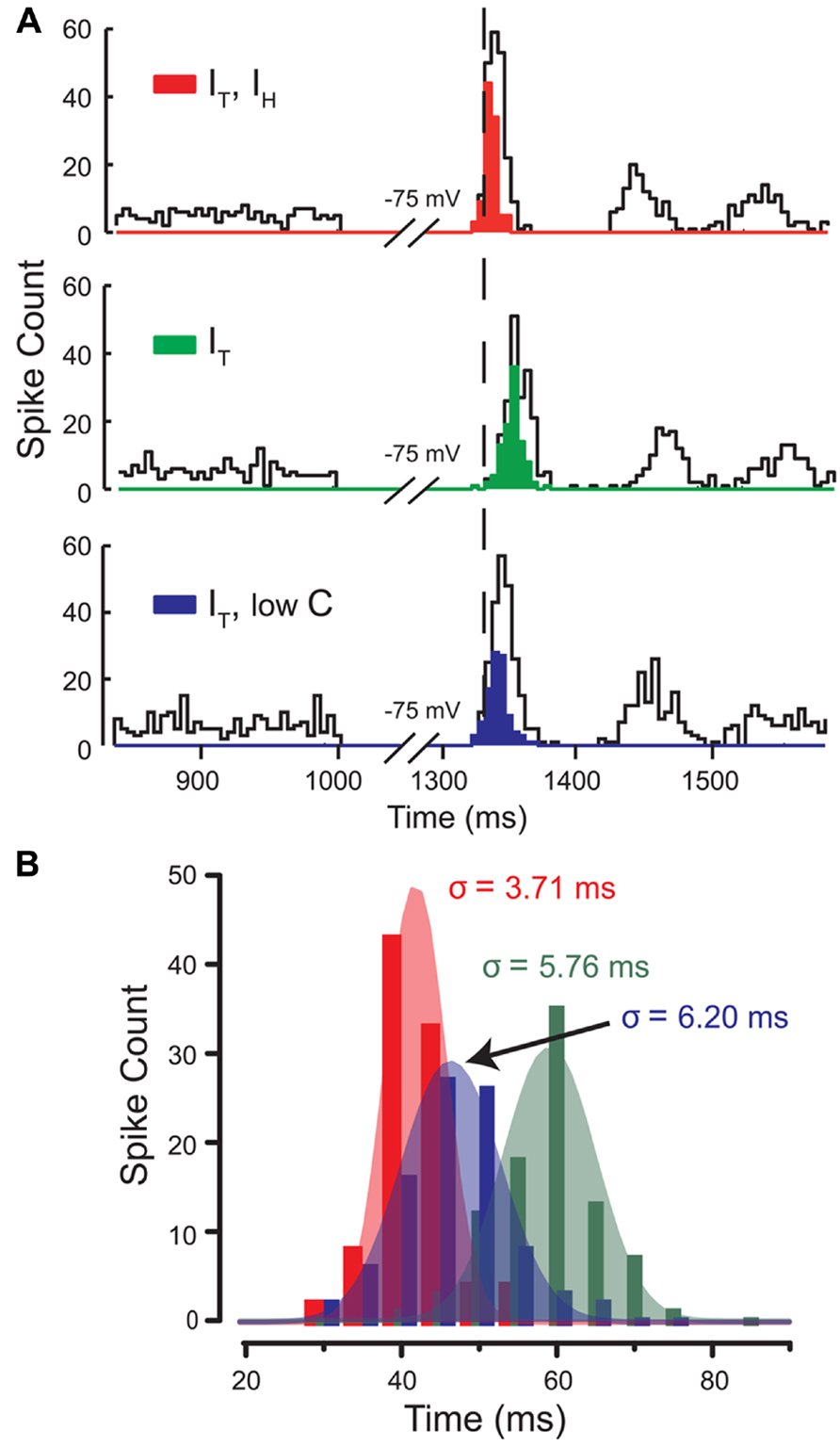
FIGURE 7. IH controls the precision of the first spike following a hyperpolarization. (A) Histograms of spike count in three different models for 100 simulations with stochastic excitatory and inhibitory synaptic conductances. Open bins denote the total spike count, while filled bins denote the first spike following release from inhibition of each of the 100 simulations in the population of rebound responses. The mean latency (relative timing) and standard deviation of FSL (spike precision) between three models indicate that the effects of IH governs both the timing and precision of first spikes in the rebound. (B) Gaussian fits of the population of first spikes for each model in (A) shows that when IH is present (red), FSL falls over a narrow distribution and with small standard deviation (increased spike precision) compared to models lacking IH (green) or with lowC (blue). Modified from Engbers et al. (2011).
Concluding Remarks
The processing of synaptic input requires the activation of ion channels in a low voltage range in order to modify subthreshold potentials. Cav3 calcium channels are low voltage-activated and are unique among calcium channels in exhibiting fast activation and voltage-dependent inactivation, leading to only a transient current when activated. These characteristics suggest that T-type current would have rapid but only short lasting effects on membrane excitability given the extent to which membrane depolarizations will inactivate the channels. In addition, unlike HVA calcium channels, Cav3 channels were long believed not to associate with any particular potassium or ligand-gated channel. However, in recent years this outlook has changed with identification of functional and even molecular associations between Cav3 calcium and other channels. Some of the first reports revealed that Cav3 channels can at least functionally couple to activation of BK and SK potassium channels (Smith et al., 2002; Cueni et al., 2008). More recently Cav3 channels were shown to associate at the molecular level with BK and IKCa channels and even Kv4 A-type potassium channels (Anderson et al., 2010a, b; Engbers et al., 2012, 2013; Rehak et al., 2013). These associations prove capable of greatly expanding the role for Cav3 calcium channels in processing input signals and modifying cell output.
