 Igor Medina1,2*
Igor Medina1,2* Perrine Friedel1,2
Perrine Friedel1,2 Claudio Rivera1,2,3
Claudio Rivera1,2,3 Kristopher T. Kahle4,5
Kristopher T. Kahle4,5 Nazim Kourdougli1,2
Nazim Kourdougli1,2 Pavel Uvarov6
Pavel Uvarov6 Christophe Pellegrino1,2
Christophe Pellegrino1,2- 1INSERM, Institut de Neurobiologie de la Méditerranée (INMED), Marseille, France
- 2Aix-Marseille Université, UMR901, Marseille, France
- 3Neuroscience Center, University of Helsinki, Helsinki, Finland
- 4Department of Cardiology, Manton Center for Orphan Disease Research, Howard Hughes Medical Institute, Boston Children's Hospital, Boston, MA, USA
- 5Department of Neurosurgery, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA
- 6Institute of Biomedicine, Anatomy, University of Helsinki, Helsinki, Finland
In the mammalian central nervous system (CNS), the inhibitory strength of chloride (Cl−)-permeable GABAA and glycine receptors (GABAAR and GlyR) depends on the intracellular Cl− concentration ([Cl−]i). Lowering [Cl−]i enhances inhibition, whereas raising [Cl−]i facilitates neuronal activity. A neuron's basal level of [Cl−]i, as well as its Cl− extrusion capacity, is critically dependent on the activity of the electroneutral K+-Cl− cotransporter KCC2, a member of the SLC12 cation-Cl− cotransporter (CCC) family. KCC2 deficiency compromises neuronal migration, formation and the maturation of GABAergic and glutamatergic synaptic connections, and results in network hyperexcitability and seizure activity. Several neurological disorders including multiple epilepsy subtypes, neuropathic pain, and schizophrenia, as well as various insults such as trauma and ischemia, are associated with significant decreases in the Cl− extrusion capacity of KCC2 that result in increases of [Cl−]i and the subsequent hyperexcitability of neuronal networks. Accordingly, identifying the key upstream molecular mediators governing the functional regulation of KCC2, and modifying these signaling pathways with small molecules, might constitute a novel neurotherapeutic strategy for multiple diseases. Here, we discuss recent advances in the understanding of the mechanisms regulating KCC2 activity, and of the role these mechanisms play in neuronal Cl− homeostasis and GABAergic neurotransmission. As KCC2 mediates electroneutral transport, the experimental recording of its activity constitutes an important research challenge; we therefore also, provide an overview of the different methodological approaches utilized to monitor function of KCC2 in both physiological and pathological conditions.
Introduction
Since the discovery of the dichotomic action of GABAergic neurotransmission more than two decades ago (Mueller et al., 1984; Ben-Ari et al., 1989), important progress has been made in the understanding of the role of intraneuronal Cl− concentration ([Cl−]i) in determining the action of GABA and glycine on post-synaptic neurons. It is now clear that [Cl−]i is critical for proper functioning of the CNS in physiological conditions, and alterations of [Cl−]i are observed in multiple neurological disorders that are characterized by GABAergic disinhibition (reviewed by Ben-Ari et al., 2007; Kahle et al., 2008)
One of the key molecules responsible for determining [Cl−]i in mature mammalian neurons, and also impaired in numerous pathological conditions resulting in deranged [Cl−]i homeostasis, is the K+-Cl− cotransporter KCC2, a neuron-specific Cl− extruder of the cation-Cl− cotransporter gene family SLC12. KCC2 plays a critical role in setting neuronal [Cl−]i; its deficiency (Rivera et al., 1999) or absence (Hübner et al., 2001; Woo et al., 2002; Balakrishnan et al., 2003; Zhu et al., 2005) raises [Cl−]i and thereby decreases the inhibitory strength of GABA and glycine, whose cognate receptors are ligand-gated ion channels permeable to Cl− and HCO−3 ions. Impaired KCC2 activity and consequent increases of [Cl−]i have been documented in epilepsy (Palma et al., 2006; Huberfeld et al., 2007), neuropathic pain (Coull et al., 2003), and spasticity following spinal cord injury (Cramer et al., 2008; Shulga et al., 2008; Boulenguez et al., 2010). These and other works (see Kahle et al., 2008; Blaesse et al., 2009; Ben-Ari et al., 2012 for detailed reviews) have suggested that decreased KCC2 activity is a principal cause of the rise in [Cl−]i, that leads to the depolarizing action and enhancement of neuronal network activity in these paradigms.
Recent findings have provided novel insights into the dynamics of KCC2 activity and [Cl−]i homeostasis in different pathology-related experimental conditions, revealing the existence of complex responses depending on the type of neuronal activity and the timing of observation (e.g., Khirug et al., 2010; Pellegrino et al., 2011; Shulga et al., 2012). In parallel, other studies strongly contributed to the understanding of the signaling pathways controlling KCC2 activity (e.g., Puskarjov et al., 2012; Bos et al., 2013; Gagnon et al., 2013; Ivakine et al., 2013; Markkanen et al., 2013; De Los Heros et al., 2014). In the present work we review recent compelling findings, highlight unresolved questions, and discuss the technical challenges that underlie the study of KCC2.
Overview of KCC2 Basic Properties
Several recent reviews have provided detailed analysis of the KCC2, its expression profile, physiological roles and implications in different neurological disorders (e.g., Gamba, 2005; Medina and Chudotvorova, 2006; Kahle et al., 2008, 2013; Blaesse et al., 2009; Chamma et al., 2012; Gagnon and Delpire, 2013). Here, to avoid repeating these reviews, we highlight briefly only some of KCC2 properties required as background for further discussion on the molecular determinants of the activity-dependent control of the transporter.
KCC2 is one of nine cation-chloride cotransporters (CCCs) encoded by SLC12 family genes. It shares high homology with three other K+-Cl− cotransporters (KCC1, KCC3, and KCC4), however it is distinct from these and other CCCs transporters by its unique importance for functioning of CNS: (i) KCC2 is the only CCC transporter expressed preferentially in neurons; (ii) it is characterized by a progressive increase in expression during development; (iii) KCC2 shows activity in isotonic conditions; (iv) KCC2 plays a critical role in establishing neuronal [Cl−]i and (v) it is involved in the control of numerous neuronal processes including migration, dendritic outgrowth, formation of synaptic connections, and spine morphology.
KCC2 was cloned in 1996 by homology with the KCC1 cotransporter from a rat brain cDNA library (Payne et al., 1996). This, and numerous other studies have characterized KCC2 as a transporter expressed exclusively in neurons of the CNS (reviewed by Blaesse et al., 2009), however some recent works showed also KCC2 expression in human fetal lens epithelial cell line (Lauf et al., 2008, 2012), chicken cardiomyocytes (Antrobus et al., 2012) and cancer cells (Wei et al., 2011) suggesting that functioning of KCC2 extends beyond the CNS. The silencing of KCC2 in mice (Hübner et al., 2001; Woo et al., 2002; Zhu et al., 2008; Khalilov et al., 2011) as well as KCC2-like genes in worms (CE-KCC-2, Tanis et al., 2009; Bellemer et al., 2011) and flies (dm-kcc, Hekmat-Scafe et al., 2006, 2010) produces severe changes of the neuronal network properties leading to appearance of seizure activity, increased neuron hyperexcitability and modifyied formation of synaptic connections. The silencing of other members of SLC12 gene family expressed in brain (NKCC1, KCC1, KCC3, and KCC4) lead to much less pronounced changes of the phenotype (see Gagnon and Delpire, 2013 for most recent review on genetically engineered CCC knockouts).
The secondary and tertiary structure of KCC2 and the other KCCs is unknown. Based on hydropathy analysis of KCC2 protein, Payne et al. (1996) proposed a putative model of KCC2, including 12 transmembrane domains and intracellular N- and C-termini. The authors predicted four glycosylation sites on the 3rd putative extracellular loop and highlighted a potential tyrosine phosphorylation site at position 1087. So far, this is the only model proposed. It is widely explored in drawings illustrating putative structure of the KCC2 in many reviews and original data publications (e.g., Li et al., 2007; Zhao et al., 2008). The KCC2 scheme drawn in the present review (Figure 1) has been also inspired by the original drawing by Payne et al. (1996). The functional importance of different structural elements of the KCC2 is also little studied. The region forming ion-transporting element of the KCC2 and mechanism controlling intrinsic activity of the transporter remain unclear (see below for more details). An important aspect of KCC2 that may affect activity of this transporter is its ability to oligomerize, and this issue may be one of the most challenging in the elucidation of KCC2 function and regulation.
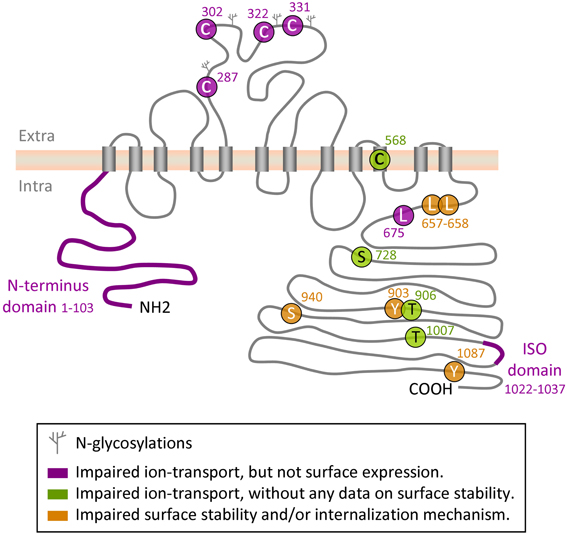
Figure 1. Regulatory sites on KCC2 protein. Depicted is a schematic representation of the amino acid residues and domains in KCC2 that have been implicated in the control of intrinsic KCC2 transport activity or in its cell surface stability and/or membrane internalization.
KCC2 is a secondary-active electroneutral transporter that carries K+ and Cl− ions in a 1:1 stoichiometry. The direction of the ion transport depends on the sum of chemical gradients of K+ and Cl− (Payne, 1997). When close to the chemical equilibrium, even slight changes in the concentration of either ion lead to a change of the direction of the ion transport (Payne, 1997). This property of the KCC2 is largely used during measurements of the KCC2 activity by mean of 86Rb+, Tl+ or NH+4 influx (see inset Box for details).
Box 1. How to Measure KCC2 Ion-Transport Activity.
Due to its electroneutrality, the KCC2 transport does not generate ion current. Therefore the electrophysiological approaches used to directly monitor opening of ion channels are not applicable to the analysis of KCC2, as well as of other members of CCC family. To bypass this problem, researchers measure consequences of transporter activity, i.e., the dynamic changes of the intracellular concentration of either K+ or Cl−.
Flame photometry. The first studies characterizing activity of the endogenous cation-Cl−-dependent ion transporters determined changes in the intracellular content of Na+, K+, and Cl− by flame photometry (Geck et al., 1980) or by analysing 24Na+ and 40K+ radioactivity (Kregenow, 1971). Amazingly, these and other pioneer works studying ion fluxes through endogenous transporters determined the exact stoichiometry of Na+-K+-Cl− (1:1:2) and K+-Cl− (1:1) co-transport, characterized the ion specificity, and described the requirement of Cl− for operation of Na+-K+-Cl− and K+-Cl− transporters (Haas, 1994). These properties were confirmed many times and are considered presently as landmarks for the NKCC and KCC subfamilies of transporters (Blaesse et al., 2009).
86RB+ flux assay. A more modern approach to analyse the NKCC and KCC activity consists in measurement of the 86Rb+ flux through the membrane. This approach was used successfully to clone and characterize NKCC1 (Gamba et al., 1994; Xu et al., 1994), KCC1 (Gillen et al., 1996) and KCC2 (Payne, 1997), and continues to be extensively utilized for characterization of different mutants and variants of CCC.
Although the above approaches strongly contributed to study the functional properties of CCC in homogenous cell preparations (erythrocytes, oocytes, different heterologous cell lines), they have limited applications for studying tissues composed of different cell types (i.e., brain slices, neuronal cultures). For example, using 86Rb+ it is difficult to discriminate in brain slices between the activity of NKCC1 in neuronal cells and surrounding glial cells. Moreover, the radioactive approach does not allow resolution of ion concentration changes at the subcellular level. Finally, the use of radioactive isotope raises the questions of potential toxicity and health hazard and imposes important security restriction.
TL+ flux assay. This method is based on the use of Thallium (Tl+) as the K+ tracer. Recent reports have shown that Tl+ binds with high affinity to the K+ ion site on the KCC2 (Zhang et al., 2010) and NKCC2 (Carmosino et al., 2013) and, once having crossed the plasma membrane, could be efficiently detected using metal-sensitive fluorescent dyes (Delpire et al., 2009; Hartmann et al., 2010; Zhang et al., 2010; Carmosino et al., 2013).
NH4+ flux assay. NH+4 uptake measures indirectly the activity of KCCs and NKCCs, which are highly-efficient carriers of NH+4 at their K+ site (see Williams and Payne, 2004 for detailed review and analysis of KCC2 selectivity). Several groups have used this property of CCC proteins to estimate KCC2 activity by measuring NH+4-dependent changes of pHi (Williams and Payne, 2004; Titz et al., 2006; Hershfinkel et al., 2009). This approach allows the analysis of KCC2 activity on the cellular and subcellular level as well as the estimation of KCC2 activity in real time. The inconveniences of the approach are related to the complex changes of the pHi in response to the application of the solution containing NH+4 (KCC2-independent alkalinisation followed by KCC2-dependent acidification) and possible contamination of pHi changes by functioning of other transporters, channels and ion exchangers. In addition, similarly to Tl+ method, the NH+4 approach measures KCC2 activity in reverse mode that increases [Cl−]i and places recorded cells in non-physiological conditions.
Thus, the methods described above allow measurement of the intrinsic transport carried by KCC transporters. These methods are designed to measure cation influx, i.e., to analyse KCC2 activity in reverse mode that is not its physiological function and will cause an increase of [Cl−]i. Accordingly, the potency of Tl+ and NH+4 methods to detect the ion flux mediated by endogenous KCC transporters under physiological conditions remains to be demonstrated.
A recording of Cl− is a highly efficient alternative to the cation methods of CCC activity measurements under conditions close to physiological. Numerous works have shown that KCC2 activity leads to strong changes in the intracellular Cl− concentration (reviewed by Ben-Ari et al., 2007; Blaesse et al., 2009). The estimate ion of [Cl−]i in neuronal cells could be achieved via different methods. Here we provide a brief description of different ways of estimating [Cl−]i and emphasize their relative advantages and limitations.
