 Ben Aviner
Ben Aviner Gideon Gradwohl
Gideon Gradwohl Merav Mor Aviner1
Merav Mor Aviner1 Yoram Grossman
Yoram Grossman- 1Department of Physiology and Neurobiology, Ben Gurion University of the Negev, Beer Sheva, Israel
- 2Department of Physics, Jerusalem College of Technology, Jerusalem, Israel
Professional deep sea divers experience motor and cognitive impairment, known as High Pressure Neurological Syndrome (HPNS), when exposed to pressures of 100 msw (1.1 MPa) and above, considered to be the result of synaptic transmission alteration. Previous studies have indicated modulation of presynaptic Ca2+ currents at high pressure. We directly measured for the first time pressure effects on the currents of voltage dependent Ca2+ channels (VDCCs) expressed in Xenopus oocytes. Pressure selectivity augmented the current in CaV1.2 and depressed it in CaV3.2 channels. Pressure application also affected the channels' kinetics, such as ƮRise, ƮDecay. Pressure modulation of VDCCs seems to play an important role in generation of HPNS signs and symptoms.
Introduction
HPNS
Every chemical reaction contributing to the continued existence of an organism is a potential target for pressure effects on biological processes. The major neurological problems associated with hyperbaric environments include N2 narcosis (inert-gas narcosis); O2 toxicity, which occurs due to increased oxidative stress (Allen et al., 2009); and HPNS (Halsey, 1982; Talpalar and Grossman, 2006). By controlling partial-pressures of absorbed tissue gases while under pressure, these neurological problems, excluding HPNS, can be alleviated and even eliminated, leading to the notion that HPNS occurs due to effects of pressure per se (Abraini, 1997; Bennett, 1997). Deep sea divers (approximately >100 m), as well as animals exposed to hyperbaric pressure (HP), may experience HPNS, which in humans includes dizziness, nausea, tremors, vision, and auditory disturbances, decrements in locomotor activity (Tarasiuk and Grossman, 1990; Darbin et al., 2000) and intellectual performance (Logue et al., 1986; Vaernes et al., 1988; Overman et al., 1989; Abraini, 1997; Steevens et al., 1999), confusion, changes in EEG and sleep disorders (Rostain et al., 1997), myoclonus (Darbin et al., 2000), convulsions, and loss of consciousness.
Synaptic transmission
Changes in synaptic transmission properties is a possible explanation for this constellation of signs and symptoms (for review see Daniels and Grossman, 2003). Indeed, it has previously been shown that synaptic transmission is suppressed by HP whether in vertebrate CNS synapse (Parmentier et al., 1981; Schleifstein-Attias et al., 1994), squid giant synapse (Parmentier et al., 1981), or crustacean neuromuscular junction (NMJ) (Campenot, 1975; Grossman and Kendig, 1990), either in excitatory (Grossman and Kendig, 1988; Golan et al., 1994) or inhibitory (Golan et al., 1994) synapses. Postsynaptic reduction in receptor sensitivity, observed in glycine receptors (Shelton et al., 1993), will exacerbate pressure-induced suppression in specific synapses (Daniels and Grossman, 2003). Various mechanisms for HP effects on synaptic transmission have been suggested over the past few decades, including modulation of ionotropic receptors activity (Heinemann et al., 1987; Shelton et al., 1993), decreased action potential (AP) amplitude (Aviner et al., 2013) and slowed kinetics (Grossman and Kendig, 1986; Etzion and Grossman, 1999), generally observed depression of neurotransmitter release (Parmentier et al., 1981; Ashford et al., 1982; Gilman et al., 1987; Etzion et al., 2008), decreased vesicle fusion (Ashford et al., 1982; Heinemann et al., 1987), and the reduction of Ca2+ currents (Talpalar and Grossman, 2003; Aviner et al., 2013). HP also causes a decrease in the frequency of spontaneous miniature excitatory postsynaptic potentials (Ashford et al., 1982), which may also suggest that [Ca2+]i could be involved in manifesting these HP effects. Indeed, it was also found that HP mimics the effects of low [Ca2+]o (Grossman and Kendig, 1990; Etzion et al., 2008), and in contrast, high [Ca2+]o can antagonize HP effects (Golan and Grossman, 1992; Talpalar et al., 2010; Aviner et al., 2013). Taken together, we may postulate that one of the major mechanisms by which HP suppresses synaptic transmission is a depression of Ca2+ influx into the presynaptic terminal through voltage-dependent Ca2+ channels (VDCC), which is the natural trigger of this process. Indeed, Grossman and his colleagues have presented indirect (Grossman and Kendig, 1990; Grossman et al., 1991; Etzion and Grossman, 2000) and semi-direct (Aviner et al., 2013) evidence for this HP effect on voltage-dependent Ca2+ currents.
Pressure- and voltage-dependent ca2+ channels
Various VDCC types exist, characterized by their electrophysiological and pharmacological traits (CaV1.1-4, CaV2.1-3, CaV3.1-3) and comprising the α1, α2δ, β, and γ subunits (Meir et al., 1999; Catterall, 2000). The major difference between the channels results from the variation in the α1 subunit, which holds the ion conducting pore, the voltage sensor, the channel gating section, and the known sites of channel regulation by second messengers, drugs, and toxins (Ertel et al., 2000). Evidence has accumulated, mostly indirectly, for HP effects on VDCC (Aviner et al., 2010). Pressure inhibited the brief reversal of swimming direction in the Paramecium, suggesting that an unclassified Ca2+ current that is associated with the reflex is impaired (Otter and Salmon, 1985). Depolarization-dependent Ca2+ influx into rat brain synaptosomes was depressed by pressure (Gilman et al., 1986). Membrane retrieval, another Ca2+-dependent process (Vogel et al., 1999; Wu et al., 2009), was also inhibited by pressure (Heidelberger et al., 2002).
Analysis of studies on crustacean neuromuscular synapses that examined the relationship between [Ca2+]o, EPSC amplitude, and facilitation (Grossman and Kendig, 1988, 1990; Golan and Grossman, 1992) suggests that pressure is acting to reduce Ca2+ influx, rather than to affect intracellular removal of Ca2+ or the release process.
To date, no attempt was made to directly measure isolated Ca2+ currents at pressure. The work detailed in this manuscript aims to do so for the first time.
Alteration in the properties of these channels at pressure can significantly influence one's cognition, sensual perception, and ability to control posture and muscle activity in a manifestation that could depict the HPNS.
Methods
Oocytes Extraction and cRNA Injection
Oocytes of a Xenopus laevis mature female frog were surgically extracted from its ovary and treated with 1.5 mg/ml collagenase for 30–60 min in order to remove connecting tissue. Suitable oocytes were sorted out by size, quality, and developmental stage (VI), and kept in NDE96 solution containing (in mM): 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, 2.5 sodium pyruvate; 50 μg/ml gentamycin; 5 HEPES pH 7.5.
cRNAs of the subunits of L or T type Ca2+ channels (LTCC or TTCC, respectively) were synthesized from rabbit cDNA by in vitro transcription with T7 or SP6 Amplicap High-Yield Message Maker Kit (Epicentre Technologies, Madison, WI). Oocytes were then injected with the specific cRNA mix (2.5 ng) encoding for the pertinent subunits to express LTCC or TTCC and were kept in an incubator for 4 days at 18°C in NDE96 solution. The following subunits were used: α1C+β2A+α2δ, comprising the LTCC CaV1.2; and α1H+α2δ, comprising the TTCC CaV3.2, whose gene code numbers are α2δ—M21948, β2—X64297, α1C—X15539, and α1H—AF051946.
Electrophysiological Recordings
Four days post-injection the oocytes were placed in a specially designed bath, and two-electrode voltage clamp (TEVC) experiments with 10 mV increments and 5–10 s interval between −70 and 40 mV were performed inside a compression chamber, utilizing an AXOCLAMP 2B amplifier (Molecular Devices, Axon Instruments, Inc., CA, USA), Master-8-cp Pulse Generator (A.M.P.I.), and AxoScope 9.2 software. While in the chamber, each oocyte was continuously perfused with a Ba2+ solution containing (in mM): 40 Ba(OH)2, 50 NaOH, 2 KOH, and 5 HEPES, titrated to pH 7.5 with methanesulfonic acid. Ba2+ was used as charge carrier, replacing the Ca2+ ions, in order to avoid Ca2+-dependent inactivation and the activation of Ca2+-activated Cl− channels (Cl−Ca), known to be endogenously expressed in oocyte membrane (Miledi and Parker, 1984). The channels also have higher conductance to Ba2+ (Tsien et al., 1987), allowing measurement of minute currents that otherwise would have been unnoticed. The solution, saturated with air at atmospheric pressure, was introduced to the chamber by the use of a high pressure pump (Minipump, LDC Analytical Inc., Riviera Beach, FL, USA) at room temperature (24–25°C), at a rate of 1.5–2 ml/min. Temperature was constantly monitored throughout the experiments by the use of a thermistor submerged in the solution in the vicinity of the oocyte groove. Deviation of only ±0.5°C was allowed from the control temperature for later measurements.
Typical recording traces are shown in Figures 1A–D. For the oocytes expressing CaV1.2, holding potential was −70 mV (Figure 1A). The duration of each depolarizing step was 500 ms, which was preconditioned by a 100 ms hyperpolarizing step to −80 mV in order to release the VDCC from partial inactivation. The latter was also used to calculate and monitor the oocytes' instantaneous input resistance, which was accounted for at each recorded trace separately. For the oocytes expressing CaV3.2 holding potential was −80 mV (Figure 1B). The duration of each depolarizing step was 100 ms, preconditioned by a 150 ms hyperpolarizing step to −90 mV, for similar reasons.
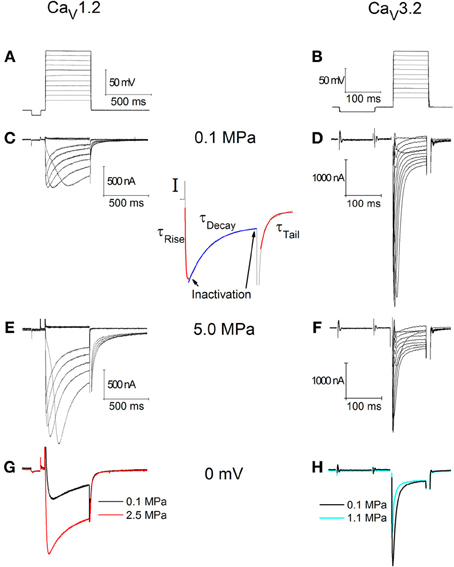
Figure 1. Currents recorded in CaV1.2 (C,E,G) and CaV3.2 (D,F,H) channels. (A) Depolarization steps for (C,E). (B) Depolarization step for (D,F). (C,D) Ba2+ currents at 0.1 MPa. (E,F) Ba2+ currents at 5.0 MPa. Note current increase in (E) at HP and decrease in (F). (G,H) Superimposed single current traces under normo- and hyperbaric conditions, in response to identical depolarization to 0 mV. (I) Indication of the corresponding sections of the currents used to calculate the inactivation and kinetics parameters.