A critical determinant of Cav3 channel activity is the extent of window current defined by the overlap of activation and inactivation curves. We now recognize that the extent of this overlap can be far greater than expected and to extend well above spike threshold, allowing evoked spikes to incorporate a significant component of T-type calcium current (Chemin et al., 2002; Swensen and Bean, 2003). More recently it was shown that activation of Cav3 calcium current can be detected electrophysiologically from potentials as low as -90 mV in cerebellar Purkinje cells (Engbers et al., 2012) and -70 mV in MVN neurons (Rehak et al., 2013). This is important in indicating that inward T-type current can be activated well within the range of subthreshold depolarizations or hyperpolarizations. While activation of inward current should provide a depolarizing drive, the influx of calcium can secondarily alter the net effect of calcium influx in ways we are only beginning to appreciate. In addition, other channels not known to physically associate with Cav3 channels but which affect voltage transitions can secondarily affect the functional role of Cav3 channels. The current review focused on two recently identified interactions between Cav3 and IKCa channels and Cav3 and HCN channels as a means of highlighting two very different forms of interactions involving Cav3 channels.
Cav3-IKCa Modulation of Temporal Summation
In cerebellar Purkinje cells a direct association between Cav3 and IKCa channels enables the activation of outward potassium current well into the subthreshold voltage range. Thus, even small amplitude PF EPSPs are capable of activating Cav3 channels and subsequently IKCa outward current to control the rate of decay of the EPSP and a subsequent AHP. The effect this has on temporal summation is dramatic in that only the first few stimuli of a train of EPSPs prove capable of summating, with marked suppression of any further summation even during a long stimulus train. This interaction is important in acting as a high pass filter to reduce the inherent level of excitability of Purkinje cells that would result from the constant barrage of even spontaneous PF EPSPs. Instead, only high frequency bursts of PF input representative of sensory input can exceed the barrier put in place by the Cav3-IKCa association to produce spike output (Engbers et al., 2012). The effects of the Cav3-IKCa complex can be compared to hippocampal and cortical pyramidal cells, where Cav3 channels activated during EPSPs act to amplify EPSP amplitude (Markram and Sakmann, 1994; Magee and Johnston, 1995; Gillessen and Alzheimer, 1997; Urban et al., 1998). In the case of the Cav3-IKCa complex, however, trains of EPSPs are suppressed and excitability is reduced. Similar results have been seen for BK channel activation in MVN neurons, where activation of BK channels by Cav3 calcium influx reduces the gain of spike output, thus reducing cell excitability (du Lac and Lisberger, 1995; Smith et al., 2002; Rehak et al., 2013). Therefore, Cav3 channels can play either an excitatory or a net inhibitory role in signal processing depending on the ion channels with which they interact.
HCN Channels Indirectly Modify Cav3 Functional Influence
Both IH and IT act within the subthreshold range and often play complementary roles. Early recordings in thalamocortical relay neurons demonstrated that an interplay of IH and IT generates oscillatory burst output (McCormick and Pape, 1990). In DCN neurons, IH and IT interact to play distinct yet complementary roles. The direct depolarizing effects of IH performs two functions. The first is to drive a graded reduction in FSL as hyperpolarizations are increased, creating an inverse relationship between the strength of the hyperpolarization and the time to burst output (Sangrey and Jaeger, 2010; Engbers et al., 2011). Increased activation of IH during hyperpolarization also increases the precision of the rebound burst, improving temporal fidelity. However, activation of IH also improves the efficacy of IT. By reducing the membrane time constant, IH decreases the inactivation of IT during repolarization, resulting in more Ca2+ influx and higher burst frequency. IH thus acts synergistically with IT to increase rebound frequency by at least 60% of the total response. As short-term facilitation and depression are known to be highly frequency-dependent (Dittman et al., 2000), the ability for IH to increase the range of frequencies generated during rebound responses (compare Figures 5A,C) could have important physiological consequences at downstream postsynaptic targets of DCN cells (Babalyan and Fanardzhyan, 1991).
Level of Interaction between Cav3 and IKCa/HCN Channels
It is clear that very different outcomes can arise from the expression of Cav3 calcium channels when their conductance or actions on the membrane potential are combined with IKCa or HCN channels. It would thus seem advantageous to employ mechanisms that could coexpress, colocalize, or otherwise ensure the pairing of these channels to mediate their effects in a given cell if not on a regional and subcellular basis. It is interesting to compare our current state of knowledge on the nature of interactions between Cav3 and either IKCa or HCN channels in terms of distribution and mechanisms of co-localization.
All protein biochemistry conducted thus far on the Cav3-IKCa complex has focused on the Cav3.2 channel isoform (Engbers et al., 2012), one of three known members of this family (Cav3.1, Cav3.2, Cav3.3; Perez-Reyes et al., 1998). Western blot analysis indicated coimmunoprecipitation between Cav3.2 and IKCa proteins from cerebellar lysates. This association also appears to be specific to the low voltage Cav3.2 isoform in finding no coimmunoprecipitation between IKCa protein and Cav2.1 (P/Q-type) channels from cerebellar lysates (Engbers et al., 2012), one of the principle HVA calcium channels expressed in Purkinje cells. The physiological interaction between Cav3 calcium influx and IKCa activation in Purkinje cells is also inferred to reflect activity of the Cav3.2 isoform given the ability to block the PF-evoked AHP with 100 μM Ni2+, a dose that preferentially blocks Cav3.2 (Zamponi et al., 1996; Lee et al., 1999). Dual immunolabel experiments also revealed a remarkable correspondence in colocalization of both Cav3.2 and IKCa protein to the cell body region and specific patches of proximal dendritic membrane of Purkinje cells. A close proximity of these channels was further indicated by the ability of internal BAPTA, but not EGTA, to block a TRAM-34-sensitive outward current evoked in outside-out recordings from Purkinje cells. This test is argued to support an interaction between channels that function within a calcium nanodomain (<50 nM distance; Berkefeld and Fakler, 2008; Fakler and Adelman, 2008). Together these data indicate that Cav3 and IKCa channels can be associated as a physical complex, although the specific sites for interaction between the subunits have not been reported thus far. We also do not know if there are mechanisms in place to establish this association at the level of the Golgi complex or if the channels are translocated to the membrane (or endocytosed) in any coordinated fashion. This is particularly the case for IKCa channels that can be highly regulated in terms of stimulus-induced transcription, translation, and translocation to and from the plasma membrane (Ghanshani et al., 2000; Toyama et al., 2008; Wulff and Castle, 2010; Bouhy et al., 2011; Schwab et al., 2012). The current evidence only identifies a coimmunoprecipitation between Cav3.2 and IKCa from brain lysates, and a functional interaction at the plasma membrane level with minimal physical separation. Further work will thus be required to establish where and how the Cav3.2-IKCa interaction is formed and the extent to which it might influence functions of the complex.
