 Osama F. Harraz
Osama F. Harraz Christophe Altier
Christophe Altier- 1Department of Physiology and Pharmacology, Hotchkiss Brain Institute, Libin Cardiovascular Institute, University of Calgary, Calgary, AB, Canada
- 2Department of Pharmacology and Toxicology, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt
- 3Department of Physiology and Pharmacology, Snyder Institute for Chronic Diseases, Inflammation Research Network, University of Calgary, Calgary, AB, Canada
The spatial and temporal regulation of cellular calcium signals is modulated via two main Ca2+ entry routes. Voltage-gated Ca2+ channels (VGCC) and Ca2+-release activated channels (CRAC) enable Ca2+ flow into electrically excitable and non-excitable cells, respectively. VGCC are well characterized transducers of electrical activity that allow Ca2+ signaling into the cell in response to action potentials or subthreshold depolarizing stimuli. The identification of STromal Interaction Molecule (STIM) and Orai proteins has provided significant insights into the understanding of CRAC function and regulation. This review will summarize the current state of knowledge of STIM-Orai interaction and their contribution to cellular Ca2+ handling mechanisms. We will then discuss the bidirectional actions of STIM1 on VGCC and CRAC. In contrast to the stimulatory role of STIM1 on Orai channel activity that facilitates Ca2+ entry, recent reports indicated the ability of STIM1 to suppress VGCC activity. This new concept changes our traditional understanding of Ca2+ handling mechanisms and highlights the existence of dynamically regulated signaling complexes of surface expressed ion channels and intracellular store membrane-embedded Ca2+ sensors. Overall, STIM1 is emerging as a new class of regulatory proteins that fine-tunes Ca2+ entry in response to endoplasmic/sarcoplasmic reticulum stress.
Ca2+ Handling Mechanisms
The second messenger calcium (Ca2+) plays a crucial role in a broad range of eukaryotic cellular functions. Regulation of its intracellular concentration ([Ca2+]i) represents a major determinant that controls signal transduction pathways such as secretion, excitation/contraction coupling, motility, transcription, growth, cell division or apoptosis (Berridge et al., 2003; Catterall, 2011). Precise neural circuit formation and control of neuronal excitability necessitate the tight handling of Ca2+. Further, Ca2+ signals are crucial for synaptic transmission and plasticity (Berridge, 1998). In addition, pathophysiological neural insults such as cerebral ischemia can evoke an unwanted rise in Ca2+ leading to Ca2+ overload toxicity and neuronal cell death (Berridge, 1998; Arundine and Tymianski, 2004).
Strict handling of intracellular Ca2+ is necessary to maintain optimized cellular functions. In general, Ca2+ signals are modified by the control of Ca2+ flux in (entry) and out (efflux) of the cell through plasma membrane (PM) channels and transporters that facilitate Ca2+ movement between the extracellular milieu and cytoplasm across a Ca2+ concentration gradient (Berridge et al., 2003). In addition, integral proteins localized in the membranes of intracellular stores allow Ca2+ release (to the cytoplasm) and reuptake (into the Ca2+ store). The main intracellular Ca2+ stores are the endoplasmic reticulum (ER) and sarcoplasmic reticulum (SR). A number of regulatory mechanisms have been proposed to mediate the cellular influx, efflux, release and reuptake of Ca2+, thus achieving Ca2+ homeostasis within the cell (Berridge et al., 2003; Stutzmann and Mattson, 2011). Accumulated data suggest that this homeostasis involves the concerted action of Ca2+ entry channels at the PM and Ca2+ release channels in intracellular ER/SR stores (Figure 1).