Gramicidin perforated patch clamp (GPPC) recording. The method is based on the use of the antibiotic gramicidin that makes cation-selective pores in the membrane; the patch pipette is filled with gramicidin and thus, preserves [Cl−]i (Ebihara et al., 1995; Kyrozis and Reichling, 1995). The concentration of Cl− is estimated using the Nernst equation after recording the reversal potential of GABAA or glycine receptor-channels that are Cl−-permeable. Using the GPPC approach, multiple studies have characterized KCC2-dependent changes in [Cl−]i (Balakrishnan et al., 2003; Chudotvorova et al., 2005; Lee et al., 2005; Nardou et al., 2011; Ivakine et al., 2013). The main advantage to GPPC recording is the estimation of [Cl−]i with high precision at single cell level. Unfortunately, this method has numerous limitations that should be taken into consideration: (i) it affects [K+]i, given that KCC2 uses K+ as main co-carrier, this should be taken into consideration when interpreting the result obtained with this method; (ii) most of the measurements using GPPC are made in voltage clamp mode of patch clamp recording, and the imposed holding potential provides an additional driving force for Cl− ions, affecting the kinetics of Cl− influx or efflux, and thus possibly confounding interpretations of [Cl−]i; (iii) the GPPC method has limited application to record [Cl−]i in immature neurons, which harbors high input resistance (Tyzio et al., 2003).
Single GABAA channel recording (SGCR). This method is a non-invasive way to estimate the driving force of GABAA using cell-attached recording of single GABAA receptor currents. This approach was used successfully to characterize the developmental shift of GABA from depolarizing to hyperpolarizing (Tyzio et al., 2006, 2008). The main advantage of this method is that it is a non-invasive way of estimating the exact difference between the levels of the resting membrane potential and the reversal potential of GABAAR-mediated currents. Limiting points of the method are that there is a labor intensive aspect to data recording and analysis, and there is a requirement of simultaneous single-channel-based recording of resting membrane potential (Tyzio et al., 2003, 2008).
Soma-to-dendrite Cl− gradient. To assess Cl− homeostasis and the dynamics of chloride removal in cortical neurons, Jarolimek et al. (1999) and Kelsch et al. (2001) proposed an elegant approach based on whole cell recording of the differences between EGABA in soma and dendrites. The EGABA in the soma is determined by the high [Cl−]i diffused from the patch pipette, whereas EGABA in dendrites is more negative (i.e., [Cl−]i is lower due to active extrusion by KCC2) (Kelsch et al., 2001). This approach was successfully used by several groups to monitor KCC2 activity in different types of neurons (Khirug et al., 2005; Li et al., 2007; Chamma et al., 2013) and neuronal compartments (Khirug et al., 2008). The advantage of the approach is that it allows a determination of “pure” Cl−-extrusion capacity under given experimental conditions. The inconvenience is that the values could be strongly influenced by efficiency of neuron perfusion with intracellular solution (serial resistance, dendrites diameter). In addition, the cell perfusion with intracellular solution may affect KCC2 phosphorylation and, thus, modify its activity.
Fluorescent probes (DYES). The recent achievements in cell biology and microscopy have promoted the use of different Cl−-sensitive fluorescent probes for analysis of neuronal Cl− homeostasis. The first set of markers were Cl−-sensitive dyes (quinolinium Cl− indicators) exploiting the property of halides to quench the fluorescence of heterocyclic organic compounds with quaternary nitrogen (Chen et al., 1988; Verkman, 1990). These compounds have relatively good sensitivity and selectivity to Cl−, and have been used in a variety of preparations. A disadvantage is that a ratiometric recording cannot be performed and their use is strongly limited due to a strong bleaching and the presence of a significant leakage rate (see review Bregestovski et al., 2009). The use of two-photon microscopy has largely resolved bleaching-related problems, and has caused a rediscovery in the use of the quinolinium Cl− indicators (Kovalchuk and Garaschuk, 2012).
YFP-based markers. A more recent and promising method for non-invasive analysis of Cl− is based on the halide-binding properties of yellow fluorescent protein (YFP) and its derivatives (Wachter and Remington, 1999; Jayaraman et al., 2000). Based on Cl−-sensitive variants of YFP, ratiometric probes allowing estimation of the approximate level of [Cl−]i have been created. The first ratiometric Cl− indicator was designed by fusion of YFP with cyan fluorescent protein (CFP) through a polypeptide linker (Kuner and Augustine, 2000). In this construct, called Clomeleon, CFP served as a reference point for normalizing expression levels. The apparent EC50 was 167 mM. Later a more sensitive probe was developed, termed “Cl-Sensor,” composed of CFP and a mutated form of YFP with higher Cl− sensitivity (YFPCl, the apparent EC50~ 30–50 mM) (Markova et al., 2008; Waseem et al., 2010). Finally, recent work reported engineering of one more Cl− sensitive ratiometric construct called SuperClomeleon that apparently has five-fold higher Cl− sensitivity than Clomeleon (Grimley et al., 2013). Transgenic mice encoding Clomeleon (Berglund et al., 2006) and Cl-Sensor (Batti et al., 2013) have been successfully created.
Both Clomeleon and Cl-Sensor have been effectively used to characterize changes in [Cl−]i driven by KCC2 and/or NKCC1 in neuronal cells (Dzhala et al., 2005; Duebel et al., 2006; Pellegrino et al., 2011; Chamma et al., 2013). These YFP-based Cl−-sensitive biosensors have a number of advantages: (i) they show much more stable fluorescence at long-lasting monitoring as compared to fluorescent dyes; (ii) they allow excitation in the visible range of wavelength; (iii) they can be targeted to specific cell types either using plasmid transfection (Waseem et al., 2010; Pellegrino et al., 2011; Chamma et al., 2013) or by transgenic creation of animals with knocked-in genes (Berglund et al., 2006; Batti et al., 2013); (iv) they have a high molecular weight, which prevents the diffusion of the indicators from cells and (v) they are suitable for ratiometric measurements. The limiting points of both Clomeleon and Cl-Sensor are: (i) the relatively high rate of bleaching of the fluorescent signal (Kuner and Augustine, 2000; Berglund et al., 2011), (ii) the sensitivity of the probe to intracellular pH (pHi) and some organic anions (Jayaraman et al., 2000).
To avoid bleaching of Clomeleon, several groups have successfully used two-photon microscopy (reviewed by Berglund et al., 2011). We reported an alternative method that allows a stable ratiometric recording of Cl-Sensor fluorescence using a conventional epifluorescent microscope (Friedel et al., 2013). The method is based on the discovery of transient inactivation of Cl-Sensor by short wavelength (440 nm) blue light and the corresponding modification of excitation light pass and fluorescence recording procedure. The bypassing of the problem of pH sensitivity of YFP-derived Cl− markers is more problematic as many cellular processes, including activation of GABAA receptors, often involve concomitant changes in both Cl− and H+ ion concentration (Russell and Boron, 1976; Kaila et al., 1989). Therefore, a researcher using YFP-based sensors as Cl- indicators should be aware that, even after precise calibration of the set-up, the obtained values of [Cl−]i are only indicative.
E2GFP-based Cl− markers. To circumvent the problem of the pHi sensitivity, Arosio et al. (2010) developed a new ratiometric biosensor (ClopHensor) composed of the Cl− and pH sensitive GFP mutant, E2GFP, fused to monomeric DsRed fluorescent protein and allowing simultaneous measurement of Cl− and pH. Some new modifications of this sensor allowing its membrane targeting and improving pH sensitivity have also been described (Mukhtarov et al., 2013). Another recent work (Raimondo et al., 2013) developed one more ClopHensor derivative designated for expression into neuronal cells (ClopHensorN). Authors found that, when expressed in neurons, DsRed component of ClopHensor formed dense intracellular aggregations that compromised correct ratiometric measurement. The problem has been resolved by replacing DsRed with a tandem dimer tomato (TdTomato). Raimondo et al. (2013) performed also detailed analysis of the dynamic of neuronal [Cl−]i and pHi following activation of synaptic and extrasynaptic GABAA receptors that could serve as manual for future works.
In summary, there is a large number of methodological approaches successfully used to assay indirectly the KCC2 activity. So far, each approach has some advantages, but has also limitations that must be taken into account when interpreting results. This is why many groups use a combination of at least two different approaches to corroborate results (i.e., Williams and Payne, 2004; Li et al., 2007; Hershfinkel et al., 2009; Pellegrino et al., 2011; Chamma et al., 2013).
Diversity of Activity- and Pathology-Dependent Changes of KCC2 Function
KCC2 Changes Related to Physiological Patterns of Neuronal Activity
Whether physiological patterns of neuronal activity control developmental enhancement of KCC2 function remains controversial. At least three studies have provided data supporting the idea that chronic blockade of GABAergic (Ganguly et al., 2001; Leitch et al., 2005) or glycinergic (Shibata et al., 2004) inputs in developing neuronal networks inhibits the developmental increase in KCC2 functional expression. In contrast, other studies have not found significant changes in KCC2 functional expression after prolonged inhibition of GABAergic neurotransmission (Ludwig et al., 2003; Titz et al., 2003). The later conclusion does not exclude, however, that the activity might contribute to fine adjustments of the post-translational regulation (such as phosphorylation) and activation of plasmalemmal KCC2 function. Indeed, a critical role of the brain-derived neurotrophic factor (BDNF) and other trophic factors in the control of the developmental expression of KCC2 has been shown (Kelsch et al., 2001; Ludwig et al., 2011a,b). Given that neuronal activity regulates release of BDNF from both pre- and post-synaptic terminals (Kuczewski et al., 2008, 2010; Matsuda et al., 2009), it is plausible that BDNF and/or other trophic factors contribute to activity-dependent modulation of KCC2 in a context-dependent manner.
An important process in the maturation of glutamatergic networks is the specific form of plasticity resulting from co-incident pre- and post-synaptic spiking (Debanne and Poo, 2010). Similarly, spike-time-dependent plasticity has been shown to exist for GABAergic synapses (Woodin et al., 2003). Most intriguingly, within a sub-time domain, this new form of plasticity is dependent on changes of [Cl−]i. A suggested critical step in the mechanism is a rapid postsynaptic change in the efficiency of chloride extrusion by KCC2 (Fiumelli et al., 2005).
Pathology-Related Changes of the KCC2 Expression
Currently there is a large number of observations describing a strong decrease in KCC2 expression in different pathologies, including temporal lobe epilepsies (Palma et al., 2006; Huberfeld et al., 2007; Bragin et al., 2009; Barmashenko et al., 2011), focal cortical dysplasia (Munakata et al., 2007; Shimizu-Okabe et al., 2011; Talos et al., 2012), ischemia (Galeffi et al., 2004; Papp et al., 2008; Jaenisch et al., 2010), as well as pathologies associated with different types of axonal injury (Coull et al., 2003; Shulga et al., 2008; Boulenguez et al., 2010). Although the primary mechanisms initiating down-regulation of KCC2 during these pathologies are not known, the common point between them is a strong decrease of mRNA and protein expression levels. The exact mechanisms triggering inactivation of the KCC2 function are not yet well understood. Several groups have shown that epileptic-like neuronal activity itself could serve as triggering event. For instance, Nardou et al. (2009) showed suppression of the KCC2 function after induction of the epileptiform-like activity in the entire immature (P5-P7) rat hippocampal preparation. Rivera et al. (2004) found out that induction of epileptic-like activity to acute hippocampal slices rapidly (30–60 min) reduced expression of KCC2 in the CA1 region of the hippocampus and suggested a critical role of the glutamate and TrkB receptors in the processes. These results, however, were updated recently by Puskarjov et al. (2012) who showed that the half–life of plasma membrane KCC2 under control conditions might be significantly longer than previously reported. The difference between the two publications may be mainly attributed to differences in slice preparation. In the work by Puskarjov and collaborators, great care was taken to minimize damage of slices during preparation. In addition, they found that the mechanism for activity-dependent degradation of plasma membrane KCC2 requires Ca2+-dependent activation of the protease calpain. To achieve similar levels of KCC2 down-regulation as Rivera et al. (2004), the authors had to incubate slices with a higher concentration of NMDA (100 μM) and for a longer time (4 h). At these doses, NMDA triggers several neurotoxic signaling cascades (Hardingham et al., 2002; Ivanov et al., 2006), reviewed by Medina (2007) and Hardingham and Bading (2010). A rapid NMDA receptor-dependent KCC2 inactivation was also reported in two other studies performed on cultured hippocampal neurons (Lee et al., 2011; Pellegrino et al., 2011).
Many studies reporting a global pathology-dependent down-regulation of KCC2 expression mentioned also no-change or up-regulation of KCC2 in a sub-population of neurons surviving excitotoxic or ischemic insults. For example, Conti et al. (2011) demonstrated an up-regulation of KCC2 cotransporter in human peritumoral epileptic cortex (Conti et al., 2011). Huberfeld et al. (2007) have shown robust KCC2 staining in some subicular pyramidal cells in human temporal lobe epilepsy (TLE) brain sections. Jaenisch et al. (2010) have found a high level of KCC2 expression in infarct core neurons surviving a focal ischemic insult. Papp et al. (2008) found a delayed pattern of down-regulation in interneurons in the same model. Consistent with these data, Pellegrino et al. (2011) showed that all neurons surviving excitotoxic treatment with NMDA restored their normal Cl− extrusion ability and increased their expression of KCC2.
While robust pathology-like activation of a neuronal network may lead primarily to inactivation of KCC2, short activity episodes might induce functional up-regulation. For instance, Khirug et al. (2010) found that a single seizure episode in vivo, as well as brief seizure-like activity in vitro in neonates, induced a potent increase in KCC2 plasma membrane expression (with unchanged total KCC2 expression) that correlated with enhancement in the efficacy of GABAergic inhibition. The up-regulated KCC2 activity was reported also in several other studies (Khirug et al., 2005; Banke and Gegelashvili, 2008; Chorin et al., 2011; Bos et al., 2013); however, it remains unclear how the described modulations of KCC2 activity are related to physiological or pathology conditions.
Since many of the discussed above reports show that restoration of the physiological level of Cl− results in normal neuronal functioning, this suggests that targeting of the KCC2 and/or NKCC1 could be an efficient therapeutic strategy (reviewed by Kahle et al., 2008; Löscher et al., 2013). Regarding this, recent studies concentrated on high-throughput screening of the compounds capable to selectively inhibit the NKCC1 (Delpire et al., 2009, 2012) and activate KCC2 (Gagnon et al., 2013). Further optimization of the reported compounds as well as creation of new agents targeting specific signaling pathways upstream of KCC2 and/or NKCC1 will make in nearest future an important breakthrough for KCC2-related therapy.
Cell-Inherent Levels of KCC2 Control
Transcriptional Regulation of KCC2 Expression
Most mature CNS neurons exhibit high levels of KCC2 expression, while low expression is detected in immature neurons (reviewed in Blaesse et al., 2009; Chamma et al., 2012). KCC2 mRNA levels are increased with neuronal maturation, KCC2 protein expression shows concordance with the mRNA level (Lu et al., 1999; Li et al., 2002; Stein et al., 2004). RNA interference approaches, such as anti-sense oligonucleotides (Rivera et al., 1999) or specific siRNAs (Bortone and Polleux, 2009; Pellegrino et al., 2011; Gauvain et al., 2011; Succol et al., 2012), lead to a reduction of KCC2 protein and a resulting loss of KCC2 activity. Thus, the control at the mRNA level is an efficient way to regulate the long-term activity of KCC2.