The ability to identify a Cav3.2-IKCa interaction also does not guarantee that all cells expressing Cav3.2 and IKCa proteins will form these ion channel complexes. Precedence for this can found in previous work showing that activation of BK channels by calcium influx through N-type channels can be detected in close association in a given cell (as recognized by paired activation of N-type and BK channels; Marrion and Tavalin, 1998; Berkefeld et al., 2006; Loane et al., 2007; Berkefeld and Fakler, 2008). L-type calcium channels can complex with SK potassium channels and still be recorded in adjacent membrane in isolation (Marrion and Tavalin, 1998). These factors must therefore be considered when one attempts to predict where a Cav3-IKCa complex might be active in cerebellum or other brain regions. The reported expression pattern of Cav3 channels differs according to available reports on mRNA expression through in situ hybridization or protein distribution by immunocytochemistry. Indeed, the most thorough in situ hybridization work predicted that Purkinje cells express Cav3.1 mRNA (Craig et al., 1999). In contrast, immunolocalization suggests that all three isoforms are expressed in Purkinje cells, but differentially across the soma-dendritic axis (Craig et al., 1999; McKay et al., 2006; Molineux et al., 2006; Hildebrand et al., 2009). The Cav3.2 isoform was primarily localized to the somatic region and restricted segments of the primary trunk of apical dendrites, and a lower detectable level on some secondary branches. As indicated above, this distribution proves to be extremely well matched to that of IKCa immunolabel in these cells (Engbers et al., 2012). However, attempting to generalize or predict what other cells might use this complex physiologically is difficult at this time given that the single report of IKCa channels in Purkinje cells represents the first demonstration of this channel type in any central neuron. Comparisons between the pattern of Cav3.2 and IKCa protein in other cells thus awaits a study on the localization of IKCa channels in other brain regions, at which time tentative predictions could be made as to how generalized this interaction might be in controlling cell output.
Much less is know of any potential relationship between Cav3.2 and HCN channels in DCN cells. In situ hybridization suggested that the primary mRNA expressed in DCN cells is for Cav3.1. As indicated, a detailed comparison of Cav3 channel immunolabel and rebound burst phenotypes in DCN cells found evidence for a correlation between the expression of Cav3.1 immunolabel and Transient Burst neurons, and Cav3.3 to a Weak Burst phenotype. While Cav3.2 immunolabel could be detected in some group of neurons in the DCN, none of the Transient or Weak Burst cells examined in that study tested positive for this Cav3 isoform. DCN cells have been shown to express immunolabel (Notomi and Shigemoto, 2004) and mRNA (Monteggia et al., 2000; Santoro et al., 2000) for at least HCN1, HCN2, and HCN4 isoforms. However, no comparisons have been made between the pattern of Cav3 and HCN channel labeling to detect any potential colocalization.
Interestingly, at least a functional coupling has been detected between calcium influx and HCN channels in some systems. Thus, calcium influx has been found to augment IH in both sino-atrial node cells (Hirano and Hagiwara, 1989) and thalamocortical cells (Luthi and McCormick, 1998b, 1999a, b) by right shifting the voltage-dependence for IH activation. In thalamocortical cells the increase in IH promotes a slow depolarization of seconds duration that depends on preceding calcium influx from rebound calcium spikes, a response attributed primarily to T-type calcium channels. The increase in IH then decreases input resistance and the magnitude of calcium spikes, slowing if not blocking the generation of oscillatory activity (Luthi and McCormick, 1998b). An IH-dependent process has also been shown to generate a slow depolarization that depends on prior membrane hyperpolarizations in a subset (23%) of cortical neurons, although the calcium dependence of this process was not definitively established (Winograd et al., 2008). Also, this effect was not found in all cortical cell types including those that otherwise expressed IH, indicating that calcium-dependent regulation of IH is not a property of all cells (Winograd et al., 2008).
The calcium dependent increase of IH in thalamocortical cells could be blocked by external application of 2–5 mM Ni2+ or internal perfusion of BAPTA or EGTA (Budde et al., 1997; Luthi and McCormick, 1998b). While the effects of Ni2+ were interpreted as reflecting a block of T-type calcium influx, this concentration is not selective enough to provide strong support for a discrete action on Cav3 channels (Zamponi et al., 1996). The best evidence of a role for T-type channels is the dependence of IH augmentation in thalamocortical cells on the generation of rebound calcium spikes (Luthi and McCormick, 1998b). Calcium-dependent regulation of IH in these cells was also concluded to be indirect through a calcium-dependent increase of PKA that modifies these cyclic nucleotide-dependent channels (Luthi and McCormick, 1999b). Therefore, at this time, there is evidence that calcium influx can modify IH in a subset of neurons, but a direct interaction, like that shown for Cav3 and IKCa channels, has not been demonstrated, with no further work carried out in DCN cells.