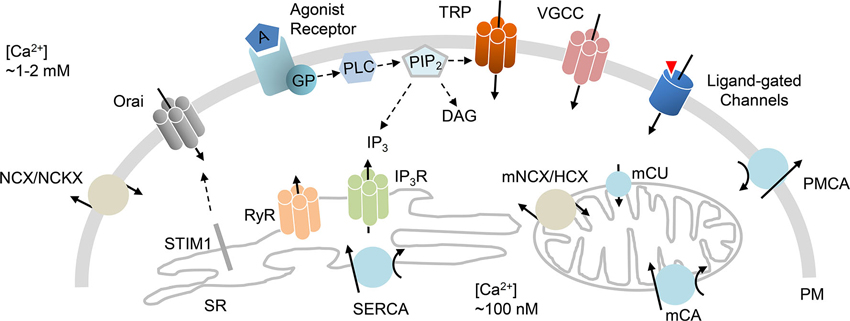
Figure 1. Ca2+ handling mechanisms. Schematic diagram that highlights ion channels and transporters directly implicated in Ca2+ homeostasis. Influx of Ca2+ is primarily mediated by VGCC, receptor-mediated Ca2+ entry, transient receptor potential channels (TRP), ligand-gated channels, and store-operated Orai channels that are activated by STIM1 protein. Efflux of Ca2+ is achieved by PM Ca2+ ATPase (PMCA), Na+/Ca2+ exchanger (NCX) or Na+/Ca2+/K+ exchanger (NCKX). Release of Ca2+ from the ER/SR is mediated through IP3 (IP3R) or ryanodine (RyR) receptors. The reuptake of Ca2+ into the ER/SR is primarily mediated by sarcoplasmic/ER Ca2+ ATPase (SERCA). Mitochondrial Ca2+ handling incorporates mitochondrial Ca2+ uniporter (mCU), Ca2+ ATPase (mCA) or Na+/Ca2+ or H+/Ca2+ exchangers (mNCX, mHCX). GP (G proteins); PIP2 (phosphatidylinositol 4,5-bisphosphate); PLC (phospholipase C).
Over the past decades, it has been recognized that Ca2+ influx into neuronal subcellular compartments (e.g., dendrites, somata, spines, axons) is mediated by two principal means of Ca2+ entry. These routes are voltage-gated Ca2+ channels (VGCC) and ionotropic neurotransmitter receptors (Berridge, 1998; Catterall, 2011), both routes elicit crucial rises in cytosolic Ca2+ in response to different stimuli. VGCC are widely expressed in excitable cells and they trigger Ca2+ influx over specific ranges of membrane potentials. Activation of VGCC generates fast neurotransmission at nerve terminals (Bezprozvanny et al., 1995), or excitation-contraction coupling in cardiac, skeletal and smooth muscle cells (Catterall, 2011; Tuluc and Flucher, 2011; Navedo and Santana, 2013). Neurons along with other cell types display an alternative Ca2+ entry mode that is coupled to intracellular Ca2+ stores (Gemes et al., 2011). This alternative type of entry, known as capacitative calcium entry, is triggered upon the depletion of Ca2+ stores to facilitate store-operated Ca2+ entry (SOCE). The latter, SOCE, would in turn replenish the intracellular ER/SR stores (Soboloff et al., 2012). Extensive work on this route of calcium influx has established its functional importance in neurons and its ability to supplement cytosolic Ca2+ required for neurotransmission (Berna-Erro et al., 2009; Gemes et al., 2011).
Store-Operated Ca2+ Entry (SOCE): a Still-Developing Story
About three decades ago, Putney first described the concept of capacitative Ca2+ entry (Putney, 1986). According to this concept, the concerted control of both Ca2+ influx and Ca2+ release from intracellular stores orchestrates Ca2+ homeostasis. In other words, Ca2+ influx is modulated by the capacity of the cell to hold Ca2+. Several studies showed that stimulus-evoked ER/SR depletion can trigger subsequent influx of extracellular Ca2+ into the cytoplasm as a means to replenish Ca2+ in intracellular stores (Takemura and Putney, 1989; Muallem et al., 1990). These findings led Putney’s model to be revised by indicating that the activation of PM Ca2+ channels was a direct consequence of ER/SR depletion (Putney, 1990). Entry of extracellular Ca2+ upon store depletion was later suggested to be mediated by Ca2+-release activated channels (CRAC) in a process referred to as SOCE (Hoth and Penner, 1992; Patterson et al., 1999).
The CRAC was first described in 1992 (Hoth and Penner, 1992), but its mechanism of activation was not revealed until 2005 when Cahalan and coworkers identified the STromal Interaction Molecule 1 (STIM1) as the intracellular CRAC component that acts as the Ca2+ sensor. Upon store depletion, this sensor STIM1 aggregates and activates the PM Ca2+ channel that is necessary for SOCE (Roos et al., 2005; Zhang et al., 2005). First physiological description of STIM1 as a key component of CRAC was in Drosophila S2 cells in which SOCE is the predominant Ca2+ entry mechanism. Using RNA interference screens of candidate genes, they reported that Stim loss altered SOCE (Roos et al., 2005; Zhang et al., 2005). It was 1 year after the intracellular STIM1 was discovered that the PM component of CRAC was identified. The Orai gene, named after the mythological keepers of heaven’s gate, was determined as a result of genetic mapping of mutations linked to impaired lymphocyte function (Zhang et al., 2006). The SOCE mechanism was then revised to involve the two key players: (1) STIM, a transmembrane Ca2+ sensor protein that is primarily embedded into the SR/ER membrane; and (2) Orai, an integral PM protein being the pore-forming subunit of the CRAC channel (Figures 1, 2A; Soboloff et al., 2012).