Every series of depolarizing pulses was used to construct an I-V curve and repeated at least three times in order to verify stability of the currents. Recorded traces with voltage fluctuation greater than 2 mV during depolarization were disregarded. We studied HP effects (examples in Figures 1C,D) on I-V curve, maximal currents, activation and inactivation functions, channel kinetics such as time to peak and time constants (Ʈ), and voltage dependency. Maximal currents were measured at the minimal point of the current curve (Figure 1I, left arrow). Inactivation (I/Imax) was measured toward the end of the depolarizing step (Figure 1I, right arrow) in comparison to the measured maximal current (as above). A fit was calculated for each decaying section of the current in every recorded trace (Figure 1I, blue fit) according to a biexponential equation defining two time constants for decay:
For the rising phase and the tail currents a single exponential fit was performed (Figure 1I, red fits). All fits were calculated between the curves' normalized values of 0.1 and 0.9.
Activation volume (ΔV‡) was calculated for time constants of channel activation, inactivation, and deactivation under normobaric and hyperbaric conditions, following the known equation: ΔV‡ = RT(ƏlnƮ/ƏP)T.
Truncated K+ Channel
The current measurement using TEVC method was carried out for long durations and under HP conditions. Since it was our first attempt, verification of the recording stability was needed. As a first approximation, a truncated variant of a Drosophila K+ channel (KCNKØ) missing its carboxy-terminal tail (700 residues) was injected into oocytes (n = 4), causing a shift of their normal resting potential from about −40 to −50 mV to approximately −80 mV (Zilberberg et al., 2000). The membrane potential of these oocytes was then measured for 1–2 h and demonstrated stable resting potential of −80 ± 2 mV (not shown). This result showed that the setup measurements are accurate and do not drift throughout the duration of the experiment.
Ca2+-Activated Cl− Channel Blocker
In an attempt to clarify whether Ba2+ currents measured in the VDCCs over-expressed in the oocyte membrane are subjected to artifacts caused by currents via endogenous channels, we performed a series of experiments in CaV1.2 channels using 9-Anthracene carboxylic acid (9-AC), a pharmacological blocker of the Cl−Ca (Boton et al., 1989). The use of Ca2+ chelators, e.g., BAPTA or EGTA, was not feasible due to their short time-span effectiveness in relation to the duration of our experiments (see Results, Time stability of HP effect). 9-AC was dissolved at 0.2–0.5 M in a 1 M solution of NaOH. This stock solution was then added to the physiological measurement solution to a final 2–4 mM concentration (Boton et al., 1989). Titration to pH 7.5 was then done using Methanesulfonic acid.
Helium Compression
After control measurement taken at 0.1 MPa, compression steps to 0.5, 1.0, 2.5, and 5.0 MPa were performed by compressed helium. Since the cDNA used to express these channels was of rodent origion, which is generally more resistant to HP than human, compressions greater than 1.0 MPa were also performed. Compression was done manually from a tank through a set of valves into the pressure chamber. Compression rate was approximately 0.25–0.5 MPa/min, and never exceeded 1.0 MPa/min. Helium was used instead of air due to its inert quality and the need to avoid known nitrogen narcosis and oxygen toxicity related effects (Dean et al., 2003). All pressure units are absolute.
Statistical Analysis
The full set of parameters was calculated off-line for each recorded trace separately, considering the instantaneous input resistance and leak currents where appropriate, using a dedicated self-designed Matlab software program. The data were exported to Microsoft Excel software, by which averaging and binning were performed. Each oocyte was used as its own control, thus values were normalized to 0.1 MPa. When data from more than one oocyte were pooled, binning was performed relative to the voltage generating the maximal current in the I-V curve (VImax); hence in figures representing these data (Figures 2 to 9C–F) the X-axis title is ΔV. Paired sample t-test was used to analyze the significance of the results.
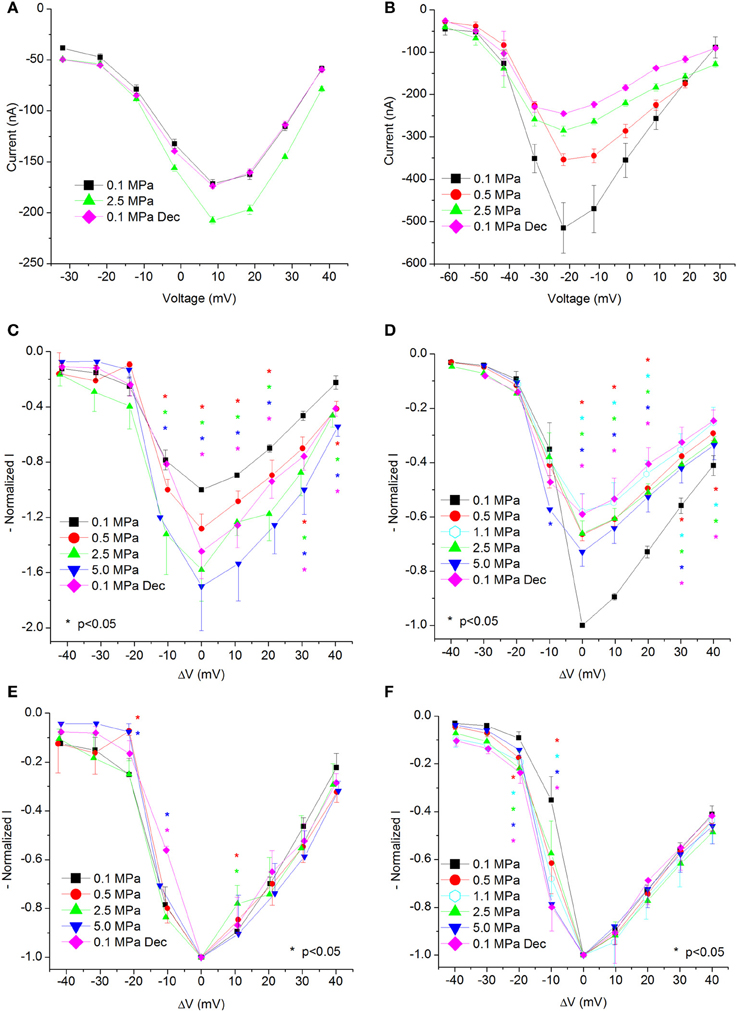
Figure 2. I-V curves of maximal currents. (A,C,E) CaV1.2, (B,D,F) CaV3.2 channels. (A,B) I-V curve of a single oocyte. (C,D) Pooled data from 9 to 17 (C) and 7 to 9 (D) oocytes exposed to 0.5–5.0 MPa pressure (color indicated). Holding potential is adjusted so that 0 indicates the potential at which maximal current is obtained (VImax). (E,F) Normalized-to-max I-V curves. Each curve is normalized to its own maximal value and corresponds to its pertinent curve in (C,D). Holding potential is adjusted as in (C,D). Statistical significance for each point on the curve is indicated by corresponding color asterisks (p < 0.05). Dec indicates decompression.
Results
Increased Current in CaV1.2
Generally, HP was expected to suppress currents of VDCCs (see Introduction). Surprisingly, Ba2+ currents in CaV1.2 were significantly increased in oocytes exposed to HP (0.5–5.0 MPa, see example in Figures 1E,G and 2A) in a dose-dependent manner, throughout the channels' voltage range of activity. Compression to 0.5, 2.5, and 5.0 MPa augmented the maximal currents by 28 ± 10, 58 ± 22, and 70 ± 32%, respectively (Figure 2C, Average ± SEM, p < 0.01, n = 9 − 17). Decompression back to 0.1 MPa only partially reversed the HP effect, leaving the maximal current 44 ± 19% increased. Neither the threshold voltage nor the depolarization (VImax) that generated the largest currents (negative peak in IV curve) was affected.
Normalizing each IV curve under the various pressures to its own maximal (negative) peak (Figure 2E) shows almost identical curves, and therefore suggests that the channel's sensitivity to the membrane voltage did not alter as a result of the compression.
Adiabatic Temperature Change and Time Stability
Elevation of ambient pressure in these experiments was achieved by gaseous (helium) compression; hence an adiabatic temperature rise always occurred (typically 0.5–2°C, but never more than 4°C), which was controlled by the rate of compression. Only measurements within 0.5°C or less of control temperature were taken under consideration. Therefore, it was necessary to wait for the chamber's atmosphere (bath fluid) to cool down (lasting 10–30 min), resulting in 1–2 h long experiments. Figure 3A shows IV curves in an oocyte expressing CaV1.2 recorded at 5.0 MPa (post-compression) while cooling back to control temperature (24°C). As expected, elevated temperature (in addition to the HP) led to an increase in the currents. However, even after the control temperature was regained, the augmented current persisted. This strongly indicates that the temperature fluctuation cannot solely explain the increase in the channel's current and it should reflect a direct effect of HP (as also shown in Figure 2A). In order to verify that, and to rule out the possibility that the surprising augmentation of CaV1.2 current at pressure is only the result of the adiabatic temperature change or a transient HP effect, currents were measured for 1–2 h post-compression (different experiments n = 2, Figure 3B). As can be seen, currents remained stable at HP for long durations after cooling back to control temperature, thus suggesting that the augmentation effect is stable and pressure induced.
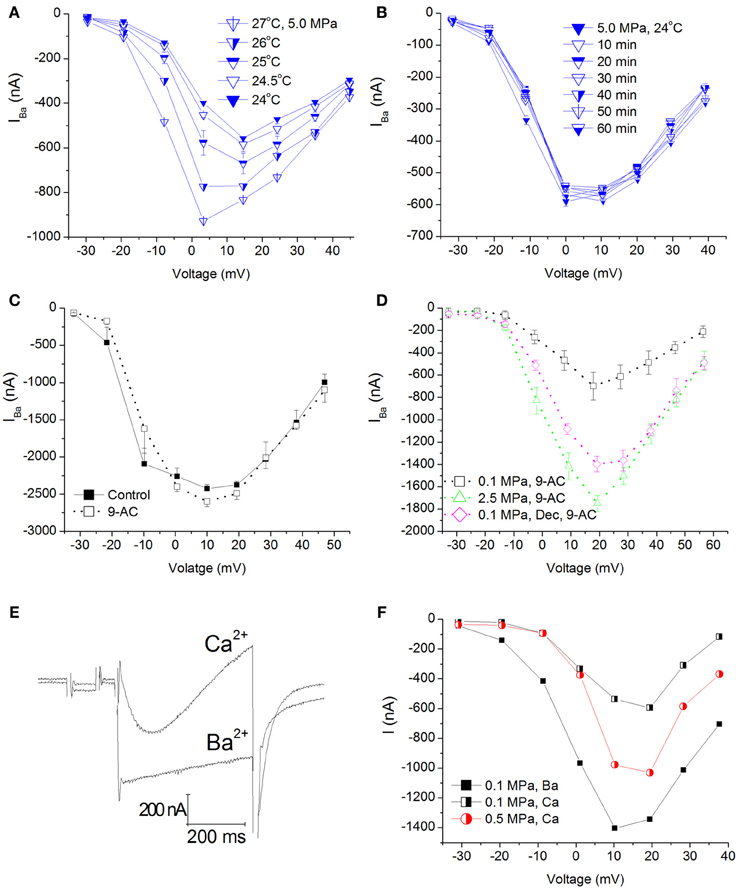
Figure 3. Temperature, time, Cl−Ca channel blocker, and Ca2+ ion control experiments in CaV1.2 channel. (A) I–V curves measured at 5.0 MPa repeatedly while the preparation's adiabatic temperature change is subsiding. (B) I-V curves measured in a different oocyte for 1 h at 5.0 MPa after control temperature was regained. (C) I-V curves measured in a solution containing 9-AC. (D) I-V curves of compression and decompression performed with 9-AC. The expected HP effect (augmentation) is evident. (E) Example of current traces recorded with Ca2+ or Ba2+ ions in the solution at the same depolarization to 0 mV. Note the stronger and faster inactivation with Ca2+. (F) Compression to 0.5 MPa when Ba2+ ions were replaced by Ca2+ ions. Replacement of ions caused a decrease of the currents at 0.1 MPa, but compression to 0.5 MPa still augmented the Ca2+ current in a similar manner to Ba2+ experiments.