It is interesting to note that in cortical layer III pyramidal cells, HCN1 channels colocalize with Cav3.2 channels in the presynaptic terminals of glutamatergic inputs, where they act to increase Cav3 inactivation by depolarizing the terminal (Huang et al., 2011). Similar effects of IH increasing IT inactivation have been reported for dendrites of hippocampal CA1 pyramidal neurons (Tsay et al., 2007). However, these effects are different from the results summarized here for DCN cells in which the effect of IH on membrane time constant instead decreases Cav3 inactivation (Figure 6). Thus, at this time we have no reason to suspect that the interplay between Cav3- or HCN-mediated components of the rebound response in DCN cells reflects anything more than coexpressing these channels in isolation in the membrane, with concomitant effects on the membrane time constant and degree of Cav3 channel inactivation.
In summary, work in this area is converging to a growing realization that Cav3 calcium channels do not operate in isolation as long believed, but in fact can interact with other ion channels or receptors indirectly or as functional channel complexes to determine the final influence on membrane excitability. All of this work emphasizes the need for a detailed knowledge of the biophysical properties of ion channels, but also underscores the fact that their functional role depends on what other ion channels they interact with in the membrane simply through coexpression or by direct interaction in a growing number of ion channel complexes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge previous and current laboratory members who assisted in some aspects of data collection in the work summarized here, including M. Molineux, R. Rehak, R. Tadayonnejad, and M. Kruskic. This work was supported by grants from the Canadian Institutes of Health Research (CIHR) by Ray W. Turner and Gerald W. Zamponi and studentships through Alberta Innovates – Health Solutions (Jordan D. T. Engbers, Dustin Anderson), T. Chen Fong awards (Jordan D. T. Engbers, Dustin Anderson), the Killam Foundation (Jordan D. T. Engbers, Dustin Anderson), an AIMs award (Dustin Anderson) and a CIHR-CGS PhD studentship (Jordan D. T. Engbers). Ray W. Turner and Gerald W. Zamponi are AI-HS Scientists and Gerald W. Zamponi holds a Canada Research Chair.
Author Contributions
Ray W. Turner and Gerald W. Zamponi received grant support for the study; Jordan D. T. Engbers, Dustin Anderson, and Ray W. Turner designed the experiments; Jordan D. T. Engbers and Dustin Anderson conducted the experiments and analyzed the data; Ray W. Turner and Jordan D. T. Engbers wrote the manuscript.
References
Aizenman, C. D., and Linden, D. J. (1999). Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J. Neurophysiol. 82, 1697–1709.
Alvina, K., Ellis-Davies, G., and Khodakhah, K. (2009). T-type calcium channels mediate rebound firing in intact deep cerebellar neurons. Neuroscience 158, 635–641. doi: 10.1016/j.neuroscience.2008.09.052
Alvina, K., and Khodakhah, K. (2008). Selective regulation of spontaneous activity of neurons of the deep cerebellar nuclei by N-type calcium channels in juvenile rats. J. Physiol. 586, 2523–2538. doi: 10.1113/jphysiol.2007.148197
Alvina, K., Walter, J. T., Kohn, A., Ellis-Davies, G., and Khodakhah, K. (2008). Questioning the role of rebound firing in the cerebellum. Nat. Neurosci. 11, 1256–1258. doi: 10.1038/nn.2195
Anderson, D., Engbers, J. D., Heath, N. C., Bartoletti, T. M., Mehaffey, W. H., Zamponi, G. W., et al. (2013). The Cav3-Kv4 complex acts as a calcium sensor to maintain inhibitory charge transfer during extracellular calcium fluctuations. J. Neurosci. 33, 7811–7824. doi: 10.1523/JNEUROSCI.5384-12.2013
Anderson, D., Mehaffey, W. H., Iftinca, M., Rehak, R., Engbers, J. D., Hameed, S., et al. (2010a). Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat. Neurosci. 13, 333–337. doi: 10.1038/nn.2493
Anderson, D., Rehak, R., Hameed, S., Mehaffey, W. H., Zamponi, G. W., and Turner, R. W. (2010b). Regulation of the K(V)4.2 complex by Ca(V)3.1 calcium channels. Channels (Austin) 4, 163–167. doi: 10.4161/chan.4.3.11955
Angelo, K., London, M., Christensen, S. R., and Hausser, M. (2007). Local and global effects of I(h) distribution in dendrites of mammalian neurons. J. Neurosci. 27, 8643–8653. doi: 10.1523/JNEUROSCI.5284-06.2007
Atherton, J. F., Kitano, K., Baufreton, J., Fan, K., Wokosin, D., Tkatch, T., et al. (2010). Selective participation of somatodendritic HCN channels in inhibitory but not excitatory synaptic integration in neurons of the subthalamic nucleus. J. Neurosci. 30, 16025–16040. doi: 10.1523/JNEUROSCI.3898-10.2010
Awatramani, G. B., Price, G. D., and Trussell, L. O. (2005). Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48, 109–121. doi: 10.1016/j.neuron.2005.08.038
Babalyan, A. L., and Fanardzhyan, V. V. (1991). Electrophysiologic properties of red nucleus neurons in the rat brain slices. Neurophysiology 23, 454–461. doi: 10.1007/BF01052452
Bengtsson, F., Ekerot, C. F., and Jorntell, H. (2011). In vivo analysis of inhibitory synaptic inputs and rebounds in deep cerebellar nuclear neurons. PLoS ONE 6:e18822. doi: 10.1371/journal.pone.0018822
Berkefeld, H., and Fakler, B. (2008). Repolarizing responses of BKCa-Cav complexes are distinctly shaped by their Cav subunits. J. Neurosci. 28, 8238–8245. doi: 10.1523/JNEUROSCI.2274-08.2008
Berkefeld, H., Sailer, C. A., Bildl, W., Rohde, V., Thumfart, J. O., Eble, S., et al. (2006). BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314, 615–620. doi: 10.1126/science.1132915