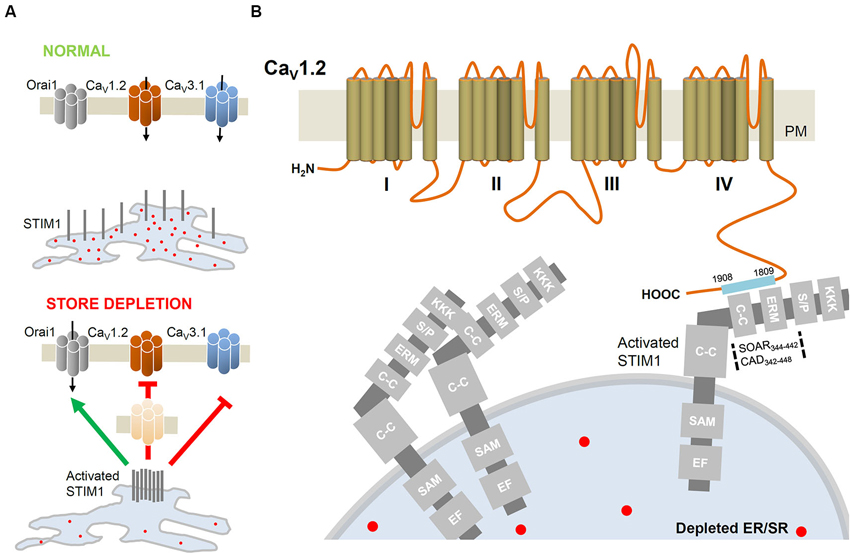
Figure 2. STIM1-mediated regulation of VGCC. (A) In basal conditions, the intraluminal EF-hand of STIM1 is occupied by Ca2+. Upon ER/SR depletion, STIM1 molecules aggregate closer to PM to activate Orai channels but inhibit CaV1.2 (L-type; Park et al., 2010; Wang et al., 2010) and CaV3.1 (T-type; Nguyen et al., 2013) channels. STIM1-induced internalization of CaV1.2 removes functional channels from the cell surface. (B) Detailed interacting sites of STIM1 and the C-terminus of CaV1.2. The STIM1 segments CAD (342–448; Park et al., 2010) or SOAR (344–442; Wang et al., 2010) interacts with the C-terminus (1809–1908; Park et al., 2010) of CaV1.2. C-C, coiled-coil domain; EF, EF hand motif; ERM, Ezrin-Radixin-Moesin domain; KKK, lysine rich domain; SAM, sterile-α motif; S/P, serine/proline rich domain.
Once the key genes governing SOCE were identified, the interplay between STIM1 and Orai was extensively examined. Investigators reported that Orai protein monomers multimerize to form a Ca2+ channel whose activity is triggered by interaction with STIM1 (Penna et al., 2008). Further, STIM-Orai Activating Region (SOAR) and CRAC Activation Domain (CAD) were identified as active STIM1 sites necessary to trigger the CRAC current (Park et al., 2009; Yuan et al., 2009). In addition, high-resolution crystal structures of the CAD and the N-terminal region of STIM1 as well as the full-length Orai channel were recently characterized (Stathopulos et al., 2008; Hou et al., 2012; Yang et al., 2012). These latter discoveries represent major landmarks towards the elucidation of the conformational changes of STIM1-Orai complexes as well as the possible interactions with key proteins involved in Ca2+ handling mechanisms.