Selective decrease in KCC2 mRNA has been observed during neurological disorders (Palma et al., 2006; Huberfeld et al., 2007), after sustained interictal-like activity (Rivera et al., 2004), and after axonal injury (Nabekura et al., 2002; Toyoda et al., 2003; Shulga et al., 2008, 2009). Activity-dependent decreases in KCC2 mRNA might therefore be an important factor contributing to the down-regulation of KCC2 activity in certain pathological conditions. At least 10 putative transcription factor binding sites have been identified in the SLC12A5 gene encoding KCC2 promoter and proximal intron-1 regions (Uvarov et al., 2006; Uvarov, 2010). These sites are highly conserved elements among promoter regions of other mammalian KCC2 genes. Transcription factor early growth response 4 (Egr4) has been identified as a potent regulator of KCC2 expression (Uvarov et al., 2006), which mediates BDNF-dependent KCC2 transcription in immature neurons (Ludwig et al., 2011b). Another regulatory element in the KCC2 promoter is the E-box, that binds the ubiquitously expressed upstream stimulating factors USF1 and 2, thus contributing KCC2 up-regulation in the developing cortex (Markkanen et al., 2008). In addition, two neuron-restrictive silencing elements (NRSE) were found in SLC12A5; however, the data on whether they are involved in the developmental up-regulation of KCC2 are controversial (Karadsheh and Delpire, 2001; Uvarov et al., 2005; Yeo et al., 2009). The role of other putative transcription factor binding elements in the KCC2 promoter remains obscure. One important task will be to characterize in more detail the regulatory elements controlling SLC12A5 transcription.
KCC2 Assembly
Numerous studies suggest that, by analogy with other types of transporters and Cl− channels, the CCC family members function as oligomers (McKee et al., 2000; Moore-Hoon and Turner, 2000; Casula et al., 2001; De Jong et al., 2003; Starremans et al., 2003; Simard et al., 2004; Blaesse et al., 2006; Parvin et al., 2007; Simard et al., 2007; Pedersen et al., 2008; Casula et al., 2009; Warmuth et al., 2009). With regard to KCC2, the ability of this molecule to form molecular complexes corresponding to mono-, di-, tri-, and tetrameric complexes was first demonstrated by Blaesse et al. (2006). The authors showed also a clear age-dependence of the oligomerization: the immature brains were characterized by higher content of the monomers, whereas adult tissue produced mostly oligomeric forms of the KCC2. An additional set of data demonstrating the ability of KCC2 to form dimers in vivo comes from studies of the distribution (Markkanen et al., 2013) and interaction (Uvarov et al., 2009) between KCC2a and KCC2b isoforms (Uvarov et al., 2007). Using the native perfluorooctanoate (PFO)-PAGE system, as well as a co-immunoprecipitation assay, Uvarov et al. (2009) have shown that KCC2a and KCC2b isoforms form homo- and heterodimers in vivo and in heterologous HEK-293 expression system. Despite multiple evidence describing oligomerization of KCC2, the functional context of this protein complex remains unclear. Is it critical for translocation to plasma membrane? What is the number of subunits that form the ion-transport unit? Is oligomerization involved in KCC2 surface expression and/or internalization? These and other questions remain to be answered and should be a subject for future studies.
By reviewing results of the KCC2 oligomerization studies, we have to be aware of some technical problems that one may encounter during analysis of the KCC2 using Western blot. Due to yet unidentified reasons, KCC2 is prone to forming SDS-resistant high molecular aggregates during standard protein extraction and solubilization procedures that are normally used for denaturation and dissociation of protein complexes (Gallagher, 2012). By performing Western blot analysis of the KCC2, we have found that in addition to the age-dependent changes in KCC2 oligomeric/monomeric ratio described in the above study (Blaesse et al., 2006), the ability to form oligomers strongly depends on the experimental procedure and is different for endogenously expressed KCC2 and for KCC2 overexpressed in heterologous systems (Figure 2). A very rapid protein extraction reduces the formation of the KCC2 oligomeric complexes under SDS-denaturating conditions, whereas long extraction (more than 4 min) increases the probability to get dimers. Interestingly, once the KCC2 dimer complexes are formed, we could not dissociate them to monomeric structure using boiling with SDS or in combination with high concentration (8 M) of urea at high temperature, a treatment that works efficiently to disrupt such strong molecular complexes as NMDA receptors-associated postsynaptic densities (Krapivinsky et al., 2003, 2004).
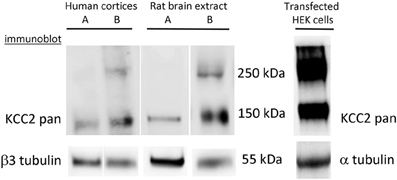
Figure 2. Complexity of interpretation of KCC2 oligomerization. Formation of KCC2 dimer-like complexes depends on the protein extraction procedure and is different for endogenous and over-expressed KCC2. KCC2 extraction using a rapid procedure (<1 min) either from human cortices or rat brains generates a single band of ~140 kDa. The extracts prepared from similar brain samples using the same solutions but with a longer overall time of extraction (> 4 min) always give at least two bands corresponding to monomer (140 kDa) and dimer (280–300 kDa) of KCC2. The extraction of eGFP-KCC2 over-expressed in HEK-293 cells resulted in the appearance of two bands independently on the procedure of extraction. Similar two-band expression profile of KCC2 was obtained after over-expression of different KCC2 forms (with or without additional tags) into different cell lines (PC-12 and Neuro2a).
Surface Expression, Turnover
Plasma membrane insertion, surface stabilization, and internalization of the molecule constitute one more important mechanism of KCC2 regulation. Numerous studies have found that pathological-like patterns of neuronal activity initiate rapid withdrawal of the KCC2 from plasma membrane that leads to a reduction of KCC2-mediated ion-transport (Rivera et al., 2004; Lee et al., 2007, 2010, 2011; Wake et al., 2007; Puskarjov et al., 2012; Chamma et al., 2013). Using an elegant approach of visualizing KCC2 internalization harboring an extracellular tag, Zhao and colleagues have shown a critical role of the cytoplasmic C- and not N-termini for KCC2 trafficking (Zhao et al., 2008). The authors also localized in the KCC2 C-terminus a novel non-canonical di-leucine motif (657LLXXEE662) that seems to be essential for the constitutive internalization of the transporter and for binding to the clathrin-binding adaptor protein-2 complex. Several phosphorylation sites on the C-terminus have been implicated in mechanisms controlling KCC2 turnover (see below “phosphorylation” chapter for details and Figure 1).
The internalization of overexpressed KCC2 is controlled by the clathrin-mediated endocytosis pathway in HEK-cells (Zhao et al., 2008) and can be prevented by an inhibitor of the clathrin pathway in cultured neuronal cells (Lee et al., 2011). However, no systematic work was performed to characterize the recycling mechanism involved or the signaling pathways controlling the KCC2 membrane insertion. A recent study (Puskarjov et al., 2012) launched the discussion on the timing of the KCC2 turnover in neuronal membrane. Previous work (Rivera et al., 2004), which was performed using a biotinylation approach and hippocampal slices prepared using a slice chopper (i.e., resulting in a significant number of damaged cells), suggested that half of the KCC2 molecules are renewed in neuronal plasma membrane within 30–40 min. Using a different experimental approach and slices prepared using a vibratome (i.e., with a lower rate of neuron damage), Puskarjov et al. (2012) estimated the half time of KCC2 turnover to be much longer (several hours or even days). On the other hand, a study performed using cultured hippocampal neurons showed that 45-min treatment of neurons with dynasore, an inhibitor of the endocytosis, produced twofold increase of the surface expression of the KCC2 (Lee et al., 2010), indicating the existence of a relatively rapid constitutive KCC2 endocytosis in this model. A further evidence supporting the high rate of the KCC2 turnover was obtained in a heterologous expression system (HEK-293 cells) where over-expressed KCC2 showed a half-time turnover rate close to 10 min (Lee et al., 2007; Zhao et al., 2008). Thus, the timing and mechanisms controlling KCC2 surface expression remain to be fully elucidated.
Regulation of the Intrinsic KCC2 Activity
The structural elements of KCC2's ion-transport unit are not known. Unlike other members of the KCC family (KCC1, KCC3, and KCC4), which become active only after osmotic stress (reviewed by Flatman, 2008; Blaesse et al., 2009), KCC2 is able to extrude Cl− in isotonic conditions. The over-expression of this transporter in heterologous expression systems (Payne, 1997; Williams and Payne, 2004) or neuronal cells (Chudotvorova et al., 2005; Lee et al., 2005; Pellegrino et al., 2011; Acton et al., 2012) produces a clearly detectable ion flux that could be rapidly (within few seconds) and reversibly inhibited using high concentrations of furosemide, a sulfamoylbenzoic acid “loop” diuretics (Payne, 1997; Williams and Payne, 2004; Friedel et al., 2013). These data suggest that, when expressed in the plasma membrane, KCC2 operates as constitutively active K+-Cl− transporter.
Several groups have identified in the KCC2 molecule different regions and amino acid residues that regulate the ion-transport activity. Hartmann et al. (2010) found that mutation of four evolutionarily conserved cysteine residues in the large extracellular loop of KCC2 (C287, C302, C322, C331, Figure 1) inhibited 86Rb+ flux mediated by the transporter without affecting its surface expression. Interestingly, mutation of the corresponding cysteines in KCC4, which is the closest to KCC2 member of the SLC12 family, did not affect the ion transport. These data suggest a distinct organization of ion-transport mechanism in KCC2 and KCC4, thus further enhancing exclusiveness of the KCC2. An important role in KCC2-mediated ion transport plays also C568, located inside the 10th putative transmembrane domain (Reynolds et al., 2008). Mutation of this residue (C568A) makes KCC2 inactive, but it remains unclear whether C568A mutation inhibits intrinsic activity or affects membrane targeting of the molecule. A study performed by Horn et al. (2010), described a decreased interaction of the KCC2C568A mutant with cytoskeleton-associated 4.1N protein, and suggested that the C568A mutation causes changes in the tertiary structure of the molecule. Additional analysis is necessary to clarify the mechanism of how the C568A mutation silences KCC2 activity.
As illustrated in Figure 1, the N- and C- terminal domains of KCC2 and other members of KCC subfamily, are both located intracellularly. Several sets of data suggest that both domains can regulate intrinsic activity of the transporter. Thus, by expression in Xenopus oocytes of different KCC1 constructs with truncated intracellular regions, Casula et al. (2001) found that deletion of either N- or C- termini abolished ion transport activity of KCC1 without affecting transporter expression at the oocyte surface. Consistent with this finding, Li et al. (2007) showed that truncation of the N-terminus of KCC2 inhibited its ion-transport activity and did not affect the surface expression of the protein (Figure 1). A further evidence highlighting the importance of the KCC C-terminus in controlling the intrinsic property of ion transport was found in a study by Rinehart et al. (2009). Authors showed that phosphorylation of KCC3 at two newly identified sites (T991 and T1048) strongly inhibited the ion-transport activity of the transporter without modifying its surface expression. Age-dependent changes of the phosphorylation status of equivalent threonine sites in the KCC2 molecule (T906 and T1007) were reported in the same study; however it remains unclear whether phosphorylation of the KCC2 at these residues modifies its intrinsic activity or surface expression.
The leucine 675 (L675) identified by Döding et al. (2012) is one more residue critically involved in KCC2 activity: its mutation to alanine (L675A) strongly reduced 86Rb+ flux mediated by the KCC2 without affecting the surface expression of the transporter in mammalian cell lines.
It had been known for a long time that KCC2 could be constitutively active in isotonic conditions in HEK293 cells (Payne, 1997), CHO cells (Strange et al., 2000) and in Xenopus oocytes (Strange et al., 2000; Song et al., 2002; Gagnon et al., 2006a). In contrast to KCC2, several studies revealed no significant cotransporter activity of KCC4 under isotonic conditions, but found that KCC4 could be strongly (about 20-fold) activated by hypotonic swelling (Mercado et al., 2000; Song et al., 2002). To identify molecular mechanisms underlining this difference, Mercado and coauthors used Xenopus oocytes and multiple KCC2-KCC4 chimeric constructs, and found a C-terminal region unique to KCC2 (ISO-domain, amino acids 1021–1035) that is both necessary and sufficient for the isotonic KCC2 activity (Mercado et al., 2006). A recent study confirmed important role of ISO domain by illustrating in transfected cultured hippocampal neurons that KCC2 mutant with deleted ISO domain showed reduced ion transport activity, whereas insertion of the ISO domain into KCC4 transporter, which is inactive under isotonic conditions, rendered it fully operational (Acton et al., 2012). Of notice, by using different KCC2-KCC4 chimera constructs overexpressed in Xenopus oocytes, Bergeron et al. (2006) obtained almost identical to Mercado et al. (2006) results showing that the deletion of distal KCC2 region strongly reduced KCC2 activity under isotonic conditions, whereas insertion of this domain to KCC4 potently activated transporter. However, after analysis of many other KCC2 and KCC4 mutants authors were not convinced that the ISO segment is “a prerequisite for K+-Cl− cotransport to occur under isotonic conditions.” Clearly, a characterization of the mechanism of ISO domain action is one more challenge in the KCC2 field.
Taken together, the above data suggest that both N- and C-terminal domains of KCC might be directly involved in the control of the ion-transport unit; however, the mechanisms of their regulatory actions are not known. An important task for future studies will be to answer these questions by analysis of the three-dimensional structure of KCC proteins.
Signaling Pathways That Control KCC2 Activity
Many signaling molecules have been implicated in the control of KCC2 function (Figure 3). Some of these molecules and pathways have been explored in detail, whereas many others remain to be characterized. Below, we overview these pathways based on their major regulatory functions.