Biel, M., Wahl-Schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885. doi: 10.1152/physrev.00029.2008
Bloodgood, B. L., and Sabatini, B. L. (2007). Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53, 249–260. doi: 10.1016/j.neuron.2006.12.017
Boehme, R., Uebele, V. N., Renger, J. J., and Pedroarena, C. (2011). Rebound excitation triggered by synaptic inhibition in cerebellar nuclear neurons is suppressed by selective T-type calcium channel block. J. Neurophysiol. 106, 2653–2661. doi: 10.1152/jn.00612.2011
Bouhy, D., Ghasemlou, N., Lively, S., Redensek, A., Rathore, K. I., Schlichter, L. C., et al. (2011). Inhibition of the Ca(2)(+)-dependent K(+) channel, KCNN4/KCa3.1, improves tissue protection and locomotor recovery after spinal cord injury. J. Neurosci. 31, 16298–16308. doi: 10.1523/JNEUROSCI.0047-11.2011
Budde, T., Biella, G., Munsch, T., and Pape, H. C. (1997). Lack of regulation by intracellular Ca2+ of the hyperpolarization-activated cation current in rat thalamic neurones. J. Physiol. 503(Pt 1), 79–85. doi: 10.1111/j.1469-7793.1997.079bi.x
Cain, S. M., and Snutch, T. P. (2010). Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 4, 475–482. doi: 10.4161/chan.4.6.14106
Cain, S. M., and Snutch, T. P. (2013). T-type calcium channels in burst-firing, network synchrony, and epilepsy. Biochim. Biophys. Acta 1828, 1572–1578. doi: 10.1016/j.bbamem.2012.07.028
Chadderton, P., Margrie, T. W., and Hausser, M. (2004). Integration of quanta in cerebellar granule cells during sensory processing. Nature 428, 856–860. doi: 10.1038/nature02442
Chemin, J., Monteil, A., Perez-Reyes, E., Bourinet, E., Nargeot, J., and Lory, P. (2002). Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J. Physiol. 540, 3–14. doi: 10.1113/jphysiol.2001.013269
Cheong, E., and Shin, H. S. (2013). T-type Ca(2)(+) channels in absence epilepsy. Biochim. Biophys. Acta 1828, 1560–1571. doi: 10.1016/j.bbamem.2013.02.002
Craig, P. J., Beattie, R. E., Folly, E. A., Banerjee, M. D., Reeves, M. B., Priestley, J. V., et al. (1999). Distribution of the voltage-dependent calcium channel alpha1G subunit mRNA and protein throughout the mature rat brain. Eur. J. Neurosci. 11, 2949–2964. doi: 10.1046/j.1460-9568.1999.00711.x
Cueni, L., Canepari, M., Adelman, J. P., and Luthi, A. (2009). Ca(2+) signaling by T-type Ca(2+) channels in neurons. Pflugers Arch. 457, 1161–1172. doi: 10.1007/s00424-008-0582-6
Cueni, L., Canepari, M., Lujan, R., Emmenegger, Y., Watanabe, M., Bond, C. T., et al. (2008). T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat. Neurosci. 11, 683–692. doi: 10.1038/nn.2124
D’Angelo, E., and De Zeeuw, C. I. (2009). Timing and plasticity in the cerebellum: focus on the granular layer. Trends Neurosci. 32, 30–40. doi: 10.1016/j.tins.2008.09.007
Dittman, J. S., Kreitzer, A. C., and Regehr, W. G. (2000). Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J. Neurosci. 20, 1374–1385.
Dreyfus, F. M., Tscherter, A., Errington, A. C., Renger, J. J., Shin, H. S., Uebele, V. N., et al. (2010). Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J. Neurosci. 30, 99–109. doi: 10.1523/JNEUROSCI.4305-09.2010
du Lac, S., and Lisberger, S. G. (1995). Cellular processing of temporal information in medial vestibular nucleus neurons. J. Neurosci. 15, 8000–8010.
Ekerot, C. F., and Jorntell, H. (2001). Parallel fibre receptive fields of Purkinje cells and interneurons are climbing fibre-specific. Eur. J. Neurosci. 13, 1303–1310. doi: 10.1046/j.0953-816x.2001.01499.x
Ekerot, C. F., and Jorntell, H. (2008). Synaptic integration in cerebellar granule cells. Cerebellum 7, 539–541. doi: 10.1007/s12311-008-0064-6
Engbers, J. D., Anderson, D., Asmara, H., Rehak, R., Mehaffey, W. H., Hameed, S., et al. (2012). Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. U.S.A. 109, 2601–2606. doi: 10.1073/pnas.1115024109
Engbers, J. D., Anderson, D., Tadayonnejad, R., Mehaffey, W. H., Molineux, M. L., and Turner, R. W. (2011). Distinct roles for I(T) and I(H) in controlling the frequency and timing of rebound spike responses. J. Physiol. 589, 5391–5413. doi: 10.1113/jphysiol.2011.215632
Engbers, J. D., Zamponi, G. W., and Turner, R. W. (2013). Modeling interactions between voltage-gated Ca2+ channels and KCa1.1 channels. Channels (Austin) 7, 24–25.
Faber, E. S. (2010). Functional interplay between NMDA receptors, SK channels and voltage-gated Ca2+ channels regulates synaptic excitability in the medial prefrontal cortex. J. Physiol. 588, 1281–1292. doi: 10.1113/jphysiol.2009.185645
Fakler, B., and Adelman, J. P. (2008). Control of K(Ca) channels by calcium nano/microdomains. Neuron 59, 873–881. doi: 10.1016/j.neuron.2008.09.001
Fernandez, F. R., Engbers, J. D., and Turner, R. W. (2007). Firing dynamics of cerebellar purkinje cells. J. Neurophysiol. 98, 278–294. doi: 10.1152/jn.00306.2007
Gastrein, P., Campanac, E., Gasselin, C., Cudmore, R., Bialowas, A., Carlier, E., et al. (2011). The role of hyperpolarization-activated cationic current (Ih) in spike-time precision and intrinsic resonance in cortical neurons in vitro. J. Physiol. 589, 3753–3773. doi: 10.1113/jphysiol.2011.209148
Ghanshani, S., Wulff, H., Miller, M. J., Rohm, H., Neben, A., Gutman, G. A., et al. (2000). Upregulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 275, 37137–37149. doi: 10.1074/jbc.M003941200
Gillessen, T., and Alzheimer, C. (1997). Amplification of EPSPs by low Ni(2+)- and amiloride-sensitive Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J. Neurophysiol. 77, 1639–1643.