Eliminating Ca2+-Activated Cl− Channels' Current Contamination
Xenopus oocytes are known to endogenously express the Ca2+-activated Cl− channels. In order to avoid their activation (leading to inaccurate current measurement), we have used a Ba2+ solution with no added Ca2+ in our experiments. In order to avoid any current flowing through these channels in the unlikely event of their activation, the solution was also Cl− free. Performing a TEVC experiment on naïve oocytes generated maximal currents in the 10–30 nA range (not shown), considered negligible compared to 1000 nA and more measured in oocytes expressing the CaV1.2 channel. Nevertheless, we performed two sets of experiments aimed to verify that the theoretical Cl−Ca current does not play a role in the HP-related effect, by adding 9-AC (a Ca2+-activated Cl− channel blocker, see Methods) to the solution before or after compression (Figure 3C). Neither adding 9-AC pre-compression (Figure 3D) nor 9-AC added post-compression altered the HP-induced augmentation of CaV1.2 current, indicating that the HP effect is real.
Ba2+ −Ca2+ Substitution
Since Ca2+ was substituted for Ba2+ due to the reasons mention in Methods, we wanted to verify that the HP effect is still valid when using the native Ca2+ ion. As expected, changing the bath solution to contain Ca2+ (using the CaV1.2 channel) resulted in a decrease in the measured current and a faster and stronger inactivation (Figures 3E,F, black curves). Application of pressure augmented this current as well (Figure 3F, red curve).
Decreased Current in CaV3.2
Ba+ currents in CaV3.2 were significantly decreased at HP (0.5–5.0 MPa) in a dose-dependent manner (see example in Figures 1F,H and 2B), in contrast to the findings of CaV1.2. Preliminary results showed that the HP effect did not change between 2.5 and 5.0 MPa; therefore another pressure step to 1.1 MPa was added in subsequent experiments in order to reveal the HP effect saturation. Neither the threshold voltage nor the VImax were affected. Compression to 0.5, 1.1, 2.5, and 5.0 MPa depressed the maximal currents by 33 ± 2, 42 ± 6, 34 ± 5, and 27 ± 5%, respectively (Figure 2D, Average ± SEM, p < 0.01, n = 4–9). Decompression to 0.1 MPa failed to recover the current, which remained depressed by 40 ± 7%. Normalizing each IV curve under the various pressure conditions to its own maximal (negative) peak (Figure 2F) reveals an increase of the currents in CaV3.2 at sub-maximal membrane potentials (ΔV −10 mV), but not at VImax and above. We hypothesize that this might occur due to a greater probability for transition to the open state of the channel in this voltage range.
Channel Conductance
Calculating the channel conductance in relation to the membrane potential shows similar results to the general findings in the IV curves (see examples in Figures 4A,B). HP decreased the conductance in the CaV3.2 channel (Figure 4D) and increased it in the CaV1.2 channel (Figure 4C). On average, the change from threshold to maximal normalized response occurred within a 20 mV depolarization range for the CaV3.2 channel and 30 mV for the CaV1.2 channel. Normalizing each curve to its own maximal value (Figures 4E,F) reveals in the CaV3.2 channel the same tendency seen in the normalized IV curve (Figure 2F) for greater conductance at HP in the ΔV −10 mV membrane potential, whereas the CaV1.2 channel did not show such trait (Figure 4E). For depolarizations above VImax only compression to 0.5 MPa caused a consistent small reduction in normalized conductance in both channels.
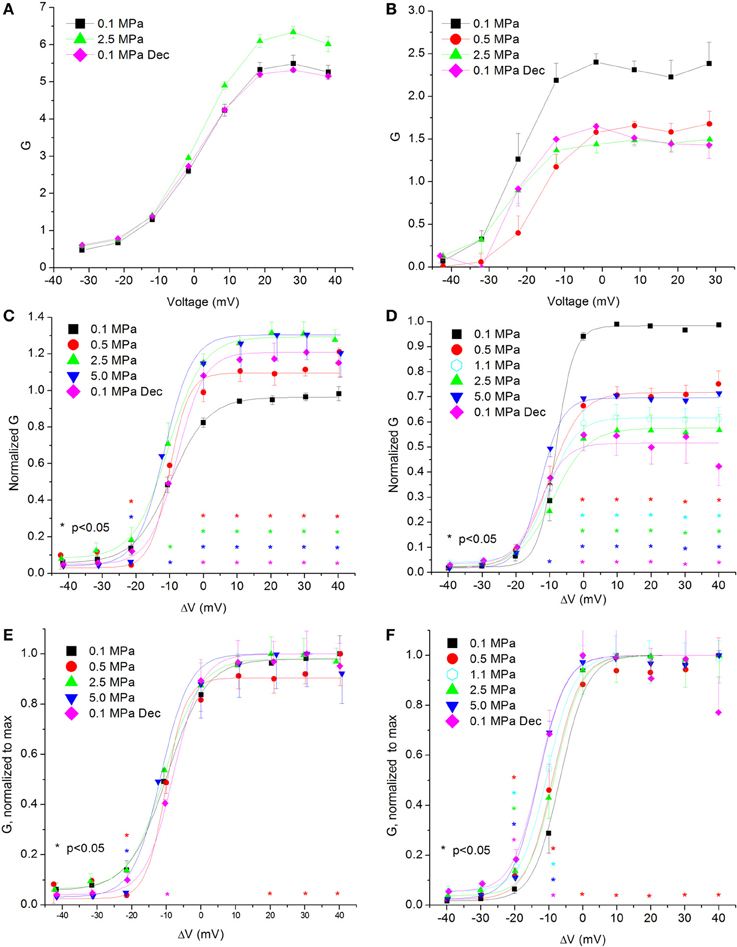
Figure 4. Channels' conductance at various pressures. (A,C,E) In CaV1.2 and (B,D,F) in CaV3.2 channels. (A,B) Conductance measured in a single oocyte. (C,D) Pooled data of the channels. (E,F) Normalized conductance: each curve is normalized to its own maximal value and corresponds to its pertinent curve in (C,D). A Boltzmann fit was used in (C–F). Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
Current Inactivation
The significant effect of HP on these channels manifested in the current and conductance parameters has led us to ask why pressure changes the total flux through them during supra-threshold depolarizations. A possible explanation could be that the channels' kinetics is affected by pressure. Previous findings have correlated HP to slowed inactivation in Na+ channels (Henderson and Gilbert, 1975; Conti et al., 1982b) and compared the effect of higher pressure to lower temperature (Grossman and Kendig, 1984, 1986). We therefore tested a few of the channels' kinetic properties. We first examined channel inactivation, measured as the ratio between the remaining fraction of the current at the end of the depolarizing voltage step and its maximal value (Iend/Imax; see examples in Figures 5A,B). The lack of Ca2+ ions in the experimental solution has eliminated the Ca2+-dependent inactivation and most likely guaranteed that only the voltage- and time-dependent inactivation will be measured. To verify this assumption, and in order to serve as a reference, a few experiments were conducted (n = 5) using a solution in which the Ba2+ was replaced with an equivalent concentration of Ca2+ (example in Figure 3E). As expected, in the presence of Ca2+ a stronger (2-fold) and faster (4-fold) inactivation was observed.
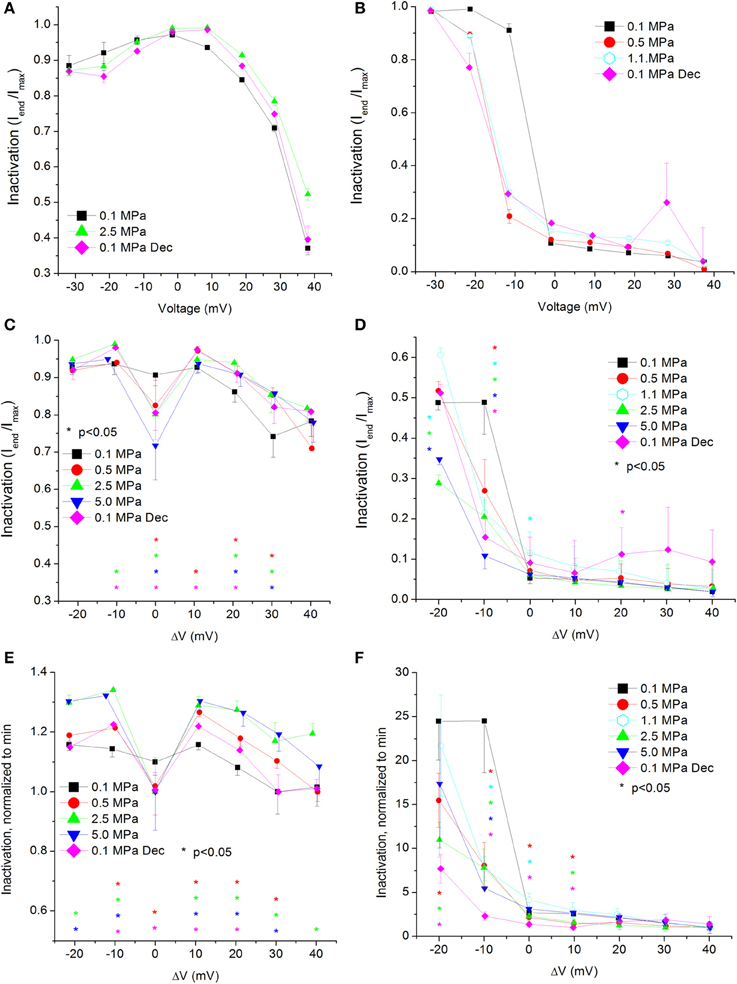
Figure 5. Voltage- and time-dependent current inactivation (Iend/Imax) at various pressures. (A,C,E) In CaV1.2 and (B,D,F) in CaV3.2 channels. (A,B) Inactivation measured in a single oocyte. (C,D) Pooled data of the channels. (E,F) Normalized inactivation: each curve is normalized to its own minimal value and corresponds to its pertinent curve in (C,D). Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
For the CaV1.2 channel, inactivation was stronger at VImax, but was weakened at stronger depolarizations (ΔV 10–40 mV, Figure 5C). Decompression did not relieve the HP effect. For the CaV3.2 channel, the inactivation was stronger around the threshold voltage (ΔV −20 to −10 mV) at HP, but was weakened at VImax at lower HP (1.1 MPa).