STromal Interaction Molecule (STIM): the Ca2+ Sensor Controlling Ca2+ Entry
STIM1 was first reported as a growth modulator (Oritani and Kincade, 1996), and was not implicated in cellular Ca2+ dynamics until 2005 (Roos et al., 2005). It became evident that STIM1 proteins function as Ca2+-sensing molecules, resident in the membranes of the intracellular Ca2+ stores (ER/SR), that regulate SOCE (Soboloff et al., 2012). STIM1 molecule is ubiquitously expressed and is involved in a wide range of cellular functions. It is an essential component of SOCE in lymphocytes (Liou et al., 2005; Zhang et al., 2005), platelets (Varga-Szabo et al., 2008), neurons (Berna-Erro et al., 2009; Venkiteswaran and Hasan, 2009; Gemes et al., 2011), skeletal muscle cells (Stiber et al., 2008), and cardiomyocytes (Touchberry et al., 2011). When Ca2+ stores are depleted, STIM1 molecules aggregate and activate the Orai channels to facilitate Ca2+ influx (Soboloff et al., 2012).
The STIM2 was identified as a homologue of STIM1. Both isoforms are ubiquitously expressed in vertebrates and the expression level of STIM1 is generally higher than STIM2 in most tissues (Williams et al., 2001; Oh-Hora et al., 2008). Notably, STIM2 is primarily found in dendritic cells (Bandyopadhyay et al., 2011) and the brain (Williams et al., 2001; Berna-Erro et al., 2009). The two proteins conserve high homology region, but their C- and N-termini display substantial divergence (Soboloff et al., 2012). Interestingly, structural aspects of the two proteins engender subtle differences that are associated with significant functional implications. Although primarily localized to the intracellular ER/SR membranes, approximately 10% of STIM1 proteins are integrated in the PM. This is in stark difference to STIM2 which, due to an ER-retention sequence in its C-terminus, is exclusively localized in the ER membrane (Soboloff et al., 2006; Saitoh et al., 2011). In addition, STIM1 is a stronger activator of Orai channels when compared to STIM2 (Bird et al., 2009) and STIM2 is more sensitive to small changes in [Ca2+]. These properties of STIM2 and its poor coupling to Orai channels could in theory be essential to limit uncontrolled SOCE (Soboloff et al., 2012). In the following section, our discussion will focus on how STIM1 modulates different Ca2+ influx routes.
STromal Interaction Molecule 1 (STIM1) and Ca2+ Entry
There are two main stimulus modalities that elicit Ca2+ entry: the membrane depolarization in excitable cells versus the ER/SR calcium depletion in non-excitable cells. The VGCC differ from CRAC in being activated by depolarization in response to action potentials or subthreshold stimuli. The STIM1/Orai CRAC complex is activated in response to ER/SR calcium depletion. Noteworthy, both channels are expressed in excitable and non-excitable cells (Kotturi and Jefferies, 2005; Lyfenko and Dirksen, 2008; Stiber et al., 2008). However, VGCC predominate in excitable cells (e.g., neurons, cardiomyocytes, smooth muscle cells) while CRAC currents are prevalent in non-excitable cells (e.g., T-lymphocytes). Both Ca2+ channels have received significant attention. In this review we will focus our discussion on the mechanisms through which STIM1 interaction with CRAC and VGCC modulate Ca2+ influx.
STromal Interaction Molecule 1 (STIM1) Stimulates Ca2+-Release Activated Channels (CRAC)
In a resting cell, STIM1 exhibits a tubular distribution throughout the ER/SR (Roos et al., 2005). The N-terminus of STIM1 resides inside the ER/SR lumen and possesses an EF-hand that binds Ca2+ with low affinity (200–600 nM), and thus acts as a Ca2+ sensor. When the lumen of the ER/SR is full of Ca2+, STIM1 EF-hands are saturated with Ca2+ ions. In contrast, upon stores depletion, STIM1 molecules aggregate into oligomers (Soboloff et al., 2012) and translocate to sites where the ER/SR membrane is closer to the PM. In these microdomains, STIM1 oligomers form clusters which interact with and activate Orai channels (Figure 2A; Zhang et al., 2006; Penna et al., 2008; Soboloff et al., 2012). STIM1 activates Orai1 by the region identified as STIM1-Orai activating region (SOAR) or CAD to facilitate Ca2+ influx. Noteworthy, the small portion of the STIM1 pool integrated into the PM is not required for CRAC channel activation (Park et al., 2009; Yuan et al., 2009).
STromal Interaction Molecule 1 (STIM1) Inhibits Voltage-Gated Ca2+ Channels
SOCE upon ER/SR depletion has been extensively studied since first proposed. Recently, another mechanism of calcium influx was found to be suppressed by store depletion (Park et al., 2010; Wang et al., 2010). This led to the novel term “store-inhibited channels (SIC)” (Moreno and Vaca, 2011); in contrast to store-operated channels that activate upon Ca2+ store depletion (Soboloff et al., 2012). The first class of channels identified to be inhibited by store depletion is the voltage-gated Ca2+ channel.