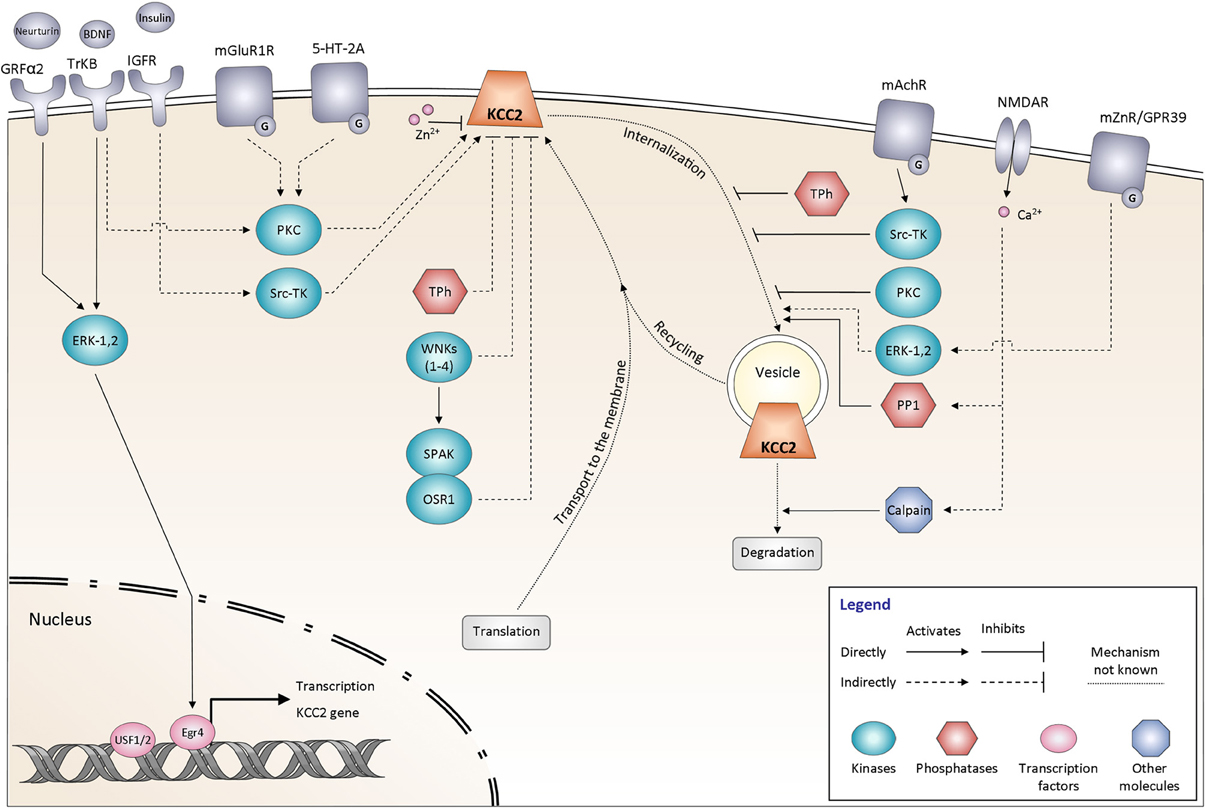
Figure 3. Signaling pathways controlling KCC2 function. The regulation of KCC2 activity is mediated by many proteins including kinases and phosphatases. It affects either the steady state protein expression at the plasma membrane or the KCC2 protein recycling. All the different pathways are explained and discussed in the main text. The schematic drawings of KCC2 as well as other membrane molecules do not reflect their oligomeric structure. GRFα2, GDNF family receptor α2; BDNF, Brain-derived neurotrophic factor; TrKB, Tropomyosin receptor kinase B; Insulin, Insulin-like growth factor 1 (IGF-1); IGFR, Insulin-like growth factor 1 receptor; mGluR1, Group I metabotropic glutamate receptor; 5-HT-2A, 5-hydroxytryptamine (5-HT) type 2A receptor; mAChR, Muscarinic acetylcholine receptor; NMDAR, N-methyl-D-aspartate receptor; mZnR, Metabotropic zinc-sensing receptor (mZnR); GPR39, G-protein-coupled receptor (GPR39); ERK-1,2, Extracellular signal-regulated kinases 1, 2; PKC, Protein kinase C; Src-TK, cytosolic Scr tyrosine kinase; WNKs1–4, with-no-lysine [K] kinase 1–4; SPAK, Ste20p-related proline/alanine-rich kinase; OSR1, oxidative stress-responsive kinase -1; Tph, Tyrosine phosphatase; PP1, protein phosphatase 1; Egr4, Early growth response transcription factor 4; USF 1/2, Upstream stimulating factor 1, 2.
BDNF and Other Trophic Factors
At least three trophic (growth) factors have been identified as efficient modulators of KCC2 functional expression: BDNF, Insulin-Like Growth Factor 1 (IGF-1), and Neurturin (Kelsch et al., 2001; Ludwig et al., 2011a; Shulga et al., 2012). Among them, the regulatory effects of BDNF on KCC2 are the most thoroughly characterized.
First evidence for the regulatory action of BDNF in developing neurons was presented by Aguado et al. (2003), who showed that in vivo transgenic over-expression of BDNF strongly increases KCC2 mRNA. Later the deletion of the receptor TrkB was shown to strongly decrease KCC2 mRNA (Carmona et al., 2006). Whether these effects are observed at the protein expression levels have so far not been reported in the literature. Consistent with this study, the application of BDNF to immature hippocampal cultures significantly increases the expression of KCC2 protein through Egr4-dependent KCC2 transcription (Ludwig et al., 2011b). Interestingly, in more mature cultures BDNF produced a decrease in KCC2 expression, thus revealing that the action of BDNF depends on the maturational state of neurons (see for review (Ferrini and De Koninck, 2013). So far, there is no evidence describing the regulatory action of the BDNF on the post-translational level at early stages of neuronal development.
In contrast to immature neurons, the exposure of adult neurons to BDNF in vivo and in vitro decreases KCC2 expression and function. BDNF decreases KCC2 mRNA levels (Rivera et al., 2002; Shulga et al., 2008), total KCC2 protein expression (Rivera et al., 2004), as well as tyrosine-phosphorylated (Wake et al., 2007) and membrane-inserted (Boulenguez et al., 2010) fractions of KCC2. Consistent with these findings, over-expression of BDNF in vivo produced a depolarizing shift of the GABA reversal potential in dorsal spinal cord lamina I (L1) neurons, presumably reflecting a decrease in KCC2 function (Coull et al., 2003, 2005).
Therefore, in mature neurons, application of exogenous BDNF induces a down-regulation of the KCC2 function. Endogenously released BDNF may therefore be a good candidate to mediate the activity-dependent down-regulation of KCC2. In support of this hypothesis, Rivera et al. (2004) showed that scavenging of endogenous BDNF, or inhibition of signaling downstream from BDNF TrkB receptors, effectively prevented activity-dependent reduction of KCC2 mRNA and protein. Several other studies have also reported a critical role of BDNF in the change of the inhibitory strength of GABA following axonal injury (Coull et al., 2005; Shulga et al., 2008; Boulenguez et al., 2010; Zhang et al., 2013). Intriguingly, BDNF application on neurons after injury, which are characterized by a decreased level of KCC2 expression, produced effects opposite to those observed in naive tissue, namely up-regulation of KCC2 surface expression (Shulga et al., 2008; Boulenguez et al., 2010). One plausible explanation is that injured neurons acquire properties of immature ones in order to launch a repair program (Kahle et al., 2008; Shulga et al., 2008, 2012).
Both during development (Westerholz et al., 2013) and in axotomized tissue (Shulga et al., 2009; Shulga and Rivera, 2013), the regulatory action of BDNF is tightly related to the thyroid hormone metabolism. The fact that thyroxine can bi-directionally regulate the expression of KCC2 in the same manner as BDNF would indicate that this effect could be mediated through BDNF signaling (Friauf et al., 2008; Shulga et al., 2009). Interestingly, the effect of thyroxin on KCC2 appears to be insensitive to BDNF signaling blockage indicating yet another pathway for regulation of KCC2 expression.
Phosphorylation
It has been long time appreciated that K+-Cl− cotransport in non-excitable cells can be potently regulated by phosphorylation (Russell, 2000; Blaesse et al., 2009). Extrapolating these data to neuronal K+-Cl− transport, several studies have made an attempt to characterize the role of protein phosphorylation in the functioning of the KCC2 (Kahle et al., 2013). Large number of putative KCC2 phosphorylation sites were shown to contribute into the control of transporter activity and/or its surface expression (Figure 1). Numerous studies have highlighted also different phosphorylation-related signaling pathways contributing to the control of KCC2 activity (Figure 3). Below we review the recent evidence and make an attempt to classify them with regard to the phosphorylation type (serine/threonine and tyrosine) and kinases implicated into the post-translational regulation of the KCC2. We emphasize, in particular, the discrepancies between different works and highlight unsolved questions.
Serine-threonine phosphorylation
To date, serine 940 (S940), localized in the cytoplasmic C-terminal domain of KCC2, is the only known residue whose phosphorylation enhances KCC2 activity. This residue has been identified using in vitro screening of the KCC2-derived peptides directly phosphorylated by protein kinase C (PKC) (Lee et al., 2007). Dephosphorylation of S940 leads to inactivation of KCC2-mediated ion transport and increases endocytosis of the transporter. A high level of S940 phosphorylation is seen under resting conditions in heterologous expression systems and cultured hippocampal neurons, as revealed using an antibody raised against the phosphorylated form of KCC2 S940 (Lee et al., 2011). Thus, the PKC-dependent phosphorylation of S940 in the C-terminus of KCC2 is critical for maintenance of the KCC2 activity under physiological conditions. The mechanisms controlling S940 are not identified yet. At least three studies have described the PKC dependent regulation of KCC2 during physiologically-relevant processes including: (i) activity-dependent attenuation of the KCC2 function in immature cultured neuronal cells (Fiumelli et al., 2005), (ii) tonic activation of the KCC2 by group I metabotropic glutamate receptors (mGluR1s) (Banke and Gegelashvili, 2008), and (iii) activation of the KCC2 via serotonin type 2A receptors (Bos et al., 2013). However, it remains unclear whether PKC-dependent regulation of KCC2 activity in the above-mentioned studies, was direct or via some intermediate signaling molecules. Treatment of cultured hippocampal neurons with glutamate provoked a rapid dephosphorylation of S940 residue that was NMDA receptor and protein phosphatase-1 dependent (Lee et al., 2011). An important task would be to determine whether the described signaling pathway contributes to decrease of the KCC2 activity following neurological pathologies. Lee et al. (2007) also identified another serine residue (S728) whose mutation to alanine (S728A, dephosphorylated-like form) produced a constitutive increase of the KCC2 activity. Unfortunately, the authors did not study how S728 phosphorylation modifies the surface expression of the transporter and did not identify the pathway controlling this phosphorylation site.
Rinehart et al. (2009) identified two threonine residues conserved in other KCC members (T906 and T1007 on the isoform b of rat KCC2 (accession number NP_599190)) whose phosphorylation strongly inhibited the transporter activity, whereas dephosphorylation rendered transporters constitutively active. T906 is partially phosphorylated in neonatal mouse brain and dephosphorylated in adult brain, suggesting a potential contribution of T906/T1007 dephosphorylation to functional KCC2 up-regulation during development. However, a systematic analysis of KCC2 phosphorylation at these sites in discrete anatomic regions during brain development is required. Engineering KCC2 T906A (or E)/T1007A (or E) double knock-in mice would be valuable in this regard.
The WNK (with-no-lysine [K]) family of serine-threonine kinases comprises four members (WNK1, WNK2, WNK3, and WNK4) that together with SPAK, a Ste20p-related proline/alanine-rich kinase (Johnston et al., 2000) and OSR1, an oxidative stress-responsive kinase -1 (Tamari et al., 1999) form a powerful signaling cascade involved in control of swelling-induced regulation of CCC family protein members (see for review Delpire and Gagnon, 2008; Kahle et al., 2010). All these kinases are expressed also in the CNS, but their functional role in neurons is not known. Importantly, the activity of the WNK-SPAK pathway is regulated by extracellular osmolality and intracellular Cl− concentration (Zagórska et al., 2007; Thastrup et al., 2012). The 4 members of WNK family have been shown to effectively inhibit the activity of the KCC2 cotransporter exogenously overexpressed in neurons (Inoue et al., 2012) or in Xenopus oocytes (Kahle et al., 2005; De Los Heros et al., 2006; Gagnon et al., 2006b; Rinehart et al., 2011). Taken together, the above evidence indicates the importance of detailed evaluation of the WNK-SPAK/OSR1 phosphorylation cascade in control of the KCC2 function.
Toward this goal, the role of KCC2 T906 and T1007 phosphorylation during brain development was recently studied by Inoue et al. (2012) using an elegant in utero electroporation approach and over-expression of KCC2 mutants mimicking hyper- and de-phosphorylated forms of transporter (T906E + T1007E and T906A + T1007A, respectively) in rat cortex. Inoue et al. (2012) suggested that the phosphorylation of these residues in immature brains is maintained by taurine acting through the WNK-SPAK/OSR1 phosphorylation cascade. The proposed scheme was based, however, exclusively on evidence obtained using application of exogenous taurine and exogenous over-expression of wild type and mutant KCC2 and WNK1 kinase constructs.
An important future task would be to verify the proposed hypothesis using models involving silencing of the endogenous molecules (WNK and SPAK), and to biochemically demonstrate how inhibition of WNK-SPAK alters KCC2 phosphorylation. Very recently de Los Heros et al. revealed that the WNK-regulated SPAK/OSR1 kinases directly phosphorylate KCC2 at T1007 (site 2), but not at T906 (site 1), and inhibition of WNK-SPAK signaling with a chemical inhibitor activates K-Cl cotransporter activity in cells by decreasing T1007 > T906 inhibitory phosphorylation in ES cells (De Los Heros et al., 2014). SPAK/OSR1 is also necessary for KCC2 T1007 but not T906 phosphorylation in ES cells. This is of high interest, given efforts aimed at finding chemical activators of KCC2 to restore ionotropic inhibition. Perhaps a novel way of activating KCC2 in certain cellular contexts would be to oppose the action of inhibitory kinases, such as WNK/SPAK (Kahle et al., 2013). While there are no classic knockout mouse models available for WNK1 or OSR1 due to their requirement for embryogenesis (Xie et al., 2013), exploring the roles of SPAK (and other WNKs, like WNK3) in KCC2 regulation in vivo should be possible using knockout mice (Yang et al., 2010; Mederle et al., 2013) along with other transgenic mice which express SPAK harboring a point mutation that renders them unable to be activated by WNK kinases (Rafiqi et al., 2010).
Tyrosine phosphorylation
KCC2 phosphorylation at tyrosine residues (TP, tyrosine phosphorylation) was shown in several studies (Stein et al., 2004; Wake et al., 2007; Watanabe et al., 2009; Lee et al., 2010), however its importance for KCC2 functioning remains obscure. While some studies showed the existence of the KCC2 TP under physiological conditions in rodent's cortex (Stein et al., 2004) and primary hippocampal cultures (Wake et al., 2007; Watanabe et al., 2009), others did not observe the basal TP under control conditions in cultured hippocampal neurons, although strong TP was observed after cultures treated with phosphatase inhibitors (Lee et al., 2010). Wake et al. (2007) clearly showed that neuronal network activity, oxidative stress, and treatment with BDNF all lead to a strong decrease of KCC2 TP. In contrast, Lee et al. (2010) observed a robust increase of KCC2 TP in brain slices obtained from rats after pilocarpine-induced status epilepticus, as well as in primary neuronal cultures treated with carbachol - an agonist of acetylcholine receptors. Finally, by comparing the influence of different experimental conditions on the KCC2 TP level, EGABA, and distribution of the KCC2 protein levels, Wake et al. (2007) and Watanabe et al. (2009) concluded that “direct tyrosine phosphorylation of KCC2 results in membrane clusters and functional transport activity” (Watanabe et al., 2009). However, based on the comparison of KCC2 TP levels, surface expression of the biotinylated KCC2 fraction, and total KCC2 expression, Lee et al. (2010) provided an opposite conclusion stating that KCC2 tyrosine phosphorylation promotes KCC2 internalization and lysosomal degradation.