Normalizing each curve to its own minimal value stresses the tendency for the strongest inactivation to occur in the CaV1.2 channel at VImax at HP, whereas at normobaric pressure and post-decompression it occurs at the strongest depolarization, as expected for this VDCC (Figure 5E). For the CaV3.2 channel, although the normalized curves converge at high depolarization, in most supra-threshold membrane potentials inactivation remained stronger at HP (Figure 5F).
Currents Time to Peak
If the rate of activation of the VDCCs is affected by HP, that may change the time window for the ionic flow through them before the voltage- and time-dependent inactivation takes place. In case activation is as slow as inactivation, that will affect the Imax in each depolarization. We have therefore measured the time passing from the stimulating depolarizing step to the development of Imax (time to peak, TTP). Examples can be seen in Figures 6A,B.
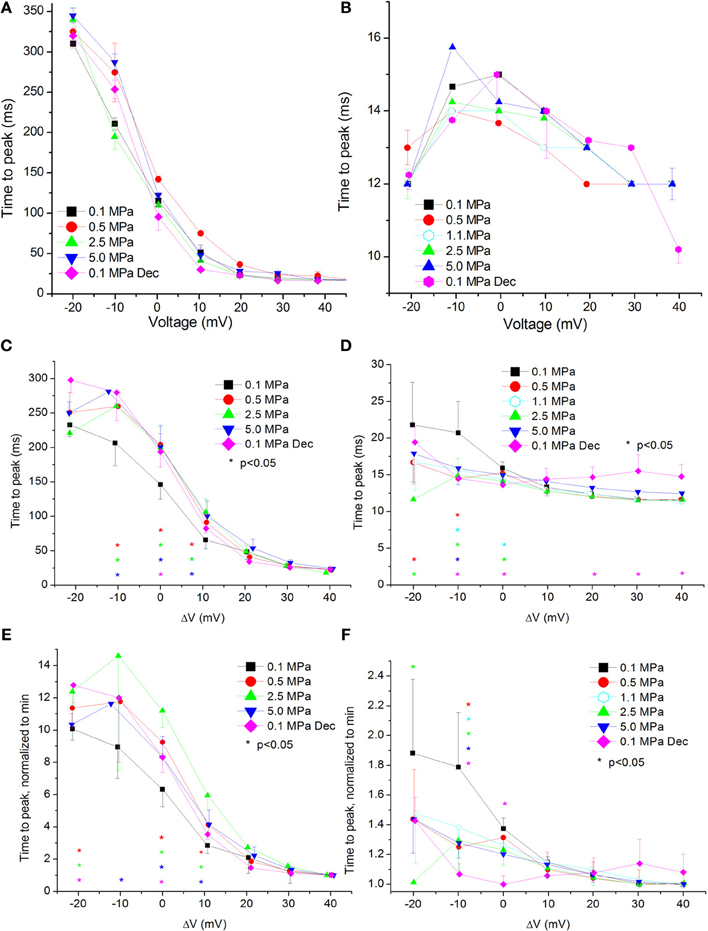
Figure 6. Time to current peak (TTP, from stimulus onset) at various pressures. (A,C,E) in CaV1.2 and (B,D,F) in CaV3.2 channels. (A,B) TTP measured in a single oocyte. (C,D). Pooled data of the channels. (E,F) Normalized TTP: each curve is normalized to its own minimal value and corresponds to its pertinent curve in (C,D) Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
For the CaV1.2 channel the TTP was increased at HP near VImax (ΔV −10 to 10 mV, Figure 6C). Decompression did not relieve this effect.
For the CaV3.2 channel TTP was decreased in the range between of ΔV −20 to 0 mV (Figure 6D). An increase of TTP was observed after decompression above VImax.
Normalizing each TTP curve to its own minimal value (normally achieved at strong depolarization) showed the same tendencies: an increased TTP in the CaV1.2 channel (Figure 6E) and a decreased TTP in the CaV3.2 channel (Figure 6F).
Currents ƮRise
A complementary trait for the TTP in regard to the opening of the channel is the time constant of the rising phase of the current, ƮRise. Slower TTP should mean greater ƮRise, and vice versa. Indeed, in the CaV1.2 HP caused an increase in the ƮRise values at VImax and 10 mV above (Figure 7A). Greater depolarizations led to convergence of ƮRise to control values (Figure 7B). Decompression only partially recovered ƮRise, which remained elevated at VImax. Normalizing each curve to its own minimal value demonstrates a similar rise in ƮRise at VImax and ΔV 10 mV (Figure 7C), which might support the notion that HP interferes with the opening mechanism of this channel, i.e., slows its kinetics. ΔV‡ of current activation for CaV1.2 at VImax was calculated to be 454 ml/mole at 5.0 MPa.
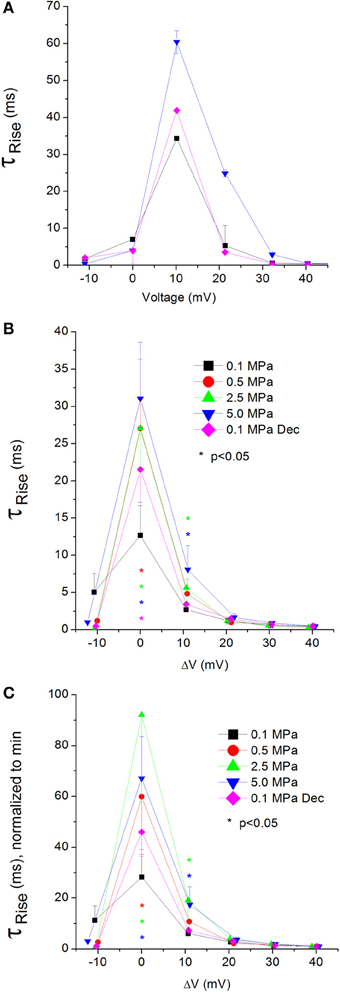
Figure 7. Time constant of current activation (ƮRise) in CaV1.2 channel. (A) ƮRise measured in a single oocyte. (B) Pooled data of ƮRise from 7 to 16 oocytes. (C) Normalized ƮRise: each curve is normalized to its own minimal value and corresponds to its pertinent curve in (B). Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
Unfortunately, relatively large currents measured in the CaV3.2 channel were accompanied by an artifact during their rising phase, which prevented accurate measurement of their ƮRise.
Currents Fast ƮDecay
The changes in inactivation (Iend/Imax) found in both channels at HP could arise from its effect on their rate of decay, since Iend was measured here after a specific time and not under steady state conditions. The analysis revealed fast and slow time constants for the decay (see examples in Figures 8A,B, 9A,B), the first being shortened by stronger depolarizations and the latter being elongated.
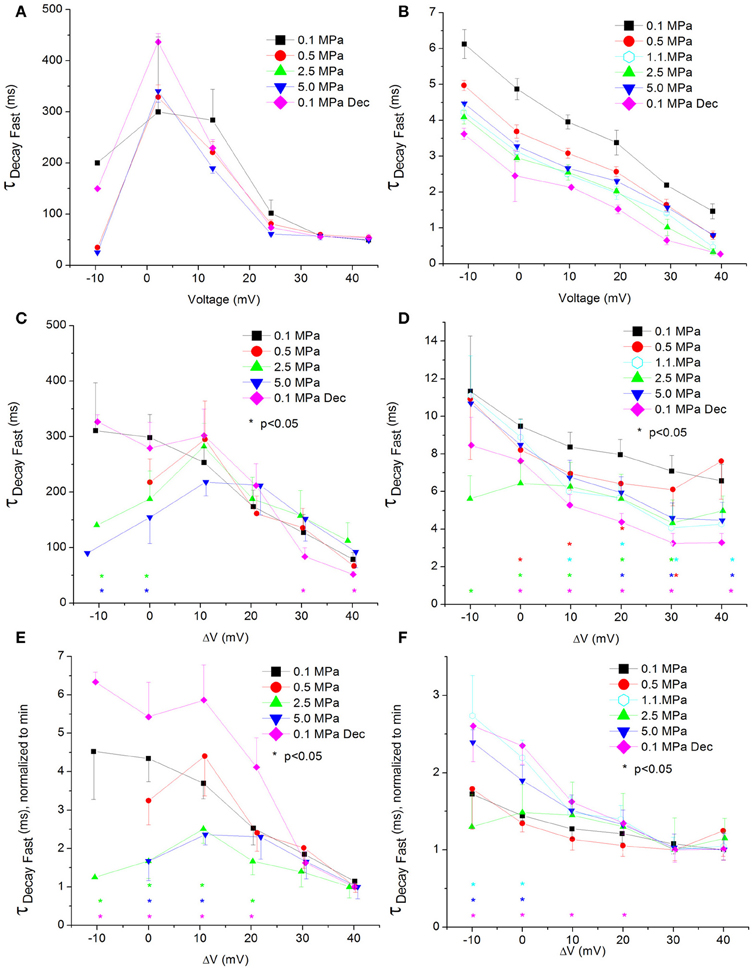
Figure 8. Fast time constant of voltage- and time-dependent current inactivation (ƮDecay Fast). (A,C,E) in CaV1.2 and (B,D,F) in CaV3.2 channels. (A,B) ƮDecay Fast measured in a single oocyte. (C,D) Pooled data of the channels. (E,F) Normalized ƮDecay Fast: each curve is normalized to its own minimal value and corresponds to its pertinent curve in (C,D). Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
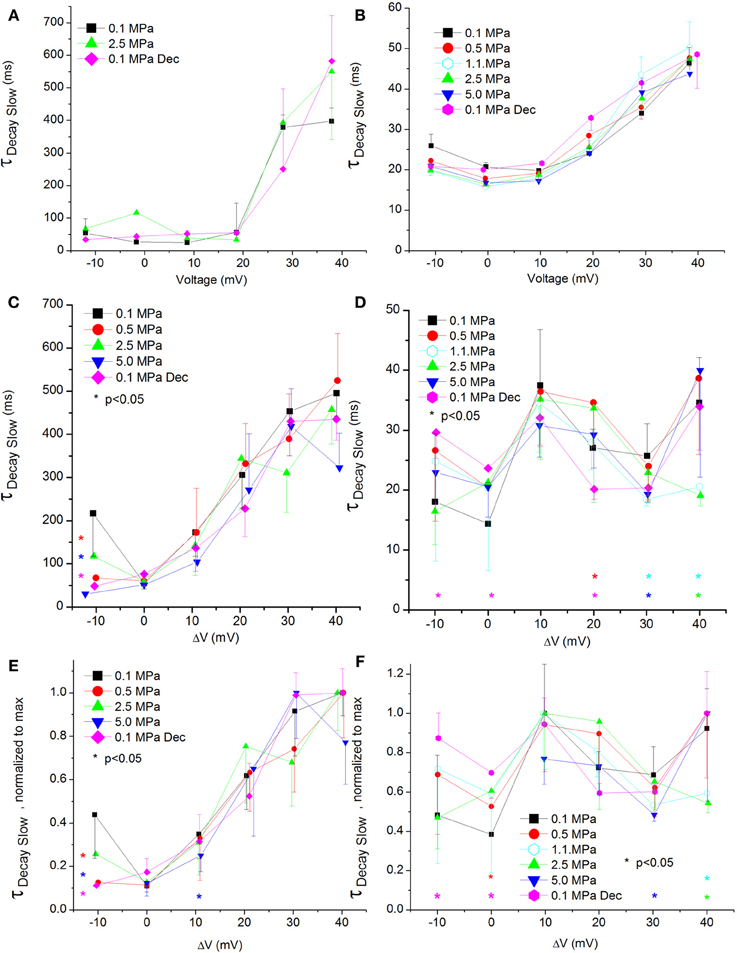
Figure 9. Slow time constant of voltage- and time-dependent current inactivation (ƮDecay Slow). (A,C,E) in CaV1.2 and (B,D,F) in CaV3.2 channels. (A,B) ƮDecay Slow measured in a single oocyte. (C,D) Pooled data of the channels ƮDecay Slow. (E,F) Normalized ƮDecay Slow: each curve is normalized to its own maximal value and corresponds to its pertinent curve in (C,D). Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
For the CaV1.2, HP caused a decrease in the fast ƮDecay (ƮDecay Fast) at VImax and 10 mV below (Figure 8C). Stronger depolarizations (ΔV 10–40 mV) at HP did not change ƮDecay Fast, but after decompression its values were slightly smaller (ΔV 30–40 mV). In the rest of the depolarizing range, decompression fully recovered ƮDecay Fast. For the CaV3.2, the ruling trend was a reduction of ƮDecay Fast at HP, mainly from VImax and above (Figure 8D), while decompression did not relieve this effect. Normalizing each curve to its own minimal value showed for the CaV1.2 channel a depression at 2.5 and 5.0 MPa, but not at 0.5 MPa (Figure 8E), while decompression increased the normalized ƮDecay Fast. In the CaV3.2 channel at ΔV −10 to 0, compression to 1.1 and 5.0 MPa (but not to 0.5 and 2.5 MPa) increased the normalized ƮDecay Fast (Figure 8F), which remained elevated after decompression in this activity range.