Gittis, A. H., and du Lac, S. (2007). Firing properties of GABAergic versus non-GABAergic vestibular nucleus neurons conferred by a differential balance of potassium currents. J. Neurophysiol. 97, 3986–3996. doi: 10.1152/jn.00141.2007
Grunnet, M., and Kaufmann, W. A. (2004). Coassembly of big conductance Ca2+-activated K+ channels and L-type voltage-gated Ca2+ channels in rat brain. J. Biol. Chem. 279, 36445–36453. doi: 10.1074/jbc.M402254200
Hausser, M., and Clark, B. A. (1997). Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19, 665–678. doi: 10.1016/S0896-6273(00)80379-7
Hildebrand, M. E., Isope, P., Miyazaki, T., Nakaya, T., Garcia, E., Feltz, A., et al. (2009). Functional coupling between mGluR1 and Cav3.1 T-type calcium channels contributes to parallel fiber-induced fast calcium signaling within Purkinje cell dendritic spines. J. Neurosci. 29, 9668–9682. doi: 10.1523/JNEUROSCI.0362-09.2009
Hirano, T., and Hagiwara, S. (1989). Kinetics and distribution of voltage-gated Ca, Na and K channels on the somata of rat cerebellar Purkinje cells. Pflugers Arch. 413, 463–469. doi: 10.1007/BF00594174
Hoebeek, F. E., Witter, L., Ruigrok, T. J., and De Zeeuw, C. I. (2010). Differential olivo-cerebellar cortical control of rebound activity in the cerebellar nuclei. Proc. Natl. Acad. Sci. U.S.A. 107, 8410–8415. doi: 10.1073/pnas.0907118107
Huang, Z., Lujan, R., Kadurin, I., Uebele, V. N., Renger, J. J., Dolphin, A. C., et al. (2011). Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat. Neurosci. 14, 478–486. doi: 10.1038/nn.2757
Huguenard, J. R. (1996). Low-threshold calcium currents in central nervous system neurons. Annu. Rev. Physiol. 58, 329–348. doi: 10.1146/annurev.ph.58.030196.001553
Ishii, T. M., Silvia, C., Hirschberg, B., Bond, C. T., Adelman, J. P., and Maylie, J. (1997). A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. U.S.A. 94, 11651–11656. doi: 10.1073/pnas.94.21.11651
Isope, P., and Barbour, B. (2002). Properties of unitary granule cell – Purkinje cell synapses in adult rat cerebellar slices. J. Neurosci. 22, 9668–9678.
Isope, P., and Murphy, T. H. (2005). Low threshold calcium currents in rat cerebellar Purkinje cell dendritic spines are mediated by T-type calcium channels. J. Physiol. 562, 257–269. doi: 10.1113/jphysiol.2004.074211
Jahnsen, H. (1986a). Electrophysiological characteristics of neurones in the guinea-pig deep cerebellar nuclei in vitro. J. Physiol. 372, 129–147.
Jahnsen, H. (1986b). Extracellular activation and membrane conductances of neurones in the guinea-pig deep cerebellar nuclei in vitro. J. Physiol. 372, 149–168.
Joiner, W. J., Wang, L. Y., Tang, M. D., and Kaczmarek, L. K. (1997). hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. U.S.A. 94, 11013–11018. doi: 10.1073/pnas.94.20.11013
Kaushal, V., Koeberle, P. D., Wang, Y., and Schlichter, L. C. (2007). The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J. Neurosci. 27, 234–244. doi: 10.1523/JNEUROSCI.3593-06.2007
Kim, D., Song, I., Keum, S., Lee, T., Jeong, M. J., Kim, S. S., et al. (2001). Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 31, 35–45. doi: 10.1016/S0896-6273(01)00343-9
Kole, M. H., Hallermann, S., and Stuart, G. J. (2006). Single Ih channels in pyramidal neuron dendrites: properties, distribution, and impact on action potential output. J. Neurosci. 26, 1677–1687. doi: 10.1523/JNEUROSCI.3664-05.2006
Lee, J. H., Gomora, J. C., Cribbs, L. L., and Perez-Reyes, E. (1999). Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys. J. 77, 3034–3042. doi: 10.1016/S0006-3495(99)77134-1
Loane, D. J., Lima, P. A., and Marrion, N. V. (2007). Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain. J. Cell Sci. 120, 985–995. doi: 10.1242/jcs.03399
Luthi, A., and McCormick, D. A. (1998a). H-current: properties of a neuronal and network pacemaker. Neuron 21, 9–12. doi: 10.1016/S0896-6273(00)80509-7
Luthi, A., and McCormick, D. A. (1998b). Periodicity of thalamic synchronized oscillations: the role of Ca2+-mediated upregulation of Ih. Neuron 20, 553–563. doi: 10.1016/S0896-6273(00)80994-0
Luthi, A., and McCormick, D. A. (1999a). Ca(2+)-mediated up-regulation of Ih in the thalamus. How cell-intrinsic ionic currents may shape network activity. Ann. N. Y. Acad. Sci. 868, 765–769. doi: 10.1111/j.1749-6632.1999.tb11354.x
Luthi, A., and McCormick, D. A. (1999b). Modulation of a pacemaker current through Ca(2+)-induced stimulation of cAMP production. Nat. Neurosci. 2, 634–641. doi: 10.1038/10189
Magee, J. C. (1999). Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat. Neurosci. 2, 848. doi: 10.1038/12229
Magee, J. C., Christofi, G., Miyakawa, H., Christie, B., Lasser-Ross, N., and Johnston, D. (1995). Subthreshold synaptic activation of voltage-gated Ca2+ channels mediates a localized Ca2+ influx into the dendrites of hippocampal pyramidal neurons. J. Neurophysiol. 74, 1335–1342.