STromal Interaction Molecule 1 (STIM1) inhibits CaV1.2 channels
The predominant expression and function of CRAC in non-excitable cells is well reported. The key components of CRAC, STIM and Orai, are also expressed in excitable cells (Stiber et al., 2008; Berna-Erro et al., 2009; Venkiteswaran and Hasan, 2009; Gemes et al., 2011; Touchberry et al., 2011) where VGCC predominate as the main route of Ca2+ entry in response to depolarizing stimuli. One subtype of VGCC, the CaV1.2 channel, is ubiquitously expressed in neuronal, cardiac and smooth muscle cells. The CaV1.2 L-type channel is involved in specific cellular functions and has been long considered as an important target for therapeutic agents such as antiarrhythmic and antihypertensive drugs (Catterall, 2011). In order to understand the coordinated interaction between CRAC and the L-type calcium channel, two studies have examined the role of STIM1 in regulating CaV1.2 and Orai1 function. Interestingly, by employing a divergent array of approaches, these studies reported an inhibitory interaction between STIM1 and CaV1.2 (Figures 2A, B). This functional crosstalk may explain the predominance of either CRAC or VGCC activity in different tissue types (Park et al., 2010; Wang et al., 2010).
Using excitable cortical neurons and vascular smooth muscle cells, Park et al. (2010) and Wang et al. (2010) assessed VGCC and CRAC functions by monitoring cytoplasmic Ca2+. Unexpectedly, depletion of ER/SR stores attenuated depolarization-induced CaV1.2 activity. Further, depolarization of non-excitable STIM1-rich T-lymphocytes could not evoke a rise in [Ca2+]i. The modulatory interaction between STIM1 and CaV1.2 was emphasized by the observations that: (1) STIM1 overexpression attenuated CaV1.2 activity; while (2) CaV1.2-mediated responses were enhanced when STIM1 function or expression was impaired. Direct interaction between STIM1 and CaV1.2 proteins was ascertained and a set of experiments, using truncated forms of STIM1, documented that SOAR domain (STIM-Orai activating region, 344–442; Wang et al., 2010) directly interacts with the C-terminus of CaV1.2 α1 subunit (1809–1908; Park et al., 2010). Co-immunoprecipitation analysis confirmed the STIM1-CaV1.2 interaction and functional studies revealed that the SOAR domain was necessary and sufficient to suppress CaV1.2 current (Figure 2B). A slower inhibitory interaction was further suggested by Park and coworkers, in which the surface expression of CaV1.2 decreased as a consequence of long-term internalization of the channel from the PM (Figure 2A; Park et al., 2010). Despite having the same core conclusion, the two reports highlight different perspectives. While Park and coworkers proposed an inhibitory mechanism that attenuates channel expression, Wang et al. found a potential role for Orai1 in the STIM1-CaV1.2 inhibitory interaction. Preventing STIM1 expression alone did not abolish CaV1.2 channel suppression while the simultaneous inhibition of both STIM1 and Orai1 was necessary to mask CaV1.2 inhibition by store depletion. In summary, the two groups reported for the first time that STIM1 effectively attenuates CaV1.2 activity.
The description of Ca2+ conductances inhibited upon store depletion has major implications for Ca2+ signaling in excitable and non-excitable cells. In excitable neuronal cells, VGCC are expressed at higher level than CRAC components and are the predominant Ca2+ influx route. In contrast, non-excitable cells display reduced VGCC expression and CRAC represents the main Ca2+ entry pathway due to the high expression of STIM1 and Orai (Liou et al., 2005; Zhang et al., 2005; Park et al., 2010). While STIM1 interacts with Orai to facilitate Ca2+ influx, a new modulatory role for STIM1 has emerged through which it inhibits Ca2+ entry mediated by VGCC. Noteworthy, the ability of STIM1 to reciprocally regulate Orai and CaV1.2 would imply that the mode of action of STIM1 is tissue specific. In other words, STIM1 would typically stimulate CRAC in non-excitable cells and inhibit VGCC in excitable cells (Park et al., 2010; Wang et al., 2010; Moreno and Vaca, 2011). This reciprocal modulation of Ca2+ entry by STIM1 seems to play a critical role in Ca2+ homeostasis by fine-tuning Ca2+ entry in cells that simultaneously express both channel types. By providing this missing piece of evidence, these studies resolved the predominant function of one channel over the other despite their co-expression in excitable and non-excitable tissues. That being said, whether other Ca2+ channels exhibit analogous or distinct modulation by STIM1 remains unknown.