Many explanations for these discrepancies are possible; however the most reasonable one seems to be that the treatments utilized to activate/inhibit TP in the mentioned studies were different. All employed treatments (H2O2, BDNF, 0 Mg2+, carbachol, pilocarpine, blockers of kinases and phosphatases) are able to modify the activity of different signaling pathways, including differential modulation of tyrosine kinases and phosphatases, as well as serine/threonine kinases, which itself can produce dual modulation of the KCC2 when acting through S940 or T906/T1007 (see above). Clearly, additional studies are required to clarify the question.
So far, two amino acid residues (Y903 and Y1087) were identified on KCC2 as potential carriers of tyrosine phosphorylation signal (Lee et al., 2010). Authors found that mutations of these residues to phenylalanine (Y903/1087F), which mimic dephosphorylated state, almost fully abolished the tyrosine phosphorylation state of the KCC2 protein exogenously overexpressed in HEK-293 cells. Using biotinylation assays, the authors showed that the Y903/1087F KCC2 mutant strongly increases the surface expression of KCC2, providing an additional argument for the TP-dependent internalization and degradation of KCC2. While the Y903 residue was characterized only recently, the Y1087 amino acid has been a subject of several studies that have resulted in contradictory conclusions. The Y1087 residue was first highlighted on a widely used KCC2 scheme presented by Payne et al. (1996) as the tyrosine residue conserved through evolution in all members of KCC and NKCC family transporters. Strange et al. (2000) showed that mutation of this residue to aspartate (Y1087D, mimicking a hyperphosphorylated state) fully abolished activity of the KCC2 transporter, whereas Y1087F mutation did not modify the KCC2-mediated ion transport. The authors, however, did not observe the change in the surface expression of the mutants in the Xenopus oocyte expression system, and they also could not find any experimental evidence to support the importance of tyrosine phosphorylation in the control of KCC2 activity. A full inactivity of KCC2 construct carrying Y1087D mutation and overexpressed in brain-derived GT1-7 cell line was also reported by Watanabe et al. (2009). Consistent with observation by Strange et al. (2000), authors did not find any changes in the surface expression of the Y1087D mutant compared to the wild type KCC2 (biotinylation assay). Interestingly, in their study, Watanabe et al. (2009) considered the Y1087D mutant of KCC2 as non-phosphorylated mutant. This was surprising as aspartate has totally different structure as compared to the tyrosine. Usually, to mimic the non-phosphorylated state of tyrosine researches use phenylalanine, an amino acid identical to tyrosine, with the exception of the absence of the phosphorylatable hydroxyl group (see Anthis et al., 2009 for detailed discussion on the strategy of tyrosine residues replacement). Given that Strange et al. (2000) found a strong difference in the activity of Y1087F and Y1087D mutants, the Y1087D mutation should be rather considered as phosphorylation-like mutation. Unfortunately, in their work Watanabe et al. (2009) did not compare the action of Y1087D with alternative Y1087F mutant. The inactivity of Y1087D mutant was confirmed several other times in other heterologous systems and cultured neurons (Chudotvorova et al., 2005; Akerman and Cline, 2006; Pellegrino et al., 2011). The above mentioned studies, however, used the Y1087D mutation as an experimental tool and did not analyse the mechanisms leading to the inactivation of KCC2 activity.
Thus, there is a good agreement between all existing studies that Y1087D mutation, that potentially mimics the phosphorylated-like state of the residue, inactivates KCC2 overexpressed in oocytes (Strange et al., 2000), mammalian cell lines (Watanabe et al., 2009) and neurons (Chudotvorova et al., 2005; Akerman and Cline, 2006; Pellegrino et al., 2011). In opposite, the mutation Y1087F, mimicking de-phosphorylated-like state of KCC2 residue, does not affect KCC2 transport in oocytes (Strange et al., 2000) and potentiates it in HEK-293 cells (Lee et al., 2010). Taken together, these data strongly suggest that Y1087 phosphorylation reduces KCC2 activity, whereas its de-phosphorylation potentiates the transporter. Due to discrepancies in the results characterizing the surface expression of Y1087D mutants, the mechanism controlling KCC2 activity (change in the surface expression vs. change of the intrinsic activity) remains unclear.
What is the relationship between Y903/Y1087 phosphorylation and endogenous KCC2 tyrosine phosphorylation? The Y903/Y1087 residues were detected in HEK-293 expression system as unique phosphorylation residues whose mutation fully abolished exogenous KCC2 phosphorylation. These data do not exclude an existence of additional residues accessible for phosphorylation by tyrosine kinases in native neuronal environment. The phosphorylation of these other residues might produce opposite effects than mutations in Y903/1087. Therefore, one cannot exclude differential effects of TP on endogenous KCC2 and on KCC2 mutated at one or two tyrosine amino acids.
Clearly, more scrupulous quantitative analysis is required to answer the following questions: (i) what is the basal level of KCC2 tyrosine phosphorylation in different in vitro preparations (acute slices, organotypic slices, primary neuronal cultures) compared to maximal phosphorylation level achieved in presence of the activators of the TP and inhibitors of the phosphatases, (ii) what is the basal level of the KCC2 TP in vivo (compared to in vitro models), (iii) how many tyrosine residues could be phosphorylated in native neuronal environment, (iv) what are the functional consequences (ion-transport activity, surface expression, protein degradation) of the phosphorylation/dephosphorylation of the KCC2 at distinct tyrosine residues?
Finally, numerous studies provided evidence of the KCC2 control via at least two distinct TP-dependent pathways involving cytosolic c-Scr kinase (Kelsch et al., 2001) and BDNF-dependent TrkB receptor tyrosine kinase (Rivera et al., 2002, 2004; Coull et al., 2005; Boulenguez et al., 2010). Each of these kinases might regulate large number of signaling cascades. An important task would be to uncover the exact regulatory pathways controlling KCC2 activity.
ZN2+-Dependent Control
Interesting data from groups of Hershfinkel and Aizenman (Hershfinkel et al., 2009; Chorin et al., 2011; Saadi et al., 2012) describe bi-directional Zn2+-dependent control of KCC2 (Figure 3). On one hand, intracellular Zn2+ in micromolar concentrations rapidly inhibits KCC2 activity (Hershfinkel et al., 2009). Although the mechanism of Zn2+ action is not clear, the finding indicates existence of a mechanism allowing inhibition of the intrinsic activity of KCC2. On the other hand, extracellular Zn2+ released from mossy fiber terminals, strongly activates KCC2 by increasing its surface expression (Chorin et al., 2011). Putatively, metabotropic zinc-sensing receptor (mZnR) is encoded by orphan Gq-coupled receptor (GPR39) and is coupled to phospholipase C (PLC)/extracellular-signal-regulated kinases (ERKs) pathway; the silencing of GPR39 or inhibition of PLC or ERKs abolished the Zn2+-dependent activation of KCC2 (Chorin et al., 2011). Consistent with a high rate of KCC2 turnover (Rivera et al., 2004), the authors have suggested that the increased surface expression of KCC2 is due to the inhibition of transporter internalization and/or degradation (Chorin et al., 2011). However, given recent evidence (see below chapter “Surface expression, turnover” and Puskarjov et al., 2012), one cannot exclude that Zn2+ potentiates the plasmalemmal insertion of the KCC2. The Zn2+-dependent control of KCC2 activity is one more subject in the KCC2 field requiring detailed analysis.
Protein-Protein Interaction
One more regulatory mechanism of KCC2 activity is through its direct interaction with other proteins (Figure 4). The first such interaction was described by Inoue et al. (2004) showing that brain-type creatine kinase (CKB) interacts with KCC2 and activates the transporter in HEK-293 cells (Inoue et al., 2004, 2006). Ikeda et al. (2004) found an intriguing interaction between the α2 subunit of the Na-K-ATPase pump and KCC2 that may stand for the breathing malfunction found in mice lacking the Na-K-ATPaseα2 subunit (Atp1a2−/−). These mice display decreased functional activity of KCC2 and have abnormal spontaneous respiratory rhythm activity, similarly to the KCC2−/− mice. KCC2 interacts also with other unrelated proteins such as the protein associated with Myc (PAM) via its RCC1 (Regulator of Chromatin Condensation) domain, resulting in the functional regulation of KCC2 activity in HEK-293 cells (Garbarini and Delpire, 2008), or with the cation-chloride cotransporter interacting protein 1 (CIP1) (Wenz et al., 2009), but the functional significance of this latter link in neurons remains unknown.
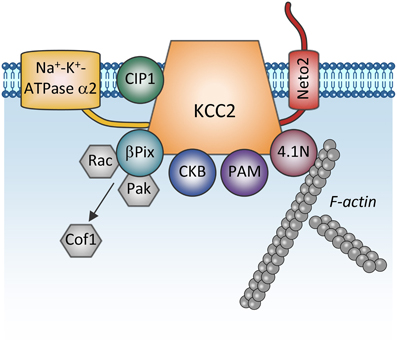
Figure 4. Proteins directly interacting with KCC2. The scheme shows membrane and cytoplasmic proteins that have been found to directly interact with KCC2. It is currently unclear whether these proteins form one big KCC2 containing complex or whether they all are combined in multiple distinct KCC2 containing pools (e.g., plasmalemmal vs. sub-membrane vesicular pools). Na+-K+-ATPase α2, subunit α2 of the Na+-K+-ATPase pump; CIP1, the cation-chloride cotransporter interacting protein 1; Neto2, neuropilin and tolloid like-2; β Pix, beta isoform of the Rac/Cdc42 guanine nucleotide exchange factor; Rac, small GTPase of the Rho family; Pak, p21-activated serine/threonine-protein kinase kinase; Cof1, cofilin-1; CKB: brain-type creatine kinase; PAM, protein associated with Myc; 4.1N, cytoskeleton-associated protein.
Furthermore, the cytoskeleton-associated protein 4.1N interacts with the cytoplasmic carboxy-terminus of KCC2 and is involved in dendritic spine formation (Li et al., 2007) and the plasticity of AMPA receptors (Gauvain et al., 2011). Interrupting the KCC2:4.1N interactions enhanced lateral diffusion of KCC2 away from excitatory synapses (Chamma et al., 2013). These results indicate a role for 4.1N in the local stabilization of KCC2 at the plasma membrane. Although lateral KCC2 diffusion was enhanced by NMDA receptor activity, the pathophysiological relevance of this is unknown.
Another molecule interacting with KCC2 is the single-pass transmembrane protein neuropilin and tolloid like-2 (Neto2) (Ivakine et al., 2013). Neto2 is required to maintain the normal abundance of KCC2 and specifically associates with the active oligomeric form of the transporter. Loss of the Neto2:KCC2 interaction reduced KCC2-mediated Cl− extrusion, resulting in decreased synaptic inhibition in hippocampal neurons. Presently, the activity-dependent pathways controlling KCC2 via interaction with Neto2 are unknown, but consistent with Neto2 being part of a neuronal scaffolding platform (Tang et al., 2012), other studies will likely characterize the role of the KCC2:Neto2 interaction in neuronal plasticity or during excitotoxic events that affect the scaffold protein organization.
A recent finding describes a direct interaction between KCC2 and the beta isoform of the Rac/Cdc42 guanine nucleotide exchange factor (betaPIX). The synergetic action of KCC2 and betaPIX leads to decreased cofilin phosphorylation and change of glutamatergic synapse properties (Ludwig et al., 2013) but the importance of this interaction for activity-dependent signal transfer needs to be determined.
Lipid Rafts
At least two studies have suggested an important role of the lipid rafts in the control of KCC2 activity (Hartmann et al., 2009; Watanabe et al., 2009). Lipid rafts are specialized membrane microdomains, enriched in glycosphingolipids and glycoproteins, serving as organising centers for the assembly of signaling molecules and control of their activity (for recent review see Lingwood and Simons, 2010). Both, Hartmann et al. (2009) and Watanabe et al. (2009) illustrated that in neurons approximately 50% of KCC2 is associated with lipid raft markers, whereas remaining transporter molecules are linked to non-rafts markers. However, author's conclusions on the importance of the function of the rafts-like organization for KCC2 were opposite: Hartmann et al. (2009) claimed that, when in rafts, KCC2 is in an inactive form, whereas Watanabe et al. (2009) were convinced that functional KCC2 forms rafts-like clusters. These contradictory assumptions, however, were made based on results obtained using different experimental paradigms, by employing different and wide-range acting pharmacological agents, and by exploring KCC2 mutants whose properties require more detailed analysis. Therefore, after careful and detailed critical analysis of both studies we propose that KCC2 might be membrane raft-associated protein, but additional experimental works are required from independent groups to clarify the role of lipid rafts in KCC2 functioning.
In summary, KCC2 is an important transporter controlling neuronal ion homeostasis and involved in several neurological disorders. The activity of this transporter could be rapidly modulated via large number of signaling pathways. The majority of these pathways remain to be characterized. In the Box 1, we review the existing methods used to study KCC2 activity in an attempt to help investigators in designing experiments to answer the many fundamental questions regarding KCC2 functional regulation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acton, B. A., Mahadevan, V., Mercado, A., Uvarov, P., Ding, Y., Pressey, J., et al. (2012). Hyperpolarizing GABAergic transmission requires the KCC2 C-terminal ISO domain. J. Neurosci. 32, 8746–8751. doi: 10.1523/JNEUROSCI.6089-11.2012
Aguado, F., Carmona, M. A., Pozas, E., Aguiló, A., Martínez-Guijarro, F. J., Alcantara, S., et al. (2003). BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development (Cambridge, England) 130, 1267–1280. doi: 10.1242/dev.00351
Akerman, C. J., and Cline, H. T. (2006). Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26, 5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006
Anthis, N. J., Haling, J. R., Oxley, C. L., Memo, M., Wegener, K. L., Lim, C. J., et al. (2009). Beta integrin tyrosine phosphorylation is a conserved mechanism for regulating talin-induced integrin activation. J. Biol. Chem. 284, 36700–36710. doi: 10.1074/jbc.M109.061275
Antrobus, S. P., Lytle, C., and Payne, J. A. (2012). K+-Cl- cotransporter-2 KCC2 in chicken cardiomyocytes. Am. J. Physiol. Cell Physiol. 303, C1180– C1191. doi: 10.1152/ajpcell.00274.2012
Arosio, D., Ricci, F., Marchetti, L., Gualdani, R., Albertazzi, L., and Beltram, F. (2010). Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat. Methods 7, 516–518. doi: 10.1038/nmeth.1471
Balakrishnan, V., Becker, M., Löhrke, S., Nothwang, H. G., Güresir, E., and Friauf, E. (2003). Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J. Neurosci. 23, 4134–4145.