ΔV‡ of fast decay inactivation at VImax was calculated to be −332 and −55 ml/mole at 5.0 MPa for CaV1.2 and CaV3.2, respectively.
Currents Slow ƮDecay
The slow ƮDecay (ƮDecay Slow) in both channels was increased by stronger depolarizations at 0.1 MPa (Figures 9A,B).
For the CaV1.2 channel ƮDecay Slow does not seem to be sensitive to HP, as no consistent significant difference was found in its values after compression or decompression (Figure 9C). For the CaV3.2 channel ƮDecay Slow was shortened only by strong depolarization (ΔV 30–40 mV) at HP, while decompression eliminated this effect (Figure 9D). The normalized curves of ƮDecay Slow for both channels did not reveal a behavior different than that described above (Figures 9E,F).
ΔV‡ of slow decay inactivation at VImax was calculated to be −32 and 179 ml/mole at 5.0 MPa for CaV1.2 and CaV3.2, respectively.
Currents ƮTail
The tail current time constant (ƮTail), representing the kinetics of the channels' deactivation, was shortened by increasing depolarization in the CaV1.2 channel (see example in Figure 10A). This trend persisted with the application of HP, but at 5.0 MPa ƮTail remained minimal throughout the activity range of the channel (Figure 10B). Strong depolarization (ΔV 10–40 mV) at HP (0.5 and 5.0, but not 2.5 MPa) shortened ƮTail. The normalized curves of ƮTail stress the absence of a significant slope at 5.0 MPa (Figure 10C). ΔV‡ of deactivation for CaV1.2 at VImax was calculated to be −432 ml/mole at 5.0 MPa.
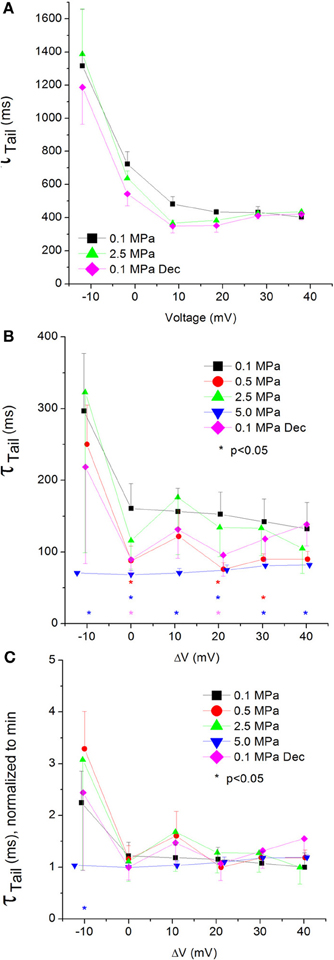
Figure 10. Tail current time constant (ƮTail) in CaV1.2 channel. (A) ƮTail measured in a single oocyte. (B) Pooled data of ƮTail from 7 to 16 oocytes. Only 5.0 MPa has affected (reduced) ƮTail throughout the channels' activity range. (C) Normalized ƮTail: each curve is normalized to its own minimal value and corresponds to its pertinent curve in (B). Pressures are color indicated. Statistical significance for each point on the curve is indicated by corresponding color asterisks. Holding potential is expressed as in Figure 2. Dec indicates decompression.
Unfortunately, the reciprocal artifact for the one interfering with measuring ƮRise in the CaV3.2 channel also prevented an accurate measurement of ƮTail.
Discussion
Establishing the Methodology
The use of a truncated K+ channel proved the stability of measurements in our setup in these unusually long oocyte experiments, which were necessary due to adiabatic temperature fluctuations. Furthermore, there was no indication of long-term (1 h) effects of the fluctuating adiabatic temperature on barium currents once control temperature was restored (Figure 3B), suggesting that it cannot account for the much greater pressure-induced changes in maximal currents. Due to the relatively quick response of the preparation (Figure 3A), and in comparison to previous studies (Grossman and Kendig, 1984), it is likely that the time course of recovery from temperature transients is no longer than a few minutes, and therefore at the time of current measurements, when the ambient temperature is within 0.5°C deviation from control value, the membrane does not exhibit properties of temperature a few degrees higher or lower.
Additional support for this assumption arises from experiments in lobster (Panulirus interruptus) bifurcating axon, which showed that 10–15 min were sufficient to allow the responses to recover to the original levels and remain stable (Grossman and Kendig, 1986).
In the present experiments, cooling back to control temperature after compression steps lasted 12–15 min, with at least an additional 5–6 min required for verifying stability of currents. Thus, the total time of 17–21 min seems pertinent to allow the preparation to “relax” to its new pressure condition without temperature change interference. However, we cannot completely rule out the possibility that transient adiabatic temperature fluctuation contributes to the current change at pressure per se. In order to do so, conditions of compression need to allow a constant temperature during pressure changes, i.e., preventing or minimizing adiabatic temperature fluctuations. Only liquid compression can achieve this stipulation to a high degree, but unfortunately cannot be employed in our current setting. Nevertheless, Schmalwasser et al. (1998) have managed to design and operate a hydrostatic (oil) pressure chamber, in which adiabatic temperature fluctuation in compressions steps of 10 MPa was only 0.5°C or less, and demonstrated a reversible reduction of currents in the Shaker potassium channel mutant Shaker BD6-46 T449K. These results show that high pressure induced its effects on the channel even when temperature fluctuation was minimized, suggesting that our results were not distorted by the small temperature change.
The use of the 9-AC blocker both in normobaric and HP conditions strongly suggested that the endogenous Cl−Ca currents are not of significant importance in our experiments (Figures 3C,D). Furthermore, the use of Ba2+ as a charge carrier proved to be efficient, as currents were stronger and the Ca2+ inactivation was eliminated (Figure 3E) as expected (Lyford et al., 2002).
Taken together, these control experiments show that the changes demonstrated after compression are indeed pressure related.
The HP-induced depression demonstrated in the CaV3.2 channel further strengthened our findings in the CaV1.2 channel, ruling out the possibility of it being a setup-related artifact.
Currents' Activation
Currents' amplitude
We show here, for the first time by direct measurement, that pressure effect can be selective: currents through CaV1.2 are increased, whereas currents through the CaV3.2 channel are depressed, at HP. It is noteworthy that the CaV3.2 is more sensitive to pressure changes than the CaV1.2 channel, as the HP effect is already saturated at 1.1 MPa. Furthermore, currents in the CaV1.2 were partially recovered on return to atmospheric pressure, whereas in the CaV3.2 currents did not recover. Although the CaV3.2 channel is found in neuronal somata and much less in the nerve terminals, the findings demonstrate the possibility of selective pressure effects on other types of Ca2+ channels (P/Q, N, R) that are believed to be present at the presynaptic nerve terminals (Uchitel et al., 1992; Etzion and Grossman, 2000). Such a selectivity was already indirectly suggested in our experiments on frog motor nerve terminals (Aviner et al., 2013).
The effect on the CaV1.2 is apparently in contrast to previous reports that suggested reduction in Ca2+ influx through CaV2.2 (Grossman et al., 1991), and a lesser (Etzion and Grossman, 2000; Aviner et al., 2013) or no effect (Heinemann et al., 1987) in L-type VDCC (CaV1) at pressure. The CaV3.2 current reduction conforms to the above notion. However, in part of the studies (Grossman et al., 1991; Etzion and Grossman, 2000) CaV1.2 involvement was excluded pharmacologically, while in others it was assumed absent based on information obtained from similar or identical preparations (Robitaille et al., 1990; French et al., 2002). In the chromaffin cells the channels were denoted as CaV1 simply due to their similar behavior to these channels in neuronal cells, and have not been characterized pharmacologically. Lastly, the one change observed in Ca2+ currents in the bovine chromaffin cells was a slight increase at 40 MPa, which might support our finding of augmentation of currents in the CaV1.2 (probably a different isoform) at lower pressure. Thus, the HP-induced increase of current in CaV1.2 could be specific and excludes other types of channel of the same family (CaV1).
The augmentation of the currents in CaV1.2 channel at HP demonstrated here is reminiscent of another member of this protein superfamily behavior, the K+ channels. The non-inactivating “delayed rectifier” K+ channels pass greater steady-state currents at pressure in invertebrates such as squid (Henderson and Gilbert, 1975; Shrivastav et al., 1981; Conti et al., 1982a), snail (Harper et al., 1981), and lobster (Grossman and Kendig, 1984). Enhanced K+ currents at pressure were also proposed as the basis for the firing patterns of Ca2+ APs in guinea pig Purkinje cells (Etzion and Grossman, 1999), and for slowing of the sinus-node pacemaker activity in various mammalian hearts (Ornhagen and Hogan, 1977). On the other hand, the reduction of currents in the CaV3.2 shown here is in agreement with other experiments concerned with an inactivating type of the K+ channel, demonstrating a depression of currents at pressure in snail (Harper et al., 1981) and in mouse Shaker B K+ channels expressed in Xenopus oocytes (Meyer and Heinemann, 1997; Schmalwasser et al., 1998).