Magee, J. C., and Johnston, D. (1995). Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science 268, 301–304. doi: 10.1126/science.7716525
Markram, H., and Sakmann, B. (1994). Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage-activated calcium channels. Proc. Natl. Acad. Sci. U.S.A. 91, 5207–5211. doi: 10.1073/pnas.91.11.5207
Marrion, N. V., and Tavalin, S. J. (1998). Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905. doi: 10.1038/27674
McCormick, D. A., and Bal, T. (1997). Sleep and arousal: thalamocortical mechanisms. Annu. Rev. Neurosci. 20, 185–215. doi: 10.1146/annurev.neuro.20.1.185
McCormick, D. A., and Pape, H. C. (1990). Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J. Physiol. 431, 291–318.
McDonough, S. I., and Bean, B. P. (1998). Mibefradil inhibition of T-type calcium channels in cerebellar purkinje neurons. Mol. Pharmacol. 54, 1080–1087.
McKay, B. E., McRory, J. E., Molineux, M. L., Hamid, J., Snutch, T. P., Zamponi, G. W., et al. (2006). Cav3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur. J. Neurosci. 24, 2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x
Mittmann, W., Koch, U., and Hausser, M. (2005). Feed-forward inhibition shapes the spike output of cerebellar Purkinje cells. J. Physiol. 563, 369–378. doi: 10.1113/jphysiol.2004.075028
Molineux, M. L., McRory, J. E., McKay, B. E., Hamid, J., Mehaffey, W. H., Rehak, R., et al. (2006). Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 5555–5560. doi: 10.1073/pnas.0601261103
Molineux, M. L., Mehaffey, W. H., Tadayonnejad, R., Anderson, D., Tennent, A. F., and Turner, R. W. (2008). Ionic factors governing rebound burst phenotype in rat deep cerebellar neurons. J. Neurophysiol. 100, 2684–2701. doi: 10.1152/jn.90427.2008
Monteggia, L. M., Eisch, A. J., Tang, M. D., Kaczmarek, L. K., and Nestler, E. J. (2000). Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res. Mol. Brain Res. 81, 129–139. doi: 10.1016/S0169-328X(00)00155-8
Muri, R., and Knopfel, T. (1994). Activity induced elevations of intracellular calcium concentration in neurons of the deep cerebellar nuclei. J. Neurophysiol. 71, 420–428.
Ngo-Anh, T. J., Bloodgood, B. L., Lin, M., Sabatini, B. L., Maylie, J., and Adelman, J. P. (2005). SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 8, 642–649. doi: 10.1038/nn1449
Notomi, T., and Shigemoto, R. (2004). Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J. Comp. Neurol. 471, 241–276. doi: 10.1002/cne.11039
Oliver, D., Klocker, N., Schuck, J., Baukrowitz, T., Ruppersberg, J. P., and Fakler, B. (2000). Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26, 595–601. doi: 10.1016/S0896-6273(00)81197-6
Pedarzani, P., and Stocker, M. (2008). Molecular and cellular basis of small – and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell. Mol. Life Sci. 65, 3196–3217. doi: 10.1007/s00018-008-8216-x
Pedroarena, C. M. (2010). Mechanisms supporting transfer of inhibitory signals into the spike output of spontaneously firing cerebellar nuclear neurons in vitro. Cerebellum 9, 67–76. doi: 10.1007/s12311-009-0153-1
Perez-Reyes, E. (2003). Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 83, 117–161.
Perez-Reyes, E. (2006). Molecular characterization of T-type calcium channels. Cell Calcium 40, 89–96. doi: 10.1016/j.ceca.2006.04.012
Perez-Reyes, E., Cribbs, L. L., Daud, A., Lacerda, A. E., Barclay, J., Williamson, M. P., et al. (1998). Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 391, 896–900. doi: 10.1038/36110
Person, A. L., and Raman, I. M. (2012). Purkinje neuron synchrony elicits time-locked spiking in the cerebellar nuclei. Nature 481, 502–505. doi: 10.1038/nature10732
Raman, I. M., and Bean, B. P. (1999). Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. 19, 1663–1674.
Rancz, E. A., Ishikawa, T., Duguid, I., Chadderton, P., Mahon, S., and Hausser, M. (2007). High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature 450, 1245–1248. doi: 10.1038/nature05995
Rehak, R., Bartoletti, T. M., Engbers, J. D. T., Berecki, G., Turner, R. W., and Zamponi, G. W. (2013). Low voltage activation of KCa1.1 current by Cav3-KCa1.1 complexes. PLoS ONE 8:e61844. doi: 10.1371/journal.pone.0061844
Robinson, R. B., and Siegelbaum, S. A. (2003). Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65, 453–480. doi: 10.1146/annurev.physiol.65.092101.142734
Sangrey, T., and Jaeger, D. (2010). Analysis of distinct short and prolonged components in rebound spiking of deep cerebellar nucleus neurons. Eur. J. Neurosci. 32, 1646–1657. doi: 10.1111/j.1460-9568.2010.07408.x
Santoro, B., Chen, S., Luthi, A., Pavlidis, P., Shumyatsky, G. P., Tibbs, G. R., et al. (2000). Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J. Neurosci. 20, 5264–5275.
Schneider, E. R., Civillico, E. F., and Wang, S. S. (2013). Calcium-based dendritic excitability and Its regulation in the deep cerebellar nuclei. J. Neurophysiol. 109, 2282–2292. doi: 10.1152/jn.00925.2012
Schwab, A., Nechyporuk-Zloy, V., Gassner, B., Schulz, C., Kessler, W., Mally, S., et al. (2012). Dynamic redistribution of calcium sensitive potassium channels (hK(Ca)3.1) in migrating cells. J. Cell. Physiol. 227, 686–696. doi: 10.1002/jcp.22776
Smith, M. R., Nelson, A. B., and Du Lac, S. (2002). Regulation of firing response gain by calcium-dependent mechanisms in vestibular nucleus neurons. J. Neurophysiol. 87, 2031–2042.