Banke, T. G., and Gegelashvili, G. (2008). Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J. Physiol. 586, 4925–4934. doi: 10.1113/jphysiol.2008.157024
Barmashenko, G., Hefft, S., Aertsen, A., Kirschstein, T., and Köhling, R. (2011). Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia 52, 1570–1578. doi: 10.1111/j.1528-1167.2011.03247.x
Batti, L., Mukhtarov, M., Audero, E., Ivanov, A., Paolicelli, O., Zurborg, S., et al. (2013). Transgenic mouse lines for non-invasive ratiometric monitoring of intracellular chloride. Front. Mol. Neurosci. 6:11. doi: 10.3389/fnmol.2013.00011
Bellemer, A., Hirata, T., Romero, M. F., and Koelle, M. R. (2011). Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J. 30, 1852–1863. doi: 10.1038/emboj.2011.83
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325.
Ben-Ari, Y., Gaiarsa, J.-L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. doi: 10.1177/1073858412438697
Bergeron, M. J., Gagnon, E., Caron, L., and Isenring, P. (2006). Identification of key functional domains in the C terminus of the K+-Cl- cotransporters. J. Biol. Chem. 281, 15959–15969. doi: 10.1074/jbc.M600015200
Berglund, K., Kuner, T., Feng, G., and Augustine, G. J. (2011). Imaging synaptic inhibition with the genetically encoded chloride indicator Clomeleon. Cold Spring Harb. Protoc. 2011, 1492–1497. doi: 10.1101/pdb.prot066985
Berglund, K., Schleich, W., Krieger, P., Loo, L. S., Wang, D., Cant, N. B., et al. (2006). Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 35, 207–228. doi: 10.1007/s11068-008-9019-6
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. doi: 10.1016/j.neuron.2009.03.003
Blaesse, P., Guillemin, I., Schindler, J., Schweizer, M., Delpire, E., Khiroug, L., et al. (2006). Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J. Neurosci. 26, 10407–10419. doi: 10.1523/JNEUROSCI.3257-06.2006
Bortone, D., and Polleux, F. (2009). KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62, 53–71. doi: 10.1016/j.neuron.2009.01.034
Bos, R., Sadlaoud, K., Boulenguez, P., Buttigieg, D., Liabeuf, S., Brocard, C., et al. (2013). Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 110, 348–353. doi: 10.1073/pnas.1213680110
Boulenguez, P., Liabeuf, S., Bos, R., Bras, H., Jean-xavier, C., Brocard, C., et al. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nature Med. 16, 302–307. doi: 10.1038/nm.2107
Bragin, A., Azizyan, A., Almajano, J., and Engel, J. (2009). The cause of the imbalance in the neuronal network leading to seizure activity can be predicted by the electrographic pattern of the seizure onset. J. Neurosci. 29, 3660–3671. doi: 10.1523/JNEUROSCI.5309-08.2009
Bregestovski, P., Waseem, T., and Mukhtarov, M. (2009). Genetically encoded optical sensors for monitoring of intracellular chloride and chloride-selective channel activity. Front. Mol. Neurosci. 2:15. doi: 10.3389/neuro.02.015.2009
Carmona, M. A., Pozas, E., Martínez, A., Espinosa-Parrilla, J. F., Soriano, E., and Aguado, F. (2006). Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb. Cortex 16, 47–63. doi: 10.1093/cercor/bhi083
Carmosino, M., Rizzo, F., Torretta, S., Procino, G., and Svelto, M. (2013). High-throughput fluorescent-based NKCC functional assay in adherent epithelial cells. BMC Cell Biol. 14:16. doi: 10.1186/1471-2121-14-16
Casula, S., Shmukler, B. E., Wilhelm, S., Stuart-Tilley, A. K., Su, W., Chernova, M. N., et al. (2001). A dominant negative mutant of the KCC1 K-Cl cotransporter: both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J. Biol. Chem. 276, 41870–41878. doi: 10.1074/jbc.M107155200
Casula, S., Zolotarev, A. S., Stuart-Tilley, A. K., Wilhelm, S., Shmukler, B. E., Brugnara, C., et al. (2009). Chemical crosslinking studies with the mouse Kcc1 K-Cl cotransporter. Blood Cells Mol. Dis. 42, 233–240. doi: 10.1016/j.bcmd.2009.01.021
Chamma, I., Chevy, Q., Poncer, J. C., and Lévi, S. (2012). Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front. Cell. Neurosci. 6:5. doi: 10.3389/fncel.2012.00005
Chamma, I., Heubl, M., Chevy, Q., Renner, M., Moutkine, I., Eugene, E., et al. (2013). Activity-dependent regulation of the K/Cl Transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J. Neurosci. 33, 15488–15503. doi: 10.1523/JNEUROSCI.5889-12.2013
Chen, P. Y., Illsley, N. P., and Verkman, A. S. (1988). Renal brush-border chloride transport mechanisms characterized using a fluorescent indicator. Am. J. Physiol. 254, F114–F120.
Chorin, E., Vinograd, O., Fleidervish, I., Gilad, D., Herrmann, S., Sekler, I., et al. (2011). Upregulation of KCC2 Activity by Zinc-Mediated Neurotransmission via the mZnR/GPR39 Receptor. J. Neurosci. 31, 12916–12926. doi: 10.1523/JNEUROSCI.2205-11.2011
Chudotvorova, I., Ivanov, A., Rama, S., Hübner, C. A, Pellegrino, C., Ben-Ari, Y., et al. (2005). Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physio. 566, 671–679. doi: 10.1113/jphysiol.2005.089821
Conti, L., Palma, E., Roseti, C., Lauro, C., Cipriani, R., de Groot, M., et al. (2011). Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 52, 1635–1644. doi: 10.1111/j.1528-1167.2011.03111.x
Coull, J. A. M., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Coull, J. A. M., Boudreau, D., Bachand, K., Prescott, S. A., Nault, F., Sík, A., et al. (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. doi: 10.1038/nature01868
Cramer, S. W., Baggott, C., Cain, J., Tilghman, J., Allcock, B., Miranpuri, G., et al. (2008). The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol. Pain 4, 36. doi: 10.1186/1744-8069-4-36
De Jong, J. C., Willems, P. H. G. M., Mooren, F. J. M., van den Heuvel, L. P. W. J., Knoers, N. V. A. M., and Bindels, R. J. M. (2003). The structural unit of the thiazide-sensitive NaCl cotransporter is a homodimer. J. Biol. Chem. 278, 24302–24307. doi: 10.1074/jbc.M303101200
De Los Heros, P., Alessi, D. R., Gourlay, R., Campbell, D. G., Deak, M., Macartney, T. J., et al. (2014). The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem. J. doi: 10.1042/BJ20131478. [Epub ahead of print].
De Los Heros, P., Kahle, K. T., Rinehart, J., Bobadilla, N. A., Vázquez, N., San Cristobal, P., et al. (2006). WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 1976–1981. doi: 10.1073/pnas.0510947103
Debanne, D., and Poo, M.-M. (2010). Spike-timing dependent plasticity beyond synapse - pre- and post-synaptic plasticity of intrinsic neuronal excitability. Front. Synaptic Neurosci. 2:21. doi: 10.3389/fnsyn.2010.00021
Delpire, E., Baranczak, A., Waterson, A. G., Kim, K., Kett, N., Morrison, R. D., et al. (2012). Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: development of a highly selective and more potent in vitro probe. Bioorg. Med. Chem. Lett. 22, 4532–4535. doi: 10.1016/j.bmcl.2012.05.126
Delpire, E., Days, E., Lewis, L. M., Mi, D., Kim, K., Lindsley, C. W., et al. (2009). Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 106, 5383–5388. doi: 10.1073/pnas.0812756106
Delpire, E., and Gagnon, K. B. E. (2008). SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem. J. 409, 321–331. doi: 10.1042/BJ20071324
Duebel, J., Haverkamp, S., Schleich, W., Feng, G., Augustine, G. J., Kuner, T., et al. (2006). Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron 49, 81–94. doi: 10.1016/j.neuron.2005.10.035
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
Döding, A., Hartmann, A.-M., Beyer, T., and Nothwang, H. G. (2012). KCC2 transport activity requires the highly conserved L(675) in the C-terminal β 1 strand. Biochem. Biophys. Res. Commun. 420, 492–497. doi: 10.1016/j.bbrc.2012.02.147
Ebihara, S., Shirato, K., Harata, N., and Akaike, N. (1995). Gramicidin-perforated patch recording: GABA response in mammalian neurones with intact intracellular chloride. J. Physiol. 484(Pt 1), 77–86.
Ferrini, F., and De Koninck, Y. (2013). Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013, 429815. doi: 10.1155/2013/429815
Fiumelli, H., Cancedda, L., and Poo, M. (2005). Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron 48, 773–786. doi: 10.1016/j.neuron.2005.10.025
Flatman, P. W. (2008). Cotransporters, WNKs and hypertension: an update. Curr. Opin. Nephrol. Hypertens. 17, 186–192. doi: 10.1097/MNH.0b013e3282f5244e
Friauf, E., Wenz, M., Oberhofer, M., Nothwang, H. G., Balakrishnan, V., Knipper, M., et al. (2008). Hypothyroidism impairs chloride homeostasis and onset of inhibitory neurotransmission in developing auditory brainstem and hippocampal neurons. Eur. J. Neurosci. 28, 2371–2380. doi: 10.1111/j.1460-9568.2008.06528.x
Friedel, P., Bregestovski, P., and Medina, I. (2013). Improved method for efficient imaging of intracellular Cl(-) with Cl-Sensor using conventional fluorescence setup. Front. Mol. Neurosci. 6:7. doi: 10.3389/fnmol.2013.00007
Gagnon, K. B. E., England, R., and Delpire, E. (2006a). Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol. Cell. Biol. 26, 689–698. doi: 10.1128/MCB.26.2.689-698.2006
Gagnon, K. B. E., England, R., and Delpire, E. (2006b). Volume sensitivity of cation-Cl- cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am. J. Physiol. Cell Physiol. 290, C134–C142. doi: 10.1152/ajpcell.00037.2005
Gagnon, K. B., and Delpire, E. (2013). Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol. Cell Physiol. 304, C693–C714. doi: 10.1152/ajpcell.00350.2012
Gagnon, M., Bergeron, M. J., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R. P., et al. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528. doi: 10.1038/nm.3356
Galeffi, F., Sah, R., Pond, B. B., George, A., and Schwartz-Bloom, R. D. (2004). Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: effect of diazepam. J. Neurosci. 24, 4478–4488. doi: 10.1523/JNEUROSCI.0755-04.2004
Gallagher, S. R. (2012). One-dimensional SDS gel electrophoresis of proteins. Curr. Protoc. Protein Sci. Chapter 10, Unit 10.1.1–44. doi: 10.1002/0471140864.ps1001s68
Gamba, G. (2005). Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493. doi: 10.1152/physrev.00011.2004
Gamba, G., Miyanoshita, A., Lombardi, M., Lytton, J., Lee, W. S., Hediger, M. A., et al. (1994). Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 269, 17713–17722.
Ganguly, K., Schinder, A. F., Wong, S. T., and Poo, M. (2001). GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105, 521–532. doi: 10.1016/S0092-8674(01)00341-5
Garbarini, N., and Delpire, E. (2008). The RCC1 domain of protein associated with Myc (PAM) interacts with and regulates KCC2. Cell. Physiol. Biochem. 22, 31–44. doi: 10.1159/000149781
Gauvain, G., Chamma, I., Chevy, Q., Cabezas, C., Irinopoulou, T., Bodrug, N., et al. (2011). The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 108, 15474–15479. doi: 10.1073/pnas.1107893108
Geck, P., Pietrzyk, C., Burckhardt, B. C., Pfeiffer, B., and Heinz, E. (1980). Electrically silent cotransport on Na+, K+ and Cl- in Ehrlich cells. Biochim. Biophys. Acta 600, 432–447. doi: 10.1016/0005-2736(80)90446-0
Gillen, C. M., Brill, S., Payne, J. A., and Forbush, B. (1996). Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem. 271, 16237–16244. doi: 10.1074/jbc.271.27.16237
Grimley, J. S., Li, L., Wang, W., Wen, L., Beese, L. S., Hellinga, H. W., et al. (2013). Visualization of synaptic inhibition with an optogenetic sensor developed by cell-free protein engineering automation. J. Neurosci. 33, 16297–16309. doi: 10.1523/JNEUROSCI.4616-11.2013
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature Neurosci. 5, 405–414. doi: 10.1038/nn835
Hartmann, A.-M., Blaesse, P., Kranz, T., Wenz, M., Schindler, J., Kaila, K., et al. (2009). Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J. Neurochem. 111, 321–331. doi: 10.1111/j.1471-4159.2009.06343.x
Hartmann, A.-M., Wenz, M., Mercado, A., Störger, C., Mount, D. B., Friauf, E., et al. (2010). Differences in the large extracellular loop between the K(+)-Cl(-) cotransporters KCC2 and KCC4. J. Biol. Chem. 285, 23994–24002. doi: 10.1074/jbc.M110.144063
Hekmat-Scafe, D. S., Lundy, M. Y., Ranga, R., and Tanouye, M. A. (2006). Mutations in the K+/Cl- cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J. Neurosci. 26, 8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006
Hekmat-Scafe, D. S., Mercado, A., Fajilan, A. A., Lee, A. W., Hsu, R., Mount, D. B., et al. (2010). Seizure sensitivity is ameliorated by targeted expression of K+-Cl-cotransporter function in the mushroom body of the Drosophila brain. Genetics 184, 171–183. doi: 10.1534/genetics.109.109074
Hershfinkel, M., Kandler, K., Knoch, M. E., Dagan-Rabin, M., Aras, M. A., Abramovitch-Dahan, C., et al. (2009). Intracellular zinc inhibits KCC2 transporter activity. Nat. Neurosci. 12, 725–757. doi: 10.1038/nn.2316
Horn, Z., Ringstedt, T., Blaesse, P., Kaila, K., and Herlenius, E. (2010). Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur. J. Neurosci. 31, 2142–2155. doi: 10.1111/j.1460-9568.2010.07258.x
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., et al. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007
Hübner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524. doi: 10.1016/S0896-6273(01)00297-5
Ikeda, K., Onimaru, H., Yamada, J., Inoue, K., Ueno, S., Onaka, T., et al. (2004). Malfunction of respiratory-related neuronal activity in Na+, K+-ATPase alpha2 subunit-deficient mice is attributable to abnormal Cl- homeostasis in brainstem neurons. J. Neurosci. 24, 10693–10701. doi: 10.1523/JNEUROSCI.2909-04.2004
Inoue, K., Furukawa, T., Kumada, T., Yamada, J., Wang, T., Inoue, R., et al. (2012). Taurine inhibits K+-Cl-cotransporter KCC2 to regulate embryonic Cl- homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem. 287, 20839–20850. doi: 10.1074/jbc.M111.319418
Inoue, K., Ueno, S., and Fukuda, A. (2004). Interaction of neuron-specific K+-Cl- cotransporter, KCC2, with brain-type creatine kinase. FEBS Lett. 564, 131–135. doi: 10.1016/S0014-5793(04)00328-X
Inoue, K., Yamada, J., Ueno, S., and Fukuda, A. (2006). Brain-type creatine kinase activates neuron-specific K+-Cl- co-transporter KCC2. J. Neurochem. 96, 598–608. doi: 10.1111/j.1471-4159.2005.03560.x
Ivakine, E. A., Acton, B. A., Mahadevan, V., Ormond, J., Tang, M., Pressey, J. C., et al. (2013). Neto2 is a KCC2 interacting protein required for neuronal Cl- regulation in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 3561–3566. doi: 10.1073/pnas.1212907110
Ivanov, A., Pellegrino, C., Rama, S., Dumalska, I., Salyha, Y., Ben-Ari, Y., et al. (2006). Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol. 572, 789–798. doi: 10.1113/jphysiol.2006.105510
Jaenisch, N., Witte, O. W., and Frahm, C. (2010). Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 41, e151–e159. doi: 10.1161/STROKEAHA.109.570424
Jarolimek, W., Lewen, A., and Misgeld, U. (1999). A furosemide-sensitive K+-Cl- cotransporter counteracts intracellular Cl- accumulation and depletion in cultured rat midbrain neurons. J. Neurosci. 19, 4695–4704.