STromal Interaction Molecule 1 (STIM1) inhibits T-type Ca2+ channels
Recent published experiments revealed that CaV1.2 channel is not the only voltage-gated Ca2+ channel suppressed by STIM1. Interestingly, a study by Nguyen et al. (2013) showed that STIM1 attenuates the activity of T-type Ca2+ channel. Using cardiomyocyte-derived HL-1 cells, an inhibitory association between STIM1 and CaV3.1 was reported and was suggested to limit excessive Ca2+ entry. Pathological Ca2+ influx in cardiac myocytes can trigger Ca2+ overload in the SR and cardiac arrhythmias (Sedej et al., 2010), and STIM1-dependent modulation of CaV3.1 could, in theory, restrain such unwanted entry. Indeed, the authors showed that knocking down STIM1 increases T-type Ca2+ current density. They further suggested that STIM1 may regulate the expression of T-type α1 subunits as knocking down STIM1 augmented the surface expression of CaV3.1 channel. Whether CaV3.1 trafficking or stability at the PM is implicated remains unknown and requires further investigation. Intriguingly, these findings represent a novel regulatory pathway for Ca2+ handling in cardiac myocytes. This regulation may be implicated in the modulation of rhythmicity and excitability of native cardiac pacemaker cells where T-type Ca2+ channel expression and function is evident (Mangoni et al., 2006; Nguyen et al., 2013).
Conclusions
The interaction of STIM1 with CRAC or VGCC extends our understanding of the role of STIM1 in cellular Ca2+ handling. Recent published work (Park et al., 2010; Wang et al., 2010; Nguyen et al., 2013) described a novel regulatory function for STIM1. Distinct from its role as an ER/SR Ca2+ sensor to facilitate Ca2+ entry and replenish the stores, STIM1 suppresses the activity of voltage-gated Ca2+ channel in excitable cells. This inhibitory association prevents excessive cellular Ca2+ influx by mechanisms including direct protein-protein interaction and reduced VGCC surface expression (Park et al., 2010; Wang et al., 2010; Nguyen et al., 2013). This novel concept raises numerous questions that pertain to the dynamic regulation of cellular Ca2+ in health and disease. Since STIM1 bi-directionally regulates VGCC and CRAC, SR/ER stress may lead to pathologies related to downstream Ca2+-dependent pathways. In fact, mutations in STIM1 elicit severe immunodeficiency syndromes associated with compromised SOCE (Feske et al., 2010). Impaired STIM-dependent regulation of VGCC and subsequent alterations in Ca2+ entry remain to be investigated; as it could be linked to diseases such as epilepsy, cardiac arrhythmia or hypertension. Finally, it will be fundamental to elucidate the cooperated interaction of PM Ca2+ channels and their associated subunits and how those signaling complexes respond to ER/SR stress through Ca2+ sensors such as STIM1 proteins.
Author Contributions
Osama F. Harraz and Christophe Altier wrote the manuscript. Osama F. Harraz produced the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in the Altier lab is supported by the Heart and Stroke Foundation of Canada and the Canadian Institutes of Health Research (CIHR). Christophe Altier holds a Canada Research Chair (Tier2). Osama F. Harraz is a Vanier Scholar (CIHR) and is supported by salary studentships from Alberta Innovates (AIHS) and Achievers in Medical Sciences (AIMS).