Previous studies in our laboratory in rat dentate gyrus corticohippocampal slices suggested that high pressure increases the transfer function between synaptic inputs and somatic spike generation by granule cells, despite the observed reduction of single field excitatory post-synaptic potential (fEPSP) amplitude and slope by nearly 50% (Talpalar and Grossman, 2004). This suggests that high pressure depresses synaptic activity while increasing excitability in the neuronal dendrites but not in the axons. The CaV1.2 channels that were studied here are known to be present in the cell bodies and proximal dendrites of neurons in the dentate gyrus (Hell et al., 1993). It is suggested that pressure-potentiated CaV1.2 currents in the dendrite may boost subthreshold synaptic potentials to generate APs. Such a dendritic hyperexcitability could explain the increased transfer function and is a good example of another way through which pressure selective effects on VDCC might impact neuronal networks, other than synaptic transmission. Such an increased transfer function mechanism may conform to the hyperexcitability manifested in HPNS.
It is interesting to note that the CaV1.2 is also expressed in the cardiac muscle, and the CaV3.2 in the cardiac Purkinje fibers. Both changes in the maximal current amplitude described here correlate with previous findings: an increase in the contractility force of isolated rat hearts (Ornhagen and Sigurdsson, 1981; Gennser and Ornhagen, 1989a) and a decrease in the contraction pace in rat (Gennser and Ornhagen, 1989b) and humans (Linnarsson et al., 1999; Kurita et al., 2002).
A positive ionotropic pressure effect at steady-state that was reported for skeletal and cardiac muscles was previously explained by increase of cytosolic Ca2+ that may occur due to inhibition of Ca2+ removal from the cytosol, into either cellular or extracellular compartments (Daniels and Grossman, 2003). Our study may offer a different or additional mechanism for these changes in inotropy.
The augmented CaV1.2 currents may increase neuronal dendritic excitability and therefore contribute to the generation of HPNS. The contribution to HPNS of the reduction of CaV3.2 currents is less obvious. These low threshold activated channels are especially involved in generating bursting behavior and rhythmic activity in pace-maker neurons in the reticular thalamic nucleus, thalamus, striatum, and cortex, where they are involved in controlling sleep, awareness, executive function, movement planning and modulation, and sensory inputs, respectively. We may speculate that the current reduction will slow this activity and impair these neuronal “clocks” function. This may interfere with time processing and coincidence detection as well as motor and sensory functions in a manner similar to HPNS signs and symptoms. A slower rhythm at HP was indeed found in humans, where EEG waves shifted from α to θ patterns (Rostain et al., 1997). The same study also demonstrated sleep disturbances that were more prominent during the beginning of the compression, subsided later, and were abolished at decompression, suggesting both transient and reversible HP effect, similar to the results shown in our experiments.
The amplitude of slow after-hyperpolarization (sAHP) in rat CA1 was reduced by HP (Southan and Wann, 1996). This reduction could be explained by a depression of the SK potassium channel, responsible for the sAHP. But since this channel is activated by the rise of [Ca2+]i during each AP, it can also be pointing to a reduction in Ca2+ influx through VDCCs, e.g., CaV3.2, not only in the synaptic terminal but also along the axon and possibly in the soma and dendrites as well.
It is worth noting that our pressure effect on CaV3.2 is saturating at about 1.1 MPa. This is well correlated with the pressure experienced by professional divers in the depth of seawater in which they begin to complain of the HPNS signs and symptoms. Therefore, this might be an indication that the CaV3.2 is also involved in the underlying mechanism of this phenomenon. It is postulated that the mild symptoms observed at lower pressures such as impaired cognitive capabilities and motor dexterity are the consequence of pacemaker malfunction as a result of reduction in CaV3.2 maximal currents.
Channels' conductance
Overall, the calculated conductance behavior relative to the membrane potential (Figure 4) is in accordance with the I-V curves described in Figure 2. Although generally the HP effect for CaV1.2 and CaV3.2 channels is opposite, at ΔV −10 mV both demonstrate an increase of conductance at HP (5.0 MPa, Figures 4C,D). The CaV3.2 normalized-to-max conductance curve better shows the channels' postulated tendency to shift to an open state at HP in membrane potentials just over the threshold. However, once depolarization is increased the maximal currents and the conductance are depressed by HP. If HP raises the probability to shift to an open state, why would the currents depress? A possible explanation could be a stronger and faster inactivation of the channel at HP throughout the channels' activity range. Indeed, an indication for that can be seen by a decrease in both Iend/Imax and ƮDecay Fast at HP (Figures 5D,F and 8D,F, respectively).
The fact that only compression to 0.5 MPa caused a reduction in the normalized-to-max conductance curve above VImax in both channels (Figures 4E,F) may suggest a transient sensitivity of their “voltage sensor” to HP.
Currents TTP and ƮRise
Both TTP and ƮRise elongate at HP for the CaV1.2 channel, strongly suggesting a slower activation (Figures 6C, 7C, respectively). A similar HP effect was reported in previous studies on VDCCs in guinea pig single cerebellar Purkinje cells (probably CaV2.1) (Etzion and Grossman, 1999) and frog motor nerve (possibly CaV2.2) (Aviner et al., 2013). The velocity of an AP was also reduced at HP after a transient increase (Grossman and Kendig, 1986).
On the other hand, TTP was shortened in a narrow voltage range (−20 mV up to VImax) in the CaV3.2 (Figure 6D), suggesting a faster activation of the channel at HP. Nevertheless, shortening of TTP may also point to a faster inactivation process, which will make the maximal current appear earlier. This indeed was found in the CaV3.2, where ƮDecay Fast has shortened at HP (Figure 8D). Unfortunately, the inability to measure ƮRise for CaV3.2 in this case prevents distinguishing the two options. However, the opposite HP effect on TTP, conductance, and maximal currents for these channels, together with the fact that ƮDecay Fast was not significantly elongated for the CaV1.2 at HP (Figure 8C) although TTP was increased, may indicate that for the CaV3.2 ƮRise may be shortened at HP.
Current Inactivation
As expected for a fast inactivating channel, the CaV3.2 shows a lower value of Iend/Imax (stronger inactivation) than the CaV1.2 for any given membrane potential (Figures 5A–D). At VImax the Iend/Imax value has decreased for the CaV1.2 at HP, and the same happened for the CaV3.2 at ΔV −10 to −20 mV. This correlates well with smaller ƮDecay Fast (faster inactivation) in both channels in this voltage range at HP (Figures 8C,D). On the other hand, stronger depolarization (ΔV 10–40 mV) at HP has led to weaker inactivation in the CaV1.2 but not in the CaV3.2 channel (Figures 5C,D). These findings are not supported by the measured ƮDecay Fast in both channels in the same voltage range: it was not changed in the CaV1.2 and it was still reduced in the CaV3.2. For the latter it seems that since the inactivation has managed to reach its maximal values at the end of the 100 ms time window (end of depolarizing step), the faster ƮDecay Fast at HP has only facilitated the already strong inactivation and did not change its values. For the CaV1.2 channel the ƮDecay Fast averages are generally higher at HP for ΔV 10 and above, although not statistically significant, but probably enough to weaken the measured inactivation. A slower inactivation at HP was also reported in Na+ channel in bovine chromaffin cells (Heinemann et al., 1987), but not in the Helix snail (Harper et al., 1981).
The flatter normalized-to-minimum curve of inactivation in CaV1.2 at 0.1 MPa relative to HP curves (Figure 5E) and the linear behavior of ƮDecay Fast at 0.1 MPa and after decompression as opposed to the behavior at HP (Figure 8C) both suggest that the molecular mechanism controlling the voltage-dependent inactivation is affected by HP.
Notwithstanding, no significant changes were observed in the ƮDecay Slow at HP in both channels, supporting the concept of different mechanisms for the fast and slow inactivation (Hering et al., 2000; Sokolov et al., 2000), which can also react differently to external treatment (Livneh et al., 2006). It was also demonstrated that the molecular structures responsible for these two types of inactivation are differently located in the VDCC's protein (Berjukow et al., 1999) and that the fast inactivation may act similarly to the “ball and chain” mechanism in the K+ channel (Cens et al., 1999), while the slow inactivation seems to be at least partially dependent on the interaction between α1 and β subunits (Sokolov et al., 2000). We suggest that the selective HP effect between the two channels' inactivation (Iend/Imax and ƮDecay) may arise from different conformational changes at HP, resulting from a different basic spatial organization of the channels, specifically their inactivation controlling regions.
Currents Deactivation
Currents ƮTail
Compression to 0.5 and 5.0 MPa, but not 2.5 MPa, shortened ƮTail for CaV1.2, implying a faster deactivation of the channel at HP (Figure 10B). The lack of consistency in the HP effect on ƮTail may suggest a transient effect caused by the initial compression (0.5 MPa), and the inability of the deactivating mechanism to compensate for the higher HP (5.0 MPa).
The normalized curves show that in the CaV1.2 the reduction of ƮTail has saturated at VImax at normobaric and HP (Figure 10C). However, at 5.0 MPa saturation was achieved already at ΔV −10 mV, which may indicate that the closure of the channel is facilitated by high HP.
Summary
HP affected the behavior of both CaV1.2 and CaV3.2, whether throughout their membrane potential activity range (maximal current, conductance, ƮTail at 5.0 MPa), or just at a confined voltage range (ƮRise, TTP, inactivation, ƮDecay Fast). The HP effect on the two channels was generally opposite (maximal currents, conductance, TTP), but some kinetic traits shared the same HP-induced tendencies (inactivation at VImax, ƮDecay Fast). Some of the effects may indicate a transient nature (conductance and partially ƮTail), while other suggested that the HP effect can be reversible (mostly for CaV1.2; ƮDecay Fast, ƮTail, and partially also maximal current and ƮRise). A summary of the major HP-induced findings is given in Table 1.
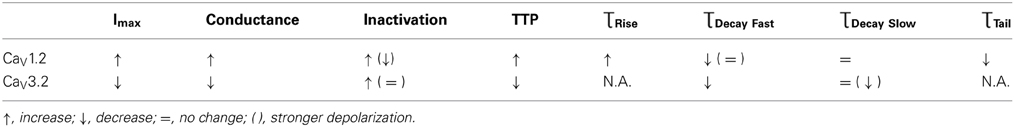
Table 1. General qualitative effect of HP found in our study on channel characteristics.
These changes in the response to depolarization, in both magnitude and kinetics, would undoubtedly influence these channels' functionality in neurons. Decrements in locomotor activity, myoclonus, tremors, changes in EEG, and sleep disorders, all part of the HPNS phenomenon, may be the manifestation of these HP-induced changes.