Jayaraman, S., Haggie, P., Wachter, R. M., Remington, S. J., and Verkman, A. S. (2000). Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J. Biol. Chem. 275, 6047–6050. doi: 10.1074/jbc.275.9.6047
Johnston, A. M., Naselli, G., Gonez, L. J., Martin, R. M., Harrison, L. C., and DeAizpurua, H. J. (2000). SPAK, a STE20/SPS1-related kinase that activates the p38 pathway. Oncogene 19, 4290–4297. doi: 10.1038/sj.onc.1203784
Kahle, K. T., Deeb, T. Z., Puskarjov, M., Silayeva, L., Liang, B., Kaila, K., et al. (2013). Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–737. doi: 10.1016/j.tins.2013.08.006
Kahle, K. T., Rinehart, J., and Lifton, R. P. (2010). Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta 1802, 1150–1158. doi: 10.1016/j.bbadis.2010.07.009
Kahle, K. T., Rinehart, J., de Los Heros, P., Louvi, A., Meade, P., Vazquez, N., et al. (2005). WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788. doi: 10.1073/pnas.0508307102
Kahle, K. T., Staley, K. J., Nahed, B. V., Gamba, G., Hebert, S. C., Lifton, R. P., et al. (2008). Roles of the cation-chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 4, 490–503. doi: 10.1038/ncpneuro0883
Kaila, K., Pasternack, M., Saarikoski, J., and Voipio, J. (1989). Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. J. Physiol. 416, 161–181.
Karadsheh, M. F., and Delpire, E. (2001). Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J. Neurophysiol. 85, 995–997.
Kelsch, W., Hormuzdi, S., Straube, E., Lewen, A., Monyer, H., and Misgeld, U. (2001). Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J. Neurosci. 21, 8339–8347.
Khalilov, I., Chazal, G., Chudotvorova, I., Pellegrino, C., Corby, S., Ferrand, N., et al. (2011). Enhanced synaptic activity and epileptiform events in the embryonic KCC2 deficient hippocampus. Front. Cell. Neurosci. 5:23. doi: 10.3389/fncel.2011.00023
Khirug, S., Ahmad, F., Puskarjov, M., Afzalov, R., Kaila, K., and Blaesse, P. (2010). A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J. Neurosci. 30, 12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010
Khirug, S., Huttu, K., Ludwig, A., Smirnov, S., Voipio, J., Rivera, C., et al. (2005). Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur. J. Neurosci. 21, 899–904. doi: 10.1111/j.1460-9568.2005.03886.x
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008
Kovalchuk, Y., and Garaschuk, O. (2012). Two-photon chloride imaging using MQAE in vitro and in vivo. Cold Spring Harb. Protoc. 2012, 778–785. doi: 10.1101/pdb.prot070037
Krapivinsky, G., Krapivinsky, L., Manasian, Y., Ivanov, A., Tyzio, R., Pellegrino, C., et al. (2003). The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40, 775–784. doi: 10.1016/S0896-6273(03)00645-7
Krapivinsky, G., Medina, I., Krapivinsky, L., Gapon, S., and Clapham, D. E. (2004). SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron 43, 563–574. doi: 10.1016/j.neuron.2004.08.003
Kregenow, F. M. (1971). The response of duck erythrocytes to nonhemolytic hypotonic media. Evidence for a volume-controlling mechanism. J. Gen. Physiol. 58, 372–395. doi: 10.1085/jgp.58.4.372
Kuczewski, N., Porcher, C., and Gaiarsa, J.-L. (2010). Activity-dependent dendritic secretion of brain-derived neurotrophic factor modulates synaptic plasticity. Eur. J. Neurosci. 32, 1239–1244. doi: 10.1111/j.1460-9568.2010.07378.x
Kuczewski, N., Porcher, C., Ferrand, N., Fiorentino, H., Pellegrino, C., Kolarow, R., et al. (2008). Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J. Neurosci. 28, 7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008
Kuner, T., and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459. doi: 10.1016/S0896-6273(00)00056-8
Kyrozis, A., and Reichling, D. B. (1995). Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J. Neurosci. Methods 57, 27–35. doi: 10.1016/0165-0270(94)00116-X
Lauf, P. K., Di Fulvio, M., Srivastava, V., Sharma, N., and Adragna, N. C. (2012). KCC2a expression in a human fetal lens epithelial cell line. Cell. Physiol. Biochem. 29, 303–312. doi: 10.1159/000337611
Lauf, P. K., Misri, S., Chimote, A. A., and Adragna, N. C. (2008). Apparent intermediate K conductance channel hyposmotic activation in human lens epithelial cells. Am. J. Physiol. Cell Physiol. 294, C820–C832. doi: 10.1152/ajpcell.00375.2007
Lee, H., Chen, C. X.-Q., Liu, Y.-J., Aizenman, E., and Kandler, K. (2005). KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur. J. Neurosci. 21, 2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x
Lee, H. H. C., Deeb, T. Z., Walker, J. A., Davies, P. A, and Moss, S. J. (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABA(A) receptor-mediated currents. Nat. Neurosci. 14, 736–743. doi: 10.1038/nn.2806
Lee, H. H. C., Jurd, R., and Moss, S. J. (2010). Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179. doi: 10.1016/j.mcn.2010.06.008
Lee, H. H. C., Walker, J. A., Williams, J. R., Goodier, R. J., Payne, J. A., and Moss, S. J. (2007). Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 282, 29777–29784. doi: 10.1074/jbc.M705053200
Leitch, E., Coaker, J., Young, C., Mehta, V., and Sernagor, E. (2005). GABA type-A activity controls its own developmental polarity switch in the maturing retina. J. Neurosci. 25, 4801–4805. doi: 10.1523/JNEUROSCI.0172-05.2005
Li, H., Khirug, S., Cai, C., Ludwig, A., Blaesse, P., Kolikova, J., et al. (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033. doi: 10.1016/j.neuron.2007.10.039
Li, H., Tornberg, J., Kaila, K., Airaksinen, M. S., and Rivera, C. (2002). Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 16, 2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x
Lingwood, D., and Simons, K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46–50. doi: 10.1126/science.1174621
Lu, J., Karadsheh, M., and Delpire, E. (1999). Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J. Neurobiol. 39, 558–568. doi: 10.1002/(SICI)1097-4695(19990615)39:4<558::AID-NEU9>3.0.CO;2-5
Ludwig, A., Li, H., Saarma, M., Kaila, K., and Rivera, C. (2003). Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur. J. Neurosci. 18, 3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x
Ludwig, A., Llano, O., Smirnov, S., Golubtsov, A., Medina, I., Nothwang, H. G., et al. (2013). KCC2 regulates actin dynamics in spines via interaction with betaPIX. in Society for neuroscience meeting, San Diego, CA.
Ludwig, A., Uvarov, P., Pellegrino, C., Thomas-Crusells, J., Schuchmann, S., Saarma, M., et al. (2011a). Neurturin evokes MAPK-dependent upregulation of Egr4 and KCC2 in developing neurons. Neural Plast. 2011, 1–8. doi: 10.1155/2011/641248
Ludwig, A., Uvarov, P., Soni, S., Thomas-Crusells, J., Airaksinen, M. S., and Rivera, C. (2011b). Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 31, 644–649. doi: 10.1523/JNEUROSCI.2006-10.2011
Löscher, W., Puskarjov, M., and Kaila, K. (2013). Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74. doi: 10.1016/j.neuropharm.2012.05.045
Markkanen, M., Karhunen, T., Llano, O., Ludwig, A., Rivera, C., Uvarov, P., et al. (2013). Distribution of neuronal KCC2a and KCC2b isoforms in mouse CNS. J. Comp. Neurol. doi: 10.1002/cne.23510. Avilable online at: http://doi.wiley.com/10.1002/cne.23510
Markkanen, M., Uvarov, P., and Airaksinen, M. S. (2008). Role of upstream stimulating factors in the transcriptional regulation of the neuron-specific K-Cl cotransporter KCC2. Brain Res. 1236, 8–15. doi: 10.1016/j.brainres.2008.08.007
Markova, O., Mukhtarov, M., Real, E., Jacob, Y., and Bregestovski, P. (2008). Genetically encoded chloride indicator with improved sensitivity. J. Neurosci. Methods 170, 67–76. doi: 10.1016/j.jneumeth.2007.12.016
Matsuda, N., Lu, H., Fukata, Y., Noritake, J., Gao, H., Mukherjee, S., et al. (2009). Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J. Neurosci. 29, 14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009
McKee, J. A., Kumar, S., Ecelbarger, C. A., Fernández-Llama, P., Terris, J., and Knepper, M. A. (2000). Detection of Na(+) transporter proteins in urine. J. Am. Soc. Nephrol. 11, 2128–2132.
Mederle, K., Mutig, K., Paliege, A., Carota, I., Bachmann, S., Castrop, H., et al. (2013). Loss of WNK3 is compensated for by the WNK1/SPAK axis in the kidney of the mouse. Am. J. Physiol. Renal Physiol. 304, F1198–F1209. doi: 10.1152/ajprenal.00288.2012
Medina, I. (2007). Extrasynaptic NMDA receptors reshape gene ranks. Signal Transduct. Knowledge Environ. 2007, pe23. doi: 10.1126/stke.3862007pe23
Medina, I., and Chudotvorova, I. (2006). GABA neurotransmission and neural cation-chloride co-transporters: actions beyond ion transport. Crit. Rev. Neurobiol. 18, 105–112. doi: 10.1615/CritRevNeurobiol.v18.i1-2.110
Mercado, A., Broumand, V., Zandi-Nejad, K., Enck, A. H., and Mount, D. B. (2006). A C-terminal domain in KCC2 confers constitutive K+-Cl- cotransport. J. Biol. Chem. 281, 1016–1026. doi: 10.1074/jbc.M509972200
Mercado, A., Song, L., Vazquez, N., Mount, D. B., and Gamba, G. (2000). Functional comparison of the K+-Cl- cotransporters KCC1 and KCC4. J. Biol. Chem. 275, 30326–30334. doi: 10.1074/jbc.M003112200
Moore-Hoon, M. L., and Turner, R. J. (2000). The structural unit of the secretory Na+-K+-2Cl- cotransporter (NKCC1) is a homodimer. Biochemistry 39, 3718–3724. doi: 10.1021/bi992301v
Mueller, A. L., Taube, J. S., and Schwartzkroin, P. A. (1984). Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J. Neurosci. 4, 860–867.
Mukhtarov, M., Liguori, L., Waseem, T., Rocca, F., Buldakova, S., Arosio, D., et al. (2013). Calibration and functional analysis of three genetically encoded Cl(-)/pH sensors. Front. Mol. Neurosci. 6:9. doi: 10.3389/fnmol.2013.00009
Munakata, M., Watanabe, M., Otsuki, T., Nakama, H., Arima, K., Itoh, M., et al. (2007). Altered distribution of KCC2 in cortical dysplasia in patients with intractable epilepsy. Epilepsia 48, 837–844. doi: 10.1111/j.1528-1167.2006.00954.x
Nabekura, J., Ueno, T., Okabe, A., Furuta, A., Iwaki, T., Shimizu-okabe, C., et al. (2002). Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J. Neurosci. 22, 4412–4417.
Nardou, R., Ben-Ari, Y., and Khalilov, I. (2009). Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J. Neurophysiol. 101, 2878–2888. doi: 10.1152/jn.90761.2008
Nardou, R., Yamamoto, S., Chazal, G., Bhar, A., Ferrand, N., Dulac, O., et al. (2011). Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain 134, 987–1002. doi: 10.1093/brain/awr041
Palma, E., Amici, M., Sobrero, F., Spinelli, G., Di Angelantonio, S., Ragozzino, D., et al. (2006). Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. U.S.A. 103, 8465–8468. doi: 10.1073/pnas.0602979103
Papp, E., Rivera, C., Kaila, K., and Freund, T. F. (2008). Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience 154, 677–689. doi: 10.1016/j.neuroscience.2008.03.072
Parvin, M. N., Gerelsaikhan, T., and Turner, R. J. (2007). Regions in the cytosolic C-terminus of the secretory Na(+)-K(+)-2Cl(-) cotransporter NKCC1 are required for its homodimerization. Biochemistry 46, 9630–9637. doi: 10.1021/bi700881a
Payne, J. A. (1997). Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am. J. Phys. 273, C1516–C1525.