References
Arundine, M., and Tymianski, M. (2004). Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 61, 657–668. doi: 10.1007/s00018-003-3319-x
Bandyopadhyay, B. C., Pingle, S. C., and Ahern, G. P. (2011). Store-operated Ca(2)+ signaling in dendritic cells occurs independently of STIM1. J. Leukoc. Biol. 89, 57–62. doi: 10.1189/jlb.0610381
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2, ra67. doi: 10.1126/scisignal.2000522
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Berridge, M. J. (1998). Neuronal calcium signaling. Neuron 21, 13–26. doi: 10.1016/s0896-6273(00)80510-3
Bezprozvanny, I., Scheller, R. H., and Tsien, R. W. (1995). Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature 378, 623–626. doi: 10.1038/378623a0
Bird, G. S., Hwang, S. Y., Smyth, J. T., Fukushima, M., Boyles, R. R., and Putney, J. W. Jr. (2009). STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 19, 1724–1729. doi: 10.1016/j.cub.2009.08.022
Catterall, W. A. (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3:a003947. doi: 10.1101/cshperspect.a003947
Feske, S., Picard, C., and Fischer, A. (2010). Immunodeficiency due to mutations in ORAI1 and STIM1. Clin. Immunol. 135, 169–182. doi: 10.1016/j.clim.2010.01.011
Gemes, G., Bangaru, M. L., Wu, H. E., Tang, Q., Weihrauch, D., Koopmeiners, A. S., et al. (2011). Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J. Neurosci. 31, 3536–3549. doi: 10.1523/jneurosci.5053-10.2011
Hoth, M., and Penner, R. (1992). Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356. doi: 10.1038/355353a0
Hou, X., Pedi, L., Diver, M. M., and Long, S. B. (2012). Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313. doi: 10.1126/science.1228757
Kotturi, M. F., and Jefferies, W. A. (2005). Molecular characterization of L-type calcium channel splice variants expressed in human T lymphocytes. Mol. Immunol. 42, 1461–1474. doi: 10.1016/j.molimm.2005.01.014
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi: 10.1016/j.cub.2005.05.055
Lyfenko, A. D., and Dirksen, R. T. (2008). Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J. Physiol. 586(Pt. 20), 4815–4824. doi: 10.1113/jphysiol.2008.160481
Mangoni, M. E., Traboulsie, A., Leoni, A. L., Couette, B., Marger, L., Le Quang, K., et al. (2006). Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 98, 1422–1430. doi: 10.1161/01.res.0000225862.14314.49
Moreno, C., and Vaca, L. (2011). SOC and now also SIC: store-operated and store-inhibited channels. IUBMB Life 63, 856–863. doi: 10.1002/iub.547
Muallem, S., Khademazad, M., and Sachs, G. (1990). The route of Ca2+ entry during reloading of the intracellular Ca2+ pool in pancreatic acini. J. Biol. Chem. 265, 2011–2016.
Navedo, M. F., and Santana, L. F. (2013). CaV1.2 sparklets in heart and vascular smooth muscle. J. Mol. Cell. Cardiol. 58, 67–76. doi: 10.1016/j.yjmcc.2012.11.018
Nguyen, N., Biet, M., Simard, E., Beliveau, E., Francoeur, N., Guillemette, G., et al. (2013). STIM1 participates in the contractile rhythmicity of HL-1 cells by moderating T-type Ca(2+) channel activity. Biochim. Biophys. Acta 1833, 1294–1303. doi: 10.1016/j.bbamcr.2013.02.027
Oh-Hora, M., Yamashita, M., Hogan, P. G., Sharma, S., Lamperti, E., Chung, W., et al. (2008). Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 9, 432–443. doi: 10.1038/ni1574
Oritani, K., and Kincade, P. W. (1996). Identification of stromal cell products that interact with pre-B cells. J. Cell Biol. 134, 771–782. doi: 10.1083/jcb.134.3.771
Park, C. Y., Hoover, P. J., Mullins, F. M., Bachhawat, P., Covington, E. D., Raunser, S., et al. (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. doi: 10.1016/j.cell.2009.02.014
Park, C. Y., Shcheglovitov, A., and Dolmetsch, R. (2010). The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105. doi: 10.1126/science.1191027
Patterson, R. L., van Rossum, D. B., and Gill, D. L. (1999). Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell 98, 487–499. doi: 10.1016/s0092-8674(00)81977-7
Penna, A., Demuro, A., Yeromin, A. V., Zhang, S. L., Safrina, O., Parker, I., et al. (2008). The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456, 116–120. doi: 10.1038/nature07338
Putney, J. W. Jr. (1986). A model for receptor-regulated calcium entry. Cell Calcium 7, 1–12. doi: 10.1016/0143-4160(86)90026-6
Putney, J. W. Jr. (1990). Capacitative calcium entry revisited. Cell Calcium 11, 611–624. doi: 10.1016/0143-4160(90)90016-n
Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445. doi: 10.1083/jcb.200502019
Saitoh, N., Oritani, K., Saito, K., Yokota, T., Ichii, M., Sudo, T., et al. (2011). Identification of functional domains and novel binding partners of STIM proteins. J. Cell. Biochem. 112, 147–156. doi: 10.1002/jcb.22910
Sedej, S., Heinzel, F. R., Walther, S., Dybkova, N., Wakula, P., Groborz, J., et al. (2010). Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc. Res. 87, 50–59. doi: 10.1093/cvr/cvq007
Soboloff, J., Rothberg, B. S., Madesh, M., and Gill, D. L. (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565. doi: 10.1038/nrm3414
Soboloff, J., Spassova, M. A., Hewavitharana, T., He, L. P., Xu, W., Johnstone, L. S., et al. (2006). STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr. Biol. 16, 1465–1470. doi: 10.1016/j.cub.2006.05.051
Stathopulos, P. B., Zheng, L., Li, G. Y., Plevin, M. J., and Ikura, M. (2008). Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 135, 110–122. doi: 10.1016/j.cell.2008.08.006
Stiber, J., Hawkins, A., Zhang, Z. S., Wang, S., Burch, J., Graham, V., et al. (2008). STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 10, 688–697. doi: 10.1038/ncb1731
Stutzmann, G. E., and Mattson, M. P. (2011). Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol. Rev. 63, 700–727. doi: 10.1124/pr.110.003814
Takemura, H., and Putney, J. W. Jr. (1989). Capacitative calcium entry in parotid acinar cells. Biochem. J. 258, 409–412.