General Considerations
The effect of HP can be targeted either at the channel (and its subunits) or at any external modulator. Some of the characteristics examined in this study exhibited sensitivity (sometimes opposite) to HP that was also dependent on the membrane potential, suggesting that HP affects the channel itself. More specifically, HP may target the channels' “voltage sensor” and thus affect voltage-dependent mechanisms of activation, inactivation, and deactivation.
Several studies have demonstrated that replacing a section within a subunit (Tang et al., 1993; Zhang et al., 1994) or even a single amino acid (Bourinet et al., 1999; Hans et al., 1999) can dramatically change its reaction to depolarization. Furthermore, it was suggested that these changes are caused by different spatial organization of the subunits, influencing their interactions (Hering et al., 2000).
Similarly, the HP-induced effects demonstrated in this study may indicate that the conformational changes involved in the channels' activity are facilitated (e.g., activation in the CaV1.2, fast inactivation in the CaV3.2) or opposed (e.g., activation in the CaV3.2, rate of activation in the CaV1.2) by an elevated ambient pressure. Indeed, this notion is supported by the calculated activation volumes corresponding to these processes, probably affecting the total ionic flux through the channels at HP. Furthermore, even the basic (i.e., not activated) spatial organization of the subunits and their interactions may be altered by the HP application (e.g., a segment within S4 transmembrane region, holding the positively charged amino acids sequence that serve as a voltage sensor), thus leading to the HP sensitivity shown here. For example, the human and rat glycine receptor affinity to its ligand was reduced at HP (Roberts et al., 1996). The reduced activity of this inhibitory mechanism may explain the tendency to hyperexcitability upon exposure to HP (HPNS manifestations). A genetic sequencing of the receptor demonstrated a few amino acid switches between the mammals mentioned above and the Pilot whale, one of them in the ligand binding site from the non-polar cysteine (human and rat) to the polar arginine (C41R). This switch may protect the whale from hyperexcitability at HP and provide it with the ability to dive to extreme depths.
It can therefore be postulated that performing HP experiments on a specific VDCC with altered subunits or a VDCC chimera might further elucidate the HP targeted site. Preliminary results from experiments on the NMDA receptor at HP may suggest that even changes in the extracellular N terminal domain can influence the magnitude and direction of the pressure effect (Mor et al., 2012).
Conclusions
(a) HP Modulation of various VDCCs is part of HPNS mechanisms and may explain some of its signs and symptoms.
(b) HP effects could be quite selective for the type of channel and/or various mechanisms underlying the channels' activity.
(c) HP effects on the channels' kinetics should be extensively studied in order to reveal small functional differences under HP conditions.
(d) Our study emphasizes the importance of pressure modulation of the membrane potential “sensor,” which may determine the extent of pressure effects on the channels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraini, J. H. (1997). Inert gas and raised pressure: evidence that motor decrements are due to pressure per se and cognitive decrements due to narcotic action. Pflugers Arch. 433, 788–791. doi: 10.1007/s004240050346
Allen, B. W., Demchenko, I. T., and Piantadosi, C. A. (2009). Two faces of nitric oxide: implications for cellular mechanisms of oxygen toxicity. J. Appl. Physiol. 106, 662–667. doi: 10.1152/japplphysiol.91109.2008
Ashford, M. L., MacDonald, A. G., and Wann, K. T. (1982). The effects of hydrostatic pressure on the spontaneous release of transmitter at the frog neuromuscular junction. J. Physiol. 333, 531–543.
Aviner, B., Gnatek, Y., Gradwohl, G., and Grossman, Y. (2010). Hyperbaric pressure effects on voltage-dependent Ca+2 channels: relevance to HPNS. Undersea Hyperb. Med. 37, 245–258.
Aviner, B., Gradwohl, G., Moore, H. J., and Grossman, Y. (2013). Modulation of presynaptic Ca2+ currents in frog motor nerve terminals by high pressure. Eur. J. Neurosci. 38, 2716–2729. doi: 10.1111/ejn.12267
Bennett, P. B. (1997). “Inert gas narcosis and high pressure nervous syndrome,” in Bove and Davis' Diving Medicine, eds A. A. Bove and J. C. Davis (Philadelphia, PA: Saunders), 117–130.
Berjukow, S., Gapp, F., Aczel, S., Sinnegger, M. J., Mitterdorfer, J., Glossmann, H., et al. (1999). Sequence differences between Alpha 1c and Alpha 1s Ca2+ channel subunits reveal structural determinants of a guarded and modulated benzothiazepine receptor. J. Biol. Chem. 274, 6154–6160. doi: 10.1074/jbc.274.10.6154
Boton, R., Dascal, N., Gillo, B., and Lass, Y. (1989). Two calcium-activated chloride conductances in xenopus laevis oocytes permeabilized with the ionophore A23187. J. Physiol. 408, 511–534.
Bourinet, E., Soong, T. W., Sutton, K., Slaymaker, S., Mathews, E., Monteil, A., et al. (1999). Splicing of alpha 1a subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 2, 407–415. doi: 10.1038/8070
Campenot, R. B. (1975). The effects of high hydrostatic pressure on transmission at the crustacean neuromuscular junction. Comp. Biochem. Physiol. B 52, 133–140. doi: 10.1016/0305-0491(75)90128-5
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16: 521–555. doi: 10.1146/annurev.cellbio.16.1.521
Cens, T., Restituito, S., Galas, S., and Charnet, P. (1999). Voltage and Calcium Use the Same Molecular Determinants to Inactivate Calcium channels. J. Biol. Chem. 274, 5483–5490. doi: 10.1074/jbc.274.9.5483
Conti, F., Fioravanti, R., Segal, J. R., and Stuhmer, W. (1982a). Pressure dependence of the potassium currents of squid giant axon. J. Membr. Biol. 69, 35–40. doi: 10.1007/BF01871239
Conti, F., Fioravanti, R., Segal, J. R., and Stuhmer, W. (1982b). Pressure dependence of the sodium currents of squid giant axon. J. Membr. Biol. 69, 23–34. doi: 10.1007/BF01871238
Daniels, S., and Grossman, Y. (2003). “Biological effects of pressure,” in Bennett and Elliott's Physiology and Medicine of Diving, 5th Edn, eds A. O. Brubakk and T. S. Neuman (London: Saunders), 265–299.
Darbin, O., Risso, J. J., and Rostain, J. C. (2000). High pressure enhanced nmda activity in the striatum and the globus pallidus: relationships with myoclonia and locomotor and motor activity in rat. Brain Res. 852, 62–67. doi: 10.1016/S0006-8993(99)02195-2
Dean, J. B., Mulkey, D. K., Garcia, A. J. III., Putnam, R. W., and Henderson, R. A. I. I. I. (2003). Neuronal sensitivity to hyperoxia, hypercapnia, and inert gases at hyperbaric pressures. J. Appl. Physiol. 95, 883–909. doi: 10.1152/japplphysiol.00920.2002
Ertel, E. A., Campbell, K. P., Harpold, M. M., Hofmann, F., Mori, Y., Perez-Reyes, E., et al. (2000). Nomenclature of voltage-gated calcium channels. Neuron 25, 533–535. doi: 10.1016/S0896-6273(00)81057-0
Etzion, Y., Mor, A., and Grossman, Y. (2008). Differential modulation of cerebellar climbing fiber and parallel fiber synaptic responses at high pressure. J. Appl. Physiol. 106, 729–736. doi: 10.1152/japplphysiol.90853.2008
Etzion, Y., and Grossman, Y. (1999). Spontaneous Na+ and Ca2+ spike firing of cerebellar purkinje neurons at high pressure. Pflugers Arch. 437, 276–284. doi: 10.1007/s004240050780
Etzion, Y., and Grossman, Y. (2000). Pressure-induced depression of synaptic transmission in the cerebellar parallel fibre synapse involves suppression of presynaptic N-Type Ca2+ channels. Eur. J. Neurosci. 12, 4007–4016. doi: 10.1046/j.1460-9568.2000.00303.x
French, L. B., Lanning, C. C., and Harris-Warrick, R. M. (2002). The localization of two voltage-gated calcium channels in the pyloric network of the lobster stomatogastric ganglion. Neuroscience 112, 217–232. doi: 10.1016/S0306-4522(01)00621-2
Gennser, M., and Ornhagen, H. C. (1989a). Effects of hydrostatic pressure and inert gases on twitch tension. Undersea Biomed. Res. 16, 415–426.
Gennser, M., and Ornhagen, H. C. (1989b). Effects of hydrostatic pressure, H2, N2, and He, on beating frequency of rat atria. Undersea Biomed. Res. 16, 153–164.
Gilman, S. C., Colton, J. S., and Dutka, A. J. (1987). Effect of pressure on the release of radioactive glycine and gamma-aminobutyric acid from spinal cord synaptosomes. J. Neurochem. 49, 1571–1578. doi: 10.1111/j.1471-4159.1987.tb01029.x
Gilman, S. C., Kumaroo, K. K., and Hallenbeck, J. M. (1986). Effects of pressure on uptake and release of calcium by brain synaptosomes. J. Appl. Physiol. 60, 1446–1450.
Golan, H., and Grossman, Y. (1992). Synaptic transmission at high pressure: effects of [Ca2+]O. Comp. Biochem. Physiol. Comp. Physiol. 103, 113–118. doi: 10.1016/0300-9629(92)90249-P
Golan, H., Moore, H. J., and Grossman, Y. (1994). Quantal analysis of presynaptic inhibition, low [Ca2+]0, and high pressure interactions at crustacean excitatory synapses. Synapse 18, 328–336. doi: 10.1002/syn.890180408
Grossman, Y., Colton, J. S., and Gilman, S. C. (1991). Interaction of Ca-channel blockers and high pressure at the crustacean neuromuscular junction. Neurosci. Lett. 125, 53–56. doi: 10.1016/0304-3940(91)90129-H
Grossman, Y., and Kendig, J. J. (1984). Pressure and temperature: time-dependent modulation of membrane properties in a bifurcating axon. J. Neurophysiol. 52, 693–708.
Grossman, Y., and Kendig, J. J. (1986). Pressure and temperature modulation of conduction in a bifurcating axon. Undersea Biomed. Res. 13, 45–61.
Grossman, Y., and Kendig, J. J. (1988). Synaptic integrative properties at hyperbaric pressure. J. Neurophysiol. 60, 1497–1512.
Grossman, Y., and Kendig, J. J. (1990). Evidence for reduced presynaptic Ca2+ entry in a lobster neuromuscular junction at high pressure. J. Physiol. 420, 355–364.
Halsey, M. J. (1982). Effects of high pressure on the central nervous system. Physiol. Rev. 62, 1341–1377.
Hans, M., Urrutia, A., Deal, C., Brust, P. F., Stauderman, K., Ellis, S. B., et al. (1999). Structural elements in domain iv that influence biophysical and pharmacological properties of human alpha1a-containing high-voltage-activated calcium channels. Biophys. J. 76, 1384–1400. doi: 10.1016/S0006-3495(99)77300-5
Harper, A. A., Macdonald, A. G., and Wann, K. T. (1981). The action of high hydrostatic pressure on the membrane currents of helix neurones. J. Physiol. 311, 325–339.