Steuber, V., Schultheiss, N. W., Silver, R. A., De Schutter, E., and Jaeger, D. (2011). Determinants of synaptic integration and heterogeneity in rebound firing explored with data-driven models of deep cerebellar nucleus cells. J. Comput. Neurosci. 30, 633–658. doi: 10.1007/s10827-010-0282-z
Swensen, A. M., and Bean, B. P. (2003). Ionic mechanisms of burst firing in dissociated Purkinje neurons. J. Neurosci. 23, 9650–9663.
Tadayonnejad, R., Anderson, D., Molineux, M. L., Mehaffey, W. H., Jayasuriya, K., and Turner, R. W. (2010). Rebound discharge in deep cerebellar nuclear neurons in vitro. Cerebellum 9, 352–374. doi: 10.1007/s12311-010-0168-7
Tadayonnejad, R., Mehaffey, W. H., Anderson, D., and Turner, R. W. (2009). Reliability of triggering postinhibitory rebound bursts in deep cerebellar neurons. Channels (Austin) 3, 149–155. doi: 10.4161/chan.3.3.8872
Talavera, K., and Nilius, B. (2006). Biophysics and structure-function relationship of T-type Ca2+ channels. Cell Calcium 40, 97–114. doi: 10.1016/j.ceca.2006.04.013
Toyama, K., Wulff, H., Chandy, K. G., Azam, P., Raman, G., Saito, T., et al. (2008). The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Invest. 118, 3025–3037. doi: 10.1172/JCI30836
Tsay, D., Dudman, J. T., and Siegelbaum, S. A. (2007). HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron 56, 1076–1089. doi: 10.1016/j.neuron.2007.11.015
Urban, N. N., Henze, D. A., and Barrionuevo, G. (1998). Amplification of perforant-path EPSPs in CA3 pyramidal cells by LVA calcium and sodium channels. J. Neurophysiol. 80, 1558–1561.
Wahl-Schott, C., and Biel, M. (2009). HCN channels: structure, cellular regulation and physiological function. Cell. Mol. Life Sci. 66, 470–494. doi: 10.1007/s00018-008-8525-0
Weiss, N., and Zamponi, G. W. (2013). Control of low-threshold exocytosis by T-type calcium channels. Biochim. Biophys. Acta 1828, 1579–1586. doi: 10.1016/j.bbamem.2012.07.031
Wetmore, D. Z., Mukamel, E. A., and Schnitzer, M. J. (2008). Lock-and-key mechanisms of cerebellar memory recall based on rebound currents. J. Neurophysiol. 100, 2328–2347. doi: 10.1152/jn.00344.2007
Williams, S. R., Christensen, S. R., Stuart, G. J., and Hausser, M. (2002). Membrane potential bistability is controlled by the hyperpolarization-activated current I(H) in rat cerebellar Purkinje neurons in vitro. J. Physiol. 539, 469–483. doi: 10.1113/jphysiol.2001.013136
Winograd, M., Destexhe, A., and Sanchez-Vives, M. V. (2008). Hyperpolarization-activated graded persistent activity in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 105, 7298–7303. doi: 10.1073/pnas.0800360105
Wolfart, J., Neuhoff, H., Franz, O., and Roeper, J. (2001). Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J. Neurosci. 21, 3443–3456.
Wolfart, J., and Roeper, J. (2002). Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J. Neurosci. 22, 3404–3413.
Womack, M. D., Chevez, C., and Khodakhah, K. (2004). Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J. Neurosci. 24, 8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004
Wulff, H., and Castle, N. A. (2010). Therapeutic potential of KCa3.1 blockers: recent advances and promising trends. Exp. Rev. Clin. Pharmacol. 3, 385–396. doi: 10.1586/ecp.10.11
Yanovsky, Y., Zhang, W., and Misgeld, U. (2005). Two pathways for the activation of small-conductance potassium channels in neurons of substantia nigra pars reticulata. Neuroscience 136, 1027–1036. doi: 10.1016/j.neuroscience.2005.08.026
Yunker, A. M., and McEnery, M. W. (2003). Low-voltage-activated (“T-Type”) calcium channels in review. J. Bioenerg. Biomembr. 35, 533–575. doi: 10.1023/B:JOBB.0000008024.77488.48
Zamponi, G. W., Bourinet, E., and Snutch, T. P. (1996). Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J. Membr. Biol. 151, 77–90. doi: 10.1007/s002329900059
Zhang, W., Shin, J. H., and Linden, D. J. (2004). Persistent changes in the intrinsic excitability of rat deep cerebellar nuclear neurons induced by EPSPs or IPSP bursts. J. Physiol. (Lond.) 561.3, 703–719. doi: 10.1113/jphysiol.2004.071696
Keywords: Cav3, HCN, T-type, IKCa, KCa3.1, channel complex
Citation: Engbers JDT, Anderson D, Zamponi GW and Turner RW (2013) Signal processing by T-type calcium channel interactions in the cerebellum. Front. Cell. Neurosci. 7:230. doi: 10.3389/fncel.2013.00230
Received: 10 September 2013; Accepted: 06 November 2013;
Published online: 27 November 2013.
Edited by:
Leigh Anne Swayne, University of Victoria, CanadaReviewed by:
Terrance P. Snutch, The University of British Columbia, CanadaPhilippe Isope, Centre National Pour la Recherche Scientifique, France
Copyright © 2013 Engbers, Anderson, Zamponi and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ray W. Turner, Department of Cell Biology and Anatomy, Hotchkiss Brain Institute, University of Calgary, HRIC 1AA14, 3330 Hospital Dr. N.W., Calgary, AB T2N 4N1, Canada e-mail:cnd0dXJuZXJAdWNhbGdhcnkuY2E=