Touchberry, C. D., Elmore, C. J., Nguyen, T. M., Andresen, J. J., Zhao, X., Orange, M., et al. (2011). Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium. Biochem. Biophys. Res. Commun. 416, 45–50. doi: 10.1016/j.bbrc.2011.10.133
Tuluc, P., and Flucher, B. E. (2011). Divergent biophysical properties, gating mechanisms, and possible functions of the two skeletal muscle Ca(V)1.1 calcium channel splice variants. J. Muscle Res. Cell Motil. 32, 249–256. doi: 10.1007/s10974-011-9270-9
Varga-Szabo, D., Braun, A., Kleinschnitz, C., Bender, M., Pleines, I., Pham, M., et al. (2008). The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J. Exp. Med. 205, 1583–1591. doi: 10.1084/jem.20080302
Venkiteswaran, G., and Hasan, G. (2009). Intracellular Ca2+ signaling and store-operated Ca2+ entry are required in Drosophila neurons for flight. Proc. Natl. Acad. Sci. U S A 106, 10326–10331. doi: 10.1073/pnas.0902982106
Wang, Y., Deng, X., Mancarella, S., Hendron, E., Eguchi, S., Soboloff, J., et al. (2010). The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109. doi: 10.1126/science.1191086
Williams, R. T., Manji, S. S., Parker, N. J., Hancock, M. S., Van, S. L., Eid, J. P., et al. (2001). Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem. J. 357(Pt. 3), 673–685. doi: 10.1042/0264-6021:3570673
Yang, X., Jin, H., Cai, X., Li, S., and Shen, Y. (2012). Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. U S A 109, 5657–5662. doi: 10.1073/pnas.1118947109
Yuan, J. P., Zeng, W., Dorwart, M. R., Choi, Y. J., Worley, P. F., and Muallem, S. (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343. doi: 10.1038/ncb1842
Zhang, S. L., Yeromin, A. V., Zhang, X. H., Yu, Y., Safrina, O., Penna, A., et al. (2006). Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. U S A 103, 9357–9362. doi: 10.1073/pnas.0603161103
Keywords: STIM1, Orai, VGCC, CRAC, L-type, T-type, calcium channels, store-operated Ca2+ entry
Citation: Harraz OF and Altier C (2014) STIM1-mediated bidirectional regulation of Ca2+ entry through voltage-gated calcium channels (VGCC) and calcium-release activated channels (CRAC). Front. Cell. Neurosci. 8:43. doi: 10.3389/fncel.2014.00043
Received: 23 December 2013; Accepted: 29 January 2014;
Published online: 24 February 2014.
Edited by:
Leigh Anne Swayne, University of Victoria, CanadaReviewed by:
Francois Rassendren, Centre National de la Recherche Scientifique, FranceJ. David Spafford, University of Waterloo, Canada
Copyright © 2014 Harraz and Altier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Altier, Department of Physiology and Pharmacology, Snyder Institute for Chronic Diseases, Inflammation Research Network, University of Calgary, 3330 Hospital Dr. NW, Calgary, AB T2N-4N1, Canada e-mail:YWx0aWVyQHVjYWxnYXJ5LmNh