Payne, J. A., Stevenson, T. J., and Donaldson, L. F. (1996). Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 271, 16245–16252. doi: 10.1074/jbc.271.27.16245
Pedersen, M., Carmosino, M., and Forbush, B. (2008). Intramolecular and intermolecular fluorescence resonance energy transfer in fluorescent protein-tagged Na-K-Cl cotransporter (NKCC1): sensitivity to regulatory conformational change and cell volume. J. Biol. Chem. 283, 2663–2674. doi: 10.1074/jbc.M708194200
Pellegrino, C., Gubkina, O., Schaefer, M., Becq, H., Ludwig, A., Mukhtarov, M., et al. (2011). Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J. Physiol. 589, 2475–2496. doi: 10.1113/jphysiol.2010.203703
Puskarjov, M., Ahmad, F., Kaila, K., and Blaesse, P. (2012). Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J. Neurosci 32, 11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012
Rafiqi, F. H., Zuber, A. M., Glover, M., Richardson, C., Fleming, S., Jovanović, S., et al. (2010). Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2, 63–75. doi: 10.1002/emmm.200900058
Raimondo, J. V., Joyce, B., Kay, L., Schlagheck, T., Newey, S. E., Srinivas, S., et al. (2013). A genetically-encoded chloride and pH sensor for dissociating ion dynamics in the nervous system. Front. Cell. Neurosci. 7:202. doi: 10.3389/fncel.2013.00202
Reynolds, A., Brustein, E., Liao, M., Mercado, A., Babilonia, E., Mount, D. B., et al. (2008). Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J. Neurosci. 28, 1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008
Rinehart, J., Maksimova, Y. D., Tanis, J. E., Stone, K. L., Caleb, A., Zhang, J., et al. (2009). Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536. doi: 10.1016/j.cell.2009.05.031
Rinehart, J., Vázquez, N., Kahle, K. T., Hodson, C. A., Ring, A. M., Gulcicek, E. E., et al. (2011). WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J. Biol. Chem. 286, 30171–30180. doi: 10.1074/jbc.M111.222893
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., et al. (2002). BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J. Cell Biol. 159, 747–752. doi: 10.1083/jcb.200209011
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipilä, S., et al. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004
Russell, J. M. (2000). Sodium-potassium-chloride cotransport. Physiol. Rev. 80, 211–276. doi: 10.1016/B978-0-12-374373-2.00027-3
Russell, J. M., and Boron, W. F. (1976). Role of choloride transport in regulation of intracellular pH. Nature 264, 73–74. doi: 10.1038/264073a0
Saadi, R. A., He, K., Hartnett, K. A., Kandler, K., Hershfinkel, M., and Aizenman, E. (2012). SNARE-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro. Neuroscience 210, 38–46. doi: 10.1016/j.neuroscience.2012.03.001
Shibata, S., Kakazu, Y., Okabe, A., Fukuda, A., and Nabekura, J. (2004). Experience-dependent changes in intracellular Cl- regulation in developing auditory neurons. Neurosci. Res. 48, 211–220. doi: 10.1016/j.neures.2003.10.011
Shimizu-Okabe, C., Tanaka, M., Matsuda, K., Mihara, T., Okabe, A., Sato, K., et al. (2011). KCC2 was downregulated in small neurons localized in epileptogenic human focal cortical dysplasia. Epilepsy Res. 93, 177–184. doi: 10.1016/j.eplepsyres.2010.12.008
Shulga, A., and Rivera, C. (2013). Interplay between thyroxin, BDNF and GABA in injured neurons. Neuroscience 239, 241–252. doi: 10.1016/j.neuroscience.2012.12.007
Shulga, A., Blaesse, A., Kysenius, K., Huttunen, H. J., Tanhuanpää, K., Saarma, M., et al. (2009). Thyroxin regulates BDNF expression to promote survival of injured neurons. Mol. Cell. Neurosci. 42, 408–418. doi: 10.1016/j.mcn.2009.09.002
Shulga, A., Magalhaes, A. C., Autio, H., Plantman, S., di Lieto, A., Nykjaer, A., et al. (2012). The loop diuretic bumetanide blocks posttraumatic p75NTR upregulation and rescues injured neurons. J. Neurosci. 32, 1757–1770. doi: 10.1523/JNEUROSCI.3282-11.2012
Shulga, A., Thomas-Crusells, J., Sigl, T., Blaesse, A., Mestres, P., Meyer, M., et al. (2008). Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci. 28, 6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008
Simard, C. F., Bergeron, M. J., Frenette-Cotton, R., Carpentier, G. A., Pelchat, M.-E., Caron, L., et al. (2007). Homooligomeric and heterooligomeric associations between K+-Cl- cotransporter isoforms and between K+-Cl- and Na+-K+-Cl- cotransporters. J. Biol. Chem. 282, 18083–18093. doi: 10.1074/jbc.M607811200
Simard, C. F., Brunet, G. M., Daigle, N. D., Montminy, V., Caron, L., and Isenring, P. (2004). Self-interacting domains in the C terminus of a cation-Cl- cotransporter described for the first time. J. Biol. Chem. 279, 40769–40777. doi: 10.1074/jbc.M406458200
Song, L., Mercado, A., Vázquez, N., Xie, Q., Desai, R., George, A. L., et al. (2002). Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Brain Res. 103, 91–105. doi: 10.1016/S0169-328X(02)00190-0
Starremans, P. G. J. F., Kersten, F. F. J., Van Den Heuvel, L. P. W. J., Knoers, N. V. A. M., and Bindels, R. J. M. (2003). Dimeric architecture of the human bumetanide-sensitive Na-K-Cl Co-transporter. J. Am. Soc. Nephrol. 14, 3039–3046. doi: 10.1097/01.ASN.0000097370.29737.5B
Stein, V., Hermans-Borgmeyer, I., Jentsch, T. J., and Hübner, C. A (2004). Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J. Comp. Neurol. 468, 57–64. doi: 10.1002/cne.10983
Strange, K., Singer, T. D., Morrison, R., and Delpire, E. (2000). Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am. J. Physiol. Cell Physiol. 279, C860–C867.
Succol, F., Fiumelli, H., Benfenati, F., Cancedda, L., and Barberis, A. (2012). Intracellular chloride concentration influences the GABAA receptor subunit composition. Nature Commun. 3, 738. doi: 10.1038/ncomms1744
Talos, D. M., Sun, H., Kosaras, B., Joseph, A., Folkerth, R. D., Poduri, A., et al. (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. doi: 10.1002/ana.22696
Tamari, M., Daigo, Y., and Nakamura, Y. (1999). Isolation and characterization of a novel serine threonine kinase gene on chromosome 3p22-21.3. J. Hum. Genet. 44, 116–120. doi: 10.1007/s100380050121
Tang, M., Ivakine, E., Mahadevan, V., Salter, M. W., and McInnes, R. R. (2012). Neto2 interacts with the scaffolding protein GRIP and regulates synaptic abundance of kainate receptors. PLoS ONE 7:e51433. doi: 10.1371/journal.pone.0051433
Tanis, J. E., Bellemer, A., Moresco, J. J., Forbush, B., and Koelle, M. R. (2009). The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J. Neurosci. 29, 9943–9954. doi: 10.1523/JNEUROSCI.1989-09.2009
Thastrup, J. O., Rafiqi, F. H., Vitari, A. C., Pozo-Guisado, E., Deak, M., Mehellou, Y., et al. (2012). SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem. J. 441, 325–337. doi: 10.1042/BJ20111879
Titz, S., Hans, M., Kelsch, W., Lewen, A., Swandulla, D., and Misgeld, U. (2003). Hyperpolarizing inhibition develops without trophic support by GABA in cultured rat midbrain neurons. J. Physiol. 550, 719–730. doi: 10.1113/jphysiol.2003.041863
Titz, S., Hormuzdi, S., Lewen, A., Monyer, H., and Misgeld, U. (2006). Intracellular acidification in neurons induced by ammonium depends on KCC2 function. Eur. J. Neurosci. 23, 454–464. doi: 10.1111/j.1460-9568.2005.04583.x
Toyoda, H., Ohno, K., Yamada, J., Ikeda, M., Okabe, A., Sato, K., et al. (2003). Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J. Neurophysiol. 89, 1353–1362. doi: 10.1152/jn.00721.2002
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hübner, C. A., Represa, A., et al. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792. doi: 10.1126/science.1133212
Tyzio, R., Ivanov, A., Bernard, C., Holmes, G. L., Ben-Ari, Y., and Khazipov, R. (2003). Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J. Neurophysiol. 90, 2964–2972. doi: 10.1152/jn.00172.2003
Tyzio, R., Minlebaev, M., Rheims, S., Ivanov, A., Jorquera, I., Holmes, G. L., et al. (2008). Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur. J. Neurosci. 27, 2515–2528. doi: 10.1111/j.1460-9568.2008.06234.x
Uvarov, P. (2010). Neuronal K-Cl Cotransporter: Transcriptional Mechanisms of KCC2 Gene Regulation. Helsinki: Helsinki University Press. Available online at: https://helda.helsinki.fi/handle/10138/22378
Uvarov, P., Ludwig, A., Markkanen, M., Pruunsild, P., Kaila, K., Delpire, E., et al. (2007). A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 282, 30570–30576. doi: 10.1074/jbc.M705095200
Uvarov, P., Ludwig, A., Markkanen, M., Rivera, C., and Airaksinen, M. S. (2006). Upregulation of the neuron-specific K+/Cl- cotransporter expression by transcription factor early growth response 4. J. Neurosci. 26, 13463–13473. doi: 10.1523/JNEUROSCI.4731-06.2006
Uvarov, P., Ludwig, A., Markkanen, M., Soni, S., Hübner, C. A., Rivera, C., et al. (2009). Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J. Biol. Chem. 284, 13696–13704. doi: 10.1074/jbc.M807366200
Uvarov, P., Pruunsild, P., Timmusk, T., and Airaksinen, M. S. (2005). Neuronal K+/Cl- co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J. Neurochem. 95, 1144–1155. doi: 10.1111/j.1471-4159.2005.03434.x
Wachter, R. M., and Remington, S. J. (1999). Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr. Biol. 9, R628–R629. doi: 10.1016/S0960-9822(99)80408-4
Wake, H., Watanabe, M., Moorhouse, A. J., Kanematsu, T., Horibe, S., Matsukawa, N., et al. (2007). Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 27, 1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007
Warmuth, S., Zimmermann, I., and Dutzler, R. (2009). X-ray structure of the C-terminal domain of a prokaryotic cation-chloride cotransporter. Structure 17, 538–546. doi: 10.1016/j.str.2009.02.009
Waseem, T., Mukhtarov, M., Buldakova, S., Medina, I., and Bregestovski, P. (2010). Genetically encoded Cl-Sensor as a tool for monitoring of Cl-dependent processes in small neuronal compartments. J. Neurosci. Methods 193, 14–23. doi: 10.1016/j.jneumeth.2010.08.002
Watanabe, M., Wake, H., Moorhouse, A. J., and Nabekura, J. (2009). Clustering of neuronal K+-Cl- cotransporters in lipid rafts by tyrosine phosphorylation. J. Biol. Chem. 284, 27980–27988. doi: 10.1074/jbc.M109.043620
Wei, W.-C., Akerman, C. J., Newey, S. E., Pan, J., Clinch, N. W. V., Jacob, Y., et al. (2011). The potassium-chloride cotransporter 2 promotes cervical cancer cell migration and invasion by an ion transport-independent mechanism. J. Physiol. 589, 5349–5359. doi: 10.1113/jphysiol.2011.214635
Wenz, M., Hartmann, A.-M., Friauf, E., and Nothwang, H. G. (2009). CIP1 is an activator of the K+-Cl- cotransporter KCC2. Biochem. Biophys. Res. Commun. 381, 388–392. doi: 10.1016/j.bbrc.2009.02.057
Verkman, A. S. (1990). Development and biological applications of chloride-sensitive fluorescent indicators. Am. J. Physiol. 259, C375–C388.
Westerholz, S., de Lima, A. D., and Voigt, T. (2013). Thyroid hormone-dependent development of early cortical networks: temporal specificity and the contribution of trkB and mTOR pathways. Front. Cell. Neurosci. 7:121. doi: 10.3389/fncel.2013.00121
Williams, J. R., and Payne, J. A. (2004). Cation transport by the neuronal K(+)-Cl(-) cotransporter KCC2: thermodynamics and kinetics of alternate transport modes. Am. J. Physiol. Cell Physiol. 287, C919–C931. doi: 10.1152/ajpcell.00005.2004
Woo, N.-S., Lu, J., England, R., McClellan, R., Dufour, S., Mount, D. B., et al. (2002). Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 12, 258–268. doi: 10.1002/hipo.10014
Woodin, M. A., Ganguly, K., and Poo, M. (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transporter activity. Neuron 39, 807–820. doi: 10.1016/S0896-6273(03)00507-5
Xie, J., Yoon, J., Yang, S.-S., Lin, S.-H., and Huang, C.-L. (2013). WNK1 protein kinase regulates embryonic cardiovascular development through the OSR1 signaling cascade. J. Biol. Chem. 288, 8566–8574. doi: 10.1074/jbc.M113.451575
Xu, J. C., Lytle, C., Zhu, T. T., Payne, J. A., Benz, E., and Forbush, B. (1994). Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. U.S.A. 91, 2201–2205. doi: 10.1073/pnas.91.6.2201
Yang, S.-S., Lo, Y.-F., Wu, C.-C., Lin, S.-W., Yeh, C.-J., Chu, P., et al. (2010). SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 21, 1868–1877. doi: 10.1681/ASN.2009121295
Yeo, M., Berglund, K., Augustine, G., and Liedtke, W. (2009). Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 29, 14652–14662. doi: 10.1523/JNEUROSCI.2934-09.2009
Zagórska, A., Pozo-Guisado, E., Boudeau, J., Vitari, A. C., Rafiqi, F. H., Thastrup, J., et al. (2007). Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J. Cell Biol. 176, 89–100. doi: 10.1083/jcb.200605093
Zhang, D., Gopalakrishnan, S. M., Freiberg, G., and Surowy, C. S. (2010). A thallium transport FLIPR-based assay for the identification of KCC2-positive modulators. J. Biomol. Screen. 15, 177–184. doi: 10.1177/1087057109355708
Zhang, Z., Wang, X., Wang, W., Lu, Y.-G., and Pan, Z. Z. (2013). Brain-derived neurotrophic factor-mediated downregulation of brainstem K+-Cl- cotransporter and cell-type-specific GABA impairment for activation of descending pain facilitation. Mol. Pharmacol. 84, 511–520. doi: 10.1124/mol.113.086496
Zhao, B., Wong, A. Y. C., Murshid, A., Bowie, D., Presley, J. F., and Bedford, F. K. (2008). Identification of a novel di-leucine motif mediating K(+)/Cl(-) cotransporter KCC2 constitutive endocytosis. Cell. Signal. 20, 1769–1779. doi: 10.1016/j.cellsig.2008.06.011
Zhu, L., Lovinger, D., and Delpire, E. (2005). Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J. Neurophysiol. 93, 1557–1568. doi: 10.1152/jn.00616.2004
Keywords: KCC2, intracellular chloride, GABA, neurons
Citation: Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P and Pellegrino C (2014) Current view on the functional regulation of the neuronal K+-Cl− cotransporter KCC2. Front. Cell. Neurosci. 8:27. doi: 10.3389/fncel.2014.00027
Received: 28 October 2013; Accepted: 18 January 2014;
Published online: 06 February 2014.
Edited by:
Laura Cancedda, Istituto Italiano di Tecnologia, ItalyReviewed by:
Junichi Nabekura, National Instritute for Physiological Sciences, JapanHans G. Nothwang, University of Oldenburg, Germany
Copyright © 2014 Medina, Friedel, Rivera, Kahle, Kourdougli, Uvarov and Pellegrino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Igor Medina, INSERM, Institut de Neurobiologie de la Méditerranée (INMED), Unité 901, 163 Route de Luminy, 13273 Marseille, France e-mail:aWdvci5tZWR5bmFAaW5zZXJtLmZy