Heidelberger, R., Zhou, Z.-Y., and Matthews, G. (2002). Multiple components of membrane retrieval in synaptic terminals revealed by changes in hydrostatic pressure. J. Neurophysiol. 88, 2509–2517. doi: 10.1152/jn.00267.2002
Heinemann, S. H., Conti, F., Stuhmer, W., and Neher, E. (1987). Effects of hydrostatic pressure on membrane processes. Sodium channels, Calcium channels, and Exocytosis. J. Gen. Physiol. 90, 765–778. doi: 10.1085/jgp.90.6.765
Hell, J. W., Westenbroek, R. E., Warner, C., Ahlijanian, M. K., Prystay, W., Gilbert, M. M., et al. (1993). Identification and differential subcellular localization of the neuronal class C and Class D L-Type calcium channel alpha 1 subunits. J. Cell Biol. 123, 949–962. doi: 10.1083/jcb.123.4.949
Henderson, J. V., and Gilbert, D. L. (1975). Slowing of ionic currents in the voltage-clamped squid axon by helium pressure. Nature 258, 351–352. doi: 10.1038/258351a0
Hering, S., Berjukow, S., Sokolov, S., Marksteiner, R., Weiss, R. G., Kraus, R., et al. (2000). Molecular determinants of inactivation in voltage-gated Ca2+ channels. J. Physiol. 528, 237–249. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x
Kurita, A., Nagayoshi, H., Okamoto, Y., Takase, B., Ishizuka, T., and Oiwa, H. (2002). Effects of severe hyperbaric pressure on autonomic nerve functions. Mil. Med. 167, 934–938.
Linnarsson, D., Ostlund, A., Lind, F., and Hesser, C. M. (1999). Hyperbaric bradycardia and hypoventilation in exercising men: effects of ambient pressure and breathing gas. J. Appl. Physiol. 87, 1428–1432.
Livneh, A., Cohen, R., and Atlas, D. (2006). A novel molecular inactivation determinant of voltage-gated Cav1.2 L-Type Ca2+ channel. Neuroscience 139, 1275–1287. doi: 10.1016/j.neuroscience.2006.01.028
Logue, P. E., Schmitt, F. A., Rogers, H. E., and Strong, G. B. (1986). Cognitive and emotional changes during a simulated 686-M deep dive. Undersea Biomed. Res. 13, 225–235.
Lyford, G. L., Strege, P. R., Shepard, A., Ou, Y., Ermilov, L., Miller, S. M., et al. (2002). Alpha 1c (Cav1.2) L-Type calcium channel mediates mechanosensitive calcium regulation. Am. J. Physiol. Cell Physiol. 283, C1001–C1008. doi: 10.1152/ajpcell.00140.2002
Meir, A., Ginsburg, S., Butkevich, A., Kachalsky, S. G., Kaiserman, I., Ahdut, R., et al. (1999). Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol. Rev. 79, 1019–1088.
Meyer, R., and Heinemann, S. H. (1997). Temperature and pressure dependence of shaker K+ Channel N- and C-Type inactivation. Eur. Biophys. J. 26, 433–445. doi: 10.1007/s002490050098
Miledi, R., and Parker, I. (1984). Chloride current induced by injection of calcium into xenopus oocytes. J. Physiol. 357, 173–183.
Mor, A., Kuttner, Y. Y., Levy, S., Mor, M., Hollmann, M., and Grossman, Y. (2012). Pressure-selective modulation of nmda receptor subtypes may reflect 3d structural differences. Front. Cell Neurosci. 6:37. doi: 10.3389/fncel.2012.00037
Ornhagen, H. C., and Hogan, P. M. (1977). Hydrostatic pressure and mammalian cardiac-pacemaker function. Undersea Biomed. Res. 4, 347–358.
Ornhagen, H. C., and Sigurdsson, S. B. (1981). Effects of high hydrostatic pressure on rat atrial muscle. Undersea Biomed. Res. 8, 113–120.
Otter, T., and Salmon, E. D. (1985). Pressure-Induced changes in Ca2+-channel excitability in paramecium. J. Exp. Biol. 117, 29–43.
Overman, W. H., Brauer, R. W., and Burke, E. R. (1989). Failure to find residual memory deficits in monkeys after repeated Hpns. Undersea Biomed. Res. 16, 115–127.
Parmentier, J. L., Shrivastav, B. B., and Bennett, P. B. (1981). Hydrostatic pressure reduces synaptic efficiency by inhibiting transmitter release. Undersea Biomed. Res. 8, 175–183.
Roberts, R. J., Shelton, C. J., Daniels, S., and Smith, E. B. (1996). Glycine activation of human homomeric alpha 1 glycine receptors is sensitive to pressure in the range of the high pressure nervous syndrome. Neurosci. Lett. 208, 125–128. doi: 10.1016/0304-3940(96)12550-7
Robitaille, R., Adler, E. M., and Charlton, M. P. (1990). Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron 5, 773–779. doi: 10.1016/0896-6273(90)90336-E
Rostain, J. C., Gardette-Chauffour, M. C., and Naquet, R. (1997). Eeg and sleep disturbances during dives at 450 msw in helium-nitrogen-oxygen mixture. J. Appl. Physiol. 83, 575–582.
Schleifstein-Attias, D., Tarasiuk, A., and Grossman, Y. (1994). “High pressure effects on modulation of mammalian spinal monosynaptic reflex,” in Basic and Applied High Pressure Biology, eds P. B. Bennett and R. E. Marquis (Rochester, NY: Rochester University Press), 193–199.
Schmalwasser, H., Neef, A., Elliott, A. A., and Heinemann, S. H. (1998). Two-electrode voltage clamp of xenopus oocytes under high hydrostatic pressure. J. Neurosci. Methods 81, 1–7. doi: 10.1016/S0165-0270(98)00030-2
Shelton, C. J., Doyle, M. G., Price, D. J., Daniels, S., and Smith, E. B. (1993). The effect of high pressure on glycine- and kainate-sensitive receptor channels expressed in xenopus oocytes. Proc. Biol. Sci. 254, 131–137. doi: 10.1098/rspb.1993.0137
Shrivastav, B. B., Parmentier, J. L., and Bennett, P. B. (1981). “A quantitative description of pressure-induced alterations in ionic channels of the squid giant axon,” in 7th Symposium on Underwater Physiology, Undersea Society Inc. (Athens), 611–619.
Sokolov, S., Weiss, R. G., Timin, E. N., and Hering, S. (2000). Modulation of slow inactivation in class a Ca2+ channels by {Beta}-subunits. J. Physiol. 527, 445–454. doi: 10.1111/j.1469-7793.2000.t01-1-00445.x
Southan, A. P., and Wann, K. T. (1996). Effects of high helium pressure on intracellular and field potential responses in the Ca1 region of the in vitro rat hippocampus. Eur. J. Neurosci. 8, 2571–2581. doi: 10.1111/j.1460-9568.1996.tb01551.x
Steevens, C. C., Russell, K. L., Knafelc, M. E., Smith, P. F., Hopkins, E. W., and Clark, J. B. (1999). Noise-induced neurologic disturbances in divers exposed to intense water-borne sound: two case reports. Undersea Hyperb. Med. 26, 261–265.
Talpalar, A. E., Giugliano, M., and Grossman, Y. (2010). Enduring medial perforant path short-term synaptic depression at high pressure. Front. Cell Neurosci. 4:128. doi: 10.3389/fncel.2010.00128
Talpalar, A. E., and Grossman, Y. (2003). Modulation of rat corticohippocampal synaptic activity by high pressure and extracellular calcium: single and frequency responses. J. Neurophysiol. 90, 2106–2114. doi: 10.1152/jn.00894.2002
Talpalar, A. E., and Grossman, Y. (2004). Enhanced excitability compensates for high-pressure-induced depression of cortical inputs to the hippocampus. J. Neurophysiol. 92, 3309–3319. doi: 10.1152/jn.00178.2004
Talpalar, A. E., and Grossman, Y. (2006). CNS manifestations of HPNS: revisited. Undersea Hyperb. Med. 33, 205–210.
Tang, S., Yatani, A., Bahinski, A., Mori, Y., and Schwartz, A. (1993). Molecular localization of regions in the l-type calcium channel critical for dihydropyridine action. Neuron 11, 1013–1021. doi: 10.1016/0896-6273(93)90215-D
Tarasiuk, A., and Grossman, Y. (1990). Pressure-induced tremor-associated activity in ventral roots in isolated spinal cord of newborn rats. Undersea Biomed. Res. 17, 287–296.
Tsien, R. W., Hess, P., McCleskey, E. W., and Rosenberg, R. L. (1987). Calcium channels: mechanisms of selectivity, permeation, and block. Annu. Rev. Biophys. Biophys. Chem. 16, 265–290. doi: 10.1146/annurev.bb.16.060187.001405
Uchitel, O. D., Protti, D. A., Sanchez, V., Cherksey, B. D., Sugimori, M., and Llinas, R. (1992). P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc. Natl. Acad. Sci. U.S.A. 89, 3330–3333. doi: 10.1073/pnas.89.8.3330
Vaernes, R. J., Bergan, T., and Warncke, M. (1988). Hpns effects among 18 divers during compression to 360 Msw on heliox. Undersea Biomed. Res. 15, 241–255.
Vogel, S. S., Smith, R. M., Baibakov, B., Ikebuchi, Y., and Lambert, N. A. (1999). Calcium Influx is required for endocytotic membrane retrieval. Proc. Natl. Acad. Sci. U.S.A. 96, 5019–5024. doi: 10.1073/pnas.96.9.5019
Wu, X. S., McNeil, B. D., Xu, J., Fan, J., Xue, L., Melicoff, E., et al. (2009). Ca(2+) and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat. Neurosci. 12, 1003–1010. doi: 10.1038/nn.2355
Zhang, J. F., Ellinor, P. T., Aldrich, R. W., and Tsien, R. W. (1994). Molecular determinants of voltage-dependent inactivation in calcium channels. Nature 372, 97–100. doi: 10.1038/372097a0
Keywords: high pressure neurological syndrome, hyperbaric helium pressure, high pressure, voltage dependent calcium channels, synaptic transmission, conformational change, voltage dependency
Citation: Aviner B, Gradwohl G, Mor Aviner M, Levy S and Grossman Y (2014) Selective modulation of cellular voltage-dependent calcium channels by hyperbaric pressure—a suggested HPNS partial mechanism. Front. Cell. Neurosci. 8:136. doi: 10.3389/fncel.2014.00136
Received: 11 September 2013; Accepted: 30 April 2014;
Published online: 27 May 2014.
Edited by:
Egidio D'Angelo, University of Pavia, ItalyReviewed by:
Emilio Carbone, Department of Drug Science, ItalyJay B. Dean, University of South Florida, USA
Copyright © 2014 Aviner, Gradwohl, Mor Aviner, Levy and Grossman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Aviner, Department of Physiology and Neurobiology, Ben Gurion University of the Negev, Building M6, Beer Sheva 84105, Israel e-mail:YXZpbmVyQGJndS5hYy5pbA==